
Factors implicated in the generation and persistence of long-lived plasma cell-mediated autoimmunity
8
Review Factors implicated in the generation and persistence of long-lived plasma cell-mediated autoimmunity Marisa Neves a , José Delgado Alves a,b, ⁎ a Autoimmune Diseases Department, Medicina IV, Fernando Fonseca Hospital, Amadora, Portugal b CEDOC, Pharmacology Department, Faculty of Medical Sciences, New University of Lisbon, Lisbon, Portugal abstract article info Article history: Received 27 November 2010 Accepted 20 December 2010 Available online 9 January 2011 Keywords: Long-lived plasma cells Survival niches Therapeutic targets Autoimmune diseases are frequently associated with the production of autoantibodies by cells that escaped the protective mechanisms that control self-tolerance. Some of these cells develop into long-lived plasma cells which are predominantly located in the bone marrow. The generation of this particular type of cell requires specific migration, differentiation, and survival signals. The identification of some of the factors involved in these pathways has permitted the development of specific therapeutic approaches and may even provide investigators with further new therapeutic targets, particularly in autoimmune diseases associated with persistent autoantibody production. We reviewed the existing evidence for the mechanisms implicated in the perpetuation of long-lived plasma cells and the most recent therapeutic proposals in this context. © 2011 Elsevier B.V. All rights reserved. Contents 1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 375 2. Long-lived plasma cells: what are they? . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 375 3. Location . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 376 4. Differentiation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 377 5. Survival . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 378 6. Therapeutic targets . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 379 7. Final considerations/conclusions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 380 8. Take-home messages . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 380 References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 380 1. Introduction B cells are important characters in the autoimmune theatre since they can develop into antibody secreting cells (ASCs), when activated. These can be short-lived or long-lived plasma cells (PCs). The first disappear within a few days after a primary immune response, which is reflected in the rapid decrease in antibody titres found in this context. The latter are a poorly understood subset of B cells, capable of living for years or decades. These long-lived PCs may explain why, in secondary immune responses, a basal level of antibodies is maintained after the steep initial fall in antibody titres [1].They are capable of secreting antibodies in a continuous fashion and simultaneously escape from apoptotic signals: a ‘professional antibody secreting cell, with an eternal life’. This fact is extremely important for the understanding of autoimmunity as many autoimmune diseases may be specifically associated with autoantibodies produced by long-lived PCs. These cells live attached to stromal niches in bone marrow, spleen, and inflamed tissues. They lose their migration and proliferation capac- ities, becoming non-dividing fixed cells. Furthermore, they lose some typical markers of B cells (e.g. CD20), and therefore they are not directly affected by some of the current therapies. 2. Long-lived plasma cells: what are they? Long-lived PCs are non-proliferating cells, insensitive to antigen or antigen-antibody complexes [2] but still with a great capacity for producing immunoglobulins. They have an exclusive surface marker Autoimmunity Reviews 10 (2011) 375–382 ⁎ Corresponding author. Serviço de Medicina IV, Hospital Fernando Fonseca, 2720- 276 Amadora, Portugal. E-mail address: [email protected] (J.D. Alves). 1568-9972/$ – see front matter © 2011 Elsevier B.V. All rights reserved. doi:10.1016/j.autrev.2010.12.007 Contents lists available at ScienceDirect Autoimmunity Reviews journal homepage: www.elsevier.com/locate/autrev
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Factors implicated in the generation and persistence of long-lived plasma cell-mediated autoimmunity

Autoimmunity Reviews 10 (2011) 375–382
Contents lists available at ScienceDirect
Autoimmunity Reviews
j ourna l homepage: www.e lsev ie r.com/ locate /aut rev
Review
Factors implicated in the generation and persistence of long-lived plasmacell-mediated autoimmunity
Marisa Neves a, José Delgado Alves a,b,⁎a Autoimmune Diseases Department, Medicina IV, Fernando Fonseca Hospital, Amadora, Portugalb CEDOC, Pharmacology Department, Faculty of Medical Sciences, New University of Lisbon, Lisbon, Portugal
⁎ Corresponding author. Serviço de Medicina IV, Hos276 Amadora, Portugal.
E-mail address: [email protected] (J.D. Alves).
1568-9972/$ – see front matter © 2011 Elsevier B.V. Aldoi:10.1016/j.autrev.2010.12.007
a b s t r a c t
a r t i c l e i n f oArticle history:Received 27 November 2010Accepted 20 December 2010Available online 9 January 2011
Keywords:Long-lived plasma cellsSurvival nichesTherapeutic targets
Autoimmune diseases are frequently associated with the production of autoantibodies by cells that escapedthe protective mechanisms that control self-tolerance. Some of these cells develop into long-lived plasma cellswhich are predominantly located in the bone marrow. The generation of this particular type of cell requiresspecific migration, differentiation, and survival signals.The identification of some of the factors involved in these pathways has permitted the development of specifictherapeutic approaches andmay even provide investigators with further new therapeutic targets, particularlyin autoimmune diseases associated with persistent autoantibody production. We reviewed the existingevidence for the mechanisms implicated in the perpetuation of long-lived plasma cells and the most recenttherapeutic proposals in this context.
pital Fernando Fonseca, 2720-
l rights reserved.
© 2011 Elsevier B.V. All rights reserved.
Contents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3752. Long-lived plasma cells: what are they? . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3753. Location . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3764. Differentiation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3775. Survival . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3786. Therapeutic targets. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3797. Final considerations/conclusions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3808. Take-home messages . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 380References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 380
1. Introduction
B cells are important characters in the autoimmune theatre since theycan develop into antibody secreting cells (ASCs), when activated. Thesecan be short-lived or long-lived plasma cells (PCs). The first disappearwithin a fewdays after a primary immune response,which is reflected inthe rapid decrease in antibody titres found in this context. The latter are apoorly understood subset of B cells, capable of living for years or decades.These long-lived PCsmay explain why, in secondary immune responses,a basal level of antibodies is maintained after the steep initial fall inantibody titres [1].They are capable of secreting antibodies in a
continuous fashion and simultaneously escape from apoptotic signals:a ‘professional antibody secreting cell, with an eternal life’.
This fact is extremely important for the understanding ofautoimmunity as many autoimmune diseases may be specificallyassociated with autoantibodies produced by long-lived PCs. Thesecells live attached to stromal niches in bone marrow, spleen, andinflamed tissues. They lose their migration and proliferation capac-ities, becoming non-dividing fixed cells. Furthermore, they lose sometypical markers of B cells (e.g. CD20), and therefore they are notdirectly affected by some of the current therapies.
2. Long-lived plasma cells: what are they?
Long-lived PCs are non-proliferating cells, insensitive to antigen orantigen-antibody complexes [2] but still with a great capacity forproducing immunoglobulins. They have an exclusive surface marker

376 M. Neves, J.D. Alves / Autoimmunity Reviews 10 (2011) 375–382
profile and need specific survival signals to stay alive. These factors areencountered in survival niches mainly located in the bone marrow(BM), and to a lesser extent in the spleen and inflamed tissues.Provided long-lived PCs are in continued contact with those survivalsignals, it seems possible that they may live for as long as thisinteraction persists. Long-lived PCs thus have an extended andundetermined lifespan, associated with the loss of B cell differenti-ation markers, increased maturation and the persistence of survivalfactors (including cytokines, adhesion molecules, transcriptionalregulators, and modulators of apoptosis) [3]. Bone marrow plasmacells express reduced levels of CD19, CD20, CD22, CD45 and HLA-DR,and increased levels of Bcl-2 and CD138 (syndecan1). These are theonly cells expressing CD138 in humans. However, it is still not clearwhether they are long-lived PCs that reached the end of theirdifferentiation, precursors of long-lived PCs, or perhaps just anotherlevel of intermediate cells [4].
3. Location
B cells are categorized into three types: B1, marginal zone, and B2.B1 and marginal zone cells participate in innate immunity. B1 cellslocate predominately in peritoneal and pleural cavities, and marginalzone B cells are found in the spleen, outside the marginal sinus. B2cells or follicular B cells are the majority and are responsible foradaptive humoral immunity, being essentially encountered in thespleen and lymph nodes [5]. B1 cells are long-lived and constitutivelyproduce high levels of surface IgM that constitute unspecific ‘natural’antibodies, and also mucosal IgA, very useful as a first immune barrier.Mature B2 cells express high levels of IgD and are capable ofproducing specific antibodies through extensive somatic hypermuta-tion, usually when stimulated by a unique antigen. They give origin toplasmablasts and short-lived PCs, or tomemory plasma cells and long-lived PCs, after germinal-center (GC) reactions [6].
Long-lived PCs live in survival niches sustained by stromal cells,mainly in the BM. There are also some niches in the spleen and inchronically inflamed tissues, like the kidneys in patients withSystemic Lupus Erythematosus (SLE), or the synovia in patientswith rheumatoid arthritis. It seems that plasmablasts migrate to theseniches and, in the presence of survival signals, can differentiate intomature PCs, even in the absence of a specific antigen [7].
The great production of autoantibodies in autoimmune diseases isthen attributed to B2 cell successors as the high degree of affinity ofthe antibodies produced by BM PCs suggests a role for GCs in thegeneration of these cells. Nevertheless, affinity maturation is notlimited to the GC reaction. B1 cells are less understood but also appearto have a role in autoimmune diseases [8], namely in SLE [9], but alsoin Sjögren's syndrome and rheumatoid arthritis. Some suggest that B1cells are able to originate BM PCs, given the necessary conditions [10].
B cell survival or death is decided based on antigen-specificity andthe avidity of the B cell-antigen interaction. Normally, protectivesignals determine the preservation of the response towards foreignantigens, and the annihilation of self-antigen responses. The B cellreceptor (BCR) stimulates the expression of anti-apoptotic moleculesin B cells which respond to foreign antigens (mitochondria-associatedBcl-XL, Fas death effector domain-associated FLIPL, Fas apoptosisinhibitory molecule — FAIM), protecting CD40-activated B cells fromcell-death mediated by Fas (CD95). In self-antigen reactive B cells, theBCR engagement is compromised, allowing them to die. Thisprotective mechanism is sometimes corrupted, due to the action ofthe B cell co-stimulatory complex. C3-binding CD21, CD19 and CD81decrease the affinity threshold of antigen-BCR interaction, facilitatingthe maintenance of self-reactive B cells. The colligation BCR:CD21decreases type I Fas-mediated apoptosis. Type II Fas-mediatedapoptosis is not directly affected by this colligation, but can also becompromised if an increase in the anti-apoptotic molecule Bcl-2occurs, which in turn is stimulated by CD19 engagement. The
colligation of CD81 with CD21 and CD19 further decreases Fas-mediated apoptosis. In some autoimmune diseases there are self-antigens (e.g. apoptotic cells expressing DNA, nucleoproteinases onsurface blebs), and antigen-antibody complexes that can easily bindC3, inducing a decrease in Fas-mediated apoptosis. However,inhibition of apoptosis can also occur independently of the reductionin membrane Fas. In fact, the role of antigen-coated complementengagement in complement receptors for determining long-lived PCsprecursors' fate was further demonstrated with CD21–CD35 [11].Thus, the innate immune system appears to have an important role infavouring the differentiation of GC B cells into BM PCs [12].
A subset of B cells present in the follicular mantle zone can expressauto-reactive antibodies encoded by the VH4-34 heavy chain variableregion gene in an intrinsic fashion. These cells are normally contained,but may be induced in the presence of CD70, IL-2, and IL-10 [13]. Inpatients with autoimmune diseases these cells can differentiate intoPCs. Actually, high levels of VH4-34 encoded antibodies have beendescribed in many autoimmune diseases, being encountered in 55% ofSLE, 89% of multiple sclerosis and virtually all cold agglutinin diseases[14], as this gene is essential for pathogenic cold agglutinin activity[15]. Serum levels of VH4-34 encoded antibodies are increased inpatients with active SLE, seeming to relate to disease activity and theextent to which organs are affected [13]. VH4-34-expressing B cellscan also originate hematologic malignancies, including those relatedto autoimmune processes. Some authors suggest that these cells tendto be resistant to tolerance mechanisms like clonal deletion [14].
Once the differentiation of any given B cell into a long-lived PC isestablished, responsiveness to germinal-center mediators is lost.Plasmablasts have less chemokine receptors associated with thecontrol of GC traffic (CXC-chemokine receptor 5 — CXCR5, CC-chemokine receptor 7 — CCR7) and therefore cannot return to GCs.Plasmablasts express CXCR4 (CXC-chemokine receptor 4), which hasCXCL12 (chemokine ligand 12, also named stroma-derived factor —
SDF1) as its ligand. CXCL12 is expressed mostly in the bone marrowstroma, but can also be present in the spleen red pulp and in themedullary cords of the lymph nodes [16], creating concentrationgradients that attract plasmablasts. Nevertheless, it is known thatCXCR4 is not essential for PC precursors homing to the BM. In fact,there is evidence that BM PCs also express CXCR6 and CXCR10,suggesting a possible additional role of other chemokine receptors[17]. When plasmablasts get close to the stromal cells, they are able toreplace old PCs that have lost their migration capacity [7]. ThesedislodgedPCs are prone to diewithindays as they lose contactwith thesurvival niche, which constitutes away of keeping the number of long-lived PCs constant. CXCL12 also induces adhesion associated with theincreased expression of VCAM-1 (vascular cell-adhesion molecule 1),hence controlling the fixation of PC precursors in the niche. Since PCsresiding in the survival niches have lost theirmigration ability, CXCL12then becomes a survival signal [2]. CXCL12+VCAM-1+stromal cellsare 1% of total BM cells and BM PCs constitute a similar portion,suggesting an individual relationship between them [18]. VCAM-1 isexpressed at the surface of bone marrow epithelial cells, and bindsα4β1-integrin (very late antigen-4 — VLA-4), which is highlyexpressed in the plasma membranes of PCs homing to the BM [19].
BM stromal cells also produce BLyS (B Lymphocyte Stimulator)and APRIL (a proliferation-inducing ligand), members of the TNFsuperfamily of cytokines [20]. Both participate in the survival of BMPCs, but APRIL also seems to be a migration factor, as it is known tobind glysosamyn-rich syndecans, like the PC marker CD138, thusfunctioning in PCs homing to the BM [21].
In addition to CD138, there are other molecules expressed by PCsthat seem to be involved in adhesion to BM stroma, like CD44, CD22,endothelial-cell selectin (E-selectin) ligand, platelet selectin (P-selectin)ligand, CD11a and CD18 [7,16]. CD44 and CD22 are type-I transmem-brane sialoglycoproteins of the immunoglobulin superfamily. CD44participates in adhesion to extracellular matrix through its ligand,
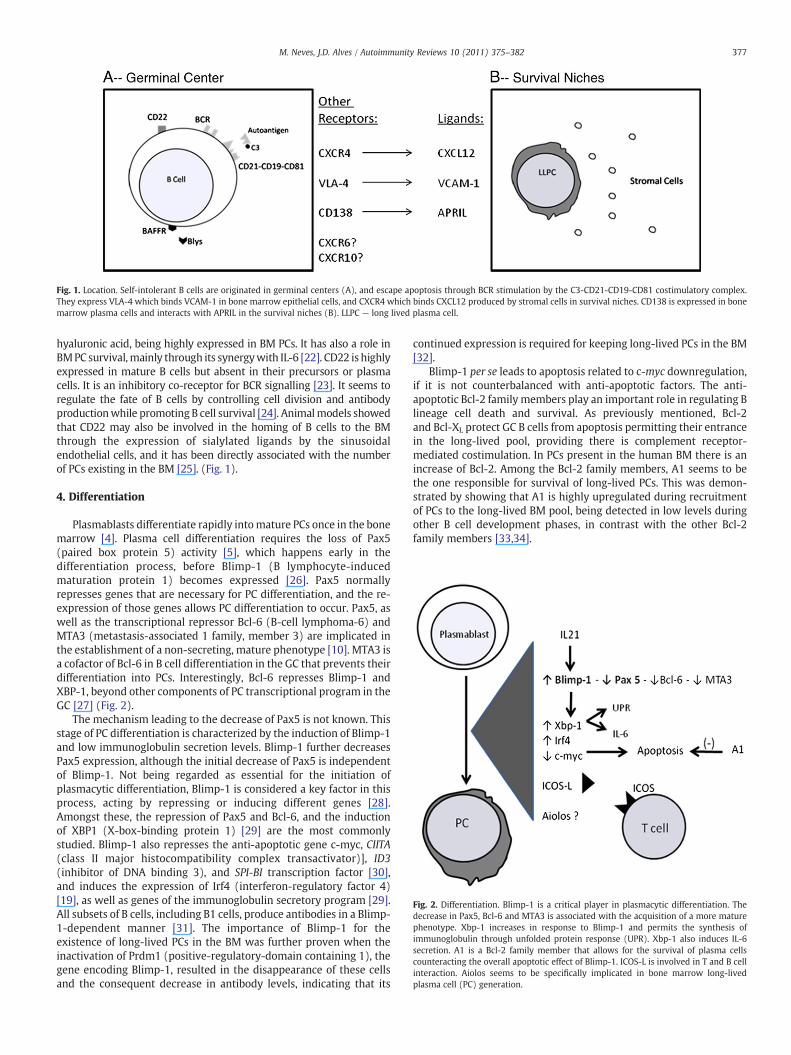
Fig. 1. Location. Self-intolerant B cells are originated in germinal centers (A), and escape apoptosis through BCR stimulation by the C3-CD21-CD19-CD81 costimulatory complex.They express VLA-4 which binds VCAM-1 in bone marrow epithelial cells, and CXCR4 which binds CXCL12 produced by stromal cells in survival niches. CD138 is expressed in bonemarrow plasma cells and interacts with APRIL in the survival niches (B). LLPC — long lived plasma cell.
Fig. 2. Differentiation. Blimp-1 is a critical player in plasmacytic differentiation. Thedecrease in Pax5, Bcl-6 and MTA3 is associated with the acquisition of a more maturephenotype. Xbp-1 increases in response to Blimp-1 and permits the synthesis ofimmunoglobulin through unfolded protein response (UPR). Xbp-1 also induces IL-6secretion. A1 is a Bcl-2 family member that allows for the survival of plasma cellscounteracting the overall apoptotic effect of Blimp-1. ICOS-L is involved in T and B cellinteraction. Aiolos seems to be specifically implicated in bone marrow long-livedplasma cell (PC) generation.
377M. Neves, J.D. Alves / Autoimmunity Reviews 10 (2011) 375–382
hyaluronic acid, being highly expressed in BM PCs. It has also a role inBMPC survival,mainly through its synergywith IL-6 [22]. CD22 is highlyexpressed in mature B cells but absent in their precursors or plasmacells. It is an inhibitory co-receptor for BCR signalling [23]. It seems toregulate the fate of B cells by controlling cell division and antibodyproductionwhile promoting B cell survival [24]. Animalmodels showedthat CD22 may also be involved in the homing of B cells to the BMthrough the expression of sialylated ligands by the sinusoidalendothelial cells, and it has been directly associated with the numberof PCs existing in the BM [25]. (Fig. 1).
4. Differentiation
Plasmablasts differentiate rapidly intomature PCs once in the bonemarrow [4]. Plasma cell differentiation requires the loss of Pax5(paired box protein 5) activity [5], which happens early in thedifferentiation process, before Blimp-1 (B lymphocyte-inducedmaturation protein 1) becomes expressed [26]. Pax5 normallyrepresses genes that are necessary for PC differentiation, and the re-expression of those genes allows PC differentiation to occur. Pax5, aswell as the transcriptional repressor Bcl-6 (B-cell lymphoma-6) andMTA3 (metastasis-associated 1 family, member 3) are implicated inthe establishment of a non-secreting, mature phenotype [10]. MTA3 isa cofactor of Bcl-6 in B cell differentiation in the GC that prevents theirdifferentiation into PCs. Interestingly, Bcl-6 represses Blimp-1 andXBP-1, beyond other components of PC transcriptional program in theGC [27] (Fig. 2).
The mechanism leading to the decrease of Pax5 is not known. Thisstage of PC differentiation is characterized by the induction of Blimp-1and low immunoglobulin secretion levels. Blimp-1 further decreasesPax5 expression, although the initial decrease of Pax5 is independentof Blimp-1. Not being regarded as essential for the initiation ofplasmacytic differentiation, Blimp-1 is considered a key factor in thisprocess, acting by repressing or inducing different genes [28].Amongst these, the repression of Pax5 and Bcl-6, and the inductionof XBP1 (X-box-binding protein 1) [29] are the most commonlystudied. Blimp-1 also represses the anti-apoptotic gene c-myc, CIITA(class II major histocompatibility complex transactivator)], ID3(inhibitor of DNA binding 3), and SPI-BI transcription factor [30],and induces the expression of Irf4 (interferon-regulatory factor 4)[19], as well as genes of the immunoglobulin secretory program [29].All subsets of B cells, including B1 cells, produce antibodies in a Blimp-1-dependent manner [31]. The importance of Blimp-1 for theexistence of long-lived PCs in the BM was further proven when theinactivation of Prdm1 (positive-regulatory-domain containing 1), thegene encoding Blimp-1, resulted in the disappearance of these cellsand the consequent decrease in antibody levels, indicating that its
continued expression is required for keeping long-lived PCs in the BM[32].
Blimp-1 per se leads to apoptosis related to c-myc downregulation,if it is not counterbalanced with anti-apoptotic factors. The anti-apoptotic Bcl-2 family members play an important role in regulating Blineage cell death and survival. As previously mentioned, Bcl-2and Bcl-XL protect GC B cells from apoptosis permitting their entrancein the long-lived pool, providing there is complement receptor-mediated costimulation. In PCs present in the human BM there is anincrease of Bcl-2. Among the Bcl-2 family members, A1 seems to bethe one responsible for survival of long-lived PCs. This was demon-strated by showing that A1 is highly upregulated during recruitmentof PCs to the long-lived BM pool, being detected in low levels duringother B cell development phases, in contrast with the other Bcl-2family members [33,34].

378 M. Neves, J.D. Alves / Autoimmunity Reviews 10 (2011) 375–382
XBP-1 is induced by Blimp-1 and consequently acts downstream inrelation to it. However, the initial trigger for the expression of XBP-1seems to be the loss of Pax5-mediated suppression and it has beenproposed that the unfolded protein response (UPR) and immuno-globulin production are the main factors responsible for highexpression of XBP-1, and not the direct effect of Blimp-1 [26]. Theactive form of XBP-1 is a spliced isoform that mediates the UPR andallows the endoplasmic reticulum (ER) to resist the stress caused byan enhanced production of immunoglobulins, by physically expand-ing it. In turn, UPR induces XBP-1 splicing. UPR itself is a controlledmodification of expressed genes to a secretory profile, initiated bychanges in the ER function. XBP-1 also promotes other cellularchanges required for the secretion of antibodies, like inducingsecretory genes and increasing cell dimensions, organelles andprotein synthesis [35]. Without the XBP-1-induced UPR, PCs die as aconsequence of ER stress and accumulation of unfolded protein. Theregulation of XBP-1 in PC differentiation is complex and many othermediators seem to be involved. IL-4 controls XBP-1 transcription,while XBP-1 and IL-6 seem to mutually enhance each other'sproduction, ensuring PC differentiation and survival [36]. IRF4 is atranscriptional activator that also appears to be involved in PCdifferentiation, being related to Blimp-1 and XBP-1 in a poorlyunderstood manner [26].
IL-21 is a cytokine produced by activated CD4+ T cells and itsreceptor is present in T, B, NK and dendritic cells. In B cells, IL-21R isexpressed at high levels on follicular B cells, also being detected innaive and GC B cells, but not being expressed on memory B cells andPCs [37,38]. As such, major IL-21 effects are seen in naive B cells, but itis less influential on memory B cell associated-responses [39]. In thecontext of CD40 stimulation, it has an important influence on B cellresponse to antigen and immunoglobulin secretion, and is involved inseveral aspects of B cell proliferation and differentiation into PC [40],but it is not known whether its actions are predominately inside oroutside the GC. Some allelic variations of the IL-21 gene are associatedwith SLE [41]. Its participation in PC differentiation has been proven tobe much greater than the effects of other cytokines, like IL-10 and IL-2[42]. It is a strong inducer of transcription factors expression, likeBlimp-1, XBP1 and IRF4. More recently, it has been shown to be acomplex mediator, as it also induces the expression of Bcl-6, whichprotects B cells from IL-21-induced apoptosis [43,44]. IL-21 has asynergistic effect with BLyS, an important B cell survival factor [45]that also provides T cell costimulation [46], in differentiating subsetsof memory B cells into PCs in an antigen-independent manner [47].The importance of this synergism was demonstrated in patients withSLE who presented high levels of both factors [48]. IL-2 seems to be anenhancer of IL-21, whilst IL-4 inhibits IL-21-induced PC differentiation[49]. Other cytokines like IL-5, IL-6 and IL-10 also seem to be a part ofthe PC differentiation process as theymay induce Prdm1 transcription[19].
IL-21 was shown to be involved in the development of an SLE-likedisease in mice [50–52], as well as in murine models of rheumatoidarthritis [53]. Furthermore, patients with rheumatoid arthritis wereshown to have increased levels of IL-21R in synovial fibroblasts andmacrophages [54]. A role for IL-21 in the pathogenesis of primarySjögren's syndrome has also been suggested as high plasma levelshave been found in these patients [55].
A new mechanism for chronic autoimmunity was elucidated byidentification of a subset of murine extrafollicular CD4+ T cellsexpressing the inducible costimulator (ICOS) that activate B cellsthrough IL-21 overexpression [52,56]. ICOS is a T-cell specificmolecule which has a ligand (ICOS-L) that is constitutively expressedin antigen-presenting cells. Thus, ICOS has an important role in T- andB-cell interaction. It was shown that patients with SLE had increasedlevels of ICOS-expressing T cells in the peripheral blood. ICOS-L wasshown to be reduced after interaction with ICOS, and this is probablyrelated to memory B cells and PC differentiation in SLE. It was
demonstrated that the subsets of B cells most associated with thisphenomenon are CD27+, CD20+, CD23−, and CD38−. When theyreacquire ICOS-L expression, they lose CD20 and differentiate into PCs.ICOS-L blockade was shown to be beneficial in a murine model oflupus nephritis [57]. IL-10 induced in CD4+ T cells expressing ICOSalso has a significant role in PC development, together with otherinterleukins [58].
Aiolos, a member of the Ikaros family of zinc-finger-containingtranscriptional regulators, is specifically required for the generation oflong-lived PCs in the BM, unlike other transcription factors [59]. Itsmechanism of action is not known, or even whether it promotesdifferentiation and/or survival of long-lived PCs. However, as amember of the Ikaros family, it is thought to act as a chromatinmodifier, maybe interacting with Blimp-1 and therefore with littleprobability of influencing survival of non-dividing PCs [7]. Interest-ingly, it was demonstrated that Blimp-1, XBP-1 and Bcl-6 were notaffected in Aiolos-deficient mice [6].
5. Survival
A significant range of different molecules has been proved to beinvolved in the survival and maintenance of long-lived PCs in the BM.The tumor necrosis factor (TNF) superfamily of cytokines includesBLyS and APRIL, and these are implicated in the activation and survivalof B lymphocytes [60,61]. They exist in both soluble and membrane-bound forms, being ligands for three receptors on the membrane of Bcells: BLyS receptor 3 (BR3 or BAFF-R), B cell maturation antigen(BCMA) and transmembrane activator and calcium modulator andcyclophilin ligand interactor (TACI). BLyS binds to BR3, TACI andBCMA. BR3 is selective for BLyS and BLyS binds more strongly withBR3 than with the other receptors. BLyS signalling through BR3 and inthe dependence of BCR is essential to B cell differentiation [62]. APRILhas a reverse affinity as it also binds to BCMA and TACI, but not to BR3.In addition to BCMA and TACI, APRIL binds specifically to proteogly-cans in cells of the extracellular matrix. BCMA is upregulated on PCs,whilst BR3 and TACI are downregulated [35,59].
BLyS has a preponderant role in early B cell differentiation and isresponsible for the survival of autoreactive B cells in GCs, as BR3signalling antagonizes apoptosis through Bcl-2 family members [63–65]. At this stage TACI and BCMA appear to be irrelevant. BCMA andTACI signalling through APRIL are more relevant to antigen-experienced B cells, and it has been shown that APRIL is responsiblefor the establishment of the BM PC reservoir [65]. APRIL-proteoglycaninteraction on the surface of CD138-expressing cells induces the anti-apoptotic gene (Bcl-XL) expression and triggers TACI/BCMA-mediatedactivation and survival, a crucial step in the transition to long-lived PC[66]. Actually, BCMA is essential for survival of long-lived PCs [63],while TACI have both positive and negative effects on B cellhomeostasis. Thus, BLyS and APRIL are implicated in autoimmuneprocesses, and although some redundant effects exist, BLyS-BR3interaction is essential for viability of B cells, while APRIL-BCMA andAPRIL-TACI interactions are implicated more in later processes thatculminate in antibody production, despite APRIL appearing not to benecessary for normal B cell development [67]. Conversely, it has beendemonstrated that BLyS has little effect on the long-lived PC pool andsecondary immune response [68].
BLyS was demonstrated to be increased in patients with systemicautoimmune diseases, and abnormal APRIL levels were also found[13,69]. In mice models, BLyS was shown to induce a lupus nephritis-like phenotype. In patients with SLE, anti-dsDNA antibody titers areassociated with BLyS levels, even before the establishment of clinicaldisease, suggesting that BLyS may be a trigger for the autoimmuneresponse in some SLE patients. This mechanism appears to be T-cellindependent [65]. An association between BLyS levels and SLE activitywas further demonstrated [70,71], but this correlation was proved tobe stronger with BLyS mRNA [13,72]. BLyS is also increased in patients

Fig. 3. Survival. Long-lived plasma cells live in niches constituted by stromal cells andare dependent on survival signals. These include the APRIL/BLyS-BCMA interaction, andCXCR4/CXCL12 colligation. IL-6 and IL-10 also seem to have an important role in thesurvival of these cells. IL-6 is produced after plasma cell-stromal cell interaction. IL-10precedes the action of CXCL12. CD93 is also important for plasma cell survival. LLPC —
long lived plasma cell.
379M. Neves, J.D. Alves / Autoimmunity Reviews 10 (2011) 375–382
with rheumatoid arthritis and Sjögren's Syndrome and an associationwith the respective antibody titers was reported [13,73–78]. Otherautoimmune disorders that present elevated BLyS levels are systemicsclerosis [79], multiple sclerosis [80], Wegener's granulomatosis [81],immune thrombocytopenic purpura [82], autoimmune hepatitis [83],primary biliary cirrhosis and type I diabetes [84]. Curiously, BLyS wasfound to be augmented in patients with chronic hepatitis C virusinfection with autoimmune manifestations [85].
BLyS and APRIL are potentially produced by the same cells inresponse to the same stimulus [86]. BLyS can be produced by myeloidcells (monocytes, macrophages, dendritic cells, astrocytes) andneutrophils, also being detected in T cells, stromal cells and mastcells. However, BLyS is predominantly produced by stromal cells,controlling the size of the peripheral B cell pool through itsconstitutive expression. In the absence of stromal cells, hematopoi-etic-derived BM cells are also capable of producing BLyS, although inlevels just sufficient for follicular B cell differentiation. There areseveral stimulators of BLyS production that can increase its expressionabove the constitutive standing levels. Myeloid cells are sensitive toCD40L, IL-10, IFN-α, IFN-γ [87], and microbial molecules [88]. IFN-γexpressed by peripheral T cells is increased in SLE and is associatedwith elevated BLyS production by monocytes [89]. Neutrophils arestimulated mainly by G-CSF [90].
ΔBLyS is a splice variant of BLyS due to inefficient cleavage [91,92]. Itforms inactive heteromerswith BLyS and antagonizes its activity.ΔBLySexpression appears to be increased in CD68 cells [93]. BLyS and APRILcan also form heterotrimers when coexpressed. These BLyS-APRILcomplexes seem to influence mature B cells through TACI and weredetected in high levels in some patientswith autoimmune diseases [86].
IL-10 is a regulatory/tolerogenic cytokine, with a predominantlyanti-inflammatory activity, but it is also capable of stimulating someimmune pathways, mainly associated with B cells, suggesting apossible role for the pathogenesis of at least some autoimmunediseases. It is elevated in patients with SLE and is correlated withdisease activity. It is thought to increase serum BLyS levels in thesepatients. In F1 (NZB/W) hybrid mice, IL-10 seems to precede theCXCL12 action in autoreactive B cells [94]. It was suggested that IL-10contributes towards maintaining B cell hyperactivity and antibodysecretion [23].
Plasma cells lose the migratory capacity of plasmablasts as themigratory stimulus shifts to survival signals. This is the case withCXCL12 that is produced by stromal cells. It attracts plasmablastsexpressing CXCR4 and maintains resident PCs expressing the samereceptor in the survival niches, as well as participating in differentdevelopmental pathways of the B lineage.
IL-6 is a complex cytokine, known for both pro-inflammatory andanti-inflammatory actions, and is a key player in many cellular andmolecular pathways associated with autoimmunity. It is increased inmost of the systemic diseases and SLE is no exception. Presence of IL-6is an important survival factor, although not essential, and its mainrole is to support PC differentiation [66]. IL-6 appears to be producedin response to interaction between PCs and stromal cells [95], and thisseems to be dependent on the adhesion between VLA-4 and probablyfibronectin. There is synergism between IL-6 and CD44, and it hasbeen suggested that CD44 may stimulate IL-6 production as well.Some regulators of IL-6 expression are C/EBP-β, NF-κB and XBP-1 [36],and IL-6 appears to induce in turn Blimp-1 and Bcl-2 expression [19].
IL-5 is involved in B cell growth and differentiation, and seems tohave a slight influence on PC survival [5].
CD93 is a transmembrane protein thought to function in theadhesion and elimination of apoptotic cells. It is induced in the earlierphase of B cell differentiation and during PC differentiation, and ishighly expressed in long-lived PCs. In contrast, GC B cells andmemoryB cells do not express CD93. There is evidence that CD93 is a criticalsurvival factor for PCs in the BM, as CD93-deficient mice have adecrease in antibody secretion and less PCs in the BM [96] (Fig. 3).
6. Therapeutic targets
With the increasing knowledge of immune mediators implicatedin the genesis and persistence of autoimmune diseases, new potentialtherapeutic targets have been developed [97]. Investigators arelooking at important molecules and key cells, paying particularattention to plasmablasts [98]. We now know that there are twosources of antibodies, short-lived and long-lived PCs, and greatattention has been given to long-lived PCs as potential therapeutictargets in refractory autoimmune diseases.
Rituximab is an anti-CD20 chimeric mouse–human monoclonalantibody used for the treatment of rheumatoid arthritis as well asother autoimmune conditions. It depletes B cells from the early to themature stages of development, leaving B cell precursors and PCs(CD20-cells) unaffected. In SLE, rituximab has produced conflictingresults, with several reasons being put forward to explain therefractoriness present in some cases. One of them is the continuedsecretion of autoantibodies by long-lived CD20-PCs [99]. Rituximabalso appears to have an effect in inducing tolerance by reducing thepool of VH4.34-bearing anti-dsDNA-expressing B cells in GCs of SLEpatients. It has been demonstrated that depletion of B cells byrituximab increases the level of free BLyS, inducing persistence orrelapsing of autoreactive B cells. As previously mentioned, some cells(e.g. stromal cells) produce BLyS constitutively. When B cells that aresustained by BLyS are depleted, circulating levels of BLyS are increased,as it is not retained in those cells [100]. This may be a mechanismexplaining elevated BLyS levels in patients treated with rituximab[101]. In turn, elevated BLyS levels contribute to recrudescence of self-reactive B cells [102]. Furthermore, it has been shown that rituximabtreatment inducedmore autoimmune phenomena associatedwith thewithdrawal of the drug in SLE patients with high BLyS levels. Thus,rituximab should probably be used with caution in patients with SLEand increased levels of free plasma BLyS, and an anti-BLyS treatmentafter rituximab could be beneficial [100]. Restriction of BLyS levelsmayalso be beneficial because autoreactive B cells are more dependent onthis survival signal than regular B cells [103].
Belimumab is a fully human monoclonal antibody that inhibits thebinding of soluble BLyS to BAFF-R, TACI and BCMA, affecting young Bcells in the periphery, without the loss of memory B cells and BM PCs,hence preserving secondary immunity [65]. Belimumab has recentlybeen approved for the treatment of SLE. Belimumab (1 and 10 mg/Kg/day) reduced disease activity and flares, as well as allowing for thereduction of corticoid dosages (independently of belimumab dose

380 M. Neves, J.D. Alves / Autoimmunity Reviews 10 (2011) 375–382
used). BR3-Fc, AMG-623, and anti-BR3 antibodies are other treatmentoptions against BLyS, and are currently under study.
Atacicept is a recombinant fusion protein of TACI receptor andhuman IgG that binds and inhibits both BLyS and APRIL effects on Bcells, hence being (in theory)more potent than belimumab alone [23].In animal models of SLE, it was associated with a global reduction inthe B cell count and an inhibition of B cell responsiveness, with areduction of the anti-DNA antibody titres, proteinuria, and improve-ment in survival. It has now been submitted to phase II/III trials in SLE,although in a former phase II trial it had to be suspended because ofaugmented risk of severe infections [65].
Indirect ways to decrease BLyS levels may also include the use ofan IFN-γ biologic antagonist [89].
Epratuzumab is a fully human monoclonal antibody anti-CD22 thatprevents CD22 adhesive function [97], and leads to B cell apoptosis. Ithas beenshownto improve clinical parameters inpatientswithSLE [23].
Anti-IL-10 murine antibodies led to clinical improvement inpatients with SLE, but all of them developed neutralizing antibodies[23]. IL-12 exists in an inverse relation to IL-10, being decreased inSLE. Some authors suggest that administration of IL-12 may havebeneficial effects by decreasing IL-10 levels [97].
Anti-IL-6 monoclonal antibody (tocilizumab) has proven efficacyin several immune-mediated diseases, including SLE [104].
A monoclonal antibody anti-CXCL12 has been demonstrated toameliorate lupus nephritis in NZB/W mice, thus proving a potentialtherapeutic target. Other suggested therapeutic targets are the C3-binding costimulatory complex and ICOS-L. Aiolos is also a potentialtarget for new alternative therapies, as it is specifically used in thegeneration of long-lived BM PCs [105].
7. Final considerations/conclusions
The mechanisms underlying the establishment and maintenanceof long-lived PCs in the BM are being progressively elucidated. Theincreasing knowledge of these cells will hopefully allow the creationof alternative therapies for autoimmune diseases associated with PC-dependent autoantibodies.
8. Take-home messages
• Long-lived plasma cells are nonproliferating cells capable ofcontinuously secreting antibodies.
• The survival of long-lived plasma cells is dependent on signalspresent in the stromal niches.
• New therapies are being developed to overcome the difficultiesimposed by long-lived plasma cells in the treatment of autoimmunediseases.
References
[1] Hoyer BF, Mumtaz IM, Yoshida T, Hiepe F, Radbruch A. How to cope withpathogenic long-lived plasma cells in autoimmune diseases. Ann Rheum Dis2008;67:iii87–9.
[2] Moser K, Muehlinghaus G, Manz R, Mei H, Voigt C, Yoshida T, et al. Long-livedplasma cells in immunity and immunopathology. Immunol Lett 2006;103:83–5.
[3] Tarlinton DM, Hodgkin PD. Targeting plasma cells in autoimmune diseases. J ExpMed 2004;199:1451–4.
[4] Moser K, Tokoyoda K, Radbruch A, McLennan I, Manz R. Stromal niches, plasmacell differentiation and survival. Curr Opin Immunol 2006;18:265–70.
[5] Fairfax KA, Kallies A, Nutt SL, Tarlinton DM. Plasma cell development: from B-cellsubsets to long-term survival niches. Semin Immunol 2008;20:49–58.
[6] Browning JL. B cells move to centre stage: novel opportunities for autoimmunedisease treatment. Nature 2006;5:564–76.
[7] Radbruch A, Muehlinghaus G, Luger EO, Inamine A, Smith KGC, Dörner T, et al.Competence and competition: the challenge of becoming a long-lived plasmacell. Nature 2006;6:741–50.
[8] Duan B, Morel L. Role of B-1a cells in autoimmunity. Autoimmun Rev 2006;6:403–8.
[9] Milner EC, Anolik J, Cappione A, Sanz I. Human innate B cells: a link between hostdefence and autoimmunity? Springer Semin Immunopathol 2005;26:433–52.
[10] Fairfax K, Corcoran LM, Pridans C, Huntington ND, Kallies A, Nutt SL, et al.Different Kinetics of Blimp-1 Induction in B Cell Subsets Revealed by ReporterGene. J Immunol 2007;178:4104–11.
[11] Gatto D, Pfister T, Jegerlehner A, Martin SW, Kopf M, Bachmann MF. Complementreceptors regulate differentiation of bone marrow plasma cell precursorsexpressing transcription factors Blimp-1 and XBP-1. J Exp Med 2005;201:993–1005.
[12] Mongini PKA, Jackson AE, Tolani S, Fattah RJ, Inman JK JK. Role of complement-binding CD21/CD19/CD81 in enhancing human B cell protection from Fas-mediated apoptosis. J Immunol 2003;171:5244–54.
[13] Pugh-Bernard AE, Silverman GJ, Cappione AJ, Villano ME, Ryan DH, Insel RA, et al.Regulation of inherently autoreactive VH4-43 B cells in the maintenance ofhuman B cell tolerance. J Clin Invest 2001;108:1061–70.
[14] Arons E, Suntum T, Stetler-Stevenson M, Kreitman RJ. VH4-34+ hairy cellleukemia, a new variant with poor prognosis despite standard therapy. Blood2009;114:4687–95.
[15] Pascual V, Victor K, Spellerberg M, Hamblin TJ, Stevenson FK, Capra JD. VHrestriction among human cold agglutinins. The VH4-21 gene segment is requiredto encode anti-I and anti-i specificities. J Immunol 1992;149:2337–44.
[16] Hargreaves DC, Hyman PL, Lu TT, Ngo VN, Bidgol A, Suzuki G, et al. A coordinatedchange in chemokine responsiveness guides plasma cell movements. J Exp Med2001;194:45–56.
[17] Muehlinghaus G, Cigliano L, Huehn S, Peddinghaus A, Leyendeckers H, Hauser AE,et al. Regulation of CXCR3 and CXCR4 expression during terminal differentiationof memory B cells into plasma cells. Blood 2005;105:3965–71.
[18] Tokoyoda K, Zehentmeier S, Chang H, Radbruch A. Organization andmaintenanceof immunological memory by stroma niches. Eur J Immunol 2009;39:2095–9.
[19] Shapiro-Shelef M, Calame K. Regulation of plasma-cell development. Nature2005;5:230–42.
[20] Schaumann DHS, Tuischer J, Ebell W, Manz RA, Lauster R. VCAM-1-positivestromal cells from human bone marrow producing cytokines for B lineageprogenitors and for plasma cells: SDF-1, flt3L, and BAFF. Mol Immunol 2007;44:1606–12.
[21] Ingold K, Zumsteg A, Tardivel A, Huard B, Steiner Q, Cachero TG, et al.Identification of proteoglycans as the APRIL-specific binding partners. J ExpMed 2005;201:1375–83.
[22] Cassesse G, Arce S, Hauser AE, Lehnert K, Moewes B, MostaracM, et al. Plasma cellsurvival is mediated by synergistic effects of cytokines and adhesion-dependentsignals. J Immunol 2003;171:1684–90.
[23] Diamanti AP, Rosado MM, Carsetti R, Valesini G. B cells in SLE: different biologicdrugs for different pathogenic mechanisms. Autoimmun Rev 2007;7:143–8.
[24] Onodera T, Poe JC, Tedder TF, Tsubata T. CD22 regulates time course of both B celldivision and antibody response. J Immunol 2008;180:907–13.
[25] Nitschke L, Flyd H, Ferguson DJP, Crocker PR. Identification of CD22 ligands onbone marrow sinusoidal endothelium implicated in CD22-dependent homing ofrecirculating B Cells. J Exp Med 1999;189:1513–8.
[26] Kallies A, Hasbold J, Fairfax K, Pridans C, Emslie D, McKenzie BS, et al. Initiation ofplasma-cell differentiation is independent of the transcription factor Blimp-1.Immunity 2007;26:555–66.
[27] Fujita N, Jaye DL, Geigerman C, Akyildiz A, Mooney R, Boss JM, et al. MTA3 and theMi/NuRD complex regulate cell fate during B lymphocyte differentiation. Cell2004;119:75–86.
[28] Sciammas R, Davis MM. Modular nature of Blimp-1 in the regulation of geneexpression during B cell maturation. J Immunol 2004;172:5427–40.
[29] Shaffer AL, Lin K, Kuo TC, Yu X, Hurt EM, Rosenwald A, et al. Blimp-1 orchestratesplasma cell differentiation by extinguishing the mature B cell gene expressionprogram. Immunity 2002;17:51–62.
[30] Lin F, Kuo H, Ying H, Yang F, Lin K. Induction of apoptosis in plasma cells by Blymphocyte-induced maturation Proein-1 knockdown. Cancer Res 2007;67:1914–23.
[31] Fairfax KA, Corcoran LM, Pridans C, Huntington ND, Kallies A, Nutt SL, et al.Different kinetics of Blimp-1 induction in B cells subsets revealed by reportergene. J Immunol 2007;178:4104–11.
[32] Shapiro-Shelef M, Lin K, Savitsky D, Liao J. Blimp-1 is required for maintenance oflong-lived plasma cells in the bone marrow. J Exp Med 2005;202:1471–6.
[33] Knödel M, Kuss AW, Lindemann D, Berberich I, Schimpl A. Reversal of Blimp-1-mediated apoptosis by A1, a member of the Bcl-2 family. Eur J Immunol 1999;29:2988–98.
[34] Tomayko MM, Cancro MP. Long-lived B cells are distinguished by elevatedexpression of A1. J Immunol 1998;160:107–11.
[35] Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee A, Qian S, Zhao H, et al. XBP1,downstreamof Blimp-1, expands the secretory apparatus and other organelles, andincreases protein synthesis in plasma cell differentiation. Immunity 2004;21:81–93.
[36] Iwakoshi NN, Lee A, Vallabhajosyula P, Otipoby KL, Rajewsky K, Glimcher LH.Plasma cell differentiation and the unfolded protein response intersect at thetranscription factor XBP-1. Nature 2003;4:321–9.
[37] Good KL, Bryant VL, Tangye SG. Kinetics of human B cell behaviour andamplification of proliferative responses following stimulation with IL-21. JImmunol 2006;177:5236–47.
[38] Brandt K, Bulfone-Paus S, Foster DC, Ruckert R. Interleukin-21 inhibits dendriticcell activation and maturation. Blood 2003;102:4090–8.
[39] Kuchen S, Robbins R, Sims GP, Sheng C, Phillips TM, Lipsky PE, et al. Essential roleof IL-21 in B cell activation, expansion, and plasma cell generation during CD4+ Tcell–B cell collaboration. J Immunol 2007;179:5886–96.
[40] Konforte D, Simard N, Paige CJ. IL-21: an executor of B cell fate. J Immunol2009;182:1781–7.

381M. Neves, J.D. Alves / Autoimmunity Reviews 10 (2011) 375–382
[41] Sawalha AH, Kaufman KM, Kelly JA, Adler AJ, Aberle T, Kilpatrick J, et al. Geneticassociation of interleukin-21 polymorphisms with systemic lupus erythemato-sus. Ann Rheum Dis 2008;67:458–61.
[42] Bryant VL, Ma CS, Avery DT, Li Y, GoodL KL, Corcoran LM, et al. Cytokine-mediatedregulation of human B cell differentiation into Ig-secreting cells: predominant role ofIL-21 produced by CXCR5+ T follicular helper cells. J Immunol 2007;179:8180–90.
[43] Arguni E, Tsuruoka N, Arima M, Sakamoto A, Hatano M, Tokuhisa T. Induction ofhigh Bcl6 expression and its roles in germinal center B cells. Int Congr 2005;1285:130–6.
[44] Linterman MA, Beaton L, Yu D, Ramiscal RR, Srivastava M, Hogan JJ, et al. IL-21acts directly on B cells to regulate Bcl-6 expression and germinal centerresponses. J Exp Med 2010;207:353–63.
[45] Mackay F, Browning JL. BAFF: a fundamental survival factor for B cells. Nature2002;2:465–75.
[46] Huard B, Schneider P, Mauri D, Tschopp J, French LA. T cell costimuation by theTNF ligand BAFF. J Immunol 2001;167:6225–31.
[47] Ettinger R, Sims GP, Robbins R, Withers D, Fischer RT, Grammer AC, et al. IL-21and BAFF/BLyS synergize in stimulating plasma cell differentiation from a uniquepopulation of human splenic memory B cells. J Immunol 2007;178:2872–82.
[48] Wang XF, Yuan SL, Jiang L, Zhang XL, Li SF, Guo Y, et al. Changes of serum BAFFand IL-21 levels in patients with systemic lupus erythematosus and their clinicalsignificance. Chin J Cell Mol Immunol 2007;23:1041–2.
[49] Ettinger R, Sims GP, Fairhurst AM, Robbins R, Silva YS, Spolski R, et al. IL-21induces differentiation of human naive and memory B cells into antibody-secreting plasma cells. J Immunol 2005;176:7867–79.
[50] Herber D, Brown TP, Liang S, Young DA, Collins M, Dunussi-Joannopoulus K. IL-21has a pathogenic role in a lupus-prone mouse model and its blockade with IL-21R.Fc reduces disease progression. J Immunol 2007;178:3822–30.
[51] Bubier JA, Bennett SM, Sproule TJ, Lyons BL, Olland S, Young DA, et al. Treatmentof BXSB-Yaa mice with IL-21R.Fc fusion protein minimally attenuates systemiclupus erythematosus. Ann NY Acad Sci 2007;1110:590–601.
[52] Bubier JA, Sproule TJ, Foreman O, Spolski R, Shaffer DJ, Morse HC, et al. A criticalrole for IL-21 receptor signalling in the pathogenesis of systemic lupuserythematosus in BXSB-Yaa mice. Proc Natl Acad Sci USA 2009;106:1518–23.
[53] Young DA, Hegen M, Ma HL, Whitters MJ, Albert LM, Lowe L, et al. Blockade of theinterleukin-21/interleukin-21 receptor pathway ameliorates disease in animalmodels of rheumatoid arthritis. Arthritis Rheum 2007;56:1152–63.
[54] Jungel A, Distler JHW, Kurowska-Stolarska M, Seemayer CA, Seibl R, Forster A,et al. Expression of interleukin-21 receptor, but not interleukin-21, in synovialfibroblasts and synovial macrophages of patients with rheumatoid arthritis.Arthritis Rheum 2004;50:1468–76.
[55] Yuan SL, Jiang L, Zhang XL, Li SF, Duan HM,Wang XF. Serum IL-21 level in patientswith primary Sjögren's syndrome and clinical significance of IL-21. Chin J CellMol Immunol 2007;23:124–6.
[56] Odegard JM, Marks BR, DiPlacido LD, Poholek AC, Kono DH, Dong C, et al. ICOS-dependent extrafollicular helper T cells elicit IgG production via IL-21 in systemicautoimmunity. J Exp Med 2008;205:2873–86.
[57] Iwai H, Abe M, Hirose S, Tsushima F, Tezuka K, Akiba H, et al. Involvement ofinducible costimulator-B7 homologous protein costimulatory pathway inmurinelupus nephritis. J Immunol 2003;171:2848–54.
[58] Hutloff A, Bϋchner K, Reiter K, Baelde HJ, Odendahl M, Jacobi A, et al. Involvementof inducible costimulator in the exaggerated memory B cell and plasma cellgeneration in systemic lupus erythematosus. Arthritis Rheum 2004;50:3211–20.
[59] Cortés M, Georgopoulos K. Aiolos Is required for the generation of high affinitybone marrow plasma cells responsible for long-term immunity. J Exp Med2004;199:209–19.
[60] Baker KP. BLyS — an essential survival factor for B cells: basic biology, links topathology and therapeutic target. Autoimmun Rev 2004;3:368–75.
[61] Schneider P. The role of APRIL and BAFF in lymphocyte activation. Curr OpinImmunol 2005;17:282–9.
[62] Cancro MP. The BLyS/BAFF family of ligands and receptors: key targets in thetherapy and understanding of autoimmunity. Ann Rheum Dis 2006;65:34–6.
[63] O'Connor BP, Raman VS, Erickson LD, CookWJ, Weaver LK, Ahonen C, et al. BCMAis essential for the survival of long-lived bone marrow plasma cells. J Exp Med2004;199:91–7.
[64] Avery DT, Kalled SL, Ellyard JI, Ambrose C, Bixler SA, Thien M, et al. BAFFselectively enhances the survival of plasmablasts generated from humanmemory B cells. J Clin Invest 2003;112:286–97.
[65] Cancro MP, Cruz DP, Khamashta A. The role of B lymphocyte stimulator (BLyS) insystemic lupus erythematosus. J Clin Invest 2009;119:1066–73.
[66] Belnoue E, Pihlgren M, McGaha TL, Tougne C, Rochat A, Bossen C, et al. APRIL iscritical for plasmablast survival in bone marrow and poorly expressed in early-life bone marrow stromal cells. Blood 2008;111:2755–64.
[67] Varfolomeev E, Kischkel F, Martin F, Seshasayee D, Wang H, Lawrence D, et al.APRIL-deficient mice have normal immune system development. Mol Cell Biol2004;24:997–1006.
[68] Scholz JL, Crowley JE, Tomayko MM, Steinel N, O'Neill PJ, Quinn III WJ, et al. BLySinhibition eliminates primary B cells but leaves natural and acquired humoralimmunity intact. Proc Natl Acad Sci USA 2008;105:15517–22.
[69] Vallerskog T, Heimbϋrger M, Gunnarsson I, Zhou W, Wahren-Herlenius M,Trollmo C, et al. Differential effects on BAFF and APRIL levels in rituximab-treatedpatients with systemic lupus erythematosus and rheumatoid arthritis. ArthritisRes Ther 2006;8:R167.
[70] Petri M, Stohl W, ChathamW, McCuneWJ, Chevrier M, Ryel J, et al. Association ofplasma B lymphocyte stimulator levels and disease activity in systemic lupuserythematosus. Arthritis Rheum 2008;58:2453–9.
[71] Zhao LD, Li Y, Smith Jr MF, Wang JS, Zhang W, Tang FL, et al. Expressions of BAFF/BAFF receptors and their correlation with disease activity in Chinese SLE patients.Lupus 2010;19:1534–49.
[72] Collins CE, Gavin AL, Migone T, Hilbert DM, Nemazee D, Stohl W. B lymphocytestimulator (BLyS) isoforms in systemic lupus erythematosus: disease activitycorrelates better with blood leukocyte BLyS mRNA levels than with plasma BLySprotein levels. Arthritis Res Ther 2006;8:R6.
[73] Cheema GS, Roschke V, Hilbert DM, Stohl W. Elevated serum B lymphocytestimulator levels in patients with systemic immune-based rheumatic diseases.Arthritis Rheum 2001;44:1313–9.
[74] Seyler TM, Park YW, Takemura S, Bram RJ, Kurtin PJ, Goronzy JJ, et al. BLyS andAPRIL in rheumatoid arthritis. J Clin Invest 2005;115:3083–92.
[75] Groom J, Kalled SL, Cutler AH, Olson C, Woodcock SA, Schneider P, et al.Association of BAFF/BLyS overexpression and altered B cell differentiation withSjögren's syndrome. J Clin Invest 2002;109:59–68.
[76] Szodoray P, Jellestad S, Teague MO, Jonsson R. Attenuated apoptosis of B cellactivating factor-expressing cells in primary Sjögren's syndrome. Lab Invest2003;83:357–65.
[77] Mariette X, Roux S, Zhang J, Bengoufa D, Lavie F, Zhou T, et al. The level of BLyS(BAFF) correlates with the titer of autoantibodies in human Sjögren's syndrome.Ann Rheum Dis 2003;62:168–71.
[78] Varin M, Le Pottier L, Youinou P, Saulep D, Mackay F, Pers J. B-cell tolerancebreakdown in Sjögren's Syndrome: Focus on BAFF. Autoimmun Rev 2010;9:604–8.
[79] Matsushita T, Hasegawa M, Yanaba K, Kodera M, Takehara K, Sato S. Elevatedserum BAFF levels in patients with systemic sclerosis. Arthritis Rheum 2006;54:192–201.
[80] Thangarajh M, Gomes A, Masterman T, Hillert J, Hjelmström P. Expression of B-cell-activating factor of the TNF family (BAFF) and its receptors in multiplesclerosis. J Neuroimmunol 2004;152:183–90.
[81] Krumbholz M, Specks U, Wick M, Kalled SL, Jenne D, Meinl E. BAFF is elevated inserum of patients with Wegener's granulomatosis. J Autoimmun 2005;25:298–302.
[82] Emmerich F, Bal G, Barakat A, Milz J, Mϋhle C, Martinez-Gamboa L, et al. High-level serum B-cell activating factor and promoter polymorphisms in patientswith idiopathic thrombocytopenic purpura. Br J Haematol 2006;136:309–14.
[83] Migita K, Abiru S, Maeda Y, Nakamura M, Komori A, Ito M, et al. Elevated serumBAFF levels in patients with autoimmune hepatitis. Hum Immunol 2007;68:586–91.
[84] Mackay IR, Groom J, Mackay CR. Levels of BAFF in serum in primary biliarycirrhosis and autoimmune diabetes. Autoimmunity 2002;35:551–3.
[85] Toubi E, Gordon S, Kessel A, Rosner I, Rozenbaum M, Shoenfeld Y, et al. Elevatedserum B-lymphocyte activating factor (BAFF) in chronic hepatitis C virusinfection: Association with autoimmunity. J Autoimmun 2006;27:134–9.
[86] Roschke V, Sosnovtseva S, Ward CD, Hong JS, Smith R, Albert V, et al. BLyS andAPRIL form biologically active heterodimers that are expressed in patients withsystemic immune-based rheumatic diseases. J Immunol 2002;169:4314–21.
[87] Tangye S, Bryant VL, Cuss AK, Good KL. BAFF, APRIL and human B cell disorders.Semin Immunol 2006;18:305–17.
[88] Katsenelson N, Kanswal S, Puig M, Mostowski H, Verthelyi D, Akkoyunlu M.Synthetic CpG oligodeoxynucleotides augment BAFF- and APRIL-mediatedimmunoglobulin secretion. Eur J Immunol 2007;37:1785–95.
[89] Harigai M, Kawamoto M, Hara M, Kubota T, Kamatani N, Miyasaka N. Excessiveproduction of IFN-{gamma} in patients with systemic lupus erythematosus andits contribution to induction of B lymphocyte stimulator/B cell-activating factor/TNF ligand superfamily-13B. J Immunol 2008;181:2211–9.
[90] Scapini P, Nardelli B, Nadali G, Calzetti F, Pizzolo G, Montecucco C, et al. G-CSFstimulated neutrophils are a prominent source of functional BLyS. J Exp Med2003;197:297–302.
[91] Gavin AL, Aït-Azzouzene D,Ware CF, Namazee D. {Delta}BAFF, an alternate spliceisoform that regulates receptor binding and biopresentation of the B cell survivalcytokine, BAFF. J Biol Chem 2003;278:38220–8.
[92] Youinou P, Pers J. The late news on BAFF in autoimmune diseases. AutoimmunRev 2010;9:804–6.
[93] Gavin AL, Duong B, Skog P, Aït-Azzouzene D, Greaves DR, Scott ML, et al. {Delta}BAFF, a splice isoform of BAFF, opposes full-length BAFF activity in vivo intransgenic mouse models. J Immunol 2005;175:319–28.
[94] Emilie D, Mariette X. Interleukin 10— a new therapeutic target in systemic lupuserythematosus? Joint Bone Spine 2001;68:4–5.
[95] Wols MHA, Underhill GH, Kansas GS, Witte PL. The role of bone marrow-derivedstromal cells in the maintenance of plasma cell longevity. J Immunol 2002;169:4213–21.
[96] Chevrier S, Genton C, Kallies A, Karnowski A, Otten LA, Malissen B, et al. CD93 isrequired for maintenance of antibody secretion and persistence of plasma cells inthe bone marrow niche. Proc Natl Acad Sci USA 2009;106:3895–900.
[97] Blank M, Shoenfeld Y. B cell targeted therapy in autoimmunity. J Autoimmun2007;28:62–8.
[98] Stichweh D, Pascual V, Banchereau J. Recent advances in therapeutic strategiesfor SLE. Drug Discov Today Ther Strat 2006;3:5–10.
[99] García-Carrasco M, Jiménez-Hernández M, Escárcega RO, Mendoza-Pinto C,Galarza-Maldonado C, Sandoval-Cruz M, et al. Use of rituximab in patients withsystemic lupus erythematosus: an update. Autoimmun Rev 2009;8:343–8.
[100] Schneider P. The role of APRIL and BAFF in lymphocyte activation. Curr OpinImmunol 2005;17:282–9.
[101] Levesque MC, Clair EW. B cell-directed therapies for autoimmune disease andcorrelates of disease response and relapse. J Allergy Clin Immunol 2008;121:13–21.

382 M. Neves, J.D. Alves / Autoimmunity Reviews 10 (2011) 375–382
[102] Cornec D, Avouac J, Youinou P, Saraux A. Critical analysis of rituximab-inducedserological changes in connective tissue diseases. Autoimmun Rev 2009;8:515–9.
[103] Lesley R, Xu Y, Kalled SL, Hess DM, Schwab SR, Shu HB, et al. Reducedcompetitiveness of autoantigen-engaged B cells due to increased dependence onBAFF. Immunity 2004;20:441–53.
Switching multiple sclerosis patients with breakthrough disease to seco
Multiple sclerosis (MS) patients with breakthrough disease on immunomimmunosuppressants. The effect of natalizumab monotherapy in patien(PLoS One 2011;6:16664) performed an open-label retrospective cohoCalifornia San Francisco MS Center, 95 had breakthrough disease oimmunosuppressants and 13 declined the switch [non-switchers]). Poiscompare the relapse rate within and across groups before and after the70% (95% CI 50,82%; pb0.001) in switchers to natalizumab and by 77relapse rate in non-switchers did not decrease (6%, p=0.87). Relative tby 68% among natalizumab switchers (95% CI 19,87%; p=0.017) andp=0.004). Switching to natalizumab or immunosuppressants in patiactivity of relapsing MS. However, authors suggest that the magniturandomized clinical trials and prospective cohort studies.
Sun exposure and vitamin D are independent risk factors for CNS demy
To examine whether past and recent sun exposure and vitamin D statusrisk of first demyelinating events (FDEs) and to evaluate the contributAustralia, Lucas RM, et al. (Neurology 2011;76:540-8) performed a mu59 years with a FDE and resident within one of 4 Australian centers (fro2006. Controls (n=395) were matched to cases on age, sex, and studyself-reported sun exposure by life stage, objective measures of skin phpast, recent, and accumulated leisure-time sun exposure were each assexposure (age 6 years to current), adjusted odds ratio (AOR) =0.70 (95increment of 1,000 kJ/m(2) (range 508-6,397 kJ/m(2)). Higher actiniclowest) and higher serum vitamin D status (AOR =0.93 [95% CI 0.8associated with decreased FDE risk. Differences in leisure-time sun expo32.4% increase in FDE incidence from the low to high latitude regions. Aindependent roles in the risk of CNS demyelination. However, bothprevention.
Reorganization in cognitive networks with progression of multiple scle
Cognitive dysfunction (CD) is frequent in multiple sclerosis (MS) anddisease progression has been described for the motor system in MS usingM, et al. (Neurology 2011;76:526-33) attempted to assess the concept oNo-go" fMRI paradigm. Patients with a clinically isolated syndrome (Cprogressive MS (SPMS) (n=10), and 28 healthy controls (HC), undexamination, structural imaging, and an fMRI Go/No-go discriminationsustained attention and concentration, and information processing. Thelicited activation in a widespread network including bilateral mesial anregions. Task performance was similar between phenotypes, but deviatiincreased with disease progression. Patients with RRMS showed increaand the right fusiform gyrus, and recruited the hippocampus with increanetwork function, including recruitment of pre-SMA, bilateral superbilateral postcentral, and right temporal brain areas. Using a cognitiveneuronal activation with progressing MS and to provide strong eviden
[104] Liang B, Gardner DB, Griswold DE, Bugelski PJ, Song XYR. Anti-interleukin-6monoclonal antibody inhibits autoimmune responses in a murine model ofsystemic lupus erythematosus. Immunology 2006;119:296–305.
[105] Chong BF, Mohan C. Targeting the CXCR4/CXCL12 axis in systemic lupuserythematosus. Expert Opin Ther Targets 2009;13:1147–53.
nd-line therapy
odulatory drugs are frequently offered to switch to natalizumab orts with breakthrough disease is unknown. Castillo-Trivino T, et al.rt study of 993 patients seen at least four times at the University ofn first-line therapy (60 patients switched to natalizumab, 22 toson regression was used to adjust for potential confounders and toswitch. In the within-group analyses, the relapse rate decreased by% (95% CI 59,87%; pb0.001) in switchers to immunosuppressants;o the reduction among non-switchers, the relapse rate was reducedby 76% among the immunosuppressant switchers (95% CI 36,91%;ents with breakthrough disease was effective in reducing clinicalde of the effect and the risk-benefit ratio should be evaluated in
elination
(serum 25-hydroxyvitamin D [25(OH)D] levels) are associated withion of these factors to the latitudinal gradient in FDE incidence inlticenter incident case-control study. Cases (n=216) were aged 18-m latitudes 27°S to 43°S), from November 1, 2003, to December 31,region, without CNS demyelination. Exposures measured included
enotype and actinic damage, and vitamin D status. Higher levels ofociated with reduced risk of FDE, e.g., accumulated leisure-time sun% confidence interval [CI] 0.53-0.94) for each ultraviolet (UV) doseskin damage (AOR =0.39 [95% CI 0.17-0.92], highest grade vs the6-1.00] per 10 nmol/L increase in 25(OH)D) were independentlysure, serum 25(OH)D level, and skin type additively accounted for authors concluded that sun exposure and vitamin D status may havewill need to be evaluated in clinical trials for multiple sclerosis
rosis: Insights from fMRI
can occur at early stages. Whereas functional reorganization withfMRI, no such studies exist for cognition. In this concern, Loitfelder
f functional reorganization concerning cognition using a simple "Go/IS, n=10), relapsing-remitting MS (RRMS) (n=10), or secondaryerwent a comprehensive neuropsychological test battery, clinicaltask at 3 T. Patients performed worse than HC regarding memory,ese differences were driven by patients with SPMS. The fMRI taskd dorsolateral frontal, parietal, insular, basal ganglia, and cerebellaron from the activation pattern observed in HC and patients with CISsed brain activation in the precuneus, both superior parietal lobes,sing demands. Patients with SPMS demonstrated the most abnormalior and inferior parietal, dorsolateral prefrontal, right precentral,fMRI paradigm, authors were able to confirm adaptive changes ofce for their compensatory nature, at least partially.




















