
Amyloid precursor protein-induced axonopathies are independent of amyloid- peptides
13
Amyloid precursor protein-induced axonopathies are independent of amyloid-b peptides Gorazd B. Stokin 1, { , Angels Almenar-Queralt 1 , Shermali Gunawardena 1 , Elizabeth M. Rodrigues 1 , Toma ´s Falzone 1 , Jungsu Kim 3 , Concepcio ´ n Lillo 2 , Stephanie L. Mount 1 , Elizabeth A. Roberts 1 , Eileen McGowan 3 , David S. Williams 2 and Lawrence S.B. Goldstein 1, 1 Department of Cellular and Molecular Medicine, Howard Hughes Medical Institute and 2 Department of Pharmacology, School of Medicine, University of California, San Diego, 9500 Gilman Drive, La Jolla, CA 92093-0683, USA and 3 Department of Neuroscience, Mayo Clinic, Jacksonville, FL 32224, USA Received June 15, 2008; Revised July 21, 2008; Accepted August 8, 2008 Overexpression of amyloid precursor protein (APP), as well as mutations in the APP and presenilin genes, causes rare forms of Alzheimer’s disease (AD). These genetic changes have been proposed to cause AD by elevating levels of amyloid-b peptides (Ab), which are thought to be neurotoxic. Since overexpression of APP also causes defects in axonal transport, we tested whether defects in axonal transport were the result of Ab poisoning of the axonal transport machinery. Because directly varying APP levels also alters APP domains in addition to Ab, we perturbed Ab generation selectively by combining APP transgenes in Drosophila and mice with presenilin-1 (PS1) transgenes harboring mutations that cause familial AD (FAD). We found that combining FAD mutant PS1 with FAD mutant APP increased Ab42/Ab40 ratios and enhanced amyloid deposition as previously reported. Surprisingly, however, this combination suppressed rather than increased APP-induced axonal transport defects in both Drosophila and mice. In addition, neuronal apopto- sis induced by expression of FAD mutant human APP in Drosophila was suppressed by co-expressing FAD mutant PS1. We also observed that directly elevating Ab with fusions to the Familial British and Danish Dementia-related BRI protein did not enhance axonal transport phenotypes in APP transgenic mice. Finally, we observed that perturbing Ab ratios in the mouse by combining FAD mutant PS1 with FAD mutant APP did not enhance APP-induced behavioral defects. A potential mechanism to explain these find- ings was suggested by direct analysis of axonal transport in the mouse, which revealed that axonal transport or entry of APP into axons is reduced by FAD mutant PS1. Thus, we suggest that APP-induced axonal defects are not caused by Ab. INTRODUCTION Alzheimer’s disease (AD) is characterized by an insidious and progressive decline of cognitive functions eventually culminating in dementia (1). Post-mortem, AD brains are distinguished by amyloid deposits and neurofibrillary changes, enriched in amyloid-b peptides (Ab) and microtu- bule-associated protein tau, respectively, in a parenchyma reflecting synaptic and neuronal loss in several brain regions, including the hippocampi, the entorhinal and the association cortices and the basal nuclei (BN), which all play a role in cog- nition (2 – 4). These pathological changes produce impairments in several neurotransmitter systems such as cholinergic depletion of the cortices owing to inadequate cholinergic input from the BN (5). Recent evidence suggests that cholin- ergic disconnection and the amyloid deposition observed in † Present address: Institute of Clinical Neurophysiology, Division of Neurology, University Medical Center, Zalos ˇka cesta 7 and Gerontopsychiatric Unit, University Psychiatric Hospital, Studenec 48, SI-1000 Ljubljana, Slovenia. To whom correspondence should be addressed at: Department of Cellular and Molecular Medicine, Howard Hughes Medical Institute, 414 Leichtag Biomedical Research Building, School of Medicine, University of California, San Diego, 9500 Gilman Drive, La Jolla, California 92093-0683. Tel: þ1 8585349700; Fax: þ1 8585349701; Email: [email protected] # The Author 2008. Published by Oxford University Press. All rights reserved. For Permissions, please email: [email protected] Human Molecular Genetics, 2008, Vol. 17, No. 22 3474–3486 doi:10.1093/hmg/ddn240 Advance Access published on August 11, 2008 at UNIVERSIDAD DE SALAMANCA on April 13, 2010 http://hmg.oxfordjournals.org Downloaded from
-
Upload
mayoclinic -
Category
Documents
-
view
3 -
download
0
Transcript of Amyloid precursor protein-induced axonopathies are independent of amyloid- peptides

Amyloid precursor protein-induced axonopathiesare independent of amyloid-b peptides
Gorazd B. Stokin1,{, Angels Almenar-Queralt1, Shermali Gunawardena1,
Elizabeth M. Rodrigues1, Tomas Falzone1, Jungsu Kim3, Concepcion Lillo2,
Stephanie L. Mount1, Elizabeth A. Roberts1, Eileen McGowan3,
David S. Williams2 and Lawrence S.B. Goldstein1,�
1Department of Cellular and Molecular Medicine, Howard Hughes Medical Institute and 2Department of
Pharmacology, School of Medicine, University of California, San Diego, 9500 Gilman Drive, La Jolla, CA 92093-0683,
USA and 3Department of Neuroscience, Mayo Clinic, Jacksonville, FL 32224, USA
Received June 15, 2008; Revised July 21, 2008; Accepted August 8, 2008
Overexpression of amyloid precursor protein (APP), as well as mutations in the APP and presenilin genes,causes rare forms of Alzheimer’s disease (AD). These genetic changes have been proposed to cause ADby elevating levels of amyloid-b peptides (Ab), which are thought to be neurotoxic. Since overexpressionof APP also causes defects in axonal transport, we tested whether defects in axonal transport were theresult of Ab poisoning of the axonal transport machinery. Because directly varying APP levels also altersAPP domains in addition to Ab, we perturbed Ab generation selectively by combining APP transgenes inDrosophila and mice with presenilin-1 (PS1) transgenes harboring mutations that cause familial AD (FAD).We found that combining FAD mutant PS1 with FAD mutant APP increased Ab42/Ab40 ratios and enhancedamyloid deposition as previously reported. Surprisingly, however, this combination suppressed rather thanincreased APP-induced axonal transport defects in both Drosophila and mice. In addition, neuronal apopto-sis induced by expression of FAD mutant human APP in Drosophila was suppressed by co-expressing FADmutant PS1. We also observed that directly elevating Ab with fusions to the Familial British and DanishDementia-related BRI protein did not enhance axonal transport phenotypes in APP transgenic mice.Finally, we observed that perturbing Ab ratios in the mouse by combining FAD mutant PS1 with FADmutant APP did not enhance APP-induced behavioral defects. A potential mechanism to explain these find-ings was suggested by direct analysis of axonal transport in the mouse, which revealed that axonal transportor entry of APP into axons is reduced by FAD mutant PS1. Thus, we suggest that APP-induced axonal defectsare not caused by Ab.
INTRODUCTION
Alzheimer’s disease (AD) is characterized by an insidiousand progressive decline of cognitive functions eventuallyculminating in dementia (1). Post-mortem, AD brains aredistinguished by amyloid deposits and neurofibrillarychanges, enriched in amyloid-b peptides (Ab) and microtu-bule-associated protein tau, respectively, in a parenchyma
reflecting synaptic and neuronal loss in several brain regions,including the hippocampi, the entorhinal and the associationcortices and the basal nuclei (BN), which all play a role in cog-nition (2–4). These pathological changes produce impairmentsin several neurotransmitter systems such as cholinergicdepletion of the cortices owing to inadequate cholinergicinput from the BN (5). Recent evidence suggests that cholin-ergic disconnection and the amyloid deposition observed in
†Present address: Institute of Clinical Neurophysiology, Division of Neurology, University Medical Center, Zaloska cesta 7 and GerontopsychiatricUnit, University Psychiatric Hospital, Studenec 48, SI-1000 Ljubljana, Slovenia.
�To whom correspondence should be addressed at: Department of Cellular and Molecular Medicine, Howard Hughes Medical Institute, 414 LeichtagBiomedical Research Building, School of Medicine, University of California, San Diego, 9500 Gilman Drive, La Jolla, California 92093-0683.Tel: þ1 8585349700; Fax: þ1 8585349701; Email: [email protected]
# The Author 2008. Published by Oxford University Press. All rights reserved.For Permissions, please email: [email protected]
Human Molecular Genetics, 2008, Vol. 17, No. 22 3474–3486doi:10.1093/hmg/ddn240Advance Access published on August 11, 2008
at UN
IVE
RS
IDA
D D
E S
ALA
MA
NC
A on A
pril 13, 2010 http://hm
g.oxfordjournals.orgD
ownloaded from

AD and AD models could be related to defects in axonaltransport (6–8). In fact, the identification of axonal defects inearly AD and in AD models is consistent with previouslyreported cytoskeletal and neuritic abnormalities in AD (9–14)and supports the hypothesis that impaired axonal transportand cytoskeletal alterations play a critical role in the pathogen-esis of AD (15–17). It is also possible that defective axonaltransport and axonal defects are related to Ab exposure. Datareported so far support a role for Ab in the axonal defect for-mation, in particular in the formation of dystrophic neuritesassociated with cored amyloid plaques (6,18) and provide evi-dence of amyloid deposits without axonal defects as well asof axonal defects either preceding amyloid deposition or inareas devoid of amyloid (7,19–23).
Familial AD (FAD) can be caused by mutations in genesencoding amyloid precursor protein (APP) and presenilins(PS1 and PS2). These mutations appear to act by alteringthe proteolytic processing of APP. Additional clues to thepathogenesis of AD are provided by the existence ofAD-like pathology, possibly caused by APP overexpression,in Down’s syndrome in which chromosome 21, harboringthe APP gene, is trisomic (24). Tenet that excess APP cancause AD is corroborated by the recent discovery of APPduplications in some forms of hereditary AD (25–27). Thus,some types of AD may be caused by overexpression ofwild-type (WT) APP (28).
Intriguingly, recent work revealed that overexpression of WTAPP or APP bearing FAD mutations in Drosophila and micecan cause impaired axonal transport and axonal defects(7,8,29,30). In this regard, it has also been reported that PS1not only has a role in regulated intramembrane proteolysis(RIP) (31) of proteins such as APP (32) and Notch (33–35),but it may also play roles in the regulation of kinesin-I-mediatedintracellular transport (36,37). These findings suggest that APPand PS1 functions may be intimately linked to the molecularmotor kinesin-I during axonal transport (38–41). In fact, inboth Drosophila and mice, APP-mediated axonal transportdefects can be enhanced by genetic reductions in kinesin-I(7,29,30). These reductions also enhanced aberrant Ab gener-ation and amyloid deposition (7).
The finding that genetic manipulations of APP and preseni-lins can cause defects in axonal transport, coupled to reportsthat axonal transport may be defective both early and late inAD (7,13,42,43) led to the suggestion that defects in axonaltransport could play a major role in the cause or progressionof AD (16,17). Unresolved, however, is the issue of whetheraxonal transport can be directly poisoned by Ab, or whethermutations that enhance aberrant Ab generation can causeaxonal transport defects independent of Ab production.Although intentionally increasing or decreasing APPexpression can alter Ab levels, such manipulations also alterthe amount of other critical APP domains that may playroles in axonal transport. To test the effects of perturbingAb on axonal transport in vivo, we altered Ab42/Ab40ratios by combining well-characterized APP transgenes withtransgenic FAD PS1 mutations or with fusions of Ab40 orAb42 to the BRI protein linked to Familial British andDanish Dementia (44–47) and tested whether axonal transportand other phenotypes induced by APP were enhanced bychanges in Ab.
RESULTS
Increasing Ab peptides does not enhance APP-inducedaxonal blockages in mice
Axonal blockages in Drosophila and mice are thought tocorrespond to aberrant accumulations of proteins, vesicles andorganelles within axons that block or otherwise inhibit normalaxonal transport (7,29,48–51). Similar axonal morphologieshave been reported in AD and may be an early event in thepathogenic progression, possibly preceding amyloid deposition(7). Axonal blockages have been observed in WT and FADmutant APP-overexpressing Drosophila larval neurons and inmouse models of AD (7,29). In the mouse models, axonalblockages were observed long before amyloid deposition.Formally, APP overexpression could cause axonal blockagesand axonal transport defects by directly increasing the amountof Ab peptides. Alternatively, axonal transport defects couldbe induced by perturbing the proteolytic generation of otherregions of APP in the intact protein (38). Since intentionallyincreasing or decreasing APP expression alters levels of Aband other critical APP domains simultaneously, we sought totest the effects of aberrant Ab generation on axonal transportby increasing Ab42/Ab40 ratios in the absence of changes inAPP expression. To achieve this goal, we first increasedAb42/Ab40 ratios by combining well-characterized APPtransgenes with transgenic FAD PS1 mutations, and testedwhether axonal transport and other phenotypes induced byAPP overexpression were enhanced. Thus, we first analyzedaxonal blockage formation in 4-month-old WT, single trans-genic FAD mutant APP overexpressing (Tg-swAPPPrp), singletransgenic FAD mutant PS1 (Tg-A246EPS1Prp) and doubletransgenic FAD mutant APP and FAD mutant PS1(Tg-swAPPPrp; Tg-A246EPS1Prp) mice in both the C57BL/6J/C3H/HeJ and C57BL/6J genetic backgrounds.
Since previous work on cholinergic fibers in the BN foundthat large diameter axonal varicosities correspond to axonalswellings, a bona fide equivalent of axonal blockages at thelight microscope level (7), we measured diameters andlengths of varicosities and inter-varicosity shafts (Supplemen-tary Material, Fig. S1). These measurements were made‘blind’ to the genotypes of the sections, which were coded.Codes were broken only after completion of the datacollection. As reported previously (7), single transgenicTg-swAPPPrp mice had a significantly higher percentage ofcholine acetyltransferase-immunoreactive (ChAT-IR) fiberswith varicosities having diameters between 2.7 and 3.6 mm,3.6 and 4.5 mm and larger than 4.5 mm compared with WTlittermates in both genetic backgrounds examined (Fig. 1Aand D and Supplementary Material, Fig. S2). Single transgenicTg-A246EPS1Prp mice displayed a slight elevation in the per-centage of ChAT-IR fibers with large diameter varicosities inthe 2.7–3.6 mm category compared with WT littermates inthe C57BL/6J genetic background. To our surprise, however,there were no differences in the percentage of ChAT-IRfibers with varicosities in any of the diameter intervals whendouble transgenic Tg-swAPPPrp; Tg-A246EPS1Prp were com-pared to WT littermates in either of the genetic backgrounds.In agreement with these measurements are the average totalsizes of the ChAT-IR varicosities and inter-varicosity shafts,which revealed a significant increase in the diameter and
Human Molecular Genetics, 2008, Vol. 17, No. 22 3475
at UN
IVE
RS
IDA
D D
E S
ALA
MA
NC
A on A
pril 13, 2010 http://hm
g.oxfordjournals.orgD
ownloaded from

Figure 1. Mutant PS1 suppresses APP-induced axonal swellings or blockages in the baso-cortical cholinergic fibers of mice. (A) Representative light microscopeimages of cholinergic fibers in BN showing varicosities (black arrows) in all genotypes, while large axonal swellings (red arrows) were seen primarily inTg-swAPPPrP (b), but not WT (a), Tg-A246EPS1PrP (c) or Tg-swAPPPrP; Tg-A246EPS1PrP (d) littermates (bar 15 mm). Perikarya (a and c) are marked withgreen asterisks. (B) Representative electron microscope images of cholinergic profiles in BN showing axonal blockages in Tg-swAPPPrP (b) andTg-A246EPS1PrP (c), but not WT (a) or Tg-swAPPPrP; Tg-A246EPS1PrP (d) mice; axonal profiles are circled in red (bar 300 nm). (C) Representative light micro-scope images of cholinergic fibers in deep layers of the SC showing swellings (red arrows) in Tg-swAPPPrP (b), to a lesser extent in Tg-A246EPS1PrP (c) andTg-swAPPPrP; Tg-A246EPS1PrP (d), but not in WT (a) mice (bar 15 mm). (D) (a) Increased percentage of cholinergic fibers in BN with varicosities of diametersbetween 2.7 and 3.6 mm, 3.6 and 4.5 mm and over 4.5 mm (swellings) in Tg-swAPPPrP (n ¼ 3, � means P � 0.05) and between 2.7 and 3.6 mm (swellings) inTg-A246EPS1PrP (n ¼ 3, � means P � 0.05), but not WT (n ¼ 3) or Tg-swAPPPrP; Tg-A246EPS1PrP mice (n ¼ 3). (b) Increased percentage of cholinergic fibersin deep layers of the SC with varicosities of diameters between 1.4 and 2.0 mm and 2.0 and 2.6 (swellings) in Tg-swAPPPrP (n ¼ 3, � means P � 0.05), but notWT (n ¼ 3), Tg-A246EPS1PrP (n ¼ 3) or Tg-swAPPPrP; Tg-A246EPS1PrP mice (n ¼ 3). (E) No significant difference in the percentage of cholinergic fibers inthe BN with varicosities of diameters between 1.8 and 2.7 mm, 2.7–3.6 mm, 3.6–4.5 and over 4.5 mm between 10- and 11-month-old Tg-swAPPPrP (n ¼ 6) andTg- BRI-Ab40Prp (n ¼ 3), Tg-BRI-Ab42Prp (n ¼ 5), Tg-swAPPPrP; BRI-Ab40Prp (n ¼ 4) and Tg-swAPPPrP; BRI-Ab42Prp (n ¼ 4) littermates.
3476 Human Molecular Genetics, 2008, Vol. 17, No. 22
at UN
IVE
RS
IDA
D D
E S
ALA
MA
NC
A on A
pril 13, 2010 http://hm
g.oxfordjournals.orgD
ownloaded from

length of the varicosities in the Tg-swAPPPrp compared with theWT, Tg-A246EPS1Prp and Tg-swAPPPrp; Tg-A246EPS1Prp
littermates (Supplementary Material, Table S1).To confirm the nature of axonal swellings, and to test for
additional fiber abnormalities, we examined cholinergicfibers in the BN by immuno-labeling for choline acetyltrans-ferase (ChAT) followed by electron microscopy. ChAT-IRprofiles of diameters less than 2.7 mm containing scarcevesicles and mitochondria were found in the BN of allgenotypes. ChAT-IR profiles of large diameters containingnumerous haphazardly distributed vesicles and mitochondriawere identified almost exclusively in Tg-swAPPPrp, but not inWT and Tg-swAPPPrp; Tg-A246EPS1Prp, littermates (Fig. 1B,Supplementary Material, Fig. S3). As previously reported (7),Tg-swAPPPrp commonly exhibited axonal blockages that werenot immunoreactive for ChAT and occasionally displayedelectron dense axoplasm reminiscent of axonal degeneration(Supplementary Material, Figs S4 and S5).
We reasoned that if cholinergic axonal abnormalities wereoccurring close to the cell bodies in the BN, then we shouldalso observe changes in cholinergic fibers at their termini.Thus, we investigated cholinergic terminals in the deep layersof the sensory cortex (SC). These data were also collected‘blind’ to the genotypes of the sections by two independentobservers (G.B.S. and E.M.R.) and produced comparable finalresults. At the level of the SC, fewer cholinergic fibers exhibitedvaricosities compared to the BN (Supplementary Material,Table S1). These ChAT-IR varicosities were substantiallysmaller compared to those sampled close to their origin inthe BN. Again, however, only Tg-swAPPPrp mice containedsignificantly increased percentage of ChAT-IR fibers havingvaricosities of diameters between 1.4 and 2.0 microns, 2.0and 2.6 microns and larger than 2.6 microns compared withWT, Tg-A246EPS1Prp and Tg-swAPPPrp; Tg-A246EPS1Prp
littermates (Fig. 1C and D). Surprisingly, in contrast to fibersin the BN, we found that the length of cholinergic fibers perfield at their termini was significantly increased in Tg-swAPPPrp
and significantly decreased in Tg-swAPPPrp; Tg-A246EPS1Prp
compared with WT and Tg-A246EPS1Prp littermates (Sup-plementary Material, Fig. S6). These data parallel thoseobtained for cholinergic synapses (52) and disclose possibleaberrancies in the metabolism or transport of ChAT or in thestructure of the termini. Taken together, however, and contraryto expectation, these data reveal that double transgenicTg-swAPPPrp; Tg-A246EPS1Prp animals, which have perturbedAb generation, particularly increased Ab42, have less severeaxonal phenotypes than single transgenic Tg-swAPPPrp animals.
To corroborate these findings and to clarify whethersuppression of axonal blockage phenotypes by transgenicFAD PS1 mutations was due to some other aspect of presenilinfunction, we turned to an alternative approach. Previouswork revealed that fusions of BRI-Ab42 chimeras lead tosynthesis and secretion of Ab42 and amyloid deposition(45). In particular, when Tg-BRI-Ab42Prp mice werecrossed with Tg-swAPPPrp (53), the double transgenic Tg-swAPPPrp; Tg-BRI-Ab42Prp mice had significantly increasedRIPA-insoluble formic acid extracted Ab40 and Ab42 aswell as enhanced amyloid deposition in several brain areasincluding BN compared with single transgenic Tg-swAPPPrp,Tg-BRI-Ab40Prp and BRI-Tg-Ab42Prp or Tg-swAPPPrp;
Tg-BRI-Ab40Prp double transgenic littermates (45). Thus,to test whether Ab40 or Ab42 enhance axonal pathologyobserved in Tg-swAPPPrp, we crossed heterozygousTg-swAPPPrp mice in the B6/SJL genetic background (53)with either heterozygous Tg-BRI-Ab40Prp or heterozygousTg-BRI-Ab42Prp mice in the B6/C3 genetic background(45). Resulting mice were examined for Ab production aswell as amyloid deposition and scored for axonal defects.Ab levels and amyloid deposition observed in WT,Tg-swAPPPrp, Tg-BRI-Ab40Prp, Tg-BRI-Ab42Prp,Tg-swAPPPrp; Tg-BRI-Ab40Prp and Tg-swAPPPrp; Tg-BRI-Ab42Prp littermates were similar to what previously reported(44,45). Specifically, 10–11-month-old Tg-swAPPPrp micehad 2935.4+ 575.35 and 1561.2+ 166.76 fmols/mg of wetbrain of formic acid extractable Ab40 and Ab42, respectively.Tg-swAPPPrp; Tg-BRI-Ab40Prp littermates had 603+ 161.41and 266.5+ 129.93 fmols/mg of wet brain formic acid extrac-table Ab40 and Ab42, respectively, while Tg-swAPPPrp;Tg-BRI-Ab42Prp littermates had 24499.67+ 4841.7 and9394.67+ 2554.19 fmols/mg of wet brain formic acid extrac-table Ab40 and Ab42, respectively. All of these transgenicmice except for Tg-BRI-Ab40Prp exhibited amyloid depo-sition with Tg-swAPPPrp; Tg-BRI-Ab42Prp demonstrating themost extensive amyloid deposition. Axonal pathology wasscored ‘blind’ to the genotypes by two independent observers(G.B.S. and E.M.R.). Data collected by either of the observersrevealed comparable percentages of fibers with varicosities inall of the diameter intervals examined among Tg-swAPPPrp,Tg-BRI-Ab40Prp and Tg-BRI-Ab42Prp and no differences inthe percentage of fibers with varicosities in any of the diameterintervals among Tg-swAPPPrp, Tg-swAPPPrp; Tg-BRI-Ab40Prp and Tg-swAPPPrp; Tg-BRI-Ab42Prp littermatesdespite dramatic changes in Ab levels and amyloid deposition(Fig. 1E). Intriguingly, although BRI-Ab40 and BRI-Ab42transgenes dramatically altered Ab levels (6–9-fold increasein Ab40 and Ab42 in Tg-swAPPPrp; Tg-BRI-Ab42Prp versusTg-swAPPPrp), they had no impact on the frequency ofaxonal pathology generated by the Tg-swAPPPrp transgene.Thus, enhancing Ab accumulation per se does not lead toenhanced formation of axonal blockages, suggesting that themajor route of generation of axonal phenotypes in APP trans-genic mice is not a result of increasing levels of Ab.
Mutant PS-1 modulates APP-induced phenotypesin Drosophila
Previous work showed that overexpression of WT and FADmutant human APP and Drosophila APPL in the Drosophilalarval nervous system causes axonal blockages reminiscentof those found in mutants for molecular motors (29,30,50).To test whether FAD mutant presenilin can suppress formationof these blockages as we observed in mice, we examinedcysteine string protein stained segmental nerves of Drosophilalarvae by immunofluorescence microscopy. Specifically, weused the Gal4-UAS system to express FAD mutant humanAPP (54) alone or in combination with a transgene encodinga FAD mutant Drosophila presenilin (PsnE280A), which iscomparable to the FAD A246E PS1 transgene that we testedin mice (55). In previous work, we found that the expressionof human APP carrying the Swedish FAD mutation caused
Human Molecular Genetics, 2008, Vol. 17, No. 22 3477
at UN
IVE
RS
IDA
D D
E S
ALA
MA
NC
A on A
pril 13, 2010 http://hm
g.oxfordjournals.orgD
ownloaded from
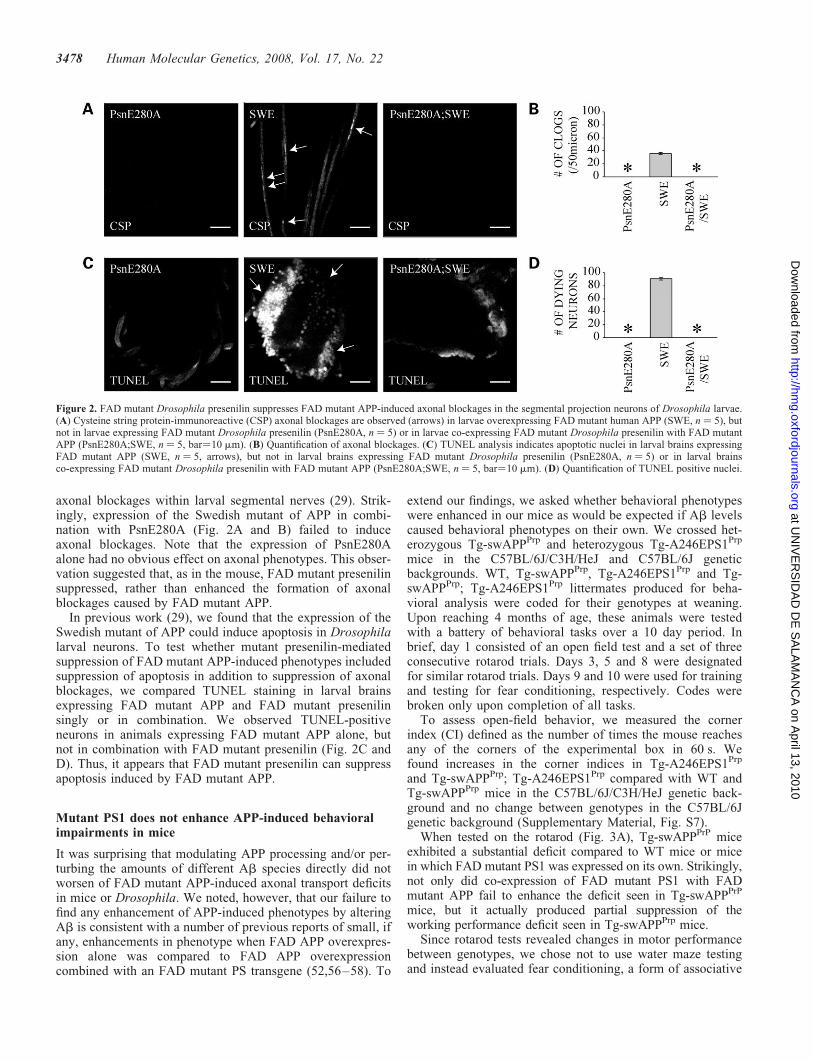
axonal blockages within larval segmental nerves (29). Strik-ingly, expression of the Swedish mutant of APP in combi-nation with PsnE280A (Fig. 2A and B) failed to induceaxonal blockages. Note that the expression of PsnE280Aalone had no obvious effect on axonal phenotypes. This obser-vation suggested that, as in the mouse, FAD mutant presenilinsuppressed, rather than enhanced the formation of axonalblockages caused by FAD mutant APP.
In previous work (29), we found that the expression of theSwedish mutant of APP could induce apoptosis in Drosophilalarval neurons. To test whether mutant presenilin-mediatedsuppression of FAD mutant APP-induced phenotypes includedsuppression of apoptosis in addition to suppression of axonalblockages, we compared TUNEL staining in larval brainsexpressing FAD mutant APP and FAD mutant presenilinsingly or in combination. We observed TUNEL-positiveneurons in animals expressing FAD mutant APP alone, butnot in combination with FAD mutant presenilin (Fig. 2C andD). Thus, it appears that FAD mutant presenilin can suppressapoptosis induced by FAD mutant APP.
Mutant PS1 does not enhance APP-induced behavioralimpairments in mice
It was surprising that modulating APP processing and/or per-turbing the amounts of different Ab species directly did notworsen of FAD mutant APP-induced axonal transport deficitsin mice or Drosophila. We noted, however, that our failure tofind any enhancement of APP-induced phenotypes by alteringAb is consistent with a number of previous reports of small, ifany, enhancements in phenotype when FAD APP overexpres-sion alone was compared to FAD APP overexpressioncombined with an FAD mutant PS transgene (52,56–58). To
extend our findings, we asked whether behavioral phenotypeswere enhanced in our mice as would be expected if Ab levelscaused behavioral phenotypes on their own. We crossed het-erozygous Tg-swAPPPrp and heterozygous Tg-A246EPS1Prp
mice in the C57BL/6J/C3H/HeJ and C57BL/6J geneticbackgrounds. WT, Tg-swAPPPrp, Tg-A246EPS1Prp and Tg-swAPPPrp; Tg-A246EPS1Prp littermates produced for beha-vioral analysis were coded for their genotypes at weaning.Upon reaching 4 months of age, these animals were testedwith a battery of behavioral tasks over a 10 day period. Inbrief, day 1 consisted of an open field test and a set of threeconsecutive rotarod trials. Days 3, 5 and 8 were designatedfor similar rotarod trials. Days 9 and 10 were used for trainingand testing for fear conditioning, respectively. Codes werebroken only upon completion of all tasks.
To assess open-field behavior, we measured the cornerindex (CI) defined as the number of times the mouse reachesany of the corners of the experimental box in 60 s. Wefound increases in the corner indices in Tg-A246EPS1Prp
and Tg-swAPPPrp; Tg-A246EPS1Prp compared with WT andTg-swAPPPrp mice in the C57BL/6J/C3H/HeJ genetic back-ground and no change between genotypes in the C57BL/6Jgenetic background (Supplementary Material, Fig. S7).
When tested on the rotarod (Fig. 3A), Tg-swAPPPrP miceexhibited a substantial deficit compared to WT mice or micein which FAD mutant PS1 was expressed on its own. Strikingly,not only did co-expression of FAD mutant PS1 with FADmutant APP fail to enhance the deficit seen in Tg-swAPPPrP
mice, but it actually produced partial suppression of theworking performance deficit seen in Tg-swAPPPrp mice.
Since rotarod tests revealed changes in motor performancebetween genotypes, we chose not to use water maze testingand instead evaluated fear conditioning, a form of associative
Figure 2. FAD mutant Drosophila presenilin suppresses FAD mutant APP-induced axonal blockages in the segmental projection neurons of Drosophila larvae.(A) Cysteine string protein-immunoreactive (CSP) axonal blockages are observed (arrows) in larvae overexpressing FAD mutant human APP (SWE, n ¼ 5), butnot in larvae expressing FAD mutant Drosophila presenilin (PsnE280A, n ¼ 5) or in larvae co-expressing FAD mutant Drosophila presenilin with FAD mutantAPP (PsnE280A;SWE, n ¼ 5, bar¼10 mm). (B) Quantification of axonal blockages. (C) TUNEL analysis indicates apoptotic nuclei in larval brains expressingFAD mutant APP (SWE, n ¼ 5, arrows), but not in larval brains expressing FAD mutant Drosophila presenilin (PsnE280A, n ¼ 5) or in larval brainsco-expressing FAD mutant Drosophila presenilin with FAD mutant APP (PsnE280A;SWE, n ¼ 5, bar¼10 mm). (D) Quantification of TUNEL positive nuclei.
3478 Human Molecular Genetics, 2008, Vol. 17, No. 22
at UN
IVE
RS
IDA
D D
E S
ALA
MA
NC
A on A
pril 13, 2010 http://hm
g.oxfordjournals.orgD
ownloaded from

learning that does not rely on motor performance. Weexcluded possible alterations in pain and sound perceptionbetween genotypes by verifying that the minimal amount ofcurrent required to elicit a reaction to foot-shock and thesound required to elicit startle response were the sameamong genotypes and genetic backgrounds (data not shown).Following training (see Materials and Methods), we foundthat all four genotypes of mice could learn to associate aneutral conditioned stimulus to an aversive unconditionedstimulus (foot-shock; Fig. 3B).
The next day, we tested mice for contextual conditioning,and 2 h later, for cued conditioning. When compared to WT,Tg-A246EPS1Prp and Tg-swAPPPrp; Tg-A246EPS1Prp litter-mates in two genetic backgrounds (C57BL/6J/C3H/HeJ andC57BL/6J), Tg-swAPPPrp mice exhibited early changes in
contextual conditioning that are not enhanced, and may evenbe mildly suppressed by expression of FAD mutant PS1(Supplementary Material, Fig. S8). When tested for cuedconditioning, all mice responded to the testing environmentas a novel context, and all mice exhibited significantconditioning in response to the conditioned stimulus.Tg-swAPPPrp, but not Tg-A246EPS1Prp or Tg-swAPPPrp;Tg-A246EPS1Prp, mice exhibited deficits in conditioningcompared to WT littermates, suggesting that these behavioralphenotypes were not enhanced by increasing Ab generation.
Mutant PS1 reduces axonal transport of APP in mice
Since recent evidence suggests that genetic manipulationof PS1 can lead to changes in the delivery of APP to cell
Figure 3. Mutant PS1 suppresses APP-induced behavioral impairments in mice. (A) Significant decrease in the time spent on the rotating rod by Tg-swAPPPrP
(n ¼ 9 and 17, � means P � 0.05) and Tg-swAPPPrP; Tg-A246EPS1PrP (n ¼ 7 and 17, � means P � 0.05) mice on trial days 1, 3, 5 and 8 and on trial day 8,respectively, in the C57BL/6J/C3H/HeJ (a) and on trial days 1, 3 and 8 and on trial days 1 and 3, respectively, in the C57BL/6J (b) genetic background comparedwith WT (n ¼ 11 and 19) littermates. No difference in the time spent on the rotating rod between WT and Tg-A246EPS1PrP (n ¼ 11 and 15) mice in either of thegenetic backgrounds. (B) No difference in the percentage of freezing between WT (n ¼ 11 and 19), Tg-swAPPPrP (n ¼ 9 and 17), Tg-A246EPS1PrP (n ¼ 11 and15) and Tg-swAPPPrP; Tg-A246EPS1PrP (n ¼ 7 and 17) littermates during fear conditioning training (a and b) in C57BL/6J/C3H/HeJ (a) and C57BL/6J (b)genetic backgrounds. Significantly reduced percentage of freezing in Tg-swAPPPrP (� means P � 0.05), but not Tg-A246EPS1PrP or Tg-swAPPPrP;Tg-A246EPS1PrP, compared with WT littermates during evaluation of cued conditioning 24 h after training (c and d) in C57BL/6J/C3H/HeJ (c) and C57BL/6J (d) genetic backgrounds. In brackets, n corresponds to the number of mice tested with the first number referring to the C57BL/6J/C3H/HeJ and second tothe C57BL/6J genetic background (EXP corresponds to 3 min exploration period, CS to 20 s conditioned stimulus and I to the 1 min interval between CS,for a description of the fear conditioning paradigm consult Behavioral analysis section of the Materials and Methods).
Human Molecular Genetics, 2008, Vol. 17, No. 22 3479
at UN
IVE
RS
IDA
D D
E S
ALA
MA
NC
A on A
pril 13, 2010 http://hm
g.oxfordjournals.orgD
ownloaded from

surfaces and synapses (41,59), we asked whether FAD mutantPS1-mediated reduction of APP axonal transport could beresponsible for reversing the observed APP-induced pheno-types. Axonal transport was evaluated in sciatic nerve ligationexperiments in which gels and western blots were run andscanned by one investigator, then coded for genotypes andanalyzed ‘blind’ by another investigator who broke thecodes only upon completion of the experiments. In4-month-old WT mice, we observed a significant increase inthe levels of APP in the proximal and distal stumps in theligated compared with unligated sciatic nerves (Fig. 4A andB). In 4-month-old Tg-swAPPPrp mice, although we observedan increase in APP accumulation at the proximal ligature site,the extent was less than in WT, suggesting a reduction in theanterograde axonal transport of APP upon its overexpression.This observation differs from the recent conclusion (40) thatoverexpressing APP does not lead to altered transport in thesciatic nerve. However, this paper did not compare Tg-APPto WT controls as we have done here. Our observation isconsistent with the many published observations (7,29,30,60)that overexpressing APP and its homologues can poisonaxonal transport. Analysis of 4-month-old Tg-A246EPS1Prp
and Tg-swAPPPrp; Tg-A246EPS1Prp ligations suggested thatthe expression of FAD mutant PS1 may reduce anterogradeaxonal transport of APP (Fig. 4C and D). Intriguingly,expression of FAD mutant APP with FAD mutant PS1 mayoperate additively in reducing anterograde axonal transportof APP. We tested the possibility that FAD mutant PS1poisons anterograde APP axonal transport by causing releaseof kinesin-I from vesicles as recently proposed (40,41). Weobserved no change in the amount of kinesin-I associatedwith brain-derived membrane preparations either by flotationor by differential centrifugation between WT and Tg-A246EPS1Prp littermates (data not shown). Control experi-ments showed increased levels of the axonal anterogrademolecular motor, kinesin-I, in the proximal stumps andincreased levels of a bidirectionally transported scaffoldprotein, SYD/JIP3, in both proximal and distal stumps of theligated compared with unligated sciatic nerves (SupplementaryMaterial, Fig. S9) (30,38,39,61). Levels of molecules residingwithin Schwann cell membranes or those thought to bemoving with the slow wave of axonal transport, such asmyelin basic protein and tubulin, were used as loading controls.Collectively, these results suggest that, in addition to increasingAb42 generation (Supplementary Material, Fig. S10),expression of FAD mutant PS1 reduces the fast anterogradeaxonal transport of endogenous and transgenic APP.
DISCUSSION
Overexpression of APP in Drosophila and mice gives rise toaxonal defects akin to those found in AD (7,29,30). Formationof these defects was shown to be enhanced by geneticreductions in kinesin-1 in Drosophila (29,30) and in mice,where increased Ab42/Ab40 ratios and enhanced amyloiddeposition were also observed (7,57). To test whether axonaldefects are the result of aberrant Ab generation, we combinedFAD mutant APP with FAD mutant PS1 or BRI-Ab chimerasin Drosophila or mice. We found that the generation of
axonal defects was unchanged or suppressed in Drosophilaand mice expressing FAD mutant PS1 combined with FADmutant APP and that significant increases in Ab produced byexpression of BRI-Ab chimeras in FAD APP mice failed toenhance formation of axonal defects. In addition, not onlywas the axonal blockage phenotype suppressed by combiningFAD mutant PS1 with FAD APP overexpression, but alsoneuronal apoptosis was suppressed in Drosophila. In parallelwith these findings, behavioral deficits were also not enhancedby perturbing Ab42 generation. In fact, in several cases, beha-vioral defects may actually have been slightly suppressed.These observations suggest that there may be different mecha-nisms by which transgenic FAD mutations in APP and PS1contribute to AD. Intriguingly, transgenic FAD PS1 mutationsappear unable to produce axonal pathology as well as amyloiddeposits on their own, while in combination with APP severalphenotypes including axonal pathology and behavior aresuppressed or unchanged despite enhanced amyloid plaqueformation.
Although our findings are in apparent contradiction to thewidely held view that phenotypic defects produced by APP over-expression are caused by elevated Ab42, we note that someprevious studies made similar observations on comparisons ofsingle versus double transgenic animals, although these werenot emphasized. For example, overexpression of mutant APPor mutant PS1, but not their combined overexpression, elevatescortical ChAT activity (62). Similarly, the density of corticalcholinergic synapses has been shown to increase in mice overex-pressing FAD mutant APP and decreased in mice expressingFAD mutant APP combined with FAD mutant PS1 (52). Ourfindings are also consistent with research showing that increas-ing Ab by expressing FAD mutant PS1 together with FADmutant APP failed to produce substantial enhancements of thebehavioral deficits observed in mice overexpressing FADmutant APP alone (56–58). Indeed, these comparisons revealedan unexpectedly subtle, if any, increase in behavioral deficitsdespite large increases in Ab42/Ab40 ratios and amyloid depo-sition in double transgenic animals. Finally, our data are consist-ent with a recent report about transgenic mice overexpressingAPP which lack a caspase cleavage site in the C-terminal cyto-plasmic region of APP. These transgenic mice generate largeamounts of Ab peptides, but they do not exhibit the significantcellular, synaptic and behavioral changes seen in matched trans-genic mice producing comparable levels of Ab, but in whichthe caspase cleavage site is intact (63). Another recent reportcompared retrograde NGF transport in mice overexpressing avariety of WT versions of mouse and human APP as well asmice that were either single transgenic Tg-swAPPPrP or doubletransgenic Tg-swAPPPrp; Tg-A246EPS1Prp (8). The resultssuggest that NGF transport deficits can be caused by overexpres-sion of APP, and that human Ab40 or Ab42 peptides are notneeded to generate NGF transport deficits, although increasinghuman Ab42 levels may minimally enhance NGF transport def-icits caused by APP overexpression. Thus, in many experimentsin addition to ours, levels of proteolytic processing of APP to Abpeptides such as Ab42 are not well correlated with the develop-ment of a number of cellular and behavioral deficits. We notethat many phenotypes present in double transgenic, but notWT animals, have been reported and attributed solely to Abor APP processing, but most such experiments do not compare
3480 Human Molecular Genetics, 2008, Vol. 17, No. 22
at UN
IVE
RS
IDA
D D
E S
ALA
MA
NC
A on A
pril 13, 2010 http://hm
g.oxfordjournals.orgD
ownloaded from

double APP and PS1 transgenic genotypes to the single trans-genic APP genotype as we have reported here.
A recent in vivo dynamics study of cortical amyloid plaqueformation found amyloid deposition prior to neuritic defects
(6). Thus, Ab is sufficient, but as we show here, and argueabove, not necessary for the formation of neuritic defects.We conclude that Ab is not the only APP- or PS1-relatedinsult that can induce axonal transport deficits and produce
Figure 4. Mutant PS1 reduces axonal transport of APP in mice. (A) Representative western blots in the 6 h sciatic nerve ligation paradigm. In 4-month-old WTmice 6 h ligation of the sciatic nerve produced significant accumulation of APP in its proximal and distal stumps, but not in the dorsal root ganglion. Proteinsundergoing slow axonal transport, such as tubulin, as well as proteins expressed by Schwann cells, such as myelin basic protein (MBP), were used as loadingcontrols. (B) Average levels of APP were significantly increased after 6 h of ligation relative to unligated controls in the proximal stump of 4-month-old WT (n ¼6, P � 0.005), Tg-swAPPPrP (n ¼ 7, P � 0.005), Tg-A246EPS1PrP (n ¼ 6, P � 0.000005) and Tg-swAPPPrP; Tg-A246EPS1PrP (n ¼ 7, P � 0.005) and in thedistal stump of 4-month-old WT (n ¼ 6, P � 0.05) and Tg-swAPPPrP (n ¼ 7, P � 0.05), but not Tg-A246EPS1PrP (n ¼ 6) and Tg-swAPPPrP; Tg-A246EPS1PrP
(n ¼ 7). (C) Representative image (a) of significantly reduced average levels of APP (b) in the proximal stump of ligated sciatic nerves in 4-month-oldTg-A246EPS1PrP (blue; n ¼ 6) compared with WT (black; n ¼ 6) littermates (P � 0.05). (D) Representative image (a) of significantly reduced averagelevels of APP (b) in the proximal stump of ligated sciatic nerves in 4-month-old Tg-swAPPPrP; Tg-A246EPS1PrP (green; n ¼ 7) compared with Tg-swAPPPrP
(red; n ¼ 7) littermates (P � 0.05). APP and tubulin bands from representative western blots (a in C and D) belong to the same membranes.
Human Molecular Genetics, 2008, Vol. 17, No. 22 3481
at UN
IVE
RS
IDA
D D
E S
ALA
MA
NC
A on A
pril 13, 2010 http://hm
g.oxfordjournals.orgD
ownloaded from

neuronal changes. A mechanism by which FAD mutant PS1might suppress APP-induced cellular defects is not yet clear,but could involve the proposed role of PS1 in gating axonalentry or axonal transport of APP (39,59,64–66). Such gatingcould lead to reduced amounts of APP entry into axons, andcould be achieved by regulating proteolytic processing ofAPP as has been proposed for BACE overexpression (67–69) and might simultaneously lead to enhanced levels ofAPP processing to Ab42. Alternatively, PS1 could controlthe association of APP with molecular motor proteins andthe transport machinery via proteins such as glycogen synthasekinase 3b, which could lead to reduced axonal transport (41).PS1 could also regulate APP transport via its putativeinteraction with proteins such as CLIP170 (70), which isthought to play a role in the formation of the dyneincomplex and possibly in retrograde axonal transport.
In conclusion, it is surprising that although expressingFAD mutant PS1 with overexpression of FAD mutantAPP enhances Ab42 production and amyloid deposition, itsuppresses axonal defects and gives mild suppression, asopposed to enhancement of other defects such as behavior.Our observation that increasing Ab in animals expressingFAD APP does not enhance axonal defects, coupled to otherreports, raise the possibility that the total amount or characterof proteolytic processing of APP to Ab is not the primarydriver of detrimental phenotypic changes in axons. Instead,we suggest that either the cellular location of APP processing,or the axonal defects generated by APP overexpression arethe major causes of neuronal and behavioral defects seen inmice overexpressing mutant APP. Further work is necessaryto discriminate between these possibilities.
MATERIALS AND METHODS
Mice and Drosophila
Transgenic mice carrying the Swedish FAD double-mutation(Tg-swAPPPrP) were crossed with mice heterozygous forPS1 carrying the A246E FAD mutation (Tg-A246EPS1PrP)to produce WT mice, Tg-swAPPPrP mice, Tg-A246EPS1PrP
mice and mice carrying both transgenes (Tg-swAPPPrP andTg-A246EPS1PrP) (71,72). Mice were initially generated andtested in the original mixed C57BL/6J/C3H/HeJ genetic back-ground. To confirm and extend preliminary findings obtainedwith mice in the C57BL/6J/C3H/HeJ genetic background,heterozygous Tg-swAPPPrP and Tg-A246EPS1PrP mice werecrossed for five generations with C57BL/6J mice and then toeach other to produce a cohort of transgenic mice in theC57BL/6J genetic background. Genetic crosses in bothgenetic backgrounds produced mice with the expectedgenotype ratios for the Mendelian mode of inheritance.Unless otherwise stated, the results shown refer to experimentsperformed with 4-month old mice in the C57BL/6J geneticbackground. Transgenic mice hemizygous for the SwedishFAD double-mutation of APP, Tg-2567 (Tg-swAPPPrP) (53),in the B6/SJL genetic background were crossed withhemizygous BRI-Ab40Prp or BRI-Ab42Prp mice in theB6/C3 genetic background (45). The resulting WT,Tg-swAPPPrP, Tg-BRI-Ab40Prp, Tg-swAPPPrP/BRI-Ab40Prp,
and WT, Tg-swAPPPrP, Tg-BRI-Ab42Prp and Tg-swAPPPrP/BRI-Ab42Prp littermates were examined at 10–11 months.
For Drosophila transgene expression, Drosophila lines carry-ing an FAD mutant human APP transgene UAS-SWE (73) or aFAD mutant Drosophila presenilin transgene UAS-PsnE280A(74) were crossed to a strain carrying the APPL-GAL4 (29)driver at 298C. Genetic interaction tests were performed aspreviously described (29). Males that were APPL-GAL4/Y;PsnE280A/B3 or APPL-GAL4/Y;SWE/B3 were crossed tofemale homozygous for UAS-SWE or UAS-PsnE280A; onlyfemales carrying APPL-GAL4 along with SWE;PsnE280A orPsnE280A;SWE were used for the analysis.
Histology and immunohistochemistry
WT, Tg-swAPPPrP, Tg-A246EPS1PrP and Tg-swAPPPrP/A246EPS1PrP littermates were transcardially perfused with0.1M phosphate solution at pH 7.4 (Sorensen’s solution) fol-lowed by buffered 4% paraformaldehyde (PFA). Cerebrawere separated from the brains, post-fixed in buffered 4%PFA, cryoprotected in 30% sucrose overnight and cut seriallyinto 50 mm thick sections to obtain sets of random systematiccoronal sections representative of the BN or of the SC. Float-ing section were quenched with 0.6% H2O2, blocked andpermeabilized in 0.3% normal donkey serum (NDS), 1%bovine serum albumin (BSA), 0.25% Triton X-100 inSorensen’s solution, incubated for 48 h with anti-cholineacetyltransferase (ChAT) antibody (AB144P, Chemicon),then with the biotinylated secondary antibody and theVectastain Elite ABC (Vector Labs) and visualized usingdiaminobenzidine (DAB) tetrachloride. Sets of sectionsadjacent to the ChAT-IR ones were stained with thionin.
Sections were imaged using an Axioplan microscope (CarlZeiss) connected to an X–Y–Z stage (MSA 001–6, RSFElektronik linked to Microcode II, Boeckler Instruments), acolor video camera (Diagnostic Instruments) and analyzedby the Bioquant image analysis software (Bioquant ImageAnalysis Corporation). ChAT-IR neurons in the BN werecounted using optical fractionator (75,76) and their volumesestimated with the isotropic rotator (77). Density ofChAT-IR fibers was approximated with a Weibel grid usingan intersection approach (78,79). Length of ChAT-IR fiberswas obtained using the strut analysis with the help of the Bio-quant imaging analysis software. Diameters and lengths ofshafts and varicosities of ChAT-IR fibers were measured bythe Bioquant image analysis software.
WT, Tg-swAPPPrP, Tg- BRI-Ab40Prp, Tg-BRI-Ab42Prp andTg-swAPPPrP/BRI-Ab40Prp, Tg-swAPPPrP/BRI-Ab42Prp brainswere formalin fixed. Five micrometer thick paraffin-embeddedrandom non-systematic coronal sections of cerebri and cerebelliwere either stained with Thioflavine S or immunostained withanti-total Ab antibody (33.1.1., courtesy of Dr T.E. Golde) onan autostainer (DAKO) or with anti-ChAT antibody anddeveloped as previously described using streptavidin biotinperoxidase methods. WT samples could not be analyzedowing to poor staining quality of the samples.
Analysis of the data was conducted using the Kruskall–Wallis non-parametric analysis of variance (ANOVA) andthe Mann–Whitney U-test for multiple comparisons at a sig-nificance of P � 0.05. Part of the results of the comparison
3482 Human Molecular Genetics, 2008, Vol. 17, No. 22
at UN
IVE
RS
IDA
D D
E S
ALA
MA
NC
A on A
pril 13, 2010 http://hm
g.oxfordjournals.orgD
ownloaded from

of the axonal blockages in WT and Tg-swAPPPrP, but notTg-A246EPS1PrP and Tg-swAPPPrP; Tg-A246EPS1PrP, micewere reported previously (7).
Drosophila immunofluorescence and tunel assay
To examine axonal blockages, larvae were dissected, fixed andstained as previously described (29,49). The antibody recog-nizing the synaptic vesicle marker cysteine string protein(anti-CSP, courtesy of Dr K. Zinsmaier) was used at a dilutionof 1:20. Anti-mouse FITC was used at a dilution of 1:200 andfixed preparations were mounted, imaged and analyzed asdescribed (29,49). Larval brains from the different genotypeswere dissected for TUNEL analysis and detection of apoptoticcells was preformed as previously described (29) using thefluorescein-based cell death kit (Roche). Quantification ofdying neuronal cells was assayed as described (29).
Immuno-electron microscopy
Mice were transcardially perfused with Sorensen’s solutionfollowed by phosphate-buffered 0.5% glutaraldehyde (GA),4% PFA. Cerebra were removed, post-fixed in 0.5% GA,4%FA and cut with a vibratome into 50 mm thick randomsystematic coronal sections representative of the entire BN.Floating sections were quenched with H2O2, permeabilizedin 0.05% Triton X-100 for 30 min at 48C, blocked in 0.1%NDS, 0.5% BSA, 1% gelatin in Sorensen’s solution, incubatedfor 48 h with anti-ChAT antibody followed by the biotinylatedsecondary antibody and the Vectastain Elite ABC (VectorLabs). Visualization of ChAT was achieved using DAB reac-tion and followed by 10 min incubation in buffered 2% GAprior to embedment into Epon 812 resin. Embedded sectionswere cut into semi-thin sections, stained with 0.25% toluidineblue in 0.1% sodium borate and 70–80 nm ultrathin sectionsthat were mounted onto copper grids and stained with uranylacetate and lead citrate. Images from semithin and ultrathinsections were collected using an Axioplan microscope (CarlZeiss) and an EM208S electron microscope (Philips), respect-ively. Comparable areas of the BN were sampled with similarnumber of semithin and ultrathin sections from each mouse.ChAT-IR profiles were analyzed using Photoshop 5.5(Adobe) and Bioquant image analysis software (BioquantImage Analysis Corporation). Part of the results of the com-parison of the axonal blockages in the WT and Tg-swAPPPrP,but not Tg-A246EPS1PrP and Tg-swAPPPrP; Tg-A246EPS1PrP,mice were reported previously (7).
Biochemical analysis
Sciatic nerves from 4-month-old WT, Tg-swAPPPrP,Tg-A246EPS1PrP and Tg-swAPPPrP; Tg-A246EPS1PrP litter-mate mice were ligated as previously described (38,39).Briefly, one sciatic nerve from each animal was ligated atthe mid thigh and the other nerve left as an unligatedcontrol. Six hours after ligation, animals were sacrificed, andapproximately 5 mm of the proximal and distal halves of thenerve flanking the ligature or from the unligated nerve weredissected and directly homogenized with 1.2x NUPAGELDS Sample buffer. Additionally, L4 to L6 dorsal root
ganglia from ligated and unligated sides were dissected andhomogenized similar to the nerves. Protein concentration ofeach sample was calculated using RC DC protein assay(Bio-Rad). The same amount of protein per sample was elec-trophoresed on NuPAGE 4–12% Bis-Tris gels with MESbuffer. Gels were transferred to nitrocellulose membranesfor 2 h at 300 mA, blocked with 5% milk and probed withantibodies against APP (22C11; Sigma), tubulin (DM1A;Sigma), tubulin beta III (Sigma), myelin basic protein(Dako), KIF5B and Syd. Secondary antibodies were conju-gated to HRP (Zymed) and developed by ECL (Pierce).
To generate a Kif5B antibody, we grew C-terminally trun-cated Kif5B-His fusion protein (pET-23B) (80) in pLysSBL21 (DE3) bacteria, induced with 0.5 mM IPTG and purifiedusing Ni-NTA agarose. Gel slabs containing approximately300 mg of Kif5B-HIS were injected into three rabbits toproduce polyclonal sera against Kif5B. Characterization ofthe Kif5B antibodies showed that these antibodies recognizeprimarily Kif5B (based on the in vitro assays with Kif5Aand Kif5C) and turned out to be exceptionally clean despitebeing crude sera (38,39). SYD antibody was generated pre-viously (61,81). Quantification of APP, Kif5B and SYD wasnormalized for a-tubulin and myelin basic protein. Experi-mental samples with signal intensities within the linearrange were quantified by two independent investigators, oneblind to the genotypes, using Image J (NIH) or Scion Imagesoftware (Scion Corporation). Almost all samples wereexamined on multiple gels and each gel on average by threeto five different exposures.
WT, Tg-swAPPPrP, Tg-A246EPS1PrP and Tg-swAPPPrP/A246EPS1PrP hemibrains were Dounce homogenized using5M guanidine hydrochloride, diluted 1 to 10 in 1X Dulbecco’sphosphate-buffered saline containing 5% BSA and proteaseinhibitors and centrifuged at 10,000 g for 20 min. Supernatantswere loaded into well-established commercially availablehuman-specific anti-Ab40 and anti-Ab42 sandwich ELISAplates (Biosource International) and processed according tothe manufacturer’s instructions (7). WT, Tg-swAPPPrP,Tg-BRI-Ab40Prp, Tg-BRI-Ab42Prp and Tg-swAPPPrP;BRI-Ab40Prp, Tg-swAPPPrP; BRI-Ab42Prp hemibrains werehomogenized in RIPA; RIPA-insoluble fractions were furthersolubilized in formic acid, neutralized with Tris base bufferand appropriately diluted. Ab levels were determined by end-specific sandwich-ELISA as previously described (45,82).
Behavioral analysis
The open field test was performed in a 50 � 35 cm box with thefloor consisting of alternative white and red 5 � 5 cm quad-rants. Upon adjusting to the new room environment for15 min, the mice were placed in the center of the box and theCI (83), defined as the number of times the mice enteredcorner quadrants with any two extremities in 1 min, established.
Rotarod apparatus (SD Instruments) consisted in ribbedplastic rotating rod separated into four sections with ‘beam-break’ sensors close to the bottom of the enclave. Therewere four testing days and during each testing day therewere three consecutive trials. In each trial, the mice wereplaced on the rod facing away from the experimenter andtowards the direction of the rod rotations. The rotation of
Human Molecular Genetics, 2008, Vol. 17, No. 22 3483
at UN
IVE
RS
IDA
D D
E S
ALA
MA
NC
A on A
pril 13, 2010 http://hm
g.oxfordjournals.orgD
ownloaded from

the rod was accelerated from 0 to 45 rpm in 2 min and thenkept rotating at 45 rpm for additional 6 min. Latenciesbefore mice fell from the rod were recorded by the controlunit of the apparatus. With rare exceptions, we tested onemouse per genotype per trial simultaneously and the positionof each mouse changed between the trial days.
For fear conditioning, we used a custom-made apparatus (SDInstruments) consisting in two open top quadrangular Plexiglasenclosures with a lid each placed in a chamber with front doorsand lateral observation window. First enclosure contained awire grid floor and ‘beam-break’ sensors and was positionedin a white chamber harboring a light bulb and a speaker. Thisenclosure was used for training and to test contextual condition-ing. The second enclosure had a different odor compared to thefirst one, contained ‘beam-break’ sensors, bedding and a lightbulb and was positioned in a black chamber harboring aspeaker, but in a location opposite to the one in the firstchamber. This enclosure was used to test for cued conditioning.Visual presentation of the room where fear conditioning tookplace changed between the training and both testing sessionwith curtains. Training started with a 3 min novel contextexploration period followed by three consecutive conditionedstimulus (CS)/unconditioned stimulus (US) pairings separatedby 1 min intervals (CS/US interval) where one CS/US pairingconsisted in a 20 s long tone (CS: 77 dB, 2.8 Hz) coupled to afoot shock during its last 2 s (US: 0.75 mA) (84). Testing tookplace 24 hours after training. To test for contextual conditioning,we measured freezing in response to a 3 min exposure to the train-ing environment. Testing cued conditioning took place 2 h later.To test for cued conditioning, we measured freezing in responseto a 3 min novel context exploration (control) and to threeconsecutive CS separated by 1 min intervals (CS interval).Freezing responses were measured by visual scoring and bycomputer-aided analysis of the video frame capture (‘beam-breaks’). These two freezing response measurement methodsare complimentary and since results from both data sets wereconsistent with each other only data obtained by visual scoringare shown.
Analysis of the data was conducted using the analysisof variance (ANOVA) and the Scheffe’s post hoc test formultiple comparisons at a significance of P � 0.05.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG Online.
FUNDING
Supported by an Ellison Medical Foundation Senior ScholarAward in Aging Research and NIH grant GM35252(L.S.B.G.), NIH grants EY13408 and EY07042 (D.S.W.),NIA grant AGO22595 (E.M.), a Boehringer-IngelheimFonds fellowship (G.B.S.), an Ellison Medical FoundationSenior postdoctoral fellowship (S.G), and a new investigatorgrant from the Alzheimer Association (S.G).
ACKNOWLEDGEMENTS
We thank Drs Fred H. Gage, Todd E. Golde, Edward H. Koo,Klara Limback-Stokin and Mark Mayford for helpful discus-
sions. L.S.B.G. is an investigator of the Howard HughesMedical Institute.
Conflict of Interest statement. None declared.
REFERENCES
1. Katzman, R. (1986) Alzheimer’s disease. N. Engl. J. Med., 314, 964–973.2. Davies, P. and Maloney, A.J. (1976) Selective loss of central cholinergic
neurons in Alzheimer’s disease. Lancet, 2, 1403.3. Hyman, B.T., Van Horsen, G.W., Damasio, A.R. and Barnes, C.L. (1984)
Alzheimer’s disease: cell-specific pathology isolates the hippocampalformation. Science, 225, 1168–1170.
4. Perry, E.K., Perry, R.H., Blessed, G. and Tomlinson, B.E. (1977)Necropsy evidence of central cholinergic deficits in senile dementia.Lancet, 1, 189.
5. Samuel, W., Terry, R.D., DeTeresa, R., Butters, N. and Masliah, E. (1994)Clinical correlates of cortical and nucleus basalis pathology in Alzheimerdementia. Arch. Neurol., 51, 772–778.
6. Meyer-Luehmann, M., Spires-Jones, T.L., Prada, C., Garcia-Alloza, M.,de Calignon, A., Rozkalne, A., Koenigsknecht-Talboo, J., Holtzman,D.M., Bacskai, B.J. and Hyman, B.T. (2008) Rapid appearance and localtoxicity of amyloid-beta plaques in a mouse model of Alzheimer’sdisease. Nature, 451, 720–724.
7. Stokin, G.B., Lillo, C., Falzone, T.L., Brusch, R.G., Rockenstein, E.,Mount, S.L., Raman, R., Davies, P., Masliah, E., Williams, D.S. et al.(2005) Axonopathy and transport deficits early in the pathogenesis ofAlzheimer’s disease. Science, 307, 1282–1288.
8. Salehi, A., Delcroix, J.D., Belichenko, P.V., Zhan, K., Wu, C., Valletta, J.S.,Takimoto-Kimura, R., Kleschevnikov, A.M., Sambamurti, K., Chung, P.P.et al. (2006) Increased App expression in a mouse model of Down’ssyndrome disrupts NGF transport and causes cholinergic neurondegeneration. Neuron, 51, 29–42.
9. Lampert, P. (1971) Fine structural changes of neurites in Alzheimer’sdisease. Acta. Neuropathol. (Berl.), 5 (Suppl. 5), 49–53.
10. Price, D.L., Altschuler, R.J., Struble, R.G., Casanova, M.F., Cork, L.C.and Murphy, D.B. (1986) Sequestration of tubulin in neurons inAlzheimer’s disease. Brain. Res., 385, 305–310.
11. Rasool, C.G., Svendsen, C.N. and Selkoe, D.J. (1986) Neurofibrillarydegeneration of cholinergic and noncholinergic neurons of the basalforebrain in Alzheimer’s disease. Ann. Neurol., 20, 482–488.
12. Terry, R.D. (1963) The fine structure of neurofibrillary tangles inAlzheimer’s disease. J. Neuropathol. Exp. Neurol., 22, 629–641.
13. Smith, K.D., Kallhoff, V., Zheng, H. and Pautler, R.G. (2007) In vivoaxonal transport rates decrease in a mouse model of Alzheimer’s disease.Neuroimage, 35, 1401–1408.
14. Minoshima, S. and Cross, D. (2008) In vivo imaging of axonal transportusing MRI: aging and Alzheimer’s disease. Eur. J. Nucl. Med. Mol.Imaging, 35 (Suppl. 1), S89–S92.
15. Stokin, G.B. and Goldstein, L.S. (2006) Linking molecular motors toAlzheimer’s disease. J. Physiol. Paris, 99, 193–200.
16. Stokin, G.B. and Goldstein, L.S. (2006) Axonal transport and Alzheimer’sdisease. Annu. Rev. Biochem., 75, 607–627.
17. Terry, R.D. (1996) The pathogenesis of Alzheimer disease: an alternativeto the amyloid hypothesis. J. Neuropathol. Exp. Neurol., 55, 1023–1025.
18. Masliah, E., Mallory, M., Deerinck, T., DeTeresa, R., Lamont, S., Miller,A., Terry, R.D., Carragher, B. and Ellisman, M. (1993) Re-evaluation ofthe structural organization of neuritic plaques in Alzheimer’s disease.J. Neuropathol. Exp. Neurol., 52, 619–632.
19. Dickson, T.C., King, C.E., McCormack, G.H. and Vickers, J.C. (1999)Neurochemical diversity of dystrophic neurites in the early and late stagesof Alzheimer’s disease. Exp. Neurol., 156, 100–110.
20. Cork, L.C., Masters, C., Beyreuther, K. and Price, D.L. (1990)Development of senile plaques. Relationships of neuronal abnormalitiesand amyloid deposits. Am. J. Pathol., 137, 1383–1392.
21. Dickson, D.W., Farlo, J., Davies, P., Crystal, H., Fuld, P. and Yen, S.H.(1988) Alzheimer’s disease. A double-labeling immunohistochemicalstudy of senile plaques. Am. J. Pathol., 132, 86–101.
22. Masliah, E., Mallory, M., Hansen, L., Alford, M., DeTeresa, R. andTerry, R. (1993) An antibody against phosphorylated neurofilaments
3484 Human Molecular Genetics, 2008, Vol. 17, No. 22
at UN
IVE
RS
IDA
D D
E S
ALA
MA
NC
A on A
pril 13, 2010 http://hm
g.oxfordjournals.orgD
ownloaded from

identifies a subset of damaged association axons in Alzheimer’s disease.Am. J. Pathol., 142, 871–882.
23. Chen, X.H., Johnson, V.E., Uryu, K., Trojanowski, J.Q. and Smith, D.H.(2008) A lack of amyloid beta plaques despite persistent accumulation ofamyloid beta in axons of long-term survivors of traumatic brain injury.Brain Pathol., Epub ahead of print.
24. Burger, P.C. and Vogel, F.S. (1973) The development of the pathologicchanges of Alzheimer’s disease and senile dementia in patients withDown’s syndrome. Am. J. Pathol., 73, 457–476.
25. Cabrejo, L., Guyant-Marechal, L., Laquerriere, A., Vercelletto, M., De laFourniere, F., Thomas-Anterion, C., Verny, C., Letournel, F., Pasquier, F.,Vital, A. et al. (2006) Phenotype associated with APP duplication in fivefamilies. Brain, 129, 2966–2976.
26. Rovelet-Lecrux, A., Hannequin, D., Raux, G., Le Meur, N., Laquerriere, A.,Vital, A., Dumanchin, C., Feuillette, S., Brice, A., Vercelletto, M. et al.
(2006) APP locus duplication causes autosomal dominant early-onsetAlzheimer disease with cerebral amyloid angiopathy. Nat. Genet., 38,24–26.
27. Sleegers, K., Brouwers, N., Gijselinck, I., Theuns, J., Goossens, D.,Wauters, J., Del-Favero, J., Cruts, M., van Duijn, C.M. and VanBroeckhoven, C. (2006) APP duplication is sufficient to cause earlyonset Alzheimer’s dementia with cerebral amyloid angiopathy. Brain,129, 2977–2983.
28. Oyama, F., Cairns, N.J., Shimada, H., Oyama, R., Titani, K. and Ihara, Y.(1994) Down’s syndrome: up-regulation of beta-amyloid proteinprecursor and tau mRNAs and their defective coordination.J. Neurochem., 62, 1062–1066.
29. Gunawardena, S. and Goldstein, L.S. (2001) Disruption of axonaltransport and neuronal viability by amyloid precursor protein mutations inDrosophila. Neuron, 32, 389–401.
30. Torroja, L., Chu, H., Kotovsky, I. and White, K. (1999) Neuronaloverexpression of APPL, the Drosophila homologue of the amyloidprecursor protein (APP), disrupts axonal transport. Curr. Biol., 9,489–492.
31. Brown, M.S., Ye, J., Rawson, R.B. and Goldstein, J.L. (2000) Regulatedintramembrane proteolysis: a control mechanism conserved from bacteriato humans. Cell, 100, 391–398.
32. De Strooper, B., Saftig, P., Craessaerts, K., Vanderstichele, H., Guhde, G.,Annaert, W., Von Figura, K. and Van Leuven, F. (1998) Deficiency ofpresenilin-1 inhibits the normal cleavage of amyloid precursor protein.Nature, 391, 387–390.
33. De Strooper, B., Annaert, W., Cupers, P., Saftig, P., Craessaerts, K.,Mumm, J.S., Schroeter, E.H., Schrijvers, V., Wolfe, M.S., Ray, W.J. et al.
(1999) A presenilin-1-dependent gamma-secretase-like protease mediatesrelease of Notch intracellular domain. Nature, 398, 518–522.
34. Struhl, G. and Greenwald, I. (1999) Presenilin is required for activity andnuclear access of Notch in Drosophila. Nature, 398, 522–525.
35. Ye, Y., Lukinova, N. and Fortini, M.E. (1999) Neurogenic phenotypes andaltered Notch processing in Drosophila Presenilin mutants. Nature, 398,525–529.
36. Naruse, S., Thinakaran, G., Luo, J.J., Kusiak, J.W., Tomita, T., Iwatsubo, T.,Qian, X., Ginty, D.D., Price, D.L., Borchelt, D.R. et al. (1998) Effects of PS1deficiency on membrane protein trafficking in neurons. Neuron, 21, 1213–1221.
37. Nishimura, M., Yu, G., Levesque, G., Zhang, D.M., Ruel, L., Chen, F.,Milman, P., Holmes, E., Liang, Y., Kawarai, T. et al. (1999) Presenilinmutations associated with Alzheimer disease cause defective intracellulartrafficking of beta-catenin, a component of the presenilin protein complex.Nat. Med., 5, 164–169.
38. Kamal, A., Stokin, G.B., Yang, Z., Xia, C.H. and Goldstein, L.S. (2000)Axonal transport of amyloid precursor protein is mediated by directbinding to the kinesin light chain subunit of kinesin-I. Neuron, 28,449–459.
39. Kamal, A., Almenar-Queralt, A., LeBlanc, J.F., Roberts, E.A. andGoldstein, L.S. (2001) Kinesin-mediated axonal transport of a membranecompartment containing beta-secretase and presenilin-1 requires APP.Nature, 414, 643–648.
40. Lazarov, O., Morfini, G.A., Pigino, G., Gadadhar, A., Chen, X., Robinson, J.,Ho, H., Brady, S.T. and Sisodia, S.S. (2007) Impairments in fastaxonal transport and motor neuron deficits in transgenic mice expressingfamilial Alzheimer’s disease-linked mutant presenilin 1. J. Neurosci., 27,7011–7020.
41. Pigino, G., Morfini, G., Pelsman, A., Mattson, M.P., Brady, S.T. andBusciglio, J. (2003) Alzheimer’s presenilin 1 mutations impairkinesin-based axonal transport. J. Neurosci., 23, 4499–4508.
42. Cash, A.D., Aliev, G., Siedlak, S.L., Nunomura, A., Fujioka, H., Zhu, X.,Raina, A.K., Vinters, H.V., Tabaton, M., Johnson, A.B. et al. (2003)Microtubule reduction in Alzheimer’s disease and aging is independent oftau filament formation. Am. J. Pathol., 162, 1623–1627.
43. Terry, R.D., Gonatas, N.K. and Weiss, M. (1964) Ultrastructural studies inAlzheimer’s presenile dementia. Am. J. Pathol., 44, 269–287.
44. Kim, J., Onstead, L., Randle, S., Price, R., Smithson, L., Zwizinski, C.,Dickson, D.W., Golde, T. and McGowan, E. (2007) Abeta40 inhibitsamyloid deposition in vivo. J. Neurosci., 27, 627–633.
45. McGowan, E., Pickford, F., Kim, J., Onstead, L., Eriksen, J., Yu, C.,Skipper, L., Murphy, M.P., Beard, J., Das, P. et al. (2005) Abeta42 isessential for parenchymal and vascular amyloid deposition in mice.Neuron, 47, 191–199.
46. Vidal, R., Frangione, B., Rostagno, A., Mead, S., Revesz, T., Plant, G. andGhiso, J. (1999) A stop-codon mutation in the BRI gene associated withfamilial British dementia. Nature, 399, 776–781.
47. Vidal, R., Revesz, T., Rostagno, A., Kim, E., Holton, J.L., Bek, T.,Bojsen-Moller, M., Braendgaard, H., Plant, G., Ghiso, J. et al. (2000) Adecamer duplication in the 30 region of the BRI gene originates anamyloid peptide that is associated with dementia in a Danish kindred.Proc. Natl Acad. Sci. USA, 97, 4920–4925.
48. Bernstein, M. and Lichtman, J.W. (1999) Axonal atrophy: the retractionreaction. Curr. Opin. Neurobiol., 9, 364–370.
49. Gunawardena, S., Her, L.S., Brusch, R.G., Laymon, R.A., Niesman, I.R.,Gordesky-Gold, B., Sintasath, L., Bonini, N.M. and Goldstein, L.S. (2003)Disruption of axonal transport by loss of huntingtin or expression ofpathogenic polyQ proteins in Drosophila. Neuron, 40, 25–40.
50. Hurd, D.D. and Saxton, W.M. (1996) Kinesin mutations cause motorneuron disease phenotypes by disrupting fast axonal transport inDrosophila. Genetics, 144, 1075–1085.
51. Lampert, P.W. (1967) A comparative electron microscopic study ofreactive, degenerating, regenerating and dystrophic axons. J. Neuropathol.
Exp. Neurol., 26, 345–368.52. Wong, T.P., Debeir, T., Duff, K. and Cuello, A.C. (1999) Reorganization
of cholinergic terminals in the cerebral cortex and hippocampus intransgenic mice carrying mutated presenilin-1 and amyloid precursorprotein transgenes. J. Neurosci., 19, 2706–2716.
53. Hsiao, K., Chapman, P., Nilsen, S., Eckman, C., Harigaya, Y., Younkin, S.,Yang, F. and Cole, G. (1996) Correlative memory deficits, Abeta elevation,and amyloid plaques in transgenic mice. Science, 274, 99–102.
54. Fossella, J., Samant, S.A., Silver, L.M., King, S.M., Vaughan, K.T.,Olds-Clarke, P., Johnson, K.A., Mikami, A., Vallee, R.B. and Pilder, S.H.(2000) An axonemal dynein at the Hybrid Sterility 6 locus: implicationsfor t haplotype-specific male sterility and the evolution of species barriers.Mamm. Genome, 11, 8–15.
55. Ye, Y. and Fortini, M.E. (1999) Apoptotic activities of wild-type andAlzheimer’s disease-related mutant presenilins in Drosophilamelanogaster. J. Cell Biol., 146, 1351–1364.
56. Dineley, K.T., Xia, X., Bui, D., Sweatt, J.D. and Zheng, H. (2002)Accelerated plaque accumulation, associative learning deficits, andup-regulation of alpha 7 nicotinic receptor protein in transgenic miceco-expressing mutant human presenilin 1 and amyloid precursor proteins.J. Biol. Chem., 277, 22768–22780.
57. Holcomb, L., Gordon, M.N., McGowan, E., Yu, X., Benkovic, S.,Jantzen, P., Wright, K., Saad, I., Mueller, R., Morgan, D. et al. (1998)Accelerated Alzheimer-type phenotype in transgenic mice carrying bothmutant amyloid precursor protein and presenilin 1 transgenes. Nat. Med.,4, 97–100.
58. Holcomb, L.A., Gordon, M.N., Jantzen, P., Hsiao, K., Duff, K. andMorgan, D. (1999) Behavioral changes in transgenic mice expressing bothamyloid precursor protein and presenilin-1 mutations: lack of associationwith amyloid deposits. Behav. Genet., 29, 177–185.
59. Cai, D., Leem, J.Y., Greenfield, J.P., Wang, P., Kim, B.S., Wang, R.,Lopes, K.O., Kim, S.H., Zheng, H., Greengard, P. et al. (2003)Presenilin-1 regulates intracellular trafficking and cell surface delivery ofbeta-amyloid precursor protein. J. Biol. Chem., 278, 3446–3454.
60. Rusu, P., Jansen, A., Soba, P., Kirsch, J., Lower, A., Merdes, G., Kuan,Y.H., Jung, A., Beyreuther, K., Kjaerulff, O. et al. (2007) Axonalaccumulation of synaptic markers in APP transgenic Drosophila depends
Human Molecular Genetics, 2008, Vol. 17, No. 22 3485
at UN
IVE
RS
IDA
D D
E S
ALA
MA
NC
A on A
pril 13, 2010 http://hm
g.oxfordjournals.orgD
ownloaded from

on the NPTY motif and is paralleled by defects in synaptic plasticity.Eur. J. Neurosci., 25, 1079–1086.
61. Cavalli, V., Kujala, P., Klumperman, J. and Goldstein, L.S. (2005)Sunday Driver links axonal transport to damage signaling. J. Cell Biol.,168, 775–787.
62. Hernandez, D., Sugaya, K., Qu, T., McGowan, E., Duff, K. andMcKinney, M. (2001) Survival and plasticity of basal forebraincholinergic systems in mice transgenic for presenilin-1 and amyloidprecursor protein mutant genes. Neuroreport, 12, 1377–1384.
63. Galvan, V., Gorostiza, O.F., Banwait, S., Ataie, M., Logvinova, A.V.,Sitaraman, S., Carlson, E., Sagi, S.A., Chevallier, N., Jin, K. et al. (2006)Reversal of Alzheimer’s-like pathology and behavior in human APPtransgenic mice by mutation of Asp664. Proc. Natl Acad. Sci. USA, 103,7130–7135.
64. Kaether, C., Lammich, S., Edbauer, D., Ertl, M., Rietdorf, J., Capell, A.,Steiner, H. and Haass, C. (2002) Presenilin-1 affects trafficking andprocessing of betaAPP and is targeted in a complex with nicastrin to theplasma membrane. J. Cell Biol., 158, 551–561.
65. Kim, S.H., Leem, J.Y., Lah, J.J., Slunt, H.H., Levey, A.I., Thinakaran, G.and Sisodia, S.S. (2001) Multiple effects of aspartate mutant presenilin 1on the processing and trafficking of amyloid precursor protein. J. Biol.Chem., 276, 43343–43350.
66. Leem, J.Y., Saura, C.A., Pietrzik, C., Christianson, J., Wanamaker, C.,King, L.T., Veselits, M.L., Tomita, T., Gasparini, L., Iwatsubo, T. et al.(2002) A role for presenilin 1 in regulating the delivery of amyloidprecursor protein to the cell surface. Neurobiol. Dis., 11, 64–82.
67. Cai, D., Zhong, M., Wang, R., Netzer, W.J., Shields, D., Zheng, H.,Sisodia, S.S., Foster, D.A., Gorelick, F.S., Xu, H. et al. (2006)Phospholipase D1 corrects impaired betaAPP trafficking and neuriteoutgrowth in familial Alzheimer’s disease-linked presenilin-1 mutantneurons. Proc. Natl Acad. Sci. USA, 103, 1936–1940.
68. Lee, E.B., Zhang, B., Liu, K., Greenbaum, E.A., Doms, R.W.,Trojanowski, J.Q. and Lee, V.M. (2005) BACE overexpression alters thesubcellular processing of APP and inhibits Abeta deposition in vivo.J. Cell Biol., 168, 291–302.
69. Yu, H., Saura, C.A., Choi, S.Y., Sun, L.D., Yang, X., Handler, M.,Kawarabayashi, T., Younkin, L., Fedeles, B., Wilson, M.A. et al. (2001)APP processing and synaptic plasticity in presenilin-1 conditionalknockout mice. Neuron, 31, 713–726.
70. Tezapsidis, N., Merz, P.A., Merz, G. and Hong, H. (2003) Microtubularinteractions of presenilin direct kinesis of Abeta peptide and itsprecursors. FASEB J., 17, 1322–1324.
71. Borchelt, D.R., Thinakaran, G., Eckman, C.B., Lee, M.K., Davenport, F.,Ratovitsky, T., Prada, C.M., Kim, G., Seekins, S., Yager, D. et al. (1996)Familial Alzheimer’s disease-linked presenilin 1 variants elevate Abeta1–42/1–40 ratio in vitro and in vivo. Neuron, 17, 1005–1013.
72. Borchelt, D.R., Ratovitski, T., van Lare, J., Lee, M.K., Gonzales, V.,Jenkins, N.A., Copeland, N.G., Price, D.L. and Sisodia, S.S. (1997)Accelerated amyloid deposition in the brains of transgenic micecoexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron,
19, 939–945.
73. Fossgreen, A., Bruckner, B., Czech, C., Masters, C.L., Beyreuther, K. andParo, R. (1998) Transgenic Drosophila expressing human amyloidprecursor protein show gamma-secretase activity and a blistered-wing
phenotype. Proc. Natl Acad. Sci. USA, 95, 13703–13708.
74. Ye, Y. and Fortini, M.E. (2000) Proteolysis and developmental signaltransduction. Semin. Cell Dev. Biol., 11, 211–221.
75. Gundersen, H.J. (1986) Stereology of arbitrary particles. A review ofunbiased number and size estimators and the presentation of some newones, in memory of William R. Thompson. J. Microsc, 143, 3–45.
76. West, M.J., Slomianka, L. and Gundersen, H.J. (1991) Unbiasedstereological estimation of the total number of neurons in the subdivisionsof the rat hippocampus using the optical fractionator. Anat. Rec., 231,482–497.
77. Jensen, E.B. and Gundersen, H.J. (1993) The rotator. J. Microsc., 170,35–44.
78. Geula, C. and Mesulam, M.M. (1996) Systematic regional variations inthe loss of cortical cholinergic fibers in Alzheimer’s disease. Cereb.
Cortex, 6, 165–177.
79. Geula, C. and Mesulam, M.M. (1989) Cortical cholinergic fibers in aging
and Alzheimer’s disease: a morphometric study. Neuroscience, 33,469–481.
80. Xia, C., Rahman, A., Yang, Z. and Goldstein, L.S. (1998) Chromosomallocalization reveals three kinesin heavy chain genes in mouse. Genomics,
52, 209–213.
81. Bowman, A.B., Kamal, A., Ritchings, B.W., Philp, A.V., McGrail, M.,Gindhart, J.G. and Goldstein, L.S. (2000) Kinesin-dependent axonal
transport is mediated by the Sunday driver (SYD) protein. Cell, 103,583–594.
82. Kawarabayashi, T., Younkin, L.H., Saido, T.C., Shoji, M., Ashe, K.H. andYounkin, S.G. (2001) Age-dependent changes in brain, CSF, and plasma
amyloid (beta) protein in the Tg2576 transgenic mouse model ofAlzheimer’s disease. J. Neurosci., 21, 372–381.
83. Hsiao, K.K., Borchelt, D.R., Olson, K., Johannsdottir, R., Kitt, C., Yunis, W.,Xu, S., Eckman, C., Younkin, S., Price, D. et al. (1995) Age-related CNS
disorder and early death in transgenic FVB/N mice overexpressing Alzheimeramyloid precursor proteins. Neuron, 15, 1203–1218.
84. Limback-Stokin, K., Korzus, E., Nagaoka-Yasuda, R. and Mayford, M.
(2004) Nuclear calcium/calmodulin regulates memory consolidation.J. Neurosci., 24, 10858–10867.
3486 Human Molecular Genetics, 2008, Vol. 17, No. 22
at UN
IVE
RS
IDA
D D
E S
ALA
MA
NC
A on A
pril 13, 2010 http://hm
g.oxfordjournals.orgD
ownloaded from




















