
Recombinase, chromosomal translocations and lymphoid neoplasia: Targeting mistakes and repair...
13
dna repair 5 ( 2 0 0 6 ) 1246–1258 available at www.sciencedirect.com journal homepage: www.elsevier.com/locate/dnarepair Recombinase, chromosomal translocations and lymphoid neoplasia: Targeting mistakes and repair failures Rodrig Marculescu a,1 , Katrina Vanura a , Bertrand Montpellier b , Sandrine Roulland b , Trang Le a , Jean-Marc Navarro b , Ulrich J ¨ ager a , Fraser McBlane c , Bertrand Nadel b,∗ a Department of Internal Medicine I, Division of Hematology, Medical University of Vienna, W¨ ahringer G ¨ urtel 18-20, A-1090 Vienna, Austria b Centre d’Immunologie de Marseille-Luminy, INSERM, CNRS, Universit´ e de la M´ editerran´ ee, Parc Scientifique de Luminy, Case 906, 13288 Marseille Cedex 9, France c European Institute of Oncology, Via Ripamonti 435, Milan 02141, Italy article info Article history: Published on line 23 June 2006 Keywords: Illegitimate V(D)J recombination Cryptic recombination site Double-strand break repair Non-homologous end-joining Chromosomal translocation T-cell acute lymphoblastic leukemia Follicular lymphoma Abbreviations: BE, broken end; BL, Burkitt’s lymphoma; B-NHL, B-cell Non-Hodgkin’s lymphoma; CE, coding end; CJ, coding joint; CSR, class-switch recombination; DSB, double-strand break; NHEJ, non-homologous end-joining; SHM, somatic hypermutation; HJ, hybrid joint; RSS, recombination signal sequence; SE, signal end; SJ, signal joint; SDSA, synthesis-dependant strand annealing; RAG, recombination activating gene; T-ALL, T-cell acute lymphoblastic leukemia; TdT, terminal deoxynucleotidyl transferase abstract A large number of lymphoid malignancies is characterized by specific chromosomal translo- cations, which are closely linked to the initial steps of pathogenesis. The hallmark of these translocations is the ectopic activation of a silent proto-oncogene through its relocation at the vicinity of an active regulatory element. Due to the unique feature of lymphoid cells to somatically rearrange and mutate receptor genes, and to the corresponding strong activ- ity of the immune enhancers/promoters at that stage of cell development, B- and T-cell differentiation pathways represent propitious targets for chromosomal translocations and oncogene activation. Recent progress in the understanding of the V(D)J recombination pro- cess has allowed a more accurate definition of the translocation mechanisms involved, and has revealed that V(D)J-mediated translocations result both from targeting mistakes of the recombinase, and from illegitimate repair of the V(D)J recombination intermediates. Sur- prisingly, V(D)J-mediated translocations turn out to be restricted to two specific sub-types of lymphoid malignancies, T-cell acute lymphoblastic leukemias, and a restricted set of mature B-cell Non-Hodgkin’s lymphomas. © 2006 Elsevier B.V. All rights reserved. ∗ Corresponding author. Tel.: +33 4 91 26 94 73; fax: +33 4 91 26 94 30. E-mail address: [email protected] (B. Nadel). 1 Present address: Department of Laboratory Medicine, Medical University of Vienna, W¨ ahringer G ¨ urtel 18-20, A-1090 Vienna, Austria. 1568-7864/$ – see front matter © 2006 Elsevier B.V. All rights reserved. doi:10.1016/j.dnarep.2006.05.015
-
Upload
independent -
Category
Documents
-
view
4 -
download
0
Transcript of Recombinase, chromosomal translocations and lymphoid neoplasia: Targeting mistakes and repair...

d n a r e p a i r 5 ( 2 0 0 6 ) 1246–1258
avai lab le at www.sc iencedi rec t .com
journa l homepage: www.e lsev ier .com/ locate /dnarepai r
Recombinase, chromosomal translocations and lymphoidneoplasia: Targeting mistakes and repair failures
Rodrig Marculescua,1, Katrina Vanuraa, Bertrand Montpellierb, Sandrine Roullandb,Trang Lea, Jean-Marc Navarrob, Ulrich Jagera, Fraser McBlanec, Bertrand Nadelb,∗
a Department of Internal Medicine I, Division of Hematology, Medical University of Vienna,Wahringer Gurtel 18-20, A-1090 Vienna, Austriab Centre d’Immunologie de Marseille-Luminy, INSERM, CNRS, Universite de la Mediterranee,Parc Scientifique de Luminy, Case 906, 13288 Marseille Cedex 9, Francec European Institute of Oncology, Via Ripamonti 435, Milan 02141, Italy
a r t i c l e i n f o
Article history:
Published on line 23 June 2006
Keywords:
Illegitimate V(D)J recombination
Cryptic recombination site
Double-strand break repair
Non-homologous end-joining
Chromosomal translocation
T-cell acute lymphoblastic leukemia
Follicular lymphoma
Abbreviations:
BE, broken end; BL, Burkitt’s
lymphoma; B-NHL, B-cell
Non-Hodgkin’s lymphoma; CE,
coding end; CJ, coding joint; CSR,
class-switch recombination; DSB,
double-strand break; NHEJ,
non-homologous end-joining; SHM,
somatic hypermutation; HJ, hybrid
joint; RSS, recombination signal
sequence; SE, signal end; SJ, signal
a b s t r a c t
A large number of lymphoid malignancies is characterized by specific chromosomal translo-
cations, which are closely linked to the initial steps of pathogenesis. The hallmark of these
translocations is the ectopic activation of a silent proto-oncogene through its relocation at
the vicinity of an active regulatory element. Due to the unique feature of lymphoid cells to
somatically rearrange and mutate receptor genes, and to the corresponding strong activ-
ity of the immune enhancers/promoters at that stage of cell development, B- and T-cell
differentiation pathways represent propitious targets for chromosomal translocations and
oncogene activation. Recent progress in the understanding of the V(D)J recombination pro-
cess has allowed a more accurate definition of the translocation mechanisms involved, and
has revealed that V(D)J-mediated translocations result both from targeting mistakes of the
recombinase, and from illegitimate repair of the V(D)J recombination intermediates. Sur-
prisingly, V(D)J-mediated translocations turn out to be restricted to two specific sub-types
of lymphoid malignancies, T-cell acute lymphoblastic leukemias, and a restricted set of
mature B-cell Non-Hodgkin’s lymphomas.
© 2006 Elsevier B.V. All rights reserved.
joint; SDSA, synthesis-dependant
strand annealing; RAG,
recombination activating gene;
T-ALL, T-cell acute lymphoblastic
leukemia; TdT, terminal
deoxynucleotidyl transferase
∗ Corresponding author. Tel.: +33 4 91 26 94 73; fax: +33 4 91 26 94 30.E-mail address: [email protected] (B. Nadel).
1 Present address: Department of Laboratory Medicine, Medical Univ1568-7864/$ – see front matter © 2006 Elsevier B.V. All rights reserved.doi:10.1016/j.dnarep.2006.05.015
ersity of Vienna, Wahringer Gurtel 18-20, A-1090 Vienna, Austria.

0 0 6 ) 1246–1258 1247
1
Lwonayildroiimtfmtmmrrtaseiridc
2
DfracTirraaotDontssrta[
Fig. 1 – V(D)J recombination model. (a) Each V, D and J genesegment (rectangle) from the IG/TCR loci is flanked by a RSS(triangle). The RSS allows the recruitment, binding andproper positioning of the RAG proteins (pink ellipses). TheRAG1/RAG2 proteins perform a first single strand nick at theexact border between the coding segment and the RSS, andcapture a second RSS. (b) This leads to the formation of asynaptic complex in which the two DNA/protein structuresare held in close juxtaposition, and in which a nick at thesecond RSS occurs. Within this complex, RAG1/RAG2catalyze a trans-esterification reaction in which eachhydroxyl group attacks the opposite DNA strands. (c) Thisprocess generates four broken ends held in a post-cleavagesynaptic complex (PCSC): two blunt RSSs or signal ends (SE)and two covalently sealed hairpin coding ends (CE). Thecomponents of the NHEJ pathway are recruited in thesynapse. These consist of the DNA-dependent proteinkinase (DNA-PK) complex, which includes DNA-PKcs, theKu70/Ku80 nuclear antigens (involved in the binding ofdistorted DNA structures such as DSB and DNA hairpin),and Artemis. (d) The second phase of the process isasymmetric: DNA hairpins are nicked by Artemis, resolvedinto free ends, and extensively “processed” before ligation.Hairpins opened at bases nearby the apex generateoverhanging flaps, which, if filled-in by a DNA polymeraseactivity, form palindromic (P) stretches. Also, nucleotidesmay be added de novo by the TdT (N), or may be deletedfrom the CEs (�). (e) The XRCC4/DNA ligase IV complexcarries out the ligation step. The two SEs are generallyjoined to form a signal joint (SJ), while the ligation of theprocessed CEs form a highly diversified coding joint (CJ). (f)Following ligation, both SJ and CJ dissociate from thesynaptic complex. The CJ corresponds to the functional
d n a r e p a i r 5 ( 2
. Introduction
ymphoid neoplasms are cancers of the immune system,hich afflict both adults and children, and account for 6–10%f all neoplastic diseases [1]. The development of lymphoideoplasia is a complex multistep process of genetic alterationsnd cellular transformations, which can take more than 20ears to progress into invasive and metastatic tumor. Alarm-ngly, and despite indisputable success achieved during theast decades in the treatment of these malignancies, their inci-ence in industrialized countries has been increasing moreapidly than that of most other tumors. In parallel to the needf further improving therapy, this underscores the increasing
mportance of developing preventive programs based on thedentification of risk factors, “at risk” individuals, and on the
onitoring of patients at early stages of the disease. However,his approach requires prior in-depth understanding of theundamental nature of the disease process, and of the actual
echanisms by which lymphoid cells undergo neoplastic ini-iation and progression. The complexity of the pathogenic
echanism in lymphoid neoplasia is related to the funda-ental strategy of the immune system, in which gene rear-
angement ensures diversity and optimal function of the B-celleceptor and T-cell receptor (TCR) for the antigen. Inadver-ently, these mechanisms cause genetic instability, imposingconstant threat of malignant transformation mostly throughpecific and recurrent chromosomal translocations [2]. Unrav-ling the molecular mechanisms by which illegitimate eventsn the normal immune system cause lymphoid cancer, and theole played by genetic, environmental and iatrogenic factorsn this process, are central to the development of preventive,iagnostic, therapeutic and disease monitoring programs oflinical significance.
. V(D)J recombination
uring B-cell differentiation in the bone marrow and T-cell dif-erentiation in the thymus, V(D)J recombination generates theearrangement of non-contiguous V (variable), D (diversity)nd J (joining) gene segments, to form a complete VDJ exonoding for the variable region of the Immunoglobulin (IG) and-cell receptor (TCR) (see [3] and references therein for details,llustrated in Fig. 1). This unique mechanism of somaticecombination generates the extraordinary diversity of theeceptors to the antigens, which stands at the heart of thedaptive strategy of the immune system. This process requiresspecialized recombinase activity, catalyzed by the products
f the recombination activating gene 1&2 (RAG1 and RAG2),he recognition of specific target motifs flanking each of the V,
and J gene segments (the recombination signal sequencesr RSSs), and the recruitment of the proteins of the ubiquitouson-homologous end-joining (NHEJ) repair complex. It is clearhat such a mechanism, generating chromosomal double-trand breaks (DSBs), rearrangement between sequencesometimes located megabases apart, and loss of genetic mate-
ial, must be regulated at the highest level. V(D)J recombina-ion is indeed a highly orchestrated mechanism, regulated intissue-, lineage-, and developmental stage-specific manner4,5]. Regulatory elements located within each IG/TCR locus
product of the V(D)J recombination, encoding the variableregion of the B- and T-cell receptors. In contrast, the SJcorresponds to the by-product of the recombination, and isgenerally deleted out on an episomal circle.

( 2 0
1248 d n a r e p a i r 5act in cis to modulate the chromatin accessibility to the recom-binase, depending on the type and on the stage of lymphoiddifferentiation. Factors like nucleosomal positioning andhigher order chromatin structure are believed to play criticalroles in enforcing these choices, but relatively little is knownabout the forces regulating the fidelity of V(D)J recombination.
One obvious subversion of the normal V(D)J recombina-tion process is the possibility of rearrangements between seg-ments belonging to different IG/TCR loci. Normally, V(D)J join-ing involves gene segments located on the same chromosome,within a given immune receptor locus. Yet, three distinct IGand four distinct (or interspersed) TCR loci located on fourdifferent chromosomes in humans rely on the same recombi-nation machinery for normal intramolecular rearrangementof their gene segments. Despite hierarchical orders in recep-tor gene rearrangements, there are significant overlaps in therearrangement timing of several IG/TCR, and hybrid recom-bination between gene segments from different loci locatedeither on the same (e.g. �/�) or on different chromosomes (e.g.�/�, �/�) can be detected both ex vivo and in vivo [6–8]. Whenthe recombination takes place between RSSs located on dis-tinct chromosomes, a balanced V(D)J-mediated translocationensues, generating two derivative chromosomes. The recom-bination reaction in trans is believed to occur similarly as incis, including coupled cleavage, formation of a post-cleavagesynaptic complex (PCSC), and generation of the same proto-typical signal joints (SJ) and coding joints (CJ). Notably, sev-eral lines of evidence, including recurrent base loss from thesignal ends (SE) in the SJ [9,10] and reduced rates of CJ for-mation [11–13], suggest a possible destabilization of the trans-chromosomal PCSC, which might partly explain the drasticreduction in the rate of trans-V(D)J recombination comparedto the normal cis-reaction [6,8,11,12,14]. Nevertheless, trans-TCR/IG translocations are observed with surprisingly highrecurrence and frequency in vivo. In normal human and mousethymocytes, for example, it can be estimated that ∼1 cell outof 10,000 carries the D�3-J�2.7 trans-TCR rearrangement [7,15].A similar frequency can be found for the D�1-J�1 trans-TCRrearrangement. If one considers the large number of possiblecombinations of trans-IG/TCR rearrangements, and providingthat most of those translocations occur in distinct cells, theabsolute rate of V(D)J-mediated trans-IG/TCR rearrangementper cell in normal thymocytes might in fact be considerablyhigher. One could ask why the system would consent to sucha high rate of potentially damaging chromosomal transloca-tions. Interestingly, some of these rearrangements have beenshown not only to produce transcripts but also to generatefunctional chimeric receptor chains, both in T-cells [16] and inB-cells [8]. Thus, from the evolutionary perspective, the bene-fit of increasing the repertoire diversity might outbalance thedanger of genomic instability.
3. V(D)J-mediated translocations at crypticsites (type 1 translocations)
3.1. Cryptic site mis-targeting by the RAGs
The pathological counterpart of this capability of the V(D)Jrearrangement process to occur in trans, arises with another
0 6 ) 1246–1258
flexibility of the recombination system: the capacity ofthe RAG proteins to recognize, bind, synapse and cleavesequences which diverge from the “consensus” RSS. The RSSsnormally consist of a highly conserved heptamer motif (con-sensus sequence: 5′-CACAGTG-3′) and a conserved nonamersequence (consensus sequence: 5′-ACAAAAACC-3′) separatedby a poorly conserved spacer sequence of 12 or 23 ± 1 bp[17–19]. Efficient recombination occurs only between a RSScontaining a spacer of 12 bp and an RSS containing a spacer of23 bp. Nevertheless, most RSSs flanking V, D and J segmentscontain some degree of polymorphism in their sequence, andthis has been shown to be a major factor affecting the relativerepresentation of gene segments in the primary repertoire[17,18,20–22]. Depending on the position, alterations in theheptamer, nonamer or spacer sequences can be favorableor deleterious for the recombination. Furthermore, multiplevariations from consensus can result in synergistic effects[22], and the presence of the heptamer’s 5′-CAC-3′, which isoften considered as a sine qua non condition for a functionalRSS, is in fact not mandatory [17,18]. Paradoxically, thisflexibility of the RAG proteins to recognize a large panel ofvariant RSSs presents a considerable threat to the joiningfidelity and to genomic stability, as it increases as muchthe chance that fortuitous DNA sequences in the genome(also called “cryptic sites”) might be potentially targetedby mistake. In ex vivo functional studies, Lewis et al. [23]have demonstrated that cryptic sites with recombinationfrequency as low as 2 × 10−5 that of a consensus RSS can havea physiological impact in vivo, and that a substantial fractionof the estimated 10 million fortuitous sites in the diploidgenome could potentially mis-target the V(D)J recombinationprocess at ∼1% the range of canonical frequency. Subsequentbioinformatics studies have confirmed and extended theseresults, suggesting that such cryptic RSSs could be presentin the genome every 1–2 Kb in average. Cowell et al. [24]developed the ’recombination information content’ (RIC)algorithm to predict the functionality of RSS sequences(http://bioinformatics.duke.edu/dulci/rss/ricInterface.pl). Inthis model, a functional threshold score was set based on thesequence distribution of RSS associated with rearranging IGand TCR sequences, and correlated well with the functionalityof RSS tested in an extrachromosomal VDJ recombinationassay [25]. To provide a searchable database of cryptic RSSin mouse and human, we incorporated the RIC filter intogenome-wide searches performed using physiologic RSSmotifs http://bio.ifom-ieo-campus.it/rss. The total number ofcryptic RSS recovered was close to the density of 1 RSS/0.6 Kbpredicted by Lewis et al. [23] with no obvious selection for oragainst the presence of RSS in any genomic region (Fabbri,FMB et al., manuscript in preparation). Incredibly, the finalnumber of functional RAG recombinase targets may be evenhigher since both the genome searches and the RIC filteringwere performed using fairly stringent conditions. Using thepoorest RIC score of functionally rearranging IG/TCR loci asa more relaxed RIC threshold, the total number increasesto 8–10 million cryptic RSS in mouse and human genomes.
It should be noted that some of the cryptic RSS associatedwith oncogenic translocations fail even this relaxed RICthreshold (Table 1 and see below), suggesting that they appearin lymphoid neoplasia as rare and highly selected events. The
d n a r e p a i r 5 ( 2 0 0 6 ) 1246–1258 1249
Table 1 – V(D)J-mediated translocations resulting from a targeting mistake of the recombinase (type 1)
Translocation Tumorsub-type
IG/TCRpartner
Oncogene Cryptic site Cases # RIC score References
t(1;7)(q34;q34) T-ALL TCR� LCK CACACAC 12 GCCAAAACC 1 −35.23(P) [34,46]t(1;14)(p34;q11) T-ALL TCR� TAL1 CACACCG 22 CGAAAAAGG 3 −66.48(F) [15,69,70]t(7;9)(q34;q32) T-ALL TCR� TAL2 CACTGTG 13 ATAAAAATA Recurrent −39.28(F) [9,15,46]t(7;11)(q34;p13) T-ALL TCR� LMO2 CACAGTA 12 GCAATAATT Recurrent −38.45(P) [10,15,42]t(11;14)(p13;q11) T-ALL TCR� LMO2 CACAGTA 12 GCAATAATT Recurrent −38.45(P) [15,33,42,71]a
t(11;14)(p13;q11) T-ALL TCR� LMO2 CACGGTG 23 CGCTGGTTG 1 −61.60(F) [72–74]b
t(11;14)(p15;q11) T-ALL TCR� LMO1 CACAGTG 23 ACTCTGGCA 2 −61.18(F) [42,75,76]t(X;14)(q28;q11) T-PLL TCR� MTCP1 CACCGCC 23 CCCCAAACC 1 −54.68(P) [77]t(2;18)(p11;q21) B-CLL IGK BCL2vcr CACACTG 23 ACAAGTAAC 1 −53.40(P) [78]
Only cases with fulfilled type 1 criteria, AND sequence information on both breakpoints or with a cryptic site tested in functional assays or witha cryptic site passing the RIC score from the “RSS project database search” (http://bio.ifom-ieo-campus.it/rss) were considered. Cryptic sitesare shown, with positions divergent from consensus underlined. The number of cases is indicated when <5; use of a cryptic site is consideredrecurrent when ≥5 cases. RIC scores are indicated, P = pass, F = fail.
sctc
isbhtcam(eFtwtfcsoaRanRrta
3t
Ctbcp
a Dik, BN, Langerak et al. (manuscript in preparation).b Our unpublished results.
heer density of cryptic RSS therefore make it impossible toonfidently predict novel genes involved in V(D)J transloca-ions and lymphoid neoplasia merely by the presence of aryptic RSS.
With so many potentially hazardous recombinase targetsn their genome, how do lymphocytes ever survive their pas-age through RAG-expressing developmental stages? Accessi-ility of RAGs to their target sites in nuclei is thought to beighly regulated by controlled changes in chromatin struc-ure. However, hundreds of different loci will be in an ‘open’hromatin configuration at the time of RAG expression andll of these contain cryptic RSS. Using LMPCR to detect chro-osomal breaks, we found that a surprisingly large fraction
up to one-third) of cryptic RSS we tested had been cut byndogenous RAGs in a pool of primary thymocyte DNA (Furia,MB et al., unpublished). No correlation was seen with loca-ion relative to genes, nor the expression level of those genesithin the RAG-positive thymocyte pool. It remains possible
hat many sites are transiently cut and resealed, or that only araction of RAG-induced genome instability is harmful to theell and leads to cell death or transformation. The immuneystem may also be able to afford the loss of large numbersf developing lymphocytes following aberrant DNA breaks. Inddition, specific chromatin tags probably preferentially directAG proteins to the correct antigen receptor locus, favoringntigen receptor gene recombination despite the predomi-ance of cryptic RSS. Compact chromatin severely inhibitsAG cutting at RSS [26,27], and growing evidence suggests aole for specific patterns of histone modification and associa-ion of chromatin remodeling complexes in targeting RAGs tosubset of accessible loci [28,29].
.2. Two different types of oncogenic V(D)J-mediatedranslocations
onsidering the high frequency of potentially functional cryp-
ic sites, it is actually not surprising that many of them cane found nearby proto-oncogenes, and that, in certain cir-umstances, some of them might be mis-targeted by the RAGroteins. V(D)J recombination has long been thought to beinvolved in genomic instability leading to cancer [2]. Indeed,chromosomal translocations between IG/TCR loci and proto-oncogenes are a hallmark of lymphoid malignancies [30,31].In a large number of cases, a proto-oncogene is juxtaposedto a strong and active regulatory element from the antigenreceptor loci (the promoter from a gene segment or the anti-gen receptor enhancer), and the ectopic/over-expression ofthe proto-oncogene generally constitutes the initial step ofpathogenesis. The observation of such recurrent transloca-tions in B- and T-cell neoplasia, led to a prescient model ofV(D)J-mediated translocation in which an authentic IG/TCRRSS mistakenly recombines with a cryptic RSS located nearbya proto-oncogene on a distinct chromosome [2,32]. A largenumber of translocations in human hemopathies have beensubsequently proposed to be due to this process, mainly basedon the involvement of an IG/TCR gene segment as one ofthe translocation partners, and/or the identification of a RSS-like motif near the germline proto-oncogene sequence. How-ever, these criteria are as such not sufficient, and althoughin few cases the involvement of this model was very likely[9,10,33,34], most cases presented features difficult to recon-cile with cryptic site mis-targeting [35,36]. A striking exampleis the t(8;14)(q24;q32) in Burkitt’s lymphoma (BL), in whichgene segments from the IGH locus are joined with variousregions around and within the C-MYC proto-oncogene. Itis now clear that the cryptic RSSs initially proposed to beinvolved in t(8;14)(q24;q32) are very poorly fitting what wenow know to be essential motifs. Furthermore, many junc-tions show the presence of short nucleotide duplications atthe C-MYC break site. Duplications are typically generated bystaggered breaks resulting in single stranded overhangs sub-sequently filled-in by DNA polymerase activity, and are incom-patible with the trans-esterification step of RAG-mediatedbreakage. Finally, neither the MYC breaks, nor the IGH (VH
or JH) breaks are located at the exact border of the heptamerand the coding end (CE), suggesting that t(8;14)(q24;q32) does
not involve V(D)J recombination at all. Interestingly, however,a number of such breaks previously proposed to result fromV(D)J recombination, now turns out to be very likely due totwo other lymphoid specific mechanisms implicating DSB and
1250 d n a r e p a i r 5 ( 2 0 0 6 ) 1246–1258
Table 2 – V(D)J-mediated translocations resulting from illegitimate repair of the V(D)J intermediates (type 2)
Translocation Tumor sub-type IG/TCR partner Oncogene Breakpoint region# References
t(1;7)(p34;q34) T-ALL TCR� TAL1 1 [79]t(1;7)(q34;q34) T-ALL TCR� LCK 1 [80]a
t(1;14)(p32;q11) T-ALL TCR� TAL1 2 [51,81]a
t(7;8)(q34;q24) T-ALL TCR� C-MYC 1 a
t(7;9)(q34;q32) T-ALL TCR� TAL2 1 [9]t(8;14)(q24;q11) T-ALL TCR�/� C-MYC Scattered [82] and ref. thereina
t(10;14)(q24;q11) T-ALL TCR�/� HOX11 Scattered [42,83–85]a
t(11;14)(p13;q11) T-ALL TCR� LMO2 Scattered [10,72]b
t(14;21)(q11;q22) T-ALL TCR� BHLHB1 1 [86]inv(14)(q11;q32) T-ALL TCR� BCL11B 1 [87]t(11;14)(q13;q32) MCL IGH BCL1 mtc [58,88]t(14;18)(q32;q21) FL, DLBCL IGH BCL2 mbr, mcr [15,41,43] and ref. therein
Only cases with fulfilled type 2 criteria, AND sequence information on both breakpoints or with a breakpoint region tested in func-tional assays were considered. The number of cases is indicated when <5; recurrent (≥5 cases) breakpoint regions <10 Kb are indi-cated; “scattered” indicates a recurrent (>5cases) breakpoint region >10 Kb; several additional translocations could not be unambigu-ously assigned either as V(D)J-mediated, type 1 or type 2, in large part due to the lack of sequence information at both breakpoints,and include t(X;7)(q28;q35), t(X;14)(q28;q11), t(5;14)(q35;q32), t(7;9)(q34;q34), t(7;10)(q34;q24), t(7;12)(q34;p13), t(7;14)(q34;q11), t(7;19)(q35;p13),
18;22
t(14;14)(q11;q32), t(2;18)(p11;q21), t(3;22)(q27;q11), t(14;18)(q32;q21), t(a Our unpublished results.b Dik, BN, Langerak et al. (manuscript in preparation).somatic DNA remodeling: class switch recombination (CSR)and somatic hypermutation (SHM) [30]. This is the case forthe t(8;14)(q24;q32) in BL, which in fact seems to result fromeither of these two mechanisms. A more ambiguous casewas the t(14;18)(q32;q21) in follicular lymphoma, in which thepresence of cryptic sites on the BCL2 breakpoint region wasuncertain, but the involvement of the V(D)J recombinase inIGH breaks seemed very likely [37–41].
To unambiguously assess which of the oncogenic translo-cations involved RAG-mis-targeting of a cryptic site, we andothers have determined their ability to recombine with aIG/TCR segment in a functional ex vivo recombination sub-strate assay ([15,42,43]; Dik, BN, Langerak et al., manuscriptin preparation). As previously anticipated on the basis of invivo translocation breakpoints analysis, these data confirmedthat some of the cryptic sites identified at the proto-oncogenebreakpoints are indeed functional, while some others are not(Tables 1 and 2). Interestingly, breakpoint regions devoid offunctional cryptic sites did not only concern translocationssuch as t(8;14)(q24;q32) in which V(D)J recombination is notinvolved, but also translocations such as t(14;18)(q32;q21) inwhich breaks at the antigen receptor locus are clearly gener-ated by V(D)J recombination. These data functionally demon-strated that V(D)J recombination is involved in at least twodistinct mechanisms leading to translocation: in the firstcategory (“type 1”, Fig. 2), illegitimate V(D)J recombinationoccurs between a legitimate IG/TCR gene segment and anillegitimate proto-oncogene locus bearing a fortuitous butfunctional RSS, as previously proposed. In a second cate-gory of translocation (“type 2”, Fig. 3), only the IG/TCR locibreaks are mediated by V(D)J recombination. Breaks at thelocus bearing the proto-oncogene (which we will here referto as broken ends or BE) are generated by other mecha-
nisms (see Lieber, this issue), and might subsequently invadethe V(D)J synaptic complex during the rearrangement pro-cess [8,37,44,45]. Thus, while type 1 translocations clearlyresult from a targeting mistake of the recombinase, type)(q21;q11).
2 translocations result from a repair mistake of the V(D)Jintermediates.
3.3. Cryptic site mis-targeting by the RAG and SJreactivity
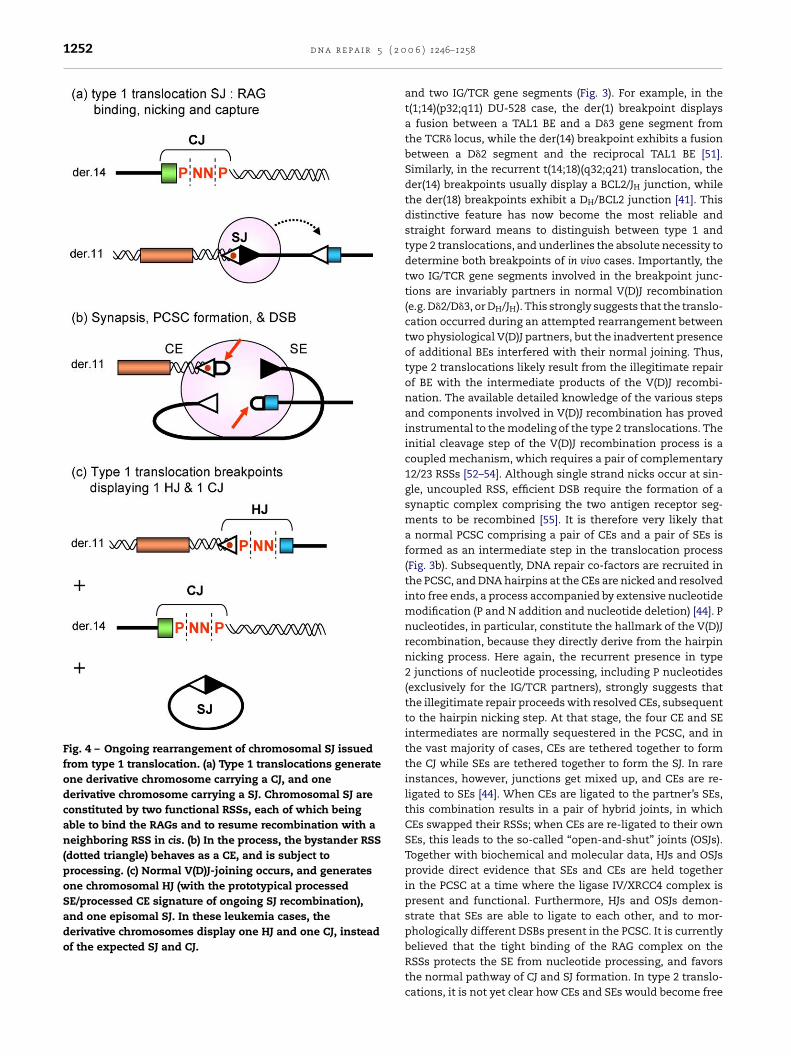
A considerable conceptual difficulty arose from these models.Indeed, one of the translocation displaying the most recur-rent and convincing use of a cryptic RSS, the t(7;9)(q34;q32)TCR�/TAL2 fusion [9], did not show the expected junctionfeatures of a type 1 translocation. Similarly to trans-TCR/IGtranslocations, the reciprocal exchange in type 1 translo-cations is expected to give rise to one derivative chromo-some bearing a CJ, and one derivative chromosome bear-ing a SJ (Fig. 2c). However, while the expected D�1/TAL2 CJwere observed on the derivative chromosome 7, the junc-tions on the derivative chromosome 9 did not consist ofthe expected reciprocal (D�1RSS/TAL2RSS) SJ, but rather of anapparent (D�1RSS/J�2CE) hybrid joint (HJ; Fig. 4c). This led to athird model involving both a targeting mistake of the recom-binase and a repair mistake of the V(D)J intermediates [2]. Inother words, RAG-mediated breaks at a cryptic site on chro-mosome 9 would have joined with the V(D)J intermediates ofan attempted D� to J� joining. Intriguingly, we have recentlyshown that the t(7;9)(q34;q32) translocation can also be foundin a large proportion of healthy individual thymocytes [46].However, contrary to T-ALL patients, derivative 9 breakpointsfrom these healthy individuals contained the expected SJ(Fig. 4a). This observation, together with reports providing evi-dence that SJ retained on the chromosome are not inert, buton the contrary continue to rearrange with neighboring RSS[47–50], led us to propose an alternative explanation for thepeculiar derivative 9 breakpoints observed in T-ALL: HJ junc-
tions in T-ALL might derive from the breakpoints observed inhealthy individuals by ongoing rearrangement involving the(D�1RSS/TAL2RSS) SJ (Fig. 4). Using recombination substrates,we demonstrated that the (D�1RSS/TAL2RSS) SJ were extremely
d n a r e p a i r 5 ( 2 0 0 6 ) 1246–1258 1251
Fig. 2 – “Type 1” translocation (targeting mistake) model. (a)A sequence fortuitously resembling a RSS (cryptic RSS,dotted triangle) is mis-targeted by the RAG1/RAG2. Whenthe cryptic RSS is located on a distinct chromosome, theV(D)J recombination proceeds in trans (b), and leads to areciprocal chromosomal translocation generating 2derivative breakpoints: one carrying a CJ, and the othercarrying a SJ (c). Ectopic expression of neighboringproto-oncogenes can ensue and lead to lymphoidn
riopmemTtapftrt
Fig. 3 – “Type 2” translocation (repair failure) model. (a) RAGbinding, nicking, capture, synapsis and DSBs occurnormally between a pair of IG/TCR gene segments; anotherDSB generated by other means occurs on a distinct location.(b) BEs are recruited/captured in the PCSC, leading to atripartite synapse comprising the four V(D)J intermediates(SEs and CEs) and the two BEs. (c) Illegitimate repair occursusing the NHEJ repair pathway. When the break at the“fragile site” takes place on a distinct chromosome, theillegitimate repair leads to a reciprocal chromosomaltranslocation. (d) In the most frequent pathway of endligation, the two BEs are joined with the two CEs andgenerate a (CE/BE) “mixed joint” (MJ) at the derivativechromosome breakpoints. (e) In an alternative pathway,one of the BEs joins with a CE and generates a derivativechromosome carrying a (CE/BE) MJ, while the other BE isjoined to a SE and displays a (SE/BE) MJ at the otherderivative chromosome breakpoint. In both cases, ectopic
eoplasia.
eactive, and generated junctions similar to those observedn T-ALL patients, confirming the SJ reactivity pathway. Thus,ur data indicated that T-ALL derivative chromosome 9 break-oints are generated in two steps: 1) a standard type 1 V(D)J-ediated t(7;9)(q34;q32) generating a SJ, a relatively frequent
vent found in healthy individuals; 2) further V(D)J rearrange-ent between this SJ and a J�2 segment, a rare event leading to
-ALL development. Importantly, the SJ reactivity step, and nothe t(7;9)(q34;q32) translocation, would lead to TAL2 activationnd therefore represent the actual oncogenic event [46]. Theresence of other T-ALL translocations displaying breakpoint
eatures similar to t(7;9)(q34;q32) [34], as well as reactivity ofheir SJ intermediates in ex vivo assays [46], suggests that SJeactivity constitutes a general pathway of oncogene activa-ion following V(D)J-mediated translocations.expression of neighboring proto-oncogenes can ensue andlead to lymphoid neoplasia.
4. Type 2 V(D)J-mediated oncogenictranslocations
4.1. Mechanistic features
While normal V(D)J recombination and type 1 translocationstypically involve two joining partners (Fig. 2), one impor-tant distinctive feature of type 2 translocations is that threejoining partners are invariably involved: one proto-oncogene
1252 d n a r e p a i r 5 ( 2
Fig. 4 – Ongoing rearrangement of chromosomal SJ issuedfrom type 1 translocation. (a) Type 1 translocations generateone derivative chromosome carrying a CJ, and onederivative chromosome carrying a SJ. Chromosomal SJ areconstituted by two functional RSSs, each of which beingable to bind the RAGs and to resume recombination with aneighboring RSS in cis. (b) In the process, the bystander RSS(dotted triangle) behaves as a CE, and is subject toprocessing. (c) Normal V(D)J-joining occurs, and generatesone chromosomal HJ (with the prototypical processedSE/processed CE signature of ongoing SJ recombination),and one episomal SJ. In these leukemia cases, thederivative chromosomes display one HJ and one CJ, insteadof the expected SJ and CJ.
0 0 6 ) 1246–1258
and two IG/TCR gene segments (Fig. 3). For example, in thet(1;14)(p32;q11) DU-528 case, the der(1) breakpoint displaysa fusion between a TAL1 BE and a D�3 gene segment fromthe TCR� locus, while the der(14) breakpoint exhibits a fusionbetween a D�2 segment and the reciprocal TAL1 BE [51].Similarly, in the recurrent t(14;18)(q32;q21) translocation, theder(14) breakpoints usually display a BCL2/JH junction, whilethe der(18) breakpoints exhibit a DH/BCL2 junction [41]. Thisdistinctive feature has now become the most reliable andstraight forward means to distinguish between type 1 andtype 2 translocations, and underlines the absolute necessity todetermine both breakpoints of in vivo cases. Importantly, thetwo IG/TCR gene segments involved in the breakpoint junc-tions are invariably partners in normal V(D)J recombination(e.g. D�2/D�3, or DH/JH). This strongly suggests that the translo-cation occurred during an attempted rearrangement betweentwo physiological V(D)J partners, but the inadvertent presenceof additional BEs interfered with their normal joining. Thus,type 2 translocations likely result from the illegitimate repairof BE with the intermediate products of the V(D)J recombi-nation. The available detailed knowledge of the various stepsand components involved in V(D)J recombination has provedinstrumental to the modeling of the type 2 translocations. Theinitial cleavage step of the V(D)J recombination process is acoupled mechanism, which requires a pair of complementary12/23 RSSs [52–54]. Although single strand nicks occur at sin-gle, uncoupled RSS, efficient DSB require the formation of asynaptic complex comprising the two antigen receptor seg-ments to be recombined [55]. It is therefore very likely thata normal PCSC comprising a pair of CEs and a pair of SEs isformed as an intermediate step in the translocation process(Fig. 3b). Subsequently, DNA repair co-factors are recruited inthe PCSC, and DNA hairpins at the CEs are nicked and resolvedinto free ends, a process accompanied by extensive nucleotidemodification (P and N addition and nucleotide deletion) [44]. Pnucleotides, in particular, constitute the hallmark of the V(D)Jrecombination, because they directly derive from the hairpinnicking process. Here again, the recurrent presence in type2 junctions of nucleotide processing, including P nucleotides(exclusively for the IG/TCR partners), strongly suggests thatthe illegitimate repair proceeds with resolved CEs, subsequentto the hairpin nicking step. At that stage, the four CE and SEintermediates are normally sequestered in the PCSC, and inthe vast majority of cases, CEs are tethered together to formthe CJ while SEs are tethered together to form the SJ. In rareinstances, however, junctions get mixed up, and CEs are re-ligated to SEs [44]. When CEs are ligated to the partner’s SEs,this combination results in a pair of hybrid joints, in whichCEs swapped their RSSs; when CEs are re-ligated to their ownSEs, this leads to the so-called “open-and-shut” joints (OSJs).Together with biochemical and molecular data, HJs and OSJsprovide direct evidence that SEs and CEs are held togetherin the PCSC at a time where the ligase IV/XRCC4 complex ispresent and functional. Furthermore, HJs and OSJs demon-strate that SEs are able to ligate to each other, and to mor-phologically different DSBs present in the PCSC. It is currently
believed that the tight binding of the RAG complex on theRSSs protects the SE from nucleotide processing, and favorsthe normal pathway of CJ and SJ formation. In type 2 translo-cations, it is not yet clear how CEs and SEs would become free
0 0 6
tptpCbBu(tlottottihvLceJAmptjprgpfsbp
4
Drtaeiseesipnlsotpfs
d n a r e p a i r 5 ( 2
o repair with the unrelated BEs from the oncogene locus. Oneossibility is that the CEs intermediates would escape fromhe PCSC. This would be in agreement with a previously pro-osed release/capture model of V(D)J recombination, in whichEs undergo repeated cycles of release from and recapturey the PCSC before ligation [12]. Alternatively, the oncogeneEs could invade (or be recruited into) the PCSC and form annusual “tripartite” synapse containing the six DNA ends [45]
Fig. 3c). One of the predictions of this tripartite synapse is thathe simultaneous presence of the six ends in the PCSC mightead to alternative combinations of end-joining in which onef the SE is fused to one of the BE, while the other BE is fusedo a CE (Fig. 3e). We have identified such alternative junc-ion combinations both in vivo and ex vivo. In vivo, 2 casesut of a library of 45 characterized t(14;18)(q32;q21) transloca-ions displayed breakpoints in which the BCL2 BEs were joinedo the CE and the SE of one JH segment (instead of display-ng standard BCL2/JH and DH/BCL2 CE/BE junctions) [56]. Weave also confirmed that BE/SE junctions can be formed in exivo assays, using recombination substrates mimicking type 2MO2/D�3 translocation (unpublished observations). This is inontrast with a recent report from Raghavan et al. [57] usingx vivo recombination substrates mimicking type 2 BCL2/DH-
H translocation, who observed BE/CE but no BE/SE formation.likely explanation for this discrepancy is that in the plas-id design from Raghavan et al., the V(D)J synapse occurred
referentially in cis favoring SJ formation, while in our designhe V(D)J synapse occurred exclusively in trans, favoring BE/SEunction formation. The relative rarity of BE/SE junctions com-ared to BE/CE both in vivo and ex vivo, goes along with theeported tighter protection of the SEs by the RAGs, and sug-ests a lower propensity of the SEs to join to unrelated BEsresent in the synapse. Finally, additional evidence arguingor the simultaneous presence of the six ends in the tripartiteynapse is provided by the insertion in type 2 translocationsreakpoints of short copies of surrounding sequences tem-lated from any of the six partners (see below) [41,58,59].
.2. T Nucleotides
e novo nucleotide additions are present in most direct andeciprocal breakpoints of type 2 translocations, and such inser-ions were at first interpreted as N nucleotides. N regionsre non-templated nucleotides randomly added to free 3′OHnds by the terminal deoxynucleotidyl transferase (TdT) dur-ng the V(D)J recombination process. Although N nucleotideynthesis is not completely random, displaying a marked pref-rence for Gs, TdT does not use any template for polymerasextension, and cannot therefore generate copies of a givenequence. Surprisingly, our detailed analysis of the breakpointnsertions revealed that in some of the t(14;18)(q32;q21) break-oints, the de novo insertions did not correspond to randomucleotide additions. Rather, such insertions contained what
ooked like copies of adjacent IGH or BCL2 sequences [41]. Weubsequently found many cases of such templated insertionsf various lengths in the t(14;18) junctions. In most cases,
he “template/insert” pair contained mismatches, includingoint mutations, insertions and deletions. Templates wereound in the adjacent DH/JH segments and BCL2 loci, and inimilar proportions. Moreover, one of the breakpoints could) 1246–1258 1253
also constitute a template for copy and insertion in the otherbreakpoint. Genealogical relationship between one templateand two copies could tentatively be reconstituted in somecases. Finally, the whole breakpoint insertion region couldconsist of a patchwork of templated nucleotides, generatedfrom different templates and inserted in both direct andreverse-complement orientations. Altogether, the multiplic-ity of copies for one template together with the occurrenceof mismatches in the template/copy pair, strongly suggestedthe presence of a short patch error-prone templated DNA syn-thesis, followed by insertion of the copy in one breakpoint(Fig. 5). The long stretches of homology in the short break-point de novo nucleotides were very unlikely to be coincidental.However, the shorter the identity between the sequences, theless obvious the identification of a real stretch of homology,and the higher the risk of fortuitous comparisons. Togetherwith Dr J. Koziol, bio-statistician at the Scripps Research Insti-tute, we therefore set up a statistical test to distinguish whichof these regions were really due to chance, and which ofthem presented enough sequence identity with the adjacentflanking sequences to exclude their concomitant presenceby chance alone. The test was designed with a conservativeapproach and was therefore very stringent, i.e. more likelyto accept than to reject the null hypothesis that the obser-vation is due to chance only. For example, significant homolo-gies typically excluded regions of less than five nucleotideshomology even if it encompassed the whole insertion. Even-tually, most of the significant observations found using thistest identified regions ranging from at least 8, and up to22 nucleotides. All together, we found that 20–30% of thet(14;18)(q32;q21) breakpoints present such highly significantregions [41,59]. This might, however, still be an underestima-tion of the real frequency due to the stringency of the testused, and others have used alternative, less stringent, statis-tical tests [43] to find homologies as low as three nucleotides[57]. We called these regions “T nucleotides”, for templatednucleotides.
To find out if T nucleotides were specific fort(14;18)(q32;q21) breakpoints or a more general featureof V(D)J-mediated translocations, we investigated thet(11;14)(q13;q32) type 2 translocation in mantle cell lymphoma[15]. T nucleotides were also present in t(11;14)(q13;q32)breakpoints, and in similar proportions, indicating that theirformation is not specific to t(14;18) [58]. Furthermore, theabsence of T nucleotides both in normal V(D)J recombinationjunctions and in type 1 translocation breakpoints stronglysuggests that T nucleotides are not specifically linked to theV(D)J recombination process. This point is also supported bythe presence of T nucleotides in cases of t(8;22)(q24;q11) invariant BL [60], which do not involve V(D)J recombination.Importantly, T nucleotide formation is also not limited tochromosomal translocation, as shown by the presence ofsignificant examples of T nucleotides in junctions from exvivo recombination substrates undergoing cis-recombination(Lieber et al., this issue). Interestingly, T nucleotides havealso been reported in DSB repair junctions from Drosophila
[61,62]. In Drosophila, DSB created by excision of a P elementare primarily repaired by a gene conversion pathway calledsynthesis-dependent strand annealing (SDSA). In SDSA,processing of a DSB generates 3′ single strand tails, which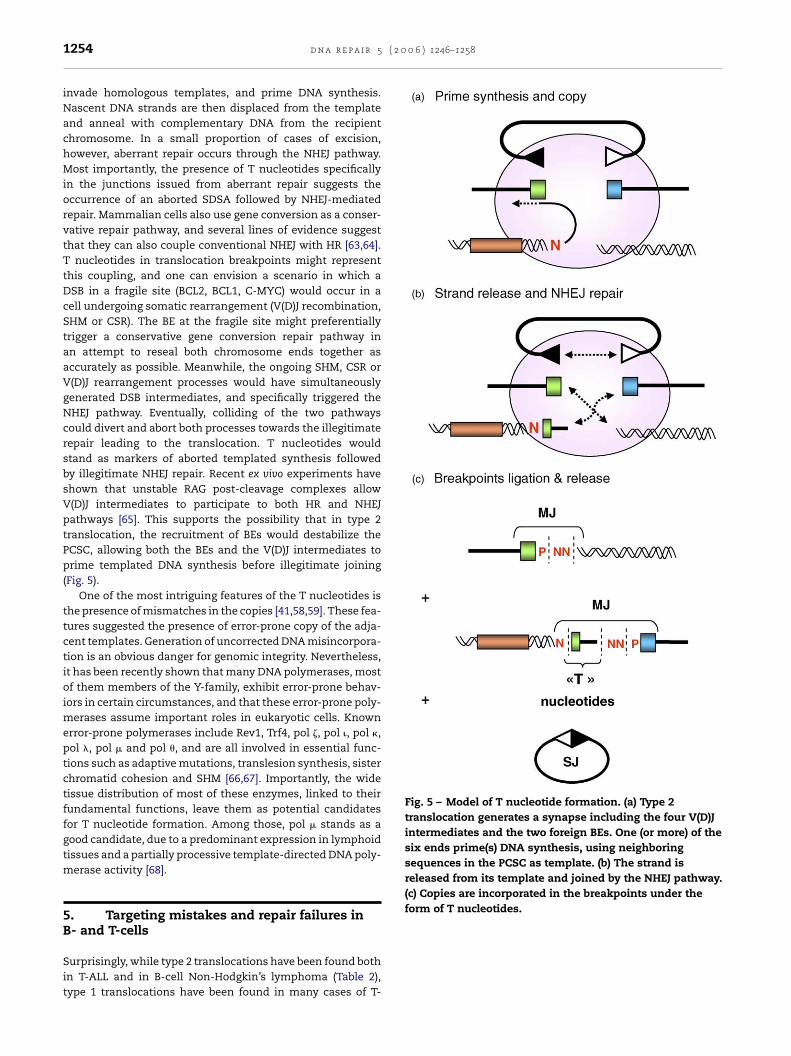
( 2 0 0 6 ) 1246–1258
Fig. 5 – Model of T nucleotide formation. (a) Type 2translocation generates a synapse including the four V(D)Jintermediates and the two foreign BEs. One (or more) of thesix ends prime(s) DNA synthesis, using neighboringsequences in the PCSC as template. (b) The strand isreleased from its template and joined by the NHEJ pathway.(c) Copies are incorporated in the breakpoints under theform of T nucleotides.
1254 d n a r e p a i r 5
invade homologous templates, and prime DNA synthesis.Nascent DNA strands are then displaced from the templateand anneal with complementary DNA from the recipientchromosome. In a small proportion of cases of excision,however, aberrant repair occurs through the NHEJ pathway.Most importantly, the presence of T nucleotides specificallyin the junctions issued from aberrant repair suggests theoccurrence of an aborted SDSA followed by NHEJ-mediatedrepair. Mammalian cells also use gene conversion as a conser-vative repair pathway, and several lines of evidence suggestthat they can also couple conventional NHEJ with HR [63,64].T nucleotides in translocation breakpoints might representthis coupling, and one can envision a scenario in which aDSB in a fragile site (BCL2, BCL1, C-MYC) would occur in acell undergoing somatic rearrangement (V(D)J recombination,SHM or CSR). The BE at the fragile site might preferentiallytrigger a conservative gene conversion repair pathway inan attempt to reseal both chromosome ends together asaccurately as possible. Meanwhile, the ongoing SHM, CSR orV(D)J rearrangement processes would have simultaneouslygenerated DSB intermediates, and specifically triggered theNHEJ pathway. Eventually, colliding of the two pathwayscould divert and abort both processes towards the illegitimaterepair leading to the translocation. T nucleotides wouldstand as markers of aborted templated synthesis followedby illegitimate NHEJ repair. Recent ex vivo experiments haveshown that unstable RAG post-cleavage complexes allowV(D)J intermediates to participate to both HR and NHEJpathways [65]. This supports the possibility that in type 2translocation, the recruitment of BEs would destabilize thePCSC, allowing both the BEs and the V(D)J intermediates toprime templated DNA synthesis before illegitimate joining(Fig. 5).
One of the most intriguing features of the T nucleotides isthe presence of mismatches in the copies [41,58,59]. These fea-tures suggested the presence of error-prone copy of the adja-cent templates. Generation of uncorrected DNA misincorpora-tion is an obvious danger for genomic integrity. Nevertheless,it has been recently shown that many DNA polymerases, mostof them members of the Y-family, exhibit error-prone behav-iors in certain circumstances, and that these error-prone poly-merases assume important roles in eukaryotic cells. Knownerror-prone polymerases include Rev1, Trf4, pol �, pol �, pol �,pol �, pol and pol , and are all involved in essential func-tions such as adaptive mutations, translesion synthesis, sisterchromatid cohesion and SHM [66,67]. Importantly, the widetissue distribution of most of these enzymes, linked to theirfundamental functions, leave them as potential candidatesfor T nucleotide formation. Among those, pol stands as agood candidate, due to a predominant expression in lymphoidtissues and a partially processive template-directed DNA poly-merase activity [68].
5. Targeting mistakes and repair failures inB- and T-cells
Surprisingly, while type 2 translocations have been found bothin T-ALL and in B-cell Non-Hodgkin’s lymphoma (Table 2),type 1 translocations have been found in many cases of T-

0 0
AsBitbiwspt[t1ttmnbbtioemsantd[
iamtcrFin
utitp
A
WfatRBICI
d n a r e p a i r 5 ( 2
LL, but unambiguously assigned in only one case of B-CLLo far (Table 1). Thus, while repair mistakes are found in both- and T-cell neoplasia, RAG targeting errors seem very rare
n B-cells. Why is cryptic site mis-targeting leading to so fewranslocations in B-cell malignancies? One possibility coulde the difference in recombinogenic potential between RSSs
n TCR and in IG loci. Indeed, recombination of pairs of RSSsith variant nonamers providing low RAG binding efficiency
uch as those generally found in the TCR loci, might be morerone to be competed out with cryptic sites located outsidehe locus than the more conserved RSSs found in the IG loci17,25]. In line with this, we have recently found that one ofhe cryptic sites targeted in LMO2/TCR� t(11;14)(p13;q11) type
translocations has a recombinogenic potential only threeimes lower than the authentic J�1 RSS, and that such D�/LMO2ranslocations are as frequent as trans-TCR D�/J� rearrange-
ents in thymocytes from healthy individuals [15]. Anotheron-exclusive possibility would be a more general differenceetween B- and T-cells towards regulation of locus accessi-ility, with a tighter restriction to the IG loci in B-cells thano the TCR loci in T-cells. In this scenario, the accessibil-ty of the recombinase to non-immune loci such as proto-ncogenes would be inhibited in B-cells, and would provide anfficient protection against RAG targeting mistakes. In agree-ent with a more relaxed accessibility in T-cells, we have
hown that oncogenic translocations such as t(11;14)(p13;q11)nd t(7;9)(q34;q32) are present at strikingly high frequency inormal human thymus, and that the recombinogenic poten-ials conferred by the cryptic sites involved are directly pre-ictive of the in vivo level of translocation at that locus
15,46].For type 2 translocations, as T nucleotide formation seems
ntimately linked to the repair mechanism involved, thebsence of reported T nucleotides in T-ALL breakpoints so faright support the notion that pre-B- and pre-T-cells handle
he repair of fragile sites differently. As such, type 2 translo-ations might represent a heterogeneous group, with distinctepair pathways linked to different types of DNA breakage.urther investigation of the mechanism and enzymatic activ-ties involved might provide important clues on the mecha-ism and timing of such type 2 translocations.
Hopefully, the better understanding of the regulatory forcesnderlying each subclass of chromosomal translocations, andhe identification of the possible genetic or exogenous factorsnvolved in their modulation, should significantly contributeo the development of urgently needed programs of cancerrevention.
cknowledgments
e are grateful to Drs W.A. Dik and A.W. Langerak for help-ul comments and suggestions. BN laboratory is supported byn AVENIR program from INSERM, and grants from the Fonda-ion pour la Recherche Medicale (FRM), the Association pour la
echerche contre le Cancer (ARC), and the Conseil General desouches du Rhone. B.N. is a recipient of a Contrat d’InterfaceNSERM/AP-HM; B.M. is a recipient from the Ligue Nationaleontre le Cancer (la Ligue). S.R. is a recipient from the Young
nvestigator INSERM Program.
6 ) 1246–1258 1255
r e f e r e n c e s
[1] S.G. Fisher, R.I. Fisher, The epidemiology of non-Hodgkin’slymphoma, Oncogene 23 (2004) 6524–6534.
[2] B. Tycko, J. Sklar, Chromosomal translocations in lymphoidneoplasia: a reappraisal of the recombinase model, CancerCells 2 (1990) 1–8.
[3] D.G. Schatz, V(D)J recombination, Immunol. Rev. 200 (2004)5–11.
[4] M.S. Schlissel, Regulating antigen-receptor gene assembly,Nat. Rev. Immunol. 3 (2003) 890–899.
[5] G.D. Yancopoulos, F.W. Alt, Developmentally controlled andtissue-specific expression of unrearranged VH genesegments, Cell 40 (1985) 271–281.
[6] B. Tycko, J.D. Palmer, J. Sklar, T cell receptor genetrans-rearrangements: chimeric gamma-delta genes innormal lymphoid tissues, Science 245 (1989) 1242–1246.
[7] Y. Kobayashi, B. Tycko, A.L. Soreng, J. Sklar,Transrearrangements between antigen receptor genes innormal human lymphoid tissues and in ataxiatelangiectasia, J. Immunol. 147 (1991) 3201–3209.
[8] S.N. Bailey, N. Rosenberg, Assessing the pathogenic potentialof the V(D)J recombinase by interlocus immunoglobulinlight-chain gene rearrangement, Mol. Cell Biol. 17 (1997)887–894.
[9] B. Tycko, T.C. Reynolds, S.D. Smith, J. Sklar, Consistentbreakage between consensus recombinase heptamers ofchromosome 9 DNA in a recurrent chromosomaltranslocation of human T cell leukemia, J. Exp. Med. 169(1989) 369–377.
[10] I.S. Garcia, Y. Kaneko, R. Gonzalez-Sarmiento, K. Campbell,L. White, T. Boehm, T.H. Rabbitts, A study of chromosome11p13 translocations involving TCR beta and TCR delta inhuman T cell leukaemia, Oncogene 6 (1991) 577–582.
[11] J.O. Han, S.B. Steen, D.B. Roth, Intermolecular V(D)Jrecombination is prohibited specifically at the joining step,Mol. Cell 3 (1999) 331–338.
[12] A. Tevelev, D.G. Schatz, Intermolecular V(D)J recombination,J. Biol. Chem. 275 (2000) 8341–8348.
[13] E.A. Agard, S.M. Lewis, Postcleavage sequence specificity inV(D)J recombination, Mol. Cell Biol. 20 (2000) 5032–5040.
[14] S. Lipkowitz, M.H. Stern, I.R. Kirsch, Hybrid T cell receptorgenes formed by interlocus recombination in normal andataxia-telangiectasis lymphocytes, J. Exp. Med. 172 (1990)409–418.
[15] R. Marculescu, T. Le, P. Simon, U. Jaeger, B. Nadel,V(D)J-mediated translocations in lymphoid neoplasms: afunctional assessment of genomic instability by crypticsites, J. Exp. Med. 195 (2002) 85–98.
[16] F. Davodeau, M.A. Peyrat, J. Gaschet, M.M. Hallet, F. Triebel,H. Vie, D. Kabelitz, M. Bonneville, Surface expression offunctional T cell receptor chains formed by interlocusrecombination on human T lymphocytes, J. Exp. Med. 180(1994) 1685–1691.
[17] S. Akira, K. Okazaki, H. Sakano, Two pairs of recombinationsignals are sufficient to cause immunoglobulin V-(D)-Jjoining, Science 238 (1987) 1134–1138.
[18] J.E. Hesse, M.R. Lieber, K. Mizuuchi, M. Gellert, V(D)Jrecombination: a functional definition of the joining signals,Genes Dev. 3 (1989) 1053–1061.
[19] Y. Akamatsu, N. Tsurushita, F. Nagawa, M. Matsuoka, K.Okazaki, M. Imai, H. Sakano, Essential residues in V(D)J
recombination signals, J. Immunol. 153 (1994) 4520–4529.[20] L. Fanning, A. Connor, K. Baetz, D. Ramsden, G.E. Wu, MouseRSS spacer sequences affect the rate of V(D)J recombination,Immunogenetics 44 (1996) 146–150.

( 2 0
1256 d n a r e p a i r 5[21] B. Nadel, A. Tang, G. Escuro, G. Lugo, A.J. Feeney, Sequence ofthe spacer in the recombination signal sequence affectsV(D)J rearrangement frequency and correlates withnonrandom Vkappa usage in vivo, J. Exp. Med. 187 (1998)1495–1503.
[22] B. Nadel, A. Tang, G. Lugo, V. Love, G. Escuro, A.J. Feeney,Decreased frequency of rearrangement due to thesynergistic effect of nucleotide changes in the heptamer andnonamer of the recombination signal sequence of the Vkappa gene A2b, which is associated with increasedsusceptibility of Navajos to Haemophilus influenzae type bdisease, J. Immunol. 161 (1998) 6068–6073.
[23] S.M. Lewis, E. Agard, S. Suh, L. Czyzyk, Cryptic signals andthe fidelity of V(D)J joining, Mol. Cell Biol. 17 (1997)3125–3136.
[24] L.G. Cowell, M. Davila, T.B. Kepler, G. Kelsoe, Identificationand utilization of arbitrary correlations in models ofrecombination signal sequences, Genome Biol. 3 (2002),RESEARCH0072.
[25] L.G. Cowell, M. Davila, K. Yang, T.B. Kepler, G. Kelsoe,Prospective estimation of recombination signal efficiencyand identification of functional cryptic signals in thegenome by statistical modeling, J. Exp. Med. 197 (2003)207–220.
[26] J. Kwon, K.B. Morshead, J.R. Guyon, R.E. Kingston, M.A.Oettinger, Histone acetylation and hSWI/SNF remodeling actin concert to stimulate V(D)J cleavage of nucleosomal DNA,Mol. Cell 6 (2000) 1037–1048.
[27] F. McBlane, J. Boyes, Stimulation of V(D)J recombination byhistone acetylation, Curr. Biol. 10 (2000) 483–486.
[28] M.A. Oettinger, How to keep V(D)J recombination undercontrol, Immunol. Rev. 200 (2004) 165–181.
[29] M.S. Krangel, J. Carabana, I. Abbarategui, R. Schlimgen, A.Hawwari, Enforcing order within a complex locus: currentperspectives on the control of V(D)J recombination at themurine T-cell receptor alpha/delta locus, Immunol. Rev. 200(2004) 224–232.
[30] R. Kuppers, R. Dalla-Favera, Mechanisms of chromosomaltranslocations in B cell lymphomas, Oncogene 20 (2001)5580–5594.
[31] P.D. Aplan, Causes of oncogenic chromosomal translocation,Trends Genet. (2005) 46–55.
[32] L.C. Showe, C.M. Croce, The role of chromosomaltranslocations in B- and T-cell neoplasia, Annu. Rev.Immunol. 5 (1987) 253–277.
[33] J.T. Cheng, C.Y. Yang, J. Hernandez, J. Embrey, R. Baer, Thechromosome translocation (11;14) (p13;q11) associated withT cell acute leukemia. Asymmetric diversification of thetranslocational junctions, J. Exp. Med. 171 (1990) 489–501.
[34] R.C. Burnett, M.J. Thirman, J.D. Rowley, M.O. Diaz, Molecularanalysis of the T-cell acute lymphoblasticleukemia-associated t(1;7)(p34;q34) that fuses LCK andTCRB, Blood 84 (1994) 1232–1236.
[35] Y. Tsujimoto, J. Gorham, J. Cossman, E. Jaffe, C.M. Croce, Thet(14;18) chromosome translocations involved in B-cellneoplasms result from mistakes in VDJ joining, Science 229(1985) 1390–1393.
[36] F.G. Haluska, S. Finver, Y. Tsujimoto, C.M. Croce, The t(8;14)chromosomal translocation occurring in B-cell malignanciesresults from mistakes in V-D-J joining, Nature 324 (1986)158–161.
[37] A. Bakhshi, J.P. Jensen, P. Goldman, J.J. Wright, O.W. McBride,A.L. Epstein, S.J. Korsmeyer, Cloning the chromosomalbreakpoint of t(14;18) human lymphomas: clustering around
JH on chromosome 14 and near a transcriptional unit on 18,Cell 41 (1985) 899–906.[38] Y. Tsujimoto, E. Louie, M.M. Bashir, C.M. Croce, Thereciprocal partners of both the t(14;18) and the t(11;14)
0 6 ) 1246–1258
translocations involved in B-cell neoplasms arerearranged by the same mechanism, Oncogene 2 (1988)347–351.
[39] F. Cotter, C. Price, E. Zucca, B.D. Young, Direct sequenceanalysis of the 14q+ and 18q− chromosome junctions infollicular lymphoma, Blood 76 (1990) 131–135.
[40] R.T. Wyatt, R.A. Rudders, A. Zelenetz, R.A. Delellis, T.G.Krontiris, BCL2 oncogene translocation is mediated by achi-like consensus, J. Exp. Med. 175 (1992) 1575–1588.
[41] U. Jager, S. Bocskor, T. Le, G. Mitterbauer, I. Bolz, A. Chott, M.Kneba, C. Mannhalter, B. Nadel, Follicular lymphomas’BCL-2/IgH junctions contain templated nucleotideinsertions: novel insights into the mechanism of t(14;18)translocation, Blood 95 (2000) 3520–3529.
[42] S.C. Raghavan, I.R. Kirsch, M.R. Lieber, Analysis of the V(D)Jrecombination efficiency at lymphoid chromosomaltranslocation breakpoints, J. Biol. Chem. 276 (2001)29126–29133.
[43] S.C. Raghavan, P.C. Swanson, X. Wu, C.L. Hsieh, M.R. Lieber,A non-B-DNA structure at the Bcl-2 major breakpoint regionis cleaved by the RAG complex, Nature 428 (2004) 88–93.
[44] S.M. Lewis, The mechanism of V(D)J joining: lessons frommolecular, immunological, and comparative analyses, Adv.Immunol. 56 (1994) 27–150.
[45] B. Nadel, R. Marculescu, T. Le, M. Rudnicki, S. Bocskor, U.Jager, Novel insights into the mechanism of t(14;18)(q32;q21)translocation in follicular lymphoma, Leuk. Lymphoma 42(2001) 1181–1194.
[46] R. Marculescu, K. Vanura, T. Le, P. Simon, U. Jager, B. Nadel,Distinct t(7;9)(q34;q32) breakpoints in healthy individualsand individuals with T-ALL, Nat. Genet. 33 (2003) 342–344.
[47] S. Lewis, A. Gifford, D. Baltimore, Joining of V kappa to Jkappa gene segments in a retroviral vector introduced intolymphoid cells, Nature 308 (1984) 425–428.
[48] S. Lewis, A. Gifford, D. Baltimore, DNA elements areasymmetrically joined during the site-specificrecombination of kappa immunoglobulin genes, Science 228(1985) 677–685.
[49] J.E. Hesse, M.R. Lieber, M. Gellert, K. Mizuuchi,Extrachromosomal DNA substrates in pre-B cells undergoinversion or deletion at immunoglobulin V-(D)-J joiningsignals, Cell 49 (1987) 775–783.
[50] A.W. Langerak, B. Nadel, A. De Torbal, I.L. Wolvers-Tettero,E.J. Gastel-Mol, B. Verhaaf, U. Jager, J.J. van Dongen,Unraveling the consecutive recombination events in thehuman IGK locus, J. Immunol. 173 (2004) 3878–3888.
[51] L.R. Finger, J. Kagan, G. Christopher, J. Kurtzberg, M.S.Hershfield, P.C. Nowell, C.M. Croce, Involvement of the TCL5gene on human chromosome 1 in T-cell leukemia andmelanoma, Proc. Natl. Acad. Sci. U.S.A. 86 (1989) 5039–5043.
[52] D.C. van Gent, D.A. Ramsden, M. Gellert, The RAG1 and RAG2proteins establish the 12/23 rule in V(D)J recombination, Cell85 (1996) 107–113.
[53] Q.M. Eastman, T.M. Leu, D.G. Schatz, Initiation of V(D)Jrecombination in vitro obeying the 12/23 rule, Nature 380(1996) 85–88.
[54] S.B. Steen, L. Gomelsky, D.B. Roth, The 12/23 rule is enforcedat the cleavage step of V(D)J recombination in vivo, GenesCells 1 (1996) 543–553.
[55] J.D. Curry, J.K. Geier, M.S. Schlissel, Single-strandrecombination signal sequence nicks in vivo: evidence for acapture model of synapsis, Nat. Immunol. (2005) 1272–1279.
[56] R. Marculescu, T. Le, S. Bocskor, G. Mitterbauer, A. Chott, C.Mannhalter, U. Jaeger, B. Nadel, Alternative end-joining infollicular lymphomas’ t(14;18) translocation, Leukemia 16(2002) 120–126.

0 0 6
d n a r e p a i r 5 ( 2[57] S.C. Raghavan, C.L. Hsieh, M.R. Lieber, Both V(D)J codingends but neither signal end can recombine at the bcl-2major breakpoint region, and the rejoining is ligase IVdependent, Mol. Cell Biol. 25 (2005) 6475–6484.
[58] N. Welzel, T. Le, R. Marculescu, G. Mitterbauer, A. Chott, C.Pott, M. Kneba, M.Q. Du, R. Kusec, J. Drach, M. Raderer, C.Mannhalter, K. Lechner, B. Nadel, U. Jaeger, Templatednucleotide addition and immunoglobulin JH-gene utilizationin t(11;14) junctions: implications for the mechanism oftranslocation and the origin of mantle cell lymphoma,Cancer Res. 61 (2001) 1629–1636.
[59] S. Roulland, P. Lebailly, P. Gauduchon, Correspondence re:Welzel et al., Cancer Res. 61:1629–1636, Cancer Res. 63 (2003)1722–1723.
[60] C.T. Denny, G.F. Hollis, I.T. Magrath, I.R. Kirsch, Burkittlymphoma cell line carrying a variant translocation createsnew DNA at the breakpoint and violates the hierarchy ofimmunoglobulin gene rearrangement, Mol. Cell Biol. 5 (1985)3199–3207.
[61] M.D. Adams, M. McVey, J.J. Sekelsky, Drosophila BLM indouble-strand break repair by synthesis-dependent strandannealing, Science 299 (2003) 265–267.
[62] M. McVey, D. Radut, J.J. Sekelsky, End-joining repair ofdouble-strand breaks in Drosophila melanogaster is largelyDNA ligase IV independent, Genetics 168 (2004) 2067–2076.
[63] C. Richardson, M. Jasin, Coupled homologous andnonhomologous repair of a double-strand break preservesgenomic integrity in mammalian cells, Mol. Cell Biol. 20(2000) 9068–9075.
[64] A.J. Pierce, P. Hu, M. Han, N. Ellis, M. Jasin, Ku DNAend-binding protein modulates homologous repair ofdouble-strand breaks in mammalian cells, Genes Dev. 15(2001) 3237–3242.
[65] G.S. Lee, M.B. Neiditch, S.S. Salus, D.B. Roth, RAG proteinsshepherd double-strand breaks to a specific pathway,suppressing error-prone repair, but RAG nicking initiateshomologous recombination, Cell 117 (2004) 171–184.
[66] M.F. Goodman, Error-prone repair DNA polymerases inprokaryotes and eukaryotes, Annu. Rev. Biochem. 71 (2002)17–50.
[67] M. Seki, C. Masutani, L.W. Yang, A. Schuffert, S. Iwai, I.Bahar, R.D. Wood, High-efficiency bypass of DNA damageby human DNA polymerase Q, EMBO J. 23 (2004) 4484–4494.
[68] O. Dominguez, J.F. Ruiz, T.L. de Lera, M. Garcia-Diaz, M.A.Gonzalez, T. Kirchhoff, A. Martinez, A. Bernad, L. Blanco,DNA polymerase mu (Pol mu), homologous to TdT, could actas a DNA mutator in eukaryotic cells, EMBO J. 19 (2000)1731–1742.
[69] Q. Chen, J.T. Cheng, L.H. Tasi, N. Schneider, G. Buchanan, A.Carroll, W. Crist, B. Ozanne, M.J. Siciliano, R. Baer, The talgene undergoes chromosome translocation in T cellleukemia and potentially encodes a helix-loop-helix protein,EMBO J. 9 (1990) 415–424.
[70] Q. Chen, C.Y. Yang, J.T. Tsan, Y. Xia, A.H. Ragab, S.C. Peiper,A. Carroll, R. Baer, Coding sequences of the tal-1 gene aredisrupted by chromosome translocation in human T cellleukemia, J. Exp. Med. 172 (1990) 1403–1408.
[71] G. Yoffe, N. Schneider, L. Van Dyk, C.Y. Yang, M. Siciliano, G.Buchanan, J.D. Capra, R. Baer, The chromosometranslocation (11;14)(p13;q11) associated with T-cell acutelymphocytic leukemia: an 11p13 breakpoint cluster region,Blood 74 (1989) 374–379.
[72] T. Boehm, L. Buluwela, D. Williams, L. White, T.H. Rabbitts, Acluster of chromosome 11p13 translocations found viadistinct D-D and D-D-J rearrangements of the human T cellreceptor delta chain gene, EMBO J. 7 (1988) 2011–2017.
) 1246–1258 1257
[73] E. Champagne, Y. Takihara, U. Sagman, J. de Sousa, S. Burrow,W.H. Lewis, T.W. Mak, M.D. Minden, The T-cell receptor deltachain locus is disrupted in the T-ALL associatedt(11;14)(p13;q11) translocation, Blood 73 (1989) 1672–1676.
[74] T. Boehm, L. Mengle-Gaw, U.R. Kees, N. Spurr, I. Lavenir, A.Forster, T.H. Rabbitts, Alternating purine–pyrimidine tractsmay promote chromosomal translocations seen in a varietyof human lymphoid tumours, EMBO J. 8 (1989) 2621–2631.
[75] E.A. McGuire, R.D. Hockett, K.M. Pollock, M.F. Bartholdi,S.J. O’Brien, S.J. Korsmeyer, The t(11;14)(p15;q11) in aT-cell acute lymphoblastic leukemia cell line activatesmultiple transcripts, including Ttg-1, a gene encoding apotential zinc finger protein, Mol. Cell Biol. 9 (1989)2124–2132.
[76] T. Boehm, L. Foroni, Y. Kaneko, M.F. Perutz, T.H. Rabbitts, Therhombotin family of cysteine-rich LIM-domain oncogenes:distinct members are involved in T-cell translocations tohuman chromosomes 11p15 and 11p13, Proc. Natl. Acad. Sci.U.S.A. 88 (1991) 4367–4371.
[77] M.H. Stern, J. Soulier, M. Rosenzwajg, K. Nakahara, N.Canki-Klain, A. Aurias, F. Sigaux, I.R. Kirsch, MTCP-1: a novelgene on the human chromosome Xq28 translocated to the Tcell receptor alpha/delta locus in mature T cellproliferations, Oncogene 8 (1993) 2475–2483.
[78] S. Tashiro, M. Takechi, H. Asou, K. Takauchi, T. Kyo, H. Dohy,M. Kikuchi, N. Kamada, Y. Tsujimoto, Cytogenetic 2; 18 and18; 22 translocation in chronic lymphocytic leukemia withjuxtaposition of bcl-2 and immunoglobulin light chaingenes, Oncogene 7 (1992) 573–577.
[79] T.J. Fitzgerald, G.A. Neale, S.C. Raimondi, R.M. Goorha, c-tal,a helix-loop-helix protein, is juxtaposed to theT-cell receptor-beta chain gene by a reciprocal chromosomaltranslocation: t(1;7)(p32;q35), Blood 78 (1991) 2686–2695.
[80] B. Tycko, S.D. Smith, J. Sklar, Chromosomal translocationsjoining LCK and TCRB loci in human T cell leukemia, J. Exp.Med. 174 (1991) 867–873.
[81] C.G. Begley, P.D. Aplan, M.P. Davey, K. Nakahara, K. Tchorz, J.Kurtzberg, M.S. Hershfield, B.F. Haynes, D.I. Cohen, T.A.Waldmann, Chromosomal translocation in a humanleukemic stem-cell line disrupts the T-cell antigen receptordelta-chain diversity region and results in a previouslyunreported fusion transcript, Proc. Natl. Acad. Sci. U.S.A. 86(1989) 2031–2035.
[82] E.A. Shima-Rich, A.M. Harden, T.W. McKeithan, J.D. Rowley,M.O. Diaz, Molecular analysis of the t(8;14)(q24;q11)chromosomal breakpoint junctions in the T-cell leukemialine MOLT-16, Genes Chromosomes Cancer 20 (1997)363–371.
[83] M. Lu, N. Zhang, S. Raimondi, A.D. Ho, S1 nucleasehypersensitive sites in an oligopurine/oligopyrimidine DNAfrom the t(10;14) breakpoint cluster region, Nucleic AcidsRes. 20 (1992) 263–266.
[84] M. Lu, I. Dube, S. Raimondi, A. Carroll, Y. Zhao, M. Minden, P.Sutherland, Molecular characterization of the t(10;14)translocation breakpoints in T-cell acute lymphoblasticleukemia: further evidence for illegitimate physiologicalrecombination, Genes Chromosomes, Cancer 2 (1990)217–222.
[85] J. Kagan, L.R. Finger, J. Letofsky, J. Finan, P.C. Nowell, C.M.Croce, Clustering of breakpoints on chromosome 10 in acuteT-cell leukemias with the t(10;14) chromosometranslocation, Proc. Natl. Acad. Sci. U.S.A. 86 (1989)
4161–4165.[86] J. Wang, S.N. Jani-Sait, E.A. Escalon, A.J. Carroll, P.J. de Jong,I.R. Kirsch, P.D. Aplan, The t(14;21)(q11.2;q22) chromosomaltranslocation associated with T-cell acute lymphoblastic

( 2 0
[88] Y. Tsujimoto, J. Yunis, L. Onorato-Showe, J. Erikson, P.C.
1258 d n a r e p a i r 5
leukemia activates the BHLHB1 gene, Proc. Natl. Acad. Sci.U.S.A. 97 (2000) 3497–3502.
[87] G.K. Przybylski, W.A. Dik, J. Wanzeck, P. Grabarczyk, S.
Majunke, J.I. Martin-Subero, R. Siebert, G. Dolken, W.D.Ludwig, B. Verhaaf, J.J. van Dongen, C.A. Schmidt, A.W.Langerak, Disruption of the BCL11B gene throughinv(14)(q11.2q32.31) results in the expression ofBCL11B-TRDC fusion transcripts and is associated with the0 6 ) 1246–1258
absence of wild-type BCL11B transcripts in T-ALL, Leukemia19 (2005) 201–208.
Nowell, C.M. Croce, Molecular cloning of the chromosomalbreakpoint of B-cell lymphomas and leukemias with thet(11;14) chromosome translocation, Science 224 (1984)1403–1406.




















