
Presence of multiple acid phosphatases activity in seedlings of cucumber, radish and rocket salad
Upload
independentCategory
view
7download
0
Determination of phosphorylated residues fromhuman respiratory syncytial virus P protein thatare dynamically dephosphorylated by cellularphosphatases: a possible role for serine 54
Ana Asenjo, Lorena Rodrıguez and Nieves Villanueva
Correspondence
Nieves Villanueva
Centro Nacional de Microbiologıa (CNM), Instituto de Salud Carlos III (ISCIII), CarreteraMajadahonda-Pozuelo Km 2, Majadahonda, E-28220 Madrid, Spain
Received 13 October 2004
Accepted 14 December 2004
The 241 aa human respiratory synctyial virus (HRSV) Long strain P protein is phosphorylated
at serines 116, 117 and/or 119, and 232. Phosphates added to these residues have slow
turnover and can be detected in the absence of protein phosphatase inhibition. Inhibition of
phosphatases PP1 and PP2A increases the level of phosphorylation at serines 116, 117 and/or
119, suggesting a more rapid turnover for phosphates added to these residues compared to
that of S232. High-turnover phosphorylation is detected in the P-protein NH2-terminal region,
mainly at S54 and, to a lesser extent, at S39, in the Long strain. When the P protein bears the T46I
substitution (in the remaining HRSV strains), phosphates are added to S30, S39, S45 and S54.
Phosphatase PP1 removes phosphate at residues in the central part of the P-protein molecule,
whereas those in the NH2-terminal region are removed by phosphatase PP2A. The significance
of the phosphorylation of the NH2-terminal region residues for some P-protein functions was
studied. The results indicated that this modification is not essential for P-protein oligomerization
or for its role in viral RNA synthesis. Nonetheless, dephosphorylation at S54 could facilitate
P–M protein interactions that probably occur during the egress of viral particles.
INTRODUCTION
Human respiratory syncytial virus (HRSV) is a pneumo-virus of the family Paramyxoviridae that causes seriousviral bronchiolitis and pneumonia in infants, children, theelderly and immunocompromised populations (Harringtonet al., 1992; Falsey et al., 1995; Collins et al., 2001). Wefocused on understanding how P-protein phosphorylationacts at the molecular level, which could aid in developinglive-attenuated vaccines and antiviral compounds to pre-vent the severe morbidity associated with HRSV infection(Dudas & Karron, 1998; Whitehead et al., 1999; Pastey et al.,2000).
This phosphorylated structural protein, the viral RNA(15 222 nt) and the N, L and M2-1 proteins form the viralribonucleoproteins (RNPs) (Yu et al., 1995; Collins et al.,1995, 1996; Hardy & Wertz, 1998). The RNPs are theinternal components of the virions and the functional unitsfor the transcription and replication processes.
The P proteins of different members of the family Para-myxoviridae have very distinct sizes and primary andsecondary structures, but all are phosphorylated (Lamb& Kolakofsky, 2001). This suggests that P-protein phos-phorylation has an important role in the viral growthcycle. This role could be to differentiate the P–L complex
that transcribes RNA (vRdRT or transcriptase) from thatinvolved in RNA replication [vRdRR or replicase, accordingto nomenclature proposed by Gubbay et al. (2001)]. The Pprotein is an essential initiation factor for the viral Lpolymerase (Collins et al., 2001), which, through inter-action with P, is bound to the viral nucleocapsid via P–Ninteractions (Horikami et al., 1992). Nucleocapsids areformed by v- or cRNA and by the N protein and act astemplates for viral RNA synthesis. In transcription, RNAsynthesis proceeds discontinuously, starting and ending atstart and stop signals of each gene. Different viral mRNAsare generated, and leader, trailer and intergenic sequences(44 first and 155 last nucleotides of vRNA, respectively) arenot read. In replication, RNA synthesis proceeds from thefirst to the last template of v- or cRNA nucleotides and acontinuous copy is made. Encapsidation of nascent RNAby the N protein is concomitant with synthesis. Only thoseN-protein molecules that previously established the P–N0
complex are functionally competent for RNA encapsidation(Collins et al., 2001). The P protein also interacts with theM2-1 protein, a transcription elongation factor (Cuestaet al., 2000; Mason et al., 2003). How the P protein iscommitted to these different interactions is unknown, butits phosphorylation state may play a role.
P-protein phosphorylation, due mainly to casein kinase II
0008-0692 G 2005 SGM Printed in Great Britain 1109
Journal of General Virology (2005), 86, 1109–1120 DOI 10.1099/vir.0.80692-0

(CKII), takes place mainly at serine residues 116, 117, 119and 232, when the P protein is expressed in HEp-2 cellsinfected by HRSV, vaccinia recombinant or by using avaccinia-based expression system, although to a differentextent (Navarro et al., 1991; Mazumder et al., 1994;Villanueva et al., 1994; Barik et al., 1995; Sanchez-Secoet al., 1995; Asenjo & Villanueva, 2000). It is not essentialfor either HRSV RNA transcription or replication(Villanueva et al., 1991, 2000), although inhibition ofextracellular viral-particle formation was observed in itsabsence (Villanueva et al., 1991). Inhibition of extracellularviral-particle formation was also found in an HRSVmutant in which these P-protein serine residues weresubstituted (Lu et al., 2002).
Here, we show that the P protein also has high-turnoverphosphorylation sites, detectable only after inhibition ofcellular protein phosphatases PP1 and PP2A. The majorityof P-protein residues to which these phosphates are addedhave been established. A large part of this phosphorylationis not essential for viral RNA synthesis or for P-proteintetramerization (Asenjo & Villanueva, 2000), although itis necessary that serine 54 is dephosphorylated to allowefficient P–M interactions that are related to the viral-particle budding process.
METHODS
Cells and viruses. HEp-2 cells were obtained from the ATCC. TheHRSV Long and A2 strains were plaque-purified and passaged atlow multiplicity in HEp-2 cells, as described previously (Villanuevaet al., 1991). The origin and handling conditions for vaccinia recom-binant vTF-3 have been reported previously (Villanueva et al., 2000).
P-protein variant construction. All plasmids containing Longstrain P-protein variant cDNAs are pGEM3 recombinants. P-proteincDNA was cloned as an HpaI–StuI fragment of the P20 plasmid(Lopez et al., 1988) in the SmaI site of the pGEM3 polylinker, torender the VP plasmid. Construction of VP3–8 and VP3–10 hasbeen described previously (Asenjo & Villanueva, 2000; Villanuevaet al., 2000). The remaining P-protein variants were obtained in VPby site-directed mutagenesis (Higuchi et al., 1988). All VP nucleo-tide sequences encoding the P protein were confirmed by automatedsequencing.
Construction of the pGEM3 recombinant containing SV40small t-antigen cDNA. Plasmid pCEP4-smt, containing SV40small t-antigen cDNA (Alberts et al., 1994), was kindly provided byDr Shenolikar (Department of Pharmacology, Duke UniversityMedical Center, Durham, NC, USA). After HindIII and BamHIdigestion, the fragment containing small t-antigen DNA was purifiedand ligated to digested pGEM3 DNA. The pGEM3 recombinantplasmid is described as smt.
Infection, transfection, cell labelling, fractionation and P-protein purification. HRSV infection and transfection experimentswere performed as indicated previously (Villanueva et al., 1991,1994, 2000). Cell labelling with [35S]methionine and [32P]orthophos-phate was done as described previously (Asenjo & Villanueva, 2000).
For protein isolation, cells were scraped into the growth medium andisolated by low-speed centrifugation. Medium from HRSV-infectedcells was removed and precipitated twice with 6 % polyethylene glycol.
Pellets were washed twice with PBS and total cell extracts or soluble-and microfilament-protein fractions were obtained by resuspendingpellets in 10 mM Tris/HCl (pH 7?5), 0?14 M NaCl, 5 mM EDTA, 1 %Triton X-100 and 1 % sodium deoxycholate, or by using the extractionconditions described previously (Ulloa et al., 1998), respectively.
P protein in the different fractions was purified by chromatographythrough Sepharose 4B containing covalently bound anti-P monoclonalantibody (mAb) 73/P or by immunoprecipitation (Villanueva et al.,1991).
Okadaic acid (OKA) treatment of HRSV-infected or trans-fected HEp-2 cells. When cytopathic effect was apparent, HRSV-infected cells were treated with 100 nM OKA in the presence of37?5 mCi [32P]orthophosphate ml21 (24 h). Transfected HEp-2 cellswere treated and labelled in the presence of 100 nM OKA at10–24 h post-transfection.
Formic acid and trypsin treatment of P protein. Treatmentconditions and phosphopeptide separation were as described pre-viously (Navarro et al., 1991; Sanchez-Seco et al., 1995).
Oligomerization, viral transcription, replication and M-inter-action capacities of P-protein variants. The conditions followedwere as described by Asenjo & Villanueva (2000), Rodrıguez et al.(2004) and Schmitt et al. (2002), respectively.
RESULTS
Inhibition of cellular protein phosphatasesPP1 and PP2A reveals new P-proteinphosphorylation sites in HRSV-infectedand transfected HEp-2 cells
To determine whether the P protein undergoes highphosphorylation turnover at specific sites, HRSV-infectedor transfected HEp-2 cells (Fig. 1a, b) were labelled with[35S]methionine or [32P]orthophosphate (upper or lowerpart), alone (2 lanes) or in the presence (+ lanes) of100 nM OKA (Villanueva et al., 1991, 2000). PP1A andPP2A are partially and totally inhibited, respectively, at thisOKA concentration (Bialojan & Takai, 1988). Extracellularviral particles (V), distinct cell-protein fractions (S, M, In)(Ulloa et al., 1998) or whole-cell extract (VP) (Asenjo &Villanueva, 2000) were isolated. P protein was immuno-purified from these samples and analysed by SDS-PAGE.
Infected-cell proteins were fractionated to improve detec-tion of P-protein isoforms, but no major differences wereobserved for untreated or OKA-treated cells for P-proteincell location, phosphorylation level or in the amount ofN protein that co-immunopurified with the P protein(Fig. 1a, lanes S, M and In, upper). No differences weredetected either in [32P]orthophosphate-labelled P proteinin transfected cells (Fig. 1b, lower) or in infected cells(Fig. 1a, lower) but, after immunochromatography, aphosphorylated protein (HP) with a slight slower mobi-lity in SDS-PAGE than that of the P protein copurifiedwith it in all fractions from OKA-treated cells (also visiblein [35S]methionine-labelled cells; Fig. 1a, b, upper). Also,two highly phosphorylated proteins (molecular massaround 45 kDa) are found, mainly in soluble (S) and in
1110 Journal of General Virology 86
A. Asenjo, L. Rodrıguez and N. Villanueva
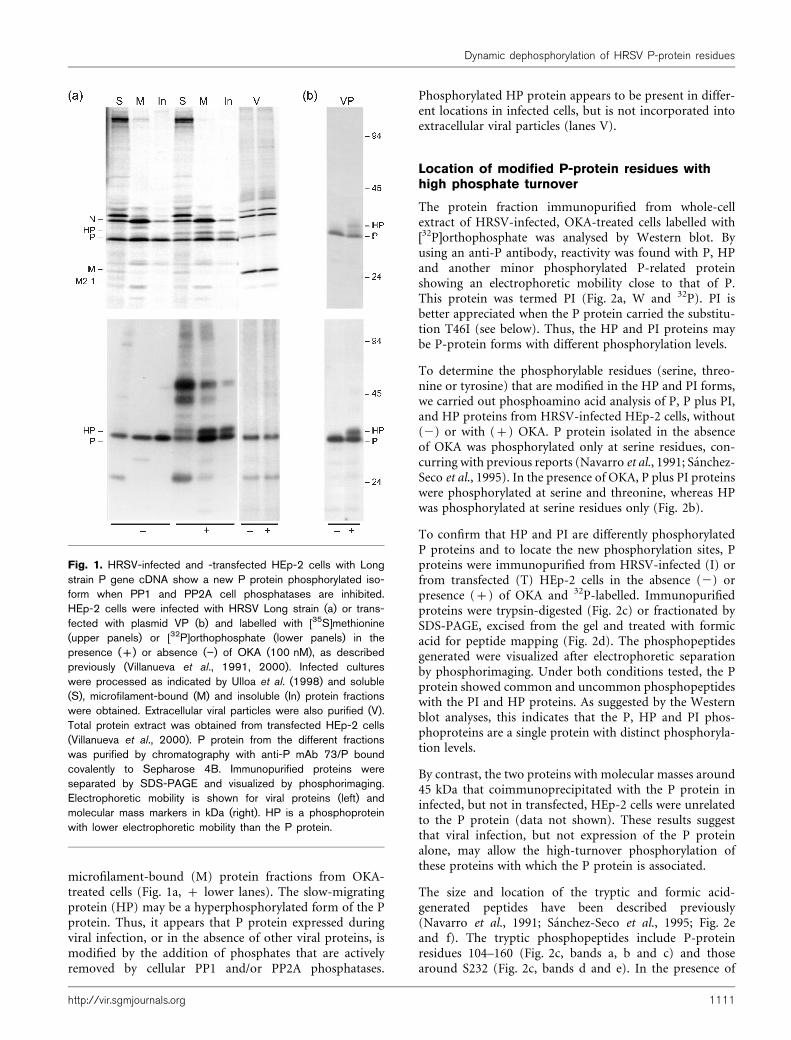
microfilament-bound (M) protein fractions from OKA-treated cells (Fig. 1a, + lower lanes). The slow-migratingprotein (HP) may be a hyperphosphorylated form of the Pprotein. Thus, it appears that P protein expressed duringviral infection, or in the absence of other viral proteins, ismodified by the addition of phosphates that are activelyremoved by cellular PP1 and/or PP2A phosphatases.
Phosphorylated HP protein appears to be present in differ-ent locations in infected cells, but is not incorporated intoextracellular viral particles (lanes V).
Location of modified P-protein residues withhigh phosphate turnover
The protein fraction immunopurified from whole-cellextract of HRSV-infected, OKA-treated cells labelled with[32P]orthophosphate was analysed by Western blot. Byusing an anti-P antibody, reactivity was found with P, HPand another minor phosphorylated P-related proteinshowing an electrophoretic mobility close to that of P.This protein was termed PI (Fig. 2a, W and 32P). PI isbetter appreciated when the P protein carried the substitu-tion T46I (see below). Thus, the HP and PI proteins maybe P-protein forms with different phosphorylation levels.
To determine the phosphorylable residues (serine, threo-nine or tyrosine) that are modified in the HP and PI forms,we carried out phosphoamino acid analysis of P, P plus PI,and HP proteins from HRSV-infected HEp-2 cells, without(2) or with (+) OKA. P protein isolated in the absenceof OKA was phosphorylated only at serine residues, con-curring with previous reports (Navarro et al., 1991; Sanchez-Seco et al., 1995). In the presence of OKA, P plus PI proteinswere phosphorylated at serine and threonine, whereas HPwas phosphorylated at serine residues only (Fig. 2b).
To confirm that HP and PI are differently phosphorylatedP proteins and to locate the new phosphorylation sites, Pproteins were immunopurified from HRSV-infected (I) orfrom transfected (T) HEp-2 cells in the absence (2) orpresence (+) of OKA and 32P-labelled. Immunopurifiedproteins were trypsin-digested (Fig. 2c) or fractionated bySDS-PAGE, excised from the gel and treated with formicacid for peptide mapping (Fig. 2d). The phosphopeptidesgenerated were visualized after electrophoretic separationby phosphorimaging. Under both conditions tested, the Pprotein showed common and uncommon phosphopeptideswith the PI and HP proteins. As suggested by the Westernblot analyses, this indicates that the P, HP and PI phos-phoproteins are a single protein with distinct phosphoryla-tion levels.
By contrast, the two proteins with molecular masses around45 kDa that coimmunoprecipitated with the P protein ininfected, but not in transfected, HEp-2 cells were unrelatedto the P protein (data not shown). These results suggestthat viral infection, but not expression of the P proteinalone, may allow the high-turnover phosphorylation ofthese proteins with which the P protein is associated.
The size and location of the tryptic and formic acid-generated peptides have been described previously(Navarro et al., 1991; Sanchez-Seco et al., 1995; Fig. 2eand f). The tryptic phosphopeptides include P-proteinresidues 104–160 (Fig. 2c, bands a, b and c) and thosearound S232 (Fig. 2c, bands d and e). In the presence of
Fig. 1. HRSV-infected and -transfected HEp-2 cells with Longstrain P gene cDNA show a new P protein phosphorylated iso-form when PP1 and PP2A cell phosphatases are inhibited.HEp-2 cells were infected with HRSV Long strain (a) or trans-fected with plasmid VP (b) and labelled with [35S]methionine(upper panels) or [32P]orthophosphate (lower panels) in thepresence (+) or absence (”) of OKA (100 nM), as describedpreviously (Villanueva et al., 1991, 2000). Infected cultureswere processed as indicated by Ulloa et al. (1998) and soluble(S), microfilament-bound (M) and insoluble (In) protein fractionswere obtained. Extracellular viral particles were also purified (V).Total protein extract was obtained from transfected HEp-2 cells(Villanueva et al., 2000). P protein from the different fractionswas purified by chromatography with anti-P mAb 73/P boundcovalently to Sepharose 4B. Immunopurified proteins wereseparated by SDS-PAGE and visualized by phosphorimaging.Electrophoretic mobility is shown for viral proteins (left) andmolecular mass markers in kDa (right). HP is a phosphoproteinwith lower electrophoretic mobility than the P protein.
http://vir.sgmjournals.org 1111
Dynamic dephosphorylation of HRSV P-protein residues
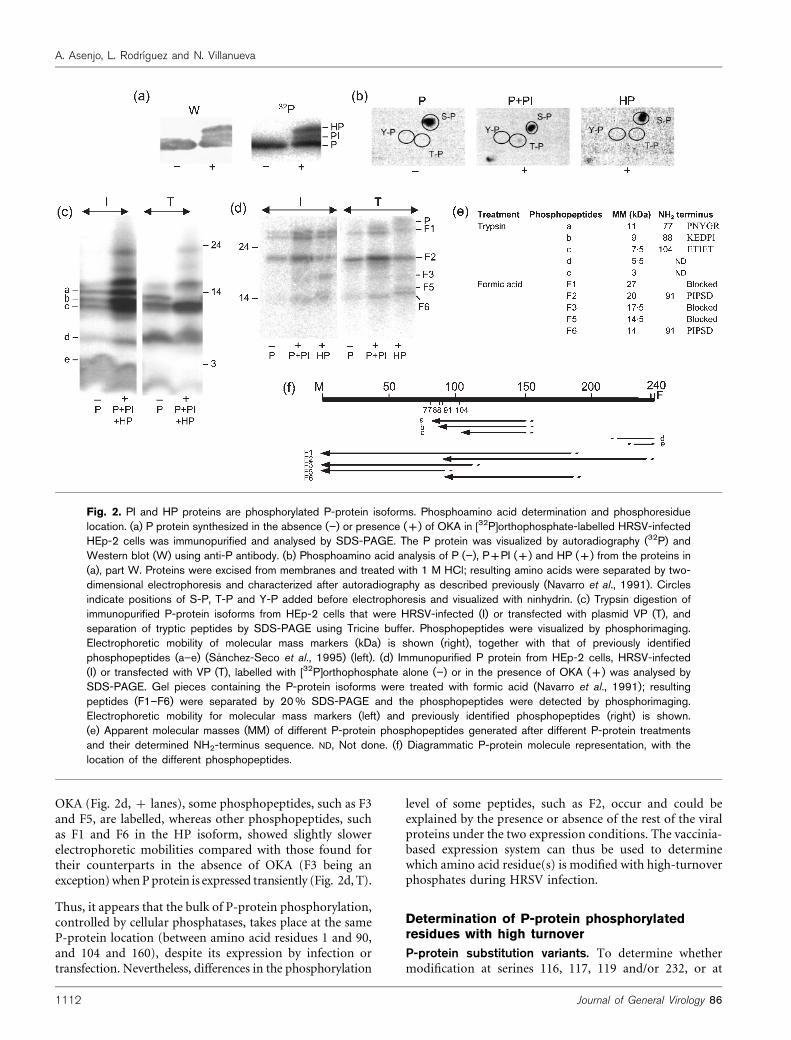
OKA (Fig. 2d, + lanes), some phosphopeptides, such as F3and F5, are labelled, whereas other phosphopeptides, suchas F1 and F6 in the HP isoform, showed slightly slowerelectrophoretic mobilities compared with those found fortheir counterparts in the absence of OKA (F3 being anexception) when P protein is expressed transiently (Fig. 2d, T).
Thus, it appears that the bulk of P-protein phosphorylation,controlled by cellular phosphatases, takes place at the sameP-protein location (between amino acid residues 1 and 90,and 104 and 160), despite its expression by infection ortransfection. Nevertheless, differences in the phosphorylation
level of some peptides, such as F2, occur and could beexplained by the presence or absence of the rest of the viralproteins under the two expression conditions. The vaccinia-based expression system can thus be used to determinewhich amino acid residue(s) is modified with high-turnoverphosphates during HRSV infection.
Determination of P-protein phosphorylatedresidues with high turnover
P-protein substitution variants. To determine whethermodification at serines 116, 117, 119 and/or 232, or at
Fig. 2. PI and HP proteins are phosphorylated P-protein isoforms. Phosphoamino acid determination and phosphoresiduelocation. (a) P protein synthesized in the absence (”) or presence (+) of OKA in [32P]orthophosphate-labelled HRSV-infectedHEp-2 cells was immunopurified and analysed by SDS-PAGE. The P protein was visualized by autoradiography (32P) andWestern blot (W) using anti-P antibody. (b) Phosphoamino acid analysis of P (”), P+PI (+) and HP (+) from the proteins in(a), part W. Proteins were excised from membranes and treated with 1 M HCl; resulting amino acids were separated by two-dimensional electrophoresis and characterized after autoradiography as described previously (Navarro et al., 1991). Circlesindicate positions of S-P, T-P and Y-P added before electrophoresis and visualized with ninhydrin. (c) Trypsin digestion ofimmunopurified P-protein isoforms from HEp-2 cells that were HRSV-infected (I) or transfected with plasmid VP (T), andseparation of tryptic peptides by SDS-PAGE using Tricine buffer. Phosphopeptides were visualized by phosphorimaging.Electrophoretic mobility of molecular mass markers (kDa) is shown (right), together with that of previously identifiedphosphopeptides (a–e) (Sanchez-Seco et al., 1995) (left). (d) Immunopurified P protein from HEp-2 cells, HRSV-infected(I) or transfected with VP (T), labelled with [32P]orthophosphate alone (”) or in the presence of OKA (+) was analysed bySDS-PAGE. Gel pieces containing the P-protein isoforms were treated with formic acid (Navarro et al., 1991); resultingpeptides (F1–F6) were separated by 20% SDS-PAGE and the phosphopeptides were detected by phosphorimaging.Electrophoretic mobility for molecular mass markers (left) and previously identified phosphopeptides (right) is shown.(e) Apparent molecular masses (MM) of different P-protein phosphopeptides generated after different P-protein treatmentsand their determined NH2-terminus sequence. ND, Not done. (f) Diagrammatic P-protein molecule representation, with thelocation of the different phosphopeptides.
1112 Journal of General Virology 86
A. Asenjo, L. Rodrıguez and N. Villanueva
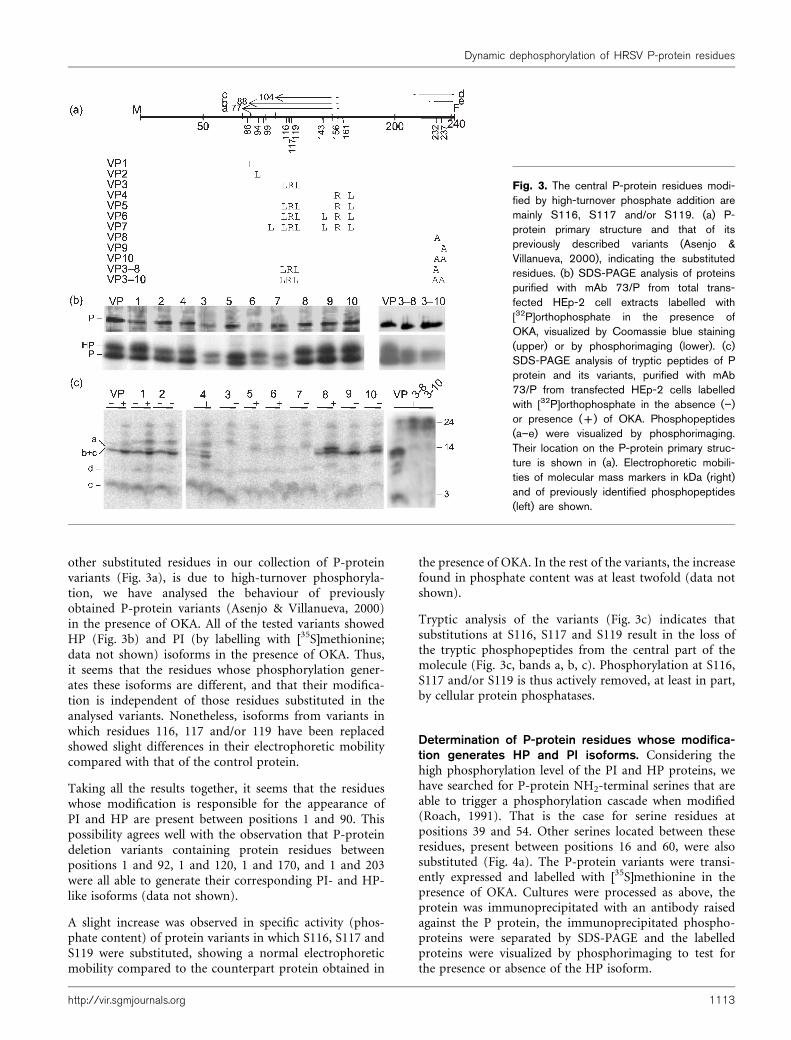
other substituted residues in our collection of P-proteinvariants (Fig. 3a), is due to high-turnover phosphoryla-tion, we have analysed the behaviour of previouslyobtained P-protein variants (Asenjo & Villanueva, 2000)in the presence of OKA. All of the tested variants showedHP (Fig. 3b) and PI (by labelling with [35S]methionine;data not shown) isoforms in the presence of OKA. Thus,it seems that the residues whose phosphorylation gener-ates these isoforms are different, and that their modifica-tion is independent of those residues substituted in theanalysed variants. Nonetheless, isoforms from variants inwhich residues 116, 117 and/or 119 have been replacedshowed slight differences in their electrophoretic mobilitycompared with that of the control protein.
Taking all the results together, it seems that the residueswhose modification is responsible for the appearance ofPI and HP are present between positions 1 and 90. Thispossibility agrees well with the observation that P-proteindeletion variants containing protein residues betweenpositions 1 and 92, 1 and 120, 1 and 170, and 1 and 203were all able to generate their corresponding PI- and HP-like isoforms (data not shown).
A slight increase was observed in specific activity (phos-phate content) of protein variants in which S116, S117 andS119 were substituted, showing a normal electrophoreticmobility compared to the counterpart protein obtained in
the presence of OKA. In the rest of the variants, the increasefound in phosphate content was at least twofold (data notshown).
Tryptic analysis of the variants (Fig. 3c) indicates thatsubstitutions at S116, S117 and S119 result in the loss ofthe tryptic phosphopeptides from the central part of themolecule (Fig. 3c, bands a, b, c). Phosphorylation at S116,S117 and/or S119 is thus actively removed, at least in part,by cellular protein phosphatases.
Determination of P-protein residues whose modifica-tion generates HP and PI isoforms. Considering thehigh phosphorylation level of the PI and HP proteins, wehave searched for P-protein NH2-terminal serines that areable to trigger a phosphorylation cascade when modified(Roach, 1991). That is the case for serine residues atpositions 39 and 54. Other serines located between theseresidues, present between positions 16 and 60, were alsosubstituted (Fig. 4a). The P-protein variants were transi-ently expressed and labelled with [35S]methionine in thepresence of OKA. Cultures were processed as above, theprotein was immunoprecipitated with an antibody raisedagainst the P protein, the immunoprecipitated phospho-proteins were separated by SDS-PAGE and the labelledproteins were visualized by phosphorimaging to test forthe presence or absence of the HP isoform.
Fig. 3. The central P-protein residues modi-fied by high-turnover phosphate addition aremainly S116, S117 and/or S119. (a) P-protein primary structure and that of itspreviously described variants (Asenjo &Villanueva, 2000), indicating the substitutedresidues. (b) SDS-PAGE analysis of proteinspurified with mAb 73/P from total trans-fected HEp-2 cell extracts labelled with[32P]orthophosphate in the presence ofOKA, visualized by Coomassie blue staining(upper) or by phosphorimaging (lower). (c)SDS-PAGE analysis of tryptic peptides of Pprotein and its variants, purified with mAb73/P from transfected HEp-2 cells labelledwith [32P]orthophosphate in the absence (”)or presence (+) of OKA. Phosphopeptides(a–e) were visualized by phosphorimaging.Their location on the P-protein primary struc-ture is shown in (a). Electrophoretic mobili-ties of molecular mass markers in kDa (right)and of previously identified phosphopeptides(left) are shown.
http://vir.sgmjournals.org 1113
Dynamic dephosphorylation of HRSV P-protein residues
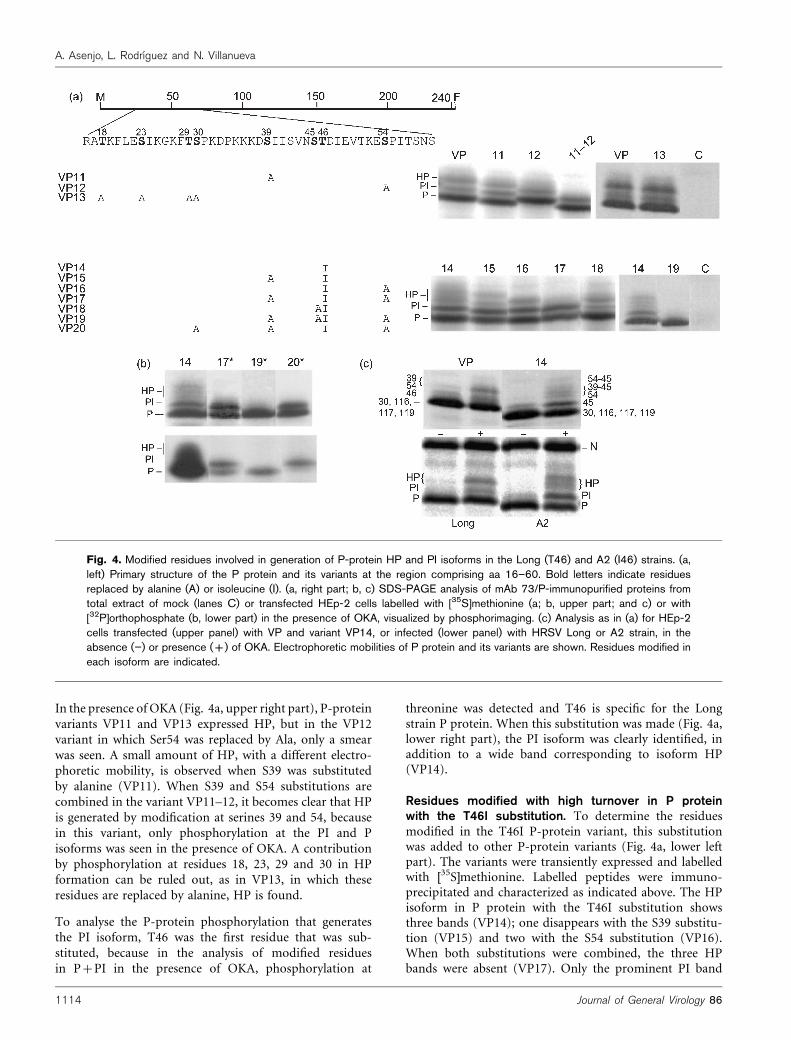
In the presence of OKA (Fig. 4a, upper right part), P-proteinvariants VP11 and VP13 expressed HP, but in the VP12variant in which Ser54 was replaced by Ala, only a smearwas seen. A small amount of HP, with a different electro-phoretic mobility, is observed when S39 was substitutedby alanine (VP11). When S39 and S54 substitutions arecombined in the variant VP11–12, it becomes clear that HPis generated by modification at serines 39 and 54, becausein this variant, only phosphorylation at the PI and Pisoforms was seen in the presence of OKA. A contributionby phosphorylation at residues 18, 23, 29 and 30 in HPformation can be ruled out, as in VP13, in which theseresidues are replaced by alanine, HP is found.
To analyse the P-protein phosphorylation that generatesthe PI isoform, T46 was the first residue that was sub-stituted, because in the analysis of modified residuesin P+PI in the presence of OKA, phosphorylation at
threonine was detected and T46 is specific for the Longstrain P protein. When this substitution was made (Fig. 4a,lower right part), the PI isoform was clearly identified, inaddition to a wide band corresponding to isoform HP(VP14).
Residues modified with high turnover in P proteinwith the T46I substitution. To determine the residuesmodified in the T46I P-protein variant, this substitutionwas added to other P-protein variants (Fig. 4a, lower leftpart). The variants were transiently expressed and labelledwith [35S]methionine. Labelled peptides were immuno-precipitated and characterized as indicated above. The HPisoform in P protein with the T46I substitution showsthree bands (VP14); one disappears with the S39 substitu-tion (VP15) and two with the S54 substitution (VP16).When both substitutions were combined, the three HPbands were absent (VP17). Only the prominent PI band
Fig. 4. Modified residues involved in generation of P-protein HP and PI isoforms in the Long (T46) and A2 (I46) strains. (a,left) Primary structure of the P protein and its variants at the region comprising aa 16–60. Bold letters indicate residuesreplaced by alanine (A) or isoleucine (I). (a, right part; b, c) SDS-PAGE analysis of mAb 73/P-immunopurified proteins fromtotal extract of mock (lanes C) or transfected HEp-2 cells labelled with [35S]methionine (a; b, upper part; and c) or with[32P]orthophosphate (b, lower part) in the presence of OKA, visualized by phosphorimaging. (c) Analysis as in (a) for HEp-2cells transfected (upper panel) with VP and variant VP14, or infected (lower panel) with HRSV Long or A2 strain, in theabsence (”) or presence (+) of OKA. Electrophoretic mobilities of P protein and its variants are shown. Residues modified ineach isoform are indicated.
1114 Journal of General Virology 86
A. Asenjo, L. Rodrıguez and N. Villanueva

remains and it can be nearly eliminated by substitution ofS45 (VP18). In addition, it is observed that the S45Amodification alone prevents the formation of two HPforms (data not shown); thus, two HP forms could arisefrom phosphorylation at S45 and S54 and at S45 and S39,respectively. To corroborate the relationship between thepresence of these P isoforms and phosphate addition tothose P-protein serines, we have analysed the VP19variant with substitutions at residues S39, S45, S54 andT46. In this variant, only the P-protein isoform withnormal electrophoretic mobility was detected.
The substitutions present in variants VP3–10 were addedto those present in variants VP17 and VP19 in an attemptto prevent the slow and medium turnover of P-proteinphosphorylation (Fig. 3, S232A, S237A and S116L, S117R,S119L, respectively). These new variants were named VP17*and VP19* and they were labelled with [35S]methionine or[32P]orthophosphate and analysed as above (Fig. 4b, upperand lower panels, respectively). Both variants showed two32P-labelled isoforms, P and PI. In the case of variant VP19*,PI is weakly labelled. The stronger band, corresponding tothe P isoform, was eliminated when the S30A substitutionwas incorporated with those already present in the VP17*variant, yielding the new variant VP20*. In this way, themajority of high-turnover phosphates added to P-proteinresidues have been identified.
The T46I substitution in the P protein from the Longstrain is sufficient to change the P-isoform pattern to thatof the A2 strain P protein after inhibition of cellular
phosphatases, as the same isoform phenotype was shownfor Long strain P-protein variant VP14 and for A2 strain Pprotein, both of which bear I46 (Fig. 4c, lower part). Basedon these results, we have deduced the residues modified togenerate each P-protein isoform (Fig. 4c, upper part).
Cellular PP2A phosphatase is implicated in thehigh-turnover phosphorylation that generatesHP
To determine which one of the cellular phosphatases inhi-bited by OKA (PP2A or PP1) is involved in removing thehigh-turnover phosphates added to P-protein residues, Pprotein was transiently coexpressed with a pGEM3 recom-binant that expresses the SV40 small t antigen under thecontrol of T7 RNA polymerase. This viral protein is aspecific PP2A inhibitor (Alberts et al., 1994). Coexpressionof the P protein and SV40 small t antigen allows the appear-ance of HP (Fig. 5a, b, lane 2) in a similar way to thatwhen the protein is transiently expressed alone and notdephosphorylated because of OKA treatment (Fig. 5a, b,lane 3). Cellular PP2A phosphatase actively removes S54and S39 phosphorylation. No phosphorylation increasewas observed in residues located in the central region ofthe P molecule, isolated after trypsin digestion (Fig. 5c,lane 2). This suggests that the high-turnover phosphatesadded, mainly at S116, S117 and S119, are probablydephosphorylated by the PP1 cellular phosphatase.
As we have suggested that high (controlled)-turnover phos-phorylation may be responsible for P-protein tetramerization
Fig. 5. Identification of cell protein phosphatases involved in dephosphorylation of residues modified at the NH2-terminal andcentral part of the P-protein molecule. VP21* variant oligomerization. (a, b) SDS-PAGE analysis of mAb 73/P-precipitatedprotein from HEp-2 cells transfected with VP (lanes 1, 2, 3), alone (lane 1) or in the presence of OKA (lane 3), orcotransfected with smt plasmid (lane 2), labelled with [35S]methionine (a) or [32P]orthophosphate (b). Electrophoretic mobilityis shown for P protein and its isoforms (right). (c) Tryptic phosphopeptides of P proteins shown in (b). Electrophoretic mobilityof molecular mass markers in kDa (right) and P-protein identified peptides (left) is shown. (d) Protein extracts of cellstransiently expressing the P protein (VP) and variant VP21* were cross-linked as described previously (Asenjo & Villanueva,2000; Rodrıguez et al., 2004). At the times indicated (min; bottom), samples were removed, separated by SDS-PAGE and P-protein oligomers (P2 and P4) were identified after Western blot using monospecific anti-P sera. Electrophoretic mobility ofmolecular mass markers (in kDa) and of P-protein oligomers is shown (right).
http://vir.sgmjournals.org 1115
Dynamic dephosphorylation of HRSV P-protein residues

(Asenjo & Villanueva, 2000), we assayed this capacity in avariant, VP21*, that lacks low-turnover (S232 and S237),medium-turnover (S116, S117 and/or S119) and high-turnover (S30, S39, S45, S46 and S54) phosphorylationresidues. Protein from an extract of HEp-2 cells express-ing VP21* was glutaldehyde cross-linked as describedpreviously (Asenjo & Villanueva, 2000) and the oligomerswere characterized.
VP21* oligomerizes, as does the normal P protein (Fig. 5d).Thus, only residual high-turnover P-protein phosphoryla-tion should be sufficient to allow homotetramer forma-tion, or unphosphorylated P protein may be able to formtetramers.
Viral RNA-synthesis capacity of P-proteinvariants with NH2-terminal substitutions
To determine whether high-turnover P-protein phosphor-ylation is related to its function as a L-polymerase cofactorin viral initiation of RNA synthesis, P-protein variants wereassayed in the system described by Hardy & Wertz (1998),which was previously used to test the role for other P-protein variants (Villanueva et al., 2000; Rodrıguez et al.,2004). In P-protein variants with the S54A (VP12) or S54D(VP12D) substitutions (Fig. 6b), it can be observed thatboth variants have viral RNA polymerase cofactor activityfor transcription and replication, and neither affects theintergenic read-through and elongation capacities descri-bed for the HRSV M2-1 protein. High-turnover P-proteinphosphorylation at S54 is thus not involved in the P-protein function concerning the formation of P–N0 or P–Lcomplexes with transcriptase or replicase specificities. Thisis also true for the VP21* variant, with substitution at allresidues with detected modifications. When the M proteinwas coexpressed in the system, we observed inhibition ofthe RNA-synthesizing capacity of the viral RNP at highM-protein concentrations in all cases, as described pre-viously (Rodrıguez et al., 2004) (data not shown).
P protein is incorporated together with Mprotein in membranous vesicles
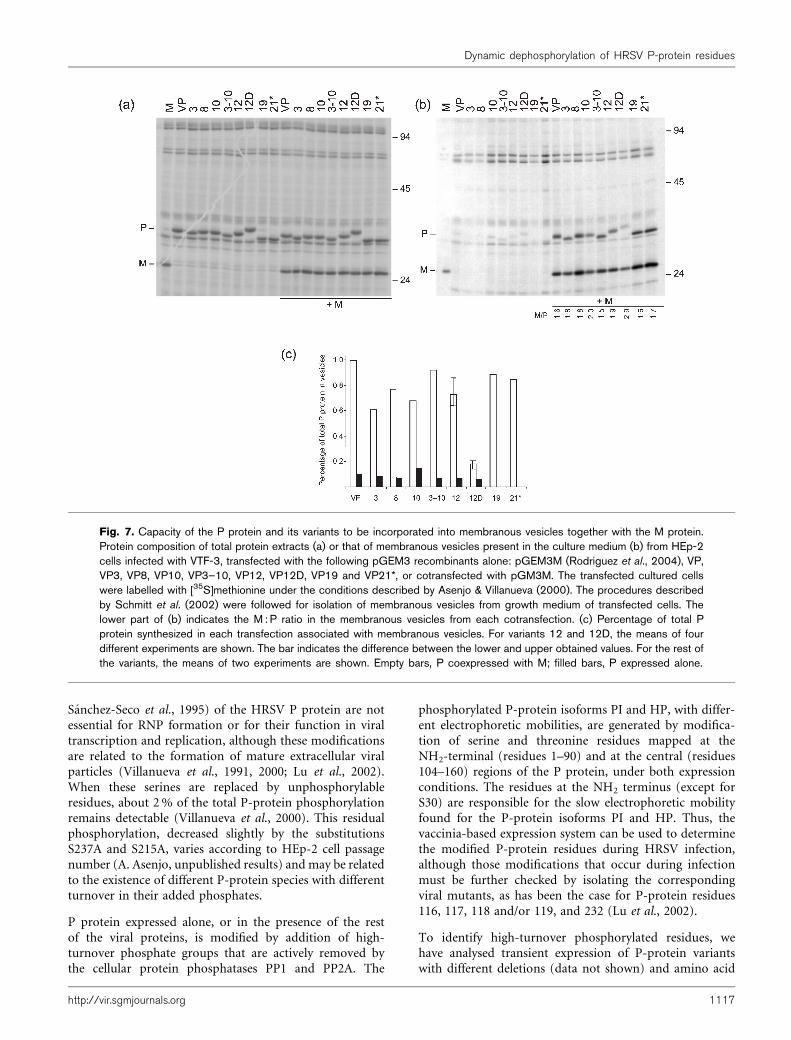
When the HRSV M protein is transiently expressed in HEp-2 cells, a small amount of the expressed protein is excretedto the growth media in membranous vesicles, as describedby Schmitt et al. (2002) (Fig. 7b, lane M). P protein,expressed in the same way, is poorly excreted in thesevesicles (lane VP), but if the M protein is coexpressed withthe P protein, a fraction of the expressed P protein isalso incorporated, together with the M protein, into themembranous vesicles (lane VP+M). It suggests an inter-action between both proteins. This interaction takes placefor all the P-protein substitution variants assayed, but isimpaired in the VP12D variant. A decrease of 5?5- andfourfold by comparison to VP and VP12, respectively, wasobserved (Fig. 7c).
These results suggest that phosphorylation at S54 could be
the signal to regulate a novel P–M protein interaction. ThisP–M interaction may play a role in the formation ofextracellular viral particles.
DISCUSSION
How paramyxovirus viral RNA polymerase, composed ofL polymerase and P protein, switches RNA synthesis fromtranscription to the replication mode remains unsolved.The idea that the amount of N protein synthesized de novoas a P–N0 complex controls the switch of viral polymerasefrom transcription to replication has been shown to beincorrect for HRSV (Fearns et al., 1997). It has beensuggested that P proteins with different phosphorylationlevels can differentiate transcriptase (vRdRT) from repli-case (vRdRR) (Collins et al., 2001). However, low-turnoverphosphate groups mainly added to S116, S117, S119 andS232 (Navarro et al., 1991; Mazumder & Barik, 1994;
Fig. 6. Viral RNA-synthesis capacity of P protein and its var-iants. (a) Scheme of the DNA construct in the pM/SH plasmidthat expresses a dicistronic HRSV RNA analogue. Sizes andpolarities are shown for the different RNAs generated duringtranscription and replication (Hardy & Wertz, 1998). (b) Separa-tion in 3?5% acrylamide gel containing 7 M urea of RNAlabelled with 25 mCi [3H]uridine ml”1 in the presence of 10 mgactinomycin D ml”1, 16 h after transfection of HEp-2 cellsgrowing on an 8 cm2 plastic surface with 1?08 mg pM/SH andpGEM3N, 0?32 mg pL, 0?062 mg pGEM3M2-1 and 0?44 mgpGEM3P (VP) or VP12, VP12D and VP21* plasmids. At 24 hpost-transfection, cytoplasmic cell extracts were processed byusing a kit (Qiagen), following the supplier’s instructions. Afterelectrophoresis, the gel was dried and autoradiographed.
1116 Journal of General Virology 86
A. Asenjo, L. Rodrıguez and N. Villanueva
Sanchez-Seco et al., 1995) of the HRSV P protein are notessential for RNP formation or for their function in viraltranscription and replication, although these modificationsare related to the formation of mature extracellular viralparticles (Villanueva et al., 1991, 2000; Lu et al., 2002).When these serines are replaced by unphosphorylableresidues, about 2 % of the total P-protein phosphorylationremains detectable (Villanueva et al., 2000). This residualphosphorylation, decreased slightly by the substitutionsS237A and S215A, varies according to HEp-2 cell passagenumber (A. Asenjo, unpublished results) and may be relatedto the existence of different P-protein species with differentturnover in their added phosphates.
P protein expressed alone, or in the presence of the restof the viral proteins, is modified by addition of high-turnover phosphate groups that are actively removed bythe cellular protein phosphatases PP1 and PP2A. The
phosphorylated P-protein isoforms PI and HP, with differ-ent electrophoretic mobilities, are generated by modifica-tion of serine and threonine residues mapped at theNH2-terminal (residues 1–90) and at the central (residues104–160) regions of the P protein, under both expressionconditions. The residues at the NH2 terminus (except forS30) are responsible for the slow electrophoretic mobilityfound for the P-protein isoforms PI and HP. Thus, thevaccinia-based expression system can be used to determinethe modified P-protein residues during HRSV infection,although those modifications that occur during infectionmust be further checked by isolating the correspondingviral mutants, as has been the case for P-protein residues116, 117, 118 and/or 119, and 232 (Lu et al., 2002).
To identify high-turnover phosphorylated residues, wehave analysed transient expression of P-protein variantswith different deletions (data not shown) and amino acid
Fig. 7. Capacity of the P protein and its variants to be incorporated into membranous vesicles together with the M protein.Protein composition of total protein extracts (a) or that of membranous vesicles present in the culture medium (b) from HEp-2cells infected with VTF-3, transfected with the following pGEM3 recombinants alone: pGEM3M (Rodrıguez et al., 2004), VP,VP3, VP8, VP10, VP3–10, VP12, VP12D, VP19 and VP21*, or cotransfected with pGM3M. The transfected cultured cellswere labelled with [35S]methionine under the conditions described by Asenjo & Villanueva (2000). The procedures describedby Schmitt et al. (2002) were followed for isolation of membranous vesicles from growth medium of transfected cells. Thelower part of (b) indicates the M : P ratio in the membranous vesicles from each cotransfection. (c) Percentage of total Pprotein synthesized in each transfection associated with membranous vesicles. For variants 12 and 12D, the means of fourdifferent experiments are shown. The bar indicates the difference between the lower and upper obtained values. For the rest ofthe variants, the means of two experiments are shown. Empty bars, P coexpressed with M; filled bars, P expressed alone.
http://vir.sgmjournals.org 1117
Dynamic dephosphorylation of HRSV P-protein residues

substitutions (Asenjo & Villanueva, 2000; Fig. 3) in thepresence of OKA. We found that S116, S117 and S119, inthe central part of the molecule, can be modified by high-turnover phosphates when the P protein is expressed in theabsence of other viral proteins, but not when the proteinis expressed during HRSV infection. This explains thedifferent phosphorylation at residues in the central and C-terminal part of the P-protein molecule, depending on theexpression system used (Sanchez-Seco et al., 1995).
Several P-protein NH2-terminal phosphorylable residueswere substituted. We found that phosphorylation at S54and, to a lesser extent, at S39 was responsible for HPgeneration. These residues have consensus sequences forglycogen synthase kinase 3 and protein kinase C, respec-tively (Pearson & Kemp, 1991). Dephosphorylation ofthese residues occurs by PP2A, as HP is found when the Pprotein is coexpressed with SV40 small t antigen, a specificPP2A phosphatase inhibitor (Alberts et al., 1994). P-proteininteraction with PP2A cellular phosphatase has been alsosuggested (Bitko & Barik, 1998).
Slight differences were seen in the HP-phosphorylationpattern for the T46I substitution, which is found in Pprotein from different RSV strains that infect mammalianand avian hosts, but is absent in the Long strain (Alansari& Potgieter, 1994). These differences were observed bycomparing the A2 and Long strain P proteins. Thus, onlyT46I induces a decreased modification at S54 and a pro-minent phosphorylation of residues S30, S39 and S45. Alarger amount of the PI isoform also results from S45modification. The meaning of these differences is unknown,but may be related to the better growth of HRSV A2 strainthan that of the Long strain in HEp-2 cells and BALB/cmice (Sudo et al., 1999). It is difficult to test whetherphosphorylated or unphosphorylated T46 has a role in theLong strain, as its substitution generates an A2 strain P-protein phosphorylation pattern.
The high-turnover phosphorylation sites could be impor-tant for certain P-protein interactions, such as P self-association (Gao & Lenard, 1995). In this way, we havesuggested that some residues with high-turnover phos-phorylation were involved in P-protein tetramerization(Asenjo & Villanueva, 2000), but other reports (Mazumderet al., 1994; Castagne et al., 2004) have indicated thatunphosphorylated P protein may form oligomers. Anexplanation of this apparent contradiction is that ourbacterially expressed P protein has a deletion of the firstNH2-terminal 20 residues that could be also responsiblefor the lack of oligomerization observed. Nevertheless, theexistence of different P-protein oligomers remains openand, in some cases, phosphorylation could play a role inthe formation of specific oligomers. Nonetheless, the VP21*variant, with substitutions at residues 30, 39, 45, 54, 116,117, 119, 232 and 237, can form tetramers (Fig. 5d).
We tested whether phosphorylation at these residues isessential for P-protein interactions that are established
during viral transcription and/or replication; negativeresults were obtained when the VP21* variant was testedin a dicistronic minireplicon replication system (Fig. 6b).Also, no effect was detected for interaction of the viralRNPs with the M protein. This interaction only takes placein the minireplicon system when a high amount of the Mprotein is expressed and, as a consequence, RNA synthesisby the viral RNPs is decreased (Rodrıguez et al., 2004). Inany case, small differences in this system cannot be ruledout. After amplification, during the growth cycle, theseeffects become crucial, especially if viral proteins are inlimited amounts, as has been described for the N proteinduring HRSV infection (Tran et al., 2004).
P-protein phosphorylation remains, although in a very lowproportion, in the variant containing all the substitutionspreviously described. As PP1 is partially inhibited in thepresence of 100 nM OKA (Ki 20–300 nM; Bialojan & Takai,1988), residual high-turnover P-protein phosphorylationstill occurs.
P-protein phosphorylation at S54 could decrease a novelP–M protein interaction, playing a role in the last viral-particle morphogenesis step in the budding process. Thispossibility explains the absence of the HP isoform inextracellular viral particles. Therefore, it can be speculatedthat RNPs with the P protein phosphorylated at S54 donot interact with M protein that is committed to formmembranous vesicles and, as a consequence, the egressprocess is abolished. Alternatively, P-protein phosphoryla-tion at S54 during adsorption or internalization at thebeginning of the viral infection could be crucial for theuncoating of the viral-particle RNP and to facilitate primarytranscription initiation. This interaction may be differentfrom that described in the minireplicon system (Rodrıguezet al., 2004).
The results indicated that all P-protein residues modifiedand characterized in this work, like the previous ones, arenot related to P-protein activities concerning viral tran-scription and replication. P-protein phosphorylation seemsto be important for regulating the formation of extracellularviral particles once functional nucleocapsids have beenformed (Villanueva et al., 1991, 2000; Lu et al., 2002; thisstudy). Among other possibilities, HRSV P-protein phos-phorylation could also be involved in a process directed tostop the antiviral cell response mediated by interferon (Luet al., 2002) or in viral cytopathogenicity (Das & Pattnaik,2004).
ACKNOWLEDGEMENTS
We are indebted to Dr G. Wertz for providing the HRSV RNA analoguereplication and transcription system and to Dr Shenolinker for hisgenerous gift of the pCEP4-smt plasmid. We thank I. Cuesta fordiscussions, Dr J. Avila for critical reading of the manuscript andmany useful suggestions and C. Mark for editorial assistance. Thiswork was supported by FIS 00/204 and SAF/2002-00070 from theSpanish Ministerio de Ciencia y Tecnologia (MCYT).
1118 Journal of General Virology 86
A. Asenjo, L. Rodrıguez and N. Villanueva

REFERENCES
Alansari, H. & Potgieter, L. N. D. (1994). Molecular cloning and
sequence analysis of the phosphoprotein, nucleocapsid protein,
matrix protein and 22K (M2) protein of the ovine respiratory
syncytial virus. J Gen Virol 75, 3597–3601.
Alberts, A. S., Montminy, M., Shenolikar, S. & Feramisco, J. R.(1994). Expression of a peptide inhibitor of protein phosphatase
1 increases phosphorylation and activity of CREB in NIH 3T3
fibroblasts. Mol Cell Biol 14, 4398–4407.
Asenjo, A. & Villanueva, N. (2000). Regulated but not constitutive
human respiratory syncytial virus (HRSV) P protein phosphoryla-
tion is essential for oligomerization. FEBS Lett 467, 279–284.
Barik, S., McLean, T. & Dupuy, L. C. (1995). Phosphorylation of
Ser232 directly regulates the transcriptional activity of the P protein
of human respiratory syncytial virus: phosphorylation of Ser237 may
play an accessory role. Virology 213, 405–412.
Bialojan, C. & Takai, A. (1988). Inhibitory effect of a marine-sponge
toxin, okadaic acid, on protein phosphatases. Specificity and kinetics.
Biochem J 256, 283–290.
Bitko, V. & Barik, S. (1998). Persistant activation of RelA by
respiratory syncytial virus involves protein kinase C, underphos-
phorylated IkBb, and sequestration of protein phosphatase 2A by
the viral phosphoprotein. J Virol 72, 5610–5618.
Castagne, N., Barbier, A., Bernard, J., Rezaei, H., Huet, J.-C.,Henry, C., Da Costa, B. & Eleouet, J.-F. (2004). Biochemical
characterization of the respiratory syncytial virus P–P and P–N
protein complexes and localization of the P protein oligomerization
domain. J Gen Virol 85, 1643–1653.
Collins, P. L., Hill, M. G., Camargo, E., Grosfeld, H., Chanock, R. M.& Murphy, B. R. (1995). Production of infectious human respiratory
syncytial virus from cloned cDNA confirms an essential role for the
transcription elongation factor from the 59 proximal open reading
frame of the M2 mRNA in gene expression and provides a capability
for vaccine development. Proc Natl Acad Sci U S A 92, 11563–11567.
Collins, P. L., Hill, M. G., Cristina, J. & Grosfeld, H. (1996).Transcription elongation factor of respiratory syncytial virus, a
nonsegmented negative-strand RNA virus. Proc Natl Acad Sci U S A
93, 81–85.
Collins, P. L., Chanock, R. M. & Murphy, B. R. (2001). Respiratory
syncytial virus. In Fields Virology, 4th edn, pp. 1443–1485. Edited by
D. M. Knipe & P. M. Howley. Philadelphia, PA: Lippincott Williams
& Wilkins.
Cuesta, I., Geng, X., Asenjo, A. & Villanueva, N. (2000). Structural
phosphoprotein M2-1 of the human respiratory syncytial virus is an
RNA binding protein. J Virol 74, 9858–9867.
Das, S. C. & Pattnaik, A. S. (2004). Phosphorylation of vesicular
stomatitis virus phosphoprotein P is indispensable for virus growth.
J Virol 78, 6420–6430.
Dudas, R. A. & Karron, R. A. (1998). Respiratory syncytial virus
vaccines. Clin Microbiol Rev 11, 430–439.
Falsey, A. R., Cunningham, C. K., Barker, W. H., Kouides, R. W.,Yuen, J. B., Menegus, M., Weiner, L. B., Bonville, C. A. & Betts, R. F.(1995). Respiratory syncytial virus and influenza A infections in the
hospitalized elderly. J Infect Dis 172, 389–394.
Fearns, R., Peeples, M. E. & Collins, P. L. (1997). Increased
expression of the N protein of respiratory syncytial virus stimulates
minigenome replication but does not alter the balance between the
synthesis of mRNA and antigenome. Virology 236, 188–201.
Gao, Y. & Lenard, J. (1995). Multimerization and transcriptional
activation of the phosphoprotein (P) of vesicular stomatitis virus by
casein kinase-II. EMBO J 14, 1240–1247.
Gubbay, O., Curran, J. & Kolakofsky, D. (2001). Sendai virus
genome synthesis and assembly are coupled: a possible mechanism
to promote viral RNA polymerase processivity. J Gen Virol 82,
2895–2903.
Hardy, R. W. & Wertz, G. W. (1998). The product of the respiratory
syncytial virus M2 gene ORF1 enhances readthrough of intergenic
junctions during viral transcription. J Virol 72, 520–526.
Harrington, R. D., Hooton, T. M., Hackman, R. C., Storch, G. A.,Osborne, B., Gleaves, C. A., Benson, A. & Meyers, J. D. (1992). An
outbreak of respiratory syncytial virus in a bone marrow transplant
center. J Infect Dis 165, 987–993.
Higuchi, R., Krummel, B. & Saiki, R. K. (1988). A general method
of in vitro preparation and specific mutagenesis of DNA frag-
ments: study of protein and DNA interactions. Nucleic Acids Res 16,
7351–7367.
Horikami, S. M., Curran, J., Kolakofsky, D. & Moyer, S. A. (1992).Complexes of Sendai virus NP-P and P-L proteins are required for
defective interfering particle genome replication in vitro. J Virol 66,
4901–4908.
Lamb, R. A. & Kolakofsky, D. (2001). Paramyxoviridae: the viruses
and their replication. In Fields Virology, pp. 1305–1340. Edited by
D. M. Knipe & P. M. Howley. Philadelphia, PA: Lippincott Williams
& Wilkins.
Lopez, J. A., Villanueva, N., Melero, J. A. & Portela, A. (1988).Nucleotide sequence of the fusion and phosphoprotein genes of the
human respiratory syncytial (RS) virus Long strain: evidence of
subtype genetic heterogeneity. Virus Res 10, 249–261.
Lu, B., Ma, C.-H., Brazas, R. & Jin, H. (2002). The major phos-
phorylation sites of the respiratory syncytial virus phospho-
protein are dispensable for virus replication in vitro. J Virol 76,
10776–10784.
Mason, S. W., Aberg, E., Lawetz, C., DeLong, R., Whitehead, P. &Liuzzi, M. (2003). Interaction between human respiratory syncytial
virus (RSV) M2-1 and P proteins is required for reconstitution of
M2-1-dependent RSV minigenome activity. J Virol 77, 10670–10676.
Mazumder, B. & Barik, S. (1994). Requirements of casein kinase II-
mediated phosphorylation for the transcriptional activity of human
respiratory syncytial viral phosphoprotein P: transdominant negative
phenotype of phosphorylation-defective P mutants. Virology 205,
104–111.
Mazumder, B., Adhikary, G. & Barik, S. (1994). Bacterial expression
of human respiratory syncytial viral phosphoprotein P and identifi-
cation of Ser237 as the site of phosphorylation by cellular casein
kinase II. Virology 205, 93–103.
Navarro, J., Lopez-Otın, C. & Villanueva, N. (1991). Location of
phosphorylated residues in human respiratory syncytial virus
phosphoprotein. J Gen Virol 72, 1455–1459.
Pastey, M. K., Gower, T. L., Spearman, P. W., Crowe, J. E., Jr &Graham, B. S. (2000). A RhoA-derived peptide inhibits syncytium
formation induced by respiratory syncytial virus and parainfluenza
virus type 3. Nat Med 6, 35–40.
Pearson, R. B. & Kemp, B. E. (1991). Protein kinase phosphorylation
site sequences and consensus specificity motifs: tabulations. Methods
Enzymol 200, 62–81.
Roach, P. J. (1991). Multisite and hierarchal protein phosphoryla-
tion. J Biol Chem 266, 14139–14142.
Rodrıguez, L., Cuesta, I., Asenjo, A. & Villanueva, N. (2004).Human respiratory syncytial virus matrix protein is an RNA-
binding protein: binding properties, location and identity of the
RNA contact residues. J Gen Virol 85, 709–719.
Sanchez-Seco, M. P., Navarro, J., Martınez, R. & Villanueva, N.(1995). C-Terminal phosphorylation of human respiratory syncytial
http://vir.sgmjournals.org 1119
Dynamic dephosphorylation of HRSV P-protein residues

virus P protein occurs mainly at serine residue 232. J Gen Virol 76,425–430.
Schmitt, A. P., Leser, G. P., Waning, D. L. & Lamb, R. A. (2002).Requirements for budding of paramyxovirus simian virus 5 virus-like particles. J Virol 76, 3952–3964.
Sudo, K., Watanabe, W., Mori, S., Konno, K., Shigeta, S. & Yokota, T.(1999). Mouse model of respiratory syncytial virus infection toevaluate antiviral activity in vivo. Antivir Chem Chemother 10, 135–139.
Tran, K. C., Collins, P. L. & Teng, M. N. (2004). Effects of alteringthe transcription termination signals of respiratory syncytial virus onviral gene expression and growth in vitro and in vivo. J Virol 78,692–699.
Ulloa, L., Serra, R., Asenjo, A. & Villanueva, N. (1998). Interactionsbetween cellular actin and human respiratory syncytial virus(HRSV). Virus Res 53, 13–25.
Villanueva, N., Navarro, J. & Cubero, E. (1991). Antiviral effects ofxanthate D609 on the human respiratory syncytial virus growthcycle. Virology 181, 101–108.
Villanueva, N., Navarro, J., Mendez, E. & Garcıa-Albert, I. (1994).
Identification of a protein kinase involved in the phosphorylation of
the C-terminal region of human respiratory syncytial virus P protein.
J Gen Virol 75, 555–565.
Villanueva, N., Hardy, R., Asenjo, A., Yu, Q. & Wertz, G. (2000). The
bulk of the phosphorylation of human respiratory syncytial virus
phosphoprotein is not essential but modulates viral RNA transcrip-
tion and replication. J Gen Virol 81, 129–133.
Whitehead, S. S., Bukreyev, A., Teng, M. N., Firestone, C.-Y.,
St Claire, M., Elkins, W. R., Collins, P. L. & Murphy, B. R.
(1999). Recombinant respiratory syncytial virus bearing a deletion
of either the NS2 or SH gene is attenuated in chimpanzees. J Virol
73, 3438–3442.
Yu, Q., Hardy, R. W. & Wertz, G. W. (1995). Functional cDNA clones
of the human respiratory syncytial (RS) virus N, P, and L proteins
support replication of RS virus genomic RNA analogs and define
minimal trans-acting requirements for RNA replication. J Virol 69,
2412–2419.
1120 Journal of General Virology 86
A. Asenjo, L. Rodrıguez and N. Villanueva
Copyright © 2022 FDOKUMEN




















