
Effects of some additives on wheat gluten solubility: A structural approach
Upload
jaidefinichonCategory
view
1download
0
Eur. J. Biochem. 230, 38-44 (1995) 0 FEBS 1995
Hydrophobicity engineering to increase solubility and stability of a recombinant protein from respiratory syncytial virus Maria MURBY ', Elisabet SAMUFLSSON', Thien Ngoc NGUYEN', Laurent MIGNARD2, Ultan POWER2, Hans BINZ2, Mathias UHLEN' and Stefan STAHL'
Department of Biochemistry and Biotechnology, Royal Institute of Technology, Stockholm, Sweden Centre d'Immunologie Pierre Fabre, Saint-Julien en Genevois, France
(Received 16 December 199419 March 1995) - EJB 94 191612
Site-directed mutagenesis has been employed to engineer the hydrophobic properties of a 101-amino- acid fragment from the human respiratory syncytial virus (RSV) major glycoprotein (G protein). When this protein was produced in Escherichia coli, more than 70% of the gene product was found as inclusion bodies, and the product recovered from the soluble fraction was severely degraded. Substitution of two cysteine residues for serine residues, did not significantly change the solubility or stability of the gene product. In contrast, a dramatic increase in both solubility and stability was achieved by multiple engineer- ing of hydrophobic phenylalanine residues. As compared to the non-engineered protein, the fraction of soluble protein in vivo could be increased from 27% to 75%. Surprisingly, this effect was accompanied by a remarkable increase in stability. The in vitro solubility of the purified gene products was similarly increased approximately fivefold. Structural studies using circular dichroism suggest that the two engi- neered fragments have a distribution of secondary-structure elements similar to the non-engineered frag- ment. In addition, the two engineered G-protein variants were demonstrated to be at least in part antigeni- cally authentic to the non-engineered gene product. These results demonstrate that engineering of hy- drophobic residues can be used as a tool to increase the solubility and proteolytic stability of poorly soluble and labile proteins.
Keywords. Protein engineering ; expression ; stabilization ; solubility ; fusion proteins.
Heterologous protein expression in recombinant hosts, such as Escherichia coli, often yields large amounts of product, up to 50% of total cellular protein (Rudolph, 1995; Hellebust et al., 1989), but the cells frequently accumulate the protein in an im- properly folded, non-active form as inclusion bodies (Marston, 1986; Schein, 1993). Since refolding schemes add to the cost and labour of product recovery during downstream processing, a major challenge in recombinant expression is to design schemes for soluble protein production. Four major routes have been investigated for this purpose; (a) lowering the growth tem- perature (Schein, 1989; Strandberg and Enfors, 1991), (b) coex- pression of chaperones (Schein, 1991), (c) fusions to soluble domains such as staphylococcal protein A (Samuelsson et al., 1991 and 1994) or thioredoxin (LaVallie et al., 1993), and (d) protein engineering of specific residues (Schein, 1993 ; Luck et al., 1992). Protein engineering as a tool to remove hydrophobic residues in attempts to make the gene product more soluble is obviously limited to applications where these changes do not disrupt the structure or destroy the desired function of the re- combinant protein. Hydrophobic residues often have important structural functions and are often found in the interior of protein domains. Thus, protein engineering, where hydrophobic residues are mutated to more hydrophilic residues, has not been exten- sively used. Few systematic studies of this strategy have been
Correspondence to S. Stbhl, Department of Biochemistry and Bio- technology, Royal Institute of Technology, S-100 44 Stockholm, Sweden
Fax: +46 8 245452. Abbreviutions. ABP, albumin-binding protein ; G protein, major gly-
coprotein; F protein, fusion glycoprotein; RSV, respiratory syncytial virus.
reported, despite the fact that dramatic differences in the struc- tural andor functional properties of recombinant proteins can be obtained by single amino acid changes (Rinas et al., 1992; Cedergren et aI., 1993; Luck et al., 1992). In certain cases, spe- cific modifications have been shown to alter the solubility andl or stability of the recombinant protein (Schein, 1993; Rinas et al., 1992; Luck et al., 1992; Hellebust et al., 1989; Strandberg and Enfors, 1991). Rinas et al. (1992) showed that a single cys- teine residue substitution caused a dramatic decrease in solubil- ity, while two other cysteine to serine substitutions did not alter the solubility properties. In contrast, Luck et al. (1992) demon- strated that by replacing cysteine residues with serine residues in bovine prolactin, a significant increase in the protein solubil- ity was achieved. Furthermore, Hellebust et al. (1989) showed that the replacement of basic amino acids in a recombinant pro- tein can dramatically enhance the proteolytic stability in vivo.
Here, we have used site-directed mutagenesis to explore the possibility of engineering hydrophobic properties of a 101- amino-acid fragment from the human respiratory syncytial virus (RSV) major glycoprotein (G protein) that constitutes part of a fusion protein with an albumin-binding protein (ABP), derived from streptococcal protein G (Nygren et al., 1988). RSV is the major cause of viral-induced lower-respiratory-tract infections in infants throughout the world (McIntosh and Chanock, 1990). The two surface glycoproteins, the major glycoprotein (G pro- tein) and the fusion glycoprotein (F), that mediate attachment and penetration, respectively, have been identified as the major viral antigens responsible for inducing protective antibodies (Olmsted et al., 1986). Therefore, the F and G proteins are both

Murby et al. (Eur: J. Biochem. 230) 39
interesting candidates for use as subunit vaccines against RSV (McIntosh and Chanock, 1990; Toms, 1991).
There are very few reports on recombinant expression of RSV F (Martin-Gallardo et al., 1991 ; Murby et al., 1994) and G proteins (Martin-Gallardo, et al., 1993) in bacteria. This is probably due to the extreme difficulties in expressing these homo-oligomeric membrane-associated proteins in a soluble and stable form (Collins and Mottet, 1991 and 1992). We recently reported on the production of different RSV F peptides as inclu- sion bodies in Escherichia coli (Murby et al., 1994). The dif- ferent F peptides were produced as fusions to the IgG-binding protein ZZ, derived from staphylococcal protein A (Nilsson et al., 1987). Relatively large amounts of fusion proteins were ob- tained, but the affinity-purified fusion proteins were found to be difficult to resolubilise after iyophilisation. Martin-Gallardo and co-workers (1993) have shown that the RSV G protein can be produced in small amounts in Salmonella typhimurium. In E. coli, however, no recombinant protein was obtained.
Here, we have produced a 101-amino-acid fragment from the RSV G protein in E. coli. Three different synthetic gene fragments were constructed, encoding a non-engineered G-pro- tein and two engineered variants, respectively. The different G- protein gene fragments were fused downstream of ABP to sim- plify recovery of the gene products. We report the effects of substitutions of cysteine and phenylalanine residues for serine residues on in vivo and in vitro solubility, as well as on the stability to proteolysis.
MATERIALS AND METHODS
Gene assembly and vector constructions. The gene frag- ment encoding the amino acid sequence (amino acids 130-230) of the RSV (Long strain) G protein (Johnson et al., 1987) was constructed by solid-phase gene assembly using synthetic oligo- nucleotides, as described by Nguyen et al. (1994). The obtained gene fragment (here denoted Gnat) was isolated from the para- magnetic solid support using EcoRI and HindIII restriction and ligated into pRIT28 (Hultman et al., 1988), previously digested with the same enzymes. By a second gene assembly using an- other set of oligonucleotides, a mutant form of G, denoted GCya, was obtained. The obtained gene fragment, which had cysteine codons 173 and 186 replaced with serine codons, was subcloned into pRIT28 via BamHI and HindIII restriction. Nucleotide se- quences were verified by solid-phase DNA sequencing (Hult- man et al., 1991). Plasmid pRIT28 containing the fragment G,,, was used as template in a PCR with mismatched PCR primers to create a second mutant form of the G fragment, Gsubr in which phenylalanine codons at positions 163, 165, 168 and 170 were replaced with serine codons. The nucleotide sequence of Gsub after insertion into pRIT28 was verified as described above.
Plasmid pRIT44 (Kohler et al., 1991) is an expression vector encoding the two synthetic IgG-binding domains ZZ, derived from staphylococcal protein A, with transcription under the con- trol of the tryptophan operon promoter. Gene expression is in- duced by addition of indole acrylic acid (25 pg/ml). This plas- mid was restricted with XbaI and EcoRI, and the small ZZ-en- coding fragment was replaced by a similarly restricted fragment encoding ABP from ptrpBB (Murby et al., 1994). The resulting vector, denoted pVAABP308, encodes ABP under the control of the tryptophan promoter.
The three generated gene fragments G,,, GCys and Gsub were isolated from their respective pRIT28 constructs by restriction with EcoRI and HindIII. The gene fragments were ligated into pVAABP308 which had previously been cleaved with the same enzymes. The obtained expression vectors, designated pVAABP-
G,,,,, pVAABPG,,, and pVAABPG,,, encode the fusion proteins ABP-G,,,, ABP-G,,, and ABP-G,,,, respectively.
Fermentation and recovery of fusion proteins. 250 ml Tryptic Soy Broth (Difco) containing 100 pg/ml tryptophan (Merck), 100 pg/ml ampicillin (Sigma) and 8 pg/ml tetracycline (Sigma) was inoculated with E. coli RV308 cells (Maurer et al., 1980) transformed with either pVAABPG,,,, pVAABPG,,, or pVAABPG,,,, respectively. The cells were grown overnight at 32°C. The overnight cultures (200ml) were used as inoculum for fermentations (2 I; Chemap CF3000, Alfa Laval). The me- dium consisted of 5 g/l glycerol, 2.5 g/l ammonium sulphate, 3 g/l potassium dihydrogenphosphate, 2 g/l dipotassium hydro- genphosphate, 0.5 gA sodium citrate, 1 g/l yeast extract, 0.1 g/l ampicillin, 8 mgA tetracycline; 0.07 g/l thiamine, 1 gA magne- sium sulphate, and 1 ml/l trace elements (Holme et al., 1970). Parameters controlled during fermentation were pH, stirrer speed, temperature, oxygen saturation, glycerol feed and aera- tion rate. The pH was maintained at 7.3 with ammonia and ace- tic acid and the temperature was kept at 32°C. The growth was controlled by feeding glycerol at a rate giving a constant dis- solved oxygen tension signal of 30%. After 27 hours cultivation (A58o =SO), protein production was induced by addition of indole acrylic acid to a final concentration of 25 mgA. 3 hours after induction, the cells from the fermentation were harvested by centrifugation. Dry mass analysis was performed at the end of the cultivation period by collecting 3 x 5 ml samples in glass test tubes, the mass of which had previously been determined. Pelleted cells were washed once in 150 mM NaCl and dried overnight at 110°C before determination of the mass. A 30-g sample of wet biomass was resuspended in 70 ml cold 50 mM Tris/HCl, pH 8.0, 200 mM NaCI, 0.05% Tween 20 and 0.5 mM EDTA (TST buffer). The cells were disintegrated by sonication (Vibracell 72401, Sonics & Materials) as previously described (Nilsson and AbrahmsCn, 1990). After centrifugation of the ly- sate, the supernatant was filtered (1.2 pm) and diluted to 500 ml with TST. The soluble fusion proteins were isolated by affinity chromatography on human-serum-albumin-Sepharose as de- scribed by StAhl et al. (1989). Eluted fractions were monitored for protein content by absorbance measurement at 280 nm and relevant fractions were lyophilised.
Renaturation and recovery of insoluble fusion proteins. The insoluble material, after sonication, was pelleted by centrif- ugation and washed once in a washing buffer containing 50 mM Tris/HCl, pH 8.5, and 5 mM MgCI,. After washing, the pellet was solubilised in 30 ml 7 M guanidine hydrochloride, 25 mM Tris/HCl, pH 8.5, and 10 mM dithiothreitol, followed by incuba- tion at 37°C for at least 2 hours. The solubilised proteins were diluted 13 times with a renaturation buffer containing 25 mM TrisMCl, pH 8.5, 150 mM NaCl and 0.05 % Tween 20 to obtain a final guanidine hydrochloride concentration of 0.5 M. This mixture was incubated at room temperature under slow stirring for 16 hours. After incubation, the mixture was clarified by cen- trifugation and the fusion proteins present in the supernatant were recovered by affinity chromatography on human-serum- albumin-Sepharose as described above.
Protein analysis. The human-serum-albumin-Sepharose- purified proteins were analysed under reducing conditions on SDSlPAGE using gradient gels (8-25%) in the Phast system (Pharmacia Biotech) or homogeneous gels (12%) in the Mini- PROTEANRII system (New England Biolabs Inc.). Protein bands were visualised with Coomassie brilliant blue R250. The isoelectric points were estimated using Phast gel IEF media 3- 9 (Pharmacia Biotech) and protein bands were visualised as de- scribed above.
The in vitro solubility of the proteins were estimated by satu- rating a small volume (0.1 -0.3 ml) 0.1 M sodium phosphate,

40 Murby et al. ( E m J. Biochem. 230)
pH 6.7 containing 150 mM NaCl, with the protein to be ana- lysed. The mixture was incubated, by end over end rotation, at room temperature for 24 hours. The insoluble fractions were pelleted by centrifugation and the amount of solubilised protein was estimated by an absorbance measurement at 280 nm. The absorption coefficients used were : ABP-G,,, 0.422 ; ABP-Gcy,, 0.426 and ABP-G,,,, 0.418 ml . mg-' . cm-'.
CD was performed in a 5-720 spectropolarimeter (Jasco) at room temperature. The scanning speed was 20 n d m i n and each spectrum was averaged from five subsequent scans. The cell path length was 1 mm and the protein concentration was approx- imately 0.1 mg/ml in 10 mM sodium phosphate, pH 6.7. The ex- act protein concentration was determined using amino acid analysis as described by Cedergren et al. (1993). The CD data were analysed for secondary structure using a variable selection method where the CD spectrum is compared to the CD spectra of 33 proteins with known three-dimensional structures (Mana- valan and Johnson, 1987). The following criteria were applied to obtain the best solutions: (a) the sum of the predicted secondary- structure elements should be in the range 0.99-1.01, (b) the lowest value allowed for any secondary-structure element was -0.05; (c) the root mean square residual should not exceed 0.15 (for ABP and fusion protein spectra) or 0.5 (for differential spectra).
Immunological assay. Human RSV-A (Long strain ; ATCC VR-26, American Type Culture Collection) was used to immu- nise 14 female BALB/c mice (IFFA Credo). Mice were immu- nised three times intraperitoneally at two-weeks intervals with approximately 1 O5 tissue-culture infectious dose,, RSV-A in 0.05 M sodium phosphate, pH 7.0 and 0.09% NaCl (NaCW,) containing 20% alum [Al(OH),] (Superfos BioSector ds) as ad- juvant. Three weeks after the last immunisation, the mice were infected intranasally with a similar dose of virus. Five-days post- infection, the mice were sacrificed and exsanguinated. The sera from individual mice were stored at -80°C until used.
Immunolon 2 microtiter plates (Dynatech Laboratories) were coated with 50 p1 NaCUP, containing 4 mg/ml either ABP-Gnat, ABP-Gcy,, ABP-G,,, or ABP. The plates were blocked with NaCW, containing 4% milk solids for 2 hours at 37°C. 50 p1 of a 1 : 30 dilution of each serum in NaCVP, containing 0.4 % milk solids were added to the first row of wells and serially diluted 1 : 3. Negative controls consisted of wells to which no serum was added. The plates were incubated at room temperature for 2 hours and washed five times with NaCVP, containing 0.05% Tween-20 (Sigma). 50 pl horse-radish-peroxidase-conjugated goat anti-mouse Ig (Southern Biotechnology Associates, Inc.), diluted 1:8000, was added to each well, and the plates were incubated at room temperature for 1 hour. The plates were washed seven times as described above and 50 pl 3,3',5,5'- tetramethylbenzidine microwell peroxidase substrate (Kirke- gaard and Perry Laboratories Inc.) was added to each well fol- lowed by a 5-min incubation at room temperature. The reaction was terminated by addition of 50 yl 1 M HCUwell. The plates were read at 450 nm. Serum titres were expressed as the recipro- cal of the last dilution with an absorbance at least twofold that of the negative controls.
RESULTS
Design of G-protein gene fragments and construction of gene fusions. The RSV G protein consists of 298 amino acids, with a predicted molecular mass of 32.6kDa (Wertz et al., 1985). Several epitopes important for viral neutralisation and host pro- tection have been identified (Trudel et al., 1991 ; Rueda et al., 1994). We designed synthetic G-protein genes, all with codons
130 TVKTNTTTT QTQPSKPTTK QRQNKPPNKP NNDF'HFEVF'N F V P ~ C S N N
230 PTCTAICKRI PNKKPGKKlT TKPTKKPTPK TTKKDHKPQT TKPKEVPTTLP PTCTAI5KRI PNKKPGKKlT TKPTKKPTPK TTKKDHKPQT TKPKEVPTTLP
A/ ILI ABP I G,", I I I I -3-
A A '
1111
. _ _ A A '
Fig. 1. Amino acid sequence of the non-engineered G-protein peptide and schematic drawing of the expressed fusion proteins. An outline of the different fusion proteins expressed in this work. L represents eight additional amino acids originating from the expression vector. The amino acid sequence of the non-engineered peptide, G.,,, is written out in full. The amino acids are numbered according to the notation of John- son et al. (1987). Filled arrows represent sites for phenylalanine to serine substitutions and open arrows represent cysteine to serine substitutions.
selected to be suitable for E. coli production, encoding amino acids 130-230 from the G protein of RSV-A (Johnson et al., 1987). The choice of fragment was based on deletion-mutant analyses of the RSV G protein (Olmsted et al., 1989) and identi- fication of protective and neutralising epitopes (Trudel et al., 1991). A synthetic gene encoding the non-engineered RSV G- protein fragment, denoted G,,,, was constructed by solid-phase gene assembly (Fig. 1 ; see Materials and Methods for details). This fragment contains four cysteine residues, and it has been suggested that two of these form a disulphide bridge, (Cysl76- Cysl82), which has to be correctly formed for the protein to react with monoclonal antibodiesowhich are able to confer pas- sive protection from challenge (Akerlind-Stopner et al., 1990; Trudel et al., 1990). The four cysteine residues are preceded by a short hydrophobic region containing four phenylalanine resi- dues.
Since it has been reported that cysteine to serine substitu- tions can lead to increased solubility of recombinant proteins (Luck et al., 1992), the two cysteine residues not included in the predicted essential loop formation were substituted for serine residues. A synthetic gene fragment encoding this peptide was assembled as described above and denoted GCys (Fig. 1). A sec- ond variant, Gsub, was also generated with the same cystein to serine mutations and, in addition, with four phenylalanine resi- dues substituted for serine residues (Fig. 1).
To simplify recovery and analysis of the recombinant pro- teins, expression vectors were constructed where the three dif- ferent versions of the RSV G protein were fused to the ABP of streptococcal protein G (Nygren et al., 1988). All three fusion proteins, ABP-G,,,, ABP-G,, and ABP-G,,, (Fig. l), are en- coded by different expression vectors and have molecular masses of approximately 39 kDa.
Protein production and purification. The three different fusion proteins (Fig. 1) were expressed intracellularly in E. coli. The fusion proteins were recovered from both the soluble and insolu-
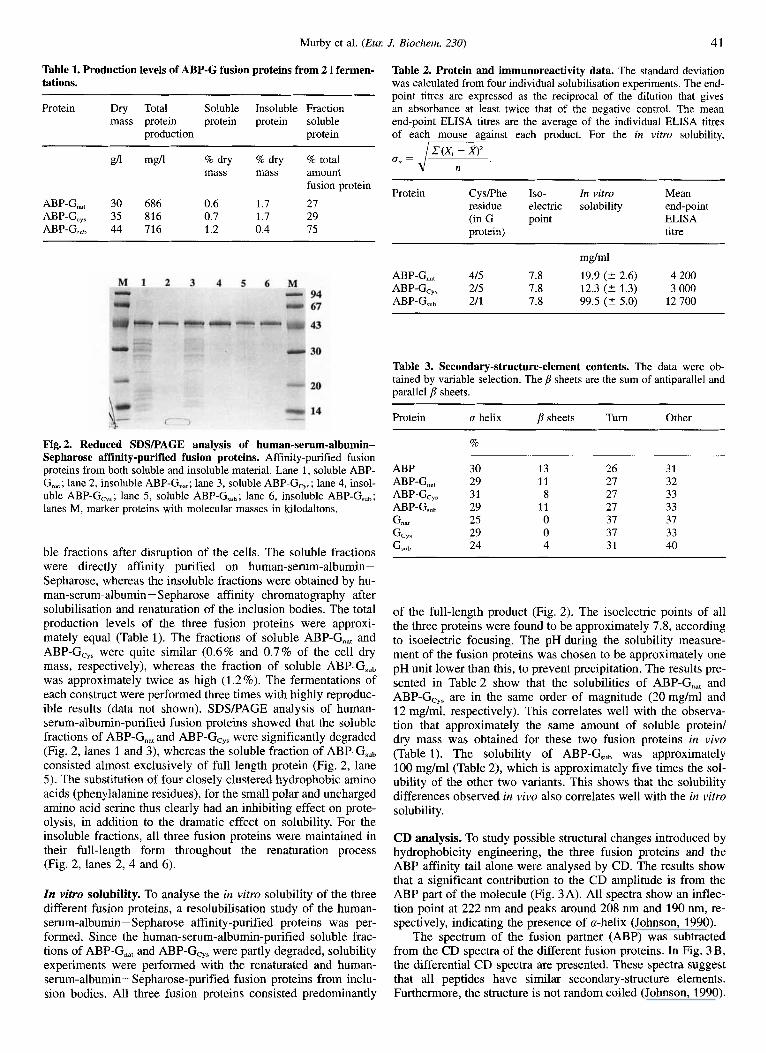
Murby et al. ( E m J. Biochem. 230) 41
Table 1. Production levels of ABP-G fusion proteins from 2 1 fermen- tations.
Protein Dry Total Soluble Insoluble Fraction mass protein protein protein soluble
production protein
a mg/l % dry % dry % total mass mass amount
fusion protein
ABP-Gnat 30 686 0.6 1.7 27 ABP-G,,, 35 816 0.7 1.7 29 ABP-G,, 44 716 1.2 0.4 75
Fig. 2. Reduced SDSPAGE analysis of human-serum-albumin- Sepharose affinity-purified fusion proteins. Affinity-purified fusion proteins from both soluble and insoluble material. Lane 1, soluble ABP- G,,,; lane 2, insoluble ABP-G,,,; lane 3, soluble ABP-G,,,; lane 4, insol- uble ABP-G,,,; lane 5, soluble ABP-G,,,; lane 6, insoluble ABP-G,,,; lanes M, marker proteins with molecular masses in kilodaltons.
ble fractions after disruption of the cells. The soluble fractions were directly affinity purified on human-serum-albumin- Sepharose, whereas the insoluble fractions were obtained by hu- man-serum-albumin - Sepharose affinity chromatography after solubilisation and renaturation of the inclusion bodies. The total production levels of the three fusion proteins were approxi- mately equal (Table 1). The fractions of soluble ABP-G,, and ABP-Gcy, were quite similar (0.6% and 0.7% of the cell dry mass, respectively), whereas the fraction of soluble ABP-G,,, was approximately twice as high (1.2%). The fermentations of each construct were performed three times with highly reproduc- ible results (data not shown). SDSPAGE analysis of human- serum-albumin-purified fusion proteins showed that the soluble fractions of ABP-G,, and ABP-G,,, were significantly degraded (Fig. 2, lanes 1 and 3), whereas the soluble fraction of ABP-G,,, consisted almost exclusively of full length protein (Fig. 2, lane 5). The substitution of four closely clustered hydrophobic amino acids (phenylalanine residues), for the small polar and uncharged amino acid serine thus clearly had an inhibiting effect on prote- olysis, in addition to the dramatic effect on solubility. For the insoluble fractions, all three fusion proteins were maintained in their full-length form throughout the renaturation process (Fig. 2, lanes 2, 4 and 6).
In vitro solubility. To analyse the in vitro solubility of the three different fusion proteins, a resolubilisation study of the human- serum-albumin- Sepharose affinity-purified proteins was per- formed. Since the human-serum-albumin-purified soluble frac- tions of ABP-G,,, and ABP-Gcys were partly degraded, solubility experiments were performed with the renaturated and human- serum-albumin- Sepharose-purified fusion proteins from inclu- sion bodies. All three fusion proteins consisted predominantly
Table 2. Protein and immunoreactivity data. The standard deviation was calculated from four individual solubilisation experiments. The end- point titres are expressed as the reciprocal of the dilution that gives an absorbance at least twice that of the negative control. The mean end-point ELISA titres are the average of the individual ELISA titres of each mouse against each product. For the in vitro solubility,
C(Xi - x)z
Protein CysPhe Iso- In vitro Mean residue electric solubility end-point (in G point ELISA protein) titre
mglml
ABP-Gnat 415 7.8 19.9 (? 2.6) 4 200 ABP-Gc,, 215 7.8 12.3 (t 1.3) 3 000 ABP-G,, 211 7.8 99.5 (t 5.0) 12700
Table 3. Secondary-structure-element contents. The data were ob- tained by variable selection. The p sheets are the sum of antiparallel and parallel p sheets.
Protein a helix psheets Turn Other
%
ABP 30 13 ABP-G,,, 29 11 ABP-Gc,, 31 8 ABP-G,,, 29 11 G", 25 0 Gc,, 29 0 G", 24 4
26 31 27 32 27 33 27 33 37 37 37 33 31 40
of the full-length product (Fig. 2). The isoelectric points of all the three proteins were found to be approximately 7.8, according to isoelectric focusing. The pH during the solubility measure- ment of the fusion proteins was chosen to be approximately one pH unit lower than this, to prevent precipitation. The results pre- sented in Table 2 show that the solubilities of ABP-G,, and ABP-Gcy, are in the same order of magnitude (20mg/ml and 12 mg/ml, respectively). This correlates well with the observa- tion that approximately the same amount of soluble protein/ dry mass was obtained for these two fusion proteins in vivo (Table 1). The solubility of ABP-GSub was approximately 100 mg/ml (Table 2), which is approximately five times the sol- ubility of the other two variants. This shows that the solubility differences observed in vivo also correlates well with the in vitro solubility.
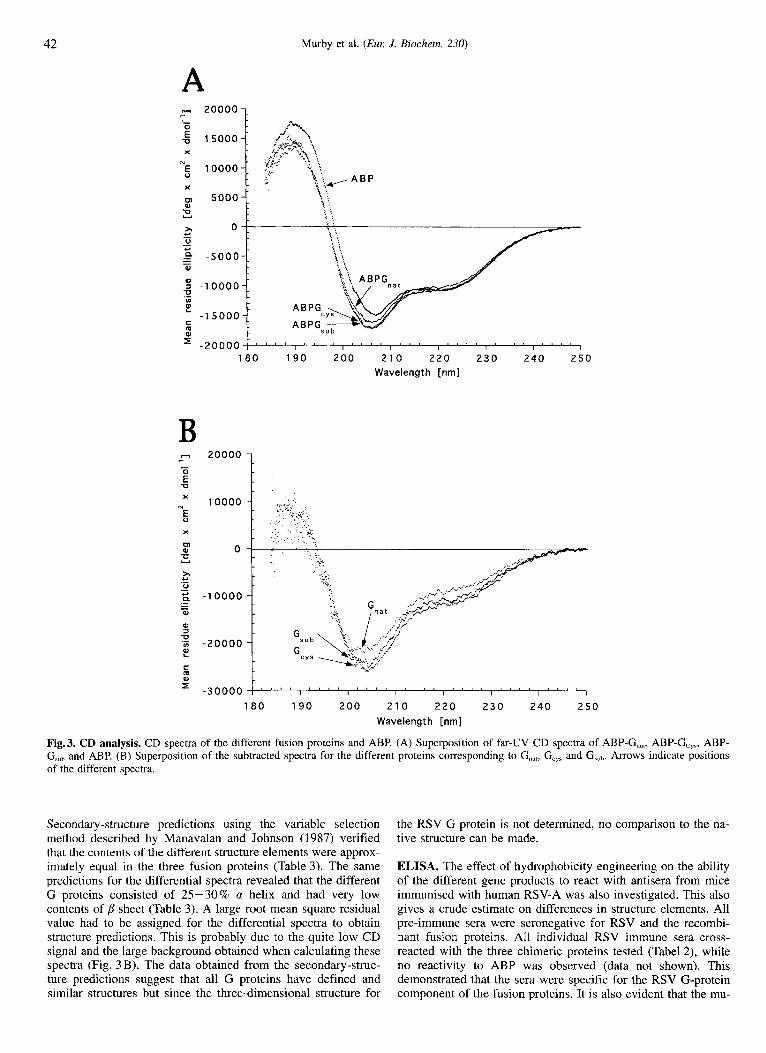
CD analysis. To study possible structural changes introduced by hydrophobicity engineering, the three fusion proteins and the ABP affinity tail alone were analysed by CD. The results show that a significant contribution to the CD amplitude is from the ABP part of the molecule (Fig. 3A). All spectra show an inflec- tion point at 222 nm and peaks around 208 nm and 190 nm, re- spectively, indicating the presence of a-helix (Johnson, 1990).
The spectrum of the fusion partner (ABP) was subtracted from the CD spectra of the different fusion proteins. In Fig. 3 B, the differential CD spectra are presented. These spectra suggest that all peptides have similar secondary-structure elements. Furthermore, the structure is not random coiled (Johnson, 1990).
42 Murby et al. (Euz J. Biochem. 230)
-20000 1 ' " ' I " ' I " " I " " I " " I " " I " " I
1 8 0 1 9 0 2 0 0 2 1 0 2 2 0 2 3 0 240 250 Wavelength [nm]
B - 20000
180 190 200 2 1 0 2 2 0 2 3 0 2 4 0 2 5 0 Wavelength [nm]
Fig. 3. CD analysis. CD spectra of the different fusion proteins and ABP. (A) Superposition of far-UV CD spectra of ABP-G,,,, ABP-G,,,, ABP- G,, and ABP. (B) Superposition of the subtracted spectra for the different proteins corresponding to G,,,, G,,, and G,ub. Arrows indicate positions of the different spectra
Secondary-structure predictions using the variable selection method described by Manavalan and Johnson (1987) verified that the contents of the different structure elements were approx- imately equal in the three fusion proteins (Table 3). The same predictions for the differential spectra revealed that the different G proteins consisted of 25530% a helix and had very low contents of sheet (Table 3). A large root mean square residual value had to be assigned for the differential spectra to obtain structure predictions. This is probably due to the quite low CD signal and the large background obtained when calculating these spectra (Fig. 3 B). The data obtained from the secondary-struc- ture predictions suggest that all G proteins have defined and similar structures but since the three-dimensional structure for
the RSV G protein is not determined, no comparison to the na- tive structure can be made.
ELISA. The effect of hydrophobicity engineering on the ability of the different gene products to react with antisera from mice immunised with human RSV-A was also investigated. This also gives a crude estimate on differences in structure elements. All pre-immune sera were seronegative for RSV and the recombi- nant fusion proteins. All individuaI RSV immune sera cross- reacted with the three chimeric proteins tested (Tabel 2), while no reactivity to ABP was observed (data not shown). This demonstrated that the sera were specific for the RSV G-protein component of the fusion proteins. It is also evident that the mu-

Murby et al. ( E m J. Biochem. 230) 43
tations required to produce ABP-Gcy, and ABP-G,,, did not eliminate RSV G-protein-specific epitopes. Thus, it is reason- able to suggest that all chimeras tested are, at least in part, anti- genically authentic relative to the native RSV G protein.
DISCUSSION Since the RSV G protein has been recognised as an impor-
tant candidate for a subunit vaccine against human RSV (Toms, 1991), considerable efforts have been made to produce RSV G proteins in E. coli. These attempts have so far failed (Martin- Gallardo et al., 1993). The reason for this is not clear but it has been suggested that certain viral glycoproteins could be detri- mental to the bacteria (Martin-Gallardo et al., 1991), though it seems more probable that the problems are related to low solu- bility and proteolytic instability.
Here, we report for the first time on the successful produc- tion in E. coli of the RSV G protein. Three variants of synthetic genes, with codon usage designed for E. coli production, have been constructed and expressed as gene fusions to ABP. The three chimeric proteins could be produced and recovered with good yields, ranging over 0.7-0.8 g/l culture. The relatively good yields might be due to the solubility effect of the fusion partner, as shown previously (Samuelsson et al., 1991) and/or be positively influenced by the use of synthetic genes designed for bacterial expression.
The three RSV G-protein variants were generated in order to investigate the effect of hydrophobicity engineering (i.e. site- specific changes of residues, such as phenylalanine, to more hy- drophilic residues) on the solubility of the RSV G-protein frag- ment, which in the non-engineered form has a very strong ten- dency to aggregate. A dramatic effect was observed by the sub- stitution of four phenylalanine residues to serine residues result- ing in a fivefold increase in the in vitro solubility and a dramatic increase from 27 % to 75 % of soluble material in the bacterial host. Interestingly, the observed effect on the solubility of the engineered fragment was also accompanied by a significant increase in proteolytic stability (Fig. 2) .
The question remains as to whether these dramatic differ- ences in solubility and stability arise due to specific changes of surface-exposed residues or if they are due to structural alter- ations of the fragment. The CD analysis provides a relatively crude estimation of structure, but the results suggest that the three fusion proteins have similar structural compositions. In ad- dition, reactivity with RSV-specific antisera was not impaired. It could be discussed whether or not the differential spectra show a true structural composition for the fused RSV G-protein frag- ments or if the fusions themselves influence the folding of ABP and thereby affect the spectra. However, it has earlier been demonstrated that ABP folds independent of C-terminal fusions (Oberg et al., 1994) and that such fusion proteins display double activities. It is important to point out that, even if a more thor- ough structural determination by nuclear magnetic resonance or X-ray crystallography show that the domains are structurally dif- ferent, the results presented here demonstrate that hydrophobi- city engineering can be used to alter the solubility and stability of an RSV G-protein fragment significantly. Obviously, a stabi- lised gene product with increased solubility has advantages for large-scale expression, purification and formulation of recombi- nant proteins. The question remains as to whether or not hydro- phobicity engineering can also be used for other recombinant proteins with poor solubility andor problems with proteolysis.
This work has been supported by the European Biotechnology pro- gramme ‘Human and veterinary vaccines’, project no. 920089. We thank Drs Lena Jendeberg, Magnus Jansson and Bjorn Nilsson at Pharniacia BioScience center for help with CD and amino acid analyses and for
fruitful discussions. Dr Per-Ake Nygren is acknowledged for his com- ments to the manuscript. We would also like to thank Marie-HCEne Gourdon, Alain Robert and Dominique Cyblat for skilful experimental assistance.
REFERENCES herlind-Stopner, B., Utter, G., Mufson, M. A., Orvell, C., Lerner, R.
A. & Norrby, E. (1990) A subgroup-specific antigenic site in the G protein of respiratory syncytial virus forms a disulphide-bonded loop, J. Virol. 64, 5143-5148.
Cedergren, L., Andersson, R., Jansson, B., UhlCn, M. & Nilsson, B. (1993) Mutational analysis of the interaction between staphylococcal protein A and human IgG,, Protein Eng. 6, 441-448.
Collins, P. L. & Mottet, G. (1991) Post-translational processing and oligomerization of the fusion glycoprotein of human respiratory syn- cytial virus, J. Gen. Virol. 72, 3095-3101.
Collins, P. L. & Mottet, G. (1992) Oligomerization and post-translational processing of glycoprotein G of human respiratory syncytial virus : altered 0-glycosylation in the presence of brefeldin A, J. Gen. Virol.
Hellebust, H., Murby, M., AbrahmsCn, L., UhlCn, M. & Enfors, S.-0. (1989) Different approaches to stabilize a recombinant fusion pro- tein, Biotechnology 7, 165 - 168.
Holme, T., Arvidson, S., Lindholm, B. & Pavlu, B. (1970) Enzymes- laboratory-scale production, Process Biochem. 5 , 62- 66.
Hultman, T., Stihl, S., Moks, T. & UhlBn, M. (1988) Approaches to solid phase DNA sequencing, Nucleoside Nucleotide 7, 629 -638.
Hultman, T., Bergh, S., Moks, T. & UhlBn, M. (1991) Bidirectional solid- phase sequencing of in vitro-amplified plasmid DNA, Biotechniques
Johnson, P. R., Spriggs, M. K., Olmsted, R. A. & Collins, P. L. (1987) The G glycoprotein of human respiratory syncytial viruses of sub- groups A and B : Extensive sequence divergence between antigeni- cally related proteins, Proc. Nut1 Acad. Sci. USA 84, 5625-5629.
Johnson, W. C. Jr (1990) Protein secondary structure and circular dichro- ism: A practical guide, Proteins Struct. Funct. Genet. 7 , 205-214.
Kohler, K., Ljungquist, C., Kondo, A,, Veide, A. & Nilsson, B. (1991) Engineering proteins to enhance their partitioning coefficients in aqueous two-phase systems, Biotechnology 9, 642- 646.
LaVallie, E. R., DiBlasio, E. A., Kovacic, S., Grant, K. L., Schendel, P. F. & McCoy, J. M. (1993) A thioredoxin gene fusion expression system that circumvents inclusion body formation in the E. coli cyto- plasm, Biotechnology 11, 187-193.
Luck, D. N., Gout, P. W., Sutherland, E. R., Fox, K., Huyer, M. & Smith, M. (1992) Analysis of disulphide bridge function in recombinant bovine prolactin using site-specific mutagenesis and renaturation un- der mild alkaline conditions : a crucial role for the central disulphide bridge in the mitogenic activity of the hormone, Prof. Eng. 5 , 559- 567.
Manavalan, P. & Johnsson, W. C. Jr (1987) Variable selection improves the prediction of protein secondary structure from circular dichroism spectra, Anal. Biochem. 167,76-85.
Marston, F. A. 0. (1986) The purification of eukaryotic polypeptides synthesized in Escherichia coli, Biochem J. 240, 1 - 12.
Martin-Gallardo, A,, Fien, K. A,, Hu, B. T., Farley, J. F., Seid, R., Col- lins, P. L., Hildreth, S. W. & Paradiso, P. R. (1991) Expression of the F glycoprotein gene from human respiratory syncytial virus in Escherichia coli: Mapping of a fusion inhibiting epitope, Virology
Martin-Gallardo, A., Fleicher, E., Doyle, S. A., Arumugham, R., Collins, P. L., Hidreth, S. W. & Paradiso, P. R. (1993) Expression of the G glycoprotein gene of human respiratory syncytial virus in Salmonella typhimuriurn, J. Gen. Virol. 74, 453-458.
Maurer, R., Meyer, B. J. & Ptashne, M. (1980) Gene regulation at the right (0,) of bacteriophage 1 I. OR3 autogenous negative control by repressor, J. Mol. Biol. 139, 147-161.
McIntosh, K. & Chanock, R. M. (1990) Respiratory Syncytial Virus, in Virology (Fields, B. N. & Knipe, D. M., ed.) 2nd edn, pp. 1045- 1072, Raven Press, Ltd, NY.
Murby, M., Nguyen, T. N., Binz, H., UhlCn, M. & Stihl, S. (1994) Pro- duction and recovery of recombinant proteins of low solubility, in Separations for biotechnology 3 (Pyle, D. L., ed.) pp. 336-344, Bookcraft Ltd., Bath.
73, 849-863.
10, 84-93.
184, 428-432.

44 Murby et al. ( E m J. Biochem. 230)
Nguyen, T. N., UhlCn, M. & Stlhl, S . (1994) De novo gene assembly using a paramagnetic solid support, in Advances in biomagnetic sep- aration (UhlCn, M., Homes, E. & Olsvik, @., eds) pp. 73-78, Eaton publishing Co., Natick.
Nilsson, B., Moks, T., Jansson, B., AbrahmsCn, L., Elmblad, A., Holm- gren, E., Henrichson, C., Jones, T. A. & UhlCn, M. (1987) A syn- thetic IgG-binding domain based on staphylococcal protein A, Prot. Eng. I , 107-113.
Nilsson, B. & AbrahmsCn, L. (1990) Fusions to staphylococcal protein A, Methods Enzymol. 185, 144-161.
Nygren, P.-A,, Eliasson, M., Palmcrantz, E., AbrahmsCn, L. & UhlCn, M. (1988) Analysis and use of the serum albumin binding domains of streptococcal protein G, J. Mol. Recogn. I , 69-74.
Oberg, U., Rundstrom, G., Gronlund, H., Uhltn, J. & Nygren, P.-A. (1994) Intracellular production and renaturation from inclusion bod- ies of a scFv fragment fused to a serum albumin binding affinity tail, in Proc. 6th Eul: Congl: Biotechnol. (Alberghina, L., Frontali, L. & Sensi, P., eds) pp. 179-182, Elsevier Science B. V., Amsterdam, The Netherlands.
Olmsted, R. A,, Elango, N., Prince, G. A., Murphy, B. R., Johnson, P. R., Moss, B., Chanock, R. M. & Collins, P. L. (1986) Expression of the F glycoprotein of respiratory syncytial virus by a recombinant vaccinia virus: Comparison of the individual contributions of the F and G glycoproteins to host immunity, Proc. Natl Acad. Sci. USA
Olmsted, R. A., Murphy, B. R., Lawrence, L. A., Elango, N., Moss, B. & Collins, P. (1989) Processing, surface expression, and immunogenic- ity of carboxy-terminally truncated mutants of G protein from human respiratory syncytial virus. J. virol. 63, 411-420.
Rinas, U., Tsai, L. B., Lyons, D., Fox, G. M., Steams, G., Fieschko, J., Fenton, D. & Bailey, J. E. (1992) Cysteine to serine substitutions in basic fibroblast growth factor: Effect on inclusion body formation and proteolytic susceptibility during in vitro refolding, Bioteclznology
Rudolph, R. (1995) Successful protein folding on an industrial scale, in Principles and practice of protein folding (Cleland, J. L. & Craik, C. S . , eds), John Wiley and Sons Inc., New York, in the press.
83, 7462-7466.
10,435-440.
Rueda, P., Garcia-Baneno, B. & Melero, J. A. (1994) Loss of conserved cysteine residues in the attachment (G) glycoprotein of two human respiratory syncytial virus escape mutants that contain multiple A-G substitutions (hypermutations) virology 198, 53 -662.
Samuelsson, E., Wadensten, H., Hartmanis, M., Moks, T. & Uhltn, M. (1991) Facilitated in vitro refolding of human recombinant insulin- like growth factor I using a solubilizing fusion partner, Biotechnol-
Samuelsson, E., Moks, T., Nilsson, B. & UhlCn, M. (1994) Enhanced in vitro refolding of insulin-like growth factor I using a solubilizing fusion partner, Biochemistry 33, 4207 -421 1.
Schein, C. H. (1989) Production of soluble recombinant proteins in bac- teria, Biotechnology 7, 1141 - 1149.
Schein, C. H. (1991) Optimizing protein folding to the native state in bacteria, Curl: Opin. Biotechnol. 2, 746-750.
Schein, C. H. (1993) Solubility and secretability, Cum Opin. Biotechnol.
Strandberg, L. & Enfors, S.-0. (1991) Factors influencing inclusion body formation in the production of a fused protein in Escherichia coli, Appl. Eviron. Microbiol. 57, 1669-1674.
Straus, D. B., Walter, W. A. & Gross, C. A. (1988) Escherichia coli heat shock gene mutants are defective in proteolysis, Genes & Dev. 2,
Stihl, S., Sjolander, A., Nygren, P.-w., Berzins, K., Perlmann, P. & Uhlen, M. (1989) A dual expression system for the generation, analysis and purification of antibodies to a repeated sequence of the Plasmodium fakiparum antigen Pfl55/RESA, J. Immunol. Meth. 124, 43-52.
Toms, G. L. (1991) Vaccination against respiratory syncytial virus: prob- lems and progress, FEMS Microbiol. Immunol. 76, 243-256.
Trudel, M., Nadon, F., Stguin, C. & Binz, H. (1991) Protection of BALB/c mice from respiratory syncytial virus infection by immuni- zation with a synthetic peptide derived from the G glycoprotein, vi- rology 185, 749-757.
Wertz, G. W., Collins. P. L., Huang, Y., Gruber, C., Levine, S. & Ball, L. A. (1985) Nucleotide sequence of the G protein of human respiratory syncytial virus reveals an unusual type of viral membrane protein, Proc. Natl Acad. Sci. USA 82,4075-4079.
ogy 9, 363-366.
4,456-461.
1851-1858.
Copyright © 2022 FDOKUMEN




















