
Lymphocide: cytokines and the control of lymphoid homeostasis
14
© 2002 Nature Publishing Group REVIEWS The peripheral T-cell pool has a constant size, despite the daily emergence of more thymic emigrants than can survive in the periphery. The mouse thymus releases 10 6 cells per day into a total naive T-cell pool that contains approximately 8 × 10 7 –10 × 10 7 cells; for example, there are 2 × 10 7 naive CD8 + T cells 1,2 . This means that approximately 10 6 cells must die or differentiate every day, independent of their age. In addition to thymic output, peripheral T-cell populations expand through proliferation, with a single cell having the potential to produce 10 15 progeny 3 . Memory T cells cycle regularly. By contrast, naive T cells are relatively quiescent, but when injected into an immunodepleted host, they pro- liferate rapidly 4 . However, despite all of this potential for growth, the size of the T-cell population is stable, and the same is true for the mature B-cell population. About 2 × 10 7 B cells are released per day from the bone mar- row of young mice, which is far in excess of peripheral demand, as a functional B-cell pool can be regenerated in mice with 30% of the normal number of pre-B cells 5 . So, as for T cells, precursor production does not limit the size of the B-cell population; rather, size is restricted by a balance between proliferation and death in the periphery (FIG. 1). The regulation of lymphocyte populations involves the principle of distinct independent ‘niches’,which supply survival signals specific for a particular cellular subset (reviewed in REF. 5). The stable maintenance of one lymphocyte population in the absence of another indicates the existence of these niches. For example, the mature B-cell niche is independent of the T-cell niche, because the number of mature B cells in T-cell-depleted mice is normal. The same is true for the number of T cells in B-cell-depleted mice 5 . Similar niches are found for T-cell subsets, such as αβ T cells compared with γδ T cells 1,5 , whereas the niches for CD4 + and CD8 + T cells seem to overlap, with the loss of either subset being partially compensated for by the remaining sub- set 1,6 . Also, there are independent niches for naive and memory T cells, as the loss of naive T cells does not result in an increased number of memory T cells 7 . It seems reasonable to surmise that the type of survival signal, which is unique to a given niche, determines the size and stability of a lymphocyte population. The lymphotrophin hypothesis Hormones, antigen receptors, regulatory cells and, in particular, cytokines are the principal signals for main- taining lymphocyte homeostasis (BOX 1). The best- characterized trophic cytokines for lymphocytes are interleukin-7 (IL-7) and IL-15, which are essential for the survival of several lymphoid subsets 8–13 and are products LYMPHOCIDE: CYTOKINES AND THE CONTROL OF LYMPHOID HOMEOSTASIS Annette R. Khaled and Scott K. Durum In a human, about 10 11 excess peripheral lymphocytes die every day. This death process maintains a constant lymphocyte population size in the face of a continuous influx of new lymphocytes and the homeostatic proliferation of old ones. Death is triggered when a lymphocyte fails to acquire signals from survival factors, the availability of which, therefore, determines the size of the pool of lymphocytes. A lymphocyte acquires survival signals through receptors for cytokines, antigens, hormones and probably other extracellular factors. Here, we discuss current concepts of the intracellular signalling pathways for survival versus death that establish cytokine-regulated lymphocyte homeostasis. NATURE REVIEWS | IMMUNOLOGY VOLUME 2 | NOVEMBER 2002 | 817 Laboratory of Molecular Immunoregulation, Center for Cancer Research, National Cancer Institute, Frederick, Maryland 21702-1201, USA. Correspondence to S.K.D. e-mail: durums@mail. ncifcrf.gov doi:10.1038/nri931
Transcript of Lymphocide: cytokines and the control of lymphoid homeostasis

© 2002 Nature Publishing Group
REVIEWS
The peripheral T-cell pool has a constant size, despitethe daily emergence of more thymic emigrants than cansurvive in the periphery. The mouse thymus releases 106
cells per day into a total naive T-cell pool that containsapproximately 8 × 107–10 × 107 cells; for example, thereare 2 × 107 naive CD8+ T cells1,2. This means thatapproximately 106 cells must die or differentiate everyday, independent of their age. In addition to thymicoutput, peripheral T-cell populations expand throughproliferation, with a single cell having the potential toproduce 1015 progeny3. Memory T cells cycle regularly.By contrast, naive T cells are relatively quiescent, butwhen injected into an immunodepleted host, they pro-liferate rapidly4. However, despite all of this potential forgrowth, the size of the T-cell population is stable, andthe same is true for the mature B-cell population. About2 × 107 B cells are released per day from the bone mar-row of young mice, which is far in excess of peripheraldemand, as a functional B-cell pool can be regeneratedin mice with 30% of the normal number of pre-B cells5.So, as for T cells, precursor production does not limitthe size of the B-cell population; rather, size is restrictedby a balance between proliferation and death in theperiphery (FIG. 1).
The regulation of lymphocyte populations involvesthe principle of distinct independent ‘niches’, which
supply survival signals specific for a particular cellularsubset (reviewed in REF. 5). The stable maintenance ofone lymphocyte population in the absence of anotherindicates the existence of these niches. For example, themature B-cell niche is independent of the T-cell niche,because the number of mature B cells in T-cell-depletedmice is normal. The same is true for the number ofT cells in B-cell-depleted mice5. Similar niches arefound for T-cell subsets, such as αβ T cells comparedwith γδ T cells1,5, whereas the niches for CD4+ and CD8+
T cells seem to overlap, with the loss of either subsetbeing partially compensated for by the remaining sub-set1,6. Also, there are independent niches for naive andmemory T cells, as the loss of naive T cells does notresult in an increased number of memory T cells7. Itseems reasonable to surmise that the type of survivalsignal, which is unique to a given niche, determines thesize and stability of a lymphocyte population.
The lymphotrophin hypothesisHormones, antigen receptors, regulatory cells and, inparticular, cytokines are the principal signals for main-taining lymphocyte homeostasis (BOX 1). The best-characterized trophic cytokines for lymphocytes areinterleukin-7 (IL-7) and IL-15, which are essential for thesurvival of several lymphoid subsets8–13 and are products
LYMPHOCIDE: CYTOKINES AND THE CONTROL OF LYMPHOIDHOMEOSTASISAnnette R. Khaledand Scott K. Durum
In a human, about 1011 excess peripheral lymphocytes die every day. This death processmaintains a constant lymphocyte population size in the face of a continuous influx of newlymphocytes and the homeostatic proliferation of old ones. Death is triggered when alymphocyte fails to acquire signals from survival factors, the availability of which, therefore,determines the size of the pool of lymphocytes. A lymphocyte acquires survival signals throughreceptors for cytokines, antigens, hormones and probably other extracellular factors. Here, wediscuss current concepts of the intracellular signalling pathways for survival versus death thatestablish cytokine-regulated lymphocyte homeostasis.
NATURE REVIEWS | IMMUNOLOGY VOLUME 2 | NOVEMBER 2002 | 817
Laboratory of MolecularImmunoregulation,Center for Cancer Research,National Cancer Institute,Frederick, Maryland 21702-1201, USA.Correspondence to S.K.D.e-mail: [email protected]:10.1038/nri931

© 2002 Nature Publishing Group
5,6-CARBOXYFLUORESCEIN
DIACETATE SUCCINIMIDYL ESTER
(CFSE). A stable green-fluorescent dye that can be usedto label populations of cellshomogeneously. When a celldivides, the fluorescenceintensity decreases by 50%, andthis allows cells that have divideda specific number of times to bevisualized by flow cytometry.This reagent can measure ~7–10cell divisions successfully.
γC CYTOKINES
Cytokines with receptors thatshare the common cytokine-receptor γ-chain (CD132). Thesecytokines are all short-chain,four-helical bundles, and theyinclude IL-2, IL-4, IL-7, IL-9,IL-15 and IL-21.
REACTIVE OXYGEN SPECIES
(ROS). Oxygen radicals that areproduced by the mitochondrialrespiratory chain. In excess, theycan cause intracellular andmitochondrial damage, whichpromotes cell death.
SMAD
(Sma and Mad proteins inCaenorhabditis elegans andDrosophila, respectively). SMADproteins are downstreameffectors of the TGF-β signallingcascade. These proteins transducesignals from the cell membraneto the nucleus, thereby initiatingthe transcription of target genes.
818 | NOVEMBER 2002 | VOLUME 2 www.nature.com/reviews/immunol
R E V I E W S
Distinct death pathways: FAS and TGF-β. There aremany paths to ‘lymphocide’ — the best understoodbeing that involving the TNF-family member FAS(CD95). This pathway is triggered during the elimina-tion of activated T cells20. However, FAS is not animportant homeostatic regulator for most lymphocytepopulations. Another means to delete cells at the endof an immune response, independent of FAS, involvesthe production of REACTIVE OXYGEN SPECIES (ROS) — whichinitiate cell death — by antigen-activated T cells21.However, these apoptotic inducers for activated cellsare clearly distinct from the intrinsic death pathwaythat maintains lymphoid homeostasis through trophicfactors (FIG. 3).
Transforming growth factor-β (TGF-β) is a complexregulator of immune responses. In addition to inhibit-ing the differentiation of helper T cells, TGF-β-mediatedapoptosis has a role in B- and T-cell homeostasis by sig-nalling through SMAD proteins. These proteins translo-cate to the nucleus and activate or repress transcription,thereby controlling the levels of pro-apoptotic and anti-apoptotic proteins22. TGF-β1-deficient mice have severe,multi-organ inflammation, which indicates a role forthis protein in the inhibition of self-reactive T cells23.Therefore, as for FAS-mediated apoptosis, TGF-β func-tions to restore normal T-cell numbers after antigen-induced activation, an activity that is distinct from thatof trophic cytokines; but, it is also essential for themaintenance of lymphocyte population size.
Intracellular signalling domains of receptors. The recep-tors for the two cytokines that are known to be crucialfor lymphoid homeostasis, IL-7 and IL-15, share thecommon cytokine-receptor γ-chain (γc), which is asso-ciated with Janus kinase 3 (JAK3), a kinase that is likelyto be involved in lymphocyte homeostasis (FIG. 4). Jak3−/−
mice have a similar thymic deficiency to mouse knock-outs of the genes encoding IL-7, IL-7 receptor α-chain(IL-7Rα) or γc, but they also accumulate excess memoryCD4+ T cells in the periphery24.
The second receptor chains of the γc cytokine recep-tors can generate different signalling pathways. The IL-2receptor (comprised of IL-2Rα and γc) induces T-cellproliferation and survival, but also sensitizes these cellsto FAS-mediated ACTIVATION-INDUCED CELL DEATH (AICD).The proliferative and FAS ligand (FASL)-susceptibilitypathways were shown to be controlled by a region of theIL-2R α-chain that binds signal transducer and activatorof transcription 5 (STAT5); survival, by contrast, is controlled by a site that binds SHC (Src-homology-2-domain-containing transforming protein), which acti-vates a downstream substrate of phosphatidylinositol 3-kinase (PI3K), AKT, and leads to synthesis of the anti-apoptotic protein BCL-2 (REF. 25).
The IL-7 receptor (comprised of IL-7Rα and γc) alsoactivates AKT (W.Q. Li, A.R.K. and S.K.D., unpublishedobservations), the general role of which in cell survival isdiscussed. The proposed direct binding site for PI3K(the upstream activator of AKT) in the cytoplasmic tailof the IL-7R α-chain is Tyr449 (REF. 26), which is requiredfor the IL-7-mediated survival signal in a thymocyte cell
of non-lymphoid cells14,15. During an immune response,lymphocytes themselves produce other γC CYTOKINES (suchas IL-2, IL-4, IL-9 and IL-21), which might account forthe temporary enlargement of the lymphocyte popula-tion during an immune response. Other lymphocytes,such as mature B cells, use different trophic signals; forexample, B-cell-activating factor (BAFF), a member ofthe tumour-necrosis factor (TNF) family, promotes B-cell survival16. In addition to cytokines, maintenancesignals might be transduced through the B-cell receptor(BCR) or T-cell receptor (TCR), promoting cell viabilityin addition to clonal expansion17,18. By contrast, the co-stimulation that is required for lymphocyte activation— for example, through the interaction of CD28 withB7 molecules — is not involved in lymphocyte homeo-stasis19. Together, these signals act to expand the lym-phocyte pool, and their availability is likely to be thelimiting factor that restricts population size (FIG. 2).Treatments or genetic manipulations of mice that targetcytokines or components of their signalling pathwayshave produced animals with either lymphopaenia orlymphocytosis, the phenotypes of which are describedin TABLE 1 and TABLE 2, respectively.
Cytokines regulate death during homeostasisSignals transduced through cytokines and their specificreceptors activate diverse pathways that promote sur-vival and inhibit cell death. Cytokine signalling throughtyrosine kinases can lead to the transcription of anti-apoptotic factors. Serine/threonine kinases induce pro-survival signalling cascades and promote cellularmetabolism. These processes are essential for homeo-stasis, but their complex mechanisms are only beginningto be defined.
JustrightDepleted
Over-filled
Div
ide
Die
Div
ide
Die
Div
ide
Die
Lymphoidcompartment
Homeostaticcorrection
Figure 1 | Forces that maintain lymphocyte homeostasis. From primary lymphoid organsand through peripheral cell division, the production of lymphocytes occurs continuously.However, except for during an immune response, the total number of lymphocytes remains fixed,a phenomenon that is known as ‘lymphoid homeostasis’. As shown, a depleted lymphocytecompartment supports more proliferation and less cell death, as would occur in AIDS patientstreated with anti-retrovirals or in an immunodepleted mouse receiving donor T cells. Conversely,an ‘over-filled’ population would not support the survival of transferred T cells. Normally, thenumber of lymphocytes is maintained at a level that is ‘just right’ by a balance of proliferation and death. The elimination of excess lymphocytes has been shown through loss of cell labelling(such as by 5,6-CARBOXYFLUORESCEIN DIACETATE SUCCINIMIDYL ESTER, CFSE; or bromodeoxyuridine,BrdU) and through genetic manipulations in mice10,11. So, in an adult mouse, it is estimated that2.5 × 107–10 × 107 lymphocytes die each day from ‘trophic’ deprivation. It is this apoptoticprocess, triggered by the loss of cytokine-mediated survival signals, that establishes the size limit of a lymphocyte population.

© 2002 Nature Publishing GroupNATURE REVIEWS | IMMUNOLOGY VOLUME 2 | NOVEMBER 2002 | 819
R E V I E W S
could have a homeostatic role30, because these micehave an increased number of peripheral CD4+ T cells(which eventually transform). However, the accumu-lating CD4+ T cells have an activated phenotype andthe mice have autoimmune manifestations, which ismore consistent with a role for PTEN in repressing auto-immunity than in homeostasis. Nevertheless, in vitro,these PTEN-deficient T cells showed considerable pro-tection from cell death caused by IL-2 withdrawal,which indicates that PTEN has potential effects on theIL-7 and IL-15 homeostatic pathways.
In contrast to the expansion of activated T-cell pop-ulations in PTEN-deficient mice, targeted deletion inmice of p85α, the regulatory subunit of PI3Kα, did notreduce the number of T cells, but instead resulted in areduced number of peripheral mature B cells anddecreased B-cell proliferative responses, which indicatesa role for p85α in homeostatic survival in the B-cell lin-eage31. Mice with a deletion of p110, the catalyticdomain of PI3Kγ, had about half the normal number ofthymocytes, but no defects in the B-cell compartment32.Hence, in thymocytes, PI3Kγ could provide a survivalsignal, whereas in B cells, this is provided by PI3Kα.However, no single isoform of PI3K has been attributeda role in peripheral T-cell homeostasis. In vitro treat-ment of T cells with PI3K inhibitors was not reported toinduce cell death, but it blocked proliferation33 and theproduction of IL-2 (REF. 34).
One of the downstream substrates of PI3K signallingis AKT (FIG. 4). Evidence for the involvement of AKT in lymphoid homeostasis is based on the retroviral
line (Q. Jiang, A.R.K. and S.K.D., unpublished observa-tions). Moreover, a transgene encoding an IL-7Rα pro-tein that lacks Tyr449 and neighbouring tyrosines failedto support the normal survival of pro-T cells, whereasthe differentiation of αβ and γδ T cells occurred inde-pendent of this region27.
Receptors for the cytokines IL-3, granulocyte–macrophage colony-stimulating factor (GM-CSF) andIL-5, although not homeostatic regulators of lympho-cytes, have provided many lessons in pro-survival sig-nals. This family of cytokine receptors uses a commonchain, βc, together with a cytokine-specific α-chain. Arecent study showed that IL-3-mediated stimulationinduces the phosphorylation of Ser585 of βc. Thisrecruits the signalling regulator 14-3-3 to the site, whichbinds to phosphoserine motifs. 14-3-3 acts as an adap-tor protein, subsequently binding the regulatory subunitof PI3K and initiating the pathway to survival28.
Signalling through the PI3K pathway. Binding of acytokine to its receptor triggers a signalling cascade thatinduces, among other survival mediators, the PI3Kpathway (FIG. 4). One line of evidence to support the roleof PI3K in lymphoid homeostasis comes from studiesof PHOSPHATASE AND TENSIN HOMOLOGUE (PTEN), a lipidphosphatase that counters the pro-survival effects ofPI3K by dephosphorylating its lipid products. PTEN isa probable tumour suppressor, in that inactivatingmutations of PTEN are found commonly in varioustumours, leukaemias and lymphomas29. Analysis ofPTEN-deficient mice indicates that the PI3K pathway
ACTIVATION-INDUCED CELL
DEATH
(AICD). A process by whichactivated T cells undergo celldeath through engagement ofdeath receptors such as FAS or the TNF receptor, or theproduction of reactive oxygenspecies.
LYMPHOPAENIA
A deficiency of lymphocytes inthe blood circulation.
14-3-3
A family of conserved proteinspresent in all eukaryoticorganisms that are involved insuch diverse cellular processes asapoptosis and stress, as well asintracellular signalling and cell-cycle regulation. 14-3-3 proteinsfunction as adaptors in proteininteractions and can regulateprotein localization andenzymatic activity.Approximately 100 bindingpartners for the 14-3-3 proteinshave been reported.
PHOSPHATASE AND TENSIN
HOMOLOGUE
(PTEN).A phosphatidylinositol3-phosphatase and tumoursuppressor that dephosphorylateslipid phosphatidylinositol-3,4,5-triphosphates and antagonizesthe activity of phosphatidyl-inositol 3-kinase.
Box 1 | Mediators of survival signals
Lymphotrophic cytokinesInterleukin-7. IL-7 is an essential lymphotrophin for naive T cells. Naive T cells do not undergo homeostatic expansion in IL-7-deficient hosts, whereas IL-4- or IL-15-deficient hosts support this process10. Exogenously administered IL-7increases the size of the naive T-cell pool110,111. IL-7 promotes the survival of memory CD8+ T cells11–13. IL-7 could also be a lymphotrophin for γδ T cells, because IL-7-receptor-knockout mice lack γδ T cells in the skin, gut, liver and spleen112.In addition, IL-7 is required for rearrangement of the T-cell receptor γ-locus113.
Interleukin-15. IL-15 is required to maintain populations of memory CD8+ T cells. Mice lacking the IL-15 receptor α-chain (IL-15Rα) are LYMPHOPAENIC and have a selective reduction in the number of memory-phenotype CD8+ T cells8,as do mice that are deficient in IL-15 (REF. 9). Treatment with IL-15 restores the lymphoid content of deficient mice9.Mice lacking IL-15 or its receptor also have a lack of natural killer (NK) cells, NKT cells and intestinal intraepitheliallymphocytes9.
Other lymphotrophic ligandsB-cell-activating factor (BAFF). Recently, several investigators have identified a ligand from the tumour-necrosisfactor (TNF) family, BAFF/BLyS/TALL1/TNFS13B/zTNF4, as an enhancer of mature B-cell survival. BAFF-deficientmice have depletion of peripheral B cells and negligible immunoglobulin-M responses after challenge with antigen16.The transgenic expression of BAFF leads to B-cell clonal expansion and autoimmune disease114.
T-cell and B-cell receptors. T-cell-receptor stimulation is required to maintain the survival of naive T cells. So, naiveCD4+ T cells require MHC class II and self-peptide, whereas naive CD8+ T cells require MHC class I and self-peptide115.Memory T cells do not seem to require survival signals from MHC and self-peptide12,116. Expression of the B-cellreceptor is required similarly to maintain the viability of mature peripheral B cells18, although it is not clear whetherextrinsic ligands are involved.
Co-stimulators and other potential lymphotrophins? Co-stimulation, for example, CD28–B7 molecules,CD40–CD40L and 4-1BB–4-1BBL is required for T-cell activation, but it is not necessary for homeostasis19. Integrinsare also potential lymphotrophins, because they are crucially anti-apoptotic for adherent cells.

© 2002 Nature Publishing Group820 | NOVEMBER 2002 | VOLUME 2 www.nature.com/reviews/immunol
R E V I E W S
NF-κB pathway. Another possible target of cytokine-mediated pro-survival signalling is the transcription factornuclear factor-κB (NF-κB), which suppresses apoptosisin diverse conditions. For example, the PRE-T-CELL RECEPTOR
signals for pro-T-cell survival through NF-κB38. NF-κBinduces the production of anti-apoptotic proteins,including cellular INHIBITORS OF APOPTOSIS (c-IAPs), FLICE-inhibitory protein (FLIP), the anti-apoptotic factor A1,and TNF-receptor-associated factor 1 (TRAF1) andTRAF2 adaptor proteins that augment NF-κB activation.Caspases target key components of the NF-κB pathway,which could accelerate apoptosis39. However, there seemsto be no direct evidence, so far, to implicate the NF-κBpathway in T-cell homeostasis, although NF-κB1 and c-Rel double-deficient mice have a reduced number ofB cells and defective B-cell function40.
Metabolic suppression — end of the beginningIn the absence of a signal from a trophic cytokine, apop-tosis is the final outcome. Although much is known
expression of constitutively active or dominant-negativeAKT in primary T cells. These experiments show thatAKT has a protective effect after the withdrawal of γccytokines35. This pro-survival activity of AKT correlatedpartly with the expression of BCL-2.An activated form ofAKT was introduced as a transgene expressed in T cells36;these mice had a slow accumulation of peripheral T cellswith an activated phenotype, which indicates a role forAKT in the survival of activated T cells, rather than in T-cell homeostasis.
In cell lines that are dependent on IL-3, the expres-sion of a constitutively active form of AKT prolongedsurvival of the cells after IL-3 withdrawal37. In the samecells, this protection could be achieved also by theforced expression of the anti-apoptotic protein BCL-X
L.
However, the survival mechanism of AKT differed fromthat of BCL-X
L, in that AKT promoted glucose uptake
and catabolism, whereas BCL-XL
prevented mitochon-drial membrane depolarization in conditions of lowmetabolic activity37.
PRE-T-CELL RECEPTOR
(pre-TCR). A receptor that isexpressed on pre-T cells formedby the TCR β-chain and theinvariant pre-Tα protein. Thisreceptor complex includes CD3proteins and transduces signalsthat allow further T-celldevelopment.
INHIBITORS OF APOPTOSIS
(IAPs). A family of proteins thatinhibit cell death by interferingwith caspase activity. IAPs, suchas XIAP and survivin, targetdistinct caspases. In addition,IAPs have other functions incell-cycle regulation, proteindegradation and signaltransduction.
Dies
Lives
a Soluble trophicfactor
b Trophic factor displayed c Trophic factor displayedon dendritic cells
T cell
Dies
Lives
Stroma
Dies
Lives
Lives
Dendriticcell
Trophicfactor
Receptor
TCRMHC
Figure 2 | The lymphotrophin hypothesis. Lymphocytes compete for limiting trophic cytokines. The best-characterizedlymphotrophins belong to the γc cytokine family, namely interleukin-7 (IL-7) and IL-15. Although they are potentially soluble (a), thesecytokines are probably present on the cell surface for presentation to dependent cells (b). IL-7 is secreted by stromal cells in thethymus and is associated with the extracellular matrix14, which indicates that it could function in peripheral lymphoid organs as ananchored cytokine. IL-15 is secreted by dendritic cells (DCs) and can stimulate homeostatic proliferation15, and it has also beensuggested that it can remain bound to the DC and stimulate T cells, together with other survival signals (Y. Takaya, personalcommunication) (c). Cytokines are limiting factors for lymphocyte proliferation, because the addition of exogenous IL-7 (REFS 110,111)
or transgenic IL-15 (REF.117) expands the T-cell pool, and excess IL-7 or IL-15 induces lymphomas118–120. Therefore, it is the quantityof a trophic factor that restricts the number of each type of lymphocyte — competitors die through deprivation. As illustrated,competition could occur for trophic factors in solution or on cell surfaces. In either case, the successful cell removes the trophicfactor from the available pool. So, a lymphocyte that migrates through a trophic niche could bind the cytokine and remove it from the cell surface, preventing the survival of successive lymphocytes. In addition, the DC surface might provide other survival andproliferative signals, including peptide–MHC and co-stimulators. γc, common cytokine-receptor γ-chain; TCR, T-cell receptor.

© 2002 Nature Publishing GroupNATURE REVIEWS | IMMUNOLOGY VOLUME 2 | NOVEMBER 2002 | 821
R E V I E W S
Mitochondria are the batteries that supply theenergy to power cellular processes. When the supply ofnutrients to a cell becomes limiting, mitochondrialstructural integrity is compromised. Decreased oxygenconsumption and alterations in the mitochondrialinner membrane potential are hallmarks of the earlyapoptotic process46. After a death-inducing insult,mitochondria change conformation, condense andeventually fragment47. Mitochondrial functions, suchas substrate transport, ion exchange and ATP synthesis,are lost48. Cells at this stage of severe reduction in mito-chondrial activity cannot be rescued by replenishingtrophic factors44.
Intracellular pH changes. The regulation of intracellularpH is severely perturbed after trophic growth-factorwithdrawal in cell lines (FIG. 5). During normal meta-bolic activity, the interior of a cell would become fatallyacidic were it not for several plasma-membrane pro-teins that extrude ions from the cell49. The best charac-terized of these belong to the SODIUM/HYDROGEN EXCHANGER
(NHE) family50. The activity of NHE proteins is stimu-lated by growth factors. This stimulatory pathway signalsthrough the mitogen-activated protein kinase (MAPK)family — MEK1 to extracellular signal-regulated kinase(ERK) and ribosomal protein S6 kinase (p90 RSK) —which phosphorylate NHE proteins at Ser703 in thecytoplasmic tail51. This phosphorylation might provide adocking site for 14-3-3 proteins, thereby preventing thedephosphorylation of Ser703. The effect is to acceleratethe exchange of protons and sodium ions by NHE pro-teins52, which stabilizes the intracellular milieu at a pointnear to neutral pH.
NHE proteins respond quite differently during theinduction of apoptosis, causing a severe, but transient,alkalinization of the cell53 (FIG. 5). Trophic-factor with-drawal activates another MAPK, the stress kinase p38MAPK53–55. p38 MAPK can phosphorylate NHE pro-teins directly56, and four p38 MAPK target sites havebeen identified in the cytoplasmic tail of NHE proteins,between amino acids 704 and 744 (REF. 53), distinct fromSer703 that is phosphorylated by ERK. Hence, trophic-factor withdrawal activates p38 MAPK, which then dys-regulates the NHE proteins. Together with the loss ofglucose uptake and glycolytic activity, this apoptoticalkalinization is a severe metabolic stress, as shown bythe blocking of ADP transport into mitochondria57 andthe triggering of pro-apoptotic factors46,58 (FIG. 5).
Apoptotic cells have also been reported to becomeacidic late into the effector stage of apoptosis58,59. Thisacidification could be a result of the breakdown of pHregulatory mechanisms or, as was proposed, a result ofmitochondrial depolarization and the release of protonsinto the cytosol60, facilitating the activity of caspases60
and DNA nucleases61.
Mitochondrial damage. In apoptotic cells, the release ofcytochrome c from mitochondria activates caspases,which, in cytokine-dependent cell lines, follows trophic-factor withdrawal by 20–48 hours, although the releaseof cytochrome c itself occurs in less than five minutes62.
about the effector stages of cell death — the activationof caspases and the fragmentation of DNA — the initi-ating metabolic events that set the stage for the laterevents of apoptosis are understood poorly. Recent find-ings, involving loss of glucose metabolism, fluctuationsin ATP levels and changes in intracellular pH, indicatethat the breakdown of normal intracellular metabolicprocesses could be the initial trigger that induces thesubsequent apoptotic cascade.
Glucose metabolism. The concept that trophic cytokinespromote glucose metabolism and ATP generation wasproposed first many years ago to explain the pro-survivalfunction of IL-3 (REF. 41). Withdrawal of IL-3 from adependent cell line caused a decrease in glucose metabo-lism42. This involved both a loss of glucose uptake,through downregulating the surface expression of theGLUCOSE TRANSPORTER PROTEINS GLUT1 and GLUT4 on theplasma membrane43, and the decreased activity of gly-colytic enzymes44. Glucose uptake is determined, inpart, by AKT-mediated regulation of GLUT1 transportand surface expression (FIG. 5), although others haveshown, in AKT1-deficient mice, that AKT1 is notrequired for glucose homeostasis45. We have observedalso that IL-7 withdrawal from an IL-7-dependentthymocyte line markedly decreases glucose import(A.R.K. and S.K.D., unpublished observations), whichindicates a possible role in lymphoid homeostasis.
GLUCOSE TRANSPORTER
PROTEINS
(GLUT). A family of GLUTproteins facilitate the transportof glucose across membranes,each member having a distincttissue distribution andbiochemical activity.
SODIUM/HYDROGEN
EXCHANGER
(NHE). The NHE-familyproteins are membrane-spanningelectroneutral ion exchangers.The family includes six isoforms(NHE1–NHE6). Diverse effectsof NHE1 include homeostasis ofthe internal pH, cell-volumeregulation and interaction withthe actin cortical network, whichregulates adhesion, cell-shapedetermination, migration andproliferation.
Table 1 | Genetic manipulations and treatments in mice: lymphopaenia
Treatment Phenotype References
Cytokines
γc knockout SCID; decreased number of lymphocytes, no NK cells 139,140
IL-7 knockout Decreased number of T and B cells 141
IL-7Rα knockout Decreased number of T and B cells, no γδ T cells 142,143
IL-15 knockout Reduction in the number of memory CD8+ 9T cells, NK cells and IELs
IL-15Rα knockout No memory CD8+ T cells, NK cells, NKT cells or IELs 8
Jak3 knockout Block in B-cell, NK-cell and γδ T-cell development 144
TNF signalling
BAFF knockout Block in B-B2 to mature B-cell development; absence of 145mature B cells; decreased development of memory T cells
FADD dominant- Depleted lymphoid pools 146negative
FADD knockout Inhibition of T-cell development 147
PI3K pathway
p85α knockout Impaired B-cell development 31,148
p110γ knockout Decreased number of T cells 32
AKT1 knockout Increased thymic apoptosis 149
Bcl-2 family
Bcl-XL knockout Bone-marrow chimeras. Reduced lymphocyte maturation 70
Bcl-2 knockout Normal lymphocyte development, but later thymic 68and splenic involution
Bad transgenic T-cell depletion, perturbed development 90
Bax transgenic Reduced number of mature T cells, increased apoptosis 150Bad, Bcl-2 antagonist of cell death; BAFF, B-cell-activating factor; Bax, Bcl-2-associated X protein;FADD, Fas-associated via death domain; γc, common cytokine-receptor γ-chain; IEL, intraepitheliallymphocyte; IL, interleukin; Jak3, Janus kinase 3; NK, natural killer; PI3K, phosphatidylinositol 3-kinase; SCID, severe combined immunodeficiency; TNF, tumour-necrosis factor.
© 2002 Nature Publishing Group822 | NOVEMBER 2002 | VOLUME 2 www.nature.com/reviews/immunol
R E V I E W S
There is more to life than BCL-2Trophic factors are well known for inducing the synthe-sis of anti-apoptotic proteins of the BCL-2 family64. Thefounding member of the family, BCL-2, was identifiedfirst through its frequent translocation to the immuno-globulin locus in follicular lymphomas (reviewed inREF. 65), and it has long been associated with the survivalof haematopoietic cells66. Other pro-survival membersof the family include BCL-X
L, myeloid-cell leukaemia
sequence 1 (MCL1), A1 and BCL-W. There is no clearconsensus as to the precise mechanism of action ofthese anti-apoptotic proteins, although, in general, theyprotect mitochondria from death proteins and influ-ence other cellular activities, such as cell cycling67.Because BCL-2 has been attributed a role in lymphoidhomeostasis so frequently, we discuss in more detail theevidence for and against this connection.
Knockout studies. Bcl-2 −/− mice have severe defects in T- and B-cell production and in the number of periph-eral lymphocytes, with both problems developing abouta month after birth68. These mice also develop severekidney failure, which complicates the issue of whetherthe requirement for Bcl-2 is intrinsic to the lymphocyte
At about this time, the MITOCHONDRIAL MEMBRANE POTENTIAL
drops, although mitochondria can continue to synthe-size ATP, even in the absence of an outer membrane.The addition of caspase inhibitors showed that theinduction of apoptosis causes mitochondria to depolar-ize transiently, and then recover the inner mitochondrialmembrane potential after loss of cytochrome c62. These‘naked’ mitochondria, resulting from swelling followedby rupture of the outer membrane, could continueoxidative phosphorylation using the cytochrome c thatis present in the cytosol, and they might provide the ATPthat is required for the activity of the effector DNAses.So, caspases contribute to the depolarization of themitochondrial membrane, generating a self-amplifyingloop that is initiated by the release of cytochrome c63.
Mitochondria also produce ROS through the elec-tron transport chain. The death of activated T cells hasbeen attributed to the ROS that are generated by theirhigh respiratory rate21. ROS damage mitochondrialenzymes and initiate lipid peroxidation, which causesmitochondrial depolarization and cell death. This formof cell death is independent of either FAS or caspases21.However, no connection between ROS and the homeo-static death of lymphocytes has been reported.
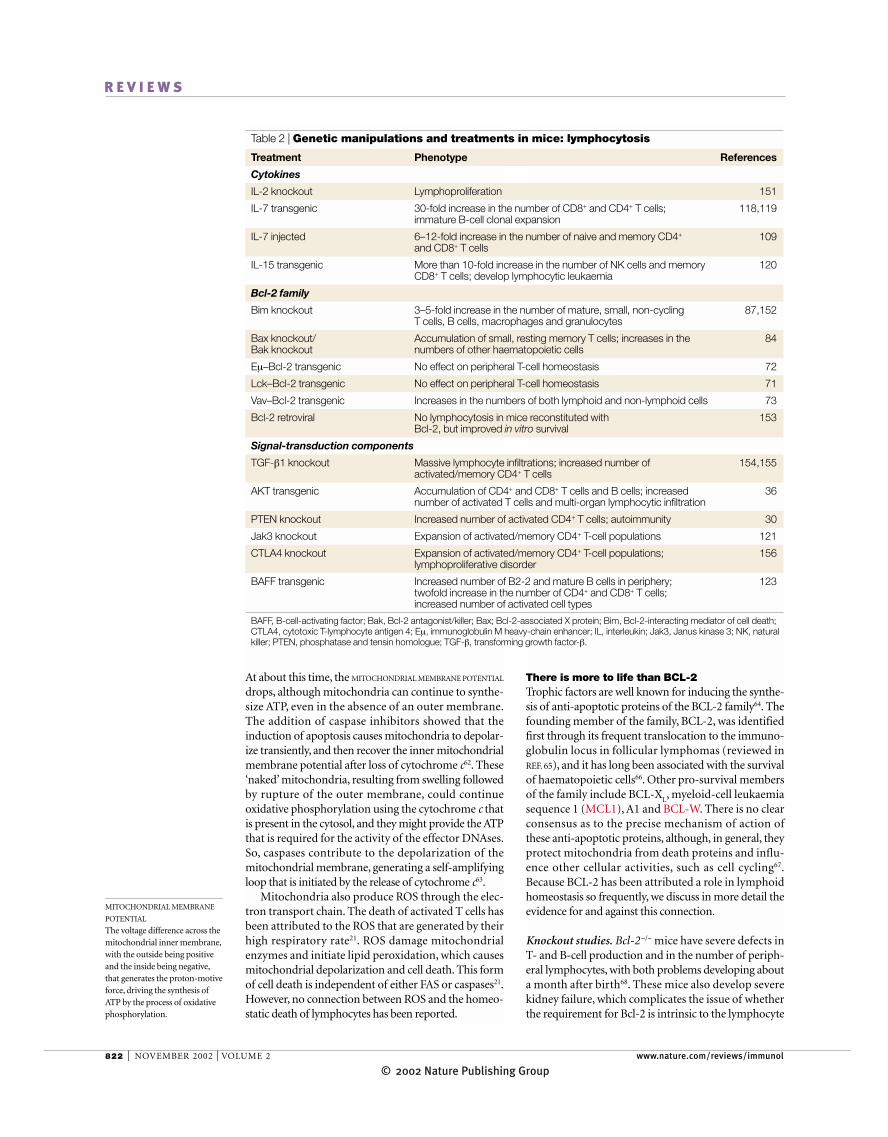
Table 2 | Genetic manipulations and treatments in mice: lymphocytosis
Treatment Phenotype References
Cytokines
IL-2 knockout Lymphoproliferation 151
IL-7 transgenic 30-fold increase in the number of CD8+ and CD4+ T cells; 118,119immature B-cell clonal expansion
IL-7 injected 6–12-fold increase in the number of naive and memory CD4+ 109and CD8+ T cells
IL-15 transgenic More than 10-fold increase in the number of NK cells and memory 120CD8+ T cells; develop lymphocytic leukaemia
Bcl-2 family
Bim knockout 3–5-fold increase in the number of mature, small, non-cycling 87,152T cells, B cells, macrophages and granulocytes
Bax knockout/ Accumulation of small, resting memory T cells; increases in the 84Bak knockout numbers of other haematopoietic cells
Eµ–Bcl-2 transgenic No effect on peripheral T-cell homeostasis 72
Lck–Bcl-2 transgenic No effect on peripheral T-cell homeostasis 71
Vav–Bcl-2 transgenic Increases in the numbers of both lymphoid and non-lymphoid cells 73
Bcl-2 retroviral No lymphocytosis in mice reconstituted with 153Bcl-2, but improved in vitro survival
Signal-transduction components
TGF-β1 knockout Massive lymphocyte infiltrations; increased number of 154,155activated/memory CD4+ T cells
AKT transgenic Accumulation of CD4+ and CD8+ T cells and B cells; increased 36number of activated T cells and multi-organ lymphocytic infiltration
PTEN knockout Increased number of activated CD4+ T cells; autoimmunity 30
Jak3 knockout Expansion of activated/memory CD4+ T-cell populations 121
CTLA4 knockout Expansion of activated/memory CD4+ T-cell populations; 156lymphoproliferative disorder
BAFF transgenic Increased number of B2-2 and mature B cells in periphery; 123twofold increase in the number of CD4+ and CD8+ T cells;increased number of activated cell types
BAFF, B-cell-activating factor; Bak, Bcl-2 antagonist/killer; Bax; Bcl-2-associated X protein; Bim, Bcl-2-interacting mediator of cell death;CTLA4, cytotoxic T-lymphocyte antigen 4; Eµ, immunoglobulin M heavy-chain enhancer; IL, interleukin; Jak3, Janus kinase 3; NK, naturalkiller; PTEN, phosphatase and tensin homologue; TGF-β, transforming growth factor-β.
MITOCHONDRIAL MEMBRANE
POTENTIAL
The voltage difference across themitochondrial inner membrane,with the outside being positiveand the inside being negative,that generates the proton-motiveforce, driving the synthesis ofATP by the process of oxidativephosphorylation.

© 2002 Nature Publishing GroupNATURE REVIEWS | IMMUNOLOGY VOLUME 2 | NOVEMBER 2002 | 823
R E V I E W S
problem or a homeostasis problem. It has also beenproposed that the stem cells that generate lymphocyteshave different requirements for Bcl-2; embryonic stemcells do not require Bcl-2, whereas adult stem cells dorequire it69. To address the issue of whether Bcl-2 isrequired to maintain homeostatic survival, it would beuseful to generate mice in which Bcl-2 can be deletedacutely in lymphocytes, as has been done for the BCRand TCR17,18.
Bcl-XL-deficient haematopoietic progenitors gener-
ated reduced numbers of T and B cells in normal hosts,but there was little effect on the lifespan of mature lym-phocytes70. Other Bcl-2-family survival proteins couldbe involved — for example, Mcl1, which has beenimplicated in the survival effect of IL-6 on plasma cells.As many of these proteins have similar functions in pro-tection from apoptosis, introducing a Bcl-2 transgenecould mimic them all, even if Bcl-2 is not the actual pro-tector of peripheral lymphocytes, and results using thisstrategy are discussed next.
Transgenic studies. Bcl-2 transgenes under the control ofdifferent promoters that are expressed specifically inlymphocytes did not cause an increase in the number ofperipheral T cells, thereby failing one of the criteria for alimiting homeostatic regulator in this compartment, butthey did result in an increase in the number of immatureB cells71,72. By contrast, a Bcl-2 transgene controlled bythe Vav promoter, which is not lymphoid specific, didinduce a large increase in the number of peripheralCD4+ and CD8+ T cells and B cells, as well as of myeloidcells73. This latter effect using the Vav promoter could bequantitative, in that the level of expression of Bcl-2might be higher than in the previous studies. It is alsoconceivable that the Vav–Bcl-2 transgene first increasesthe number of non-lymphoid cells that produce IL-7or IL-15, which then secondarily increases the numberof lymphocytes. By analogy, an increase in the number ofthymocytes results from transgenes that increase thenumber of thymic epithelial cells74.
A Bcl-2 transgene that is expressed specifically inlymphocytes could restore αβ T-cell development in IL-7R-deficient mice75. Introducing the Bcl-2 transgeneinto IL-7R-deficient mice also restored the number ofperipheral CD4+ and CD8+ T cells, polyclonal in vitrofunction and the in vivo rejection of allogeneic tumourcells76. However, it has been observed that peripheral T cells in IL-7R-deficient mice eventually accumulate,even without expression of the Bcl-2 transgene. Anotherproblem in ascribing an important trophic role to Bcl-2in lymphoid homeostasis is the failure of a Bcl-2 trans-gene to restore T-cell development and peripheral T-cellnumbers in some lines of γc-deficient mice77. So, thesuccess of the Bcl-2 transgene in restoring the numberof T cells in IL-7R-deficient mice could be to amplifyadditional survival signals from other γc cytokines, suchas IL-15. Finally, it is unclear whether the peripheral T cells that are rescued by Bcl-2 are of a naive pheno-type, which is the central question addressed by thisreview on homeostatic cell death. We conclude thatBCL-2 is required for normal lymphoid development
or whether sickness causes the loss of lymphocytes.Because lymphocyte production is markedly reducedwith age, it is also unclear whether the decline in thenumber of peripheral lymphocytes reflects a production
Trophic factor
BCL-2
Loss of:PI3K–AKT,JAKs–STATs,MEKs
BAXBAX
BAD
BAD
Receptor
Apoptosis
Cytochrome c release
Formation ofthe apoptosomeand activation ofdownstreamcaspases
FAS
Activation ofcaspase-3,-6 and -7
BID
Mitochondrion
FASLa b
Metabolicperturbations
Arming ofpro-apoptoticfactors
Loss ofanti-apoptoticfactors
FADD (adaptor)
Pro-caspase-8
Activecaspase-8
APAF1
Pro-caspase 9
Figure 3 | Death from trophic-factor loss is distinct from death through FAS. An immuneresponse results in the clonal expansion of the lymphocyte compartment to handle challenge with aforeign antigen. Once the antigen has been cleared, it is necessary to eliminate the large number ofcirculating lymphocytes and restore homeostasis. To achieve this, death receptors such as FAS(CD95) are engaged and induce apoptosis rapidly. This form of cell death is different from that whichoccurs when a trophic cytokine is limiting. a | Ligand binding trimerizes FAS, inducing the formation of a signalling complex within seconds of receptor engagement. The adaptor FADD (FAS-associateddeath domain protein) binds to the death domain in FAS. Pro-caspase-8 is recruited and cleaved,releasing active caspase-8, which activates pro-caspase-3, followed by many other death effectors.FAS can secondarily attack mitochondria through cleavage of the pro-apoptotic protein BID (BH3-interacting death domain agonist), leading to the induction of another death protein, BAX (BCL-2-associated X protein), which disrupts mitochondrial function. b | By contrast, trophic-cytokinewithdrawal does not seem to induce an initiating caspase cascade but, instead, disruptsmitochondria by other mechanisms, although this pathway also uses apoptotic inducers such asBAX. Trophic deprivation causes loss of signalling through the JAK–STAT and PI3K–AKT pathways,which are known to promote anti-apoptotic activities, and produces severe metabolic perturbationsthat arm cells for death. The eventual outcome of both death pathways is mitochondrial damage,whether through FAS activation or trophic-factor loss, which results in the release of cytochrome c,formation of the apoptosome, caspase activation and the terminal breakdown of cellular constituents.APAF1, apoptotic protease-activating factor 1; BAD, BCL-2 antagonist of cell death; JAK, Januskinase; PI3K, phosphatidylinositol 3-kinase; STAT, signal transducer and activator of transcription.

© 2002 Nature Publishing Group824 | NOVEMBER 2002 | VOLUME 2 www.nature.com/reviews/immunol
R E V I E W S
and is active in protecting lymphocytes from stressesin vitro. However, it seems that in terms of the homeo-static regulation of lymphocytes, BCL-2 and related pro-teins might account only partly for the trophic effect ofcytokine-receptor stimulation.
BAX, BAK, BIM and BAD — beginning of the endIf BCL-2 is a part of the lymphotrophic effect, what doesit do? The best-characterized action of BCL-2 is to blockthe death effectors that target mitochondria. Prominentamong these are the pro-apoptotic members of theBCL-2 family, which are classified in two categories. Thefirst category includes the multi-domain proteins thatcontain BCL-2-homology domains (BH1–BH3 andBH4) and a transmembrane domain; these include BAX(BCL-2-associated X protein) and BAK (BCL-2 antago-nist/killer), which are expressed widely, and BOK (BCL-2-related ovarian killer), which is found in reproductivetissue. These proteins are thought to damage mitochon-dria directly (FIGS 5,6). The second category of moleculescontains only a BH3 domain and, in some cases, a trans-membrane domain, and includes BAD (BCL-2 antago-nist of cell death), BID (BH3-interacting domain deathagonist), BIK (BCL-2-interacting killer), BIM (BCL-2-interacting mediator of cell death), HRK (harakiri) andBLK (B-lymphoid tyrosine kinase). These proteinsmight damage mitochondria indirectly, either by inter-fering with BCL-2 or by activating the multi-domainproteins (FIGS 5,6).
BAX and BAK: the multi-domain death proteins. BAXwas identified first as a binding partner for BCL-2 (REF. 78),but it was shown subsequently to translocate to themitochondria and oligomerize with itself or other BCL-2-family members after trophic-factor withdrawal58,79,80.Deletion of Bax in mice results in an enlarged thymus81,and it partially corrects the thymic block that resultsfrom a deficiency of Jak3. Bax deletion does not correctperipheral T-cell defects in Jak3−/− mice, which are char-acterized by the absence of CD8+ T cells and accumula-tion of CD4+ T cells82. Bax deficiency can rescue thymicdevelopment in neonatal IL-7R-deficient mice, but itdoes not restore the later peripheral T-cell defects83. So,BAX is an essential death-inducing protein in thymo-cytes that are deprived of IL-7, but the homeostasis ofperipheral T cells, which is also under the control of IL-7,must involve more than BAX.
The marked redundancy of BAX has been attributedto the death protein BAK (FIG. 5). Mice deficient in Bakare developmentally normal and do not have any age-related defects84. The redundancy between Bax and Bakis shown clearly in double-deficient mice, which havemany severe abnormalities, compared with the mildeffects of a single deletion. Most Bax−/−Bak−/− mice diedperinatally, with less than 10% reaching adulthood.They retain webs between their digits, and have neuro-logical abnormalities and an increased number ofhaematopoietic cells, including lymphocytes and granu-locytes. Large accumulations of small, resting CD44+
memory CD8+ T cells occur in the lymphoid organs,and B cells accumulate also. Bax−/−Bak−/− lymphocytes
Trophic factor
Receptor
PI3K
SRC
PtdIns3,4,5P3 PTEN
GSK3
Glucose transportand metabolism
MAPKs ??
?
AKT
BAD
FKHRL1
Anti-apoptotic activities
Caspase-9
BCL-XL genetranscription
BCL-2 genetranscription
PP
P
P
P
P
PP
P
P
14-3-3
14-3-3
Cytosol
P
NF-κBA-MybCREBE2F
Anti-apoptoticgenes
JAK
PI3K/AKT-mediated activities:regulation of gene transcription(NFAT, NF-κB, CREB) andcell cycle (E2F family, cyclin D1 and p21)
Nucleus
STAT
STAT
STAT
Figure 4 | Signal-transduction pathways promoting lymphocyte survival. Cytokinebinding to its receptor triggers a cascade of signalling events that induce cell survival. Thehomeostatic survival effects of interleukin-7 (IL-7) and IL-15 are probably mediated throughJanus kinase 3 (JAK3), because mice that express Jak3 in the thymus, but not in peripheral T cells, still have a peripheral T-cell deficiency121. Activated JAKs then activate signaltransducers and activators of transcription (STATs). IL-3 drives synthesis of the anti-apoptoticprotein BCL-XL through STAT5 (REF. 122). Expression of the BCL-2 gene is induced by severaltranscription factors, including cyclic-AMP-responsive binding protein (CREB) and nuclearfactor-κB (NF-κB)123. Cytokine binding further triggers the phosphatidylinositol 3-kinase (PI3K)pathway. PI3Ks consist of a regulatory subunit (p85α, p85β or p55γ) and a p110 catalyticsubunit. Binding of the regulatory subunit to a phosphorylated site on a cytokine receptorengages the activity of the p110 subunit. PI3Ks phosphorylate phosphatidylinositol and its derivatives, producing lipid products that activate kinases. In turn, these kinasesphosphorylate AKT, inducing its movement to membranes, where it is activated. Phosphataseand tensin homologue (PTEN) inhibits this process. There are three isoforms of AKT — AKT1,AKT2 and AKT3. Downstream targets of AKT phosphorylation include glycogen synthasekinase 3 (GSK3), which is inactivated by phosphorylation. GSK3 was identified initially as aregulator of glycogen metabolism, but it is now known to regulate gene transcription, forexample through nuclear factor of activated T cells (NFAT)124, and cell cycling. AKT alsophosphorylates and inactivates a forkhead transcription factor, FKHRL1, causing its cytosolicretention124. AKT can promote cell survival by phosphorylating the death inducer, BAD (BCL-2-antagonist of cell death), causing its sequestration by 14-3-3 proteins in the cytosol125,and by phosphorylating caspase-9, preventing its activation99. AKT is also thought to promoteglucose transport and metabolism by enabling the uptake of glucose and its metabolicbreakdown44. MAPK, mitogen-activated protein kinase; PtdIns3,4,5P3, phosphatidylinositol-3,4,5-triphosphate.

© 2002 Nature Publishing GroupNATURE REVIEWS | IMMUNOLOGY VOLUME 2 | NOVEMBER 2002 | 825
R E V I E W S
conceivable, as we discussed in the section on Bcl-2transgenes, that more cells are available to produce IL-7and/or IL-15, which then expand the memory CD8+
T-cell pool. It would help to clarify this issue to deter-mine whether the LYMPHOCYTOSIS effect is intrinsic to theT cell by transferring Bax−/−Bak−/− T cells into IL-7- orIL-15-deficient recipients.
It should be emphasized also that the numbers ofnaive CD4+ and CD8+ T cells and memory CD4+ T cellsare not increased in Bax−/−Bak−/− mice, or after expres-sion of the Bcl-2 transgene, which indicates that other
are resistant to several inducers of apoptosis, includingirradiation and cell culture, but death mediated throughengagement of the Fas pathway is still intact84,85.
So, BAX and BAK seem to mediate the default deathpathway in memory CD8+ T cells, possibly being acti-vated by the withdrawal of IL-7 or IL-15. This model isalso compatible with the effect of the Vav–Bcl-2 trans-gene in expanding the CD8+ T-cell population; forexample, expression of Bcl-2 could mimic the lack ofBax and Bak. Alternatively, because the number of non-lymphocytes is also increased in Bax−/−Bak−/− mice, it is
LYMPHOCYTOSIS
An increase in the number of lymphocytes in the blood,which is usually associated with chronic infections orinflammation.
∆Ψm ↑ATP:ADP ↓
Loss of PI3K–AKT
∆Ψm ↓
Stress
IntracellularalkalinizationADP
import
NHE1Na+
H+
BIM
BIM genetranscription
p38 MAPK
P P P
Nucleus
Cytosol
P P
Glucosemetabolism ↓
GLUT1
Glucoseuptake
Hexokinase 2Phosphofructokinase 1
BCL-2
BCL-XL
BCL-2
BAD
BAKBAD
BAXBAX
Trophic factor
Receptor
FKHRL1
FKHRL1
Apoptosis
Cytochrome c release
Figure 5 | Early death-inducing events after trophic-factor withdrawal. In the first hours after loss of cytokine binding, signaltransduction from the receptor ceases and changes in intracellular metabolism are detected. Glucose transport stops and glycolyticenzyme activity is decreased, which stalls glucose metabolism46. Stress, a consequence of lymphotrophin deprivation, induces theactivity of p38 mitogen-activated protein kinase (MAPK), which phosphorylates the pH regulatory protein, sodium/hydrogenexchanger (NHE). As a result of NHE over-activity, a transient intracellular alkalinization occurs, which inhibits the import of ADP intomitochondria, increases the mitochondrial membrane potential (∆Ψm) and disrupts ATP synthesis. Both the loss of glucose and/orthe rise in pH can promote the mitochondrial translocation of the death protein BAX (BCL-2-associated X protein)46,58. Cytokinewithdrawal also activates the apoptotic protein BAK (BCL-2 antagonist/killer). Although it is similar in structure to BAX, BAK is pre-associated with mitochondria before an apoptotic signal126. Induction of the BH3-only proteins BIM (BCL-2-interacting mediatorof cell death) and BAD (BCL-2-antagonist of cell death) also contributes to death during cytokine deprivation. BIM contains ahydrophobic carboxyl terminus, and alternative splicing gives rise to three isoforms, BIMS, BIML and BIMEL
127. In the absence ofapoptosis, BIM assoicates with LC8, a component of the microtubule-associated dynein motor complex. After cytokine withdrawal,BIM is released and translocates to mitochondria, where it targets anti-apoptotic proteins such as BCL-2 (REF. 128). Also, BIM issynthesized at a higher rate due to the activity of the forkhead transcription factor FKHRL1 (REF. 129). In the absence of cytokinesignalling, the unphosphorylated form of the death protein BAD targets mitochondria, also binding to anti-apoptotic proteins. Theresulting mitochondrial breakdown, as detected by depolarization of the mitochondrial membrane and release of cytochrome c,induces the caspase cascade, which leads to cell death. GLUT1, glucose transporter protein 1; PI3K, phosphatidylinositol 3-kinase.

© 2002 Nature Publishing Group826 | NOVEMBER 2002 | VOLUME 2 www.nature.com/reviews/immunol
R E V I E W S
BIM and BAD: BH3-only death proteins. The pro-apoptotic protein BIM was isolated from a complementary DNA library through its interactionwith BCL-2. Bim−/− mice have marked defects inhaematopoietic homeostasis, with the accumulationof both lymphoid and myeloid cells and abnormal T-cell development87, and a block in thymic negativeselection88. Small, resting memory CD8+ T cells accu-mulate in the periphery, and older mice have anincreased number of plasma cells and develop anautoimmune kidney disease. Bim−/− lymphocytes areresistant to trophic-factor withdrawal in vitro, butretain sensitivity to FasL-induced cell death87. Bimdeficiency can also compensate for the later cellularattrition that is seen in Bcl-2-deficient animals.
Of all the BH3-only proteins, BIM is the most proba-ble candidate for a death inducer (acting together withBAX and BAK) after IL-7 or IL-15 withdrawal frommemory CD8+ T cells. However, the argument invokedfor the Bcl-2-transgenic mice and the Bax/Bak double-deficient mice also applies to Bim, in that the observedlymphocyte clonal expansion could be secondary toincreased cytokine production by non-lymphoid cells87.It will be important to test whether Bim−/− T cells cansurvive after transfer into IL-7- or IL-15-deficient recipi-ents. It also remains to be explained how BIM is acti-vated after lymphotrophin withdrawal. In this regard,metabolic stresses could be important.
BAD, an additional member of the BH3-only fam-ily, is a possible apoptotic activator after trophic-factorwithdrawal. BAD was first shown to have a role in celldeath after IL-3 withdrawal from dependent cells89. Inthe presence of IL-3, BAD is phosphorylated andsequestered in the cytosol; after IL-3 withdrawal,dephosphorylated BAD translocates to mitochondria(FIG. 5). IL-7 signalling in pro-T cells and in a thymiccell line also induced the phosphorylation of BAD andblocked its translocation to the nucleus83. Althoughexpression of a Bad transgene greatly depleted thenumber of T cells and perturbed T-cell development90,Bad −/− mice had no apparent lymphocytosis91, whichindicates that, unlike Bim and Bax/Bak, Bad is not alimiting factor in peripheral lymphocyte homeostasis.
Caspases — the end of the end?A prominent feature of all forms of apoptosis is the acti-vation of caspases, a family of cysteine proteases thatcleave target proteins at specific aspartate residues.Caspases are activated by the withdrawal of lym-photrophins, but blocking caspases does not protectpro-T cells from the effects of IL-7 withdrawal in vitro92
or block the translocation of BAX to the nucleus58. Bycontrast, blocking caspase-8 protects cells from the FAS-mediated cell-death pathway, because its activation is anearly event that couples death-receptor ligation to boththe mitochondrial pathway (through cleavage of BID)and the direct cleavage of downstream caspases.
Studies in caspase-knockout mice have yet to identifya caspase that is required for peripheral lymphoidhomeostasis. Many knockouts die before lymphoiddevelopment and cannot be evaluated. Although effects
death pathways, perhaps superimposed on the Bax/Bakpathway, are involved in the homeostatic regulation ofthese cells. However, a note of caution needs to be intro-duced here regarding ‘memory’ phenotypes. After lym-phocyte depletion, the proliferation of naive T cellsinduces the reversible expression of memory-T-cell-likemarkers and function4,86, bringing into question theidentity of the abundant CD8+ ‘memory’ T cells that areobserved in some of these genetically manipulated mice.So, it should be (but has not been) tested whether theseare real or reversible memory CD8+ T cells in the Bcl-2-transgenic and Bax/Bak-deficient mice.
No mitochondrial damage
Mitochondrial damage
Presence of trophic factor
Absence of trophic factor
Apoptosisprevented
Apoptosis
Cytochrome crelease
BCL-2
BCL-2
BCL-2
BCL-2
BCL-2
BCL-2
BCL-2
BAD
BAX
BAX
BAX
BAX
BAK
BIM
Figure 6 | The pro-apoptotic BCL-2-family members. In a healthy cell, the presence of a trophicfactor induces the synthesis of BCL-2, which targets mitochondria, providing protection fromdamage. In the absence of a trophic stimulus, however, this protection is blocked by the binding ofBH3-only proteins, such as BIM (BCL-2-interacting mediator of cell death) and BAD (BCL-2-antagonist of cell death), as well as BAX (BCL-2-associated X protein), to BCL-2, which furtherenhances the mitochondria-damaging activity of BAX and BAK (BCL-2 antagonist/killer). Recently,BIM has also been found to bind to voltage-dependent anion channels (VDACs), contributing tomitochondrial breakdown130. The means by which trophic-factor withdrawal triggers BAX and BAKremain elusive. Several mechanisms have been proposed to induce the translocation of BAX to themitochondria131, including changes in cytosolic pH58,132, involvement of the phosphatidylinositol 3-kinase (PI3K)–AKT pathway133 and activation by the BH3-only proteins BID (BH3-interactingdomain death agonist)134 and BIM135. BID is an unlikely candidate for involvement in the effects oftrophic-factor withdrawal because it is activated by cleavage by caspase-8 and there is no evidencethat caspases lie upstream of BAX translocation in trophic-cytokine withdrawal. BIM might beinvolved in this pathway and, alone, was shown not to kill cells in Bax−/−Bak−/− mice136. Variousmitochondrial lesions have been attributed to BAX, including the formation of pores in the outermitochondrial membrane137 that contain the VDACs138. The combined actions of the pro-apoptoticmembers of the BCL-2 family lead to disruption of mitochondrial integrity, which promotescytochrome c release and mitochondrial fragmentation, leading to cell death.

© 2002 Nature Publishing GroupNATURE REVIEWS | IMMUNOLOGY VOLUME 2 | NOVEMBER 2002 | 827
R E V I E W S
of caspase-3 and caspase-7 (REF. 106). IAPs block a broadspectrum of apoptotic stimuli, including death-receptorengagement, irradiation and chemotherapeutic agents,and they have other activities that involve the cell cycle,protein degradation and signal transduction. XIAP(X-linked inhibitor of apoptosis), in particular, canblock the activity of caspase-9 directly and is, in turn,neutralized by SMAC/DIABLO, which is released from themitochondria107. However, XIAP-deficient mice arenot defective for caspase-dependent or -independentapoptosis, which indicates that there are probably other,compensatory mechanisms108.
ConclusionsThe size of lymphocyte pools is limited, in part, by com-petition for cytokines, including IL-7 and IL-15. Cells thatfail to acquire sufficient signalling from the receptors forthese cytokines undergo cell death. In this review, weexamine the state of knowledge regarding this deathpathway and conclude that the list of suspects is long andmuch of the evidence would not hold up in court.
The most satisfying evidence relates to the death ofone of the main T-cell subsets, memory CD8+ T cells.IL-7 and IL-15 maintain the survival of these cells, andmost of the evidence indicates that the death pathwayinvolves BIM, BAX and BAK, with protection mediatedby BCL-2. If these are the leading suspects, theirupstream and downstream accomplices have yet to bedetermined. What triggers the activation of BIM andBAX/BAK? Perhaps, metabolic stresses? How are BAXand/or BAK activated and how do they kill cells in cir-cumstances that do not always require the apoptosome?
The homeostatic death pathways for the other mainT-cell subsets and B cells have been more elusive. Thetrophic cytokine signal in naive T cells might requireBCL-2 to block the BIM–BAX/BAX pathway, becausenaive T cells are absent in Bcl-2−/− mice and Bim defi-ciency corrects this problem. However, the naive T-cellpool does not seem to increase in size if the BCL-2–BIM–BAX/BAK pathway is perturbed, which indi-cates that there is an additional limit. This could be alimitation in the peptide–MHC survival signal or prolif-erative signals, or it could be a metabolic limit owing tolimiting cytokine trophic signals.
So, the induction of BCL-2 (and repression of BIM–BAX/BAK) by cytokines might be part of the require-ment for lymphocyte survival, but cytokine deprivationmust lead to other problems, and metabolic disturbancesare leading suspects.As the TCR and BCR are involved inlymphocyte survival also, they must stimulate survivalsignals that are superimposed on the cytokine survivalpathways. Perhaps, the survival of memory CD8+ T cellshas been simpler to perturb because it is thought to beindependent of TCR signalling.
Survival is not the only effect of IL-7 and IL-15 onlymphocytes. These cytokines are also implicated insome lymphoid differentiation events, but more rele-vant to the present discussion, they can induce cellcycling. So, injecting IL-7 into a mouse induces the pro-liferation of naive and memory T cells109. This prolifera-tive activity complicates the picture arithmetically, in
of caspase knockout on thymocytes have been reported93,the transgenic expression of a caspase inhibitor had no effect on thymocyte development or selection94.Caspase-10 defects in humans are one cause of autoim-mune lymphoproliferative syndrome (ALPS), an accu-mulation of peripheral lymphocytes that is associatedwith defective FAS signalling95. Somatic mutation of thegene encoding caspase-10 is associated also with somenon-Hodgkins lymphomas96, which is also consistentwith a pro-apoptotic function for this protein, possiblyin death-receptor signalling.
It is perhaps surprising that the deletion of caspase-9or apoptotic protease-activating factor 1 (APAF1) doesnot seem to dysregulate lymphoid homeostasis. Mito-chondrial damage, which is proposed to occur duringcytokine withdrawal through BIM–BAX/BAK signalling,induces the release of cytochrome c and promotes theassembly of the APOPTOSOME, a caspase-activating com-plex. APAF1, which is part of the apoptosome97, isrequired together with dATP98 for cytochrome c to acti-vate caspase-9. Caspase-9 then cleaves and activates theexecutioner caspases caspase-3 and caspase-7, which dis-mantle the cell and induce engulfment by phagocytes99.Caspase-9 is also regulated through phosphorylation. Thekinase AKT, which is implicated in the lymphotrophicresponse, induces the phosphorylation of pro-caspase-9,which blocks its activation by cytochrome c100.
Mice that are deficient in caspase-9, although theyhave neurological defects and radiosensitivity, havenormal lymphocyte development and homeostasis101,as do mice deficient in Apaf1 (REF. 102). So, although theBCL-2–BIM–BAX/BAK pathway is proposed to controlthe number of memory CD8+ T cells, and is thought tooperate at the level of mitochondria, it seems that thedestruction of these lymphocytes does not proceedsolely through APAF1 and caspase-9. One possibility isthat the mitochondrial destruction mediated byBAX/BAK (and blocked by BCL-2) is sufficiently severethat it does not require the apoptosome to kill the cell.
Several caspase-independent death effectors havebeen described recently that, in addition to cytochrome c,are released from damaged mitochondria. APOPTOSIS-
INDUCING FACTOR (AIF) is one such protein. Activated T cells release AIF from mitochondria into the cytosol,and this has been proposed to account for their caspase-independent death103. AIF-deficient embryonic stemcells are resistant to death after serum starvation104,which indicates that AIF could participate in deathinduced by trophic-factor withdrawal in other cells,such as lymphocytes. Another caspase-independentdeath promoter is OMI, the precursor of which resides inmitochondria, where it undergoes autocatalysis to cleaveits mitochondrial-translocation sequence, producingthe active form. OMI binds the IAPs (discussed below)and blocks their caspase-suppressive activities, but italso has other activities beyond IAPs that have yet to bedetermined105.
Another family of anti-apoptotic proteins, the IAPs,are potentially involved in lymphoid homeostasis,although direct evidence for this is lacking. IAPs regu-late assembly of the apoptosome and block the activity
APOPTOSOME
An apoptotic protein complexformed from the association ofAPAF1, cytochrome c and dATPwith pro-caspase-9. Complexformation leads to the cleavageand activation of caspase-9,which activates caspase-3 andother effector caspases, leadingto cell death.
APOPTOSIS-INDUCING FACTOR
(AIF). A protein found in themitochondrial intermembrane.When released frommitochondria after loss of themitochondrial outer membrane,AIF travels to the nucleus andactivates a nuclease thatdegrades DNA.
OMI
A mitochondrial serine protease.When released from themitochondrial intermembranespace, OMI enters the cytosol,where it enhances caspase-dependent apoptosis by blockinginhibitors. Caspase-independentcell death might result from itsserine-protease activity.
SMAC/DIABLO
(second mitochondrialapoptosis-activating factor).Another protein found in themitochondrial intermembrane.When released, SMAC bindsIAPs, inhibiting their pro-survival functions. Complexes ofSMAC and IAPs have decreasedanti-apoptotic activity.

© 2002 Nature Publishing Group828 | NOVEMBER 2002 | VOLUME 2 www.nature.com/reviews/immunol
R E V I E W S
1. Tanchot, C. et al. Lymphocyte homeostasis. Semin.Immunol. 9, 331–337 (1997).
2. Tanchot, C., Fernandes, H. V. & Rocha, B. The organizationof mature T-cell pools. Philos. Trans. R. Soc. Lond. B 355,323–328 (2000).
3. Rocha, B., Dautigny, N. & Pereira, P. Peripheral T lymphocytes: expansion potential and homeostaticregulation of pool sizes and CD4/CD8 ratios in vivo. Eur. J. Immunol. 19, 905–911 (1989).
4. Kieper, W. C. & Jameson, S. C. Homeostatic expansion andphenotypic conversion of naive T cells in response to self-peptide/MHC ligands. Proc. Natl Acad. Sci. USA 96,13306–13311 (1999).
5. Freitas, A. A. & Rocha, B. Population biology oflymphocytes: the flight for survival. Annu. Rev. Immunol.18, 83–111 (2000).This article provides a thorough review ofexperimental data supporting the concept that thereare limits that control the size of lymphocytepopulations and that each lymphocyte subsetoccupies a unique environmental niche.
6. Marrack, P. et al. Homeostasis of αβ TCR+ T cells. NatureImmunol. 1, 107–111 (2000).
7. von Boehmer, H. & Hafen, K. The life span of naive αβT cells in secondary lymphoid organs. J. Exp. Med. 177,891–896 (1993).
8. Lodolce, J. P. et al. IL-15 receptor maintains lymphoidhomeostasis by supporting lymphocyte homing andproliferation. Immunity 9, 669–676 (1998).
9. Kennedy, M. K. et al. Reversible defects in natural killer andmemory CD8 T-cell lineages in interleukin-15-deficient mice.J. Exp. Med. 191, 771–780 (2000).An evaluation of IL-15-deficient mice, providingevidence that this cytokine is essential for thedevelopment of many types of immune cell.
10. Tan, J. T. et al. IL-7 is critical for homeostatic proliferationand survival of naive T cells. Proc. Natl Acad. Sci. USA98, 8732–8737 (2001).Definitive proof that IL-7 is required for themaintenance of naive T cells.
11. Schluns, K. S. et al. Interleukin-7 mediates the homeostasisof naive and memory CD8 T cells in vivo. Nature Immunol.1, 426–432 (2000).This study provides elegant experimental evidencethat IL-7 is important for the maintenance of bothnaive and memory CD8+ T cells.
12. Tan, J. T. et al. Interleukin (IL)-15 and IL-7 jointly regulatehomeostatic proliferation of memory-phenotype CD8+ cells,but are not required for memory-phenotype CD4+ cells. J. Exp. Med. 195, 1523–1532 (2002).
13. Kieper, W. C. et al. Overexpression of interleukin (IL)-7 leadsto IL-15-independent generation of memory phenotypeCD8+ T cells. J. Exp. Med. 195, 1533–1539 (2002).
14. Kitazawa, H. et al. IL-7 activates α4β1 integrin in murinethymocytes. J. Immunol. 159, 2259–2264 (1997).
15. Ge, Q. et al. Homeostatic T-cell proliferation in a T-cell–dendritic-cell coculture system. Proc. Natl Acad. Sci.USA 99, 2983–2988 (2002).
16. Schiemann, B. et al. An essential role for BAFF in the normaldevelopment of B cells through a BCMA-independentpathway. Science 293, 2111–2114 (2001).
17. Polic, B. et al. How αβ T cells deal with induced TCRαablation. Proc. Natl Acad. Sci. USA 98, 8744–8749 (2001).A series of elegant experiments designed to test therequirement for TCR signalling in T cells. Inducibledeletion of the TCR constant region enabled theauthors to evaluate the role of the TCR in both CD4+
and CD8+ T cells.18. Lam, K. P., Kuhn, R. & Rajewsky, K. In vivo ablation of surface
immunoglobulin on mature B cells by inducible gene targetingresults in rapid cell death. Cell 90, 1073–1083 (1997).
19. Prlic, M. et al. Homeostatic expansion occurs independentlyof costimulatory signals. J. Immunol. 167, 5664–5668(2001).
20. Siegel, R. M. et al. The multifaceted role of Fas signaling in immune-cell homeostasis and autoimmunity. NatureImmunol. 1, 469–474 (2000).
21. Hildeman, D. A. et al. Molecular mechanisms of activated T-cell death in vivo. Curr. Opin. Immunol. 14, 354–359(2002).
22. Schuster, N. & Krieglstein, K. Mechanisms of TGF-β-mediated apoptosis. Cell Tissue Res. 307, 1–14 (2002).
23. Gorelik, L. & Flavell, R. A. Transforming growth factor-β in T-cell biology. Nature Rev. Immunol. 2, 46–53 (2002).
24. Nosaka, T. et al. Defective lymphoid development in micelacking Jak3. Science 270, 800–802 (1995).
25. Van Parijs, L. et al. Uncoupling IL-2 signals that regulate T-cell proliferation, survival and Fas-mediated activation-induced cell death. Immunity 11, 281–288 (1999).
26. Venkitaraman, A. R. & Cowling, R. J. Interleukin-7 inducesthe association of phosphatidylinositol 3-kinase with the α-chain of the interleukin-7 receptor. Eur. J. Immunol. 24,2168–2174 (1994).
27. Porter, B. O., Scibelli, P. & Malek, T. R. Control of T-celldevelopment in vivo by subdomains within the IL-7receptor α-chain cytoplasmic tail. J. Immunol. 166,262–269 (2001).
28. Guthridge, M. A. et al. Site-specific serine phosphorylationof the IL-3 receptor is required for hemopoietic cell survival.Mol. Cell 6, 99–108 (2000).
29. Butler, M. P. et al. Analysis of PTEN mutations and deletionsin B-cell non-Hodgkin’s lymphomas. Genes ChromosomesCancer 24, 322–327 (1999).
30. Suzuki, A. et al. T-cell-specific loss of Pten leads to defectsin central and peripheral tolerance. Immunity 14, 523–534(2001).
31. Fruman, D. A. et al. Impaired B-cell development andproliferation in absence of phosphoinositide 3-kinase p85α.Science 283, 393–397 (1999).
32. Sasaki, T. et al. Function of PI3Kγ in thymocytedevelopment, T-cell activation and neutrophil migration.Science 287, 1040–1046 (2000).
33. Exley, M. & Varticovski, L. Evidence for phosphatidylinositol3-kinase-dependent T-cell antigen receptor (TCR) signaltransduction. Mol. Immunol. 34, 221–226 (1997).
34. Eder, A. M. et al. Phosphoinositide 3-kinase regulation of T-cell-receptor-mediated interleukin-2 gene expression innormal T cells. J. Biol. Chem. 273, 28025–28031 (1998).
35. Kelly, E. et al. IL-2 and related cytokines can promote T-cellsurvival by activating AKT. J. Immunol. 168, 597–603(2002).
36. Parsons, M. J. et al. Expression of active protein kinase B in T cells perturbs both T- and B-cell homeostasis andpromotes inflammation. J. Immunol. 167, 42–48 (2001).
37. Plas, D. R. et al. Akt and Bcl-xL promote growth-factor-independent survival through distinct effects onmitochondrial physiology. J. Biol. Chem. 276, 12041–12048(2001).
38. Voll, R. E. et al. NF-κB activation by the pre-T-cell receptorserves as a selective survival signal in T-lymphocytedevelopment. Immunity 13, 677–689 (2000).
39. Karin, M. & Lin, A. NF-κB at the crossroads of life and death.Nature Immunol. 3, 221–227 (2002).
40. Pohl, T. et al. The combined absence of NF-κB1 and c-Relreveals that overlapping roles for these transcription factorsin the B-cell lineage are restricted to the activation andfunction of mature cells. Proc. Natl Acad. Sci. USA 99,4514–4519 (2002).
41. Dexter, T. M., Heyworth, C. M. & Whetton, A. D. The role of haemopoietic cell growth factor (interleukin-3) in thedevelopment of haemopoietic cells. Ciba Found. Symp.116, 129–147 (1985).
42. Garland, J. M. & Halestrap, A. Energy metabolism duringapoptosis. Bcl-2 promotes survival in hematopoietic cellsinduced to apoptose by growth factor withdrawal bystabilizing a form of metabolic arrest. J. Biol. Chem. 272,4680–4688 (1997).
43. Summers, S. A. & Birnbaum, M. J. A role for theserine/threonine kinase, Akt, in insulin-stimulated glucoseuptake. Biochem. Soc. Trans. 25, 981–988 (1997).
44. Plas, D. R. & Thompson, C. B. Cell metabolism in theregulation of programmed cell death. Trends Endocrinol.Metab. 13, 75–78 (2002).
45. Cho, H. et al. Akt1/PKBα is required for normal growth butdispensable for maintenance of glucose homeostasis inmice. J. Biol. Chem. 276, 38349–38352 (2001).
46. Vander, H. M. et al. Growth factors can influence cell growthand survival through effects on glucose metabolism. Mol.Cell. Biol. 21, 5899–5912 (2001).This article provides a thorough evaluation of the role of glucose metabolism in the survival of IL-3-dependent cells, comparing the effects of glucosewithdrawal with loss of cytokine signalling in a cellline.
47. Frank, S. et al. The role of dynamin-related protein 1, amediator of mitochondrial fission, in apoptosis. Dev. Cell1, 515–525 (2001).
48. Desagher, S. & Martinou, J. C. Mitochondria as the centralcontrol point of apoptosis. Trends Cell. Biol. 10, 369–377(2000).
49. Puceat, M. pHi regulatory ion transporters: an update onstructure, regulation and cell function. Cell. Mol. Life Sci.55, 1216–1229 (1999).
50. Fliegel, L. et al. Regulation and characterization of theNa+/H+ exchanger. Biochem. Cell. Biol. 76, 735–741 (1998).
51. Takahashi, E. et al. p90(RSK) is a serum-stimulated Na+/H+
exchanger isoform-1 kinase. Regulatory phosphorylation ofserine 703 of Na+/H+ exchanger isoform-1. J. Biol. Chem.274, 20206–20214 (1999).
52. Lehoux, S. et al. 14-3-3 binding to Na+/H+ exchangerisoform-1 is associated with serum-dependent activation ofNa+/H+ exchange. J. Biol. Chem. 276, 15794–15800(2001).
53. Khaled, A. R. et al. Trophic factor withdrawal: p38 mitogen-activated protein kinase activates NHE1, which inducesintracellular alkalinization. Mol. Cell. Biol. 21, 7545–7557(2001).A dissection of the intracellular signalling pathwaythat is activated by cytokine withdrawal that induces arise in cytosolic pH.
54. Kummer, J. L., Rao, P. K. & Heidenreich, K. A. Apoptosisinduced by withdrawal of trophic factors is mediated by p38mitogen-activated protein kinase. J. Biol. Chem. 272,20490–20494 (1997).
55. Rajnavolgyi, E. et al. IL-7 withdrawal induces a stresspathway activating p38 and Jun N-terminal kinases. Cell. Signal. 14, 761–769 (2002).
56. Kusuhara, M. et al. p38 kinase is a negative regulator ofangiotensin II signal transduction in vascular smooth musclecells: effects on Na+/H+ exchange and ERK1/2. Circ. Res.83, 824–831 (1998).
57. Khaled, A. R. et al. Interleukin-3 withdrawal induces anearly increase in mitochondrial membrane potentialunrelated to the Bcl-2 family. Roles of intracellular pH, ADPtransport and F(0)F(1)-ATPase. J. Biol. Chem. 276,6453–6462 (2001).
58. Khaled, A. R. et al. Withdrawal of IL-7 induces Baxtranslocation from cytosol to mitochondria through a rise in intracellular pH. Proc. Natl Acad. Sci. USA 96,14476–14481 (1999).
59. Rebollo, A. et al. Apoptosis induced by IL-2 withdrawal isassociated with an intracellular acidification. Exp. Cell Res.218, 581–585 (1995).
the sense that increases in cell number are a function ofboth replication and survival — for example, a Bcl-2transgene might protect from cell death, but also slowreplication, and so have little net effect. The cell-cycleeffects also raise additional questions about the intracel-lular pathways that lead to proliferation versus survival.Does a low level of receptor signalling ensure survival,whereas a high level induces cycling?
In conclusion, there have been important advances inidentifying the extracellular stimuli that regulate lympho-cyte homeostasis — namely,cytokines and peptide–MHCcomplexes. Probing the intracellular pathways that con-trol the life and death of lymphocytes has yielded someimportant potential components recently, but comparedwith the FAS-mediated death pathway,much is unknown,and the metabolic effects, in particular, are unexplored.

© 2002 Nature Publishing GroupNATURE REVIEWS | IMMUNOLOGY VOLUME 2 | NOVEMBER 2002 | 829
R E V I E W S
60. Matsuyama, S. et al. Changes in intramitochondrial andcytosolic pH: early events that modulate caspase activationduring apoptosis. Nature Cell Biol. 2, 318–325 (2000).
61. Famulski, K. S. et al. Activation of a low pH-dependentnuclease by apoptotic agents. Cell Death Differ. 6, 281–289(1999).
62. Waterhouse, N. J. et al. Cytochrome c maintainsmitochondrial transmembrane potential and ATP generationafter outer mitochondrial membrane permeabilization duringthe apoptotic process. J. Cell Biol. 153, 319–328 (2001).
63. Marzo, I. et al. Caspases disrupt mitochondrial membranebarrier function. FEBS Lett. 427, 198–202 (1998).
64. Chao, D. T. & Korsmeyer, S. J. BCL-2 family: regulators ofcell death. Annu. Rev. Immunol. 16, 395–419 (1998).
65. Cory, S. Regulation of lymphocyte survival by the Bcl-2 genefamily. Annu. Rev. Immunol. 13, 513–543 (1995).
66. Vaux, D. L., Cory, S. & Adams, J. M. Bcl-2 gene promoteshaemopoietic cell survival and cooperates with c-myc toimmortalize pre-B cells. Nature 335, 440–442 (1988).
67. O’Reilly, L. A., Huang, D. C. & Strasser, A. The cell-deathinhibitor Bcl-2 and its homologues influence control of cell-cycle entry. EMBO J. 15, 6979–6990 (1996).
68. Veis, D. J. et al. Bcl-2-deficient mice demonstrate fulminantlymphoid apoptosis, polycystic kidneys and hypopigmentedhair. Cell 75, 229–240 (1993).
69. Matsuzaki, Y. et al. Role of bcl-2 in the development oflymphoid cells from the hematopoietic stem cell. Blood 89,853–862 (1997).
70. Motoyama, N. et al. Massive cell death of immaturehematopoietic cells and neurons in Bcl-X-deficient mice.Science 267, 1506–1510 (1995).
71. Sentman, C. L. et al. Bcl-2 inhibits multiple forms ofapoptosis, but not negative selection, in thymocytes. Cell 67, 879–888 (1991).
72. Strasser, A. et al. Lessons from Bcl-2-transgenic mice forimmunology, cancer biology and cell-death research.Behring Inst. Mitt. 97, 101–117 (1996).
73. Ogilvy, S. et al. Constitutive Bcl-2 expression throughout thehematopoietic compartment affects multiple lineages andenhances progenitor-cell survival. Proc. Natl Acad. Sci. USA96, 14943–14948 (1999).Expression of a Bcl-2 transgene under the control of aVav promoter enabled the authors to show the effectsof overexpression of Bcl-2 on cells of the immunesystem.
74. Robles, A. I. et al. Expression of cyclin D1 in epithelial tissuesof transgenic mice results in epidermal hyperproliferationand severe thymic hyperplasia. Proc. Natl Acad. Sci. USA93, 7634–7638 (1996).
75. Akashi, K. et al. Bcl-2 rescues T lymphopoiesis ininterleukin-7-receptor-deficient mice. Cell 89, 1033–1041(1997).
76. Maraskovsky, E. et al. Bcl-2 can rescue T-lymphocytedevelopment in interleukin-7-receptor-deficient mice but notin mutant Rag-1−/− mice. Cell 89, 1011–1019 (1997).
77. Rodewald, H. R., Waskow, C. & Haller, C. Essentialrequirement for c-kit and common γ-chain in thymocytedevelopment cannot be overruled by enforced expression of Bcl-2. J. Exp. Med. 193, 1431–1437 (2001).
78. Oltvai, Z. N., Milliman, C. L. & Korsmeyer, S. J. Bcl-2heterodimerizes in vivo with a conserved homolog, Bax, thataccelerates programmed cell death. Cell 74, 609–619(1993).
79. Wolter, K. G. et al. Movement of Bax from the cytosol tomitochondria during apoptosis. J. Cell Biol. 139, 1281–1292(1997).
80. Goping, I. S. et al. Regulated targeting of BAX tomitochondria. J. Cell Biol. 143, 207–215 (1998).
81. Knudson, C. M. et al. Bax-deficient mice with lymphoidhyperplasia and male germ-cell death. Science 270, 96–99(1995).
82. Wen, R. et al. Jak3 selectively regulates Bax and Bcl-2expression to promote T-cell development. Mol. Cell. Biol.21, 678–689 (2001).
83. Khaled, A. R. et al. Bax deficiency partially correctsinterleukin-7 receptor-α deficiency. Immunity (in the press).
84. Lindsten, T. et al. The combined functions of proapoptoticBcl-2 family members bak and bax are essential for normaldevelopment of multiple tissues. Mol. Cell 6, 1389–1399(2000).A discussion of the phenotype of Bax and Bak double-deficient mice in terms of the effects of loss of thesepro-apoptotic proteins on the development of afunctional immune system.
85. Wei, M. C. et al. Proapoptotic BAX and BAK: a requisitegateway to mitochondrial dysfunction and death. Science292, 727–730 (2001).
86. Goldrath, A. W., Bogatzki, L. Y. & Bevan, M. J. Naive T cellstransiently acquire a memory-like phenotype duringhomeostasis-driven proliferation. J. Exp. Med. 192,557–564 (2000).
87. Bouillet, P. et al. Proapoptotic Bcl-2 relative Bim required forcertain apoptotic responses, leukocyte homeostasis and topreclude autoimmunity. Science 286, 1735–1738 (1999).The phenotype of Bim-deficient mice is discussed inrelation to apoptosis and immune cells.
88. Bouillet, P. et al. BH3-only Bcl-2 family member Bim isrequired for apoptosis of autoreactive thymocytes. Nature415, 922–926 (2002).
89. Yang, E. et al. Bad, a heterodimeric partner for Bcl-XL andBcl-2, displaces Bax and promotes cell death. Cell 80,285–291 (1995).
90. Mok, C. L. et al. Bad can act as a key regulator of T-cellapoptosis and T-cell development. J. Exp. Med. 189,575–586 (1999).
91. Downward, J. How BAD phosphorylation is good forsurvival. Nature Cell Biol. 1, E33–E35 (1999).
92. Kim, K. et al. The trophic action of IL-7 on pro-T cells:inhibition of apoptosis of pro-T1,-T2 and -T3 cells correlateswith Bcl-2 and Bax levels and is independent of Fas andp53 pathways. J. Immunol. 160, 5735–5741 (1998).
93. Zheng, T. S. & Flavell, R. A. Divinations and surprises:genetic analysis of caspase function in mice. Exp. Cell. Res.256, 67–73 (2000).
94. Doerfler, P., Forbush, K. A. & Perlmutter, R. M. Caspaseenzyme activity is not essential for apoptosis duringthymocyte development. J. Immunol. 164, 4071–4079(2000).
95. Wang, J. & Lenardo, M. J. Roles of caspases in apoptosis,development and cytokine maturation revealed byhomozygous gene deficiencies. J. Cell. Sci. 113, 753–757(2000).
96. Shin, M. S. et al. Inactivating mutations of CASP10 gene innon-Hodgkin lymphomas. Blood 99, 4094–4099 (2002).
97. Zou, H. et al. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependentactivation of caspase-3. Cell 90, 405–413 (1997).
98. Hu, Y. et al. Role of cytochrome c and dATP/ATP hydrolysisin Apaf-1-mediated caspase-9 activation and apoptosis.EMBO J. 18, 3586–3595 (1999).
99. Adrain, C. & Martin, S. J. The mitochondrial apoptosome: a killer unleashed by the cytochrome seas. Trends Biochem.Sci. 26, 390–397 (2001).
100. Cardone, M. H. et al. Regulation of cell-death proteasecaspase-9 by phosphorylation. Science 282, 1318–1321(1998).
101. Hakem, R. et al. Differential requirement for caspase 9 inapoptotic pathways in vivo. Cell 94, 339–352 (1998).
102. Hara, H. et al. The apoptotic protease-activating factor-1-mediated pathway of apoptosis is dispensable for negativeselection of thymocytes. J. Immunol. 168, 2288–2295(2002).
103. Bidere, N. & Senik, A. Caspase-independent apoptoticpathways in T lymphocytes: a minireview. Apoptosis6, 371–375 (2001).
104. Joza, N. et al. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature 410,549–554 (2001).
105. Hegde, R. et al. Identification of Omi/HtrA2 as amitochondrial apoptotic serine protease that disruptsinhibitor of apoptosis protein–caspase interaction. J. Biol.Chem. 277, 432–438 (2002).
106. Deveraux, Q. L. et al. IAPs block apoptotic events inducedby caspase-8 and cytochrome c by direct inhibition ofdistinct caspases. EMBO J. 17, 2215–2223 (1998).
107. Du, C. et al. Smac, a mitochondrial protein that promotescytochrome c-dependent caspase activation by eliminatingIAP inhibition. Cell 102, 33–42 (2000).
108. Harlin, H. et al. Characterization of XIAP-deficient mice. Mol.Cell. Biol. 21, 3604–3608 (2001).
109. Geiselhart, L. A. et al. IL-7 administration alters theCD4:CD8 ratio, increases T-cell numbers and increases T-cell function in the absence of activation. J. Immunol. 166,3019–3027 (2001).
110. Komschlies, K. L. et al. Administration of recombinanthuman IL-7 to mice alters the composition of B-lineage cellsand T-cell subsets, enhances T-cell function and inducesregression of established metastases. J. Immunol. 152,5776–5784 (1994).
111. Mackall, C. L. et al. IL-7 increases both thymic-dependentand thymic-independent T-cell regeneration after bone-marrow transplantation. Blood 97, 1491–1497 (2001).
112. Maki, K. et al. Interleukin-7-receptor-deficient mice lack γδT cells. Proc. Natl Acad. Sci. USA 93, 7172–7177 (1996).
113. Candeias, S. et al. Defective T-cell receptor γ generearrangement in interleukin-7 receptor knockout mice.Immunol. Lett. 57, 9–14 (1997).
114. Gross, J. A. et al. TACI and BCMA are receptors for a TNFhomologue implicated in B-cell autoimmune disease. Nature404, 995–999 (2000).
115. Surh, C. D. & Sprent, J. Regulation of naive and memory T-cell homeostasis. Microbes Infect. 4, 51–56 (2002).
116. Sprent, J. & Surh, C. D. T-cell memory. Annu. Rev. Immunol.20, 551–579 (2002).An excellent review of recent findings on the originsand regulation of memory T cells.
117. Marks-Konczalik, J. et al. IL-2-induced activation-inducedcell death is inhibited in IL-15-transgenic mice. Proc. NatlAcad. Sci. USA 97, 11445–11450 (2000).
118. Rich, B. E. et al. Cutaneous lymphoproliferation andlymphomas in interleukin-7-transgenic mice. J. Exp. Med.177, 305–316 (1993).
119. Mertsching, E., Burdet, C. & Ceredig, R. IL-7-transgenicmice: analysis of the role of IL-7 in the differentiation ofthymocytes in vivo and in vitro. Int. Immunol. 7, 401–414(1995).
120. Fehniger, T. A. et al. Fatal leukemia in interleukin-15-transgenic mice follows early expansions in natural killer and memory phenotype CD8+ T cells. J. Exp. Med. 193,219–231 (2001).
121. Thomis, D. C. & Berg, L. J. Peripheral expression of Jak3 isrequired to maintain T-lymphocyte function. J. Exp. Med.185, 197–206 (1997).
122. Dumon, S. et al. IL-3-dependent regulation of Bcl-xL geneexpression by STAT5 in a bone-marrow-derived cell line.Oncogene 18, 4191–4199 (1999).
123. Heckman, C. A., Mehew, J. W. & Boxer, L. M. NF-κBactivates Bcl-2 expression in t(14;18) lymphoma cells.Oncogene 21, 3898–3908 (2002).
124. Cantrell, D. Protein kinase B (Akt) regulation and function in T lymphocytes. Semin. Immunol. 14, 19–26 (2002).
125. del-Peso, L. et al. Interleukin-3-induced phosphorylation ofBAD through the protein kinase Akt. Science 278, 687–689(1997).
126. Griffiths, G. J. et al. Cellular damage signals promotesequential changes at the N-terminus and BH-1 domain ofthe pro-apoptotic protein Bak. Oncogene 20, 7668–7676(2001).
127. O’Connor, L. et al. Bim: a novel member of the Bcl-2 familythat promotes apoptosis. EMBO J. 17, 384–395 (1998).
128. Puthalakath, H. et al. The proapoptotic activity of the Bcl-2family member Bim is regulated by interaction with thedynein motor complex. Mol. Cell. 3, 287–296 (1999).
129. Dijkers, P. F. et al. Expression of the pro-apoptotic Bcl-2family member Bim is regulated by the forkheadtranscription factor FKHR-L1. Curr. Biol. 10, 1201–1204(2000).
130. Sugiyama, T. et al. Activation of mitochondrial voltage-dependent anion channel by a pro-apoptotic BH3-onlyprotein Bim. Oncogene 21, 4944–4956 (2002).
131. Khaled, A. R. & Durum, S. From cytosol to mitochondria: theBax translocation story. J. Biochem. Mol. Biol. 34, 391–394(2001).
132. Belaud-Rotureau, M. A. et al. Early transitory rise inintracellular pH leads to Bax conformation change duringceramide-induced apoptosis. Apoptosis 5, 551–560 (2000).
133. Tsuruta, F., Masuyama, N. & Gotoh, Y. Thephosphatidylinositol 3-kinase (PI3K)–Akt pathwaysuppresses Bax translocation to mitochondria. J. Biol.Chem. 277, 14040–14047 (2002).
134. Desagher, S. et al. Bid-induced conformational change ofBax is responsible for mitochondrial cytochrome c releaseduring apoptosis. J. Cell Biol. 144, 891–901 (1999).
135. Marani, M. et al. Identification of novel isoforms of the BH3domain protein Bim which directly activate Bax to triggerapoptosis. Mol. Cell. Biol. 22, 3577–3589 (2002).
136. Zong, W. X. et al. BH3-only proteins that bind pro-survivalBcl-2-family members fail to induce apoptosis in the absenceof Bax and Bak. Genes Dev. 15, 1481–1486 (2001).
137. Saito, M., Korsmeyer, S. J. & Schlesinger, P. H. BAX-dependent transport of cytochrome c reconstituted in pureliposomes. Nature Cell Biol. 2, 553–555 (2000).
138. Shimizu, S., Narita, M. & Tsujimoto, Y. Bcl-2-family proteinsregulate the release of apoptogenic cytochrome c by themitochondrial channel VDAC. Nature 399, 483–487 (1999).
139. Leonard, W. J. The molecular basis of X-linked severecombined immunodeficiency: defective cytokine-receptorsignaling. Annu. Rev. Med. 47, 229–239 (1996).
140. DiSanto, J. P. et al. Lymphoid development in mice with atargeted deletion of the interleukin-2 receptor γ-chain. Proc.Natl Acad. Sci. USA 92, 377–381 (1995).

© 2002 Nature Publishing Group830 | NOVEMBER 2002 | VOLUME 2 www.nature.com/reviews/immunol
R E V I E W S
141. von-Freeden-Jeffry, U. et al. Lymphopenia in interleukin (IL)-7gene-deleted mice identifies IL-7 as a nonredundantcytokine. J. Exp. Med. 181, 1519–1526 (1995).
142. Peschon, J. J. et al. in Cytokine Knockouts (eds Durum, S. K.& Muegge, K.) 37–52 (Humana, Totowa, New Jersey, 1998).
143. He, Y. W. & Malek, T. R. Interleukin-7 receptor α is essentialfor the development of γδ+ T cells, but not natural killer cells.J. Exp. Med. 184, 289–293 (1996).
144. Thomis, D. C. et al. Defects in B-lymphocyte maturation andT-lymphocyte activation in mice lacking Jak3. Science 270,794–797 (1995).
145. Rolink, A. G. & Melchers, F. BAFFled B cells survive andthrive: roles of BAFF in B-cell development. Curr. Opin.Immunol. 14, 266–275 (2002).
146. Walsh, C. M. et al. A role for FADD in T-cell activation anddevelopment. Immunity 8, 439–449 (1998).
147. Kabra, N. H. et al. T-cell-specific FADD-deficient mice: FADDis required for early T-cell development. Proc. Natl Acad. Sci.USA 98, 6307–6312 (2001).
148. Suzuki, H. et al. Xid-like immunodeficiency in mice withdisruption of the p85α subunit of phosphoinositide 3-kinase.Science 283, 390–392 (1999).
149. Chen, W. S. et al. Growth retardation and increasedapoptosis in mice with homozygous disruption of the Akt1gene. Genes Dev. 15, 2203–2208 (2001).
150. Brady, H. J. et al. T cells from Baxα-transgenic mice showaccelerated apoptosis in response to stimuli but do notshow restored DNA damage-induced cell death in theabsence of p53. gene product in. EMBO J. 15, 1221–1230(1996).
151. Sadlack, B. et al. Development and proliferation oflymphocytes in mice deficient for both interleukins-2 and -4. Eur. J. Immunol. 24, 281–284 (1994).
152. Strasser, A. et al. The role of bim, a proapoptotic BH3-onlymember of the Bcl-2 family in cell-death control. Ann. NYAcad. Sci. 917, 541–548 (2000).
153. Innes, K. M. et al. Retroviral transduction of enrichedhematopoietic stem cells allows lifelong Bcl-2 expression inmultiple lineages but does not perturb hematopoiesis. Exp.Hematol. 27, 75–87 (1999).
154. Shull, M. M. et al. Targeted disruption of the mousetransforming growth factor-β1 gene results in multifocalinflammatory disease. Nature 359, 693–699 (1992).
155. Gorelik, L. & Flavell, R. A. Abrogation of TGFβ signaling in T cells leads to spontaneous T-cell differentiation andautoimmune disease. Immunity 12, 171–181 (2000).
156. Tivol, E. A. et al. Loss of CTLA-4 leads to massivelymphoproliferation and fatal multiorgan tissue destruction,revealing a critical negative regulatory role of CTLA-4.Immunity 3, 541–547 (1995).
Online links
DATABASESThe following terms in this article are linked online to:Cancer.gov: http://www.cancer.gov/search/leukaemia | lymphomaInterPro: http://www.ebi.ac.uk/interpro/BH1–BH3 | BH4LocusLink: http://www.ncbi.nlm.nih.gov/LocusLink/4-1BB | 4-1BBL | AIF | AKT | APAF1 | βc | B7 | BAD | BAFF | BAK | BAX | BCL-2 | BCL-W | BCL-XL | BID | BIK | BIM | BLK | BOK | caspase-3 | caspase-7 | caspase-8 | caspase-9 | caspase-10 | CD28 | CD40 | CD40L | CREB | c-Rel | ERK | FAS | FASL | FKHRL1 | FLICE | FLIP | γc | GLUT1 | GLUT4 | GM-CSF | GSK3 | HRK | IL-2 | IL-2Rα | IL-3 | IL-4 | IL-5 | IL-6 | IL-7 | IL-7Rα | IL-9 | IL-15 | IL-15Rα | IL-21 | JAK3 | LC8 | MCL1 | MEK1 | NHE | OMI | p38 MAPK | p85α | p90 RSK | p110 | PI3K | PTEN | SHC | STAT5 | TGF-β | TNF | TRAF1 | TRAF2 | Vav | XIAPOMIM: http://www.ncbi.nlm.nih.gov/Omim/ALPSAccess to this interactive links box is free online.




















