
Pore-blocking and pore-assisting factors during capillary condensation and evaporation
Upload
independentCategory
view
8download
0
Coherent anti-Stokes vibrational Raman spectra of artificialrhodopsin pigments containing ring structures blocking 11-cis
isomerization
Yidong Zhoua, Laszlo Ujja, Jihong Loub, Frank Ja¨gera, K. Nakanishib, G.H. Atkinsona,*aDepartment of Chemistry and Optical Sciences Center, University of Arizona, Tucson, AZ, USA
bDepartment of Chemistry, Columbia University, New York, NY, USA
Received 17 April 1998; accepted 29 September 1998
Abstract
The structural changes in the retinal chromophore that underlie the initial picosecond processes in the room temperaturerhodopsin (RhRT) photo-reaction (i.e., photoRT and bathoRT intermediates) are examined through the vibrational spectra of fourartificial Rh pigments (new vibrational spectra from three artificial Rh pigments are presented here). Each of these Rh pigmentscontains a retinal in which isomerization around the C11yC12 bond, thought to be the primary reaction coordinate in the RhRT
photo-reaction, is blocked. Specifically, vibrational spectra from the ground electronic states of Rh7.10 (containing a 7-membered carbon ring spanning the C10–C11yC12–C13 bonds in the retinal), Rh7.10/8D (Rh7.10 containing a deuterium atC8), Rh7.10/ND (Rh7.10 containing a deuterium at the Schiff-base nitrogen) and Rh8.10 (containing an 8-membered carbonring spanning the C10–C11yC12–C13 bonds in the retinal) are recorded by picosecond resonance coherent anti-Stokes Ramanspectroscopy (PR/CARS). These PR/CARS data are analyzed with the aid of vibrational mode, assignments originating withnative Rh used to identify the structural motions that can be associated with specific vibrational features. These PR/CARSassignments provide the spectroscopic information required to interpret the structural changes in the retinal chromophore,observed via picosecond time-resolved CARS measurements (reported elsewhere), following optical excitation of these sameartificial Rh pigments. The relationships between structural changes in these artificial Rh pigments and those occurring in nativeRh are also discussed.q 1999 Elsevier Science B.V. All rights reserved.
Keywords:Coherent anti-Stokes vibrational Raman spectra; Rhodopsin pigments; Picosecond processes
1. Introduction
The molecular mechanism underlying the energystorage/signal transduction process(es) in the roomtemperature photo-reaction of native rhodopsin(RhRT)is understood to involve the 11-cis to all-transisomer-ization of the retinal chromophore as a primary step[1]. The driving force underlying such C11yC12
isomerization originates with the optical excitationof RhRT and was characterized largely by transientabsorption studies monitoring the formation dynamicsassignable to the first observable intermediate,photoRT [2]. Since photoRT was identified onlythrough its red-shifted, transient absorption appearingwithin 200 fs after 60 fs excitation [3,4], the structuralnature of photoRT remains to be determined. It isreasonable to expect that some structural transforma-tion occurs in the RhRT retinal chromophore during theformation of photoRT, but the short (< 5 ps) lifetime of
Journal of Molecular Structure 478 (1999) 107–119
0022-2860/99/$ - see front matterq 1999 Elsevier Science B.V. All rights reserved.PII: S0022-2860(98)00621-8
* Corresponding author. Tel.: 001 520 621 6293; Fax: 001 520621 4858; e-mail: [email protected]

photoRT in the native RhRT photo-reaction hasprecluded its direct examination by time-resolvedvibrational spectroscopy. Time-resolved vibrationalspectra of the bathoRT intermediate formed fromphotoRT, measured by picosecond time-resolvedcoherent Raman spectroscopy (PTR/CARS), weremeasured and have definitively shown significantstructural differences between bathoRT and RhRT [5].Recently, PTR/CARS was used to measure thebathoRT vibrational structure from 10 ps to 100 ns
after excitation [5] and to characterize the vibrationalstructure of subsequent intermediates in the RhRT
photo-reaction such as lumiRT and BSIRT[6].The degree to which RhRT photo-reaction proceeds,
together with the kinetics with which specific inter-mediates are formed, can be controlled by altering thestructure of the retinal chromophores to form artificialRh pigments. For example, isomerization at theC11yC12 bond, thought to be the principal coordinatefor reactivity in the RhRT photo-reaction, can beprevented by incorporating saturated carbon ringsspanning the C10–C11yC12–C13 retinal bonds [7].The size of these blocking rings determines the degreeof flexibility remaining in the C10–C11yC12–C13
region of the retinal chromophore and therefore,whether structurally distinct RhRT intermediates canbe formed. The dynamical properties of three suchartificial Rh pigments, examined previously by tran-sient absorption spectroscopy over the 20 ps to 10 nstime regime [7], reveal that transient species exhi-biting red-shifted absorption spectra (relative to thatof the respective ground-state species) are presenteven when C11yC12 isomerization itself cannotoccur. Thus, these artificial Rh pigments in whichthe C11yC12 isomerization is blocked can be utilizedto elucidate the structural/steric forces which drive theRhRT energy storage process since the photoRT-likeintermediates have longer lifetimes [8] than the analo-gous photoRT. These longer lifetimes make it experi-mentally more feasible to measure vibrational spectrafrom the respective intermediates and thereby, todetermine the structural changes underlying reactivityin both artificial and native RhRT.
Transient absorption data themselves provide nodirect information on the structural changes occurringin these retinals and therefore, do not address themechanistic issues concerning the role(s) of retinaland protein structure in the appearance of thephotoRT-like species. Vibrational spectra from theseintermediates are required to determine the relation-ship(s) between structural changes and RhRT reac-tivity. Two of these artificial Rh pigments,containing either a 7-(Rh7.10) or 8-membered(Rh8.10) ring spanning the C10–C11yC12–C13 bonds(Fig. 1), together with two isotopically substitutedanalogues of the Rh7-10 (Rh7.10/8D which isRh7.10 containing a deuterium at C8 and Rh7.10/ND which is Rh7.10 containing a deuterium at the
Y. Zhou et al. / Journal of Molecular Structure 478 (1999) 107–119108
Fig. 1. Retinal structures of (a) the 11-cis retial in native Rh, (b) the11-cis retinal containing a 7-membered ring bridging the C10–C11yC12–C13 (i.e., locked 11-cis structure) in the artificial Rhpigment, Rh7.10, (c) the Rh7.10 structure with the C8 positiondeuterated to form Rh7.10/8D, (d) the Rh7.10 structure with theSchiff-base nitrogen position deuterated to form Rh7.10/ND, (e)the 11-cis retinal containing an 8-membered ring bridging theC10–C11yC12–C13 in the artificial Rh pigment, Rh8.10.

Schiff-base nitrogen) were examined by coherentvibrational spectroscopy. The time-dependent struc-tural changes occurring within a few picoseconds ofexcitation were measured by PTR/CARS methods andare described elsewhere [9].
In this paper, picosecond resonance CARS (PR/CARS) spectra from the stable, ground electronicstates of these same four artificial Rh pigments arepresented. Vibrational analyses of these PR/CARSspectra provide the vibrational mode assignmentsrequired to interpret the retinal structural changeswithin each artificial pigment and therefore, may berelated to those structural changes underlying theformation of photoRT. Thus, emphasis is placed hereon the vibrational band assignments to specific retinalnormal modes in these three artificial Rh pigments andon the comparison of assignments with those in bothRh7.10 and native Rh.
2. Materials and methods
2.1. Preparation of the structurally modified Rhpigments
The rod outer segments (ROS) from bovine retinas(W. Lawson Co., Lincoln, NE) are prepared followingstandard procedures [9,10]. Opsin is extracted frompurified ROS [9] in order to bind with the artificialretinal chromophores to form each artificial Rhpigment. The structurally modified retinals examinedhere (Fig. 1), synthesized separately by methodsdescribed previously [11], are combined with opsinin a suspension under controlled light conditionsusing an approximately 1.5 molar ratio (opsin toretinal). The artificial retinal chromophores aredissolved in ethanol. These mixed solutions are incu-bated for 2 h at 208C and subsequently for 24 h at 48Cto produce the artificial Rh pigments. The resultantartificial Rh pigments are washed five times withbuffer and re-suspended in about 50 ml of buffer forPR/CARS measurements. The total amount ofRh7.10, Rh7.10/8D and Rh8.10 (Fig. 1), estimatedfrom ultraviolet/visible absorption spectra, providesan approximately 150 OD-ml sample.
The Rh7.10/ND pigment is prepared by dissolvingRh7.10 in a D2O solvent. Specifically, after centrifu-gation of the Rh7.10 sample, the resultant pellet is
re-suspended in 50 ml of a buffer prepared with D2Oinstead of H2O [9]. All artificial Rh pigments exam-ined here are obtained by reconstitution and therefore,each sample contains only one isomer (11-cis-retinal)and is stable in normal light conditions [11].
2.2. PR/CARS experiment
Since detailed descriptions of the laser instrumenta-tion, experimental procedure, and spectral analysismethodology underlying these PR/CARS measure-ments are given elsewhere [6,12,13], only a briefsummary is presented here. Two probe lasers (withl1 � 600 nm, 2.5 nJ,, 2 cm21 HWHM and l s �640/653 nm, 3 nJ pulse, 700 cm21 HWHM) are usedto generate CARS spectra centered at 565 nm(hydrogen out-of-plane or HOOP and fingerprintregions) and 555 nm (fingerprint and CyC regions).The CARS signals originating from the Rh sampleand from a reference sample containing only thebuffer solution are dispersed with respect to bothwavelength (Spex Triplemate monochromator) andvertical position onto a multichannel detector(EG&G, model 1024, LN/CCD). The spectral resolu-tion of the overall detection system, limited by thebandwidth of the narrowband laser (v1), is,2 cm21. The spectral range of the resultant CARSspectrum is determined by the spectral bandwidth ofthe broadband laser pulse (< 700 cm21). Both lasersare operated at 400 kHz repetition rates with 5.5-psand 7-ps temporal pulse widths. The less than 2-pstiming jitter between laser pulses is determined bymeasuring cross-correlation functions during eachPR/CARS experiment.
Typically, exposure times on the detector surface of30 s and the accumulation of 5–10 spectra are suffi-cient to obtain PR/CARS spectra of the qualitypresented here. No (,1%) change (i.e., decrease) inthe Rh pigment concentration is observed during themeasurement of a PR/CARS spectrum.
The methods used to process and analyze each PR/CARS spectrum are described elsewhere [5,6]. Theseprocedures are designed to quantitatively determinethe parameters (vide infra) of each vibrational bandby fitting the measured PR/CARS spectrum with amodel function [6]. These nonlinear optical fits areperformed with Microcal-Origin 5.0 program using
Y. Zhou et al. / Journal of Molecular Structure 478 (1999) 107–119 109
the Levenberg-Marquardt nonlinear estimatormethod.
3. Results and discussion
3.1. PR/CARS spectra
PR/CARS spectra of the three artificial Rhpigments examined here (Fig. 1) are shown in Figs.2–7. These artificial Rh pigments include those
1. containing a retinal chromophore with a 7-membered ring spanning C10–C11yC12–C13 bondsin retinal (Rh7.10) with a deuterium at the C8
retinal positions (Rh7.10/8D) (Figs. 2 and 3),2. with Rh7.10 having a deuterium at the Schiff-base
N (Rh7.10/ND)(Figs. 4 and 5), and
3. containing with an 8-membered ring spanning theC10–C11yC12–C13 bonds in retinal (Rh8.10) (Figs.6 and 7).
PR/CARS spectra from Rh.7.10 were presentedearlier [9] and are reproduced here only for compar-ison purposes. Procedurally, all of these PR/CARSdata were measured over two, overlapping spectralregions, each, 700 cm21 wide.
The x (3) function used to fit these derivative PR/CARS spectra are plotted as solid lines over theirrespective PR/CARS spectra (Figs. 2–7). The corre-sponding background-free PR/CARS spectra, derivedfrom thesex (3) fits using Lorentzian lineshapes [6],are also displayed at the bottom of each of thesefigures. The correspondingx (3)-fitting parameters(i.e., band origin position (Vi), bandwidth, (Vi), and
Y. Zhou et al. / Journal of Molecular Structure 478 (1999) 107–119110
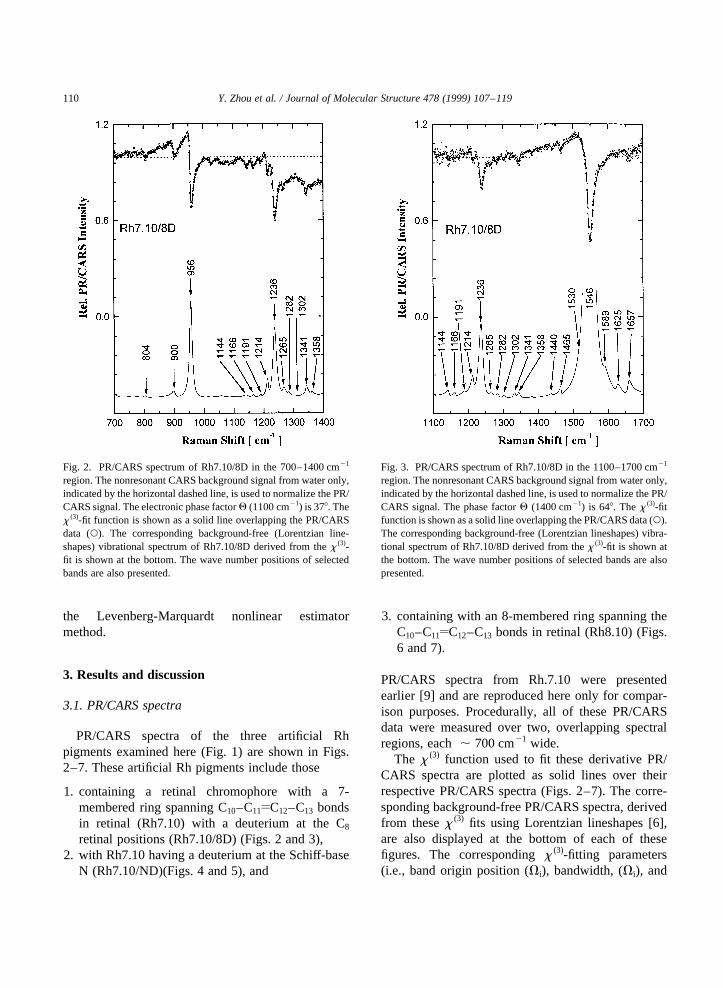
Fig. 2. PR/CARS spectrum of Rh7.10/8D in the 700–1400 cm21
region. The nonresonant CARS background signal from water only,indicated by the horizontal dashed line, is used to normalize the PR/CARS signal. The electronic phase factorQ (1100 cm21) is 378. Thex (3)-fit function is shown as a solid line overlapping the PR/CARSdata (W). The corresponding background-free (Lorentzian line-shapes) vibrational spectrum of Rh7.10/8D derived from thex (3)-fit is shown at the bottom. The wave number positions of selectedbands are also presented.
Fig. 3. PR/CARS spectrum of Rh7.10/8D in the 1100–1700 cm21
region. The nonresonant CARS background signal from water only,indicated by the horizontal dashed line, is used to normalize the PR/CARS signal. The phase factorQ (1400 cm21) is 648. Thex (3)-fitfunction is shown as a solid line overlapping the PR/CARS data (W).The corresponding background-free (Lorentzian lineshapes) vibra-tional spectrum of Rh7.10/8D derived from thex (3)-fit is shown atthe bottom. The wave number positions of selected bands are alsopresented.

amplitudes (Ai)) for Rh7.10/8D, Rh7.10/ND, andRh8.10 are presented in Tables 1, 2 and 3respectively. The error in measuring the bandorigin position of a given vibrational feature isfound typically to be ,2 cm21 throughout the700–1700 cm21 region measured here. To facilitatecomparisons, background-free, Lorentzian-band-shape PR/CARS spectra in two overlappingspectral regions from native Rh [6], Rh7.10 [9],Rh7.10/8D, Rh7.10/ND and Rh8.10 are shown inFigs. 8 and 9.
The strongest vibrational features in the 700–1700 cm21 region for all three artificial Rh pigmentsexamined here appear near 955, 1235, and 1550 cm21
which correspond generally to the HOOP, C–Cstretching (fingerprint), and CyC stretching vibra-tional modes. The discussion of these PR/CARS
results is presented in terms of data from these threespectral regions.
3.2. Effect of artificial ring size
Few differences are observed between the PR/CARS spectra for Rh7.10 and Rh8.10 (Figs. 8 and9), but there are several differences at low frequencies(i.e., HOOP region) when the PR/CARS spectrum ofeither Rh7.10 or Rh8.10 is compared to that of nativeRh [6]. These low-frequency band changes (e.g., at795, 804, and 858 cm21 for Rh7.10 [9]; and at 781,804, 824 and 899 cm21 for Rh8.10), can be attributedto the introduction of the artificial rings. The bandsthemselves can be assigned to HOOP modes asso-ciated with the polyene chain and to the –CH2 vibra-tions of the artificial ring.
Y. Zhou et al. / Journal of Molecular Structure 478 (1999) 107–119 111
Fig. 4. PR/CARS spectrum of Rh7.10/ND in the 700–1400 cm21
region. The nonresonant CARS background signal from water only,indicated by the horizontal dashed line, is used to normalize the PR/CARS signal. The phase factorQ (1100 cm21) is 418. Thex (3)-fitfunction is shown as a solid line overlapping the PR/CARS data (W).The corresponding background-free (Lorentzian lineshapes) vibra-tional spectrum of Rh7.10/ND derived from thex (3)-fit is shown atthe bottom. The wave number positions of selected bands are alsopresented.
Fig. 5. PR/CARS spectrum of Rh7.10/ND in the 1100–1700 cm21
region. The nonresonant CARS background signal from water only,indicated by the horizontal dashed line, is used to normalize the PR/CARS signal. The phase factorQ (1400 cm21) is 768. Thex (3)-fitfunction is shown as a solid line overlapping the PR/CARS data (W).The corresponding background-free (Lorentzian lineshapes) vibra-tional spectrum of Rh7.10/ND derived from thex (3)-fit is shown atthe bottom. The wave number positions of selected bands are alsopresented.
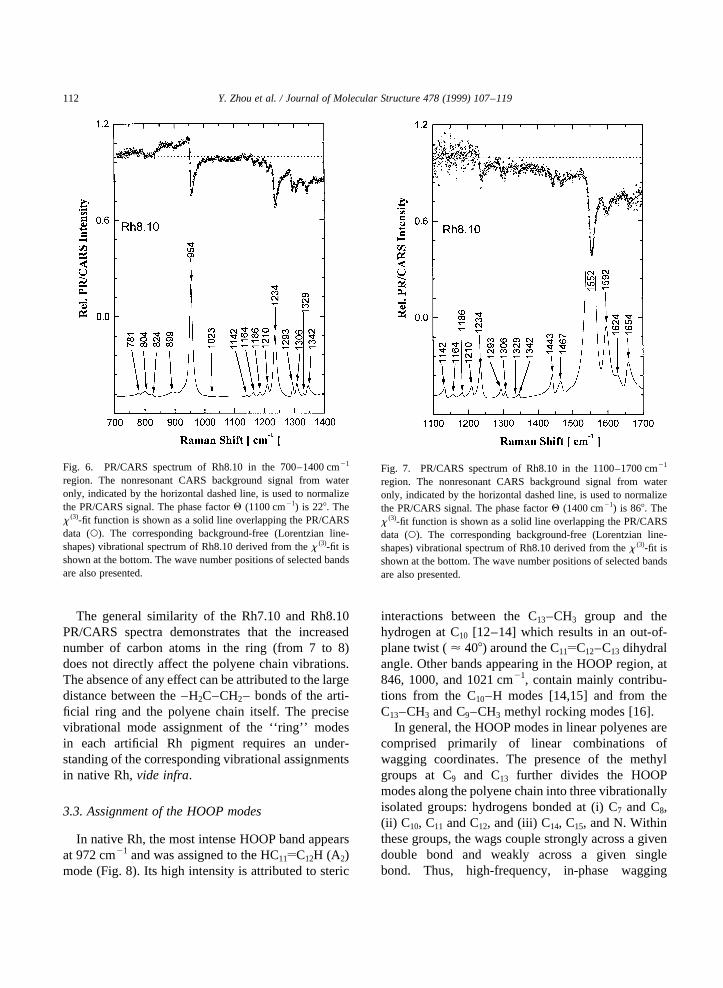
The general similarity of the Rh7.10 and Rh8.10PR/CARS spectra demonstrates that the increasednumber of carbon atoms in the ring (from 7 to 8)does not directly affect the polyene chain vibrations.The absence of any effect can be attributed to the largedistance between the –H2C–CH2– bonds of the arti-ficial ring and the polyene chain itself. The precisevibrational mode assignment of the ‘‘ring’’ modesin each artificial Rh pigment requires an under-standing of the corresponding vibrational assignmentsin native Rh,vide infra.
3.3. Assignment of the HOOP modes
In native Rh, the most intense HOOP band appearsat 972 cm21 and was assigned to the HC11yC12H (A2)mode (Fig. 8). Its high intensity is attributed to steric
interactions between the C13–CH3 group and thehydrogen at C10 [12–14] which results in an out-of-plane twist (< 408) around the C11yC12–C13 dihydralangle. Other bands appearing in the HOOP region, at846, 1000, and 1021 cm21, contain mainly contribu-tions from the C10–H modes [14,15] and from theC13–CH3 and C9–CH3 methyl rocking modes [16].
In general, the HOOP modes in linear polyenes arecomprised primarily of linear combinations ofwagging coordinates. The presence of the methylgroups at C9 and C13 further divides the HOOPmodes along the polyene chain into three vibrationallyisolated groups: hydrogens bonded at (i) C7 and C8,(ii) C10, C11 and C12, and (iii) C14, C15, and N. Withinthese groups, the wags couple strongly across a givendouble bond and weakly across a given singlebond. Thus, high-frequency, in-phase wagging
Y. Zhou et al. / Journal of Molecular Structure 478 (1999) 107–119112
Fig. 6. PR/CARS spectrum of Rh8.10 in the 700–1400 cm21
region. The nonresonant CARS background signal from wateronly, indicated by the horizontal dashed line, is used to normalizethe PR/CARS signal. The phase factorQ (1100 cm21) is 228. Thex (3)-fit function is shown as a solid line overlapping the PR/CARSdata (W). The corresponding background-free (Lorentzian line-shapes) vibrational spectrum of Rh8.10 derived from thex (3)-fit isshown at the bottom. The wave number positions of selected bandsare also presented.
Fig. 7. PR/CARS spectrum of Rh8.10 in the 1100–1700 cm21
region. The nonresonant CARS background signal from wateronly, indicated by the horizontal dashed line, is used to normalizethe PR/CARS signal. The phase factorQ (1400 cm21) is 868. Thex (3)-fit function is shown as a solid line overlapping the PR/CARSdata (W). The corresponding background-free (Lorentzian line-shapes) vibrational spectrum of Rh8.10 derived from thex (3)-fit isshown at the bottom. The wave number positions of selected bandsare also presented.
combinations in the 950–970 cm21 range are formedin which both hydrogens move out-of-plane in thesame direction (Au), thereby becoming stronglyRaman active. The corresponding low-frequency,out-of-phase vibrational modes appear in the 750–850 cm21 region have approximately Bg (local)symmetry in the planar configuration and becomeRaman activeonly if the polyene chains contains atwisted region [14,17].
The observation that some of the HOOP features inthe respective vibrational spectra from native Rh,Rh7.10 and Rh8.10 are similar strongly suggeststhat at least some of the vibrational assignmentspreviously made in native Rh have validity for bothRh7.10 and Rh8.10. This similarity reflects the factthat the degree of rigidity introduction by 7- or 8-membered rings doesnot strongly alter all theHOOP modes, but only a few.
Y. Zhou et al. / Journal of Molecular Structure 478 (1999) 107–119 113
Table 1Parameters (Vk – band origin positions,Gk – bandwidth (HWHM) andAk – amplitudes) derived fromx (3) fits to PR/CARS data (700–1700 cm21) assigned to Rh7.10/8D (Figs. 2 and 3). The values for the PR/CARS amplitudes (Ak) are normalized to the amplitude of the 1546-cm21 band
Rh7.10/8D PR/CARS dataVk (cm21) Gk (cm21) Ak Vk (cm21) Gk (cm21) Ak
700 15 0.07709 1265 7 0.06683751 10 0.0234 1282 4 0.06251779 9 0.02801 1302 4 0.05559804 5 0.04487 1332 4 0.06941900 7 0.07986 1341 4 0.08295956 5 0.40805 1358 5 0.040541023 12 0.03631 1382 2 0.041641058 10 0.03604 1440 4 0.020581087 17 0.02892 1465 2 0.069851144 4 0.05965 1546 9 11166 4 0.04868 1589 8 0.045121191 4 0.02068 1625 6 0.05151214 4 0.08695 1657 6 0.094351236 7 0.34092
Table 2Parameters (Vk – band origin positions,Gk – bandwidth (HWHM) andAk – amplitudes) derived fromx (3) fits to PR/CARS data (700–1700 cm21) assigned to Rh7.10/ND (Figs. 4 and 5). The values for the PR/CARS amplitudes (Ak) are normalized to the amplitude of the 1551-cm21 band
Rh7.10/ND PR/CARS dataVk (cm21) Gk (cm21) Ak Vk (cm21) Gk (cm21) Ak
723 10 0.04266 1246 3 0.11906793 15 0.04605 1293 3 0.13117847 7 0.00963 1305 3 0.08743954 6 0.38993 1329 2 0.023821026 10 0.03112 1342 6 0.055211067 18 0.03005 1372 8 0.002411142 3 0.03797 1441 3 0.080541164 4 0.05857 1464 8 0.11311186 4 0.06333 1551 9 11212 3 0.04841 1592 11 0.213191232 6 0.26652 1622 8 0.16363

Y. Zhou et al. / Journal of Molecular Structure 478 (1999) 107–119114
Table 3Parameters (Vk – band origin positions,Gk – bandwidth (HWHM) andAk – amplitudes) derived fromx (3) fits to PR/CARS data (700–1700 cm21) assigned to Rh8.10 (Figs. 6 and 7). The values for the PR/CARS amplitudes (Ak) are normalized to the amplitude of the 1552-cm21
band
Rh8.10 PR/CARS dataVk (cm21) Gk (cm21) Ak Vk (cm21) Gk (cm21) Ak
702 15 0.0033 1234 7 0.26351781 4 0.0182 1274 10 0.00314804 10 0.05418 1293 3 0.11796824 9 0.04256 1306 4 0.1003899 15 0.03822 1329 2 0.0373933 6 0.04288 1342 6 0.08897954 6 0.34636 1382 10 0.01858998 10 0.0104 1443 4 0.14621023 5 0.01046 1467 8 0.117911059 18 0.02198 1495 15 0.036521142 4 0.02325 1552 9 11164 5 0.05451 1592 11 0.28731186 5 0.0546 1625 12 0.081941210 6 0.08845 1654 8 0.16564
Fig. 8. Background-free (Lorentzian lineshape) vibrational spec-trum of (from top to bottom) native Rh; Rh7.10; Rh7.10/8D;Rh7.10/ND and Rh8.10 in the 700–1400 cm21 region. The vertical,dashed lines indicate the most prominent spectral changes.
Fig. 9. Background-free (Lorentzian lineshape) vibrational spec-trum of (from top to bottom) native Rh; Rh7.10; Rh7.10/8D;Rh7.10/ND and Rh8.10 in the 1100–1700 cm21 region. Thevertical, dashed lines indicate the most prominent spectral changes.

Evidence to support this general conclusion can befound in many specific parts of the respective PR/CARS spectra.
3.3.1. The 955/972 cm21 bandThe prominent 955-cm21 band can be assigned to
the HC11yC12H HOOP mode in Rh7.10 (as well as inRh7.10/8D and Rh7.10/ND) and in Rh8.10. Thisassignment is independent of the rigidity associatedwith the 7- or 8-membered rings.
1. Contribution from the HC7yC8H HOOP mode tothe 955-cm21 band can be excluded since deutera-tion of the C8 position (Rh7.10/8D) causes nochange in either its intensity or position (Fig. 8).Generally, deuterium substitution is expected touncoupled wagging modes (mass differences alterlocal symmetry) and cause shifts (decreases) ofhydrogen wagging frequencies [14,17]. Thus, theinvolvement of the HC7yC8H HOOP mode in the955-cm21 band of the Rh7.10/8D spectrum wouldhave decreased its intensity and resulted in theappearance of a strong, new band at lowerfrequency.
2. Contributions from the C15yNH HOOP mode tothe 955-cm21 assignment can be excluded sinceno changes in either its position or intensity areobserved in the PR/CARS spectrum of Rh7.10/ND (Fig. 9).
3. The steric interaction of C10–H and C13–CH3
motions, known to underlie the high intensity ofthe 972-cm21 band in native Rh, is not present inneither Rh7.10 nor Rh8.10 (eliminated by thering). In addition, neither C10–H wagging(846 cm21) nor C13–CH3 rocking modes(1000 cm21) in the native Rh are significant ineither Rh7.10 or Rh8.10 since neither band appearsin the respective PR/CARS spectra (Figs. 8 and 9).These rings, however, do provide some structuralconstraint within the retinal which alters the degreeof twisting of the chromophore (relative to the non-planar geometry in native Rh [7]) and results in thehigh intensity of the HOOP band at 955 cm21. The7- and 8-membered rings make the angles in theC10–C11yC12–C13 region of the 11-cis retinal morerigid (relative to native Rh) which significantlyaffects in-plane hydrogen wagging modes, butwhich only slightly affects out-of-plane motions.
Thus, it is reasonable to conclude that incorpora-tion of an artificial ring shifts the C11yC12 HOOPmode frequency from 972 cm21 in native Rh to955 cm21 in Rh7.10 and Rh8.10 (Figs. 8 and 9).
4. A band at 899 cm21, appearing only in the PR/CARS spectrum of Rh7.10/8D, can be assignedto HOOP of C7yC8 by considering the decouplingof the in-plane vibrations of the hydrogens at posi-tions C7 and C8, vide infra.
3.3.2. Flexibility in the C10–C11yC12–C13 regionStructurally, the 7- and 8-membered rings prevent
isomerization around the C11yC12 bond normallyconsidered to be the principle reaction coordinate innative Rh. Unlike the other artificial Rh pigmentcontaining a 5-membered ring which was examinedextensively [7,18], the 7- and 8-membered ring Rhsystems remain flexible in the C10–C11yC12–C13
region of the 11-cis retinal and therefore, are assumedto be more capable of forming (via torsional motion inthe C10–C11yC12–C13 region) a trans-like inter-mediate following optical excitation. Transientabsorption data from native Rh and artificial Rhpigments containing 5-, 7-, and 8-membered ringshave indicated that the formation of intermediates isdependent on the flexibility of the 11-ene part of theretinal containing the artificial ring. [7,8,18].
Since the retinal in Rh is spatially fixed at both endsvia the ionone-ring binding site and the Schiff-baselinkage to the lysine residue at the protein, the mostflexible part of the chromophore should be near thecis-11 ene. Restricting motion in this C10–C11yC12–C13 region by incorporation of a ring obviously altersboth the forward progress of the photo-reaction andthe degree to which any intermediate acting as aprecursor to C11yC12 isomerization can be formed.This phenomenon is reflected in the frequency shiftfrom 972 cm21 in native Rh to 955 cm21 in Rh7.10(Fig. 8) and Rh8.10 (Fig. 7).
3.4. Assignments of the fingerprint modes
The PR/CARS bands observed in the C–Cstretching (fingerprint) region for both Rh7.10 andRh8.10 are significantly different than those foundin the native Rh PR/CARS spectrum. The most promi-nent PR/CARS band appears at 1235 cm21 for theartificial pigments (Figs. 6 and 8). Other important
Y. Zhou et al. / Journal of Molecular Structure 478 (1999) 107–119 115

bands are observed at 1142, 1165, 1187, 1212, 1293,1306, and 1342 cm21 which are to be comparedrespectively with the 1191, 1214, 1238, 1267, 1317,and 1359 cm21 found in native Rh (Fig. 8).
Several specific differences are to be noted:
1. The high intensity 1267 cm21 band, assigned to theHC11yC12H hydrogen in-plane rocking mode innative Rh [16], is not present in either the Rh7.10or Rh8.10 spectra.
2. The 1238, 1214, 1190 cm21 bands, assignedrespectively to the C12–C13, C8–C9, and C14–C15
stretching modes in native Rh [16], are shifted by3 , 4 cm21 to lower energy in the Rh7.10 (i.e.,1235, 1212, 1187 cm21) and Rh8.10 spectra (i.e.,1234, 1210, 1186 cm21). Two other small intensitybands, located at 1142 and 1164 cm21, appear onlyin all artificial Rh spectra.
• The influence of changes in local symmetrythrough changes in the effective center ofmass at specific bonds is evident in the PR/CARS spectra from Rh7.10/8D (Fig. 2). Threebands found in the PR/CARS spectra fromRh7.10 and Rh8.10, comparable to the respec-tive bands in the native Rh spectrum [9] at 1238,1214, and 1191 cm21, shift to lower frequen-cies. The positions of the corresponding bandsfrom Rh7.10/8D (Fig. 8) shift back in proximityto the origins of the respective bands fromnative RhRT. These decreased frequencies canbe attributed to a weakening of the strongcoupling of the C–C stretching modes alongthe polyene chain caused by the increased mass.
• Isotopic substitutions (at the C13–CH3 position)indicate that the C12–C13 stretching coordinatecontributes to four different C–C stretchingnormal modes [16] and therefore, these modesare all extensively delocalized. Specifically, the1235-cm21 band observed in Rh7.10, Rh7.10/8D, and Rh8.10 can be assigned to the C12–C13
and C14–C15 stretching modes (comparable toassignments in native Rh). Thus, the introduc-tion of either the 7- or 8-membered ring doesnot affect these modes significantly. Theremaining bands at 1210 1 and 1190^1 cm21 can be assigned to the C8–C9 and C14–C15 stretching modes in analogy to assignmentsin native Rh [16],vide supra.
3. The 1293- and 1306-cm21 bands which appear inRh7.10 and Rh8.10 spectra are not present in theRh7.10/8D spectrum (Figs. 2, 6 and 8). These twobands can be associated with those that appear nearthe 1267 cm21 band in native Rh (i.e., 1317,1309 cm21).
4. The 1246-cm21 band appears for Rh7.10 andRh8.10 while the prominent band at 1235 cm21
in Rh7.10 shifts to 1232 cm21 in the Rh7.10/NDspectrum (Fig. 4).
• The absence of the 1267-cm21 band can beattributed to the reduction of local symmetrycaused by the incorporation of the respectivering structures. The dipole moment and polariz-ability changes from which the PR/CARSsignals originate are different when thesymmetry at specific locations within the retinalchromophore are altered. Of interest here is theHC11yC12H hydrogen in-plane, rocking mode.In native Rh, the HC11–C12H mode is locatedapproximately at the mass center of the polyene(i.e., centered between C6 and the Schiff-basenitrogen). This assumes that the ionone-ringand the Schiff-base binding sites are fixed rela-tive to the protein. Thus, the HC11yC12H modeexhibits some degree of geometrical symmetry,especially when the approximately equivalenteffective masses of the two retinal sections oneither side of the C11yC12 bond are noted. Theintroduction of a rigid ring spanning the C10–C11yC12–C13 region (i.e., Rh7.10 and Rh8.10)removes this symmetry by changing the centerof mass. Such reduction of symmetry is clearlydemonstrated in artificial Rh pigmentscontaining deuterium at the C11 and/or C12 posi-tions [14].
• The appearance of the 1267-cm21 band is espe-cially sensitive to alterations in the retinal struc-ture. Its intensity is significantly reduced inartificial Rh pigment containing deuterium ateither oneof the C11 or C12 positions, but hashigh intensity (at a position 7 cm21 higher infrequency) whenboth the C11 and C12 positionshave a deuterium attached. In the 13-demethyl[19] and 9-demethyl [14] artificial Rh pigments,the 1267-cm21 band again is not present. The1267-cm21 band also loses intensity (and shifts
Y. Zhou et al. / Journal of Molecular Structure 478 (1999) 107–119116

slightly in frequency) during the RhRT photo-reaction when intermediates such as batho [5],lumi [6], meta I, and meta II [20] are formed.For the cases of Rh7.10 and Rh8.10 describedhere, the intensity reduction or absence of the1267-cm21 band can be attributed to the struc-tural rigidity caused by the introduction of thering. In these two artificial Rh pigments, thisspecifically results in removing flexibility inthe C11yC12 in plane-modes while leaving theHOOP motions near the C11yC12 bond flexible,vide supra.
5. The 1142 and 1166 cm21 bands only appear inRh7.10, Rh7.10/8D, Rh7.10/ND and Rh8.10 andnot in PR/CARS spectrum of native Rh [5,6].There are two assignments for these bands toconsider: (i) C–C stretching modes in the artificialring or (ii) the conjugated C–CyC-skeletal modesin retinal itself which, by the introduction of thering, have increased intensities via enhancedRaman resonances. No changes occur in either ofthese two bands when the ring size, and thereforethe C–CyCy angle, changes (Fig. 8). Since bothfrequency shifts and even the appearance of newbands are anticipated if C–C stretching modes inthe rings themselves are involved, such an assign-ment cannot be reasonably made. Alterations in theretinal backbone structure involvingp -electrondelocalization along the polyene chain, however,can be anticipated when either ring is introduced.Such changes in thep -electrons can result inpolarizability changes which enhance the Ramanactivity of specific normal modes. The assignmentof the 1142 and 1166 cm21 bands, therefore, are tothe C–C stretching modes of the retinal backbone.
6. The absence of the 1293- and 1306-cm21 bands inthe Rh7.10/8D spectrum, together with the appear-ance of the 1265- and 899-cm21 bands in this spec-trum, suggests that these bands are assignable tothe C7H and C8H in-plane, rocking modes. Char-acteristically, the frequencies of in-plane modesare sensitive to the configuration of the chromo-phore (e.g., the HC11–C12H in-plane rockingmode). In this case, it appears that deuteration ofthe hydrogen at C8 position causes the shift of oneband to 899 cm21 (vide supra) and of theremaining band, via significant decoupling of the
C7H and C8H rocking vibrations, to 1265 cm21
(Fig. 2).7. The approximately equal intensities (relative to the
955-cm21 band intensity) of the 1235-cm21 band inRh7.10 and of the 1232- and 1246-cm21 bands inRh7.10/ND (Fig. 8) indicate that deuteration of theC15–N position in Rh7.10 effectively decouplesthe single bond stretching modes and therefore,the 1235-cm21 band splits into two bands. In thePR/CARS spectrum of native Rh [5,21], deutera-tion of the C15–N position results in the appearanceof two bands (at 1234 and 1240 cm21 [21]). The1235 cm21 band, therefore, has contributions fromboth the C12–C13 and C14–C15 stretching modes,vide supra.
8. The existence of electronic delocalization ofpolyene chains [16] suggests that the 1246-cm21
band is assignable to the –C15yN–D bendingmode. Thus, deuteration of the C15–N-positioncan affect the C14 2 C15 stretching mode andcause a shift of the 1235 cm21 band to1232 cm21 (assignable to the C12–C13 and C14–C15 stretching modes in Rh7.10/ND).
3.5. Assignment on the CyC stretching and Schiff-base modes
1. The most itense CyC stretching feature in both theRh7.10 and Rh8.10 PR/CARS spectra appears at1551 cm21 (Figs. 7 and 9) rather than at the1545 cm21 position found in the PR/CARS spec-trum from native Rh [5,6]. This CyC stretchingbands shifts to 1545 cm21 in the Rh7.10/8D(Figs. 3 and 9). Thex (3) analyses of these spectralregions reveal that two bands appearing at 1530and 1546 cm21 for Rh7.10/8D (Fig. 3) correspondto the two bands at 1536 and 1547 cm21 found innative RhRT. The corresponding CyC stretchingbands in Rh7.10 and Rh8.10 can be fitted wellvia x (3) analysis with only a single Lorentzianfunction and therefore, it can be concluded thatno additional bands appear in this region. Thesecharacteristics of the PR/CARS data support theconclusion that shifts in Raman band frequenciesare attributable primarily to changes in the centerof mass at specific bonds and the consequent
Y. Zhou et al. / Journal of Molecular Structure 478 (1999) 107–119 117

alteration of symmetry,vide supra. Force fieldstudies further indicate that the (CyC)sym stretchis a delocalized mode with major contributionsfrom the C7yC8, C9yC10 and C11yC12 stretchingmodes in native Rh. The introduction of a ring inRh7.10 and Rh8.10 alters the C11yC12 stretchingmotions and decouples this stretching mode fromthe other two. Only a single Lorentzian lineshape isneeded to accurately fit the 1550-cm21 CyC band.These effects also cause a 6-cm21 increase in theCyC frequency in both Rh7.10 and Rh8.10. Thedeuteration of C8 position (Rh7.10/8D) demon-strates that the C7yC8 stretching mode contributesto this band.
2. The assignment of 1595-cm21 band from native Rh(1592 cm21 in Rh7.10 and Rh8.10, and 1589 cm21
in Rh7.10/8D) involves contributions from theC9yC10 and C13yC14 stretching modes in additionto contributions from the ring itself. These twomodes can be coupled through the ring and appearwith relatively high intensity in the PR/CARSspectra from both Rh7.10 and Rh8.10 (Fig. 9).The 1592 to 1589 cm21 shift can be attributed tothe deuteration in Rh7.10/8D.
3. The 1657-cm21 band, assigned in native Rh to theSchiff-base CyN stretching mode, appears at1625 cm21 in Rh7.10/8D (Fig. 3). This mode, rela-tively isolated from the ring structure, has assign-able bands at 1659 cm21 in Rh7.10, 1654 cm21 inRh8.10, 1657 cm21 in Rh 7.10/8D, and 1622 cm21
in Rh7.10/ND.
4. Concluding remarks
The introduction of 7- and 8-membered rings intothe retinal structure of Rh rigidly fixes the 11-enestructure which results in significant changes relativeto the PR/CARS spectrum of native Rh:
1. the 972-cm21 band shifts to 955 cm21 in the HOOPregion,
2. the 1267-cm21 band is absent in the fingerprintregion, and
3. the CyC stretching mode shifts from 1545 to1551 cm21.
These artificial rings rigidly fix the angles in theC10–C11yC12–C13 in the region of the 11-locked
retinal thereby significantly altering the in-planehydrogen wagging motions e.g., the C11yC12 in-plane mode at 1267-cm21 disappears. The out-of-plane modes in the C10–C11yC12–C13 region, howeverare not fixed (the C11yC12 HOOP mode shifts from972 cm21 in native Rh to 955 cm21 in the artificialring Rh pigments). The similarity of the PR/CARSspectra from Rh7.10 and Rh8.10 demonstrates thatthe size of the artificial ring (from 7 to 8) does notdirectly affect the vibrational modes within thepolyene retinal chain. Descriptions based onsymmetry (i.e., Raman selection rules) and center ofmass concepts effectively explain essentially all theband frequency changes observed: CyC stretchingregion (1545 cm21 band in native Rh, 1551 cm21
bands in both Rh7.10 and Rh8.10, 1546 cm21 bandin Rh7.10/8D), C–C stretching region (1293 cm21/1306 cm21 bands to 1265 cm21/899 cm21 bands inRh7.10/8D involving the coupling of C7H and C8Hin-plane rocking modes), Schiff-base region(1246 cm21/1235 cm21 bands in Rh7.10/ND invol-ving the decoupling of the C15yN–D bending modeand the 1659-cm21 band in Rh7.10 and the 1622 cm21
in Rh7.10/ND involving the CyN stretching mode).
Acknowledgements
This research was supported by a grant (GM46439)to G.H.A. from the National Institutes of Health andgrant (GM36564) to K.N. from the National Instituteof Health.
References
[1] G. Wald, Science 162 (1968) 230.[2] G.G. Kochendoerfer, R.A. Mathies, Isr. J. Chem. 35 (1995)
211.[3] Q. Wang, R.W. Schoenlein, L.A. Peteanu, R.A. Mathies, C.V.
Shank, Science (Washington DC) 266 (1994) 422.[4] R.W. Schoenlein, L.A. Peteanu, R.A. Mathies, C.V. Shank,
Science 254 (1991) 412.[5] F. Jager, L. Ujj, G.H. Atkinson, J. Am. Chem. Soc. 119 (1997)
12 610.[6] L. Ujj, F. Jager, G.H. Atkinson, Biophys. J. 74 (1998) 1492–
1501.[7] K. Nakanishi, A.W. Chen, F. Derguini, P. Franklin, S. Hu, J.
Wang, Pure and Appl. Chem. 66 (1994) 981.[8] P.F. McGarry, J. Cheh, B. Ruiz-Silva, S. Hu, J. Wang, K.
Nakanishi, N.J. Turro, J. Phys. Chem. 100 (1996) 646.
Y. Zhou et al. / Journal of Molecular Structure 478 (1999) 107–119118

[9] F. Jager, J. Lou, K. Nakanishi, L. Ujj, G.H. Atkinson, J. Am.Chem. Soc. 120 (1998) 3739.
[10] D.S. Papermaster, Methods Enzymol. 81 (1982) 49.[11] H. Akita, S.P. Tanis, M. Adams, V. Balogh-Nair, K. Naka-
nishi, J. Am. Chem. Soc. 102 (1980) 6370.[12] L. Ujj, A. Popp, G.H. Atkinson, Chem. Phys. 188 (1994) 221.[13] L. Ujj, B.L. Volodin, A. Popp, J.K. Delaney, G.H. Atkinson,
Chem. Phys. 182 (1994) 291.[14] G. Eyring, B. Curry, A. Broek, J. Lugtenburg, R. Mathies,
Biochem. 21 (1982) 384.[15] G. Eyring, B. Curry, R.A. Mathies, R. Fransen, I. Paling, J.
Lugtenburg, Biochem. 19 (1980) 2410.
[16] I. Palings, J.A. Pardoen, E. Berg, C. Winkel, J. Lugtenburg,R.A. Mathies, Biochem. 26 (1987) 2544.
[17] I. Palings, E. Berg, J. Lugtenburg, R.A. Mathies, Biochem. 28(1989) 1498.
[18] H. Kandori, H. Sasabe, K. Nakamishi, T. Yoshizawa, T. Mizu-kami, Y. Shichida, J. Am. Chem. Soc. 118 (1996) 1002.
[19] U.M. Ganter, W. Gartner, F. Siebert, Eur. Biophys. J. 18(1990) 295.
[20] A.G. Doukas, R.H. Callender, T.G. Ebrey, Biochem. 17(1978) 2430.
[21] H. Deng, R.H. Callender, Biochem. 26 (1987) 7418.
Y. Zhou et al. / Journal of Molecular Structure 478 (1999) 107–119 119
Copyright © 2022 FDOKUMEN



















