
Phosphorylation and oligomerization states of native pig brain HSP90 studied by mass spectrometry
Upload
independentCategory
view
3download
0
1
Controlling β amyloid oligomerization by the use of naphthalene sulfonates: Trapping low-molecular-weight
oligomeric species Astria D. Ferrão-Gonzales# §, Bruno K. Robbs# §, Vitor Hugo Moreau
§†, Aricéle Ferreira §, Luis
Juliano NetoΨ, Ana Paula Valente§†, Fabio C. L. Almeida§†, Jerson L. Silva§†, and Debora
Foguel§* §Instituto de Bioquímica Médica, Programa de Biologia Estrutural, †Centro Nacional de Ressonância Magnética Nuclear, Universidade Federal do Rio de Janeiro, Av. Bauhínia, 400- 21941-590 - Rio de Janeiro, RJ, Brazil, and Ψ Escola Paulista de Medicina- UNIFESP, São Paulo- SP, Brazil. # These authors contributed equally as first authors to this study
Running Title: Stabilization of small oligomers of β-amyloid by anionic sulfonate molecules Address correspondence to: Debora Foguel, Tel: +55- 21- 2562-6761; FAX +55-21- 2270-8647, e-mail: [email protected] * This work was supported in part by grants from the Conselho Nacional de Desenvolvimento Científico e Tecnológico, CAPES and FAPERJ to JLS, APV and DF, and from a Grant from ICGEB to JLS. Aggregation of proteins and peptides has been shown to be responsible for several diseases known as amyloidoses, which include Alzheimer's disease (AD), prion diseases, among several others. AD is a neurodegenerative disorder caused primarily by the aggregation of β-amyloid peptide (Aβ). Here we describe the stabilization of small oligomers of Aβ by the use of sulfonated hydrophobic molecules such as AMNS (1-amino-5-naphthalene sulfonate); 1,8-ANS (1-anilinonaphthalene-8-sulfonate) and bis-ANS (4,4´-dianilino-1,1´-binaphthyl-5,5´-disulfonate). The experiments were performed with either Aβ 1-42 or with Aβ 13-23, a shorter version of Aβ that is still able to form amyloid fibrils in vitro and contains amino-acid residues 16-20, previously shown to be essential to peptide-peptide interaction and fibril formation. All sulfonated molecules tested were able to prevent Aβ aggregation in a concentration dependent fashion, in the following order of efficacy: 1,8 ANS < AMNS < bis-ANS. Size-exclusion chromatography revealed that in the presence of bis-ANS, Aβ forms a heterogeneous population of low-molecular-
weight species that proved to be toxic to cell cultures. Since the ANS compounds all have apolar rings and negative charges (sulfonate groups), both hydrophobic and electrostatic interactions may contribute to interpeptide contacts that lead to aggregation. We also performed NMR experiments to investigate the structure of Aβ 13-23 in SDS micelles, and found features of an α-helix from Lys16 to Phe20. 1H-TOCSY spectra of Aβ 13-23 in the presence of AMNS displayed a chemical-shift dispersion quite similar to that observed in SDS, which suggests that in the presence of AMNS this peptide might adopt a conformation similar to that reported in the presence of SDS. Taken together, our studies provide evidence for the crucial role of small oligomers and their stabilization by sulfonate hydrophobic compounds.
The aggregation of proteins and peptides in vivo plays a fundamental role in the onset of several human pathologies known as amyloid diseases (1, 2). Despite the lack of significant sequence homology among the proteins involved in these diseases, amyloid fibrils seem to share a common structural motif, namely the cross β-pleated sheet (3-6).
JBC Papers in Press. Published on July 22, 2005 as Manuscript M501651200
Copyright 2005 by The American Society for Biochemistry and Molecular Biology, Inc.

2
The mechanisms responsible for fibril formation have been extensively investigated but they are still poorly understood, which makes it difficult to develop new drugs against these diseases and even to select suitable targets (4). Recent studies indicate that early aggregates and fibrils have a great contribution of hydrophobic interactions whereas late amyloid fibrils are more stabilized by hydrogen bonds (7, 8).
Alzheimer’s disease (AD) is one of the most common forms of senile dementia and is believed to be caused by aggregation of β-amyloid peptide (Aβ). Aβ is a peptide of 39 to 43 amino acid that is derived from a larger, type I transmembrane protein called amyloid precursor protein (APP). During APP processing by proteases called β and γ secretases, this precursor protein generates Aβ, which includes a segment of the APP protein that is embedded in the plasma membrane (9). Thus, Aβ is highly apolar and undergoes aggregation rapidly when dissolved in an aqueous environment, especially the larger peptide with 42-43 amino acids. Aβ 1-40 and Aβ 1-42 are the most common peptides found in amyloid plaques (10). Aβ 1-42 undergoes aggregation in vitro within a few minutes (11), while the Aβ 1-40 needs several hours or even days for its complete aggregation (12).
Due to the low solubility of Aβ peptides in water, all the structures obtained thus far in liquid media deposited in the Protein Data Bank (PDB, NIH, Bethesda) were obtained by NMR in apolar solvents (DMSO, TFE) (13, 14) or in a membrane-mimicking environment such as SDS (15-17). The description of experimental conditions in which this peptide remains soluble could be crucial for a better understanding of the mechanisms involved in its fibrillogenesis (4). In addition, greater solubility would allow the design of drugs that could trap this soluble conformation, impeding its aggregation and opening new strategies for treatment.
It has been proposed that Aβ aggregation occurs in several steps, including the formation of soluble, low-molecular-weight (LMW) oligomers (18-20). The LMW species that are present prior to fibril formation include
dimers and tetramers of Aβ, which can also be extracted from neuritic and vascular amyloids (21). Stable dimers have been purified from fibril suspensions of the synthetic peptide Aβ 1-40 (18, 20, 22). Also it has been shown that there is a population of soluble oligomers in AD brains, but not in normal brains (23) or in cell cultures (24, 25).
Much evidence has shown that Aβ has to be in a fibrillar arrangement to be neurotoxic (26, 27). However, dimers produced by the synthetic Aβ 1-42 have also been shown to be highly neurotoxic in vitro (21) but only in the presence of microglia cells. On the other hand, several other reports have shown that LMW species of Aβ are bioactive either by interacting with synapses (28,29), which could explain the loss of memory in AD, either by disrupting cognitive function (30), or by deregulating calcium homeostasis (31), which ultimately leads to cell death. These observations led the researchers to investigate further the role of these LMW species in the aggregation pathway of Aβ and in the development of AD (32,33).
Several attempts have been made to block fibril formation (34, 35) or even to promote fibril solubilization (36, 37). However, it is very important to know how and when these LMW species are formed and what is their participation in AD pathogenesis. Nakagami and coworkers (38) have shown that is possible to inhibit the toxic effect of Aβ to HeLa cells without affecting its fibrillogenesis.
Tjernberg and coworkers (39) mapped the regions of Aβ that are crucial for fibrillogenesis. They showed that a pentapeptide (KLVFF) comprising the Aβ sequence between residues 16-20 blocked fibril formation and that this region in Aβ 1-40 is crucial for inter-peptide interactions involved in fibril formation. Thereafter, Li and coworkers (40) showed that the residues 17-21 were essential to amyloid core formation. Norstedt and coworkers showed that the region comprising residues 14-23 of Aβ was still capable of forming amyloid fibrils and deletions or substitutions in this region resulted in the loss of fibril formation capability (41). For these reasons, besides Aβ 1-42, we

3
selected for our studies Aβ 13-23, a derivative of Aβ that is still able to form fibrillar aggregates in solution (Figure 1A). This region includes the hydrophobic core (17LVFF20) flanked by two charged residues, 16K and 22ED23.
Since hydrophobic and ionic interactions play an important role in Aβ fibril formation, several hydrophobic and/or charged molecules have been used successfully to block Aβ aggregation (35, 37, 42). Recently, we observed that the aggregation of a peptide derived from the murine prion protein was significantly attenuated by the hydrophobic anionic disulfonate fluorescent probe 4,4´-dianilino-1,1´- binaphthyl-5,5´-disulfonate (bis-ANS) in the micromolar range (43). The dye binds in a hydrophobic pocket, which appears to be the binding site for nucleic acids (44).
In the present study, we investigate the effects of bis-ANS and other compounds of the same family, namely ANS (1-anilino-naphthalene-8-sulfonate) and AMNS (1-amino-5-naphthalene sulfonate; Figure 1B) on the aggregation of two Aβ peptides, Aβ 1-42 and Aβ 13-23. We report that this family of compounds was able to abolish the aggregation of these peptides in aqueous solution almost completely, keeping them soluble. By size-exclusion chromatography we were able to trap small oligomers that accumulate when aggregation was performed in the presence of bis-ANS. These small oligomers were toxic to RAW (mouse monocyte-macrophage) cell cultures when assayed by MTT (3-(4,5-dimethylthiazol-2-yl)- 2,5-diphenyltetrazolium bromide) reduction. These results allowed us to perform TOCSY experiments with Aβ 13-23 in the presence of AMNS, an effective inhibitor of the aggregation of this peptide. However, the peptide presented an enormous flexibility under these conditions, which prevented us from solving its structure. On the other hand, we were able to solve the structure of Aβ 13-23 in the presence of SDS, where it forms an α-helix extending from Lys16 through Phe20. The chemical shifts observed in the presence of AMNS and SDS were almost superimposable, suggesting similar structures. We postulate that ANS-related compounds inhibit fibril
formation through hydrophobic and electrostatic interactions established between these compounds (rings and sulfonate groups, respectively) and the amino-acid residues of the peptide (apolar and charged, respectively).
Materials and Methods
Material - Synthetic Aβ 1–42 was obtained from Dr. Luis Juliano (Universidade Federal do Estado de São Paulo, SP- Brazil). The fragment Aβ 13-23 was purchased from Genemed Synthesis, San Francisco, CA. They were lyophilized and stored at -80 °C. D2O and SDS-D25 were purchased from Cambridge Isotope Laboratories (Woburn, MA). Both peptides were subsequently purified to greater than 98% homogeneity by reverse-phase HPLC. The purity of the peptides was verified by electron spray mass spectrometry. All ANS-related compounds were from Molecular Probes (Eugene, OR). They were dissolved in deionized, filtered water and their concentrations measured as described in the catalogue from Molecular Probes. All other reagents were of the highest analytical grade available. Preparation of peptide stock solutions - For aggregation assays, a stock solution of Aβ was prepared as follow: 1 mg of each peptide was weighted and dissolved with 100 µL of 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) to disassemble preformed aggregates. The samples were then dried in a SpeedVac system. Then, 1 mL of 100 % DMSO was added to each sample and these stock solutions were stored at -20 °C until use. The stock concentrations of the peptides were approximately 250 µM for Aβ 1-42 and 1000 µM for Aβ 13-23. In-vitro aggregation measurements - Kinetics of aggregation were monitored in an ISS K2 spectrofluorometer (Champaign, IL) by following the increase in light scattering (LS) using a 1.0 cm quartz cuvette lightening the samples at 320 nm and collecting scattered light at 320 nm. Aggregation of Aβ 1-42 and Aβ 13-23 was accomplished by mixing an aliquot of the peptides from the DMSO stock solution into aggregation buffer (50 mM Na

4
phosphate in 100 mM NaCl, pH 7.4) at 37oC. Light scattering by the buffer was used as baseline. Negative controls for aggregation were performed by diluting the peptides in 100% DMSO. The residual DMSO concentration present in the experiments (10% for Aβ 1-42 and 2.5% for Aβ 13-23) did not interfere with the aggregation assays (data not shown). Aggregation assays in the presence of the ANS-derivatives (Figure 1) were performed by diluting the peptides into a solution already containing the ANS derivatives. The extent of inhibition caused by ANS derivatives was based on the maximum aggregation attained in the buffer without the probe. Cell cultures and MTT assay - RAW cells (mouse monocyte-macrophage, American Type Culture Collection) were routinely cultured in RPMI medium (Gibco BRL) supplemented with 10% (v/v) bovine calf serum and 3 mM glutamine, 100 U mL-1 penicillin and 10 µg mL-1 streptomycin, in a 5% CO2 humidified atmosphere at 37 oC. Cells were used after five passages and were plated at a density of 10,000 cells per well in 100 µL of fresh medium on 96-well plates. After 24 h, RPMI medium was exchanged for 100 µL of RPMI without phenol red and supplemented with 10% bovine calf serum and antibiotics. Cell-mediated reduction of 3-(4,5-dimethylthiazol- 2-yl)- 2, 5- diphenyltetrazolium bromide (MTT) was used to monitor Aβ toxicity as described by Bucciantini et al. (45). Aggregates of Aβ 1-42 and Aβ 13-23 were prepared as described above and added to cell cultures to a final concentration of 25 µM. For controls, the buffer with or without the probes was added. Reduced MTT was determined by measuring absorbance at 540 nm after 24 h using an automatic microplate reader. Thioflavin T (ThT) and Congo Red (CR) binding assays - CR and ThT binding assays were performed after fibril formation to ensure that the observed increase in light scattering was indeed due to the presence of amyloid fibrils. The amount of CR (15 µM) bound was evaluated by measuring the ratio between the absorbance values at 480 nm and at 540 nm, according to Lai et al. (46). The binding of ThT (10 µM) was monitored by the increase in ThT
fluorescence at 482 nm, setting excitation at 440 nm. Size exclusion chromatography - Size exclusion chromatography was performed on an HPLC system (Shimadzu) using a prepacked TSK G3000swxl (0.78 x 30 cm) and Synchropak GPC 60 (0.5 x 20 cm) columns. The system was equilibrated with aggregation buffer at 1.0 mL/min. Sample elution was monitored by absorption at 280 nm and by fluorescence emission at 315 nm (excitation at 275 nm, not shown). The column and buffer were maintained at room temperature. TSK G300swxl column resolves globular proteins ranging in size from 5,000-150,000 while Synchropak GPC60 resolves proteins from 250-28,000. SDS-PAGE electrophoresis - The aggregates obtained after 1 h under aggregating conditions (100 µL) were dried and 15 µL of SDS-PAGE sample buffer was added. The samples were loaded onto a 12% SDS-polyacrylamide gel (47) without boiling. Low molecular weight markers purchased from Sigma Chemical Company were used as molecular weight standard. The gels were stained with Comassie Brilliant Blue. NMR experiments and structural calculations - The Aβ 13-23 stock was prepared as described above. A 3 mM sample was diluted in 50 mM phosphate buffer, 100 mM NaCl, pH 7.4 in the presence of either D25-SDS (35 mM) or AMNS (20 mM). The pH was previously verified and 10% D2O was then added for lock. The resulting solutions were immediately centrifuged for 15 min at 13,000 g and no pellet was observed. 1H-NMR data were acquired in a Bruker DRX-600 MHz spectrometer employing pulsed field gradient probes. The experiments were performed at 310 or 298 K, when D25-SDS or AMNS were present, respectively. NOESY and TOCSY spectra were acquired with 2048 x 512 complex data points and phase-sensitive mode using States-TPPI for quadrature detection in the t1 dimension. Water signal was suppressed using watergate sequence (48). Mixing times for NOESY and TOCSY were 150 ms and 70 ms, respectively. Spectra were processed using NMRPipe (49) and analysed using NMRView (50). Structure calculations were performed

5
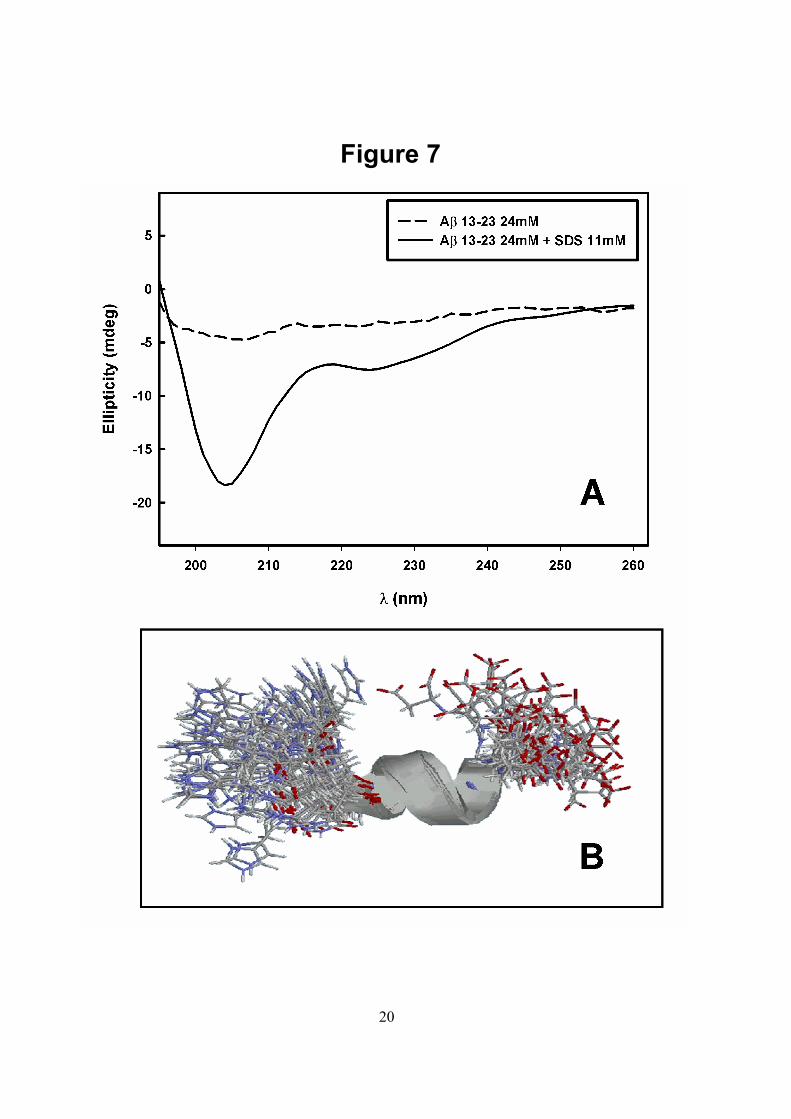
using NOE as distance restraints and 3JHNHα couplings constants as dihedral restraints in peptide. Fifty-two intraresidue and 22 inter-residue NOEs and 6 dihedral restraints from 3JHNHα coupling constants were used. One hundred structures were calculated with simulated annealing protocols using CNS program (51). Twenty structures with the lowest energies were selected for RMSD calculation and shown in Figure 7B.
RESULTS In vitro inhibition of fibril formation by anilinonaphthalene compounds - Aggregation of β 1-42 and β 13-23 in vitro is already well documented (11, 39). We performed aggregation kinetics with both peptides in stagnant solutions under optimal conditions (pH 7.4, 37 oC) by following the increment in the light scattering (LS) that takes place when the peptides are diluted out from a pure DMSO solution (Figure 2).
As seen in Figure 2, the increase in the LS for both peptides is very rapid and pronounced, taking less than 5 min for completion. The aggregation of Aβ 1-42 is slightly faster and considerably more extensive than the aggregation of Aβ 13-23 (Table 1), although both peptides present a similar aggregation profile (Figure 2). In order to confirm that the increase in LS represents the formation of amyloid fibrils, thioflavin T and Congo Red binding were also evaluated and the data are presented in Table 1. As seen by the values presented in Table 1, both peptides form real fibrils in solution.
To gain a preliminary idea of the mass of the species that coexist with the amyloid fibrils or are rescued from fibrils by the action of SDS, we carried out SDS-PAGE with the fibrils of Aβ 1-42 (inset of Figure 2, left) and Aβ 13-23 (inset of Figure 2, right) after one hour in aggregating buffer. As seen, the fibrils of Aβ 1-42 solubilized in SDS show the presence of species with ~ 4 and 16-20 kDa, which are compatible with the mass of monomers and tetramers. These species were either rescued from the fibrils by the action of SDS or were present in equilibrium with them.
The gel obtained with the fibrils of Aβ 13-23 treated with SDS also presented low-molecular-weight species, although the bands are very faint. Since the fibrils of β 13-23 are SDS-resistant (data not shown), the presence of these low-molecular-weight species (range ~10 – 16 kDa) suggests they are probably in equilibrium with the fibrils before SDS addition.
Since the light-scattering increase reflects the formation of amyloid fibrils with both two peptides, we were able to examine the effect of small, hydrophobic molecules belonging to the family of ANS compounds. The use of light scattering measurements to evaluate the extent of fibril formation is essential, since the ANS compounds fluoresce in the same range as thioflavin T.
To verify the effect of bis-ANS on the aggregation of Aβ 1-42 and Aβ 13-23, experiments similar to those in Figure 2 were performed in the presence of increasing concentrations of this probe in the aggregating solution. The final LS values attained after 20 min were compared with the LS of the control sample (absence of bis-ANS, 100% aggregation) and the results (Figure 3) shows that bis-ANS inhibits the aggregation of both peptides in a dose-dependent fashion. In the presence of 200 µM bis-ANS (peptide:bis-ANS molar ratio = 1:8), aggregation of Aβ 1-42 and Aβ 13-23 is inhibited by 80 and 95 %. However, as seen, aggregation of Aβ 13-23 is more sensitive to the probe with a half-maximal effect attained in 15 µM bis-ANS compared to 62 µM for the longer peptide.
In order to evaluate the size of the species formed during aggregation of Aβ in the presence of bis-ANS, size exclusion chromatography was performed. Figure 4 shows the chromatograms of Aβ 1-42 (panel A) and Aβ 13-23 (panel B) after aggregation in the absence (dashed line) or in the presence (solid line) of 200 µM bis-ANS. After one hour in aggregating buffer, the samples of Aβ 1-42 and Aβ 13-23 were injected directly into a G3000swxl or GPC 60 column, respectively, without previous centrifugation. In both cases there are high-molecular-weight species present in the control samples, which elute in

6
the void volume of the columns; these species are absent from the samples incubated in the presence of bis-ANS. This indicates that bis-ANS is able to prevent the formation of high-molecular-weight species in both Aβ samples. Furthermore, the main population present in the samples aggregated in the presence of bis-ANS consists of low-molecular-weight (LMW) species that elute close to or after the total volume of the columns. Although, their precise weights cannot be inferred from these data, the presence of these LMW species during the aggregation of Aβ is consistent with the data from other groups (19, 20).
To better visualize the LMW species of Aβ 1-42 stabilized by bis-ANS, the samples were briefly centrifuged to remove the large oligomers before injection into the G3000swxl (panel C) or GPC 60 (panel D) column. As seen in panel C, the high-molecular-weight species present in the aggregation of Aβ 1-42 that elute at ~ 4 min (panel A) disappeared with the centrifugation, leaving a more homogeneous population containing LMW. In the sample of Aβ 1-42 aggregated in the presence of 200 µM bis-ANS, the population is more heterogeneous, comprising species with different weights, but all of them smaller then the resolution of the column (panel C). This indicates that bis-ANS is stabilizing small oligomeric forms of Aβ (LMW species), which are present in the aggregation pathway of Aβ. This can be confirmed in panel D, which shows larger amounts of these LMW species in the sample aggregated in the presence of bis-ANS. Altogether, these data suggests that bis-ANS is able to prevent fibril formation by stabilizing a large range of LMW species of Aβ.
It has been proposed that the LMW species formed during aggregation of several amyloidogenic proteins are in fact the toxic species (27-31, 33, 45). In order to evaluate whether bis-ANS is trapping such species, MTT assays were performed to evaluate RAW cell viability (Figure 5) (52). Thus, we incubated 25 µM of Aβ 1-42 with different concentrations of bis-ANS (50 and 100 µM) during 20 min, in order to stabilize the LMW species. These suspensions were added to
RAW cells and MTT reduction was evaluated 24h hours later (Figure 5). The species trapped in the presence of 100 µM bis-ANS was more toxic (bar 6; *p < 0.01) to these cells than the fibrils of Aβ produced in the absence of any addition (bar 4) or in the presence of 50 µM bis-ANS (bar 5), a concentration that inhibited aggregation by less than 50% (Figure 3). These results are consistent with a role for the LMW species, which are more abundant in the presence of bis-ANS. We note that these concentrations of bis-ANS are not toxic to this cell line (Figure 5, bars 2 and 3), although higher concentrations of bis-ANS (> 200 µM) exhibited a toxic effect (not shown).
Since both peptides responded similarly to the inhibitory effect of bis-ANS, we chose Aβ 13-23 as a model for further experiments performed with other bis-ANS related compounds (ANS and AMNS). As seen in Figure 6, ANS (triangles) also inhibited the aggregation of β 13-23, but less effectively than bis-ANS (Figure 3). Strikingly, AMNS (Figure 6, circles) was more potent than ANS and 100-150 µM AMNS abolished completely the aggregation of this peptide. The half-maximal effects were obtained at 150 and 30 µM for ANS and AMNS, respectively. At these concentrations, the drug:peptide molar ratio was 6:1 (ANS) and 1.2:1 (AMNS). Comparing the structure of β 13-23 in the presence of SDS or in the presence of AMNS by NMR - Several laboratories have shown that Aβ remains soluble in SDS micelles and the region comprising the hydrophobic residues 17-21 tends to assume an α-helical structure (15, 53, 54). In the case of Aβ 13-23, circular dichroism (CD) measurements were performed in the presence of SDS as shown in Figure 7A. The spectrum presented two minima at 208 and 222 nm, a profile that is characteristic of α-helix structure. Although Aβ 13-23 also remains soluble in the presence of AMNS, the CD spectrum of Aβ 13-23 in this case (not shown) was too noisy to provide useful data due to the large absorption of AMNS in the UV region.
Since Aβ 13-23 remains soluble and ordered in the presence of SDS, we performed

7
1H-NMR experiments at pH 7.4 and 37 oC to determine its three-dimensional structure. Although we already knew that this peptide adopts an α –helical structure in the presence of SDS (15-17), we aimed to determine which region of the peptide is involved in this helix. For this purpose, TOCSY and NOESY experiments were performed as described under Experimental Procedures. The three-dimensional structure of Aβ 13-23 resolved in the presence of SDS micelles is presented in Figure 7B, which shows the twenty lowest energy structures calculated using NOEs and 3JHNHα coupling constants as restraints. As shown, there is a short α-helix comprising the residues from Lys16 to Phe20.
1H-NMR (TOCSY and NOESY) spectra of Aβ 13-23 in the presence of AMNS (molar ratio 4:1) were also collected. The TOCSY spectrum in the presence of AMNS presented chemical-shift dispersion very similar to that observed in the presence of SDS. Figure 8 shows the TOCSY spectra in the amide region of 4 mM Aβ 13-23 in the presence of 35 mM SDS (black) or in the presence of 20 mM AMNS (red). All spin systems are present in both samples and could be seen by changing the spectra threshold. This result suggests that the soluble form of Aβ 13-23 stabilized by AMNS may be similar to the helicoidal structure observed in presence of SDS. Aβ 13-23 NOESY spectrum in the presence of AMNS displayed fewer NOE peaks when compared with the same spectrum performed in the presence of SDS, what suggests that soluble Aβ 13-23 in AMNS must be dynamically more flexible than the peptide in SDS micelles. Due to the limited number of NOE restraints on the NOESY spectrum of Aβ 13-23 in the presence of AMNS, we were not able to perform structural calculations. We are working now to establish other conditions that will make it possible to determine the three-dimensional structure of Aβ 13-23 in aqueous solution.
DISCUSSION
The neurotoxic effects observed in AD
have been linked to fibrils present in the brain
parenchyma of the patients (“amyloid cascade” hypothesis, by 55). Several laboratories have been trying to characterize the species present in the very first steps of Aβ aggregation. From these studies, oligomeric forms of Aβ formed prior to fibril formation have been identified as neurotoxic (28-30, 38). These soluble oligomers are common to most amyloids and may be responsible for the amyloidogenic diseases. Recently, a polyclonal antibody that recognizes soluble oligomeric intermediates of Aβ was also able to recognize soluble oligomers from several other types of amyloidogenic proteins and peptides, indicating that all of them have a common architecture (56). This antibody abolished the in-vitro neurotoxic effect of these oligomers, reinforcing the idea that they are indeed the pathogenic agents in these diseases.
Several sub fragments of Aβ were tested in an effort to dissect the minimum core necessary to keep fibril formation. The region 16-20 has been shown to be essential for fibrillogenesis of Aβ peptides (11, 39, 57). Molecular modeling performed with this region using Aβ 14–23 showed that oligomers could be formed by an antiparallel β-sheet kept by favorable hydrophobic interactions and stabilized by salt bridges between charged residues (39). Besides, Aβ 1-40 and Aβ 12-28 had their three-dimensional structures determined in SDS micelles at acidic pH and presented the region 15-24 structured as an α-helix. This structure seems to be stabilized by charged groups present in the β-amyloid primary sequence (15, 53). Thus, this same sequence of amino acids could be organized in β-sheets or as α-helix, depending on the environment. In addition, Kirkitadze and coworkers (32) have already described the formation of a transient oligomer during aggregation of Aβ 1-40 and 1-42 with a high content of α-helixes. Thus, it is possible that the helix here observed by NMR is the same one observed in this transient α-helical species.
Studies performed by Montserret et al. (58) using a basic and an acidic peptide whose primary sequence was derived from the sequence of human platelet factor 4 (PF-4) showed that only the basic peptide

8
(QAPAYKKAAKKLAES) in the presence of SDS could assume an α-helical structure at neutral pH (58). This helix was stabilized by strong, electrostatic interactions established between the anionic groups of SDS and the cationic groups of the lysines. This region seems to be necessary to initiate the folding of this basic peptide. From these results, a model was proposed to explain the effects of SDS as a α-helix inductor. This model proposes that, although the folding of the peptide is mostly driven by hydrophobic effects, electrostatic interactions (salt bridges between lysine residues and SDS sulfate groups) might play a significant role in the formation and stabilization of the α-helical structure (58).
Recently, LeVine (59) has shown that bis-ANS interacts strongly with the soluble Aβ 1-40 in acidic buffer solutions but interacts poorly with the formed fibrils. In this study, however, the effects of this probe on the aggregation of Aβ were not evaluated. Interestingly, bis-ANS was also able to inhibit the aggregation of a peptide derived from the hamster prion protein (43). In the present study, we were able to show that ANS-related compounds abolish almost completely the aggregation of Aβ 1-42 and 13-23 in a dose-dependent manner. These compounds seem to interact with and stabilize soluble species present in the aggregation pathway that leads to fibril formation. These species were observed in size exclusion chromatography experiments and proved to be more toxic than the aggregates generated in the absence of ANS-related compounds. One possibility would be the formation of bis-ANS micelles with monomers inserted in them, which would elute in a SEC experiment at the same position as the oligomers described here. This possibility was discarded because a solution of 200 µM bis-ANS elutes in a size exclusion column at the same position as 2 µM (not shown). Besides, bis-ANS has no aliphatic tail to allow micelle formation.
What could be the molecular basis for the inhibition of aggregation? The ANS family of compounds might interfere with electrostatic and hydrophobic interactions that are crucial
for fibril formation and stabilization. This effect could be achieved due to the dual nature of the ANS-derived molecules, which have hydrophobic rings and charged groups. Thus, it is possible that, similarly to SDS, bis-ANS, ANS and AMNS might interact with specific, crucial regions of the Aβ peptide in the very beginning of the nucleation step, shielding them from interaction with other peptide molecules. As a result, the oligomerization reaction would be halted. This mechanism of inhibition has been proposed for other peptides and proteins solubilized in SDS micelles (58, 60).
It has been shown that the binding of bis-ANS to tubulin monomers blocks their polymerization induced by microtubule-associated proteins (61). In addition, Teschke et al. (62) showed that bis-ANS was able to inhibit the assembly of coat proteins from bacteriophage P22 into procapsids. These results suggest that bis-ANS can interfere with inter-subunit contacts regardless of whether these are related to the assembly of a microtubule, a viral particle or in our case, a fibril.
Another relevant observation is the fact that highly sulfated glycosaminoglycans (HSPG) have been found to be associated with Aβ fibrils in vivo (63). In vitro, HSPG and Congo Red (CR) compete for the same binding sites on Aβ fibrils (64). CR is a specific dye for amyloid fibrils sharing similarities with bis-ANS such as the presence of sulfonate groups. CR was able to inhibit aggregation of Aβ into fibrils (27, 65) decreasing concomitantly its neurotoxicity (66). Li et al. (40) proposed a model to explain CR effects on the fibrillogenesis of Aβ. These authors postulated that Lys 16 could interact electrostatically with the sulfonate groups of CR, in addition to any hydrophobic contacts that could be established between uncharged regions of the two molecules.
Bis-ANS and CR share structural similarities in size and number of aromatic rings as well as in chemical properties such as hydrophobicity and the presence of charged groups (sulfonates). Thus, it is possible that both compounds might act similarly in inhibiting the aggregation of Aβ. However,

9
while the LMW species trapped in the presence of bis-ANS are toxic leading to an increase in cell death, the one trapped in the presence of CR are not. We do not have any explanation for this observation, but it is possible that the LMW species trapped in the presence of these compounds are different in terms of size and consequently in toxicity. Recently, Gazit et al. (67) showed that phenol red, an aromatic compound with a sulfonate radical, could inhibit with high efficiency fibril formation of the human islet amyloid polypeptide (hIAPP). The authors propose that heteroaromatic interactions exerts a crucial role in the
inhibitory mechanism. A similar mechanism might be underlying the effect of bis-ANS describe here, especially because the Aβ 13-23 contains two adjacent phenilalanines.
The search for molecules that can interfere with Aβ aggregation could lead to the understanding of the driving forces behind inter- and intra-peptide interactions as well as the structural motifs responsible for them. These molecules could also be used as lead compounds for developing drugs against this devastating disease.
REFERENCES
1. Kelly, J. W. (1998) Curr. Opin. Struct. Biol. 8, 101–106 2. Dobson, C.M. (2004) Methods 34, 4-14 3. Kirschner, D. A., Abraham, C., and Selkoe, D. J. (1986) Proc. Natl. Acad. Sci. U.S.A. 83, 503- 507 4. Ross, C. A., and Poirier, M. A. (2004) Nat. Med. 10, S10-S17 5. Urbanc, B., Cruz, L., Yun, S., Buldyrev, S.V., Bitan, G., Teplow, D.B., and Stanley, H.E. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 17345-17350 6. Sumner, O., Atkins, E., Sikorski, P., Johansson, J., and Serpell, L.C. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 315-320 7. Foguel, D., Suarez, M.C., Ferrao-Gonzales, A.D., Porto, T.C., Palmieri, L., Einsiedler, C.M., Andrade, L.R., Lashuel, H.A., Lansbury, P.T., Kelly, J.W. and Silva, J.L. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 9831-9836 8. Foguel, D. and Silva, J.L. (2004) Biochemistry 43, 11361-11370 9. Vassar, R., and Citron, M. (2000) Neuron, 27, 419- 422 10. Joachim, C. L.., Mori, H., and Selkoe, D. J. (1989). Nature 341, 226- 230 11. Wood, S. J., Wetzel, R., Martin, J. D., and Hurle, M. R. (1995) Biochemistry 34, 724–730 12. Jarrett, J.T., Berger, E.P., and Lansbury, P.T. Jr. (1993) Ann. N. Y. Acad. Sci. 695, 144-148 13. Sticht, H., Bayer, P., Willbold, D., Dames, S., Hilbich, C., Beyreuther, K., Frank, R.W., and Rosch, P. (1995) Eur. J. Biochem. 233, 293-298 14. Serpell, L.C. (2000) Biochim. Biophys. Acta 1502, 16-30 15. Coles, M., Bicknell, W., Watson, A., Fairlie, D., and Craik, D. (1998) Biochemistry 37 11064-11077 16. Riek, R., Guntert, P., Dobeli, H., Wipf, B., and Wüthrich, K. (2001) Eur. J. Biochem. 268, 5930- 5936 17. Ippel, J.H., Olofsson, A., Schleucher, J., Lundgren, E., and Wijmenga, S.S. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 8648- 8653 18. Walsh, D. M., Lomakin, A., Benedek, G. B., Condron, D. B., and Teplow, D. B. (1997) J. Biol. Chem. 272, 22634- 22372 19. Klein, W. L, Stine, W. B., Jr., and Teplow, D. B. (2004) Neurobiol. Aging 25, 569-580 20. Klein, W. L., Krafft, G. A., and Finch, C.E. (2001) TRENDS in Neurosciences 24, 219-224 21. Roher, A. E., Chaney, M. O., Kuo, Y. M., Webster, S.D., Stine, W. B., Haverkamp, L. J., Woods, A. S., Cotter, R. J., Tuohy, J. M., Kraft, G. A., Bonnell, B. S., and Emmerling, M. R. (1996) J. Biol. Chem. 271, 20631-20635

10
22. Garzon-Rodriguez, W., Sepulveda-Becerra, M., Milton, S., and Glabe, C. G. (1997) J. Biol. Chem. 274, 21037- 21044 23. Kuo, Y.M., Emmerling, M. R., Vigo-Pelfrey, C., Kasunic, T. C.,. Kirkpatrick, J. B, Murdoch, G. H., Ball, M. J., and Roher, A. E. (1996) J. Biol. Chem. 271, 4077-4081 24. Haas, C., Schlossmacher, M.G., Hung, A. Y., Vigo-Pelfrey, C., Mellon A., Ostaszewski, B. L., Lieberburg, I., Koo, E. H., Schenk, D., and Teplow, D. B. (1992) Nature 359, 322-325 25. Seubert, P., Vigo-Pelfrey, C., Esch, F., Lee, M., Dovey, H., Davis, D., Sinha, S., Schlossmacher, M., Whaley, J., and Swindlehurst, C. (1992) Nature 359, 325- 327 26. May, P. C., Gitter, B. D., Waters, D. C., Simmons, L. K., Becker, G. W., Small, J. S., and Robison, P. M. (1992) Neurobiol. Aging 13, 605–607 27. Lorenzo, A., and Yankner, B. (1994) Proc. Natl. Acad. Sci. U.S.A. 91, 12243-12247 28. Gong, Y., Chang, L., Viola, K.L., Lacor, P. N., Lambert, M. P., Finch, C. E., Krafft, G. A., and Klein, W. L. (2003) Proc. Natl. Acad. Sci. U.S.A 100, 10417-10422 29. Lacor, P. N., Buniel, M. C., Chang, L., Fernandez, S. J., Gong, Y., Viola, K. L., Lambert, M.P., Velasco, P. T., Bigio, E. H., Finch, C. E., Krafft, G. A., and Klein, W.L. (2004) J. Neurosci. 24, 10191-10200 30. Cleary, J.P., Walsh, D.M., Hofmeister, J., Shankar, G.M., Kuskowski, A. A., Selkoe, D. J., and Ashe, K. H. (2004) Nature Neurosci. 8, 79-83 31. Demuro, A., Mina, E., Kayed, R., Milton, S.C., and Parker, I. (2005) J. Biol. Chem. 280, 17294-17300 32. Kirkitadze, M. D., Bitan, G., and Teplow, D. B. (2002) J. Neurosci. Res. 69, 567-577 33. Klein, W. L. (2002) Neurobiol. Aging 23, 231-235 34. Howlett, D. R., Perry, A. E., Godfrey, F., Swatton, J. E., Jennings, K. H., Spitzfaden, C., Wadsworth, H., Wood, S. J., and Markwell, R. E. (1999) Biochem. J. 340, 283- 289 35. Bornemann, K. D., and Staufenbiel, M. (2000) Ann. NY. Acad. Sci., 908, 260-266 36. Solomon, B., Koppel, R., Frankel, D., and Hanan-Aharon, E. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 4109-4112 37. De Felice, F. G., Houzel, J.C., Garcia-Abreu, J., Louzada, P.R. Jr., Afonso, R. C., Meirelles, M. N., Lent, R., Neto, V.M., and Ferreira, S.T. (2001) FASEB J. 15, 1297-1299 38. Nakagami, Y., Nishimura, S., Murasugi, T., Kubo, T., Kaneko, I., Meguro, M., Marumoto, S., Kogen, H., Koyama, K., and Oda, T. (2002) E. J. Pharmacol. 457, 11-17 39. Tjernberg, L., Callaway, D., Tjernberg, A., Hahne, S., Lilliehook, C., Terenius, L., Thyberg, J., and Norstedt, C. (1999) J. Biol. Chem. 274, 12619-12625 40. Li, L., Darden, T. A., Bartolotti, L., Kominos, D., and Pedersen , L. G. (1999) Biophys. J. 76, 2871–2878 41. Tjernberg, L. O., Näslund, J., Lindqvist, F., Johansson, J., Karlström, A.R., Thyberg, J., Terenius, L., and Nordstedt, C. (1996) J. Biol. Chem. 271, 8545–8548 42. Kisilevsky, R., Lemieux, L.J., Fraser, P.E., Kong, X., Hultin, P.G., and Szarek, W.A. (1995) Nat. Med. 1, 143- 148 43. Cordeiro, Y., Lima, L.M., Gomes, M.P., Foguel, D., and Silva, J.L. (2004) J. Biol. Chem. 279, 5346-5352 44. Cordeiro, Y., Machado, F., Juliano, L., Juliano, M.A., Brentani, R.R., Foguel, D. and Silva, J.L. (2001) J. Biol. Chem. 276, 49400-49409 45. Bucciantini, M., Giannoni, E., Chiti, F., Baroni, F., Formigli, L., Zurdo, J., Taddei, N., Ramponi, G., Dobson, C. M., and Stefani, M. (2002) Nature 416, 507- 511 46. Lai, Z., Colon, W., Kelly, J. (1996) Biochemistry 35, 6470- 6482 47. Laemmli, U.K. (1970) Nature 227, 680-685 48. Piotto, M., Sandek, V., and Sklenar, V. (1992) J. Biomol. NMR 2, 661-666 49. Delaglio, F., Grzesiek, S, Vuister, G. W., Zhu, G., Pfeifer, J., and Bax, A. (1995) J. Biomol. NMR. 6, 277-293 50. Johnson, B. A., and Blevins, R. A. (1994) J. Biomol. NMR 4, 603-614

11
51. Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T., and Warren, G. L. (1998) Acta Crystallogr. D54, 905-921 52. Liu, Y., and Schubert, D. (1997) J. Neurochem. 69, 2285–2293 53. Talafous, J., Marcinowski, K. J., Klopman, G., and Zagorski, M. G. (1994) Biochemistry 33, 7788-7796 54. Fletcher, F., and Keire, D. (1997) Prot. Sci. 6, 666-675 55. Hardy, J. A., and Higgins, G. A. (1992) Science 256, 184–185 56. Kayed, R., Head, E., Thompson, J.L., McIntire, T.M., Milton, S.C., Cotman, C.W., and Glabe CG. (2003) Science 300, 486-489 57. Hilbich, C., Kisters-Woike, B., Reed, J., Masters, C. L., and Beyreuther, K. (1992) J. Mol. Biol. 228, 460–473 58. Montserret, R., McLeish, M. J., Böckmann, A., Geourjon, C., and F. Penin (2001) Biochemistry 39, 8362- 8373 59. Levine, H. 3rd. (2002) Arch. Biochem. Biophys. 404, 106-115 60. Dong, M., Penin, F., and Baggetto, L. G. (1996). J. Biol. Chem. 271, 28875-28883 61. Mazumdar, M., Parrack, P.K., Mukhopadhyay, K., and Bhattacharyya, B. (1992) Biochemistry 31, 6470- 6474 62. Teschke, C. M., King, J., and Prevelige, P. E., Jr. (1993) Biochemistry 32, 10658-10665 63. Snow, A. D., R. Sekiguchi, D. Nochlin, P. Fraser, K. Kimata, A. Mizutani, M. Arai, W. A. Schreier, and D. G. Morgan. (1994) Neuron 12, 219 –234 64. Pollack, S. J., Sadler, I. I. J., Hawtin, S. R., Tailor, V. J., and Shearman, M. S. (1995a) Neurosci. Lett. 197, 211–214 65. Esler, W. P., Stimson, E. R., Ghilardi, J. R., Felix, A. M., Lu, Y.-A., Vinters, H. V., Mantyh, P. W., and Maggio, J. E. (1997) Nat. Biotechnol. 15, 258-263 66. Pollack, S. J., Sadler, I. I. J., Hawtin, S. R., Tailor, V. J., and Shearman, M. S. (1995b) Neurosci. Lett. 184, 113-116 67. Porat, Y., Mazor, Y., Efrat, S., and Gazit, E. (2004) Biochemistry 43, 14454-11462
FOOTNOTES
The authors thank Emerson Gonçalves for his competent technical assistance, Dr. Katya Sousa and Dr. Marcius Almeida for assistance in NMR structural determination. The authors acknowledge Dr. Martha M. Sorenson for her kind reading of the manuscript.
The abbreviations used are: ANS: 1-anilinonaphthalene-8-sulfonic acid, sodium salt; bis-ANS: 4,4´-dianilino-1,1´- binaphthyl-5,5´-disulfonate potassium salt ; AMNS: 1- amino-5-naphthalene sulfonate; SDS: sodium dodecyl sulphate.
TABLE I
Extent of aggregation for Aβ 1-42 and Aβ13-23 at pH 7.4, 37oC.
Sample Increase in LS at 320 nm (LS/LS0)
ThT binding (Spectral area of ThT bound/
Spectral area of free ThT)
CR binding (mmoles CR/ L fibrils)
Aβ 1-42 (25 µM) 90-120 110 0.8- 1.5 Aβ 13-23 (25 µM) 40-50 36 1.5- 2

12
FIGURE LEGENDS Fig. 1. Primary sequence of Aβ 1-42 and the structures of the compounds utilized in this study. ANS: 1-1 anilino-naphthalene-8-sulfonic acid, sodium salt; bis-ANS: 4,4´-dianilino-1,1´- binaphthyl-5,5´-disulfonate, potassium salt; AMNS: 1-amino-5-naphthalene sulfonate and SDS: sodium dodecyl sulphate. The region 13-23 is printed in gray in the primary sequence of Aβ 1-42. Fig. 2. Kinetics of aggregation of Aβ 1-42 and Aβ 13-23 as followed by the increase in light scattering (LS). Stock solutions of Aβ 1-42 (black) and Aβ 13-23 (dashed line) in 100 % DMSO were diluted to 25 µM in phosphate buffer 50 mM, NaCl 100 mM, pH 7.4 at 37oC and the increase in LS followed with time. The LS measured at each time point was divided by the initial LS value (LS/LS0). The LS was measured by exciting the sample at 320 nm and collecting the scattered light at 320 nm. Note that the ordinate values for Aβ 1-42 (right axis) are twice those for Aβ 13-23 (left axis). Inset: After 1 h under aggregating conditions, the samples were analysed by SDS-PAGE (12 % acrylamide). Lane 1: 10 µL of Aβ 1-42 sample; lane 2: 10 µL of Aβ 13-23 sample. Gels were stained with Coomassie Brilliant Blue. Fig. 3. Effect of increasing concentrations of bis-ANS on the aggregation of Aβ 1-42 (●) and Aβ 13-23 (▲) evaluated by the change in the LS. Aggregation was carried out in the presence of the concentrations of bis-ANS shown on the abscissa and the LS was recorded for 20 mim. The final LS value reached in each experiment was compared to the value attained in a control sample, which underwent aggregation for the same period of time in the absence of bis-ANS. Peptide concentration was 25 µM. Other conditions are as described for Figure 2. Continuous lines represent a non-linear regression to a single exponential. Fig. 4. Size exclusion chromatography of Aβ 1-42 and Aβ 13-23 aggregated in the absence or in the presence of bis-ANS (200 µM). Aggregates of Aβ 1-42 (A) and Aβ 13-23 (B) obtained as in Figure 3 were vortexed and applied directly onto a G3000swxl or GPC 60 column, respectively. The chromatograms of the control samples (absence of bis-ANS) are shown in dashed lines while the one with for bis-ANS-treated samples are shown in continuous lines. The same samples used in (A) were centrifuged for 15 min at 13,000 g and the supernatants were applied onto the G3000 swxl (C) or GPC 60 (D) columns. The arrows indicate the void and total volume of the columns used. The elution of proteins was followed by the absorbance at 280 nm. Fig. 5. Cytotoxic effect of the species trapped by bis-ANS. Aβ 1-42 (25 µM) was incubated under aggregating conditions for 20 min in the absence or in the presence of 50 or 100 µM bis-ANS, then added to cultures of RAW cells. MTT reduction was evaluated after 24 h. The control cells were treated with aggregating buffer alone; where no toxic effect was observed. Bars 2 and 3 show the effect of bis-ANS in aggregating buffer, without peptides. (*) denotes P value < 0.0001 and (**) denotes P value < 0.01 and was calculated by a two-tailed unpaired t test. Error bars are ± SD of triplicates. Fig. 6. Effects of AMNS (•) and 1,8 ANS (▲) on the aggregation of Aβ 13-23 (25 µM). Aggregation was performed in the presence of the concentrations of the probes shown on the abscissa. The final LS value reached in each experiment was compared to the value attained in the control sample, which underwent aggregation for the same period in the absence of these compounds. Other conditions as in Figure 3. Continuous lines represent a non-linear regression to a single exponential. Fig. 7. Determination of the secondary structure of Aβ 13-23 in SDS. (A) CD spectra of 24 µM Aβ 13-23 in the absence (black) or in the presence of 11 mM SDS (dashed line). The spectra were recorded in 50 mM Na phosphate buffer containing 100 mM NaCl, pH 7.4, 37oC. (B) The twenty lowest energy

13
structures of Aβ 13-23 in the presence of 35 mM SDS obtained by 1H-NMR. The α-helix comprises the residues from Lys16 to Phe20. Red represents the oxygen, blue the nitrogen, white the hydrogen and gray the carbon atoms. Fig. 8. Comparison of the TOCSY spectra of Aβ 13-23 in SDS and AMNS. HN region of TOCSY spectra of 3 mM Aβ 13-23 in 35 mM SDS (black spectrum) and 20 mM AMNS (red spectrum).

14

15
Figure 2

16
Figure 3

17
Figure 4

18
Figure 5

19
Figure 6
20
Figure 7

21
Figure 8
Copyright © 2022 FDOKUMEN




















