
Protective effects of glutathione on cisplatin neurotoxicity in rats

Oligomerization and neurotoxicity of the amyloid ADan peptide
implicated in familial Danish dementia
Gillian Gibson,* Nicola Gunasekera,* Maria Lee,* Victor Lelyveld,* Omar M. A. El-Agnaf,*
Andrew Wright� and Brian Austen*
*Neurodegeneration Unit, Department of Basic Medical Sciences, St George’s Hospital Medical School, London, UK
�PP19, MRIC, Northeast Wales Institute, Wrexham, UK
Abstract
Familial Danish dementia (FDD) is a rare neurodegenerative
disorder, which is pathologically characterized by widespread
cerebral amyloid angiopathy, parenchymal protein deposits
and neurofibrillary degeneration. FDD is associated with
mutation in the BRI gene. In FDD a decamer duplication
between codons 265 and 266 in the 3¢ region of the BRI gene
originates an amyloid peptide named ADan, 11 residues longer
than the wild-type peptide produced from the normal BRI gene.
ADan deposits have been found widely distributed in the CNS
of FDD cases. The deposits of ADan are predominantly non-
fibrillar aggregates. We show here that synthetic ADan forms
oligomers in vitro, seen by Tricine–PAGE and gel filtration, and
higher aggregates, which are seen by atomic force spectros-
copy and electron microscopy as carrot-shaped objects that
bunch together. Here we report that oligomeric ADan is toxic to
neuronal cell lines. We find that the soluble non-fibrillar oligo-
meric species of both the reduced and oxidized forms of ADan
are toxic. These results support the idea that the non-fibrillar
soluble aggregates are the pathogenic species, which may
play a central role in the pathogenesis of FDD, and imply that
similar mechanism may also be involved in other neurode-
generative diseases associated with amyloid deposits.
Keywords: ADan, amyloid, BRI, Danish dementia, neuro-
toxicity, oligomers.
J. Neurochem. (2004) 88, 281–290.
Familial British dementia (FBD) and familial Danish
dementia (FDD) are rare autosomal dominant neurodegen-
erative disorders that share features of Alzheimer’s disease
(AD), including a pathology of amyloid plaques surrounded
by astrocytes and microglia, neurofibrillary tangles, neuronal
loss and progressive dementia (Vidal et al. 1999, 2000). FBD
is clinically characterized by the onset of dementia in the
fourth decade, progressive spastic tetraparesis and cerebellar
ataxia. FDD also involves cataract formation and auditory
loss. FBD and FDD are distinguished from AD and other
dementing disorders by plaque deposition in the cerebellum
and the accompanying cerebellar ataxia. These conditions
were previously reported as atypical forms of familial AD
(Worster-Drought et al. 1933; Aikawa et al. 1985; Plant
et al. 1990). Histological studies show that some of the
amyloid deposits in FBD patients exhibit yellow–green
birefringence under polarized light after staining with Congo
red, indicating the presence of amyloid-like fibrils with
crossed b-sheet structure (Plant et al. 1990; Holton et al.
2002). Immunohistochemical and biochemical analysis of
plaques and vascular amyloid of FBD and FDD brains
revealed that �4 kDa peptides named ABri and ADan,
respectively, are the main components of the insoluble
amyloid deposits likely to be involved in pathogenesis (Vidal
et al. 1999, 2000).
The ABri and ADan peptides were determined to be
fragments derived from a larger, membrane-anchored
Received June 10, 2003; revised manuscript received July 29, 2003;
accepted September 9, 2003.
Address correspondence and reprint requests to Brian Austen.
E-mail: [email protected] address for Omar M. A. El-Agnaf is Department of Biological
Sciences, Lancaster University, Lancaster LA1 4YQ, UK.
Abbreviations used: AD, Alzheimer’s disease; ADDL, Ab-deriveddiffusible ligands; CAA, cerebral amyloid angiopathy; DTT, dithio-
threitol; ES-MS, electrospray mass spectroscopy; FBD, familial British
dementia; FDD, familial Danish dementia; HPLC, high-performance
liquid chromatography; LDH, lactate dehydrogenase; MALDI–TOF,
matrix-assisted laser desorption ionization time of flight; MEM, modified
essential medium; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetra-
zolium bromide; oxi-ADan, oxidized form of ADan peptides; PBS,
phosphate-buffered saline; red-ADan, reduced form of ADan peptides;
SDS, sodium dodecyl sulphate; SEC, size exclusion chromatography;
TFA, trifluoroacetic acid; WT, wild-type; zVAD-FMK, benzyloxycar-
bonyl valylalanyl aspartic acid fluoromethyl ketone .
Journal of Neurochemistry, 2004, 88, 281–290 doi:10.1046/j.1471-4159.2003.02134.x
� 2003 International Society for Neurochemistry, J. Neurochem. (2004) 88, 281–290 281

precursor protein termed BRI precursor protein, encoded by
the BRI gene on chromosome 13 (Vidal et al. 1999, 2000).
Wild-type (WT) BRI protein has 266 amino acid residues. A
decamer duplication between codons 265 and 266 in the 3¢region of the BRI gene originates the ADan peptide that is
associated with the pathology of FDD (Vidal et al. 2000;
Fig. 1).
In the mutant form, the resulting reading frame is extended
to 277 amino acid residues, and cleavage by furin releases a
peptide of 34 residues, which is identical to ABri and WT in
its N-terminal 22-residues, but contains a distinct C-terminal
10-residues composed of mainly hydrophobic residues (Vidal
et al. 2000). Staining of post-mortem sections of FDD
patients with an antibody to ADan revealed widespread
cerebral amyloid angiopathy (CAA), and widespread depos-
its of ADan in the leptomeninges, blood vessels and paren-
chyma. Congo red staining of adjacent sections revealed that
the majority of parenchymal lesions were non-fibrillar
(Holton et al. 2002), although deposits elsewhere were bi-
refringent. Previously, we have shown that solutions of the
oxidized form of ABri (oxi-ABri), which contain non-
fibrillar oligomeric species, are toxic to neuronal cells in
culture, whereas solutions of the WT peptide are not toxic to
cells (El-Agnaf et al. 2001a). In this study we investigated
the biophysical and neurotoxic properties of both reduced
(red-ADan) and oxidized forms (oxi-ADan) of ADan pep-
tides. In contrast to ABri, soluble oligomers of red-ADan are
more neurotoxic than those of oxi-ADan.
Materials and methods
Materials
The reduced form of ADan (red-ADan) peptide was synthesized
using the Fmoc/tBu chemistry recently optimized for the synthesis
of amyloid peptides (El-Agnaf et al. 2000). The crude peptide was
purified on a preparative Vydac C4 (250 · 22 mm) 214TP1022column with a gradient of acetonitrile in 0.1% trifluoroacetic acid
(TFA). Purity of red-ADan peptide was judged to be > 95% by
analytical high-performance liquid chromatography (HPLC) on a
column (250 · 4.6 mm) of Phenomenix Jupiter C8, and by
electrospray (ES-MS) and matrix-assisted laser desorption ioniza-
tion time of flight (MALDI-TOF) mass spectrometry. The mass/
charge of red-ADan was found to be 4065, identical to its calculated
mass (Fig. 2). Syntheses of the WT and ABri peptides have been
described previously (El-Agnaf et al. 2001b).
As the sequence of the BRI protein predicts the possibility of an
intramolecular disulphide bond in its C-terminal region, we
produced the cyclic form of ADan (oxi-ADan) by air oxidation.
HPLC-purified red-ADan was dissolved in 0.1 M ammonium
bicarbonate at pH 8.0 and 0.1 mg/mL and stirred for 2 days at
room temperature in contact with air, then lyophilized. The
oxidation was monitored by analytical HPLC from the disappear-
ance of the reduced form and appearance of the faster eluting
oxidized form, and by the loss of 2 mass units seen by MALDI-TOF
mass spectrometry (m/z ¼ 4062.6) (Fig. 2). Tryptic digests were
performed on 0.1 mg of the peptide in 0.1 M ammonium bicarbonate
(pH 8), in 0.2% sodium 3-[2-methyl-2-undecyl-1,3-dioxolan-4-
yl)methoxy]-1-propanesulphonate (Waters, Milford, MA, USA) as
solubilizing agent with 0.005 mg modified trypsin (Promega,
Madison, WI, USA) at 37�C for 12 h, then incubated with 20 mMHCl at 37�C for 1 h to hydrolyse the solubilizing agent, prior to
spotting on a MALDI plate. MALDI analyses were performed after
co-spotting and drying samples with an equal volume of 10 mg/mL
solution of a-cyano-4-hydroxycinnamic acid in 50% (v/v) acetonit-
rile in 0.1% TFA on a Kratos Axima MALDI mass spectrometer.
Calibration was with angiotensin I, angiotensin II and neuropeptide
Y on an adjacent sample spot.
Dimers of oxi-ADan and red-ADan were analysed in solutions in
0.01 M Tris–HCl (pH 7.4) by ES-MS. A sample (20 lL) of oxi-ADan (1 mg/mL) was diluted with 40 lL of methanol and 1 lL ofconcentrated formic acid (to improve solubility of the peptide).
Samples of red-ADan in 0.01M Tris–HCl (pH 7.4) either with
fourfold molar excess of dithiothreitol (DTT) or without added
reducing agent, were aged for 3 days at 37�C, then precipitatedmaterial was dissolved in formic acid (5 lL), methanol (200 lL)and water (100 lL) as above. Samples were centrifuged and used instatic nanospray mode on a Thermo LCQ Deka XP mass
spectrometer.
Preparation of aged solutions of ADan peptides
Solutions of red-ADan were made in sterile 0.1 M Tris–HCl
(pH 7.4) at the required concentrations. oxi-ADan was first
dissolved in sterile water, and then sterile 1 M Tris–HCl (pH 7.4)
was added to give a final 0.1 M concentration of Tris–HCl. Aging
was performed by incubation at 37�C for the number of days
recorded below.
E A SNC F A IR H F E N K F AVE TLIC S R T V KK N II EE N
E A SNC F A IR H F E N K F AVE TLIC F N L F LN S O E K HY
ABri
AD an
W T
Transmembranedomain
N
Extracellular domain100 200 244 AG A 277
C
In AD anDuplicate TTAATTGT
In ABri substituteStop TGA
Fur in
Fig. 1 Schematic presentation of the BRI precursor protein and pep-
tides ABri and ADan released in the brains of patients with FBD and
FDD. In FDD, there is a 10-base duplication occurring between
codons 265 and 266 of the BRI gene, 1 codon before the normal stop
codon, resulting in a frame-shift in the gene and a 277-residue
extended precursor protein. In patients with FBD, there is a single-
base substitution at the stop codon of the BRI gene, which again
generates a 277-residue precursor protein. The arrow indicates where
furin cleavage releases the 34-residue peptides ADan and ABri from
the mutated proteins, or the 23-residue WT-peptide.
282 G. Gibson et al.
� 2003 International Society for Neurochemistry, J. Neurochem. (2004) 88, 281–290

Size exclusion chromatography (SEC) system
At various time points, aliquots (200 lL) of solutions of ADanpeptides incubated in 0.1 M Tris–HCl at 2.5 or 1.0 mg/mL were
loaded onto a Superdex 75 column (10 · 270 mm) equilibrated andeluted with 0.1 M Tris–HCl (pH 7.4) at a flow rate of 0.45 mL/min.
Elution was monitored at 215 nm.
Congo red bi-refringence
Aged 50 lL aliquots of aged ADan solutions were added to 950 lLof Congo red solution [1% w/v solution in phosphate-buffered saline
(PBS) pH 7.4]. This mixture was incubated for 1 h at room
temperature and then centrifuged at 336 000 g in a TL-100
Beckman centrifuge (Beckman, Palo Alto, CA, USA). The resultant
pellet was placed on a slide and examined under polarizing light
using an Nikon Optiphot light microscope under a ·10 lens fittedwith polarizers.
Gel electrophoresis
Samples were electrophoresed on precast 16.5% Tris–Tricine gels
(Bio-Rad Laboratories, Hercules, CA, USA). Fresh and aged
solutions of oxi-ADan peptide samples were diluted 1 part with 2
parts of sample buffer [glycerol, 40% (w/v); sodium dodecyl
sulphate (SDS), 2% (w/v); 0.2 M Tris–HCl, pH 6.8 and Coomassie
G-250, 0.005% (w/v)], prior to electrophoresis; the concentration of
Coomassie G-250 in the sample was reduced 10-fold from that
recommended by the gel manufacturers, because co-migration with
low molecular weight peptides obstructed visualization after
staining of the gel. Red-ADan was treated with DTT (50-fold
molar excess) in sample buffer, whereas oxi-ADan was applied in
the absence of DTT. Samples were not boiled. The gels were loaded
with 150 nmole of peptides, and run in tricine buffer [Tris base,
1.31% (w/v), tricine 1.8% (w/v) and SDS 0.1% (w/v)], at 20 mA per
gel on the Bio-Rad Mini-Gel. The gels were fixed and stained in
methanol, 40% (v/v), acetic acid 10% (v/v) solution containing
Coomassie blue R-250 0.1% (w/v) for 3 h. De-staining was
performed in the methanol/acetic acid solution without Coomassie
blue.
Atomic force microscopy
Red-ADan (1 mg/mL) was dissolved in sterile semiconductor grade
water (18 MOhm), made 0.1 M in Tris–HCl (pH 7.4) by addition of
sterile 1 M Tris–HCl (pH 7.4) and agitated for several minutes. A
few drops were placed onto a freshly cleaved square of mica and a
few minutes were allowed for any materials to adsorb before it was
gently washed off with a light stream of ultra-pure water. The
sample, after removing the excess around the edges, was allowed to
dry under a cover. AFM using AC (or tapping) mode at 300 kHz
was done using a type 1620 Si tip with tip radius of 20 nm or less.
The mica was held down onto magnetic holders using conductive
double-sided adhesive discs (Agar). The Setpoint for AC was 50%,
proportional gain 1, integral 0.5 and differential ¼ 0.75.
Electron microscopy
A solution (1 mg/mL) of either red-ADan or oxi-ADan in sterile
0.1 M Tris–HCl (pH 7.4), were either analysed immediately or
incubated at 37�C for either 3 or 7 days. Solutions were subse-
quently diluted to 0.1 mg/mL in sterile distilled water. Then, 5 lL ofthe diluted solutions were spotted onto Parloidin-covered EM grids
and allowed to air dry for 5 min. Staining of the samples was
achieved using 2% uranyl acetate, with removal of excess using
filter paper (Whatman, Maidstone, UK). Grids were examined using
a Zeiss 900 transmission EM.
Cell culture
SH-SY5Y cells (European Collection of Cell Cultures, Porton
Down, UK) were cultured in Dulbecco’s modified essential medium
(MEM)/Nutrient Mix F-12 (1 : 1; Gibco–BRL, Gaithersburg, MD,
USA) containing (10 IU penicillin/mL, 100 lg streptomycin/mL,15% foetal calf serum, 1% non-essential MEM amino acid
supplement, and 2 mM freshly prepared glutamine), at 37�C in a
humidified incubator with 5% CO2/95% air.
Measurement of cell viability
The effect of ADan peptides on cell viability was assessed by
measuring lactate dehydrogenase (LDH) release, and residual
cellular redox activity with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphe-
nyltetrazolium bromide (MTT; Sigma, St Louis, MO, USA) as
previously described (El-Agnaf et al. 1998). Briefly, cells were
plated at 7500 cells/well in 96-well plates in 100 lL of fresh
medium. After 24 h, the medium was replaced with 200 lL of freshOPTI-MEM (Gibco–BRL) serum-free medium, containing freshly
prepared or aged solutions (20 days) of the peptides, alone or in the
Mass / Charge
100
90
80
70
60
50
40
30
20
10
03070 3080 3090 4000 4010 4020 4030 4040 4050 4060 4070 4080 4090 4100 4110 4120 4130
4063
.0
4066
.0
spot 2 Adan Reduced0001, spot 3Adan Redoxised0001Kratos PCAxima CFR V2.1.0
%Int.
2.6 mV 0.4 mV
Fig. 2 The mass spectrometry (MALDI-
TOF) of red-ADan (broken line) oxi-Adan
(continuous line), shows loss of two protons
upon oxidation.
Oligomerization and neurotoxicity in FDD 283
� 2003 International Society for Neurochemistry, J. Neurochem. (2004) 88, 281–290
presence of fourfold molar excess of DTT. Cells were then incubated
at 37�C in 5% CO2 for 2 days for LDH assay or 24 h for MTT
assay. An index of cell death was also obtained by measuring LDH
in cell supernatants using a cytotoxicity detection kit (Boehringer
Mannheim, Mannheim, Germany). For MTT assay, 20 lL of stockMTT (6 mg/mL) was added to each well, and the plates were
incubated at 37�C for 4.5 h. The medium-MTT solution was
carefully removed, and cell lysis buffer (100 lL per well; 15% (w/v)SDS/50% (v/v) N,N-dimethylformamide, pH 4.7) was added, and
the plate incubated overnight at 37�C, and the absorbance values at570 nm were measured.
Caspase activation
The caspase activities were measured by means of the binding of
the carboxyfluorescein derivative of benzyloxycarbonyl valylalanyl
aspartic acid fluoromethyl ketone (zVAD-FMK; Caspatag, Inter-
gen, Oxford, UK) which is a potent general inhibitor of caspase
activity. Briefly, SH-SY5Y cells were seeded in fresh media at
50 000 cells/mL, and 0.45 mL/well in eight-well chambered cover
glass (Nalgene Nunc Int., Naperville, IL, USA), and incubated for
24 h. The media were then replaced with fresh serum-free OPTI-
MEM containing ADan peptides, the incubation was continued for
8 h, and Caspatag was then added at 10 lM, and incubated for 1 h.After one wash with PBS, cells were visualized under a Carl Zeiss
LSM using a narrow pass 515–565 nm filter and 488 nm laser
irradiation.
Results
In view of our previous findings that the oxidized form of
ABri was more neurotoxic than the reduced form (El-Agnaf
et al. 2001a), some of the synthetic reduced form of ADan
peptide was converted into its oxidized form containing an
internal disulphide linkage by air oxidation. The oxidation
was monitored by HPLC and confirmed by mass spectrom-
etry. The oxidized form product gave one peak on MALDI–
TOF mass spectrometry of 4062.6 Da (Fig. 2), showing a
loss of two protons. To demonstrate that this change was not
due to intermolecular disulphide scrambling into oligomers,
the preparation was re-analysed after tryptic digestion.
The reduced form produced tryptic fragments with
reduced Cys residues at m/z of 1009.3 Da (data not shown),
corresponding to residues 1–9 (calculated 1010.5 Da), a
fragment (F/6) of m/z of 2771.9 Da corresponding to
residues 10–32 (calculated 2772.39 Da; Fig. 3), F/7 at
3072.3 Da residues 10–34 (calculated m/z of 3072.51 Da)
and a fragment F/8 of m/z of 3763.94 Da containing residues
1–32 with (calculated 3763.84 Da), the oxidized material
gave a large peak at 3125.0 Da, coincident with a peptide
containing residues 1–9 and 15–33 containing the intact
disulphide between residues 5 and 22 (calculated mass
3124.6 Da) (Fig. 3), showing that the Cys residues were
engaged in intramolecular cyclisation and not intermolecular
bonding. The same peaks were seen after trypsin digestion of
aged samples, showing that no scrambling of the disulphide
occurred during incubation at neutral pH.
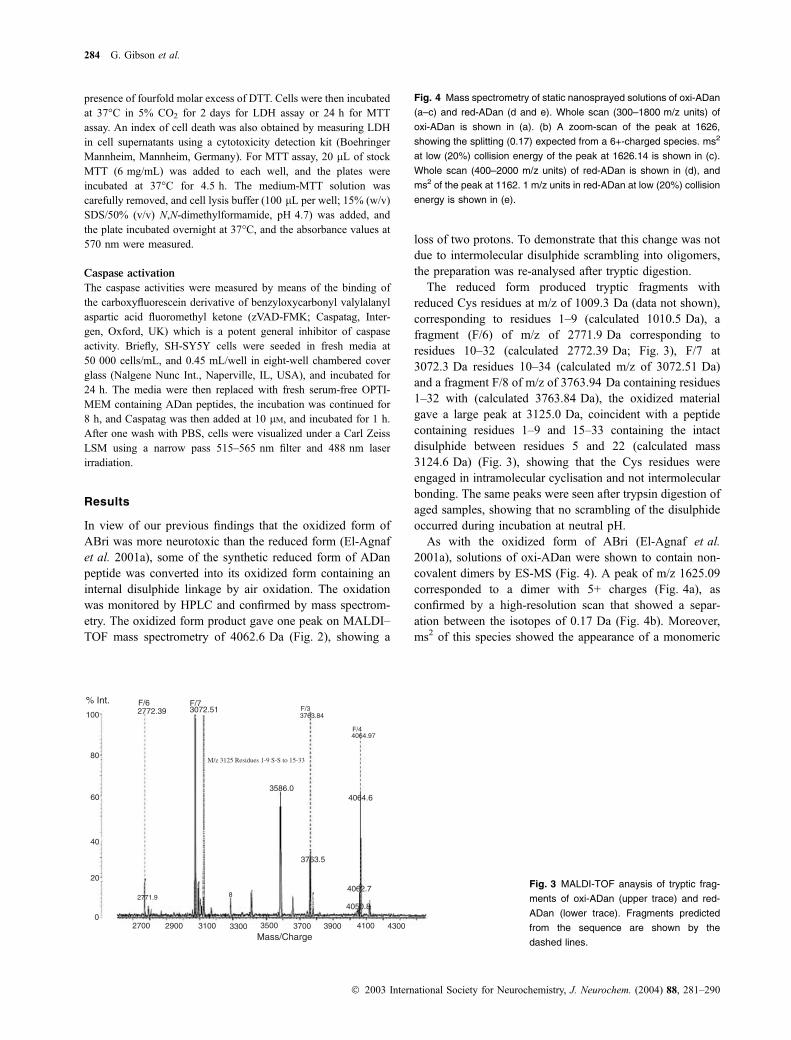
As with the oxidized form of ABri (El-Agnaf et al.
2001a), solutions of oxi-ADan were shown to contain non-
covalent dimers by ES-MS (Fig. 4). A peak of m/z 1625.09
corresponded to a dimer with 5+ charges (Fig. 4a), as
confirmed by a high-resolution scan that showed a separ-
ation between the isotopes of 0.17 Da (Fig. 4b). Moreover,
ms2 of this species showed the appearance of a monomeric
% Int.
100
F/62772.39
80
60
40
20
02700 2900 3100 3300 3500 3700 3900 4100 4300
4062.7
4050.8
3763.5
3586.04064.6
3763.84F/3
F/44064.97
M/z 3125 Residues 1-9 S-S to 15-33
F/73072.51
2771.9
Mass/Charge
8Fig. 3 MALDI-TOF anaysis of tryptic frag-
ments of oxi-ADan (upper trace) and red-
ADan (lower trace). Fragments predicted
from the sequence are shown by the
dashed lines.
Fig. 4 Mass spectrometry of static nanosprayed solutions of oxi-ADan
(a–c) and red-ADan (d and e). Whole scan (300–1800 m/z units) of
oxi-ADan is shown in (a). (b) A zoom-scan of the peak at 1626,
showing the splitting (0.17) expected from a 6+-charged species. ms2
at low (20%) collision energy of the peak at 1626.14 is shown in (c).
Whole scan (400–2000 m/z units) of red-ADan is shown in (d), and
ms2 of the peak at 1162. 1 m/z units in red-ADan at low (20%) collision
energy is shown in (e).
284 G. Gibson et al.
� 2003 International Society for Neurochemistry, J. Neurochem. (2004) 88, 281–290

tm/z = 1355.
1000 1100 1200 1300 1400 1500 1600 1700 1800 1900 2000
1000800600400 1200 1400 1600 1800 2000
m/z
05
101520253035404550556065707580859095
100
Rel
ativ
e A
bund
ance
1626
1355
Adancorrect1pptinformicdilutedfull #3RT:0.06AV:1NL:3.90E6T:+ c Full ms [ 390.00-2000.00]
m/z
05
101520253035404550556065707580859095
100
Rel
ativ
e A
bund
ance
1017.1
1355.4
1162.1
1626.31374.2
1196.21021.3904.2 1375.31220.5503.1 813.6 1636.71109.4 1229.1 1527.9793.8 1653.7678.5 1015.5 1741.9415.1 520.2 1843.9
Monomer 4+
Monomer 3+
Dimer 5+
Covalent dimer 7+
Full MS scan of Red AdanAdancorrect1pptinformicdiluted1162msms#10RT:0.20AV:1NL:5.47E4T:+ p sid=25.00 Full ms2 [email protected] [ 390.00-2000.00]
400 500 600 700 800 900 1000 1100 1200 1300 1400 1500 1600 1700 1800 1900 2000m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100 1159.5
1142.9
1186.71280.8
1318.01081.21007.9935.7 1428.2822.4447. 1366.4 1918.5878.9 1441.7 1505.6 1584.1791.5 1884.51668.9 1764.5 1934.0592.1470.1 709.1
800 1000 1200 1400 1600m/z
0
10
20
30
40
50
60
70
80
90
100
Rel
ativ
e A
bund
ance
1016.62813.66
1355.27
897.49
1625.69
Dimer5+ 5+
Monomer 3+
Monomer 4+Monomer 5+
10
20
Rel
ativ
e A
bund
ance
1623.5 1624.0 1624.5 1625.0 1625.5 1626.0 1626.5 1627.0 1627.5 1628.0 1628.5
30
40
5565
85
1626.14
1626.541625.74
1626.341625.551625.36
1626.591625.19
(a) (b)
(d)(c)
(e)
Rel
ativ
e A
bund
ance
Oligomerization and neurotoxicity in FDD 285
� 2003 International Society for Neurochemistry, J. Neurochem. (2004) 88, 281–290

triple-charged species at m/z ¼ 1355 (Fig. 4c), confirming
a non-covalent dimer. The low yield of dimers found by
this technique may have been due to the use of organic
solvents in the nanospray injector. Analyses of pre-incuba-
ted solutions of red-ADan also showed the presence of the
non-covalent dimeric peaks, and in addition showed the
presence of a covalent 7+ charged dimer of 1162.1 m/z
(Fig. 3d). Ms2 at 1162.1 did not yield a monomeric species,
but generated fragments from b-type and y-type fragmen-
tation (Fig. 3e). The same dimers were present in samples
incubated in the absence or presence of DTT; showing that
the intermolecular disulphide was not readily accessible to
DTT.
Non-covelent oligomers in solutions of both oxi-ADan
and red-ADan were also shown by electrophoresis on highly
cross-linked SDS–PAGE (Fig. 5). Fresh solutions of red-
ADan after DTT treatment gave rise to an oligomer of about
32 kDa, and a dimer of 8 kDa, plus aggregated material not
entering the separating gel, whereas oxi-ADan fresh solu-
tions showed a strong dimeric species, as well as trimer and
quadramer (depicted by arrows; Fig. 5), but no higher
aggregates.
Aged solutions showed only dimer and aggregated
material not entering the separating gel.
Gel filtration chromatography of fresh preparations of red-
ADan solutions showed oligomers of molecular weight
higher than the monomeric form (Fig. 6).
A peak eluting at 41 mins from the column was indicative
of an oligomer of about 30 kDa (Fig. 6), as well as peak with
MW > 70 kDa, eluting rapidly at 19 min from the column
were also present, in keeping with the SDS–PAGE. However,
when the red-ADan solutions were incubated at 37�C for
21 days, only peaks containing high MW oligomers
(> 70 kDa) were detected. Incubation for 9 days converted
oxi-ADan from a dimer into high MW soluble oligomers
> 70 kDa eluting at 24 min (data not shown).
Aged preparations of oxi-ADan were very weakly
bi-refringent after treatment with Congo red, whereas aged
red-ADan was not birefringent.
ADan on Tris-Tricine PAGE
Freshoxidised
Agedoxidised
Freshreduced
Agedreduced
>70K>70K
32K
8K-Dimer8K Dimer8K
Octamer
Quadrimer
Trimer
Dimer
Monomer
34.3k
26k
17.9k
8k
4k
Fig. 5 SDS–PAGE (16% acrylamide) ana-
lysis of self-oligomerization of fresh and
aged red- and oxi-ADan peptides using
Tricine buffers. Sample preparation is des-
cribed in Methods. Only the reduced sam-
ples contained DTT.
10.00 20.00 30.00 40.00 50.00 60.00Elution time (min)
OD230nm
1.00
0.50
0.00
Oligomer>75kDa Oligomer 30kDa
Monomer
–0.50
Fig. 6 Size exclusion chromatography of red-ADan, A freshly pre-
pared solution of red-ADan (1 mg/mL; continuous line) or aged or 24 h
(dotted line) or 21 days (dashed-dotted line) was eluted from a column
of Superdex 75 in 0.1 M Tris–HCl t pH 7.4 at 0.5 mL/min. Insoluble
material was removed by centrifugation prior to loading.
2000 nm
1000 nm
0 nm0 nm 1000 nm 2000 nm
13.15 nm
0.00nm
Fig. 7 Atomic force microscopy of a fresh solution of red-ADan
(1 mg/mL).
286 G. Gibson et al.
� 2003 International Society for Neurochemistry, J. Neurochem. (2004) 88, 281–290

Microscopy
When viewed by atomic force microscopy, fresh red-ADan
suspensions appeared as carrot-shaped objects each with a
head and elongated body of about 130 nm in length and
70 nm in maximum diameter (Fig. 7). The objects can be
seen stacked together in uniform orientation with heads
uppermost to form larger aggregates.
Electron microscopy of oxi-ADan preparations revealed
that after aging for 3 days, regular-shaped electron-dense
oligomers adhering to each other in arrays were formed,
whereas after 7 days the aggregated material appeared as
a matrix. Clusters of oligomeric spheroids were seen in
red-ADan preparations immediately after dissolution, which
formed three-dimensional matrices after 3–7 days (Fig. 8).
Neurotoxicity
The neurotoxicity of the reduced and oxidized forms of BRI
peptides on human neuroblastoma SH-SY5Y cells was
investigated. Cells were treated with solutions of BRI peptides
(200 lM), diluted from fresh or aged (20 day) solutions of
1 mg/mL. After incubation for 24 h, cell viability was
measured by LDH released to the media by the cells. Fresh
ABri andADan solutions were more toxic than aged solutions.
In contrast to ABri, red-ADan was more toxic than oxi-ADan.
The WT BRI peptides showed very little toxicity, either in
aged, fresh, oxidized or reduced states, as reported previously.
(Fig. 9). Similar relative results were obtained when toxicities
were measured by MTT uptake (Fig. 9, open bars) or LDH
release (Fig. 9, black shaded bars).
DTT at fourfold molar excess included with red-ADan to
retain it in the sulphydryl form did not change the levels of
toxicity recorded (data not shown).
We further investigated the mechanisms involved in the
neurotoxicity induced by ADan and ABri peptides. Using
FAM-VAD-FMK green fluorescent caspase inhibitor, we
investigated caspase induction following treatment of cells
oxi-Adan (0days aging) oxi-Adan (3 days aging)
red-Adan (3 days aging)red-Adan (0 days aging)
oxi-Adan (7 days aging)
red-Adan (7 days aging)
Fig. 8 Electron micrographs of prepara-
tions of (1 mg/mL) red-ADan and oxi-ADan;
the white tab is equivalent to 100 lm.
LDH
MTT
LDH
Peptides at 200µM
% Toxicity
100
80
60
40
20
0
Red
AB
ri
Red
Ada
n
Red
AB
riFr
esh
OxA
Bri
Fres
h
Fres
h
Fres
h
Oxi
d A
Bri
OxA
Dan
Oxi
d W
T
OxW
T
Age
d
Age
d
Age
dAge
d
MTT
Fig. 9 Neurotoxicity of aged (20 days; Ag)
and fresh (Fr) solutions of BRI peptides at
200 lM, determined by LDH release (h) or
MTT uptake (j) in SHSY-5Y cells in cul-
ture.% toxicity with the LDH assay is the
enzyme relased compared to that released
by 0.1% Triton X-100, and toxicity of the
MTT assay is the colour reduction com-
pared to that achieved with 100 lM campt-
othecin. Results are those averaged from
six readings on separate batches of cells.
Standard deviations were less than 10% of
averages recorded.
Oligomerization and neurotoxicity in FDD 287
� 2003 International Society for Neurochemistry, J. Neurochem. (2004) 88, 281–290

with fresh solutions of ADan and ABri peptides. Our results
show that at 200 lM concentration red-ADan led to panac-
tivation of caspases in cells, as shown by increased binding
of green fluorescent FAM-VAD-FMK as compared to control
cells (Fig. 10). At this concentration, red-ADan produced
more caspase activation than oxi-ABri. Altogether, these data
suggest that red-ADan oligomer-induced caspase activation
might represent a seminal event of ADan-induced neuronal
cell death.
To elucidate atomic interactions between ADan molecules
that might give rise to oligomer formation, we modelled
oxi-ADan by residue replacement in the previously mod-
elled ABri (El-Agnaf et al. 2001b) and docked two of these
molecules together using Swiss PBD Viewer. The resulting
model (Fig. 11) was subjected to energy-minimization steps.
The model shows that the monomers are held together by
electrostatic bonding between Arg (blue) and Glu (red)
residues in a largely hydrophobic environment provided by
other side-chains in ADan. Although the Cys–Cys (marked
1.42) intramolecular disulphide is involved in holding two
strands together in oxi-ADan, the triple-stranded sheet may
be stable in its absence, suggesting that red-ADan would
also form non-covalent oligomers readily. Examination of
the molecular model shows that the presence of an
intermolecular disulphide in a dimer of red-ADan, as
detected by ES-MS, would add to the hydrophobic and
(a) Control (Cells alone)
(b) 200 µM Oxi-ABri (c) 10 µM Oxi-ABri
(d) 200 µM Oxi-ADan (e) 10 µM Oxi-ADan
(f) 200 µM Red ADan (g) 10 µM Red ADan
Fig. 10 (a–g) Apoptotic effects of reduced
and oxi-ADan and oxi-ABri, detected with
FAM-VAD-FMK (Caspatag).
Fig. 11 A model of an ADan dimer mod-
elled in Swiss-PDB-viewer.
288 G. Gibson et al.
� 2003 International Society for Neurochemistry, J. Neurochem. (2004) 88, 281–290

electrostatic forces annealing the oligomeric structure and
could explain the enhanced neurotoxicity of red-ADan over
oxi-ADan.
Discussion
In this study we have investigated the biophysical and the
neurotoxic properties of ADan, the peptide which is found
deposited in the brain of patients suffering from FDD. Using
SDS-PAGE, SEC and ES-MS we found that the reduced
form of ADan red-ADan forms stable soluble oligomers
containing both covalent and non-covalent oligomers more
readily than the oxidized form, oxi-ADan. Higher molecular-
weight forms were visualized as adherent elongated sphe-
roids of about 50 lm in diameter, by atomic force microcopyand electron microscopy. In our previous report we have
shown that the oxidized form of the ABri, which is released
in the brains of a British family with dementing disease,
forms very toxic soluble oligomers, whereas the reduced
form did not form soluble oligomers and was not toxic to
neuronal cells (El-Agnaf et al. 2001a). To investigate
whether ADan is toxic to neuronal cells, we tested the
toxicity of both peptide forms the red-ADan and oxi-ADan.
Peptide solutions prepared freshly or aged for 20 days were
added to the cells and LDH released by cells to the media
were measured. Remaining cells were also measured by dye
binding (MTT). Interestingly, our results showed that the
fresh and aged preparations of red-ADan induced cell death
more than oxi-ADan as measured by LDH release. Fresh
preparation were more toxic than aged preparations. The
cell death induced by ADan involves the activation of
caspases. Interestingly, these results are in contrast with our
recent studies on ABri peptide, where we found that the
oxidized form was toxic whereas the reduced form was not
toxic to neuronal cells (El-Agnaf et al. 2001a). However, in
that study we have found that fresh solutions of the
oxidized form of ABri contained a series of soluble
oligomers, whereas the reduced form solution did not
contain a series of oligomers. The results for the ADan and
ABri peptides are in agreement with the notion that the
non-fibrillar aggregates ‘soluble oligomers’ formed by
amyloid peptides are the pathogenic species, which cause
neurodegeneration.
We hypothesize that the non-fibrillar aggregates of ADan
deposited in the brain are the cause of FDD. In support of
this hypothesis, the pathological studies of FDD brains have
shown that the hippocampal deposits can be recognized with
anti-ADan antibody but are negative to Congo red staining,
suggesting that ADan is deposited predominantly in a non-
fibrillar form (Vidal et al. 2000; Holton et al. 2002).
Interestingly, recent studies have shown that small oligomers
of many amyloid proteins, associated with neurodegenerative
disease, are the pathogenic species that drive neurodegener-
ation and neuronal cell death (Chiesa et al. 1998; Saudou
et al. 1998; Goldberg and Lansbury 2000; Conway et al.
2000; Masliah et al. 2000; Sian et al. 2000; Van der Putten
et al. 2000; El-Agnaf et al. 2000, 2001a; Walsh et al. 2002).
Ab-derived diffusible ligands (ADDLs) were shown to betoxic by Lambert et al 1998, whereas the experiments of
Bucciantini et al. (2002) suggested that some proteins,
previously considered non-pathogenic, were toxic in soluble
oligomeric form. Support for the idea that soluble aggregates
of amyloidogenic proteins are pathogenic and play a central
role in the development of several neurodegenerative
diseases also came from the detection of soluble Abaggregates in cerebrospinal fluid (CSF) from AD patients
(Pitschke et al. 1998) and prion protein aggregates in CSF
from patients with Creutzfeldt–Jakob disease (Bieschke et al.
2000; Giese et al. 2000) but not in control patients.
Identifying the molecular structure of the amyloid aggregate
species leading to neurodegeneration is an important step for
the design of novel drugs for neurodegenerative disorders
caused by amyloid deposits.
In summary, in this study we have shown for the first time
that both reduced and oxidized forms of ADan peptide
associated with FDD are toxic to neuronal cells, and that
fresh solutions containing soluble oligomers are more toxic
than aged solutions containing insoluble aggregates. Soluble
aggregates appear to activate caspases and apoptosis in
neuronal cells. Hence, an understanding of the role of ADan
in the pathogenesis of the FDD may shed light on causes of
neurodegeneration in more common neurological disorders
associated with amyloid deposition, including Alzheimer’s
disease.
Acknowledgements
We are grateful to the Wellcome Trust for their support and to
Dr Ted Tarelli for help with mass spectrometry.
References
Aikawa H., Suzuki K., Iwasaki Y. and Iiuzuka R. (1985) Atypical
Alzheimer’s disease with spastic paresis and ataxia. Ann. Neurol.
17, 297–300.
Bieschke J., Giese A., Schulz-Schaeffer W., Zerr I., Poser S., Eigen M.
and Kretzschmar H. (2000) Ultrasensitive detection of pathological
prion protein aggregates by dual-color scanning for intensely
fluorescent targets. Proc. Natl Acad. Sci. USA 97, 5468–5473.
Bucciantini M., Giannoni E., Chiti F. et al. (2002) Inherent toxicity of
aggregates implies a common mechanism for protein misfolding
diseases. Nature 416, 507–511.
Chiesa R., Piccardo P., Ghetti B. and Harris D. A. (1998) Neurological
illness in transgenic mice expressing a prion protein with an
insertional mutation. Neuron 6, 1339–1351.
Conway K. A., Lee S. J., Rochet J. C., Ding T. T., Williamson R. E.
and Lansbury P. T. Jr (2000) Acceleration of oligomerization,
not fibrillization, is a shared property of both a-synucleinmutations linked to early-onset Parkinson’s disease: implications
for pathogenesis and therapy. Proc. Natl Acad. Sci. USA 97,
571–576.
Oligomerization and neurotoxicity in FDD 289
� 2003 International Society for Neurochemistry, J. Neurochem. (2004) 88, 281–290

El-Agnaf O. M. A., Jakes R., Curran M. D., Middletone D., Ingenito F.,
Bianchi E., PessiA.,Neill D. andWallaceA. (1998)Aggregates from
mutant and wild-type a-synuclein proteins and NAC peptide induceapoptotic cell death in human neuroblastoma cells by formation of
b-sheet and amyloid-like filaments. FEBS Lett. 440, 71–75.El-Agnaf O. M. A., Mahil D. S., Patel B. P. and Austen B. A. (2000)
Oligomerization and toxicity of b-amyloid-42 implicated in Alz-heimer’s disease. Biochem. Biophy. Res. Commun. 237, 1003–
1007.
El-Agnaf O. M., Nagala S., Patel B. P. and Austen B. M. (2001a) Non-
fibrillar oligomeric species of the amyloid ABri peptide, implicated
in familial British dementia, are more potent at inducing apoptotic
cell death than protofibrils or mature fibrils. J. Mol. Biol. 310, 157–
168.
El-Agnaf O. M. A., Sheridan J. M., Sidera C., Siligardi G., Hussain R.,
Haris P. I. and Austen B. M. (2001b) Effect of the disulphide
bridge and the C-terminal extension on the oligomerisation of the
amyloid peptide ABri implicated in familial British dementia.
Biochemistry 40, 3449–3457.
Giese A., Bieschke J., Eigen M. and Kretzschmar H. A. (2000) Putting
prions into focus: application of single molecule detection to the
diagnosis of prion diseases. Arch. Virol. Suppl. 16, 161–171.
Goldberg M. S. and Lansbury P. T. (2000) Is there a cause-and-effect
relationship between a-synuclein fibrillization and Parkinson’s
disease? Nat. Cell Biol. 2, 115–119.
Holton J. L., Lashley T., Ghiso J. et al. (2002) Familial Danish dementia:
a novel form of cerebral amyloidosis associated with deposition of
both amyloid-Dan and amyloid-b. J. Neuropathol. Exp. Neurol. 61,254–267.
Lambert M. P., Barlow A. K., Chromy B. A. et al. (1998) Diffusible,
nonfibrillar ligands derived from Ab1–42 are potent central ner-vous system neurotoxins. Proc. Natl Acad. Sci. USA 95, 6448–
6453.
Masliah E., Rockenstein E., Veinbergs I., Mallory M., Hashimoto M.,
Takeda A., Sagara Y., Sisk A. and Mucke L. (2000) Dopaminergic
loss and inclusion body formation in a-synuclein mice: implica-tions for neurodegenerative disorders. Science 287, 1265–1269.
Pitschke M., Prior R., Haupt M. and Riesner D. (1998) Detection of
single amyloid b-protein aggregates in the cerebrospinal fluid ofAlzheimer’s patients by fluorescence correlation spectroscopy. Nat.
Med. 4, 832–834.
Plant G. T., Revesz T., Barnard R. O., Harding A. E. and Gautier-Smith
P. C. (1990) Familial cerebral amyloid angiopathy with nonneuritic
amyloid plaque formation. Brain 13, 721–747.
Saudou F., Finkbeiner S., Devys D. and Greenberg M. E. (1998)
Huntingtin acts in the nucleus to induce apoptosis but death does
not correlate with the formation of intranuclear inclusions. Cell 95,
55–66.
Sian A. K., Frears E. R., El-Agnaf O. M. A., Patel B. P., Manca M. F.,
Siligardi G., Hussain R. and Austen B. A. (2000) Oligomerization
of b-amyloid of the Alzheimer’s and Dutch cerebral haemorragetypes. Biochem. J. 349, 299–308.
Van der Putten H., Wiederhold K. H., Probst A. et al. (2000) Neuro-
pathology in mice expressing human a-synuclein. J. Neurosci. 20,6021–6029.
Vidal R., Frangione B., Rostagno A., Mead S., Revesz T., Plant G. and
Ghiso J. (1999) A stop-codon mutation in the BRI gene associated
with familial British dementia. Nature 399, 776–781.
Vidal R., Revesz T., Rostagno A., Kim E. et al. (2000) A decamer
duplication in the 3¢ region of the BRI gene originates an amyloidpeptide that is associated with dementia in a Danish kindred. Proc.
Natl Acad. Sci. USA 97, 4920–4925.
Walsh D. M., Klyubin I., Fadeeva J. V., Cullen W. K., Anwyl R., Wolfe
M. S., Rowan M. J. and Selkoe D. J. (2002) Naturally secreted
oligomers of amyloid b protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539.
Worster-Drought C., Hill T. R. and McMenemey W. H. (1933) Familial
cerebral amyloid angiopathy with non-neuritic plaque formation.
J. Neurol. Psychopathol. 14, 27–34.
290 G. Gibson et al.
� 2003 International Society for Neurochemistry, J. Neurochem. (2004) 88, 281–290
Copyright © 2022 FDOKUMEN




















