
Oligomerization and neurotoxicity of the amyloid ADan peptide implicated in familial Danish dementia

Hetero-oligomerization does not compromise `gain of function' oftumor-derived p53 mutants
Debabrita Deb1,2,3, Mariano Scian1,2,3, Katherine E Roth1,2, Wei Li1,2, Jane Keiger1,2,Abhay Sankar Chakraborti1,2, Swati Palit Deb1,2 and Sumitra Deb*,1,2
1Department of Biochemistry and Molecular Biophysics, Medical College of Virginia, Virginia Commonwealth University,Richmond, Virginia, VA 23298, USA; 2Massey Cancer Center, Medical College of Virginia, Virginia Commonwealth University,Richmond, Virginia, VA 23298, USA
Tumor-derived p53 mutants activate transcription frompromoters of various growth-related genes. We testedwhether this transactivation function of the mutant proteinis su�cient to induce tumorigenesis (`gain of function').Tumor-derived mutant p53-281G transactivates thepromoters of human epidermal growth factor receptor(EGFR) and human multiple drug resistance gene (MDR-1). To determine whether the C-terminal domain functionsonly as an oligomerization domain in mutant p53-mediated transactivation, we have replaced the tetramer-ization domain of p53 by a heterologous tetramerizationdomain; although this mutant protein formed tetramers insolution, it failed to transactivate signi®cantly. Therefore,for successful mutant p53-mediated transactivation,sequences near the C-terminus of mutant p53 are requiredto perform functions in addition to tetramerization. Wealso demonstrate that co-expression of a deletion mutantof p53 (p53 del 1-293), which retains the p53 oligomeriza-tion domain, inhibits this transactivation. p53 del 1-293co-immunoprecipitates with p53-281G suggesting thathetero-oligomers of p53-281G and p53 del 1-293 aredefective in transactivation. We also show that a cell linestably transfected with p53-281G expresses higher levelsof endogenous NF-kB and proliferating cell nuclearantigen (PCNA) compared to that transfected with vectoralone. On co-expression, p53 del 1-293 lowered the levelsof NF-kB and PCNA in p53-281G-expressing cells.However, on co-expression, p53 del 1-293 did not inhibitthe tumorigenicity and colony forming ability of p53-281Gexpressing cells. Our earlier work showed that a deletionof the C-terminal sequences of p53-281G overlapping theoligomerization domain obliterates `gain of function'.Taken together, the above information suggests that theC-terminal sequences have some critical role in `gain offunction' in addition to transactivation.Oncogene (2002) 21, 176 ± 189. DOI: 10.1038/sj/onc/1205035
Keywords: p53; transcriptional activation; repression;oligomerization
Introduction
Wild-type p53 is a tumor-suppressor and is found to bemutated in a large number of human cancers (Donehowerand Bradley, 1993; Greenblatt et al., 1994; Ko and Prives,1996; Lane, 1994; Levine, 1993, 1997; Lowe, 1999; Oren,1999; Ozbun and Butel, 1995; Prives and Hall, 1999;Sheikh and Fornace, 2000; Soussi et al., 2000; Vogelsteinet al., 2000). Under normal situations wild-type p53 has ashort half-life. However, under stress such as that causedby DNA damage p53 is stabilized and its intracellularlevel rises due predominantly to post-transcriptionalmodi®cations; p53 then induces G1-S arrest or apoptosissuch that the damaged cell cannot perpetuate.
The majority of p53 mutations found in human cancerare missense in nature (Greenblatt et al., 1994; Levine etal., 1991; Soussi et al., 2000). Presumably, tumor-derivedp53 mutants cannot induce the degradation pathwayactivated by wild-type p53 through induction of MDM2.The resulting mutant protein is, thus, usually present at arelatively high level inside the cell (Haupt et al., 1997;Kubbutat et al., 1997; Prives and Hall, 1999). Wild-typep53 acts as a sequence-speci®c transcriptional activatorfor a series of genes, including some that are involved incell cycle control and apoptosis (Levine, 1997; Oren,1999; Prives and Hall, 1999; Vogelstein et al., 2000).Tumor-derived p53 mutants are defective in thissequence-speci®c transactivation. The wild-type proteincan also repress promoters of a number of cellular andviral genes (Deb et al., 1992; Ginsberg et al., 1991;Lechner et al., 1992; Santhanam et al., 1991; Seto et al.,1992; Subler et al., 1992, 1994). It has been shown toinhibit the endogenous expression of a number of genes(Lee et al., 1999; Murphy et al., 1996, 1999; Zhao et al.,2000). Although the molecular mechanism behind thetranscriptional repression is not completely clear, thattumor-derived p53 mutants have in general lost thisfunction (Subler et al., 1992) is suggestive of itsfunctional importance.
Wild-type p53 has a well-de®ned domain structure(Ko and Prives, 1996). Near the N-terminus it has twotransactivation domains (amino acid 1 ± 42, 43 ± 63)(Candau et al., 1997; Subler et al., 1994; Zhu et al.,1998), followed by a proline rich segment (amino acids64 ± 91) (Sakamuro et al., 1997; Venot et al., 1998;Walker and Levine, 1996). At the center is the DNA-
Oncogene (2002) 21, 176 ± 189ã 2002 Nature Publishing Group All rights reserved 0950 ± 9232/02 $25.00
www.nature.com/onc
*Correspondence: S Deb; E-mail: [email protected] two authors contributed equally to the projectReceived 2 April 2001; revised 14 September 2001; accepted 9October 2001

binding domain (amino acids 100 ± 300) (Prives, 1994);this is followed by the presumptive nuclear localizationsignal (amino acids 316 ± 325) (Ko and Prives, 1996;Levine et al., 1991) and tetramerization domain (aminoacids 320 ± 360); (Sturzbecher et al., 1992; Subler et al.,1994; Wang et al., 1993) and ®nally near the C-terminus is the basic domain (amino acids 360 ± 393)(Ko and Prives, 1996; Levine et al., 1991). The C-terminal segment can take part in DNA strand transferand reassociation (Bakalkin et al., 1994; Brain andJenkins, 1994). It can also bind to DNA di�erentstructural complexities, including DNA damaged byionizing radiation or chemicals (Lee et al., 1995, 1997;Reed et al., 1995). The C-terminal region can bind toDNA non-sequence-speci®cally; it is also the site formodi®cations such as phosphorylation, acetylation orbinding to proteins such as 14-3-3 protein. Thesemodi®cations seem to regulate p53's transcriptionaland DNA-binding activities (Abarzua et al., 1995;Baudier et al., 1992; Fourie et al., 1997; Halazonetis etal., 1993; Hansen et al., 1996, Hupp et al., 1992,Takenaka et al., 1995; Waterman et al., 1998).
A number of tumor-derived p53 mutants that aredefective in transactivating genes normally induced bywild-type p53 can transactivate the promoters ofgrowth-related genes such as, human proliferating cellnuclear antigen, EGFR, MDR-1, vascular endothelialgrowth factor, human interleukin 6, basic ®broblastgrowth factor, human heat shock protein 70, c-fos,insulin-like growth factor II, BAG-1, 15-lipoxygenenaseand c-myc (Chin et al., 1992; Frazier et al., 1998;Kelavkar and Badr, 1999; Kieser et al., 1994; Marguliesand Sehgal, 1993; Preuss et al., 2000, Tsutsumi-Ishii etal., 1995; Ueba et al., 1994; Yang et al., 1999). Lin et al.(1995) showed that amino acids at positions 22 and 23in the N-terminal transactivation domain of the mutantprotein p53 (Arg to Gly) [p53-281G] are required fortransactivation of the MDR-1 promoter. They alsoshowed that although murine 10(3) cells (free ofendogenous p53) expressing p53-281G are tumorigenicin nude mice, cells expressing p53-281G with substitu-tions at amino acids 22 and 23 are not. This suggests adirect relationship between the transactivation ability ofmutant p53 and the `gain of function' where mutant p53procures a dominant oncogenic role.
More recently, using similar assay procedures asdescribed above, we showed that the `gain of function'phenotype of tumor-derived p53 mutants also requiresthe C-terminally located oligomerization/nucleic acid-binding domain (Lanyi et al., 1998). Whether the C-terminal region is only required to perform oligomer-ization or it has other function(s) necessary for mutantp53-mediated transactivation and/or `gain of function'is not known. Results of experiments with wild-type p53in which the tetramerization domain has been replacedby a heterologous dimerization or tetramerizationdomain have shown that the heterologous oligomeriza-tion domain preserves wild-type p53-mediated transac-tivation and growth suppression (including G1-Sinhibition) functions (Attardi et al., 1996a,b; Ishizakaet al., 1995; Pietenpol et al., 1994; Reed et al., 1993;
Waterman et al., 1996). However, no information isavailable regarding the consequence of replacement ofthe oligomerization domain of tumor-derived p53mutants by heterologous oligomerization domains.
Here, we determine whether transactivation bytumor-derived mutant p53 is responsible for its `gainof function' phenotype and describe our investigationof the importance of oligomerization in the tumor-derived p53-mediated transcriptional activation and`gain of function'. We have also replaced thetetramerization domain of p53-281G by a coiled-coildimerization motif or a modi®ed coiled-coil structurethat codes for a tetramerization domain. Although themodi®ed heterologous coiled-coil tetramerization do-main induced tetramerization of the mutant p53, itcould not successfully substitute the domain in mutantp53-mediated transactivation of the EGFR promoter.This suggests that sequences near the C-terminus ofmutant p53 are required to perform functions inaddition to tetramerization for successful mutant p53-mediated transactivation. Since wild-type p53 with itstetramerization domain replaced by a heterologousdimerization/tetramerization motif retains transcrip-tional functions (Attardi et al., 1996a,b; Ishizaka etal., 1995; Pietenpol et al., 1994; Reed et al., 1993;Waterman et al., 1996) our work suggests a mechan-istic di�erence between transactivation mediated bywild-type p53 and its tumor-derived mutants. Hetero-oligomerization of wild-type p53 by a mini-proteinwith the oligomerization domain can e�ciently inhibitwild-type p53's biological functions leading to trans-formation of rat embryo ®broblasts (Reed et al., 1993;Shaulian et al., 1992). We, therefore, tested whetherhetero-oligomerization between p53 del 1-293 and p53-281G would lead to inhibition of tumorigenicity (`gainof function') by p53-281G, and found that co-expression did not signi®cantly impact tumorigenesis.We ®nd that hetero-oligomerization between full-lengthmutant p53-281G and a p53 deletion mutant preser-ving the oligomerization domain inhibits transactiva-tion of the EGFR and MDR-1 promoters by tumor-derived mutant p53. We also show that a cell linestably transfected with p53-281G expresses higherlevels of endogenous NF-kB2 and PCNA comparedto that transfected with vector alone. On co-expression,p53 del 1-293 lowered the levels of NF-kB2 and PCNAin p53-281-expressing cells. Since sequences from 393-327 are required for tumorigenicity (Lanyi et al., 1998),the C-terminal sequences must have a crucial role in`gain of function' in addition to transactivation.
Results
Replacement of the tetramerization domain of mutant p53by a heterologous dimerization or tetramerization domainhas a strong inhibitor effect on mutant p53-mediatedtransactivation
Earlier, we have shown that several tumor-derived p53mutants could transactivate promoters of a number of
Oncogene
Mutant p53-mediated transactivationD Deb et al
177

growth-related genes including EGFR (Deb et al.,1992; Ludes-Meyers et al., 1996). However, thedeletion mutant p53-281G del 393-327 that eliminatesthe oligomerization domain failed to transactivate(Lanyi et al., 1998; Ludes-Meyers et al., 1996)suggesting that proper oligomerization of a tumor-derived p53 mutant is required for the transactivationfunction of the mutant protein. We, therefore, checkedwhether the C-terminally located oligomerization/non-sequence-speci®c nucleic acid binding domain isrequired only for oligomerization. It was earlier shownthat replacement of the tetramerization domain ofwild-type p53 by the dimerization domain consisting ofthe coiled-coil structure from the yeast GCN4 protein(or a derivative of the coiled-coil structure that resultsin a tetramerization domain) could e�ectively sustainwild-type p53's transactivation and growth suppressionfunctions (Attardi et al., 1996a; Pietenpol et al., 1994;Waterman et al., 1996). Performing similar experimentswe wanted to determine whether a p53-281G derivativewith such a replacement of the tetramerization domainwould retain the transactivation function. Therefore,we studied the e�ect of expression of p53-281G, p53-281G 343cc (the coiled-coil structure from GCN4 islaced after the amino acid 343) (Pietenpol et al., 1994)and p53-281G TZ334NR (a modi®ed GCN4 coiled-coilstructure is placed after amino acid 334 such that itbecomes a tetramerization domain) (Waterman et al.,1996) on the activity of the EGFR promoter.Transfections were carried out in Saos-2 cells.
Luciferase assay results shown in Figure 1 demon-strate that p53-281G-mediated transactivation is notpreserved by replacing its tetramerization domain by aheterologous dimerization/tetramerization motif sug-gesting that this region performs some vital function(s)required for mutant p53-mediated transactivation thatcannot be substituted by a heterologous dimerization/tetramerization domain. Amino acid residues 320 ± 360of p53 code for the tetramerization domain (Sturz-becher et al., 1992; Subler et al., 1994; Wang et al.,1993); besides substitution of this domain (startingfrom amino acid 334), C-terminal amino acids past theoligomerization domain up to 393 are missing in p53-281G TZ334NR, and this segment may also play animportant role in mutant p53-mediated transactivation.Therefore, we have tested one more mutant p53-281GTZ334NRI352, which has the sequences 352 ± 393inserted after an Isoleucine at amino acid 352 followingthe TZ moiety. This mutant transactivated the bestamong all the domain replacement mutants. TheWestern blot analysis shown with the luciferase assay®gure demonstrated equivalent amounts of expressionby p53-281G and p53-281G TZ334NR, suggesting thatexpression de®ciency cannot explain the inability ofp53-281G TZ334NR to transactivate. Thus, the resultsof our experiments suggest that the C-terminallylocated amino acids 352 ± 393 perform a vital functionfor mutant p53-mediated transactivation. Earlier, asimilar conclusion was reached by Frazier et al. (1998)demonstrating a requirement for amino acids 370 ± 380for transactivation the c-myc promoter by p53-281G.
Replacement of the tetramerization domain of mutant p53by a heterologous tetramerization domain preservestetramerization of the protein
One possible reason for the failure of p53-281GTZ334NR and p53-281G TZ334NRI352 to transacti-vate the EGFR promoter e�ciently can be a defect inoligomerization in the context of mutant p53. We,therefore, tested whether p53-281G TZ334NR and p53-281G TZ334NRI352 exist predominantly as tetramersin solution. For this purpose we have used glutar-aldehyde cross-linking assays as described earlier(Subler et al., 1994).
We have synthesized 35S-labeled p53-281G, p53-281GTZ334NR, p53-281G TZ334NRI352 and p53-281G del393-327 using an in vivo transcription-translation systemas described in Materials and methods. Syntheticproteins were incubated with glutaraldehyde at varying®nal concentrations (Figure 2). After cross-linking,proteins were analysed by gradient (4 ± 20%) sodiumdodecyl sulfate (SDS)-polyacrylamide gel electrophor-esis. The data shown in Figure 2 indicate that p53-281G,p53-281G TZ334NRI352 and p53-281G TZ334NRoligomerized, while p53-281G del 393-327 could not,although there may be some variations in tetrameriza-tion among p53-281G, p53-281G TZ334NRI352 andp53-281G TZ334NR. Since p53-281G TZ334NRI352and p53-281G TZ334NR are capable of oligomerizationinto tetramers, the results of our experiments shown inFigure 1 demonstrate that for p53-281G-mediatedtransactivation, the C-terminally located oligomeriza-tion/nucleic acid binding domain is required forfunctions in addition to oligomerization.
Transactivation of the EGFR and MDR-1 promoters bythe tumor-derived mutant p53-281G was inhibited byco-expression of p53 del 1-293
Hetero-oligomerization of wild-type p53 with minipro-teins containing its C-terminal oligomerization domainresults in inactivation of wild-type p53's function(Shaulian et al., 1992). Therefore, to investigate therole of oligomerization further we tested whether suchhetero-oligomerization of a tumor-derived mutant p53-281G would inactivate its transactivation potential.
Saos-2 cells were co-transfected with EGFR.Luc(Ludes-Meyers et al., 1996; Deb et al., 2001) or MDR-1.CAT (Chin et al., 1992; Lin et al., 1995), theexpression plasmid for p53-281G (or expression vectoralone) and the expression plasmid for p53 del 1-293 (orexpression vector alone). Transfections and CAT (orluciferase) assays were carried out as described inMaterials and methods. Figure 3a,b shows the CAT(and luciferase) assay data, it is apparent that althoughthe tumor-derived p53-mutant transactivated theEGFR and MDR-1 promoters, the transactivationability was reduced signi®cantly by the co-expression ofp53 del 1-293. Western blot analyses examiningexpression of p53-281G and p53 del 1-293 for arepresentative experiment shown in the ®gure demon-strate that proteins were adequately expressed; thus,
Mutant p53-mediated transactivationD Deb et al
178
Oncogene

Figure 2 Replacement of the tetramerization domain of mutant p53 by a heterologous tetramerization domain preservestetramerization of the protein. In vivo translated p53-281G and its derivatives (p53-281G TZ334NR, p53-281G TZ334NRI352 andp53-281G del 393-327) were incubated with the indicated concentrations of glutaraldehyde as described (Subler et al., 1994).Samples were loaded onto a gradient (4 ± 20%) SDS polyacrylamide gel after cross-linking, electrophoresed, dried andautoradiographed. Positions of presumptive monomers, dimers, trimers and tetramers of p53 derivatives are indicated
Figure 1 Replacement of the tetramerization domain of mutant p53 by a heterologous dimerization or tetramerization domain hasa drastic e�ect on mutant p53-mediated transactivation. (a) Schematic representation of p53-281G derivatives used in the domainexchange experiments. The tetramerization domain of p53 amino acid 320 ± 360 is shown by a dark box. p53-281G TZ334NR hassequences from amino acids 334 to 393 replaced by a modi®ed coiled-coil structural domain (shown by a stippled box) that makes ita tetramerization domain, p53-281G TZ334NRI352 has the sequences 352 ± 393 inserted after an Isoleucine at amino acid 352following the TZ moiety, p53-281G 343cc has sequences from amino acids 344 to 393 replaced by the coiled-coil structural domain(shown by a shaded box, coding for a dimerization motif) from yeast GCN4 protein. (b) Transactivation of the EGFR promoter byp53-281G derivatives. Saos-2 cells were co-transfected with EGFR.Luc and the expression plasmid for tumor-derived mutant p53-281G or one of its derivatives (or expression vector alone) in a 24 well plate by Lipofectamine 2000 as described in Materials andmethods. After transfection, cells were treated as described, and luciferase assays were performed. The left panel shows luciferaseassays and the right panel shows Western blot analysis carried out with equal amounts of protein from each luciferase assay extractfrom one representative experiment using a monoclonal anti-p53 antibody (PAb421)
Oncogene
Mutant p53-mediated transactivationD Deb et al
179

lack of transactivation by p53-281G in the presence ofp53 del 1-293 cannot be explained by any proteinexpression problem. It is possible that hetero-oligomersbetween p53-281G and p53 del 1-293 are inactive intransactivation.
p53 del 1-293 forms complexes with p53-281G in vivo
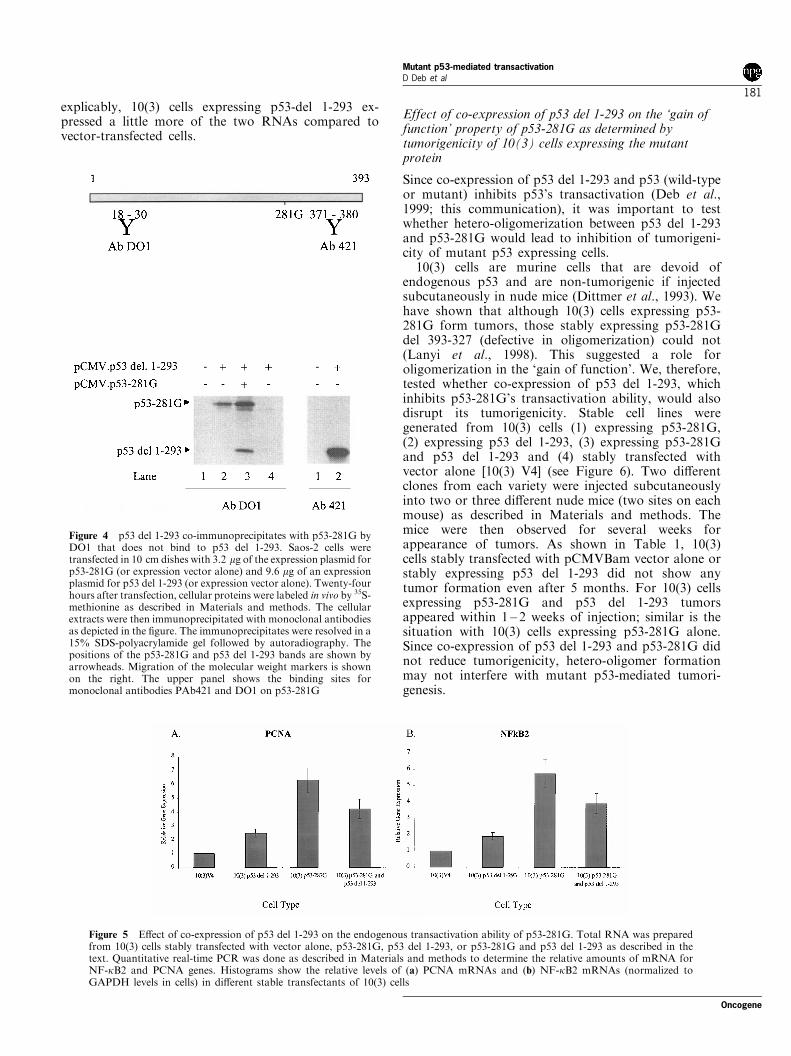
Next, we checked whether p53-281G would form hetero-oligomeric complexes with p53 del 1-293 in vivo. We co-transfected expression plasmids for p53-281G (orexpression vector alone) and p53 del 1-293 (or expressionvector alone) into Saos-2 cells. Twenty-four hours aftertransfection, cells were labeled in vivo with 35S-methionine as described in Materials and methods. Cellextracts were used for immunoprecipitation analysis(Figure 4). Although DO1 could not immunoprecipitatep53 del 1-293 alone (lane 4), it could precipitate bothp53-281G and p53 del 1-293 when both were present(lane 3). This shows that p53 del 1-293 binds with p53-281G in vivo forming a hetero-oligomeric complex. Thus,results of these experiments (Figures 3 and 4) suggestthat proper oligomeric forms of p53-281G are necessaryfor its transactivation function.
Effect of co-expression of p53 del 1-293 on theendogenous transactivation ability of p53-281G
Hetero-oligomerization of wild-type p53 with tumor-derived p53 mutants (or polypeptides covering theoligomerization domain of p53) inhibit wild-type p53's
biological functions leading to immortalization andtransformation of rat embryo ®broblasts (Shaulian etal., 1992). Since co-expression of p53 del 1-293 and p53(wild-type or mutant) inhibits p53's transactivation(Deb et al., 1999; this communication), it wasimportant to test whether hetero-oligomerizationbetween p53 del 1-293 and p53-281G would lead toinhibition of transactivation of endogenous genes byp53-281G.
To identify genes that are endogenously transacti-vated by p53-281G, we have used murine 10(3) cellsthat are devoid of endogenous p53 and havecompared gene expression pro®les of cells stablytransfected with p53-281G with that of cells stablytransfected with vector (pCMVBamNeo, Hinds et al.,1990) alone. Microarray analysis done by IncyteGenomics (to be reported separately) indicated anumber of genes that are expressed at a higher levelin cells expressing p53-281G than in cells stablytransfected with vector alone. We chose two suchgenes, PCNA and NF-kB2, to see the e�ect of co-expression of p53 del 1-293. We have used quantita-tive real-time PCR analysis of cDNA, made fromtotal RNA, to determine the level of expression ofPCNA and NF-kB2 RNAs. Figure 5 shows that thelevels of both the genes are considerably higher in10(3) cells expressing p53-281G compared to that incells stably transfected with vector alone. Co-expres-sion of p53 del 1-293 lowered the level of the RNAsto some extent, suggesting that hetero-oligomerizationindeed inhibits transactivation by mutant p53. In-
Figure 3 Transactivation of the EGFR and MDR-1 promoters by the tumor-derived mutant p53-281G is inhibited by co-expression of p53 del 1-293. Saos-2 cells were transfected with (a) EGFR.Luc or (b) MDR-1 CAT. The experiments with EGFR.Lucwere done using 24 well plates. Each well had 170 ng of the EGFR-Luc, 170 ng of pCMVBam p53-281G (or expression vectoralone) and 510 ng of pCMVBam p53 del 1-293 (or expression vector alone). The experiments with MDR1-CAT were done in 10 cmplate with 2.5 mg of MDR1.CAT, 2.5 mg of pCMVBam p53-281G (or expression vector alone) and 7.5 mg of pCMVBam p53 del 1-293 (or expression vector alone). After transfection, cells were treated as described in Materials and methods and CAT (orluciferase) assays were performed. The upper panels show the results of the CAT (or luciferase) assay and the lower panels showWestern blot analyses carried out with equal amounts of protein from each system of one representative experiment. The positionsof the p53-281G (detected by monoclonal antibody Pab1801) and p53 del 1-293 (detected by monoclonal PAb421) bands are shownby arrowheads. Migration of the molecular weight markers are shown on the right
Mutant p53-mediated transactivationD Deb et al
180
Oncogene
explicably, 10(3) cells expressing p53-del 1-293 ex-pressed a little more of the two RNAs compared tovector-transfected cells.
Effect of co-expression of p53 del 1-293 on the `gain offunction' property of p53-281G as determined bytumorigenicity of 10(3) cells expressing the mutantprotein
Since co-expression of p53 del 1-293 and p53 (wild-typeor mutant) inhibits p53's transactivation (Deb et al.,1999; this communication), it was important to testwhether hetero-oligomerization between p53 del 1-293and p53-281G would lead to inhibition of tumorigeni-city of mutant p53 expressing cells.
10(3) cells are murine cells that are devoid ofendogenous p53 and are non-tumorigenic if injectedsubcutaneously in nude mice (Dittmer et al., 1993). Wehave shown that although 10(3) cells expressing p53-281G form tumors, those stably expressing p53-281Gdel 393-327 (defective in oligomerization) could not(Lanyi et al., 1998). This suggested a role foroligomerization in the `gain of function'. We, therefore,tested whether co-expression of p53 del 1-293, whichinhibits p53-281G's transactivation ability, would alsodisrupt its tumorigenicity. Stable cell lines weregenerated from 10(3) cells (1) expressing p53-281G,(2) expressing p53 del 1-293, (3) expressing p53-281Gand p53 del 1-293 and (4) stably transfected withvector alone [10(3) V4] (see Figure 6). Two di�erentclones from each variety were injected subcutaneouslyinto two or three di�erent nude mice (two sites on eachmouse) as described in Materials and methods. Themice were then observed for several weeks forappearance of tumors. As shown in Table 1, 10(3)cells stably transfected with pCMVBam vector alone orstably expressing p53 del 1-293 did not show anytumor formation even after 5 months. For 10(3) cellsexpressing p53-281G and p53 del 1-293 tumorsappeared within 1 ± 2 weeks of injection; similar is thesituation with 10(3) cells expressing p53-281G alone.Since co-expression of p53 del 1-293 and p53-281G didnot reduce tumorigenicity, hetero-oligomer formationmay not interfere with mutant p53-mediated tumori-genesis.
Figure 5 E�ect of co-expression of p53 del 1-293 on the endogenous transactivation ability of p53-281G. Total RNA was preparedfrom 10(3) cells stably transfected with vector alone, p53-281G, p53 del 1-293, or p53-281G and p53 del 1-293 as described in thetext. Quantitative real-time PCR was done as described in Materials and methods to determine the relative amounts of mRNA forNF-kB2 and PCNA genes. Histograms show the relative levels of (a) PCNA mRNAs and (b) NF-kB2 mRNAs (normalized toGAPDH levels in cells) in di�erent stable transfectants of 10(3) cells
Figure 4 p53 del 1-293 co-immunoprecipitates with p53-281G byDO1 that does not bind to p53 del 1-293. Saos-2 cells weretransfected in 10 cm dishes with 3.2 mg of the expression plasmid forp53-281G (or expression vector alone) and 9.6 mg of an expressionplasmid for p53 del 1-293 (or expression vector alone). Twenty-fourhours after transfection, cellular proteins were labeled in vivo by 35S-methionine as described in Materials and methods. The cellularextracts were then immunoprecipitated with monoclonal antibodiesas depicted in the ®gure. The immunoprecipitates were resolved in a15% SDS-polyacrylamide gel followed by autoradiography. Thepositions of the p53-281G and p53 del 1-293 bands are shown byarrowheads. Migration of the molecular weight markers is shownon the right. The upper panel shows the binding sites formonoclonal antibodies PAb421 and DO1 on p53-281G
Oncogene
Mutant p53-mediated transactivationD Deb et al
181
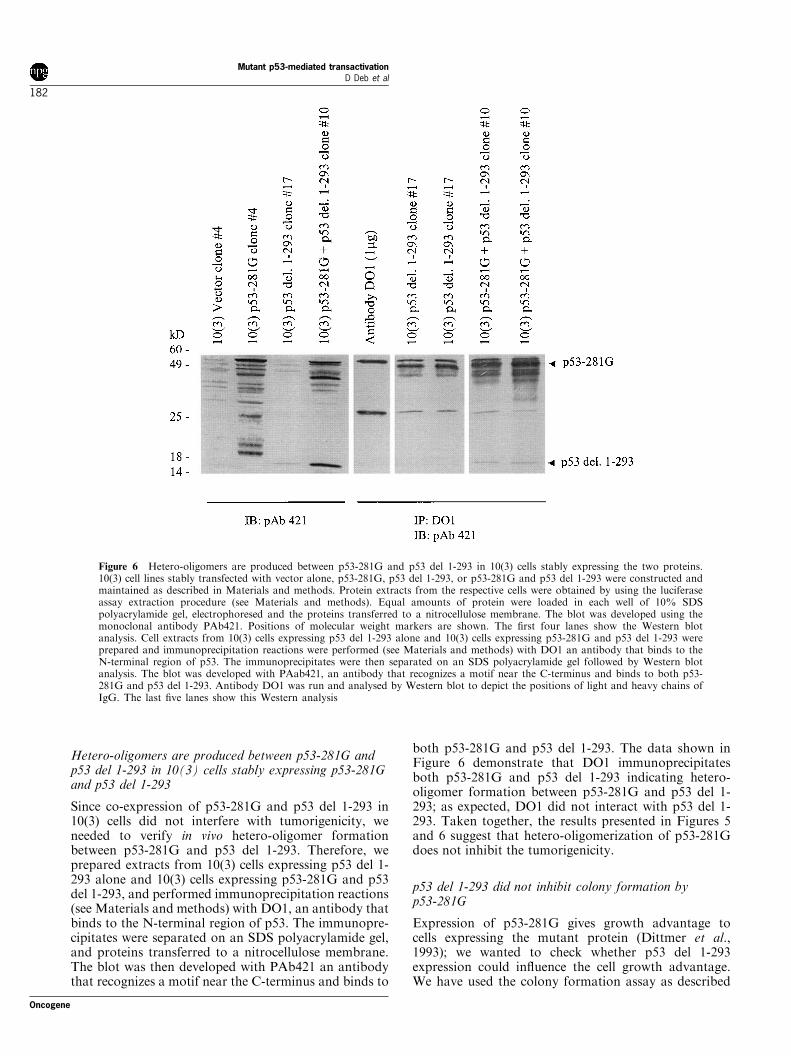
Hetero-oligomers are produced between p53-281G andp53 del 1-293 in 10(3) cells stably expressing p53-281Gand p53 del 1-293
Since co-expression of p53-281G and p53 del 1-293 in10(3) cells did not interfere with tumorigenicity, weneeded to verify in vivo hetero-oligomer formationbetween p53-281G and p53 del 1-293. Therefore, weprepared extracts from 10(3) cells expressing p53 del 1-293 alone and 10(3) cells expressing p53-281G and p53del 1-293, and performed immunoprecipitation reactions(see Materials and methods) with DO1, an antibody thatbinds to the N-terminal region of p53. The immunopre-cipitates were separated on an SDS polyacrylamide gel,and proteins transferred to a nitrocellulose membrane.The blot was then developed with PAb421 an antibodythat recognizes a motif near the C-terminus and binds to
both p53-281G and p53 del 1-293. The data shown inFigure 6 demonstrate that DO1 immunoprecipitatesboth p53-281G and p53 del 1-293 indicating hetero-oligomer formation between p53-281G and p53 del 1-293; as expected, DO1 did not interact with p53 del 1-293. Taken together, the results presented in Figures 5and 6 suggest that hetero-oligomerization of p53-281Gdoes not inhibit the tumorigenicity.
p53 del 1-293 did not inhibit colony formation byp53-281G
Expression of p53-281G gives growth advantage tocells expressing the mutant protein (Dittmer et al.,1993); we wanted to check whether p53 del 1-293expression could in¯uence the cell growth advantage.We have used the colony formation assay as described
Figure 6 Hetero-oligomers are produced between p53-281G and p53 del 1-293 in 10(3) cells stably expressing the two proteins.10(3) cell lines stably transfected with vector alone, p53-281G, p53 del 1-293, or p53-281G and p53 del 1-293 were constructed andmaintained as described in Materials and methods. Protein extracts from the respective cells were obtained by using the luciferaseassay extraction procedure (see Materials and methods). Equal amounts of protein were loaded in each well of 10% SDSpolyacrylamide gel, electrophoresed and the proteins transferred to a nitrocellulose membrane. The blot was developed using themonoclonal antibody PAb421. Positions of molecular weight markers are shown. The ®rst four lanes show the Western blotanalysis. Cell extracts from 10(3) cells expressing p53 del 1-293 alone and 10(3) cells expressing p53-281G and p53 del 1-293 wereprepared and immunoprecipitation reactions were performed (see Materials and methods) with DO1 an antibody that binds to theN-terminal region of p53. The immunoprecipitates were then separated on an SDS polyacrylamide gel followed by Western blotanalysis. The blot was developed with PAab421, an antibody that recognizes a motif near the C-terminus and binds to both p53-281G and p53 del 1-293. Antibody DO1 was run and analysed by Western blot to depict the positions of light and heavy chains ofIgG. The last ®ve lanes show this Western analysis
Mutant p53-mediated transactivationD Deb et al
182
Oncogene

in Materials and methods to assay for cell prolifera-tion. We used 10(3) cells stably transfected with theexpression vector (pCMVBamneo) alone or the expres-sion plasmid for p53-281G as starting materials andtransfected them with the following expression plas-mids: (1) pCMVBam and pHyg, (2) pCMVBam p53(wild-type) and pHyg, (3) pCMVBam FLAG p73 aand pHyg, (4) pCMVBam p53 del 1-293 and pHyg.The data given in Table 2 indicate p53-281G expressingcells have a small but discernable growth advantageover vector-transfected cells. We can see that thesystems with wild-type p53 and p73 a had substantiale�ects on the number of colonies formed on p53-281G-expressing cells compared to vector-transfected cells.p53 del 1-293 had little e�ect, if any (compared tovector alone), on the growth properties of p53-281G-expressing cells. Thus, once again we ®nd that del 1-293 could not signi®cantly change mutant p53's growthenhancing function, suggesting that hetero-oligomer-ization may not be enough to inhibit `gain of function'.Earlier, we showed that a deletion of the C-terminalsequences of p53-281G overlapping the oligomerizationdomain disrupts `gain of function', and here wedemonstrate that hetero-oligomerization disables trans-activation by p53-281G without eliminating `gain offunction' (tumorigenicity). Taken together, this infor-mation suggests that the C-terminal sequences have aseparate and critical role in `gain of function' inaddition to that in transactivation.
Discussion
Since the C-terminal region of wild-type p53 harborsthe oligomerization domain, and the oligomerizationdomain is necessary for the biological functions ofwild-type p53 (Attardi et al., 1996a; Deb et al., 1999;Ishioka et al., 1997; Ozbun and Butel, 1995, Pietenpolet al., 1994; Reed et al., 1993; Sang et al., 1994; Subleret al., 1994; Waterman et al., 1996), it was hypothe-sized that the C-terminal sequences for mutant p53would be required for oligomerization, and theoligomerization may be critical for `gain of function'(Lanyi et al., 1998). We have tested this hypothesisusing di�erent approaches. An N-terminal deletionmutant of wild-type p53 that deletes amino acids 1through 293 has been used as a tool to perform hetero-oligomerization studies. This mutant retains the entireoligomerization domain but dispenses o� the transacti-vation domain and a large portion of the sequence-speci®c DNA-binding domain inactivating the transac-tivation and DNA-binding activities of wild-type p53(Subler et al., 1994). By co-transfection experimentsfollowed by immunoprecipitation analysis we showthat p53 del 1-293 forms hetero-oligomeric complexeswith p53-281G (Figure 4). p53 del 1-293 inhibits p53-281G-mediated transactivation of the EGFR andMDR-1 promoters (Figure 3) suggesting that hetero-oligomerization inactivates transcriptional functions ofmutant p53. We have also determined that co-expression of p53 del 1-293 and p53-281G reducedtransactivation potential of p53-281G in stably trans-fected 10(3) murine cells. Although 10(3) cells stablytransfected with p53-281G express 5 ± 6-fold morePCNA and NF-kB2 mRNA compared to the cellstransfected with vector alone, co-expression of p53 del1-293 reduced this transactivation considerably (Figure5). Our data, therefore, support the idea that properoligomeric forms of mutant p53 are required for itstransactivation function.
We have used another approach to investigate theimportance of oligomerization in mutant p53-mediatedtransactivation, and swapped the tetramerization do-main of p53-281G with a coiled-coil dimerizationdomain from the yeast GCN4 protein (Pietenpol etal., 1994) or a modi®ed coiled-coil domain that acts as atetramerization motif (Waterman et al., 1996). Tran-scriptional data generated with these heterologous
Table 2 Colony formation in 10(3) cells stably transfected with vector (pCMVBamNeo) or p53-281G after transfectionwith di�erent expression plasmids
Number of colonies (1 mm or more)Cells stably transfected with Expression plasmids used for transfection formed after 3 weeks of selection
Vector alone, clone 4 pHyg and pCMVBam 0,0,2; avg. 0.67p53-281G, clone 4 pHyg and pCMVBam 137, 117, 132; avg. 129Vector alone, clone 4 pHyg and pCMVBamp53 wt 4,0,0; avg. 1.33p53-281G, clone 4 pHyg and pCMVBamp53 wt 4,21,5; avg. 10Vector alone, clone 4 pHyg and pCMVBamp73a 3,8,38; avg. 16.33p53-281G, clone 4 pHyg and pCMVBamp73a 0,8,18; avg. 8.67Vector alone, clone 4 pHyg and pCMVBamp53 del 1-293 1,4,0; avg. 1.67p53-281G, clone 4 pHyg and pCMVBamp53 del 1-293 113,125,107; avg. 115
Table 1 Tumorigenicity of 10(3) cells expressing p53 derivatives.E�ect of overexpression of p53 derivatives on the tumorigenic
potential of p53 null 10(3) murine ®broblast cells
Number of tumorsNumber of mice/ generated/
Cells stably number of sites number of sitestransfected with injected injected
Vector alone, clone 2 2/4 0/4Vector alone, clone 21 2/4 0/4p53 del 1-293, clone 17 2/4 0/4p53 del 1-293, clone 19 3/6 0/6p53-281G, clone 13 3/6 6/6p53-281G, clone 4 2/4 4/4p53-281G, clone 4, p53 del 3/6 6/6
1-293 clone 22p53-281G, clone 4, p53 del 3/6 6/6
1-293, clone 10
Oncogene
Mutant p53-mediated transactivationD Deb et al
183

constructs showed that mutant p53-281G requires theC-terminally located oligomerization/non-sequence-spe-ci®c nucleic acid-binding domain for its transactivationproperties (Figure 1). This requirement is sequence-speci®c and not met by merely allowing the protein tooligomerize, as we demonstrated also that p53-281Gwith its tetramerization domain replaced by a hetero-logous tetramerization domain preserves the tetramericstructure of the protein (see Figure 2), but disrupts thetransactivation function (Figure 1). However, replace-ment of the tetramerization domain of wild-type p53 bythe dimerization domain, consisting of the coiled-coilstructure from the yeast GCN4 protein (p53 343cc),could e�ectively sustain wild-type p53's transactivationand growth suppression functions (data not shown)(Attardi et al., 1996a; Ishioka et al., 1997; Pietenpol etal., 1994; Reed et al., 1993; Waterman et al., 1996).Interestingly, in the context of wild-type p53 thereplacement of its tetramerization domain by aderivative of the coiled-coil structure that results in atetramerization domain could not activate transcriptionfrom the human p21 promoter as well as p53 343cc(data not shown). The reason for this apparentdiscrepancy is not known at the present time, but couldbe related to the DNA concentrations used in thedi�erent transfections (Subler et al., 1994). Also, thewild-type p53 derivatives with heterologous dimeriza-tion/tetramerization domain inhibited the CMV im-mediate-early promoter signi®cantly; the extent ofinhibition is comparable to that mediated by wild-typep53 (data not shown). Thus, the functions of wild-typep53 are preserved when a heterologous oligomerizationdomain replaces the oligomerization domain of p53 asshown earlier (Attardi et al., 1996a; Ishioka et al., 1997;Pietenpol et al., 1994; Reed et al., 1993; Waterman etal., 1996). Taken together, these data strongly suggestthat the C-terminal region of wild-type p53 mainlyfunctions in oligomerization in transient-transfectiontranscription assays, whereas mutant p53 requires anadditional function. Thus, there is a clear distinction inthe requirement of the C-terminal domain for wild-typeand mutant p53-mediated transcriptional functions.
For mutant p53-mediated transactivation, sequencerequirements at the C-terminus are stringent, suggestingthat the region is needed perhaps for sequence-speci®cfunctions in addition to oligomerization. A number ofpossible explanations for this requirement can be o�ered:(1) p53 has a nucleic acid binding activity associated withits C-terminal region (Ko and Prives, 1996; Reed et al.,1995). It is possible that under appropriate conditionsmutant p53 utilizes this activity to interact with thepromoter to activate transcription. (2) A number of post-translational modi®cations, e.g. phosphorylation andacetylation (Prives, 1998; Prives and Hall, 1999;Vogelstein et al., 2000) of wild-type p53 have beenreported to occur in speci®c sites located near the C-terminus. Perhaps mutant p53 requires these modi®ca-tions for its transactivation function. (3) The C-terminalregion could be a site for protein ± protein interaction[e.g., interaction needed with c-abl (Yuan et al., 1996)]needed for mutant p53-mediated transactivation; speci®c
interactions may occur between a transcription factorand the tetrameric C-terminal region of mutant p53. It ispossible that overexpression of p53 del 1-293 would alsocompete for such modi®cations or functions with theintact protein causing an inhibition of transactivation.Analyses involving more speci®c mutations near the C-terminus along with functional assays are required beforewe can get a comprehensive idea about the function ofthe C-terminus in mutant p53-mediated transactivation.
We also investigated whether a homo-tetramericstructure of the mutant p53 protein is necessary for its`gain of function' by studying the in¯uence of hetero-oligomerization on the `gain of function' as determinedby tumorigenicity and colony forming ability of cells inwhich the mutant is expressed. We found that co-expression of p53 del 1-293 and p53-281G causedinhibition of transactivation of p53-281G, and earlier,we and others have shown that transactivation is directlyrelated to `gain of function' (Lanyi et al., 1998; Lin et al.,1995). Therefore, a likely scenario would be that co-expression of p53 del 1-293 would hinder p53-281G's`gain of function' property as determined by thetumorigenicity assay. Our results shown in Table 1,however, indicate that co-expression of p53 del 1-293does not inhibit tumorigenicity of 10(3) cells expressingp53-281G. Results from our co-immunoprecipitationexperiments (Figure 6) demonstrate that p53-281Gforms hetero-oligomers with p53 del 1-293 in 10(3) cellsstably expressing both proteins. This observation arguesthat hetero-oligomerization although disrupts transacti-vation capacity of mutant p53 cannot eliminate its `gainof function' (tumorigenicity) property noticeably. Thecolony formation assay also indicates that p53 del 1-293had no signi®cant impact on growth of p53-281Gexpressing cells; it behaved very similarly to the vectorcontrol. In these assays, however, wild-type p53 and p73a reduced colony numbers signi®cantly for cells expres-sing p53-281G, showing that the growth advantageinduced by p53-281G was countered better by the growthsuppressors. Thus, hetero-oligomerization between p53-281G and p53 del 1-293 may not inhibit p53-281G'sgrowth enhancing properties, although it does inhibit itstransactivation property. This observation goes againstthe notion that the transactivation property of thetumor-derived p53 mutants may be responsible for the`gain of function'. We must add though that before wedraw such a conclusion it must be proven that all themutant p53 protein molecules are complexed with p53del 1-293, because it is possible that any free mutant p53molecule could induce the growth enhancement.Although we have shown that p53 del 1-293 and p53-281G forms hetero-oligomeric complexes in 10(3) cellsexpressing the two proteins, it is possible that some p53-281G molecules are still free. Because of extendednumber of passages that the cells had to pass through,it is also possible that the 10(3) cells expressing p53-281Gbecame spontaneously transformed independent of theinvolvement of p53-281G; in that case neutralization ofmutant p53 would not reduce its oncogenicity. 10(3) cellsindeed have a tendency to become spontaneouslytumorigenic (data not shown).
Mutant p53-mediated transactivationD Deb et al
184
Oncogene

Interestingly, expression of `gain of function' mutantsof p53 has also been shown to enhance drug resistance,interfere with p53-independent apoptosis, and inducecentrosome abnormalities in cells expressing them(Blandino et al., 1999; Li et al., 1998; Murphy et al.,2000; Sigal and Rotter, 2000; Wang et al., 1998). It is notknown whether mutant p53-mediated transactivation isinvolved as the etiologic agent for these phenotypes.Gualberto et al. (1998) reported that some mutant p53confer a dominant `gain of function' phenotype thatdisrupts the spindle checkpoint control. They alsoshowed that for that function the mutant protein doesnot require transactivation. Thus, it is possible that some`gain of function' is achievable even in the absence of thetransactivation function of mutant p53.
How mutant p53 induces `gain of function' is still anopen question. It is possible that the newly found familymembers of p53, such as p73 and p63, may act assurrogate p53 in p53's absence and all p53 mutants do isto neutralize their activity, perhaps by hetero-oligomer-ization, and some such evidence have been presented (DiComo et al., 1999; Gaiddon et al., 2001; Marin et al.,2000; Strano et al., 2000). It has also been shown that forits neutralizing and hetero-oligomerization activitya�ecting p73 or p63 the mutant p53 protein requiresonly its central `DNA-binding domain'. However, earlierwork (Lanyi et al., 1998; Lin et al., 1995) indicate that thetransactivation domain and C-terminally located regionoverlapping the oligomerization and non-sequence-speci®c nucleic acid-binding domains are required for`gain of function'. This shows that sequences outside theregion needed for neutralizing p73 are needed for `gain offunction'. Therefore, the `gain of function' cannot befully explained by p73-mutant p53 interactions. Also,our attempts to identify endogenous p73-p53-281Gcomplexes in 10(3) cells expressing p53-281G failed sofar (data not shown). This, however, suggests that even ifp73-p53-281G complexes are formed, they are low innumber. The work presented here shows that hetero-oligomerization inhibited but not obliterated transacti-vation, and Dittmer et al. (1993) reported that the tumor-derived p53 mutants that di�er in transactivation did notdi�er in their tumorigenicity suggesting that any remnanttransactivation might be enough to induce `gain offunction'. Thus, a small fraction of free mutant p53 mayinduce oncogenic transformation by transactivatingexpression of critical growth-related genes. At this point,it is not clear whether mutant p53-mediated transactiva-tion is the cause of `gain of function'. Thus, it is necessaryto identify target genes that are a�ected by `gain offunction' directly, and to determine whether transactiva-tion of these genes lead to increased oncogenesis (`gain offunction').
Materials and methods
DNA
The p53 and p73 expression plasmids contained the humanp53 and p73 cDNAs under the regulation of the CMV
immediate-early promoter in the pCMVBam expressionvector (Hinds et al., 1990; Subler et al., 1992). The N-terminal deletion derivative p53 del 1-293 was generated inthe context of the human wild-type p53 cDNA as describedearlier (Subler et al., 1994). p73 a and p73 b cDNAs havebeen cloned in such a way that they have a FLAG epitope atthe N-terminus when converted into the respective proteins(Deb et al., 2001).p53 mutants, p53 343cc and p53 TZ334NR, have been
described earlier (Pietenpol et al., 1994; Waterman et al.,1996). p53 343cc had sequences from amino acids 344 to 393replaced with the coiled-coil structural domain from theyeast GCN4 protein, while p53 TZ334NR had sequencesfrom amino acids 334 to 393 replaced by a modi®ed coiled-coil structure de®ning a tetramerization domain. p53-281G343cc and p53-281G TZ334NR contained the amino acidsubstitution at the position 281 from Arg to Gly; theseclones were constructed by standard sub-cloning techniquesby replacing a restriction fragment of the wild-type p53cDNA overlapping the sequence coding for amino acid 281with the corresponding fragment from p53-281G cDNA. Allthe coding sequences were cloned in the pCMVBam vectorin the correct orientation downstream of the CMVimmediate-early promoter. pHyg (Invitrogen) contained thehygromycin resistance gene cloned under the herpes simplexvirus thymidine kinase promoter (Clontech).The chloramphenicol acetyltransferase (CAT) plasmids
utilized the Escherichia coli CAT gene under the transcrip-tional control of the MDR-1 (Lanyi et al., 1998) promoter.The luciferase plasmids used the ®re¯y luciferase (luc) geneunder the control of the human EGFR and p21 promoters(Deb et al., 2001; el-Deiry et al., 1993).
Cell culture and transfection
Human osteosarcoma (Saos-2) cells were cultured andtransfected by the calcium phosphate-DNA co-precipitationmethod, as described previously (Subler et al., 1992). In atypical experiment 36106 cells in a 10 cm dish weretransfected with indicated amounts of plasmids. Sometransfections were done using Lipofectamine 2000 (LifeTechnologies) using 24-well plates following manufacturer'sprotocol. For 24-well plates each well was transfected with1 mg total DNA. In general, the promoter luciferase constructand the e�ector plasmid are added at 170 ng per well, andthe rest of the 1 mg DNA came from pUC 19 DNA.
Chloramphenicol acetyltransferase assay
Cells were harvested 36 ± 40 h post-transfection and lysed bythree successive cycles of freezing and thawing. Extracts werenormalized for protein concentration and assayed for CATenzyme activity as described earlier (Ludes-Meyers et al.,1996). Multiple independent experiments (three or more) weredone to determine the standard deviations. CAT activity wasdetected by thin-layer chromatographic separation of [14C]-chloramphenicol from its acetylated derivatives followed byautoradiography. Quantitation was done using a Phosphor-imager (Molecular Dynamics).
Luciferase assay
Luciferase assays were carried out using a commercial kitfrom Promega as suggested by the vendor, and luminescencewas measured in a Luminometer made by Turner Designs.Cells were harvested 36 ± 40 h after transfection, and extractswere prepared as suggested. As described for the CAT assay,
Oncogene
Mutant p53-mediated transactivationD Deb et al
185

equal amounts of protein from each extract were used for theluciferase assay. Relative luciferase activity was plotted onthe Y-axis and the value of the vector control was set to100%.
In vivo labeling of cellular proteins and immunoprecipitation
Saos-2 cells were transfected with p53 expression plasmids (orexpression vector alone) as described in the text. Twenty-fourhour post-transfection labeling of cellular proteins was carriedout with 35S-methionine for 2 h, after which cells wereharvested and cell extracts prepared as described earlier(Subler et al., 1992). Immunoprecipitations were carried outfor 14 h at 48C using indicated monoclonal antibodies asdescribed earlier (Subler et al., 1992). PAb421 (Lane andCrawford, 1979; Vojtesek et al., 1992) was used to bind p53proteins with an intact C-terminus (intact p53-281G as well asp53 del 1-293) and DO1 (Lane and Crawford, 1979; Vojteseket al., 1992) was used to detect p53 proteins with an intactN-terminus (p53-281G but not p53 del 1-293).
Establishment of stable 10(3) cell lines expressing p53 del 1-293(or p53-281G and p53 del 1-293)
Untransfected 10(3) cells (Dittmer et al., 1993) or 10(3) cellsstably expressing p53-281G (Lanyi et al., 1998) weretransfected with pCMVBam p53 del 1-293 and pCMVBamneo (Hinds et al., 1990) or pCMVBam p53 del 1-293 andpHyg (Clontech), respectively, using the calcium phosphateprecipitation technique (Subler et al., 1992). Forty-eighthours after transfection, cells were washed, trypsinized,subcultured at 1 : 4, and plated into selective mediasupplemented with 200 mg/ml G-418 (active concentration)for previously untransfected 10(3) cells and 200 mg/ml G-418and 200 mg/ml hygromycin for p53-281G-expressing cells.Cells were maintained in the selective media with twiceweekly changes of media. After 14 ± 21 days in selectivemedia, cells from individual antibiotic-resistant colonies wereisolated and expanded into cell lines. Western blot analyseswere used to detect p53 del 1-293 or p53 del 1-293 and p53-281G expressed in these cell lines.
Immunoprecipitation followed by Western blot analysis todetermine in vivo complex formation between p53-281Gand p53 del 1-293
In vivo complex formation between p53-281G and p53 del 1-293 in 10(3) cells stably transfected to express p53-281G andp53 del 1-293 was done by immunoprecipitation of p53-281Gwith an antibody (DO1) that recognizes a motif on the N-terminal part of the protein and is absent in p53 del 1-293.We carried out the procedure following the method describedby Leng et al. (1995). Brie¯y, cells from a 10 cm dish wereharvested in 4 ml of 10 s bu�er (containing 250 mM HEPES,pH 7.2, 1.5% NP40, 0.5% Triton X-100, 0.025% SDS,50 mM sodium phosphate bu�er, pH 7.0, 5 mM NaF, 0.5 mM
phenyl methyl sulphonyl ¯uoride and 2.5 mM dithiothreitol)as described. One ml of the extract was then incubated with 1or 2 mg of p53 antibody DO1 and 16 ml of protein A agarose(Calbiochem) and shaken overnight (head-to-tail) at 48C. Thefollowing morning, the immunoprecipitate was washed threetimes with 10 s bu�er and once with a bu�er containing10 mM Tris pH 7.5, 50 mM NaCl, 0.5% sodium deoxycholateand 0.5% Triton X-100. The pellet was then suspended in20 ml of 26 Laemmlli loading bu�er, boiled and analysed by15% SDS polyacrylamide gel electrophoresis, followed byWestern blot analysis using PAb421 as the probe.
Tumorigenicity assay
The tumorigenic potential of 10(3) cells expressing p53derivatives was tested using nude mice. Cells (56106 per site)were subcutaneously injected into ¯anks of athymic nude mice(National Cancer Institute) and tumor development wasmonitored as described (Dittmer et al., 1993; Lanyi et al., 1998).
Colony formation assay
10(3) cells stably transfected with the vector pCMVBam Neo(Hinds et al., 1990) or stably expressing p53-281G (Lanyi etal., 1998) in each 10 cm dish were transfected with 1 mg ofpHyg and 5 mg (1) pCMVBam (Subler et al., 1992), (2)pCMVBam wild-type p53 (Subler et al., 1992), (3)pCMVBam FLAG p73 a (Deb et al., 2001), (4) pCMVBamFLAG p73 b Deb (Deb et al., 2001), or (5) pCMVBam p53del 1-293 (Subler et al., 1994). Transfections were done bycalcium phosphate precipitation technique as described earlier(Lanyi et al., 1998). After 48 h of transfection cells weresubcultured 1 : 4; hygromycin was added next day at 200 mg/ml to start the selection process. Neomycin (G418 at 200 mg/ml) was also added to those cells selected earlier for neomycinresistance as described before (Lanyi et al., 1998). Every 2 ± 3days media was changed. After 3 weeks, cells were ®xed withmethanol and stained with methylene blue.
Western blot analysis
Western blot analysis was carried out after SDS polyacryla-mide gel electrophoresis using monoclonal antibodies againstp53 or FLAG and a commercial kit (Promega) for labelingwith an alkaline phosphatase-conjugated secondary antibody.Manufacturer's protocol was followed for this procedure.
Oligomerization assays with in vitro-synthesized intactp53-281G and its derivatives
Analyses for the presence of oligomeric forms of p53-281Gand its derivatives were carried out as described (Subler et al.,1994). Brie¯y, human p53-281G and its derivatives weregenerated by a coupled in vivo transcription-translation system(Promega) and were incubated with glutaraldehyde at varying®nal concentrations at 378C for 15 min. After cross-linking,proteins were boiled in 26 Laemmli loading dye (Subler et al.,1994), subjected to gradient (4 ± 20%) SDS-polyacrylamide gelelectrophoresis, and visualized by autoradiography.
RNA isolation and microarray hybridization analysis
Total RNA was isolated from 10(3) cells stably transfectedwith vector alone [pCMVBamNeo (Lanyi et al., 1998)],pCMVBamNeo and expression plasmid for p53-281G,pCMVBamNeo and expression plasmid for p53 del 1-293,and pCMVBamNeo and expression plasmids for p53-2981Gand p53 del 1-293 using Trizol (Gibco) reagent followingmanufacturer's protocol. Quality of the RNAs was checkedby 1.2% agarose Tris-borate-EDTA gel electrophoresis.Microarray analysis from RNA from 10(3) cells stably
transfected with (1) vector alone [pCMVBamNeo (Lanyi etal., 1998)] and (2) pCMVBamNeo and expression plasmidfor p53-281G was done by Incyte Genomics, Inc. (PaloAlto, CA, USA) starting from the whole RNA that wesent. Brie¯y, polyadenylated RNA was isolated with theOligoTex (Qiagen) RNA kit. Polyadenylated RNA wasreverse transcribed to generate Cy3- and Cy5-labeled cDNAprobes, respectively. cDNA probes were competitively
Mutant p53-mediated transactivationD Deb et al
186
Oncogene

hybridized to a mouse Unigene 1 cDNA microarray (IncyteGenomics, Inc.) containing immobilized cDNA fragments(average cDNA length, 500 ± 5000 base pairs). Cy3 and Cy5¯uorescence were imaged individually, and the normalizedratios of Cy3/Cy5 ¯uorescence at a given spot on themicroarray were used to calculate di�erential gene expres-sion.
Quantitative real-time PCR
Relative quantitation of gene expression was performed usingquantitative real-time PCR (QPCR). The relative amount ofeach sample was calculated from a standard curve afteremploying the Fit Points algorithm for quanti®cation. Thisexpression value was normalized to a housekeeping gene,glyceraldehyde 3-phosphate dehydrogenase (GAPDH)(Krause et al., 2000). A relative gene expression wasdetermined by assigning 10(3) V4 [10(3) cells stablytransfected with the vector pCMVBam Neo (Hinds et al.,1990)] a relative value of 1.0, with all other values relative to10(3)V4. This method is described in detail by Johnson et al.(2000); Tanguay et al. (2000). Following is a brief descriptionof the entire process.
(1) cDNA synthesis Total RNA was isolated from 56106
mouse 10(3) cells stably transfected to express p53 281G,clone 4, p53D1.291, clone 10, p53 281GD1-293, clone 22, andvector (V4) alone using Trizol reagent (Gibco BRL,Gaithersburg, MD, USA) following the manufacturer'sinstructions. Ten micrograms of total RNA was treated withRA1 Rnase-free DNase (Promega, Madison, WI, USA),followed by phenol extraction, chloroform extraction andethanol precipitation. cDNA was synthesized with randomhexamer using the Thermoscript RT±PCR System (GibcoBRL) according to the manufacturer's protocol. cDNAvolume was increased twofold to decrease pipetting error.
(2) Primers Primers were designed using Oligo 5.0 Software(National Biosciences, Inc, Plymouth, MN, USA) and weresynthesized by Integrated DNA Technologies. Primeroptimization was performed to determine the optimalconcentration of MgCl2 and annealing temperature for eachprimer set.
(3) Relative standard curves Relative standard curves wereconstructed for each target gene using previously generated
PCR products of the target. Serial dilutions were made, andwere given arbitrary values corresponding to the dilutions.
(4) Quantitative real-time PCR Quantitative real-time PCRwas performed using a LightCycler (Roche). Each reaction(20 ml) contained 2 ml of the respective diluted cDNA,primers (0.5 mM), and 4 mM MgCl2, and FastStart ReactionMix SYBR Green I, (Roche) which includes FastStart TaqDNA polymerase, reaction bu�er, dNTPs, and SYBR GreenI dye. A negative control was included in each QPCR run.
Except for the composition of the standard curves, eachreaction was performed in triplicate. All samples weretreated with an initial denaturation step at 958C for10 min to activate the enzyme and denature the cDNA.Samples investigated with GAPD primers (F) 5'-CCAGCCTCGTCCCGTAGACA-3' and (R) 5'-GCCTCACCCCATTTGATGTTAGTG-3' underwent 40cycles of the following: 958C for 15 s, 568C for 5 s, and708C for 12 s. Samples checked with PCNA primers (F)5'-GATAAAGAAGAGGAGGCGGTAA-3' and (R)5'-ACACGCTGGCATCTCAGGAGCA-3' underwent 45cycles of 958C for 15 s, 518C for 5 s, 728C for 12 s. NF-kB2 samples (F) 5'-TGGCCCCTATCTGGTGATTGT-3'and (R) 5'-ACACTCCCAACTCTGAACACTGCT-3' under-went 50 cycles of 958C for 15 s, 568C for 5 s, and 728C for10 s. The presence of SYBR Green I, a non-speci®c dyewhich binds to double-stranded DNA, can be detected by¯uorescence at the end of each extension step. In this waythe synthesis of DNA is determined in real-time. Agarose gelelectrophoresis and melting curve analysis were used todemonstrate the presence of a speci®c product.
AcknowledgmentsThis work was supported by grants from NIH to SumitraDeb (CA70712) and to Swati Palit Deb (CA74172). Part ofthe work for this manuscript was done in the Departmentof Microbiology, University of Texas Health ScienceCenter at San Antonio, San Antonio, Texas and Depart-ment of Cancer Biology, Wake Forest University School ofMedicine, Winston-Salem, North Carolina. We thankArnold Levine, Bert Vogelstein, Glenn Merlino, andThanos Halazonetis for providing us with plasmids andcells.
References
Abarzua P, LoSardo JE, Gubler ML and Neri A. (1995).Cancer Res., 55, 3490 ± 3494.
Attardi LD, Lowe SW, Brugarolas J and Jacks T. (1996a).EMBO J., 15, 3693 ± 3701.
Attardi LD, Lowe SW, Brugarolas J and Jacks T. (1996b).EMBO J., 15, 3702 ± 3712.
Bakalkin G, Yakovleva T, Selivanova G, Magnusson KP,Szekely L, Kiseleva E, Klein G, Terenius L and WimanKG. (1994). Proc. Natl. Acad. Sci. USA, 91, 413 ± 417.
Baudier J, Delphin C, Grunwald D, Khochbin S andLawrence JJ. (1992). Proc. Natl. Acad. Sci. USA, 89,11627 ± 11631.
Blandino G, Levine AJ and Oren M. (1999). Oncogene, 18,477 ± 485.
Brain R and Jenkins JR. (1994). Oncogene, 9, 1775 ± 1780.Candau R, Scolnick DM, Darpino P, Ying CY, Halazonetis
TD and Berger SL. (1997). Oncogene, 15, 807 ± 816.
Chin KV, Ueda K, Pastan I and Gottesman MM. (1992).Science, 255, 459 ± 462.
Deb D, Chakraborti AS, Lanyi A, Troyer DA and Deb S.(1999). Int. J. Oncol., 15, 413 ± 422.
Deb D, Lanyi A, Scian M, Keiger J, Brown DR, Le Roith D,Deb SP and Deb S. (2001). Int. J. Oncol., 18, 401 ± 409.
Deb S, Jackson CT, Subler MA and Martin DW. (1992). J.Virol., 66, 6164 ± 6170.
Di Como CJ, Gaiddon C and Prives C. (1999). Mol. Cell.Biol., 19, 1438 ± 1449.
Dittmer D, Pati S, Zambetti G, Chu S, Teresky AK, MooreM, Finlay C and Levine AJ. (1993). Nat. Genet., 4, 42 ± 46.
Donehower LA and Bradley A. (1993). Biochim. Biophys.Acta., 1155, 181 ± 205.
el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R,Trent JM, Lin D, Mercer WE, Kinzler KW and VogelsteinB. (1993). Cell, 75, 817 ± 825.
Oncogene
Mutant p53-mediated transactivationD Deb et al
187

Fourie AM, Hupp TR, Lane DP, Sang BC, Barbosa MS,Sambrook JF and Gething MJ. (1997). J. Biol. Chem., 272,19471 ± 19479.
Frazier MW, He X, Wang J, Gu Z, Cleveland JL andZambetti GP. (1998). Mol. Cell. Biol., 18, 3735 ± 3743.
Gaiddon C, Lokshin M, Ahn J, Zhang T and Prives C.(2001). Mol. Cell. Biol., 21, 1874 ± 1887.
Ginsberg D, Mechta F, Yaniv M and Oren M. (1991). Proc.Natl. Acad. Sci. USA, 88, 9979 ± 9983.
Greenblatt MS, Bennett WP, Hollstein M and Harris CC.(1994). Cancer Res., 54, 4855 ± 4878.
Gualberto A, Aldape K, Kozakiewicz K and Tlsty TD.(1998). Proc. Natl. Acad. Sci. USA, 95, 5166 ± 5171.
Halazonetis TD, Davis LJ and Kandil AN. (1993). EMBO J.,12, 1021 ± 1028.
Hansen S, Hupp TR and Lane DP. (1996). J. Biol. Chem.,271, 3917 ± 3924.
Haupt Y, Maya R, Kazaz A and Oren M. (1997). Nature,387, 296 ± 299.
Hinds PW, Finlay CA, Quatin RS, Baker SJ, Fearon ER,Vogelstein B and Levine AJ. (1990). Cell Growth Di�er., 1,571 ± 580.
Hupp TR, Meek DW, Midgley CA and Lane DP. (1992).Cell, 71, 875 ± 886.
Ishioka C, Shimodaira H, Englert C, Shimada A, Osada M,Jia LQ, Suzuki T, Gamo M and Kanamaru R. (1997).Biochem. Biophys. Res. Commun., 232, 54 ± 60.
Ishizaka Y, Chernov MV, Burns CM and Stark GR. (1995).Proc. Natl. Acad. Sci. USA, 92, 3224 ± 3228.
Johnson MR, Wang K, Smith JB, Heslin MJ and Diasio RB.(2000). Analyt. Biochem., 278, 175 ± 184.
Kelavkar UP and Badr KF. (1999). Proc. Natl. Acad. Sci.USA, 96, 4378 ± 4383.
Kieser A, Weich HA, Brandner G, Marme D and Kolch W.(1994). Oncogene, 9, 963 ± 969.
Ko LJ and Prives C. (1996). Genes Dev., 10, 1054 ± 1072.Krause K, Wasner M, Reinhard W, Haugwitz U, Dohna CL,
Moessner J and Engeland K. (2000). Nucleic Acids Res.,28, 4410 ± 4418.
Kubbutat MH, Jones SN and Vousden KH. (1997). Nature,387, 299 ± 303.
Lane DP. (1994). Br. Med. Bull., 50, 582 ± 599.Lane DP and Crawford LV. (1979). Nature, 278, 261 ± 263.Lanyi A, Deb D, Seymour RC, Ludes-Meyers JH, Subler
MA and Deb S. (1998). Oncogene, 16, 3169 ± 3176.Lechner MS, Mack D, Finicle AB, Crook T, Vousden KH
and Laimins LA. (1992). EMBO J., 11, 3045 ± 3052.Lee KC, Crowe AJ and Barton MC. (1999). Mol. Cell. Biol.,
19, 1279 ± 1288.Lee S, Cavallo L and Gri�th J. (1997). J. Biol. Chem., 272,
7532 ± 7539.Lee S, Elenbaas B, Levine A and Gri�th J. (1995). Cell, 81,
1013 ± 1020.Leng P, Brown DR, Shivakumar CV, Deb S and Deb SP.
(1995). Oncogene, 10, 1275 ± 1282.Levine AJ. (1993). Annu. Rev. Biochem., 62, 623 ± 651.Levine AJ. (1997). Cell, 88, 323 ± 331.Levine AJ, Momand J and Finlay CA. (1991). Nature, 351,
453 ± 456.Li R, Sutphin PD, Schwartz D, Matas D, Almog N,
Wolkowicz R, Gold®nger N, Pei H, Prokocimer M andRotter V. (1998). Oncogene, 16, 3269 ± 3277.
Lin J, Teresky AK and Levine AJ. (1995). Oncogene, 10,2387 ± 2390.
Lowe SW. (1999). Endocr. Relat. Cancer, 6, 45 ± 48.
Ludes-Meyers JH, Subler MA, Shivakumar CV, MunozRM, Jiang P, Bigger JE, Brown DR, Deb SP and Deb S.(1996). Mol. Cell. Biol., 16, 6009 ± 6019.
Margulies L and Sehgal PB. (1993). J. Biol. Chem., 268,15096 ± 15100.
Marin MC, Jost CA, Brooks LA, Irwin MS, O'Nions J, TidyJA, James N, McGregor JM, Harwood CA, Yulug IG,Vousden KH, Allday MJ, Gusterson B, Ikawa S, HindsPW, Crook T and Kaelin WG. (2000). Nat. Genet., 25,47 ± 54.
Murphy KL, Dennis AP and Rosen JM. (2000). FASEB J.,14, 2291 ± 2302.
Murphy M, Ahn J, Walker KK, Ho�man WH, Evans RM,Levine AJ and George D. (1999). Genes Dev., 13, 2490 ±2501.
Murphy M, Hinman A and Levine AJ. (1996). Genes Dev.,10, 2971 ± 2980.
Oren M. (1999). J. Biol. Chem., 274, 36031 ± 36034.Ozbun MA and Butel JS. (1995). Adv. Cancer Res., 66, 71 ±
141.Pietenpol JA, Tokino T, Thiagalingam S, el-Deiry WS,
Kinzler KW and Vogelstein B. (1994). Proc. Natl. Acad.Sci. USA, 91, 1998 ± 2002.
Preuss U, Kreutzfeld R and Scheidtmann KH. (2000). Int. J.Cancer, 88, 162 ± 171.
Prives C. (1994). Cell, 78, 543 ± 546.Prives C. (1998). Cell, 95, 5 ± 8.Prives C and Hall PA. (1999). J. Pathol., 187, 112 ± 126.Reed M, Wang Y, Mayr G, Anderson ME, Schwedes JF and
Tegtmeyer P. (1993). Gene Expr., 3, 95 ± 107.Reed M, Woelker B, Wang P, Wang Y, Anderson ME and
Tegtmeyer P. (1995). Proc. Natl. Acad. Sci. USA, 92,9455 ± 9459.
Sakamuro D, Sabbatini P, White E and Prendergast GC.(1997). Oncogene, 15, 887 ± 898.
Sang BC, Chen JY, Minna J and Barbosa MS. (1994).Oncogene, 9, 853 ± 859.
Santhanam U, Ray A and Sehgal PB. (1991). Proc. Natl.Acad. Sci. USA, 88, 7605 ± 7609.
Seto E, Usheva A, Zambetti GP, Momand J, Horikoshi N,Weinmann R, Levine AJ and Shenk T. (1992). Proc. Natl.Acad. Sci. USA, 89, 12028 ± 12032.
Shaulian E, Zauberman A, Ginsberg D and Oren M. (1992).Mol. Cell. Biol., 12, 5581 ± 5592.
Sheikh MS and Fornace AJ. (2000). J. Cell Physiol., 182,171 ± 181.
Sigal A and Rotter V. (2000). Cancer Res., 60, 6788 ± 6793.Soussi T, Dehouche K and Beroud C. (2000). Hum. Mutat.,
15, 105 ± 113.Strano S, Munarriz E, Rossi M, Cristofanelli B, Shaul Y,
Castagnoli L, Levine AJ, Sacchi A, Cesareni G, Oren Mand Blandino G. (2000). J. Biol. Chem., 275, 29503 ±29512.
Sturzbecher HW, Brain R, Addison C, Rudge K, Remm M,Grimaldi M, Keenan E and Jenkins JR. (1992). Onco-gene,7, 1513 ± 1523.
Subler MA, Martin DW and Deb S. (1992). J. Virol., 66,4757 ± 4762.
Subler MA, Martin DW and Deb S. (1994). Oncogene, 9,1351 ± 1359.
Takenaka I, Morin F, Seizinger BR and Kley N. (1995). J.Biol. Chem., 270, 5405 ± 5411.
Tanguay RL, Andreasen E, Heidman W and Peterson RE.(2000). Biochim. Biophys. Acta., 1494, 117 ± 128.
Mutant p53-mediated transactivationD Deb et al
188
Oncogene

Tsutsumi-Ishii Y, Tadokoro K, Hanaoka F and Tsuchida N.(1995). Cell Growth Di�er., 6, 1 ± 8.
Ueba T, Nosaka T, Takahashi JA, Shibata F, FlorkiewiczRZ, Vogelstein B, Oda Y, Kikuchi H and Hatanaka M.(1994). Proc. Natl. Acad. Sci. USA, 91, 9009 ± 9013.
Venot C, Maratrat M, Dureuil C, Conseiller E, Bracco L andDebussche L. (1998). EMBO J., 17, 4668 ± 4679.
Vogelstein B, Lane D and Levine AJ. (2000). Nature, 408,307 ± 310.
Vojtesek B, Bartek J, Midgley CA and Lane DP. (1992). J.Immunol. Methods, 151, 237 ± 244.
Walker KK and Levine AJ. (1996). Proc. Natl. Acad. Sci.USA, 93, 15335 ± 15340.
Wang XJ, Greenhalgh DA, Jiang A, He D, Zhong L,Brinkley BR and Roop DR. (1998). Mol. Carcinog., 23,185 ± 192.
Wang Y, Reed M, Wang P, Stenger JE, Mayr G, AndersonME, Schwedes JF and Tegtmeyer P. (1993). Genes Dev., 7,2575 ± 2586.
Waterman MJ, Stavridi ES, Waterman JL and HalazonetisTD. (1998). Nat. Genet., 19, 175 ± 178.
Waterman MJ, Waterman JL and Halazonetis TD. (1996).Cancer Res., 56, 158 ± 163.
Yang X, Pater A and Tang SC. (1999). Oncogene, 18, 4546 ±4553.
Yuan ZM, Huang Y, FanMM, Sawyers C, Kharbanda S andKufe D. (1996). J. Biol. Chem., 271, 26457 ± 26460.
Zhao R, Gish K, Murphy M, Yin Y, Notterman D, Ho�manWH, Tom E, Mack D and Levine AJ. (2000). Genes Dev.,14, 981 ± 993.
Zhu J, Zhou W, Jiang J and Chen X. (1998). J. Biol. Chem.,273, 13030 ± 13036.
Oncogene
Mutant p53-mediated transactivationD Deb et al
189
Copyright © 2022 FDOKUMEN




















