
Dietary salt restriction accelerates atherosclerosis in apolipoprotein E-deficient mice
Upload
independentCategory
view
1download
0
FEBS Letters 586 (2012) 143–148
journal homepage: www.FEBSLetters .org
The yeast metacaspase is implicated in oxidative stress responsein frataxin-deficient cells
Sophie Lefevre a,b, Dominika Sliwa a, Françoise Auchère a, Caroline Brossas a, Christoph Ruckenstuhl c,Nicole Boggetto d, Emmanuel Lesuisse a, Frank Madeo c, Jean-Michel Camadro a, Renata Santos a,⇑a Mitochondria, Metals and Oxidative Stress Laboratory, Institut Jacques Monod, CNRS-Université Paris-Diderot, Sorbonne Paris Cité, 15 rue Hélène Brion, 75205 Paris Cedex 13, Franceb ED515 UPMC, 4 Place Jussieu, 75005 Paris, Francec Institute of Molecular Biology, University Graz, Humboldtstrasse 50, 8010 Graz, Austriad Institut Jacques Monod, ImagoSeine Bioimaging Core Facility, 15 rue Hélène Brion, 75205 Paris Cedex 13, France
a r t i c l e i n f o a b s t r a c t
Article history:Received 22 October 2011Revised 18 November 2011Accepted 1 December 2011Available online 7 December 2011
Edited by Vladimir Skulachev
Keywords:FrataxinFriedreich ataxiaMetacaspaseOxidative stressYca1
0014-5793/$36.00 � 2011 Federation of European Biodoi:10.1016/j.febslet.2011.12.002
Abbreviations: DC-FDA, 20 ,70-dichlorodihydrofluodihydrorhodamine 123; FA, Friedreich ataxia; FCH,glutathione; GSSG, oxidized glutathione; H2O2, hydrtime quantitative PCR; ROS, reactive oxygen speciescleotidyl transferase dUTP nick end labeling⇑ Corresponding author.
E-mail address: [email protected]
Friedreich ataxia is the most common recessive neurodegenerative disease and is caused by reducedexpression of mitochondrial frataxin. Frataxin depletion causes impairment in iron–sulfur clusterand heme biosynthesis, disruption of iron homeostasis and hypersensitivity to oxidants. Currentlyno pharmacological treatment blocks disease progression, although antioxidant therapies provedto benefit patients. We show that sensitivity of yeast frataxin-deficient cells to hydrogen peroxideis partially mediated by the metacaspase. Metacaspase deletion in frataxin-deficient cells resultsin recovery of antioxidant capacity and heme synthesis. In addition, our results suggest that meta-caspase is associated with mitochondrial respiration, intracellular redox control and genomicstability.� 2011 Federation of European Biochemical Societies. Published by Elsevier B.V. All rights reserved.
1. Introduction
Oxidative stress is a central pathological feature of Friedreichataxia (FA) and a valuable target for therapy [1,2]. FA is a hereditaryneurodegenerative disease caused by reduced expression of frataxin[3,4]. Frataxin deficiency in humans and model systems causesdecreased biosynthesis of iron–sulfur cluster and heme cofactors,reduced respiratory capacity, disturbed iron homeostasis and hyper-sensitivity to oxidants [5]. Iron accumulation in the mitochondria,which can participate in the Fenton reaction and increase reactiveoxygen species (ROS) production, is considered an important mech-anism inducing oxidative stress in frataxin-deficient cells [1,2].However, substantial evidence shows that glutathione-dependentdefenses and antioxidant enzymes are repressed or not properly reg-ulated [6–9]. In addition, patient fibroblasts and lymphoblasts un-dergo apoptosis when challenged with oxidants [10–12]. Here, we
chemical Societies. Published by E
rescein diacetate; DHR123,ferrichrome; GSH, reduced
ogen peroxide; RT-PCR, real-; TUNEL, terminal deoxynu-
rot.fr (R. Santos).
report that the yeast metacaspase Yca1 is implicated in the regula-tion of the antioxidant status of yeast frataxin-deficient cells(Dyfh1).
2. Materials and methods
2.1. Yeast strains, plasmids and growth conditions
The Saccharomyces cerevisiae strains used in this study werederived from the YPH499 cycloheximide-resistant (wild-type) andthe YPH499yfh1 shuffle strains [13]. The YPH499Dyfh1 (Dyfh1::TRP1) mutants were constructed prior to experiments in aerobiosis[13]. YCA1 was deleted using the Dyca1::kanMX4 locus of theBY4741 Dyca1 mutant (Euroscarf collection) amplified by PCR using50-CTGGATACTTAATGCCCATCGAGTC-30 and 50-GTCTGAATACATCTACCAACGTACACAT-30 primers. Successful integration was verifiedusing one external primer to the deleted genomic region and oneprimer specific of the kanMX4 cassette. The primer sets used were:50-CGCTAGCGTTATCTGAATCAG-30 and 50-CTGCAGCGAGGAGCCG-30; 50-CACTAATTGCTACTTTGGCCG-30 and 50-TGATTTTGATGACGAGCGTAAT-30. Liquid cultures were grown at 30 �C in defined YNB(yeast nitrogen base, 2% D-glucose, plus the required amino acids)or in rich YPD (1% yeast extract, 2% Bacto peptone, 2% D-glucose)
lsevier B.V. All rights reserved.

144 S. Lefevre et al. / FEBS Letters 586 (2012) 143–148
media; both supplemented with 200 mg/l adenine. Yeast cells werechallenged with H2O2 (Sigma) in YNB or YPD medium as described[14].
2.2. Detection of metacaspase activation, ROS accumulation and cellcycle
Exponentially growing cells in YNB medium were labeled with2 ll of FITC-VAD-fmk (Promega) in PBS for 20 min at 30 �C in thedark. Samples were analyzed by flow cytometry using a CyAnADP LX apparatus (Beckman Coulter) with excitation at 488 nmand emission detection through a 530/40 nm band pass filter.Intracellular ROS were detected using 25 lg/ml dihydrorhodamine123 (DHR123; Calbiochem�) and 10 lg/ml 20,70-dichlorodihydro-fluorescein diacetate (DC-FDA; Sigma–Aldrich Co.) by flow cytom-etry as described [14]. For cell cycle analysis exponentially growingcells were fixed with 70% ethanol overnight, washed and RNAsdigested overnight at 37 �C with 0.2 mg/ml RNase A in 50 mM so-dium citrate buffer, pH7.4. DNA was labeled overnight with100 lg/ml propidium iodide. Samples were analyzed by flowcytometry with excitation at 488 nm and emission detectionthrough a 610/20 nm band pass filter. For mutation rate calcula-tion, single colonies were inoculated in YNB medium, grown for48 h and appropriate dilutions were plated in YPD and YNB with-out arginine plus 100 lg/ml canavanine.
Fig. 1. Oxidative stress sensitivity and metacaspase activation in Dyfh1 cells. (A) Expondiluted and spotted onto YPD plates containing 0.5 mM diamide, 2 mM H2O2 and 2 mM patype cells treated with 1 and 2 mM H2O2 for 60 min in YNB medium. Data represent ratiofor 4 h. Data represent percentages of cells stained ± S.E. for 3 < n < 5 cultures; ⁄⁄⁄P < 0.060 min. Diluted cultures were spotted onto YPD plates. (E) Surviving fraction of exponentmeans ± S.E. (n = 3); ⁄P < 0.05, ⁄⁄P < 0.01.
2.3. Other techniques
Mitochondria from yeast cells cultured at 30 �C in YNB Raf med-ium (2% raffinose, 0.1% glucose) were prepared as described [15].Low-temperature absorption spectra, iron uptake measurements,enzymatic activities, respiration were assayed as described[16,17]. Glutathione levels were determined as previously described[8] using GSSG standard curves. GSH/GSSG ratios were calculatedusing the equation GSH/GSSG = 2 � [(total glutathione) � GSSG]/GSSG. RNA extraction, cDNA preparation and real-time quantitativePCR (RT-PCR) were carried out exactly as described [8]. The primersused are listed in Supplementary Table 1. Statistical analysis wasperformed using the paired T-test.
3. Results
3.1. Sensitivity of Dyfh1 cells to H2O2 is partially dependent onmetacaspase
One of the first phenotypes described for the Dyfh1 mutant wasits sensitivity to pro-oxidant chemicals [4,18]. However, in ourhands this mutant was highly sensitive to H2O2 but not to diamide,paraquat and tert-butyl hydroperoxide (Fig. 1A; data not shown).Treatment of Dyfh1 and wild-type cells for 60 min with 2 mMH2O2 resulted in only 4.0 ± 0.8% survival of mutant compared to
ential cultures of wild-type and Dyfh1 in YPD medium were sequentially five-foldraquat and incubated for 3 days at 30 �C. (B) Ratio of surviving of Dyfh1 versus wild-of means ± S.E. (n = 4). (C) Metacaspase activation in cells treated with 0.4 mM H2O2
01. (D) Sensitivity of exponentially growing cells in YPD medium to 2 mM H2O2 forially growing cells in YNB medium to 60 min H2O2 treatment. Data represent ratio of

S. Lefevre et al. / FEBS Letters 586 (2012) 143–148 145
wild-type cells (Fig. 1B). Since the protease metacaspase Yca1 isactivated when wild-type yeast cells are challenged with H2O2
mediating apoptotic cell death [19], we investigated whether thispathway was effective in Dyfh1 cells. The fluorescent probe FITC-VAD-fmk and flow cytometry analysis were used to assay metacas-pase activation. The number of fluorescent cells was significantlyhigher in Dyfh1 than in wild-type cells after treatment with H2O2
(Fig. 1C and Supplementary Fig. S1). This corresponds to metacas-pase induction, since the number of fluorescent cells in the doubleDyfh1Dyca1 mutant was drastically reduced compared to Dyfh1.Of note, a considerable percentage of the double mutant-cells werefluorescently labeled suggesting that another caspase-like proteaserecognizing FITC-VAD-fmk may be induced by H2O2 in Dyfh1 cells,which is consistent with previous observations [20,21]. Inagreement with the increase in metacaspase activation in Dyfh1cells, the Dyfh1Dyca1 cells were less sensitive to H2O2 treatment(Fig. 1D and E).
3.2. Metacaspase deletion increases the antioxidant status of Dyfh1cells
Glutathione is a major protective antioxidant in yeast cells. Wehave shown that Dyfh1 cells have low total glutathione contentand a reduced GSH/GSSG ratio [8]. Since the double Dyfh1Dyca1mutant was more resistant to H2O2, we tested if glutathione anti-oxidant defense was altered. The glutathione-specific content was2.5-fold higher in the Dyfh1Dyca1 mutant than in the Dyfh1 mu-tant, and similar to that observed in the Dyca1 mutant (Fig. 2A).The GSH/GSSG ratio was also slightly increased in the double mu-tant (5.3 in Dyfh1 compared to 9.3 in Dyfh1Dyca1). In addition, theexpression of 19 genes implicated in the response to oxidativestress using RT-PCR was investigated (Fig. 2B). Seven genes encod-ing key defense enzymes showed more than two-fold repression in
AB
C
0
100
200
300
400
Glu
tath
ione
(n
mol
es/m
g pr
otei
n)
WT Δyfh1Δyca1 Δyfh1Δyca1
GSH+GSSG GSSG
SOD
GSHTRRTRR
TRXTRX
GRX
GRXGRXGRXGRXHYRGPXGPXAHPCTA
CTT
SOD
TRX
Gen
es
DHR123 DC-FDA
Med
ian
of fl
uore
scen
ce (a
u)
WT Δyfh1Δyca1 Δyfh1Δyca1
454035302520151050 0
2
4
6
8
10
12
Med
ian
of fl
uore
scen
ce (a
u)
Fig. 2. Oxidative stress defenses in frataxin- and metacaspase-deficient cells. (A) Total aexpression (Dyfh1 or Dyfh1Dyca1 versus wild-type). Data represent ratio of means ± S.E. (DC-FDA probes. Data represent medians of fluorescence ± S.E. (n = 3).
Dyfh1 compared to wild-type. Transcription of all these genes wasincreased in the double Dyfh1Dyca1 mutant (Fig. 2B). Exceptingthe GRX4 gene, the expression of the other genes was at least com-parable to that observed in the wild-type. Therefore, only one geneout of seven was repressed in the Dyfh1Dyca1 mutant. A smallreduction in ROS accumulation was observed in Dyfh1Dyca1 cellscompared to Dyfh1 cells using two different fluorescent probes(Supplementary Fig. 2C). Taken together these results show thatfrataxin-deficiency in yeast cells leads to deregulation of antioxi-dant defences that may be mediated by metacaspase or an uniden-tified metacaspase substrate.
3.3. Metacaspase deletion rescues other phenotypes in Dyfh1 cells
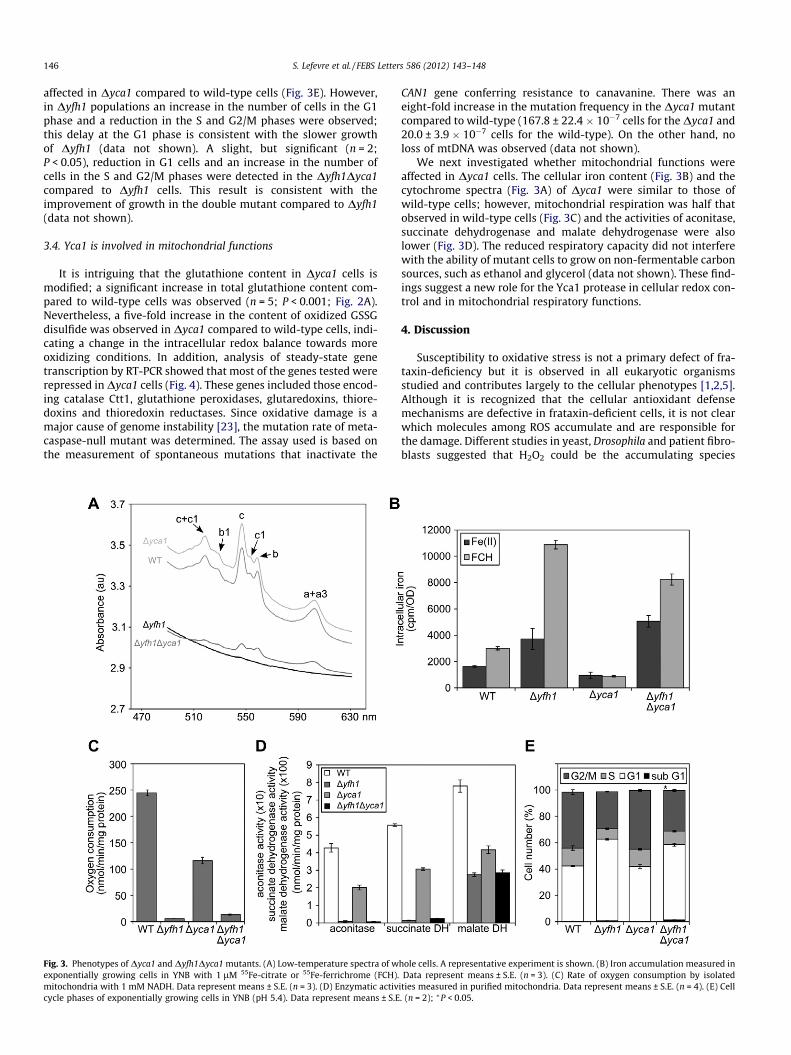
Cytochrome synthesis in vivo in Dyfh1 cells was partially re-stored by the Dyca1 deletion (Fig. 3A). For instance, cytochromesa + a3 and b levels were 10-fold increased in Dyfh1Dyca1 com-pared to the Dyfh1 cells and cytochrome c level was five-fold in-creased. However, these levels were at least four-fold lower thanthose observed in wild-type and Dyca1 cells. Total iron accumula-tion was not consistently reduced in Dyfh1Dyca1 compared to theDyfh1 mutant (Fig. 3B). Respiration was slightly but not signifi-cantly increased (Fig. 3C). This was possibly due to mitochondrialDNA loss in the Dyfh1Dyca1 mutant, which was comparable to thatobserved for the Dyfh1 mutant (98–99% in our experimental condi-tions). In addition, there was no difference in the activity of twomitochondrial iron–sulfur cluster enzymes, aconitase and succi-nate dehydrogenase, in the Dyfh1 and Dyfh1Dyca1 mutants(Fig. 3D), suggesting that iron–sulfur cluster biogenesis is not im-proved by metacaspase deletion.
A non-death function has been described for Yca1 in promotingthe G1/S transition and delaying the G2/M transition in cells grownin acidified rich media [22]. In our conditions, the cell cycle was not
Δyfh1/WTΔyfh1Δyca1/WT
Relative expression compared to the WT0 1 2 3 4 5 6
1
121
21
5
432112111
1
2
3
nd oxidized glutathione levels. Data represent means ± S.E. (n = 5). (B) Relative gene3 < n < 4). (C) ROS levels detected by flow cytometry after staining with DHR123 and
146 S. Lefevre et al. / FEBS Letters 586 (2012) 143–148
affected in Dyca1 compared to wild-type cells (Fig. 3E). However,in Dyfh1 populations an increase in the number of cells in the G1phase and a reduction in the S and G2/M phases were observed;this delay at the G1 phase is consistent with the slower growthof Dyfh1 (data not shown). A slight, but significant (n = 2;P < 0.05), reduction in G1 cells and an increase in the number ofcells in the S and G2/M phases were detected in the Dyfh1Dyca1compared to Dyfh1 cells. This result is consistent with theimprovement of growth in the double mutant compared to Dyfh1(data not shown).
3.4. Yca1 is involved in mitochondrial functions
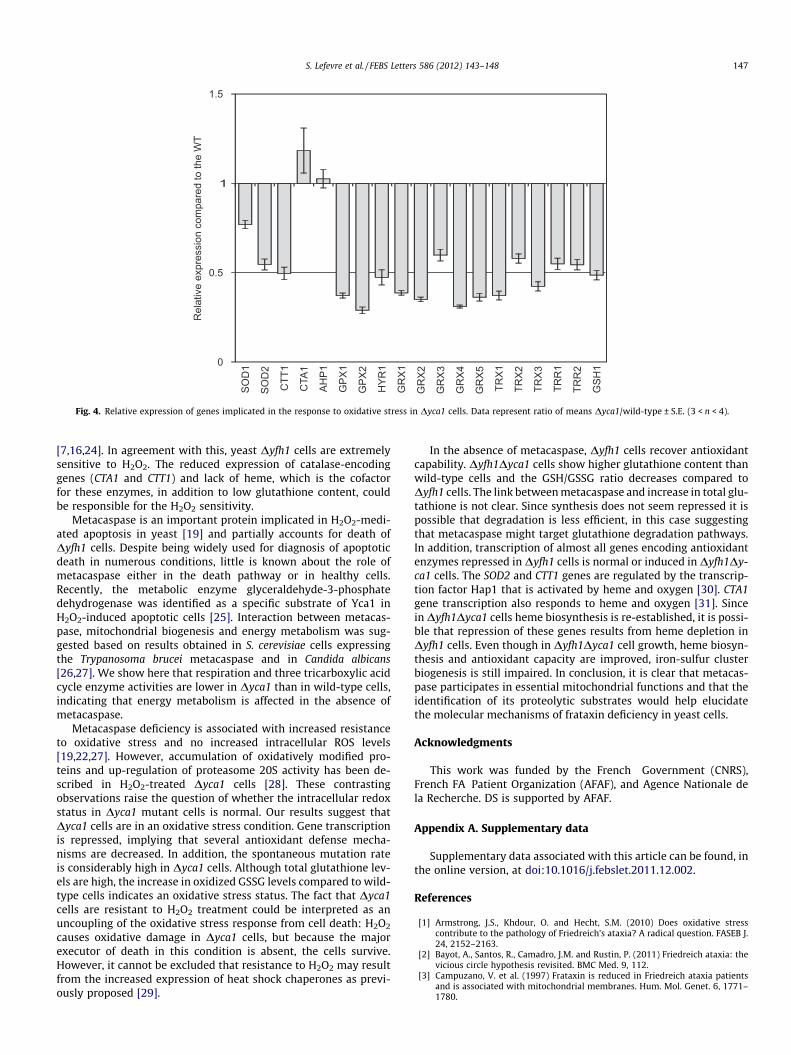
It is intriguing that the glutathione content in Dyca1 cells ismodified; a significant increase in total glutathione content com-pared to wild-type cells was observed (n = 5; P < 0.001; Fig. 2A).Nevertheless, a five-fold increase in the content of oxidized GSSGdisulfide was observed in Dyca1 compared to wild-type cells, indi-cating a change in the intracellular redox balance towards moreoxidizing conditions. In addition, analysis of steady-state genetranscription by RT-PCR showed that most of the genes tested wererepressed in Dyca1 cells (Fig. 4). These genes included those encod-ing catalase Ctt1, glutathione peroxidases, glutaredoxins, thiore-doxins and thioredoxin reductases. Since oxidative damage is amajor cause of genome instability [23], the mutation rate of meta-caspase-null mutant was determined. The assay used is based onthe measurement of spontaneous mutations that inactivate the
Fig. 3. Phenotypes of Dyca1 and Dyfh1Dyca1 mutants. (A) Low-temperature spectra of wexponentially growing cells in YNB with 1 lM 55Fe-citrate or 55Fe-ferrichrome (FCH).mitochondria with 1 mM NADH. Data represent means ± S.E. (n = 3). (D) Enzymatic activcycle phases of exponentially growing cells in YNB (pH 5.4). Data represent means ± S.E
CAN1 gene conferring resistance to canavanine. There was aneight-fold increase in the mutation frequency in the Dyca1 mutantcompared to wild-type (167.8 ± 22.4 � 10�7 cells for the Dyca1 and20.0 ± 3.9 � 10�7 cells for the wild-type). On the other hand, noloss of mtDNA was observed (data not shown).
We next investigated whether mitochondrial functions wereaffected in Dyca1 cells. The cellular iron content (Fig. 3B) and thecytochrome spectra (Fig. 3A) of Dyca1 were similar to those ofwild-type cells; however, mitochondrial respiration was half thatobserved in wild-type cells (Fig. 3C) and the activities of aconitase,succinate dehydrogenase and malate dehydrogenase were alsolower (Fig. 3D). The reduced respiratory capacity did not interferewith the ability of mutant cells to grow on non-fermentable carbonsources, such as ethanol and glycerol (data not shown). These find-ings suggest a new role for the Yca1 protease in cellular redox con-trol and in mitochondrial respiratory functions.
4. Discussion
Susceptibility to oxidative stress is not a primary defect of fra-taxin-deficiency but it is observed in all eukaryotic organismsstudied and contributes largely to the cellular phenotypes [1,2,5].Although it is recognized that the cellular antioxidant defensemechanisms are defective in frataxin-deficient cells, it is not clearwhich molecules among ROS accumulate and are responsible forthe damage. Different studies in yeast, Drosophila and patient fibro-blasts suggested that H2O2 could be the accumulating species
hole cells. A representative experiment is shown. (B) Iron accumulation measured inData represent means ± S.E. (n = 3). (C) Rate of oxygen consumption by isolated
ities measured in purified mitochondria. Data represent means ± S.E. (n = 4). (E) Cell. (n = 2); ⁄P < 0.05.
Rel
ativ
e ex
pres
sion
com
pare
d to
the
WT
1D
OS
1HS
G
2R
RT
1R
RTTRX2
TRX1
GR
X5
GR
X4
GR
X3
GR
X2
GR
X11RY
H
2XPG
1XPG
1PHA
1ATC
1TTC
2D
OS TRX3
0
0.5
1
1.5
Fig. 4. Relative expression of genes implicated in the response to oxidative stress in Dyca1 cells. Data represent ratio of means Dyca1/wild-type ± S.E. (3 < n < 4).
S. Lefevre et al. / FEBS Letters 586 (2012) 143–148 147
[7,16,24]. In agreement with this, yeast Dyfh1 cells are extremelysensitive to H2O2. The reduced expression of catalase-encodinggenes (CTA1 and CTT1) and lack of heme, which is the cofactorfor these enzymes, in addition to low glutathione content, couldbe responsible for the H2O2 sensitivity.
Metacaspase is an important protein implicated in H2O2-medi-ated apoptosis in yeast [19] and partially accounts for death ofDyfh1 cells. Despite being widely used for diagnosis of apoptoticdeath in numerous conditions, little is known about the role ofmetacaspase either in the death pathway or in healthy cells.Recently, the metabolic enzyme glyceraldehyde-3-phosphatedehydrogenase was identified as a specific substrate of Yca1 inH2O2-induced apoptotic cells [25]. Interaction between metacas-pase, mitochondrial biogenesis and energy metabolism was sug-gested based on results obtained in S. cerevisiae cells expressingthe Trypanosoma brucei metacaspase and in Candida albicans[26,27]. We show here that respiration and three tricarboxylic acidcycle enzyme activities are lower in Dyca1 than in wild-type cells,indicating that energy metabolism is affected in the absence ofmetacaspase.
Metacaspase deficiency is associated with increased resistanceto oxidative stress and no increased intracellular ROS levels[19,22,27]. However, accumulation of oxidatively modified pro-teins and up-regulation of proteasome 20S activity has been de-scribed in H2O2-treated Dyca1 cells [28]. These contrastingobservations raise the question of whether the intracellular redoxstatus in Dyca1 mutant cells is normal. Our results suggest thatDyca1 cells are in an oxidative stress condition. Gene transcriptionis repressed, implying that several antioxidant defense mecha-nisms are decreased. In addition, the spontaneous mutation rateis considerably high in Dyca1 cells. Although total glutathione lev-els are high, the increase in oxidized GSSG levels compared to wild-type cells indicates an oxidative stress status. The fact that Dyca1cells are resistant to H2O2 treatment could be interpreted as anuncoupling of the oxidative stress response from cell death: H2O2
causes oxidative damage in Dyca1 cells, but because the majorexecutor of death in this condition is absent, the cells survive.However, it cannot be excluded that resistance to H2O2 may resultfrom the increased expression of heat shock chaperones as previ-ously proposed [29].
In the absence of metacaspase, Dyfh1 cells recover antioxidantcapability. Dyfh1Dyca1 cells show higher glutathione content thanwild-type cells and the GSH/GSSG ratio decreases compared toDyfh1 cells. The link between metacaspase and increase in total glu-tathione is not clear. Since synthesis does not seem repressed it ispossible that degradation is less efficient, in this case suggestingthat metacaspase might target glutathione degradation pathways.In addition, transcription of almost all genes encoding antioxidantenzymes repressed in Dyfh1 cells is normal or induced in Dyfh1Dy-ca1 cells. The SOD2 and CTT1 genes are regulated by the transcrip-tion factor Hap1 that is activated by heme and oxygen [30]. CTA1gene transcription also responds to heme and oxygen [31]. Sincein Dyfh1Dyca1 cells heme biosynthesis is re-established, it is possi-ble that repression of these genes results from heme depletion inDyfh1 cells. Even though in Dyfh1Dyca1 cell growth, heme biosyn-thesis and antioxidant capacity are improved, iron-sulfur clusterbiogenesis is still impaired. In conclusion, it is clear that metacas-pase participates in essential mitochondrial functions and that theidentification of its proteolytic substrates would help elucidatethe molecular mechanisms of frataxin deficiency in yeast cells.
Acknowledgments
This work was funded by the French Government (CNRS),French FA Patient Organization (AFAF), and Agence Nationale dela Recherche. DS is supported by AFAF.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, inthe online version, at doi:10.1016/j.febslet.2011.12.002.
References
[1] Armstrong, J.S., Khdour, O. and Hecht, S.M. (2010) Does oxidative stresscontribute to the pathology of Friedreich’s ataxia? A radical question. FASEB J.24, 2152–2163.
[2] Bayot, A., Santos, R., Camadro, J.M. and Rustin, P. (2011) Friedreich ataxia: thevicious circle hypothesis revisited. BMC Med. 9, 112.
[3] Campuzano, V. et al. (1997) Frataxin is reduced in Friedreich ataxia patientsand is associated with mitochondrial membranes. Hum. Mol. Genet. 6, 1771–1780.

148 S. Lefevre et al. / FEBS Letters 586 (2012) 143–148
[4] Babcock, M., de Silva, D., Oaks, R., Davis-Kaplan, S., Jiralerspong, S.,Montermini, L., Pandolfo, M. and Kaplan, J. (1997) Regulation ofmitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin.Science 276, 1709–1712.
[5] Santos, R., Lefevre, S., Sliwa, D., Seguin, A., Camadro, J.M. and Lesuisse, E. (2010)Friedreich ataxia: molecular mechanisms, redox considerations andtherapeutic opportunities. Antioxid. Redox Signal. 13, 651–690.
[6] Jiralerspong, S., Ge, B., Hudson, T.J. and Pandolfo, M. (2001) Manganesesuperoxide dismutase induction by iron is impaired in Friedreich ataxia cells.FEBS Lett. 509, 101–105.
[7] Paupe, V., Dassa, E., Goncalves, S., Auchère, F., Lönn, M., Holmgren, A. andRustin, P. (2009) Impaired nuclear Nrf2 translocation undermines theoxidative stress response in Friedreich ataxia. PLoS One 4, e4253.
[8] Auchère, F., Santos, R., Planamente, S., Lesuisse, E. and Camadro, J.M. (2008)Glutathione-dependent redox status of frataxin-deficient cells in a yeastmodel of Friedreich’s ataxia. Hum. Mol. Genet. 17, 2790–2802.
[9] Pastore, A. et al. (2003) Actin glutathionylation increases in fibroblasts ofpatients with Friedreich’s ataxia: a potential role in the pathogenesis of thedisease. J. Biol. Chem. 278, 42588–42595.
[10] Pianese, L. et al. (2002) Up-regulation of c-Jun N-terminal kinase pathway inFriedreich’s ataxia cells. Hum. Mol. Genet. 11, 2989–2996.
[11] Wong, A., Yang, J., Danielson, S., Gellera, C., Taroni, F. and Cortopassi, G. (2000)Sensitivity of FRDA lymphoblasts to salts of transition metal ions. Antioxid.Redox Signal. 2, 461–465.
[12] Condò, I., Ventura, N., Malisan, F., Tomassini, B. and Testi, R. (2006) A pool ofextramitochondrial frataxin that promotes cell survival. J. Biol. Chem. 281,16750–16756.
[13] Zhang, Y., Lyver, E.R., Knight, S.A., Lesuisse, E. and Dancis, A. (2005) Frataxinand mitochondrial carrier proteins, Mrs3p and Mrs4p, cooperate in providingiron for heme synthesis. J. Biol. Chem. 280, 19794–19807.
[14] Santos, R., Buisson, N., Knight, S.A., Dancis, A., Camadro, J.M. and Lesuisse, E.(2004) Candida albicans lacking the frataxin homologue: a relevant yeastmodel for studying the role of frataxin. Mol. Microbiol. 54, 507–519.
[15] Sliwa, D., Dairou, J., Camadro, J.M. and Santos, R. (2011) Inactivation ofmitochondrial aspartate aminotransferase contributes to the respiratorydeficit of yeast frataxin-deficient cells. Biochem J., doi:10.1042/BJ20111574.
[16] Seguin, A., Bayot, A., Dancis, A., Rogowska-Wrzesinska, A., Auchère, F.,Camadro, J.M., Bulteau, A.L. and Lesuisse, E. (2009) Overexpression of theyeast frataxin homolog (Yfh1): contrasting effects on iron-sulfur clusterassembly, heme synthesis and resistance to oxidative stress. Mitochondrion 9,130–138.
[17] Englard, S. and Siegel, L. (1969) Mitochondria L-malate dehydrogenase of beefheart. Methods Enzymol. 13, 99–106.
[18] Foury, F. and Cazzalini, O. (1997) Deletion of the yeast homologue of thehuman gene associated with Friedreich’s ataxia elicits iron accumulation inmitochondria. FEBS Lett. 411, 373–377.
[19] Madeo, F. et al. (2002) A caspase-related protease regulates apoptosis in yeast.Mol. Cell 9, 911–917.
[20] Herker, E. et al. (2004) Chronological aging leads to apoptosis in yeast. J. CellBiol. 164, 501–507.
[21] Yang, H., Ren, Q. and Zhang, Z. (2008) Cleavage of Mcd1 by caspase-likeprotease Esp1 promotes apoptosis in budding yeast. Mol. Biol. Cell 19, 2127–2134.
[22] Lee, R.E., Puente, L.G., Kærn, M. and Megeney, L.A. (2008) A non-death role ofthe yeast metacaspase: Yca1p alters cell cycle dynamics. PLoS One 3, e2956.
[23] Wong, C.M., Siu, K.L. and Jin, D.Y. (2004) Peroxiredoxin-null yeast cells arehypersensitive to oxidative stress and are genomically unstable. J. Biol. Chem.279, 23207–23213.
[24] Anderson, P.R., Kirby, K., Orr, W.C., Hilliker, A.J. and Phillips, J.P. (2008)Hydrogen peroxide scavenging rescues frataxin deficiency in a Drosophilamodel of Friedreich’s ataxia. Proc. Natl. Acad. Sci. USA 105, 611–616.
[25] Silva, A., Almeida, B., Sampaio-Marques, B., Reis, M.I., Ohlmeier, S., Rodrigues,F., Vale, A.D. and Ludovico, P. (2011) Glyceraldehyde-3-phosphatedehydrogenase (GAPDH) is a specific substrate of yeast metacaspase.Biochim. Biophys. Acta 1813, 2044–2049.
[26] Szallies, A., Kubata, B.K. and Duszenko, M. (2002) A metacaspase ofTrypanosoma brucei causes loss of respiration competence and clonal deathin the yeast Saccharomyces cerevisiae. FEBS Lett. 517, 144–150.
[27] Cao, Y. et al. (2009) Candida albicans cells lacking CaMCA1-encodedmetacaspase show resistance to oxidative stress-induced death and changein energy metabolism. Fungal Genet. Biol. 46, 183–189.
[28] Khan, M.A., Chock, P.B. and Stadtman, E.R. (2005) Knockout of caspase-likegene, YCA1, abrogates apoptosis and elevates oxidized proteins inSaccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 102, 17326–17331.
[29] Lee, R.E., Brunette, S., Puente, L.G. and Megeney, L.A. (2010) Metacaspase Yca1is required for clearance of insoluble protein aggregates. Proc. Natl. Acad. Sci.USA 107, 13348–13353.
[30] Hon, T., Dodd, A., Dirmeier, R., Gorman, N., Sinclair, P.R., Zhang, L. and Poyton,R.O. (2003) A mechanism of oxygen sensing in yeast. Multiple oxygen-responsive steps in the heme biosynthetic pathway affect Hap1 activity. J. Biol.Chem. 278, 50771–50780.
[31] Skoneczny, M. and Rytka, J. (2000) Oxygen and haem regulate the synthesis ofperoxisomal proteins: catalase A, acyl-CoA oxidase and Pex1p in the yeastSaccharomyces cerevisiae; the regulation of these proteins by oxygen is notmediated by haem. Biochem J. 350 (Pt. 1), 313–319.
Copyright © 2022 FDOKUMEN




















