

Critical role of extracellular vesicles in modulating the cellular effects of cytokines

For Peer Review
Cyclin-Dependent Kinase 9 Inhibition protects cartilage from the catabolic effects of pro-inflammatory cytokines.
Journal: Arthritis and Rheumatism
Manuscript ID: ar-13-0940
Wiley - Manuscript type: Full Length
Date Submitted by the Author: 02-Jul-2013
Complete List of Authors: Yik, Jasper; UC Davis, Orthopaedic Surgery Hu, Zi'ang; Zhejang University, Orthopaedic Surgery; UC Davis, Orthopaedic Surgery Kumari, Ratna; UC Davis, Orthopaedic Surgery; KIIT University, KIIT School of Biotechnology Christiansen, Blaine; UC Davis, Orthopaedic Surgery Haudenschild, Dominik; University of California Davis Medical Center, Orthopaedic Surgery;
Keywords: CDK9, inflammatory cytokines, primary response genes, Flavopiridol, chondrocytes
John Wiley & Sons
Arthritis & Rheumatism

For Peer Review
1
Running Head: CDK9 Inhibition is Chondroprotective
Title: Cyclin-Dependent Kinase 9 Inhibition protects cartilage from
the catabolic effects of pro-inflammatory cytokines.
Authors: Jasper H. N. Yik, Ph.D. 1
Zi’ang Hu, M.D.2,1
Ratna Kumari, Ph.D. 1,3
Blaine A. Christiansen, Ph.D. 1
Dominik R. Haudenschild, Ph.D. 1,*
Grants: Arthritis Foundation 2012 IRG Award to DRH DOD PRMRP IIRA Award #PR110507 to DRH Departmental Startup Funds to BAC and DRH National Natural Science Fund of China (81101378, 81271971) to ZH No author received any financial support or other benefits from commercial sources for the work reported on in this manuscript, and no author has any other financial interest that could create a potential conflict of interest or the appearance of a conflict of interest with regard to the work.
2 Department of Orthopaedic Surgery, Sir Run Run Shaw Hospital, School of Medicine, Zhejang University, Hangzhou, 210016 PR China
3 present address: KIIT School of Biotechnology, KIIT University, Bhubaneswar, India 1,*Address correspondence to Dominik R. Haudenschild University of California Davis, Department of Orthopaedic Surgery, Lawrence J. Ellison Musculoskeletal Research Center Research Building 1 Suite 2000 4635 Second Avenue Sacramento, CA 95817 Tel: 916-734-5015 Fax: 916-734-5750
Page 1 of 44
John Wiley & Sons
Arthritis & Rheumatism

For Peer Review
3
Abstract
Objective. CDK9 controls the activation of primary inflammatory response genes. We
determined whether CDK9 inhibition could protect cartilage from the catabolic effects of pro-
inflammatory cytokines.
Methods. Human chondrocytes were challenged with different pro-inflammatory stimuli (IL-1β,
lipopolysaccharides, and TNFα), in the presence or absence of the CDK9 inhibitor Flavopiridol.
The mRNA expression of inflammatory mediators, catabolic, and anabolic genes were
determined by real-time PCR. Human cartilage explants were incubated with IL-1β, with or
without Flavopiridol, for 6 days. Cartilage matrix degradation was assessed by the release of
glycosaminoglycan (GAG) and cleaved Type II collagen (Col2a) peptides.
Results. CDK9 inhibition by Flavopiridol effectively suppressed iNOS mRNA induction by all
three pro-inflammatory stimuli. Results from NFkB-targets PCR array showed that Flavopiridol
suppressed the induction of a broad range of inflammatory mediator genes (59 out of 67 tested)
by IL-1β. Flavopiridol also inhibited the induction of catabolic genes MMP 1, 3, 9, 13, and
ADAMTS4, 5; but did not affect the basal expression of anabolic genes such as Col2a, aggrecan,
and COMP, and housekeeping genes. Flavopiridol had no apparent short-term cytotoxicity as
assessed by glucose-6-phosphate dehydrogenase activity. Finally, in IL-1β-treated cartilage
explants, Flavopiridol reduced the release of matrix degradation products GAG and cleaved
Col2a peptides, but did not affect long-term chondrocyte viability.
Conclusion. CDK9 activity is required for the primary inflammatory response in chondrocytes.
Flavopiridol effectively suppresses the induction of inflammatory mediators and catabolic genes
to protect cartilage from the deleterious effects of pro-inflammatory cytokines, without
impacting cell viability and functions.
Page 3 of 44
John Wiley & Sons
Arthritis & Rheumatism

For Peer Review
4
Key words: CDK9, inflammatory cytokines, primary response genes, Flavopiridol, chondrocytes,
cartilage
Page 4 of 44
John Wiley & Sons
Arthritis & Rheumatism

For Peer Review
5
Introduction
Osteoarthritis (OA) affects more than half of the people over age 65 in the United States.
OA is a degenerative disease of the articular joints characterized by slow but progressive lost of
cartilage. The main protein component of articular cartilage is a fibrillar network of type II
collagen (Col2a), which provides tensile strength to the cartilage. The compressive stiffness of
the cartilage is provided by the proteoglycan components, through their attraction of water
molecules. Although the etiology of OA remains incompletely understood, various inflammatory
conditions that cause damage to the collagens and proteoglycans in cartilage are suspected of
initiating OA.
Pro-inflammatory cytokines, such as interleukin 1 beta (IL-1β) and tumor necrosis factor
alpha (TNFα), can induce the catabolic destruction of the cartilage matrix. These cytokines elicit
a cascade of events that activate inflammatory mediator genes and apoptosis in chondrocytes
(reviewed by (1)). Pro-inflammatory cytokines induce the expression of proteinases that degrade
cartilage matrix, including matrix metallopeptidases (MMPs), aggrecanases, and cathepsins
(reviewed by (2)). Pro-inflammatory cytokines are induced by a variety of stress conditions in
cartilage, including joint overloading and physical damage such as occurs in sports-related
injuries. Strikingly, a disproportionally large number of people with joint injuries developed OA
(3). Since injury activates pro-inflammatory cytokines expression, we hypothesize that injury-
related activation of the inflammatory response in chondrocytes ultimately contributes to
cartilage destruction and the development of OA. Therefore, a strategy to suppress the
Page 5 of 44
John Wiley & Sons
Arthritis & Rheumatism

For Peer Review
6
inflammatory response in chondrocytes after acute tissue stress or injury may help prevent the
onset of OA.
Acute tissue stress and inflammatory signaling activate primary response genes (PRG)
that do not require de novo protein synthesis. Recent advances demonstrate that despite their
initiation by diverse signaling pathways, the transcriptional activation of most, if not all, PRG is
similarly controlled by a general transcription factor (4, 5), namely the cyclin-dependent kinase 9
(CDK9). CDK9 is ubiquitously and abundantly expressed in all cell types, but unlike the other
more well-known CDKs, it is not directly involved in regulating cell cycle progression. It was
believed for many years that the rate-limiting step in transcriptional activation of PRG is the
recruitment of transcription factors and RNA Polymerase II (Pol II) complex to the promoters.
However, recent studies show that in order for these PRG to be activated rapidly, in their basal
and unstimulated states, the Pol II complex is already pre-assembled and producing short mRNA
transcripts (4, 5). However, the Pol II is then paused ~40 base-pair downstream of the
transcription start site. Promoter-proximal pausing of Pol II is accomplished by the concerted
efforts of the negative elongation factor (NELF) and the DRB-sensitive inducible factor (DSIF)
(6)). The rapid activation of PRG is the result of signal-induced recruitment of CDK9 to the
promoters, where it phosphorylates NELF and DISF to negate their repression. CDK9 also
phosphorylates Pol II at the Serine 2 residues of the C-terminal domain heptapeptide repeats.
Phosphorylation by CDK9 induces a conformational change that allows Pol II to enter possessive
elongation to efficiently transcribe full-length mRNAs (reviewed in (7)). The recruitment of
CDK9 to activated PRG promoters is accomplished by the bromo-domain containing protein 4
(Brd4), through its binding to acetylated histones (8). Thus, CDK9 regulation represents a
Page 6 of 44
John Wiley & Sons
Arthritis & Rheumatism

For Peer Review
7
unique mechanism of activating PRG transcription and has a broad impact on many aspects of
biological functions.
Given that CDK9 controls a common mechanism of all PRG activation, it is an attractive
target for anti-inflammatory therapy (reviewed in (9)). The objective of this study is to determine
whether CDK9 inhibition effectively suppress the inflammatory response in chondrocytes and
protect cartilage from the catabolic effects of pro-inflammatory cytokines in vitro.
Methods
Articular chondrocytes and cartilage explants – Human primary chondrocytes and cartilage
explants were isolated from cartilage tissues obtained from total knee arthroplasty (15 donors,
age 44-80 years, all with end stage OA) with IRB approval and cultured in DMEM with 10%
FBS as described (10). The chondrocytes were used for experiments within 3-5 days without
passaging to avoid dedifferentiation. A total of 5 cartilage explant donors were used for matrix
degradation studies (Figure 5). All other experiments were performed with chondrocytes from at
least 3 individual donors. Full thickness bovine cartilage explants were isolated from the stifle
joints of young veal calves using 6-mm biopsy punches and maintained in DMEM with 10%
FBS.
Treatments of chondrocytes with inflammatory stimuli and small molecule inhibitors –
Primary chondrocytes grown in 12-well plates (~80% confluence) were treated with 10 ng/ml
lipopolysaccharide (LPS) (Sigma), 10 ng/ml IL-1β (R&D System), or 10 ng/ml TNFα (R&D
System) for various time, with or without pharmacological inhibitors. The inhibitors used in this
Page 7 of 44
John Wiley & Sons
Arthritis & Rheumatism

For Peer Review
8
study included Flavopiridol (Sigma), JQ-1 (a kind gift from Dr. James Bradner, Harvard
University (11)), BS-181 HCl (Selleckchem), and SNS-032 (Selleckchem). After the treatment,
the cells were washed 3 times with phosphate buffered saline and harvested for gene and protein
expression analysis.
Lentiviral siRNA constructs - The CDK9-targeting siRNA used in this study
(AGGGACATGAAGGCTGCTAAT) (8) was inserted into the AgeI and EcoRI sites of the lentiviral
vector pLKO.1 (plasmid #8453, www.Addgene.com). A siRNA targeting GFP was used as
control. Lentiviral particles were generated and tittered as described (12). Human chondrocytes
were seeded in 12-well plate at 70-80% confluency. Lentiviral particles harboring CDK9- or
GFP-targeting siRNA were then added at a multiplicity of infection of 10, in the presence of 1
µg/ml polybrene (American Bioanalytical). The media was replaced after 16 hours and the cells
were used for experiments after 5 days.
Quantitative real-time (RT) PCR – For the determination of individual mRNA expression,
cytokine/inhibitor-treated chondrocytes in 12-well plates were harvested by scraping and
transferred to Eppendorf tubes, followed by cell lysis and reverse transcription to generate cDNA
using the Cells-to-CT kit (Ambion) according to the manufacturer’s instructions. 2 ul of cDNA
was used for quantitative RT-PCR (in a final volume of 10 ul) performed in triplicate in a
7900HT RT-PCR system with gene-specific Taqman probes (Applied Biosystem Inc.) according
to the manufacturer’s conditions. Results were normalized to 18s rRNA and calculated as fold-
change in mRNA expression relative to untreated control, using the 2-∆∆CT method. The probes
used are as follow: iNOS, Hs01060345_m1; MMP1, Hs00899658_m1; MMP3,
Page 8 of 44
John Wiley & Sons
Arthritis & Rheumatism

For Peer Review
9
Hs00968305_m1; MMP9, Hs03234579_m1; MMP13, Hs0023992_m1; ADAMTS4,
Hs00192708_m1; ADAMTS5, Hs00199841_m1; ACAN, Hs00202971_m1; COMP,
Hs00154339_m1; COL2A, Hs01060345_m1; 18s rRNA, 4319413E.
For PCR array analysis, IL-1β/Flavopiridol-treated chondrocytes grown in 10 cm plates
were harvested and total RNA was isolated using the RNeasy Mini Kit (Qiagen). RNA quality
and quantity were assessed with a Nanodrop 2000 spectrophotometer (Thermo Scientific) and
~500 ng of total RNA was reverse transcribed using a Superscript First-Strand kit (Invitrogen).
RT-PCR was performed in a 7900HT PCR system using the PCR Arrays for Human NFκB
Signaling Targets (Qiagen, cat# 330231), according to the manufacturers’ protocol. PCR array
data were analyzed by the accompanying online analysis software provided by Qiagen at
www.qiagen.com.
Cytotoxicity/Viability Assays – For determining the short-term cytotoxic effects of
Flavopiridol, chondrocytes were seeded in 96-well plates at 1, 5, or 10 x 103 cells per well and
treated with 300 nM Flavopiridol for 5 hours. Cytotoxicity was assessed using the Vybrant
Cytotoxicity Assay kit (Invitrogen, cat# V23111), according to the manufacturer’s protocol to
measure soluble and total glucose 6-phosphate dehydrogenase (G6PD) activity with resazurin as
substrates. Fluorescence was detected using a Synergy HT plate reader (Bio-TEK Instruments
Inc.) with excitation and emission filters set at 528 and 635 nm, respectively.
For determining long-term effects of Flavopiridol on the viability of chondrocytes in
cartilage, bovine cartilage explants (6-mm disk) were cultured in 6-well plates in DMEM and
10% FBS, in the presence or absence of 300 nM Flavopiridol for 6 days, with media changed
every other day. The live and dead cells were stained using the LIVE/DEAD
Page 9 of 44
John Wiley & Sons
Arthritis & Rheumatism

For Peer Review
10
Viability/Cytotoxicity kit (Invitrogen, cat# L3224) according to the manufacturer’s protocol. The
percentages of live and dead cells were determined by counting the cell numbers at three random
fields of the cross-section images of explants captured using a fluorescence microscope. A total
of 3 different donors were used.
Western blot analysis – Chondrocytes grown in 12-well plates were harvested and lysed with
sample loading buffer (50 mM Tris-HCl, pH 6.8; 100 mM dithiothreitol; 4% 2-mercaptoethanol;
2% sodium dodecyl sulfate; 10% glycerol). Lysates were resolved by 4-12% SDS-
polyacrylamide gels and transferred onto nitrocellulose membranes (BioRad). The membranes
were blocked with 3% skim milk in TBST (25 mM Tris-HCl, pH 7.5; 125 mM NaCl; 0.1%
Tween 20), followed by incubation with rabbit anti-CDK9 (0.6 ug/ml) (8) mouse anti-MMP13
(1:500 dilution) (Calbiochem, cat# IM78), or mouse anti-GAPDH (1:5000) (Ambion, cat#
AM4300) at 4oC overnight. Blots were then probed with horseradish peroxidase-conjugated
secondary antibody (Santa Cruz Biotechnology), and reactive protein bands were visualized with
Western Lightning Plus-ECL (Perkin Elmer) on radiographic films.
Assessment of cartilage degradation – Human cartilage explants (~3mm cubes) were treated
with 1 ng/ml IL-1β for 6 days, in the presence or absence of 6 or 300 nM Flavopiridol (with
media change on day 3). The amount of glycosaminoglycan (GAG) released into the media was
determined by the colorimetric dimethylmethylene blue dye-assay, with chondroitin sulfate as
standard (13). The release of Col2a degradation products into the media was determined by
measuring the amount of cleaved Col2a peptides (14) with the C2C ELISA kit (IBEX
Pharmaceuticals) according to the manufacturer’s protocol.
Page 10 of 44
John Wiley & Sons
Arthritis & Rheumatism

For Peer Review
11
Statistical analysis – Values of all measurements were expressed as the mean + standard
deviation. Statistical comparison was performed by unpaired, two-tailed Student’s t test. Values
of p<0.05 were considered significant.
Results
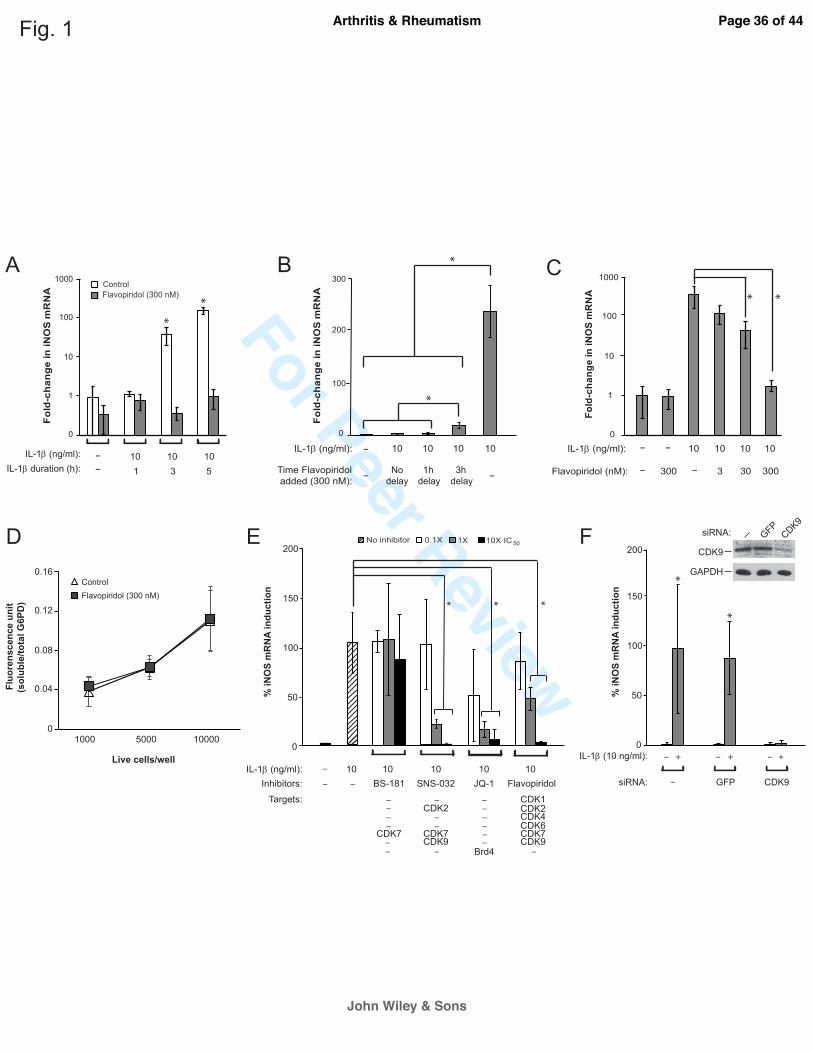
The role of CDK9 on the induction of the primary response gene iNOS. Although the rate-
limiting step for transcriptional activation of primary inflammatory response genes in
lymphocytes is controlled by CDK9 (4, 5), whether CDK9 plays a similar role in articular
chondrocytes has not been investigated. Therefore, we take advantage of a widely used and well-
characterized pharmacological CDK9 inhibitor, Flavopiridol, to determine the role of CDK9 in
the activation of PRG in chondrocytes. To activate PRG, chondrocytes in culture were treated
with IL-1β, in the presence or absence of 300 nM Flavopiridol (the effective plasma
concentration determined in clinical trials (15)). The induction of the stress response gene iNOS
(inducible nitric oxide synthase) (16) was determined at various time points. The results showed
that iNOS mRNA was unchanged after 1 hour of IL-1β treatment, but was induced significantly
after 3 and 5 hours (Figure 1A, open bars). However, co-treatment with Flavopiridol completely
suppressed the induction of iNOS by IL-1β (Figure 1A, grey bars). These results indicate a time-
dependent induction of iNOS by IL-1β that is sensitive to Flavopiridol treatment.
The above results demonstrated the effects of Flavopiridol administered concurrently
with the inflammatory cytokines. To gain insight in a clinically relevant context, we next tested
whether a delay in the addition of Flavopiridol could still suppress iNOS induction by IL-1β.
Chondrocytes were treated with IL-1β for a total of 5 hours, with either no delay, or a 1- or 3-
Page 11 of 44
John Wiley & Sons
Arthritis & Rheumatism

For Peer Review
12
hour delay in the addition of Flavopiridol. The data showed that when compared to no
Flavopiridol treatment (~235-fold iNOS induction), the addition of Flavopiridol markedly
repressed iNOS induction if it was administered at no delay (down to ~2.7-fold induction) and at
a 1-hour delay (~4-fold) (Figure 1B). Although less effective, a 3-hour delay still allowed
significant repression of iNOS induction (~18-fold) (Figure 1B). These results indicate that there
is at least a 3-hour window of opportunity for administering Flavopiridol to suppress iNOS
induction after the initial treatment of IL-1β. We next demonstrated that Flavopiridol suppressed
iNOS induction by IL-1β in a dose-dependent manner, with the most effective dose at 300 nM
(Figure 1C). Importantly, there was no apparent cytotoxicity of Flavopiridol at the highest dose,
in terms of G6PD activity, on cultured chondrocytes within the 5 hours time frame tested (Figure
1D).
Besides CDK9, Flavopiridol has off-target effects that include other CDKs (listed in
Figure 1E). While these CDKs do not affect PRG activation directly, we never the less used
additional CDK inhibitors to rule out their involvement in suppressing iNOS induction. To this
end, we tested the abilities to suppress iNOS induction by the following three small molecule
inhibitors: BS-181 HCl (specific for CDK7) (17), SNS-032 (targeting CDK2, 7, and 9) (18), and
JQ-1 (specific for Brd4, which is required for CDK9’s function (11)). The data showed that only
SNS-032, JQ-1, and Flavopiridol, but not BS-181, suppressed iNOS induction in a dose-
dependent manner (Figure 1E), thus effectively ruling out the involvement of CDK7 in IL-1β-
induced iNOS transcription. It is important to note that unlike the other CDK inhibitors tested
here, JQ-1 does not directly inhibit the kinase activity of CDK, but rather, it prevents the
association of acetylated histones with Brd4 (11), which specifically recruits CDK9 to the
Page 12 of 44
John Wiley & Sons
Arthritis & Rheumatism

For Peer Review
13
promoters for activation of transcription of PRG (4,5). Therefore, the above results strongly
suggest that CDK9 is involved in iNOS induction. To definitively prove this, we used siRNA to
specifically knockdown CDK9 expression in chondrocytes (Figure 1F, inset) and determined the
effects on iNOS induction. The results showed that in CDK9-depleted cells (confirmed by
Western blot, Figure 1F inset), IL-1β failed to induce iNOS expression (Figure 1F), thus
demonstrating the absolute requirement of CDK9. Taken together, our results clearly show the
specific involvement of CDK9, but not other CDKs, in the induction of iNOS by IL-1β. Since
Flavopiridol is the first small molecule CDK inhibitor in clinical trials with well-characterized
pharmacokinetics, it has the highest potential for translation into clinical studies, and therefore, it
is used exclusively in the remainder of this study.
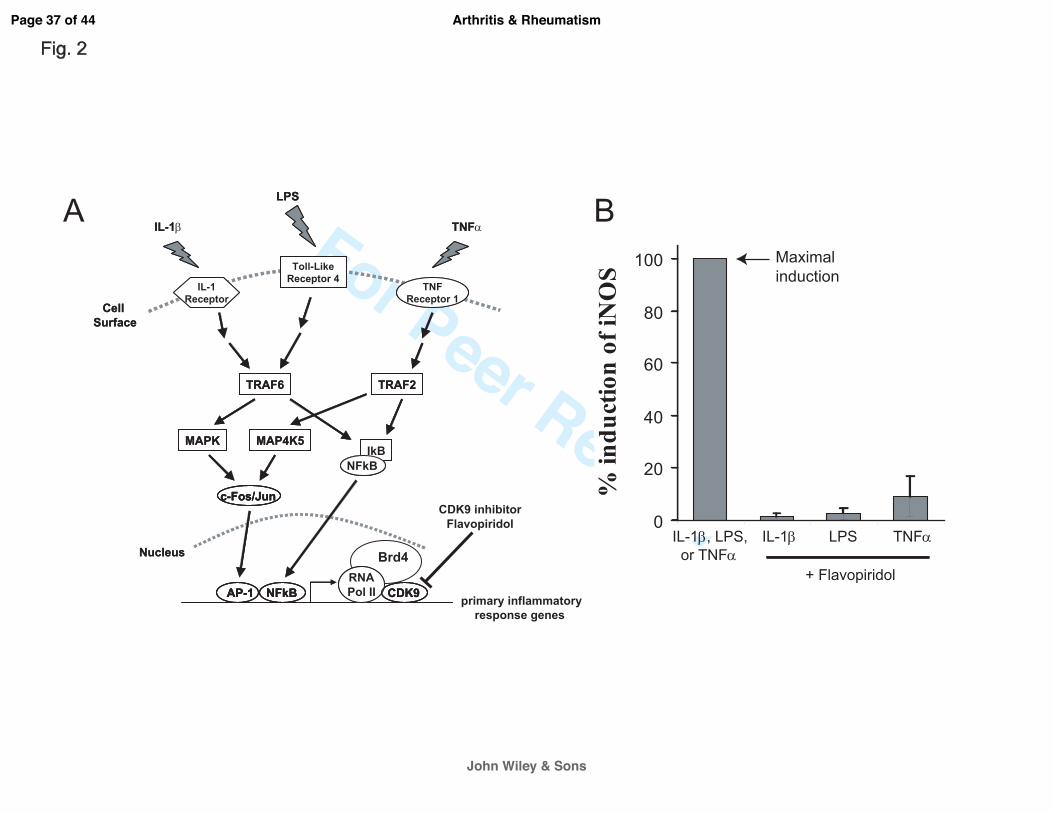
CDK9 controls the activation of inflammatory response from diverse signals. PRG are
activated by diverse inflammatory signals. However, regardless of the sources, most
inflammatory signals converge onto the rate-limiting step of transcriptional elongation of PRG
controlled by CDK9. In order to illustrate this, three different inflammatory signaling pathways
were selected; namely, IL-1β, Lipopolysaccharides (LPS), and Tumor Necrosis Factor alpha
(TNFα). Cellular response to IL-1β, LPS, or TNFα is mediated by three distinct pathways –
activation of the IL-1Receptor, Toll-Like Receptor 4, or TNF Receptor 1, respectively (Figure
2A). Chondrocytes were treated with the above three agents independently, in the presence or
absence of the CDK9 inhibitor Flavopiridol. The mRNA expression of iNOS, a common effector
gene for all three pathways (16), was then determined. The results showed that Flavopiridol
greatly suppressed the activation of iNOS expression in all three pathways (Figure 2B),
demonstrating the effectiveness and broad range of Flavopiridol in preventing inflammatory
Page 13 of 44
John Wiley & Sons
Arthritis & Rheumatism

For Peer Review
14
response from diverse signals. Thus our data confirmed previous finding in other cellular
systems (4, 5) and established CDK9 as a central regulatory point for primary inflammatory
response in chondrocytes.
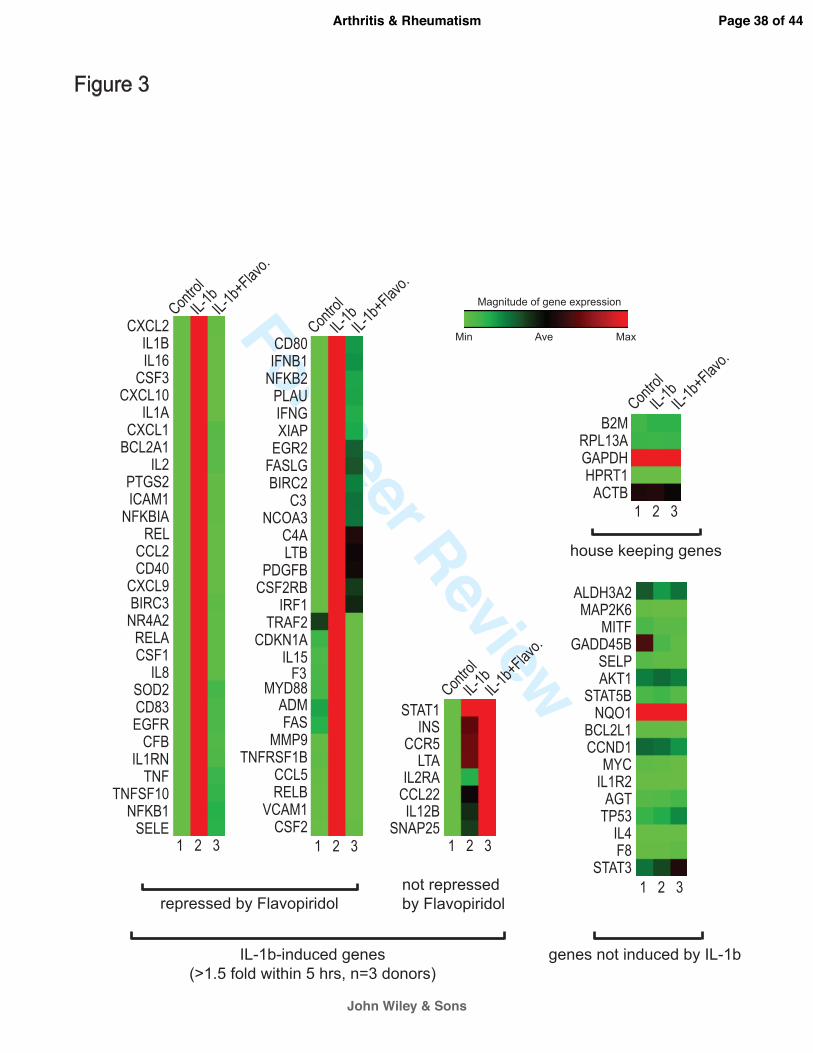
CDK9 inhibition prevents the activation of a broad spectrum of inflammatory response genes.
To further investigate the effects of CDK9 inhibition on the activation of other inflammatory
mediators, the gene expression profiles of chondrocytes treated with IL-1β for 5 hours were
determined by real-time PCR arrays. The PCR array contained 84 key genes responsive to NFκB
signal transduction (Qiagen), which is central to the regulation of multiple cellular processes
such as inflammatory, immunity, and stress responses. The gene expression profiles from three
chondrocyte donors were averaged, and are presented as heat maps in which low and high
relative expressions are represented by green and red colors, respectively (Figure 3). The array
data in numerical format is included as supplementary data. The results showed that IL-1β
strongly activated the majority of the 84 NFκB-target genes tested (Figure 3, compared lane 1&
2), while CDK9 inhibition by Flavopiridol almost completely abolished the effects of IL-1β (lane
3). On average, across three chondrocyte donors, CDK9 inhibition repressed IL-1β activity by a
magnitude of greater than 86%, and suppressed 59 out of 67 NFκB-target genes that were
activated at least 1.5-fold by IL-1β. Importantly, house-keeping genes, as well as genes not
induced by IL-1β, were not affected by Flavopiridol. These data demonstrated that CDK9 can be
targeted to effectively suppress only the activation of a cascade of downstream inflammatory
response genes, without affecting the basal expression of non-responsive genes.
Page 14 of 44
John Wiley & Sons
Arthritis & Rheumatism

For Peer Review
15
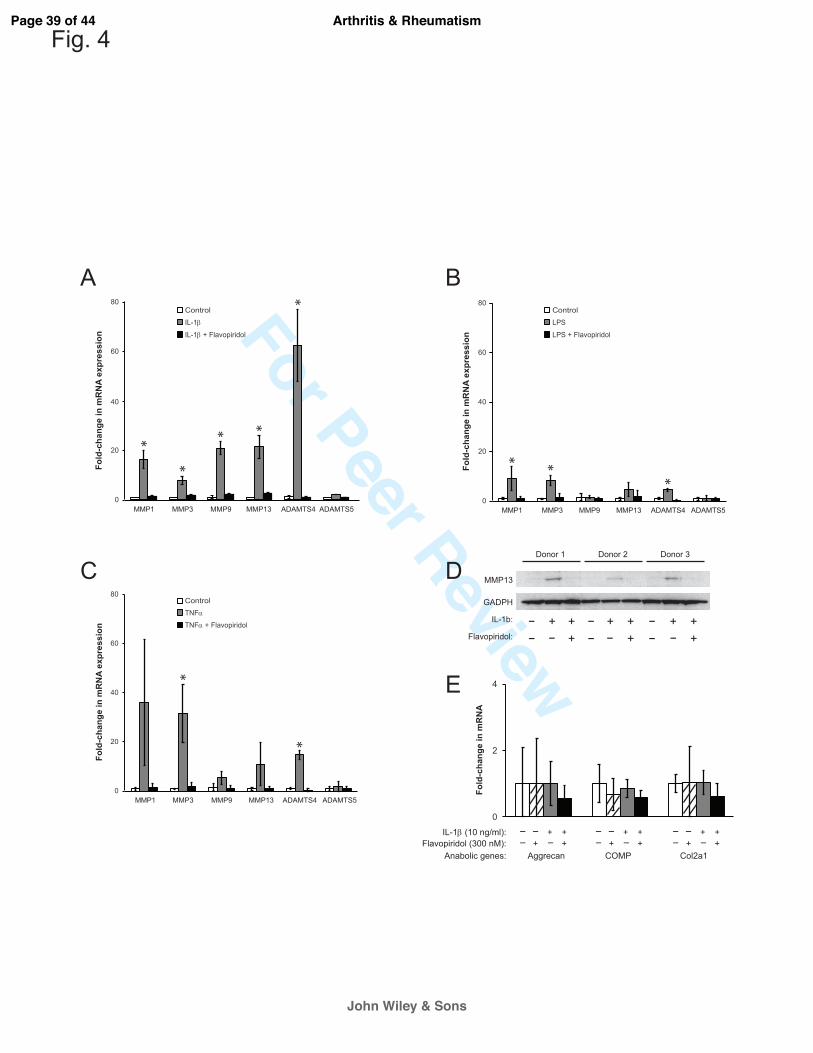
CDK9 inhibition prevents the activation of catabolic genes, but has no effects on basal
expression of anabolic genes in chondrocytes. Besides activating the acute phase inflammatory
genes, pro-inflammatory cytokines such as IL-1β and TNFα can also stimulate the expression of
catabolic genes in chondrocytes (2, 19). These catabolic genes include the various matrix MMPs
and ADAMTS (a disintegrin and metalloproteinase with a thrombospondin type 1 motif) that
degrade the cartilage matrix. Given the role of CDK9 in activation of inflammatory genes, we
next examined the effects of CDK9 inhibition on the induction of MMPs and ADAMTS in
chondrocytes treated independently with IL-1β, LPS, and TNFα for 5 hours. The results showed
that IL-1β-mediated upregulation of MMP1, 3, 9, and 13, as well as ADAMTS4 mRNAs was
markedly suppressed by co-treatment with Flavopiridol (Figure 4A). Similar trends were
observed in LPS- or TNFα-treated samples (Figure 4B & C). These data indicated that CDK9
inhibition prevents the transcriptional activation of catabolic genes in chondrocytes. Next, we
sought to confirm the upregulation of MMP13 mRNA at the protein level, which is implicated in
collagen degradation and osteoarthritis (20). Chondrocytes were treated with IL-1β, with and
without Flavopiridol, for 2 days. Cell-associated active MMP13 protein (~48 kDa) was then
detected by Western blots. The data showed that MMP13 protein expression was elevated in all
three donors treated with IL-1β, but remained at basal levels with IL-1β and Flavopiridol
treatment (Figure 4D). Thus, the results on protein expression corroborate with the mRNA
expression profiles of MMP13. On the other hand, the mRNA expression of selected anabolic
genes (Aggrecan, Cartilage Oligomeric Matrix Protein (COMP), and Collagen 2a) in
chondrocytes was not significantly affected by IL-1β or Flavopiridol within the same 5-hours
time frame (Figure 4E). Taken together, the above results demonstrate that Flavopiridol
Page 15 of 44
John Wiley & Sons
Arthritis & Rheumatism

For Peer Review
16
selectively suppresses only the induction of catabolic genes by pro-inflammatory stimuli, while
having negligible effects on the basal expression of anabolic genes.
CDK9 inhibition protects cartilage from the catabolic effects of IL-1ββββ. Since CDK9 inhibition
suppresses the activation of inflammatory genes and catabolic enzymes in chondrocytes, we next
determined whether Flavopiridol can protect cartilage from the deleterious effects of pro-
inflammatory cytokines. Human arthritic cartilage explants were isolated and cultured in media
containing 1 ng/mL IL-1β, in the presence or absence of Flavopiridol for 6 days. Note that the
concentration of IL-1β was reduced from the 10 ng/ml used in short-term monolayer culture, to a
level similar to those detected in the synovial fluids of inflamed joints (21-23). Degradation of
cartilage matrix was then assessed by measuring the release of GAG and Col2a cleavage
peptides into the culturing media. As expected, IL-1β alone increased the amount of both GAG
(Figure 5A) and Col2a peptides (Figure 5B) released into the media (compared first and second
bars). However, the concentrations of both GAG and Col2a peptides were reduced by 6 nM
Flavopiridol and returned to baseline levels by 300 nM Flavopiridol (Figure 5A & B). Thus, our
data provides evidence that CDK9 inhibition prevents the catabolic destruction of cartilage by
IL-1β. Importantly, the percentages of live/dead cells were similar in both untreated and
Flavopiridol-treated bovine cartilage explants (Figure 5C). This result indicates that prolonged
treatment of cartilage with Flavopiridol did not have an adverse effect on the viability of
chondrocytes in cartilage explants. Taken together, our data indicate that CDK9 inhibition
protects cartilage explants from the catabolic effects of IL-1β.
Discussion
Page 16 of 44
John Wiley & Sons
Arthritis & Rheumatism

For Peer Review
17
The etiology of primary OA remains incompletely understood and the involvement of the
inflammatory response is somewhat controversial. However, it is well established that damage to
the collagen network originates around chondrocytes at the cartilage matrix surface (24). Since
inflammatory response induces chondrocyte apoptosis and cartilage matrix breakdown (2), there
are several anti-OA strategies that target either specific branches of the inflammatory signaling
cascade (e.g. IL-1, IL-6, TNFα, and NFκB inhibitors) (19, 25, 26), or the downstream events
such as apoptosis with caspase inhibitors (1). However, because inflammation can be induced by
a variety of stimuli, the above individual approaches would have limited effectiveness in
handling the diverse simultaneous challenges in a biological system, as well as limited abilities
in efficiently suppressing a broad range of inflammatory mediator expression. A novel approach
to addressing these limitations is to directly target CDK9, which activates transcription of
primary inflammatory response genes. Using the pharmacological CDK9 inhibitor Flavopiridol,
we have shown that cartilage can be protected from the harmful effects of pro-inflammatory
cytokines.
Our results demonstrate for the first time in chondrocytes that Flavopiridol effectively
suppresses the acute response to multiple inflammatory stimuli (Figure 2), and prevents the
induction of a broad range of inflammatory mediators (Figure 3), as well as catabolic genes that
contribute to the degradation of the cartilage matrix (Figure 4). In most cases, Flavopiridol
almost completely abolishes the activation of inflammatory mediator expression. For example,
from the PCR array data (Fig 3 and supplemental data), IL-1β induced expression of IL-6 by
492-fold, but only 4.2-fold in the presence of Flavopiridol, representing a 99.2% repression of
IL-1ß-dependent transcription. Importantly, our data demonstrate the selectivity of Flavopiridol-
Page 17 of 44
John Wiley & Sons
Arthritis & Rheumatism

For Peer Review
18
mediated inhibition, in which only the IL-1b-inducible genes are suppressed, but not the basal
expression of non-inducible genes, housekeeping genes (Figure 3), and the anabolic genes
(Col2a, COMP, and aggrecan) in chondrocytes (Figure 4E). The gene expression profiles are
further supported by the experiments demonstrating Flavopiridol can effectively prevent
cartilage degradation induced by IL-1β (Fig 5A & B). The reduction in matrix degradation
products was not due to changes in cell viability in cartilage treated with Flavopiridol, because
live/dead staining revealed similar chondrocyte viabilities between control and Flavopiridol
treated cartilage (Figure 5C).
Flavopiridol is an ATP analog that preferentially inhibits CDK9 kinase activity (Ki=30
nM) by a high affinity interaction with its ATP-binding pocket (27). Although Flavopiridol can
potentially inhibit other CDKs, numerous studies using a combination of specific inhibitors and
siRNA have demonstrated that only CDK9 inhibition is responsible for the anti-inflammatory
action of Flavopiridol (28, 29). We have also shown that both JQ-1-mediated inhibition of Brd4,
which does not directly interact with other CDKs (8); as well as siRNA-mediated inhibition of
CDK9, lead to the loss of IL-1β-mediated induction of iNOS in chondrocytes (Figure 1E & F).
In addition, it is not documented that other CDKs involved in cell cycle regulation have a
pronounced effect on the transcriptional activation of a broad range of PRG within the 5-hour
time frame used in this study. Therefore, our results provide strong evidence that only CDK9 is
responsible for the activation of PRG in chondrocytes.
Flavopiridol was originally known for its anti-proliferation properties by suppressing
cell-cycle progression in rapidly dividing cells (e.g. cancers) or in cells with short life-span (e.g.
Page 18 of 44
John Wiley & Sons
Arthritis & Rheumatism

For Peer Review
19
neutrophils). Its pharmacological activity is well-documented over the last two decades because
of its use in clinical trials as anti-proliferation/cancer agent (reviewed by (30)). Sekine et al have
demonstrated that systemic administration of Flavopiridol reduces synovial hyperplasia, but does
not induce apoptosis, and result in preventing the development of rheumatoid arthritis in a
collagen-induced mouse model (31). However, it is not known whether the anti-arthritic activity
of Flavopiridol is due to the systematic suppression of the leukocyte-mediated immune response
to the injected collagen, or the localized suppression of the inflammatory response of
chondrocytes in cartilage. Our group has developed a non-invasive knee injury mouse model for
post-traumatic OA (PTOA) (32). Preliminary data indicate that systemic administration of
Flavopiridol effectively suppresses the production of pro-inflammatory cytokines locally at the
injured knee, thus confirming our in vitro finding detailed in this study. Future experiments are
needed to determine whether Flavopiridol treatment will prevent or delay the development of
PTOA in our mouse model, or in other existing PTOA models (33, 34).
In summary, our data for the first time demonstrate the absolute requirement of CDK9
activity in the activation of PRG in human chondrocytes. In addition, our data strongly indicate
that Flavopiridol is an effective agent to prevent the activation of acute inflammatory response
and catabolic pathways in cartilage. Our results thus may provide a new strategy to prevent or
delay the onset of OA.
Competing interest
The authors declare that they have no competing interest.
Acknowledgements
Page 19 of 44
John Wiley & Sons
Arthritis & Rheumatism

For Peer Review
20
This study was supported by an Arthritis Foundation 2012 IRG award to DRH, a DOD PRMRP
IIRA award #PR110507 to DRH, Departmental Funds to BAC and DRH, and the National
Natural Science Fund of China (81101378, 81271971) to ZH
Page 20 of 44
John Wiley & Sons
Arthritis & Rheumatism

For Peer Review
21
Figure legends
Figure 1. Characterization of the effects of CDK9 inhibition by Flavopiridol on iNOS
induction. (A) Kinetics of IL-1β-induced iNOS expression. Human chondrocytes were treated
with 10 ng/ml IL-1β, with and without 300 nM Flavopiridol, for the indicated time. The fold-
induction of iNOS mRNA relative to untreated control was determined by quantitative RT-PCR.
(B) Time window of administration of Flavopiridol for iNOS suppression. Chondrocytes were
treated with IL-1β for a total of 5 hours, with Flavopiridol added at different delayed time points
to determine the window of opportunity for effective suppression of iNOS induction. (C) Dose-
dependent suppression of iNOS induction by Flavopiridol. Chondrocytes were treated with IL-1β
for 5 hours, with co-treatments of Flavopiridol at various concentrations to determine the dose-
response for suppressing iNOS induction. (D) Cytotoxicity assays. Chondrocytes seeded in 96-
wells at the indicated cell density were treated with 300 nM Flavopiridol for 5 hours, followed
by measurement of soluble/total G6PD activity to determine the cytotoxic effects of
Flavopiridol. (E) Suppression of iNOS induction by different inhibitors. Chondrocytes were
treated with IL-1β for 5 hours in the presence of various small molecule inhibitors at the
indicated concentrations. The induction of iNOS mRNA by IL-1β was determined and the
maximum iNOS induction by IL-1β in the absence of inhibitor was set to 100%. The selected
IC50 of various drugs based on their kinase inhibition are: BS-181, 21 nM for CDK7; SNS-032,
60 nM for CDK7 (used in this experiment) and 4 nM for CDK9; Flavopiridol, 30 nM for CDK9.
JQ-1 is not a kinase inhibitor but prevents CDK9 recruitment to PRG promoters through
suppressing the binding of Brd4 to acetylated histones at an IC50 of 300 nM. (F) siRNA-mediated
depletion of CDK9 and its effect on iNOS induction. Chondrocytes were transduced with
Page 21 of 44
John Wiley & Sons
Arthritis & Rheumatism

For Peer Review
22
lentiviral particles harboring siRNA against CDK9, or GFP as control. After 5 days, cells were
treated with IL-1β for 5 hours and harvested for Western and iNOS mRNA analysis. For all the
above experiments, each data point was the mean +/- standard deviation from three different
donors. (*p<0.05)
Figure 2. The CDK9 inhibitor Flavopiridol is effective against different inflammatory
stimuli. (A) Activation of inflammatory genes by diverse signals. Multiple pro-inflammatory
stimuli, such as IL-1β, LPS, and TNFα activate their respective cell surface receptors. These
signals are then transmitted through different intracellular mediators/pathways, which ultimately
converge on CDK9-dependent transcription of inflammatory genes. Brd4 functions to recruit
CDK9 to activated promoters. (B) Flavopiridol is effective against multiple inflammatory
stimuli. Human chondrocytes (n=3 different donors) in monolayer culture were treated with
different inflammatory stimuli (10ng/ml of either IL-1β, LPS, or TNFα) with or without 300 nM
Flavopiridol for 5 hours. iNOS mRNA was quantified by real-time PCR as a measure of
inflammatory response. The induction of iNOS by each stimulus alone was arbitrarily set to
100% (first bar) and compared to the respective value obtained in sample co-treated with each
inflammatory stimulus and Flavopiridol. Results were the mean +/- standard deviation from three
different donors.
Figure 3. Flavopiridol effectively suppresses the induction of a broad range of
inflammatory mediators. Primary human chondrocytes (n=3 different donors) in monolayer
culture were treated with 10 ng/ml IL-1β with or without 300 nM Flavopiridol for 5 hours. Gene
expression was analyzed using real-time PCR Array for NFκB targets (Qiagen) and shown here
Page 22 of 44
John Wiley & Sons
Arthritis & Rheumatism

For Peer Review
23
as heat map (Green=minimum expression, Red=maximum expression). Of the 84 NFκB target
genes tested, 67 were induced greater than 1.5-fold by IL-1β (compare lanes 1 & 2).
Flavopiridol almost completely abolishes the effects of IL-1β in 59 of these 67 genes (lane 3).
Importantly, housekeeping genes and non-inducible genes are unaffected by either IL-1β or
Flavopiridol.
Figure 4. CDK9 inhibition prevents induction of MMP and ADAMTS expression by
various inflammatory stimuli. (A-C) Primary chondrocytes (n=3 different donors) were treated
with either 10 ng/ml of IL-1β, LPS, or TNFα, with or without 300 nM Flavopiridol for 5 hours,
and the relative mRNA expression of the cartilage degrading enzymes MMP-1,-3,-9,-13, and
ADAMTS4 and ADAMTS5 was determined by real-time PCR. (D) Flavopiridol suppresses
MMP13 protein expression. Human chondrocytes (n=3 different donors) grown in 6-well plates
were treated with 10 ng/ml IL-1β, with or without 300 nM Flavopiridol for 2 days. Cell-
associated active MMP13 (~48kD) was detected by Western blot. (E) Flavopiridol does not
affect basal expression of anabolic genes. Chondrocytes were treated with IL-1β and/or
Flavopiridol for 5 hours and the mRNA expression of the cartilage matrix genes aggrecan,
COMP, and Col2a were determined by RT-PCR. For all the above experiments, each data point
was the mean +/- standard deviation from three different donors. (*p<0.05)
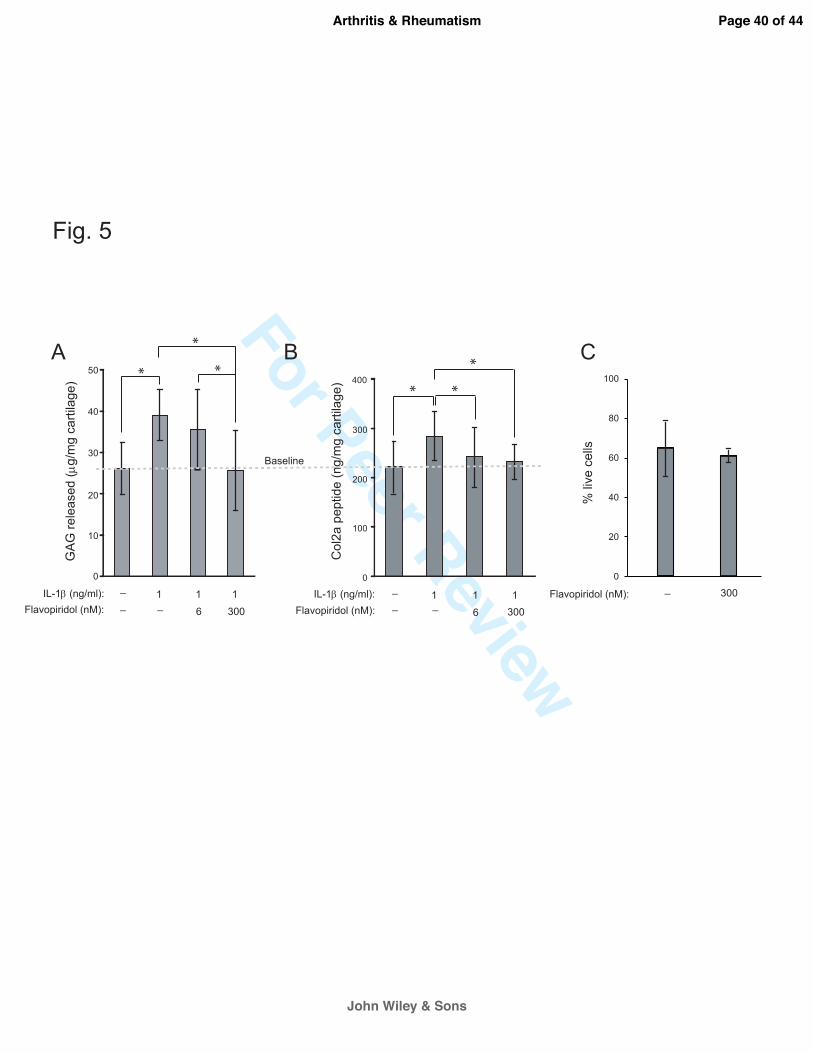
Figure 5. CDK9 inhibition protects cartilage from the catabolic effects of IL-1ß. (A) GAG
breakdown in cartilage explants. Human arthritic cartilage explants (3mm cubes) were treated
with 1 ng/ml IL-1ß and the indicated concentrations of Flavopiridol for 6 days (media change at
day 3). GAG released into the medium was measured by DMMB assays and normalized to the
Page 23 of 44
John Wiley & Sons
Arthritis & Rheumatism

For Peer Review
24
wet weight of the explants. Treatment with IL-1β alone caused cartilage degradation as indicated
by increased GAG release. In the presence of Flavopiridol, GAG release returned to baseline.
(B) Col2a degradation in cartilage explants. Human cartilage explants (n=5 donors) were treated
with 1 ng/ml IL-1ß and the indicated concentrations of Flavopiridol for 6 days (media change at
day 3). Cleaved Col2a peptides released into the medium was measured by C2C ELSIA and
normalized to the wet weight of the explants as described in the Methods. Treatment with IL-1β
alone caused cartilage degradation as indicated by increased Col2a peptides. In the presence of
Flavopiridol, Col2a peptides release returned to baseline. For all the above experiments, each
data point was the mean +/- standard deviation from five different donors. (*p<0.05). (C) Long-
term Flavopiridol treatment does not reduce chondrocyte viability. Bovine cartilage explants (6
mm full thickness disk) (n=3 different donors) were treated with 300 nM Flavopiridol for 6 days.
The explants were sliced in half and stained with LIVE/DEAD stain as described in the Methods.
The numbers of live and dead cells in three random fields were counted and the percentages of
live cells were calculated.
Page 24 of 44
John Wiley & Sons
Arthritis & Rheumatism

For Peer Review
25
References
1. Lotz MK, Kraus VB. New developments in osteoarthritis. Posttraumatic osteoarthritis:
pathogenesis and pharmacological treatment options. Arthritis Res Ther. 2007;12(3):211.
2. Goldring MB, Otero M, Tsuchimochi K, Ijiri K, Li Y. Defining the roles of inflammatory and
anabolic cytokines in cartilage metabolism. Ann Rheum Dis. 2008;67 Suppl 3:iii75-82.
3. Lohmander LS, Englund PM, Dahl LL, Roos EM. The long-term consequence of anterior
cruciate ligament and meniscus injuries: osteoarthritis. Am J Sports Med. 2007;35(10):1756-69.
4. Hargreaves DC, Horng T, Medzhitov R. Control of inducible gene expression by signal-
dependent transcriptional elongation. Cell. 2009;138(1):129-45.
5. Zippo A, Serafini R, Rocchigiani M, Pennacchini S, Krepelova A, Oliviero S. Histone
crosstalk between H3S10ph and H4K16ac generates a histone code that mediates transcription
elongation. Cell. 2009;138(6):1122-36.
6. Yamaguchi Y, Shibata H, Handa H. Transcription elongation factors DSIF and NELF:
promoter-proximal pausing and beyond. Biochimica et biophysica acta. 2013;1829(1):98-104.
7. Zhou Q, Yik JH. The Yin and Yang of P-TEFb regulation: implications for human
immunodeficiency virus gene expression and global control of cell growth and differentiation.
Microbiol Mol Biol Rev. 2006;70(3):646-59.
8. Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K, et al. Recruitment of P-TEFb for
stimulation of transcriptional elongation by the bromodomain protein Brd4. Molecular cell.
2005;19(4):535-45.
9. Krystof V, Baumli S, Furst R. Perspective of cyclin-dependent kinase 9 (CDK9) as a drug
target. Current pharmaceutical design. 2012;18(20):2883-90.
Page 25 of 44
John Wiley & Sons
Arthritis & Rheumatism

For Peer Review
26
10. Li H, Haudenschild DR, Posey KL, Hecht JT, Di Cesare PE, Yik JH. Comparative analysis
with collagen type II distinguishes cartilage oligomeric matrix protein as a primary TGFbeta-
responsive gene. Osteoarthritis Cartilage. 2011;19(10):1246-53.
11. Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition
of BET bromodomains. Nature. 2010;468(7327):1067-73.
12. Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, et al. A third-generation
lentivirus vector with a conditional packaging system. Journal of virology. 1998;72(11):8463-71.
13. Farndale RW, Buttle DJ, Barrett AJ. Improved quantitation and discrimination of sulphated
glycosaminoglycans by use of dimethylmethylene blue. Biochimica et biophysica acta.
1986;883(2):173-7.
14. Poole AR, Ionescu M, Fitzcharles MA, Billinghurst RC. The assessment of cartilage
degradation in vivo: development of an immunoassay for the measurement in body fluids of type
II collagen cleaved by collagenases. J Immunol Methods. 2004;294(1-2):145-53.
15. Fornier MN, Rathkopf D, Shah M, Patil S, O'Reilly E, Tse AN, et al. Phase I dose-finding
study of weekly docetaxel followed by flavopiridol for patients with advanced solid tumors.
Clinical cancer research : an official journal of the American Association for Cancer Research.
2007;13(19):5841-6.
16. Maier R, Bilbe G, Rediske J, Lotz M. Inducible nitric oxide synthase from human articular
chondrocytes: cDNA cloning and analysis of mRNA expression. Biochimica et biophysica acta.
1994;1208(1):145-50.
17. Ali S, Heathcote DA, Kroll SH, Jogalekar AS, Scheiper B, Patel H, et al. The development of
a selective cyclin-dependent kinase inhibitor that shows antitumor activity. Cancer research.
2009;69(15):6208-15.
Page 26 of 44
John Wiley & Sons
Arthritis & Rheumatism

For Peer Review
27
18. Heath EI, Bible K, Martell RE, Adelman DC, Lorusso PM. A phase 1 study of SNS-032
(formerly BMS-387032), a potent inhibitor of cyclin-dependent kinases 2, 7 and 9 administered
as a single oral dose and weekly infusion in patients with metastatic refractory solid tumors.
Investigational new drugs. 2008;26(1):59-65.
19. Kobayashi M, Squires GR, Mousa A, Tanzer M, Zukor DJ, Antoniou J, et al. Role of
interleukin-1 and tumor necrosis factor alpha in matrix degradation of human osteoarthritic
cartilage. Arthritis Rheum. 2005;52(1):128-35.
20. Wang M, Sampson ER, Jin H, Li J, Ke QH, Im HJ, et al. MMP13 is a critical target gene
during the progression of osteoarthritis. Arthritis Res Ther. 2013;15(1):R5.
21. Fiocco U, Sfriso P, Oliviero F, Roux-Lombard P, Scagliori E, Cozzi L, et al. Synovial
effusion and synovial fluid biomarkers in psoriatic arthritis to assess intraarticular tumor necrosis
factor-alpha blockade in the knee joint. Arthritis Res Ther. 2010;12(4):R148.
22. McNiff PA, Stewart C, Sullivan J, Showell HJ, Gabel CA. Synovial fluid from rheumatoid
arthritis patients contains sufficient levels of IL-1 beta and IL-6 to promote production of serum
amyloid A by Hep3B cells. Cytokine. 1995;7(2):209-19.
23. Deirmengian C, Hallab N, Tarabishy A, Della Valle C, Jacobs JJ, Lonner J, et al. Synovial
fluid biomarkers for periprosthetic infection. Clinical orthopaedics and related research.
2010;468(8):2017-23.
24. Hollander AP, Pidoux I, Reiner A, Rorabeck C, Bourne R, Poole AR. Damage to type II
collagen in aging and osteoarthritis starts at the articular surface, originates around chondrocytes,
and extends into the cartilage with progressive degeneration. J Clin Invest. 1995;96(6):2859-69.
Page 27 of 44
John Wiley & Sons
Arthritis & Rheumatism

For Peer Review
28
25. Attur M, Millman JS, Dave MN, Al-Mussawir HE, Patel J, Palmer G, et al. Glatiramer
acetate (GA), the immunomodulatory drug, inhibits inflammatory mediators and collagen
degradation in osteoarthritis (OA) cartilage. Osteoarthritis Cartilage. 2011;19(9):1158-64.
26. Attur MG, Dave M, Cipolletta C, Kang P, Goldring MB, Patel IR, et al. Reversal of autocrine
and paracrine effects of interleukin 1 (IL-1) in human arthritis by type II IL-1 decoy receptor.
Potential for pharmacological intervention. J Biol Chem. 2000;275(51):40307-15.
27. Ni W, Ji J, Dai Z, Papp A, Johnson AJ, Ahn S, et al. Flavopiridol pharmacogenetics: clinical
and functional evidence for the role of SLCO1B1/OATP1B1 in flavopiridol disposition. PLoS
One. 2010;5(11):e13792.
28. Wang K, Hampson P, Hazeldine J, Krystof V, Strnad M, Pechan P, et al. Cyclin-dependent
kinase 9 activity regulates neutrophil spontaneous apoptosis. PLoS One. 2012;7(1):e30128.
29. Schmerwitz UK, Sass G, Khandoga AG, Joore J, Mayer BA, Berberich N, et al. Flavopiridol
protects against inflammation by attenuating leukocyte-endothelial interaction via inhibition of
cyclin-dependent kinase 9. Arteriosclerosis, thrombosis, and vascular biology. 2011;31(2):280-8.
30. Wang LM, Ren DM. Flavopiridol, the first cyclin-dependent kinase inhibitor: recent
advances in combination chemotherapy. Mini Rev Med Chem. 2010;10(11):1058-70.
31. Sekine C, Sugihara T, Miyake S, Hirai H, Yoshida M, Miyasaka N, et al. Successful
treatment of animal models of rheumatoid arthritis with small-molecule cyclin-dependent kinase
inhibitors. J Immunol. 2008;180(3):1954-61.
32. Christiansen BA, Anderson MJ, Lee CA, Williams JC, Yik JH, Haudenschild DR.
Musculoskeletal changes following non-invasive knee injury using a novel mouse model of post-
traumatic osteoarthritis. Osteoarthritis Cartilage. 2012;20(7):773-82.
Page 28 of 44
John Wiley & Sons
Arthritis & Rheumatism

For Peer Review
29
33. Glasson SS, Blanchet TJ, Morris EA. The surgical destabilization of the medial meniscus
(DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthritis Cartilage.
2007;15(9):1061-9.
34. Furman BD, Strand J, Hembree WC, Ward BD, Guilak F, Olson SA. Joint degeneration
following closed intraarticular fracture in the mouse knee: a model of posttraumatic arthritis.
Journal of orthopaedic research : official publication of the Orthopaedic Research Society.
2007;25(5):578-92.
Page 29 of 44
John Wiley & Sons
Arthritis & Rheumatism

For Peer Review
Response to reviewers’ comments. Before we begin, we would like to thank the reviewers and editors for their time and constructive criticism, which helped us in obtaining much stronger evidence and convincing results to be submitted in the revised manuscript. Editor Comments to the Author: The reviewers raised a number of important limitations. A key issue is that all of the results presented to suggest a role for CDK9 rely on a single inhibitor. It is well known that most small molecule inhibitors, including the one used here, have off target effects. Additional experiments will be needed with other means of inhibiting CDK9 to confirm the findings as well as studies to show that in the culture system used here the inhibitor tested blocks CDK9 specifically. The other limitation is that the results are all from in vitro experiments without in vivo data needed for a complete study. Reviewer: 1 Comments to the Author Jasper et al have studied the role of CDK inhibitor on chondrocytes under inflammatory conditions. Primary chondrocyte cells were stimulated with IL-1 in the presence and absence of CDK9 inhibitor flavopiridol. Authors further demonstrated that CDK9 inhibitor suppressed many of NFkB dependent genes which are induced by IL-1 including MMPs, ADAMTS4 and iNOS and moderately induced ECM protein collagen and proteoglycan. Comments: 1) It is stated that IL-1 induced increased Pol II activity is via CDK9 activation no evidence have been provided. Is the CDK9 expression or activity is altered under inflammatory condition in chondrocytes. Furthermore, it must be clearly demonstrated the basal CDK9 expression in chondrocytes and in cartilage? We apologize for the lack of general background information on CDK9. Although it may be well-known in the transcription field, CDK9 is new to the cartilage biology field. CDK9 is a general transcription factor that is ubiquitously and constitutively expressed in all cell types (For expression in chondrocytes please see Western blot in new Figure 1F). To our knowledge, CDK9’s activity is not controlled at the mRNA nor protein expression level, but by revesible phosphorylation that leads to its dynamic association with inhibitor or activator complexes. The introduction has been expanded to provide more background on CDK9 and transcription, as well as the important role of Brd4 that recruits CDK9 to promoters during activation of inflammatory genes. In addition, several review articles have been cited in the manuscript for detail coverage of CDK9’s cellular function. 2) What is the effect of flavopiridol on chondrocyte survival and proliferation? In many studies it has been shown that flavopiridol induces apoptosis and it is not specific for CDK9? Flavopiridol inhibits many other CDKs activity at the concentration studied in this study. Now there are commercially available CDK9 specific inhibitors these can be explored. Authors should present the kinetics and different concentration effect of flavopiridol on chondrocyte toxicity and IL-1 dependent effects.
Page 30 of 44
John Wiley & Sons
Arthritis & Rheumatism

For Peer Review
Although Flavopiridol has potent anti-proliferation effects on rapidly dividing cells such as cancers, we observed that it has no noticeable effects on chondrocyte survival in bovine cartilage explants over a period of 6 days (new Figure 5C). In addition, Flavopiridol has no cytotoxic effects on monolayer chondrocyte culture over a period of 5 hours, which is the time frame tested for activation of primary response genes in this study. We have used other CDK inhibitors, siRNA against CDK9, and the Brd4 inhibitor JQ-1 (which is not a CDK inhibitor, but it inhibits Brd4, a downstream effector required for CDK9 activity, but not involved with other CDKs), to demonstrate the observation in this study is CDK9-dependent (new Figure 1). 3) What is the effect of flavopiridol on spontaneous production of mediator and matrix synthesis? Flavopiridol does not affect cartilage matrix gene expression (new Figure 4E) 4) Details of patient material should be elaborated (age, gender) and it is end stage disease cartilage used for the studies and how many patient materials were used? Chondrocytes and cartilage explants were isolated from a total of 15 donors, all with end stage OA. Five different donors were used in the experiments in Figure 5A&B. A minimum of 3 different donors were used in all other experiments. This was stated in the Method section. 5) Authors should also elaborate on PCR protocol, how much RNA was used for cDNA synthesis, is it SYBER green or Taqman probes used for analysis and how relative fold change was calculated. Details were provided in the revised manuscript in the Method section. 6) Statistical analysis, please explain what is n=3 represents is it three patient material or reaction performed from single patient material. N=3 means three different donors. This point was clarified in the revised manuscript. 7) Introduction – role of cdk in transcription can be expanded for readers to understand better the role of these kinases in transcription. The introduction has been expanded with citation to several recent review articles. 8) Figure 2 NFkB regulated genes were inhibited within 5h of drug treatment in the presence of IL-1, many of the NFkB family transcription factors. This brings question of specificity of this drug on gene expression? Specific involvement of CDK9 is addressed with additional CDK inhibitors, JQ-1, and siRNA against CDK9. (new Figure 1) 9) Flavopiridol is shown to be blocking most of the IL-1 induced and NFkB regulated gene expression. Some of these gene products should be quantified at protein level. Additionally, It is also important to show using NFkB luciferase reporter assay or inhibition of NFkB (translocation) by western analysis. In addition it is also important to know the effect of flavopiridol on chondrocyte matrix gene expression by PCR and proteins such as collagen 2, collagen 1 and aggrecan by immunoblot.
Page 31 of 44
John Wiley & Sons
Arthritis & Rheumatism

For Peer Review
We have shown MMP13 protein expression in new Figure 4D. The effects of Flavopiridol on CDK9-dependent activation of NFkB-mediated genes have been demonstrated by luciferase reporter and other assays by other groups, for example, from the following studies: -RelA Ser276 phosphorylation is required for activation of a subset of NF-kappaB-dependent genes by recruiting cyclin-dependent kinase 9/cyclin T1 complexes. Nowak DE, Tian B, Jamaluddin M, Boldogh I, Vergara LA, Choudhary S, Brasier AR. Mol Cell Biol. 2008 Jun;28(11):3623-38. doi: 10.1128/MCB.01152-07. Epub 2008 Mar 24. -Flavopiridol inhibits lipopolysaccharide-induced TNF-α production through inactivation of nuclear factor-κB and mitogen-activated protein kinases in the MyD88-dependent pathway. Haque A, Koide N, Iftakhar-E-Khuda I, Noman AS, Odkhuu E, Badamtseren B, Naiki Y, Komatsu T, Yoshida T, Yokochi T. Microbiol Immunol. 2011 Mar;55(3):160-7. doi: 10.1111/j.1348-0421.2010.00304.x. -Contribution of disruption of the nuclear factor-kappaB pathway to induction of apoptosis in human leukemia cells by histone deacetylase inhibitors and flavopiridol. Gao N, Dai Y, Rahmani M, Dent P, Grant S. Mol Pharmacol. 2004 Oct;66(4):956-63. Epub 2004 Jul 2. Finally, Flavopiridol has no effects on cartilage matrix gene expression (new Figure 4E) Reviewer: 2 Comments to the Author This study examined the effects of Flavopiridol, a cyclin-dependent kinase 9 (CDK9) inhibitor, on cartilage destruction. The authors assessed the effects of the inhibitor in inflammation-induced genes in human primary articular chondrocytes. In addition, the authors studied the effects of CDK9 inhibition in cartilage degradation, utilizing cartilage explants. Flavopiridol treatment blocked the expression of several genes induced by inflammatory cytokines, including genes involved in cartilage degradation, and it also blocked the destructive effects of IL-1b administration ex vivo, assessed measuring GAG release and Collagen 2 fragments in conditioned medium of explant cultures treated with the cytokine. The authors concluded that CDK9 inhibition prevents the induction of inflammation related genes and matrix degrading enzymes and that Flavopiridol treatment protects cartilage from the destructive, catabolic effects of inflammatory cytokines. Overall, this is a well conducted study and the findings are of interest. I have only a few comments that could be addressed in order to help to strengthen the work. 1. Figure 3 would benefit from the use of the three inflammatory cytokines utilized in Figure 1. All three inflammatory stimuli have been tested and data shown as new Figure 4.
Page 32 of 44
John Wiley & Sons
Arthritis & Rheumatism

For Peer Review
2. Time-course experiments (especially using earlier time-points) will add relevant information to the manuscript, especially since the authors deal with mechanism/s related with recruitment of CDK9 independent on chromatin remodeling events. Time-course experiments were included in the new Figure 1. 3. The authors mentioned that cell viability did not change with Flavopiridol treatment (stated as data not shown, pg.10). Perhaps they could indeed elaborate and include that data (along with data in monolayer cultures) in the manuscript since the compound could potentially lead to cytotoxicity or induce apoptosis, as shown in other cell types and settings. Hence, to show that the treatment does not impair viability is of relevance to their interpretations. Short-term cytotoxcity in monolayer chondrocytes, and long-term viability of chondrocyte in cartilage explants, were shown in new Figure 1D and 5C, respectively. 4. Related with the previous comment, while it is reasonable that the authors focused their work on inflammation-induced genes, paying especial attention to proteases or those genes with known relevant roles in cartilage destruction, it would be also important to determine whether the inhibitor affects the expression of cartilage-specific genes (eg. COMP, COL2, or ACAN) involved in anabolism or general homeostasis. Flavopiridol has no effects on the basal expression of cartilage matrix genes (Figure 4E), housekeeping genes (Figure 3), and genes not induced by IL-1b (Figure 3) 5. The authors stated in the Introduction that “injury-related” activation of innate inflammatory responses contributes to cartilage degradation. However, no injury-related model is used in this study. In that regard, perhaps the authors could elaborate in the use of the selected pro-inflammatory factors (IL-1b, TNFa and LPS) since injurious stimuli, including mechanical overload, may trigger the same signaling pathways than those activated by cytokines without necessarily relaying on the expression/release of cytokines by articular chondrocytes. This excellent point by the reviewer is elaborated in the introduction of the revised manuscript. 6. There are some spelling mistakes (e.g. “provids”, page 8). The manuscript is well written but perhaps could be fine-tuned. Spelling errors were fixed. Reviewer: 3 Comments to the Author The manuscript titled “Cyclin-dependent Kinase 9 Inhibition Protects Cartilage from the Catabolic Effects of Pro-Inflammatory Cytokines” by Yik et al addresses the effect of the CDK inhibitor, Flavopiridol, on the responses of articular chondrocytes to inflammatory stimuli. In this study, monolayer cultures of human articular chondrocytes were treated with IL-1�, TNF� or LPS, in the presence or absence of Flavopiridol. The effects of Flavopiridol on the induction of MMPs, iNOS and aggrecanase were assessed by QPCR.
Page 33 of 44
John Wiley & Sons
Arthritis & Rheumatism

For Peer Review
More global analyses of inflammatory mediator up-regulation were assessed by focused microarrays. In these assays, Flavopiridol significantly suppressed the induction of inflammatory genes. The effects of Flavopiridol on cartilage matrix degradation were assessed in a cartilage explant model, using IL-1� as the inflammatory stimulus and release of sGAGs and collagen cleavage peptides as the outcome measures. Flavopiridol significantly reduced sGAG and collagen peptide release in these experiments. The results of this study provide compelling evidence, within the limitations of the in vitro systems used, that Flavopiridol potently antagonizes the effects of inflammatory cytokines on articular chondrocytes and on cartilage matrix degradation, consistent with murine models of inflammatory arthritis. Given that these responses require, to a large extent, transcriptional activation, the outcomes are not unexpected. Accepting the compelling nature of the results, there are several concerns that need to be addressed, as follow: The major concern I have with the study is that the authors are representing Flavopiridol as a CDK9-specific inhibitor; this is implicit in the title. In fact, this agent has inhibitory effects on a wide range of CDKs and other kinases, particularly at the high dose used in these experiments. Further, several published studies demonstrate that the transcription-inhibitory effects of Flavopiridol are not restricted to primary response genes. In short, there are no data provided in the study that demonstrate specificity of action against CDK9 or, more importantly, that any transcriptional effects are restricted to primary response genes downstream of inflammatory cytokines…. these are merely the only genes looked at in the study. Ideally, the authors should use an independent and more specific method of CDK9 inhibition to corroborate the effects of Flavopiridol and also demonstrate that the transcriptional and consequent effects of Flavopiridol on articular chondrocytes are limited to those PRGs downstream of IL-1�/TNF�/LPS signaling (or not). Demonstrating a lack of any significant effect on representative chondrocytic genes would be reassuring. The concerns on inhibitor specificity and specific involvement of CDK9 were addressed (new Figure 1) using other CDK inhibitors, siRNA against CDK9, and lastly, with the Brd4 inhibitor JQ-1, which does not affect other CDKs besides CDK9. Moreover, Flavopiridol has no effects on anabolic genes and housekeeping genes as described above. More specific concerns are listed below: 1. In the Introduction and Discussion, there are several references to the “innate immune system”, the “innate immune response” and the “innate inflammatory response”. The innate immune system is generally differentiated from the adaptive or acquired immune system, but there is no innate inflammatory response per se. The terminology used to refer to these ‘systems’ and ‘responses’ should be clarified in the narrative. Although LPS is a strong stimulant for the innate immune system (being derived from
Page 34 of 44
John Wiley & Sons
Arthritis & Rheumatism

For Peer Review
bacteria) there is nothing inherently immunological about IL-1� or TNF�, nor is there much evidence to support that most cases of OA are immunologically based, innate or otherwise. We apologize for the poor choice of words. We have replaced the term “innate inflammatory/immune response” with “inflammatory response” in chondrocyte. 2. Experimentally, Flavopiridol was administered concurrently with the inflammatory cytokines, whereas, in a more clinically relevant context, Flavopiridol would more realistically be administered some time after the traumatic/inflammatory insult. Do the authors have any data on whether Flavopiridol is effective in mitigating chondrocytic “inflammatory responses” in a therapeutic, rather than prophylactic, situation? This excellent point was specifically addressed in the new Figure 1B, with results showing there is a 3-hour window of opportunity for the application of Flavopiridol after the initial treatment with inflammatory cytokines. 3. The dose of IL-1� in the cartilage explant study was only 1ng/ml; 10% of that used in the monolayer study. Not surprisingly, the catabolic effect of the Il-1� was pretty marginal; a less than 50% increase in sGAG release over 6 days and around 25% increase in collagen cleavage over the same interval. Accepting that the differences were statistically significant and did demonstrate efficacy following Flavopiridol administration, why was such a low dose of Il-1� used in these explant experiments? To more closely mimic the conditions of inflamed joints in the longer term study, the IL-1b dose was reduced to 1 ng/ml, a level more in line with that detected in the synovial fluids of inflamed joints. References were included in the revised manuscripts (ref 21-23). 4. In figure 1, accepting that the data in Fig 1B compellingly show the inhibitory effects of Flavopiridol on iNOS induction, it would be informative to know the relative potencies of IL-1�, LPS and TNF� in inducing iNOS. This information is lost in the standardization presented in the first column. We observed that when given at 10 ng/ml, LPS is the least potent in inducing iNOS and other inflammatory-related genes, while IL-1 and TNF is more potent, although the response of individual genes to different stimuli vary widely. This can be seen in the new Figure 4. For example, ADAMTS4 was induced ~60-fold by IL-1, ~15-fold by TNF, and ~3-fold by LPS.
Page 35 of 44
John Wiley & Sons
Arthritis & Rheumatism
For Peer Review
Fig. 1
Live cells/well
Control
Flavopiridol (300 nM)
tin
u e
cn
ec
sn
er
oul
F
)D
P6
G lat
ot/el
bul
os(
F
1000 5000 10000
0
0.04
0.08
0.12
0.16
IL-1E (ng/ml):
Flavopiridol (nM):
10 10
300
10 10
1000
100
10
1
0
303
Fo
ld-c
ha
ng
e in
iN
OS
mR
NA
300
*
--
- -
*
C
10 10 10 10 -
-Time Flavopiridol
added (300 nM):
*
100
200
300
0
Fo
ld-c
ha
ng
e in
iN
OS
mR
NA
B
IL-1E (ng/ml): 10 10 10
1000
100
10
1
0
-
Fo
ld-c
ha
ng
e in
iN
OS
mR
NA
Flavopiridol (300 nM)
Control
**
IL-1E duration (h): 1 3 5-
A
IL-1E�(ng/ml):
*
No
delay
1h
delay
3h
delay-
BS-181 SNS-032 JQ-1
% i
NO
S m
RN
A i
nd
uc
tio
n
IL-1E (ng/ml): 10 10 1010
Inhibitors:
Targets:
CDK7
Brd4
Flavopiridol
10
- -
CDK7CDK9
CDK2CDK4
CDK6CDK7CDK9
CDK1
CDK2
D
150
100
50
0
-
* * *
0.1X 1X 10X IC 50
No inhibitor
200
IL-1E (10 ng/ml):
siRNA:
150
100
50
0
200
% i
NO
S m
RN
A i
nd
uc
tio
n
- + - + - +
GFP CDK9-
CDK9
GAPDH
CDK9
GFP
E
*
*
siRNA:
- -----
--
--
-
---
- - -
Page 36 of 44
John Wiley & Sons
Arthritis & Rheumatism
For Peer Review0
20
40
60
80
100
%inductionofiNOS
+ Flavopiridol
IL-1E, LPS, or TNFD
IL-1E LPS TNFD
Maximalinduction
Fig. 2
A B
CDK9
TNFReceptor 1
Toll-LikeReceptor 4
IL-1Receptor
IL-1 TNF�
LPS
CellSurface
Nucleus
TRAF6 TRAF2
MAPKIkB
NFkB
NFkB
MAP4K5
c-Fos/Jun
AP-1RNAPol II CDK9CDK9
TNFReceptor 1
Toll-LikeReceptor 4
IL-1Receptor
IL-1E TNFD
LPS
CellSurface
Nucleus
primary inflammatoryresponse genes
CDK9 inhibitorFlavopiridol
TRAF6 TRAF2
MAPKIkB
NFkBIkB
NFkB
NFkBNFkB
MAP4K5
c-Fos/Junc-Fos/Jun
AP-1AP-1RNAPol II
Brd4
Page 37 of 44
John Wiley & Sons
Arthritis & Rheumatism
For Peer Review
Figure 3
IFNB1CD80
NFKB2PLAUIFNGXIAP
C3NCOA3
EGR2FASLG
C4ALTB
PDGFBCSF2RB
IRF1
MaxMin Ave
Magnitude of gene expression
TRAF2CDKN1A
IL15F3
MYD88ADMFAS
MMP9TNFRSF1B
CCL5RELB
VCAM1CSF2
1 2 3 1 2 3
B2MRPL13AGAPDHHPRT1
ACTB1 2 3
ControlIL-1
bIL-1
b+Flav
o.
repressed by Flavopiridol
IL-1b-induced genes (>1.5 fold within 5 hrs, n=3 donors)
genes not induced by IL-1b
STAT5BAKT1
IL4
STAT3
TP53AGT
F8
NQO1BCL2L1CCND1
MYC
ALDH3A2MAP2K6
MITFGADD45B
SELP
IL1R2
1 2 3 not repressed by Flavopiridol
house keeping genes
CXCL2
CSF3CXCL10
IL1A
IL1BIL16
SOD2CD83EGFR
CFBIL1RN
TNFTNFSF10
NFKB1SELE
CXCL1BCL2A1
IL2
BIRC3
ICAM1NFKBIA
PTGS2
RELANR4A2
CSF1IL8
RELCCL2CD40
CXCL9
BIRC2
ControlIL-1
bIL-1
b+Flav
o.
ControlIL-1
bIL-1
b+Flav
o.
IL2RA
IL12BSNAP25
CCL22
INSCCR5
LTA
STAT1
1 2 3
ControlIL-1
bIL-1
b+Flav
o.
Page 38 of 44
John Wiley & Sons
Arthritis & Rheumatism
For Peer Review
Fig. 4
MMP1 MMP3 MMP9 MMP13 ADAMTS4 ADAMTS5
Fold
-cha
nge
in m
RN
A ex
pres
sion
0
20
60
80Control
IL-1E
IL-1E + Flavopiridol
MMP1 MMP3 MMP9 MMP13 ADAMTS4 ADAMTS5
Fold
-cha
nge
in m
RN
A ex
pres
sion
0
20
40
60
80Control
LPS
LPS + Flavopiridol
MMP1 MMP3 MMP9 MMP13 ADAMTS4 ADAMTS5
Fold
-cha
nge
in m
RN
A ex
pres
sion
0
20
40
60
80Control
TNFD
TNFD + Flavopiridol
40
A B
C D
2
4
0
IL-1E (10 ng/ml):Flavopiridol (300 nM):
Anabolic genes: Aggrecan COMP Col2a1
+ +-+ +--
-
Fold
-cha
nge
in m
RN
A
+ +-+ +--
- + +-+ +--
-
*
**
**
*
*
* **
GADPH
MMP13
Donor 1 Donor 2 Donor 3
IL-1b: - + + - + + - + +Flavopiridol: - + - + - +- - -
E
Page 39 of 44
John Wiley & Sons
Arthritis & Rheumatism
For Peer Review
0
10
20
30
40
50
GA
G re
leas
ed (P
g/m
g ca
rtila
ge)
IL-1E (ng/ml):Flavopiridol (nM):
__
1 1 1
6 300_
A B
_ 3000
20
40
60
80
100
Flavopiridol (nM):
C
% li
ve c
ells
Fig. 5
Baseline
*
**
200
300
400
0
100
IL-1E (ng/ml):Flavopiridol (nM):
__
1 1 1
6 300_
Col
2a p
eptid
e (n
g/m
g ca
rtila
ge)
*
* *
Page 40 of 44
John Wiley & Sons
Arthritis & Rheumatism
For Peer Review
Donor 1 Donor 2Wells Untreated IL-1b IL-1b + Flavo cotreatUntreated IL-1bA01 ADM 20.05 17.94 24.81 21.46 20.22A02 AGT 24.83 24.64 25.42 28.6 28.83A03 AKT1 23.58 24.36 25.68 24.46 24.96A04 ALDH3A2 23.6 24.79 26.23 23.82 25.4A05 BCL2A1 24.15 19.54 24.89 27.72 21.5A06 BCL2L1 28.35 29.62 31.25 27.76 28.78A07 BIRC2 23.4 22.93 24.28 25.67 24.32A08 BIRC3 27.61 23.36 27.82 31.19 25.33A09 C3 27.43 26 28.43 36.32 25.61A10 C4A 26.33 26.36 27.48 28.77 28.89A11 CCL2 22.21 17.13 23.95 23.37 18.4A12 CCL22 35 33.34 35 36.13 34.98B01 CCL5 32.21 28.54 34.91 32.17 27.5B02 CCND1 22.71 24.46 26.9 23.81 23.89B03 CCR5 35 35 35 33.37 30.54B04 CD40 29.65 28.93 30.7 30.56 28.68B05 CD80 33.11 32.54 34.52 28.51 25.74B06 CD83 30.97 26.55 30.83 29.6 24.62B07 CDKN1A 22.52 21.28 25.47 21.88 20.92B08 CFB 22.95 21.45 24.02 25.69 21.96B09 CSF1 25.16 21.32 27.45 26.37 21.83B10 CSF2 35 23.92 33.08 34.13 20.12B11 CSF2RB 35 35 35 26.59 22.46B12 CSF3 35 23.54 35 37.18 20.64C01 CXCL1 26.94 17.81 23.23 31.98 20.21C02 CXCL10 35 23.67 35 34.2 24.52C03 CXCL2 28.92 16.96 25.61 32.24 18.94C04 CXCL9 32.6 30.32 35 35.3 29.3C05 EGFR 25.42 23.74 26.89 27.35 25.7C06 EGR2 31.57 33.18 33.63 31.4 30.9C07 F3 27.33 25.98 30.87 23.13 21.93C08 F8 29.23 30.1 30.25 30.52 29.82C09 FAS 26.75 25.83 29.29 25.78 24.65C10 FASLG 35 35 35 29.76 26.87C11 GADD45B 22.7 26.96 29.64 21.93 24.69C12 ICAM1 25.18 19.32 25.72 28.47 19.38D01 IFNB1 35 34.23 33.6 34.9 26.75D02 IFNG 35 32.61 35 38.6 31.45D03 IL12B 35 35 35 36.74 30.79D04 IL15 28.66 27.24 32.8 29.28 26.16D05 IL1A 33.26 28.39 35 28.76 26.12D06 IL1B 35 23.78 35 32.96 22.23D07 IL1R2 29.31 29.77 32.39 31.36 32.14D08 IL1RN 33.37 26.92 31.89 34.67 22.61D09 IL2 35 32.75 35 34.95 28.79D10 IL2RA 35 35 32.11 35.6 32.99D11 IL4 35 35 35 35 35D12 IL6 23.87 15.48 23.97 28.36 17.74E01 IL8 24.73 15.5 21.22 32.11 18.35
gene symbols
Page 41 of 44
John Wiley & Sons
Arthritis & Rheumatism
For Peer Review
E02 INS 35 35 35 33.98 23.19E03 IRF1 29.2 26.17 27.35 28.68 25.7E04 LTA 33.79 32.3 32.52 35.46 32.71E05 LTB 35 30.65 32.42 34.6 30.1E06 MAP2K6 30.23 33.64 32.93 30.49 32.83E07 MITF 26.31 27.47 29.97 25 27.1E08 MMP9 31.02 27.75 33.28 31.33 29.5E09 MYC 26.35 28.07 31.41 28.64 28.52E10 MYD88 26.61 27.25 28.5 28.13 27.38E11 NCOA3 27.21 27.5 28.63 27.74 27.39E12 NFKB1 23.48 19.46 22.92 23.72 20.15F01 NFKB2 29.73 29.2 30.93 29.83 28.8F02 NFKBIA 24.05 19.38 24.64 23.37 19.57F03 NQO1 22.22 22.7 24.81 20.64 21.22F04 NR4A2 30 27.58 31.4 28.98 25.83F05 PDGFB 35 34.49 35 38.21 25.31F06 PLAU 27.13 26.98 28.86 23.16 22.12F07 PTGS2 26.79 19.58 26.35 21.84 17.96F08 REL 30.76 28.22 32.32 30.58 26.9F09 RELA 26.01 25.07 28.32 24.98 23.47F10 RELB 31.75 27.9 33.3 29.68 25.73F11 SELE 35 26.59 30.64 31.43 22.55F12 SELP 27.45 27.93 29.51 24.3 28.63G01 SNAP25 35 32.67 35 35.03 33.68G02 SOD2 21.17 17.6 21.41 22.27 16.99G03 STAT1 25.8 25.69 26.2 23.67 23.54G04 STAT3 24.18 24.87 24.78 23.42 23G05 STAT5B 25.91 25.33 28.63 24.11 24.65G06 TNF 33.18 23.27 26.62 38.06 19.9G07 TNFRSF1B 30.1 27.29 34.01 31.52 28.87G08 TNFSF10 30.26 28.98 30.35 30.88 26.92G09 TP53 27.31 26.97 27.36 23.22 23.92G10 TRAF2 28.44 28.16 32.7 27.16 26.71G11 VCAM1 22.1 17.26 24.45 22.78 17.66G12 XIAP 27.68 27.58 29.3 24.66 23.93H01 B2M 21 21.32 22.32 19.92 19.88H02 HPRT1 24.81 25.03 27.39 22.33 22.65H03 RPL13A 20.66 21.34 22.51 19.15 19.79H04 GAPDH 16.7 17.23 19.25 17.31 17.69H05 ACTB 18 19.01 20.81 18.28 18.41H06 HGDC 35 35 35 35 35H07 RTC 22.92 23.23 23.63 23.72 22.92H08 RTC 22.9 23.16 23.65 23.97 22.81H09 RTC 22.95 23.15 23.68 23.93 22.61H10 PPC 19.15 19.63 19.32 18.78 19.21H11 PPC 19.31 19.48 19.49 18.78 19.23H12 PPC 19.17 19.44 19.56 18.85 19.28
Page 42 of 44
John Wiley & Sons
Arthritis & Rheumatism
For Peer Review
Donor 3IL-1b + Flavo cotreatUntreated IL-1b IL-1b + Flavo
25.26 23.62 20.87 29.428.63 26.72 26.59 27.7426.08 24.5 23.76 25.2125.13 24 24.56 25.2724.36 27.33 21.39 29.3130.41 29.23 28.13 29.9825.67 24.98 23.28 26.5530.12 30.64 23.76 31.59
29.1 30.78 27.9 30.2930.48 28.69 27.71 28.823.38 24 17.76 24.5735.54 35 34.73 34.5631.94 32.25 26.29 34.4824.57 25.36 24.88 26.4332.19 37.35 34.49 35.8132.45 29.39 27.24 30.4129.67 33.87 33.63 34
28.4 31.5 27.55 32.522.86 22.59 21.4 23.67
26.1 25.81 23.08 26.9626.4 26.35 21.83 26.54
25.19 35 20.36 32.5126.72 35 33.22 3530.05 35 23.45 3524.28 30.75 19.65 27.3636.25 33.98 23.65 35
25.6 34.65 19.47 30.3733.79 33.52 26.67 3528.46 26.16 23.88 26.7130.11 31.69 28.96 34.0224.02 27.56 24.75 28.62
29.8 28.17 27.71 28.5426.89 26.36 24.95 28.3532.14 35 35 3527.14 23.78 27.49 30.8725.61 25.76 18.93 26.6530.44 35 28.66 3538.56 35 31.59 3530.93 35 35 3531.16 32.37 28.14 34.530.33 32.54 27.72 33.2930.61 39.31 22.69 32.9430.36 31.14 32.58 32.6727.32 33.23 27.38 32.82
35 35 27.81 3531.89 35 32.84 33.1
35 35 35 3526.39 28.76 16.9 27.8423.06 28.88 16.33 26.18
Page 43 of 44
John Wiley & Sons
Arthritis & Rheumatism
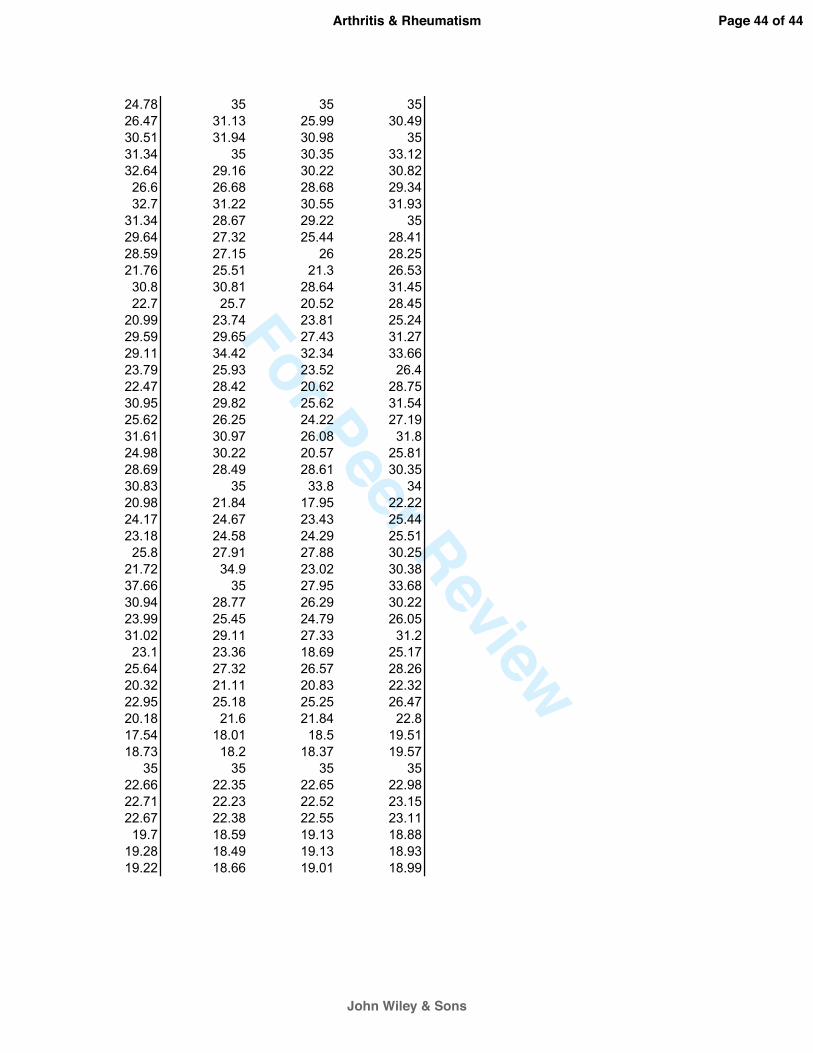
For Peer Review
24.78 35 35 3526.47 31.13 25.99 30.4930.51 31.94 30.98 3531.34 35 30.35 33.1232.64 29.16 30.22 30.82
26.6 26.68 28.68 29.3432.7 31.22 30.55 31.93
31.34 28.67 29.22 3529.64 27.32 25.44 28.4128.59 27.15 26 28.2521.76 25.51 21.3 26.53
30.8 30.81 28.64 31.4522.7 25.7 20.52 28.45
20.99 23.74 23.81 25.2429.59 29.65 27.43 31.2729.11 34.42 32.34 33.6623.79 25.93 23.52 26.422.47 28.42 20.62 28.7530.95 29.82 25.62 31.5425.62 26.25 24.22 27.1931.61 30.97 26.08 31.824.98 30.22 20.57 25.8128.69 28.49 28.61 30.3530.83 35 33.8 3420.98 21.84 17.95 22.2224.17 24.67 23.43 25.4423.18 24.58 24.29 25.51
25.8 27.91 27.88 30.2521.72 34.9 23.02 30.3837.66 35 27.95 33.6830.94 28.77 26.29 30.2223.99 25.45 24.79 26.0531.02 29.11 27.33 31.2
23.1 23.36 18.69 25.1725.64 27.32 26.57 28.2620.32 21.11 20.83 22.3222.95 25.18 25.25 26.4720.18 21.6 21.84 22.817.54 18.01 18.5 19.5118.73 18.2 18.37 19.57
35 35 35 3522.66 22.35 22.65 22.9822.71 22.23 22.52 23.1522.67 22.38 22.55 23.11
19.7 18.59 19.13 18.8819.28 18.49 19.13 18.9319.22 18.66 19.01 18.99
Page 44 of 44
John Wiley & Sons
Arthritis & Rheumatism
Copyright © 2022 FDOKUMEN




















