
Signatures of Isomerization in Photodissociation of trans - Crotonaldehyde Probed by Multiphoton...
8
Signatures of Isomerization in Photodissociation of trans- Crotonaldehyde Probed by Multiphoton Ionization Mass Spectrometry Arup K. Ghosh, Sayan Datta, Anamika Mukhopadhyay, and Tapas Chakraborty* Department of Physical Chemistry, Indian Association for the Cultivation of Science, 2A Raja S C Mullick Road, Jadavpur, Calcutta 700032, India ABSTRACT: We report the observation of new isomerization effects in the UV-photodissociation of trans-crotonaldehyde upon multiphoton excitation by the third harmonic (355 nm) pulses of a Nd:YAG laser. A time-of-flight mass spectrometric analysis reveals formation of acetaldehyde, acetyl, and methoxy radical cations as signatures of isomerization processes. A small segment of the multiphoton ionization spectrum of jet-cooled crotonaldehyde is recorded by tuning the laser frequency around 355 nm. An oxetene type transient intermediate in the ground state has been considered for acetaldehyde formation following a photochemical model suggested earlier (Reguero; et al. J. Am. Chem. Soc. 1994,116, 2101−2114) for such compounds. Likewise, for methoxy radical formation, a trans−cis isomer- ization about the CC double bond has been considered in a triplet surface. Electron ionization mass spectra of the compound are also recorded by varying the electron kinetic energy in the range 11−70 eV. Ionic fragments in the mass spectra of the two ionization processes are dramatically different. Our suggested mechanisms for isomerization and fragmentation channels are substantiated by density functional theory calculations. Combined experimental and calculated data lead us conclude that isomerization occurs in neutral potential energy surfaces prior to dissociation and photoionization. ■ INTRODUCTION Photodissociation of α,β-enones, which involves an interplay of the ground electronic state (S 0 ) with at least three of the lowest excited electronic states (S 1 ,T 1 , and T 2 ), has generated much interest in spectroscopy and nonadiabatic dynamic commun- ities over the past several decades. 1−13 To understand the fundamentals of photodissociation dynamics, the two simplest members of the acyclic α,β-unsaturated aldehydes, acrolein and crotonaldehyde, where the latter is a β-methyl derivative of the former (Figure 1) have been extensively investigated. 8−12 In these compounds, an ethylenic (CC) and a carbonyl (C O) functional groups are linked in direct conjugation. Upon electronic excitation or ionization, the bonding characters of the CC single and double bonds (Figure 1) are significantly altered. In effect, this class of molecules shows several types of rearrangement reactions upon UV excitation in addition to typical photofragmentation and photochemical reactions displayed by simple aliphatic aldehydes. Notable among the rearrangement reactions are those proposing acrolein (CH 2 CHCHO) conversion to methylketene before photodissocia- tion to produce CO and CH 3 CH; 7,11,12 isomerization via internal rotation of the molecules about the CC double bond; 12 oxetene formation in the ground state 3 and conversion of crotonaldehyde to 3-butene-1-al, ethylketene, and enol- crotonaldehyde. 13 In this report, using multiphoton ionization (MPI) time-of-flight mass spectrometry (TOF-MS), we demonstrate the occurrence of a new type of photoisomeriza- tion of crotonaldehyde (CA) upon excitation by 355 nm pulses of a Q-switched Nd:YAG laser under the collision-free conditions of a supersonic jet expansion. The photoreaction results in the formation of acetaldehyde, in addition to other fragments reported so far. A relevant photochemical process, in the context of the current study, is isomerization of α,β-enones to oxetenes upon excitation to high vibrationally excited regions of the S 1 (nπ*) surface. 3 Although oxetene formation has not been exper- imentally identified for acrolein or CA, the product has been identified for the case of 3,4-dimethyl-3-pentene-2-one when irradiated with light of wavelengths 250 nm or shorter in a pentane solution. 14 This reaction does not occur when irradiated with light of wavelengths longer than 300 nm, which indicates that the reaction barrier is quite high. Furthermore, oxetene formation was found unaffected by the presence of butadiene, a well-known triplet quencher. 15 This latter observation clearly implies that photochemical oxetene formation is the outcome of an interplay of the three singlet states (S 0 ,S 1 , and S 2 ). On the other hand, quite high quantum yield for photoisomerization across the α and β CC double Special Issue: Structure and Dynamics: ESDMC, IACS-2013 Received: March 31, 2013 Revised: July 9, 2013 Published: July 10, 2013 Article pubs.acs.org/JPCA © 2013 American Chemical Society 8710 dx.doi.org/10.1021/jp403178x | J. Phys. Chem. A 2013, 117, 8710−8717
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Signatures of Isomerization in Photodissociation of trans - Crotonaldehyde Probed by Multiphoton...

Signatures of Isomerization in Photodissociation of trans-Crotonaldehyde Probed by Multiphoton Ionization MassSpectrometryArup K. Ghosh, Sayan Datta, Anamika Mukhopadhyay, and Tapas Chakraborty*
Department of Physical Chemistry, Indian Association for the Cultivation of Science, 2A Raja S C Mullick Road, Jadavpur, Calcutta700032, India
ABSTRACT: We report the observation of new isomerization effects in theUV-photodissociation of trans-crotonaldehyde upon multiphoton excitationby the third harmonic (355 nm) pulses of a Nd:YAG laser. A time-of-flightmass spectrometric analysis reveals formation of acetaldehyde, acetyl, andmethoxy radical cations as signatures of isomerization processes. A smallsegment of the multiphoton ionization spectrum of jet-cooled crotonaldehydeis recorded by tuning the laser frequency around 355 nm. An oxetene typetransient intermediate in the ground state has been considered foracetaldehyde formation following a photochemical model suggested earlier(Reguero; et al. J. Am. Chem. Soc. 1994,116, 2101−2114) for suchcompounds. Likewise, for methoxy radical formation, a trans−cis isomer-ization about the CC double bond has been considered in a triplet surface.Electron ionization mass spectra of the compound are also recorded byvarying the electron kinetic energy in the range 11−70 eV. Ionic fragments in the mass spectra of the two ionization processes aredramatically different. Our suggested mechanisms for isomerization and fragmentation channels are substantiated by densityfunctional theory calculations. Combined experimental and calculated data lead us conclude that isomerization occurs in neutralpotential energy surfaces prior to dissociation and photoionization.
■ INTRODUCTION
Photodissociation of α,β-enones, which involves an interplay ofthe ground electronic state (S0) with at least three of the lowestexcited electronic states (S1, T1, and T2), has generated muchinterest in spectroscopy and nonadiabatic dynamic commun-ities over the past several decades.1−13 To understand thefundamentals of photodissociation dynamics, the two simplestmembers of the acyclic α,β-unsaturated aldehydes, acrolein andcrotonaldehyde, where the latter is a β-methyl derivative of theformer (Figure 1) have been extensively investigated.8−12 Inthese compounds, an ethylenic (CC) and a carbonyl (CO) functional groups are linked in direct conjugation. Uponelectronic excitation or ionization, the bonding characters of theCC single and double bonds (Figure 1) are significantlyaltered. In effect, this class of molecules shows several types ofrearrangement reactions upon UV excitation in addition totypical photofragmentation and photochemical reactionsdisplayed by simple aliphatic aldehydes. Notable among therearrangement reactions are those proposing acrolein (CH2CHCHO) conversion to methylketene before photodissocia-tion to produce CO and CH3CH;
7,11,12 isomerization viainternal rotation of the molecules about the CC doublebond;12 oxetene formation in the ground state3 and conversionof crotonaldehyde to 3-butene-1-al, ethylketene, and enol-crotonaldehyde.13 In this report, using multiphoton ionization(MPI) time-of-flight mass spectrometry (TOF-MS), wedemonstrate the occurrence of a new type of photoisomeriza-
tion of crotonaldehyde (CA) upon excitation by 355 nm pulsesof a Q-switched Nd:YAG laser under the collision-freeconditions of a supersonic jet expansion. The photoreactionresults in the formation of acetaldehyde, in addition to otherfragments reported so far.A relevant photochemical process, in the context of the
current study, is isomerization of α,β-enones to oxetenes uponexcitation to high vibrationally excited regions of the S1 (nπ*)surface.3 Although oxetene formation has not been exper-imentally identified for acrolein or CA, the product has beenidentified for the case of 3,4-dimethyl-3-pentene-2-one whenirradiated with light of wavelengths 250 nm or shorter in apentane solution.14 This reaction does not occur whenirradiated with light of wavelengths longer than 300 nm,which indicates that the reaction barrier is quite high.Furthermore, oxetene formation was found unaffected by thepresence of butadiene, a well-known triplet quencher.15 Thislatter observation clearly implies that photochemical oxeteneformation is the outcome of an interplay of the three singletstates (S0, S1, and S2). On the other hand, quite high quantumyield for photoisomerization across the α and β CC double
Special Issue: Structure and Dynamics: ESDMC, IACS-2013
Received: March 31, 2013Revised: July 9, 2013Published: July 10, 2013
Article
pubs.acs.org/JPCA
© 2013 American Chemical Society 8710 dx.doi.org/10.1021/jp403178x | J. Phys. Chem. A 2013, 117, 8710−8717
Anamika
Highlight
Anamika
Highlight
Anamika
Highlight
Anamika
Highlight
Anamika
Highlight
Anamika
Highlight
Anamika
Highlight
Anamika
Highlight
Anamika
Highlight

bond was observed for excitation in the lower energy regions ofthe S1 surface.
3 In an extensive theoretical study using completeactive space self-consistent field (CASSCF) methods, Regueroet al. presented a model for photochemical conversion of α,β-enones to oxetene.3 Using trans-acrolein as a model system, theauthors predicted a conical intersection between the S1 (nπ*)and S0 states at about 15 kcal/mol above the S1 minimum,which drives an ultrafast electronic relaxation to the groundstate. Thus, excitation of the molecule with a shorterwavelength photon is essential to overcome the surface-crossingbarrier in S1. In contrast, for the occurrence of photo-isomerization across the α and β CC double bond,intersystem crossings involving S1 (nπ*), T2 (ππ*), T1(nπ*), and S0 are essential. Both the triplet states are lowerin energy compared to S1, and the energy minimum of T2appears at a twisted geometry with respect to the CC doublebond. Thus, trans−cis isomerization becomes a facile processupon nπ* excitation and this is consistent with the very lowquantum yields of fluorescence and phosphorescence reportedfor the acyclic α, β-enones.1
The nonadiabatic photodissociation dynamics of CA andacrolein were investigated recently by Geβner et al. using time-resolved photoelectron and coincidence imaging spectroscopy.5
The authors suggested that the methyl group of CAsignificantly enhances the S0 ← S1 internal conversion rate,an effect that has been extensively discussed in the literaturewith other systems.16,17 For 193 nm photodissociation, theHCO• radical yield in the ground state produced due to α-cleavage in the T1 (nπ*) state of CA was found to be muchsmaller compared to that obtained for acrolein. Fang performeda very detailed CASSCF theoretical study using the cc-pVDZbasis set to calculate the potential energy profiles correspondingto CO loss. Two types of Norrish type-I α-cleavages of acroleinin the ground and low-lying excited electronic states wereproposed.4 HCO• formation in the ground state was shown tobe favored in the T1 (nπ*) surface. In a very recent study Suand co-workers used time-resolved infrared spectroscopy toprobe the photochemical/photodissociation dynamics ofacrolein induced by 193 nm excitation in acetonitrile solution.12
Unlike gas-phase behavior, the authors noted that 1,3-migrationof the H atom that results in methylketene formation is themost dominant reaction channel, but the branching ratio for theC−H α cleavage was found to be very small, and the differingbehavior in the solution phase was attributed to a cage effect bythe solvent molecules. In TOF mass spectra corresponding to193 nm photofragmentation of CA,11 Ahmed and co-workersnoted that dominant dissociation channels are the two α-cleavages, CH3CHCHCO + H and CH3CHCH + HCO,decarbonylation and methyl loss (CH3 + CHCHCHO). Theauthors suggested that H loss occurs in the S0 surface, but forCO loss the molecule first isomerizes to ethylketene. Theauthors also estimated the center of mass translational energydistribution corresponding to different dissociation channelsobserved. On the other hand, HCO• as a radical intermediatewas considered in 255 nm photochemistry by Coomber andPitts.13
In the present study, we have investigated the photo-dissociation of CA in the collision free environment of asupersonic jet expansion at several wavelengths around thethird harmonic (355 nm) of a pulsed (10 ns) Nd:YAG laser,and the fragments have been probed using TOF-MS. A smallsegment of the MPI spectrum around this wavelength is alsorecorded by tuning the excitation wavelength. This wavelengthregion corresponds to S1 ← S0 one photon excitation of themolecule with ∼1500 cm−1 excess vibronic energy of the lowestS1 (nπ*) surface. The wavelength also corresponds to the two-photon energy region of n → 3s and n → 3p Rydbergexcitations.18 Furthermore, as the pulse length (10 ns) is longercompared to the much faster electronic relaxation (S0 ← S1internal conversion) time scale (typically subpicosecond), thevibrationally hot molecules produced through initial excitation/relaxation cycles can reabsorb from the same laser pulseresulting to very high vibronic excitation in S1, suitable to crosshigher reaction barriers. We demonstrate here new reactions ofthis molecule, which have not been observed so far. Ourproposed mechanisms of these reactions are substantiated bythe predictions of electronic structure calculations.
2. EXPERIMENTAL AND THEORETICAL METHODSCA (99%), purchased from LobaChemie Pvt. Ltd. (India) waspurified repeatedly by vacuum distillation before use. A UVabsorption spectrum of the molecule in the vapor phase wasrecorded in a home-built gas cell with a 1 m path length. Lightfrom a Xe arc lamp (Newport, Oriel Instruments, 66901) wasdispersed by a small monochromator (Optometrics, SDMC1-01) and the selected wavelengths were passed through the gasabsorption cell after collimation by a telescopic arrangement.Light intensity after traversing through the gas cell wasmeasured with (I) and without (I0) the sample vapors by aphotomultiplier tube (IP-28). The output signal of thephotomultiplier tube was amplified and acquired into apersonal computer by use of a home-built data acquisitionsystem. The spectrum was obtained by plotting log(I0/I)against wavelength (nm) of light.The electron ionization (EI) mass spectra presented here
were obtained using a quadrupole mass spectrometer (Extrel,Model MAX500) whose details have been discussed else-where.19−21 Briefly, CA vapor was introduced in the ionizationregion via an effusive nozzle of diameter 100 μm and a workingpressure of 3.0 × 10−6 mbar was maintained in the mass filterregion. The ions were detected by an assembly that consists ofa conversion dynode and a channeltron electron multiplier,
Figure 1. Geometries of trans-CA in S0, S1, T1, and ionic statesoptimized at DFT/B3LYP-6311++G(d,p) and TDDFT/B3LYP-6311++G(d,p) levels for the excited states. The C−C and C−Obond lengths are mentioned in Å units.
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp403178x | J. Phys. Chem. A 2013, 117, 8710−87178711
Anamika
Highlight
Anamika
Highlight
Anamika
Highlight
Anamika
Highlight
Anamika
Highlight
Anamika
Highlight
Anamika
Highlight
Anamika
Highlight

both negatively biased at 5 and 2 kV, respectively. For dataacquisition and processing, the software package MerlinAutomation provided by Extrel was used.For TOF-MS measurements, CA vapor was mixed with
ultrapure (99.999%) argon gas at a nominal backing pressure(0.1 atm) and was expanded through a pulsed nozzle valve oforifice diameter 500 μm (General Valve Corp.) to produce gaspulses of width 200 μs at a repetition rate of 10 Hz. The pulsedbeam was skimmed through a skimmer of orifice diameter 2mm and the photoionized subsequently using the thirdharmonic wavelength (355 nm) of a pulsed Nd:YAG laser(Spectra Physics, Quanta Ray Lab-150 series). The laser wasoperated at 10 Hz repetition rate and fired with a delay of 600μs after the nozzle opening. A digital delay/pulse generator was
employed as a master trigger for all the instruments tosynchronize temporally. The laser beam was focused at thecollimated supersonic beam using a lens of focal length 25 cm.The laser fluences were varied from 0.1 to 0.5 mJ/pulse. Formeasurements of MPI spectrum around 355 nm, tunable UVlight pulses obtained as a doubled output of a Nd:YAG pumpeddye laser (Continuum, ND6000) was used. The signals fromthe microchannel plate (MCP) detector were fed to a boxcaraverager and the averaged output was recorded on a PC.The ions formed were mass analyzed in a home-built 1.05 m
TOF mass spectrometer, which has a typical mass resolution of300 at 100 Da. The TOF setup consists of a differentiallypumped two-stage vacuum chamber assembly made of stainlesssteel. The ionization chamber has a double walled liquid
Figure 2. 355 nm MPI mass spectrum of trans-CA (panel A) and EI mass spectra for e-KE of 11, 25, and 70 eV in panels B, C, and D, respectively.Insets show the MPI spectrum in a small energy range around 355 nm (INSET 1) and the absorption spectrum of trans-CA in the 260−390 nmregion (INSET 2).
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp403178x | J. Phys. Chem. A 2013, 117, 8710−87178712
nitrogen jacket, and during operation it is filled with liquidnitrogen to maintain an ultraclean vacuum. The ionizationchamber is separated from the detector by a gate valve. Theelectrode assembly in the ionization chamber consists ofbacking, extractor, and accelerating plates each separated fromthe other by 1 cm. Backing and extractor plates are positivelybiased with 3.3 and 2.1 kV, respectively, whereas theaccelerating plate is grounded with respect to the other twoplates. The working pressures in the expansion chamber andthe detection chamber were 1.0 × 10−5 and 1.0 × 10−6 mbar,respectively. The ions after separation in the field-free regionwere detected by a MCP (Advance Performance Detector 2APTOF, Photonis) of collection diameter 25 mm and a gain of1 × 107. The ion signal thus obtained was averaged over 1000laser shots using a 600 MHz oscilloscope (Lecroy, Wavesurfer62Xs) and displayed on an inbuilt PC of the oscilloscope.The geometric structures of the molecule in S0, S1, and T1
states and in the ground electronic state of its cation and also inseveral transition states (TS) associated with the fragmentationand isomerization reactions have been optimized using thedensity functional theory (DFT) method with the B3LYPfunctional and a 6-311++G(d,p) basis set. All structures ofclosed-shell electronic configurations were optimized usingrestricted B3LYP (RB3LYP) whereas those with open-shellconfigurations were optimized using the unrestricted B3LYP(UB3LYP) option. The excited-state geometries were opti-mized using the time-dependent density functional theory(TDDFT) method with the same functional and basis set. Theenergy minima of the various stable species as well as TS wereconfirmed by calculating the harmonic frequencies. We havealso calculated the intrinsic reaction coordinates (IRC) thatconnect the reactants and products. All the calculationspresented here were performed using Gaussian G09 suite ofprogram.22
3. RESULTS AND DISCUSSIONMPI (355 nm) and EI Mass Spectra. The TOF mass
spectrum recorded upon photoionization of CA by the thirdharmonic (355 nm) pulses of a Q-switched Nd:YAG laser isshown in the topmost panel (A) of Figure 2. The laser fluenceused to record this mass spectrum was 0.1 mJ/pulse. When thefluence is increased to 0.5 mJ/pulse, the relative intensity of thesmaller fragments increases and the molecular ion peakbecomes almost unnoticeable. The inset shows (1) a smallsegment of the MPI spectrum of the jet-cooled moleculesrecorded by tuning the laser frequency around 355 nm and (2)the UV absorption spectrum for S1 ← S0 electronic transition ofCA recorded using a low-pressure static gas cell of 1 m pathlength. On the latter, wavelength positions of the electronicorigin band and that for the photoionization laser (355 nm) areindicated using two arrows. The absorption spectrum appearsbroad due to many hot and sequence transitions. The MPIspectrum also looks congested, but all the sharp features arehighly reproducible. The excitation wavelength corresponds tothe ∼1500 cm−1 vibronic energy region of the S1 surface of CA.The congested features of the MPI spectrum originates becauseof high density of states as the molecule contains several low-frequency vibrational modes including methyl torsion. It isworth mentioning that similar congested features also show upin the S1 ← S0 electronic spectrum of relatively smaller carbonylcompounds like acetaldehyde and acetone at such (∼1500cm−1) vibronic energy region.23 Furthermore, for two-photonenergy region the wavelengths are also expected to be resonant
with n → 3p(O) Rydberg transitions, and the correspondingspectrum quickly becomes congested with excess vibronicenergy.18
The ionization energy of CA has been previously reported tobe 9.73 eV.24 Therefore, the molecule needs to absorbsimultaneously at least three photons to cross the ionizationthreshold. However, because of very low intensity of the M•+
ion peak, we could not measure directly the ion currentvariation for M•+ by varying the laser fluence. It has beendepicted in Figure 2 (panels B−D) that H atom loss from M•+
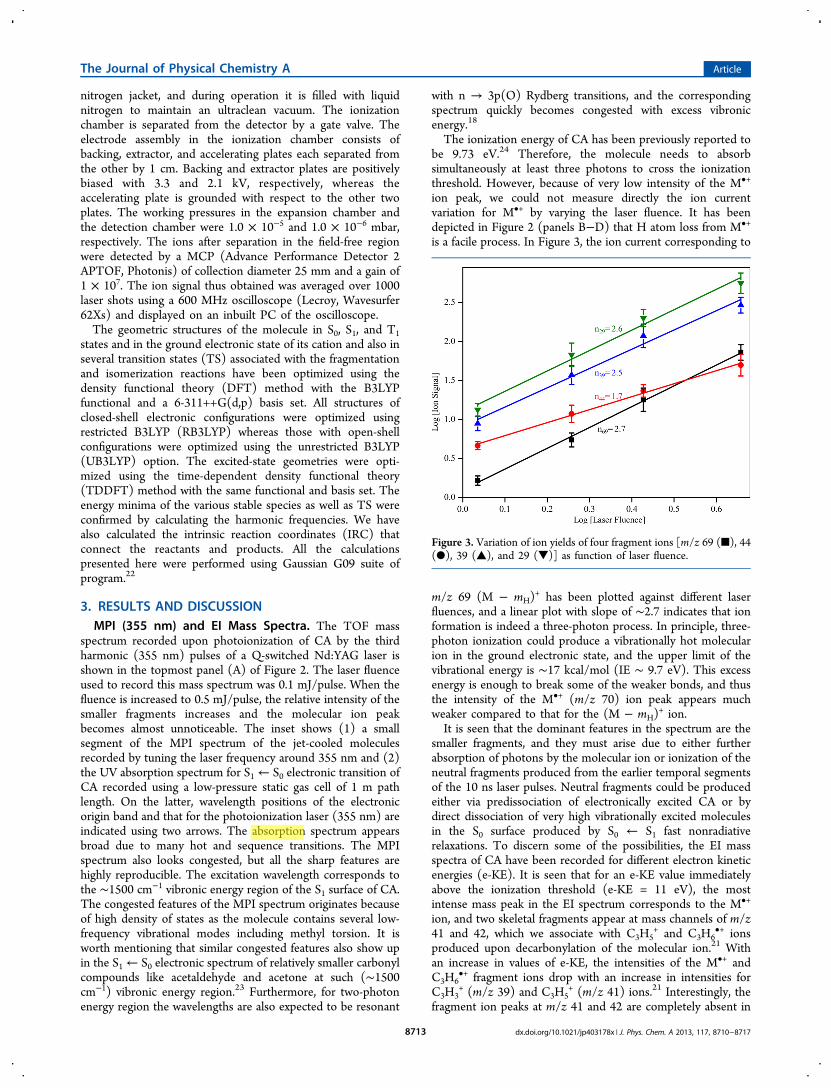
is a facile process. In Figure 3, the ion current corresponding to
m/z 69 (M − mH)+ has been plotted against different laser
fluences, and a linear plot with slope of ∼2.7 indicates that ionformation is indeed a three-photon process. In principle, three-photon ionization could produce a vibrationally hot molecularion in the ground electronic state, and the upper limit of thevibrational energy is ∼17 kcal/mol (IE ∼ 9.7 eV). This excessenergy is enough to break some of the weaker bonds, and thusthe intensity of the M•+ (m/z 70) ion peak appears muchweaker compared to that for the (M − mH)
+ ion.It is seen that the dominant features in the spectrum are the
smaller fragments, and they must arise due to either furtherabsorption of photons by the molecular ion or ionization of theneutral fragments produced from the earlier temporal segmentsof the 10 ns laser pulses. Neutral fragments could be producedeither via predissociation of electronically excited CA or bydirect dissociation of very high vibrationally excited moleculesin the S0 surface produced by S0 ← S1 fast nonradiativerelaxations. To discern some of the possibilities, the EI massspectra of CA have been recorded for different electron kineticenergies (e-KE). It is seen that for an e-KE value immediatelyabove the ionization threshold (e-KE = 11 eV), the mostintense mass peak in the EI spectrum corresponds to the M•+
ion, and two skeletal fragments appear at mass channels of m/z41 and 42, which we associate with C3H5
+ and C3H6•+ ions
produced upon decarbonylation of the molecular ion.21 Withan increase in values of e-KE, the intensities of the M•+ andC3H6
•+ fragment ions drop with an increase in intensities forC3H3
+ (m/z 39) and C3H5+ (m/z 41) ions.21 Interestingly, the
fragment ion peaks at m/z 41 and 42 are completely absent in
Figure 3. Variation of ion yields of four fragment ions [m/z 69 (■), 44(●), 39 (▲), and 29 (▼)] as function of laser fluence.
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp403178x | J. Phys. Chem. A 2013, 117, 8710−87178713
Anamika
Highlight

the MPI mass spectrum, but they appear weakly when higherlaser fluences are used. The most interesting feature of the massspectrum is the appearance of two intense peaks at masschannels of m/z 43 and 44, which we assign to CH3CO
+ andCH3CHO
•+ ions. However, significant rearrangement of themolecular structure is essential for the formation ofacetaldehyde from CA before fragmentation. Below we havepresented further discussion in support of this assignment and aprobable mechanism for its formation.A striking contrast between the two types of ionization
methods is that the most intense mass peak in the MPI massspectrum at m/z 29 is absent in the EI spectra for low values ofe-KE but becomes discernible only near 70 eV. We prefer toassign this as HCO+ over the alternative possibility of C2H5
+. Ifα-cleavage of the molecular ion occurs in an ionic channel, thecharge is expected to be retained on the heavier fragment, i.e.,at the mass of 41 amu, and this regular behavior is observed inthe EI mass spectra (panels B−D, Figure 2). On the otherhand, HCO• can easily be produced in the T1 (nπ*) surface ofthe molecule (α-cleavage) following nπ* (S1 ← S0) electronicexcitation, and the corresponding ion can be produced byfurther absorption of two 355 nm photons. For formation ofthe C2H5
+ ion, CA must isomerize first to ethylketene.Although such isomerization has been suggested, but upondissociation of ethylketene in the ionic channel, the chargeshould be retained on CHCO+. Thus, we infer that HCO•
is produced initially in the neutral channel, which undergoesionization subsequently. We also suggest that some otherfragments (e.g., CH3CO
+ and CH3CHO•+) that appeared in
the MPI mass spectrum are produced via different neutralchannels.In Figure 3, we have shown as how the ion current for
CH3CHO•+ changes with laser fluences. Although the change is
linear, the slope is very different compared to that observed forthe fragment (M − mH)
+ ion. This indicates that CH3CHO•+ is
produced via some complex pathway and not by directfragmentation of molecular ion. We propose below a pathwaythat involves rearrangement/isomerization of the neutralmolecule before fragmentation. The feasibility of the pathwayis substantiated by using the predicted data of electronicstructure calculation.Another important feature of the MPI mass spectrum is the
simultaneous appearance of C3H3+ ion and CH3O
+ ion peaks.The EI mass spectra do not show any signal for CH3O
+. TheC3H3
+ appears only at higher values of e-KE, possibly viadisintegration of C3H5
+ (H2 loss). As the mass peak at m/z 41is absent in the MPI mass spectrum for low laser fluence, weinfer that the pathway for the formation of C3H3
+ here isdifferent from that in the EI mass spectra. Below we havesuggested a rearrangement scheme and present energetic dataas obtained from electronic structure calculation for thesimultaneous formation of C3H3
+ and CH3O+ species in the
ionic channel.Assignment for m/z 44 Ion and Mechanism of Its
Formation. As mentioned above, we assign this fragment ionas CH3CHO
•+. However, for the same mass, there are twoother possibilities, CO2
•+ and C3H8•+. First we argue to discard
the last two possibilities. As the experiment has been performedin a high vacuum condition, there is no external source forcarbon dioxide. Second, from an isolated parent molecule(C4H6O), neither CO2
•+ nor C3H8•+ ions can be produced. An
alternative possibility is of course photochemical reactionswithin clusters that can be produced in a supersonic jet
expansion. Earlier, in 193 nm photodissociation studies onacrolein, El-Sayed et al. explained the observed peaks at muchhigher masses compared to acrolein in terms of clusterreactions.25 In the present study, to avoid cluster formation,we have varied expansion condition, by reducing backingpressure, using helium carrier gas at a very low backingpressure, and also reducing the sample partial pressure in thepre-expansion gas mixture. Indeed, in the mass spectra we haverecorded, no CA cluster peak is observed. Thus CH3CHO
•+ isleft as the only possibility. We propose below that for formationof this species the molecule needs to undergo prior structuralrearrangement.A reaction scheme involving the essential photophysical steps
for conversion of trans-CA to acetaldehyde in neutral form areshown in Figure 4. This mechanism is based on a reaction
model proposed for this class of compounds using very detailedtheoretical calculations.3 The first step is the conversion of CAto a rotameric form labeled RCA, which is only 2 kcal/molabove the global minimum configuration (Figure 5). Thebarrier calculated for this internal rotation (about C1−C2bond) in the ground state is 10 kcal/mol. Calculation alsoshows that this bond gains partial double bond character in S1,T1, and ionic states (Figure 1). Therefore, the barrier crossingcan occur only at higher vibrational levels in the ground state.These levels can be populated via nonradiative electronicrelaxation following nπ* (S1 ← S0) excitation.The next step is the formation of a four-membered cyclic
intermediate oxetene, which falls apart to acetaldehyde andacetylene (m/z 26). Photochemical oxetene formation from
Figure 4. Photochemical schemes for two different reaction channelsof trans-CA.
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp403178x | J. Phys. Chem. A 2013, 117, 8710−87178714
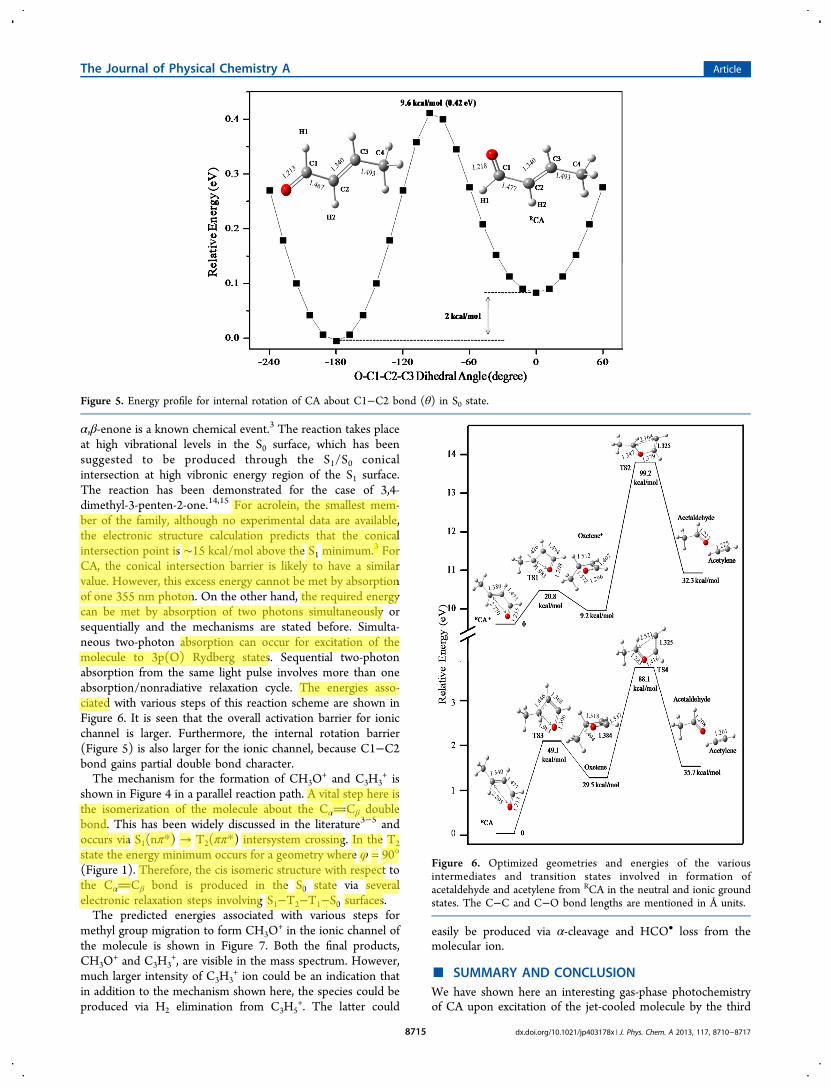
α,β-enone is a known chemical event.3 The reaction takes placeat high vibrational levels in the S0 surface, which has beensuggested to be produced through the S1/S0 conicalintersection at high vibronic energy region of the S1 surface.The reaction has been demonstrated for the case of 3,4-dimethyl-3-penten-2-one.14,15 For acrolein, the smallest mem-ber of the family, although no experimental data are available,the electronic structure calculation predicts that the conicalintersection point is ∼15 kcal/mol above the S1 minimum.3 ForCA, the conical intersection barrier is likely to have a similarvalue. However, this excess energy cannot be met by absorptionof one 355 nm photon. On the other hand, the required energycan be met by absorption of two photons simultaneously orsequentially and the mechanisms are stated before. Simulta-neous two-photon absorption can occur for excitation of themolecule to 3p(O) Rydberg states. Sequential two-photonabsorption from the same light pulse involves more than oneabsorption/nonradiative relaxation cycle. The energies asso-ciated with various steps of this reaction scheme are shown inFigure 6. It is seen that the overall activation barrier for ionicchannel is larger. Furthermore, the internal rotation barrier(Figure 5) is also larger for the ionic channel, because C1−C2bond gains partial double bond character.The mechanism for the formation of CH3O
+ and C3H3+ is
shown in Figure 4 in a parallel reaction path. A vital step here isthe isomerization of the molecule about the CαCβ doublebond. This has been widely discussed in the literature3−5 andoccurs via S1(nπ*) → T2(ππ*) intersystem crossing. In the T2
state the energy minimum occurs for a geometry where φ = 90°(Figure 1). Therefore, the cis isomeric structure with respect tothe CαCβ bond is produced in the S0 state via severalelectronic relaxation steps involving S1−T2−T1−S0 surfaces.The predicted energies associated with various steps for
methyl group migration to form CH3O+ in the ionic channel of
the molecule is shown in Figure 7. Both the final products,CH3O
+ and C3H3+, are visible in the mass spectrum. However,
much larger intensity of C3H3+ ion could be an indication that
in addition to the mechanism shown here, the species could beproduced via H2 elimination from C3H5
+. The latter could
easily be produced via α-cleavage and HCO• loss from themolecular ion.
■ SUMMARY AND CONCLUSIONWe have shown here an interesting gas-phase photochemistryof CA upon excitation of the jet-cooled molecule by the third
Figure 5. Energy profile for internal rotation of CA about C1−C2 bond (θ) in S0 state.
Figure 6. Optimized geometries and energies of the variousintermediates and transition states involved in formation ofacetaldehyde and acetylene from RCA in the neutral and ionic groundstates. The C−C and C−O bond lengths are mentioned in Å units.
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp403178x | J. Phys. Chem. A 2013, 117, 8710−87178715
Anamika
Highlight
Anamika
Highlight
Anamika
Highlight
Anamika
Highlight
Anamika
Highlight
Anamika
Highlight
Anamika
Highlight
Anamika
Highlight
Anamika
Highlight
Anamika
Highlight
Anamika
Highlight
Anamika
Highlight
Anamika
Highlight

harmonic (355 nm) of a Q-switched (10 ns) Nd: YAG laserpulses. The photochemistry is probed by measuring the TOF-MS upon multiphoton ionizations. A small segment of the MPIspectrum is also recorded by tuning the excitation wavelengtharound 355 nm to verify that the jet-cooled CA indeed haselectronic absorption at this wavelength. The mass spectrumshows intense features corresponding to acetaldehyde, acetyl,and methoxy radical cations, which are not observed in EI massspectra. We argue that acetaldehyde is produced in neutral S0potential energy surfaces and the methoxy group is produced inthe ground electronic state of the ion.Trans−cis isomerization about CαCβ double bond and
formation of an oxetene type intermediate in the ground statehave been considered to explain the observed molecularfragments. Both types of isomerizations were theoreticallyproposed earlier for this class of compounds. The barriercrossing for oxetene formation can occur at high vibrationallevels in S0 state. We have argued here that such levels could bepopulated via S1/S0 conical intersection from highly excitedvibronic region of S1. For excitations to latter levelsmultiphoton absorption is essential, and mechanisms of suchabsorption are also discussed. To elucidate that the photo-chemistry reported here for CA is common for this class ofcompounds, we are currently studying other similar molecules,and these will be reported elsewhere.
■ AUTHOR INFORMATION
Corresponding Author*E-mail: [email protected].
NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTS
The authors sincerely thank the Department of Atomic EnergyCommision (BRNS), Government of India, for the financialsupport. A.K.G. thanks CSIR, Government of India, for aSenior Research Fellowship to carry out the research presentedhere.
■ REFERENCES(1) Becker, R. S.; Inuzuka, K.; King, J. Acrolein: Spectroscopy,Photoisomerization, and Theoretical Considerations. J. Chem. Phys.1970, 52, 5164−5170.(2) Ros, M.; Groene, E. J. J.; van Hemert, M. C. Ab InitioCalculations on the Lower Excited States of the Short Chain Polyenals.J. Am. Chem. Soc. 1992, 114, 6820−6827.(3) Reguero, M.; Olivucci, M.; Bernardi, F.; Robb, M. A. Excited-State Potential Surface Crossings in Acrolein: A Model forUnderstanding the Photochemistry and Photophysics of α,β-Enones.J. Am. Chem. Soc. 1994, 116, 2101−2114.(4) Fang, W. H. A CASSCF Study on Photodissociation of Acroleinin the Gas Phase. J. Am. Chem. Soc. 1999, 121, 8376−8384.(5) Geβner, O.; Chrysostom, E. t.-H.; Lee, A. M. D.; Wardlaw, D. M.;Ho, M.-L.; Lee, S,-J.; Cheng, B.-M.; Zgierski, M. Z.; Chen, I.-C.;Shaffer, J. P.; et al. Non-adiabatic Intramolecular and Photo-dissociation Dynamics Studied by Femtosecond Time-ResolvedPhotoelectron and Coincidence Imaging Spectroscopy. FaradayDiscuss 2004, 127, 193−212.(6) Saha, B.; Ehara, M.; Nakatsuji, H. Singly and Doubly ExcitedStates of Butadiene, Acrolein, And Glyoxal: Geometries and ElectronicSpectra. J. Chem. Phys. 2006, 125, 014316(1−14).(7) Chin, C.-H.; Lee, S.-H. Theoretical Study of Isomerization andDecomposition of Propenal. J. Chem Phy. 2011, 134, 044309 (1−10).(8) Shinohara, H.; Nishi, N. Laser Photofragmentation Dynamics ofan Acrolein Supersonic Molecular Beam at 193 nm. J. Chem. Phys.1982, 77, 234−245.(9) Fujimoto, G. T.; Umstead, M. E.; Lin, M. C. CO Product EnergyDistribution in the Photodissociation of Methylketene and Acrolein at193 nm. J. Chem. Phys. 1985, 82, 3042−3044.(10) Haas, B.-M.; Minton, T. K.; Felder, P.; Huber, J. R.Photodissoclatlon of Acrolein and Propynal at 193 nm in a MolecularBeam. Primary and Secondary Reactions. J. Phys. Chem. 1991, 95,5149−5159.(11) Shu, J.; Peterka, D. S.; Lenone, S. R.; Ahmed, M. TunableSynchrotron Vacuum Ultraviolet Ionization, Time-of-Flight Inves-tigation of the Photodissociation of trans-Crotonaldehyde at 193 nm. J.Phys. Chem. A 2004, 108, 7895−7902.(12) Wu, W.; Yang, C.; Zhao, H.; Liu, K.; Su, H. Photodissociationand Photoisomerization Dynamics of CH2CHCHO in Solution. J.Chem. Phys. 2010, 132, 124510(1−8).(13) Coomber, J. W.; Pitts, J. N., Jr. Molecular Structure andPhotochemical Reactivity. XIV. The Vapor-Phase Photochemistry oftrans-Crotonaldehyde. J. Am. Chem. Soc. 1969, 91, 4955−4960.(14) Friedrich, L. E.; Schuster, G. E. Photochemical Preparation of aStable Oxetene. J. Am. Chem. Soc. 1969, 91, 7204−7205.(15) Friedrich, L. E.; Schuster, G. B. Irradiation of α,β-UnsaturatedKetones. Search for Intermediate Oxabicyclobutanes. J. Am. Chem. Soc.1972, 94, 1193−1199.(16) Hazra, M. K.; Chakraborty, T. Impact of Methyl Rotor in theExcited State Level Mixing of Doubly Hydrogen Bonded Complexesof 2-Pyridone. J. Phys. Chem. A 2008, 112, 1100−1104.(17) Biswas, P.; Panja, S. K.; Manogaron, S.; Chakraborty, T. Impactof Extended π-Conjugation on Methyl Rotor-Induced IVR in AromaticMolecules. J. Phys. Chem. A 2005, 109, 3225−3234.(18) Aquilante, F.; Barone, V.; Roos, B. O. A TheoreticalInvestigation of Valence and Rydberg Electronic States of Acrolein.J. Chem. Phys. 2003, 119, 12323−12334.(19) Mukhopadhyay, A.; Mukherjee, M.; Ghosh, A. K.; Chakraborty,T. UV Photolysis of α-Cyclohexanedione in the Gas Phase. J. Phys.Chem. A 2011, 115, 7494−7502.(20) Mukhopadhyay, A.; Ghosh, A. K.; Mukherjee, M.; Chakraborty,T. Electron Ionization Cross-Section and Fragmentation of Α-Cyclohexanedione. Int. J. Mass Spectrom. 2012, 309, 192−199.(21) Ghosh, A. K.; Chattopadhyay, A.; Mukhopadhyay, A.;Chakraborty, T. Isomeric Effects on Fragmentation of Crotonaldehydeand Methacrolein in Low-Energy Electron-Molecule Collisions. Chem.Phy. Lett. 2013, 561−562, 24−30.
Figure 7. Energies corresponding to the optimized geometries of thevarious intermediates and transition states involved in formation ofC3H3
+ and CH3O+ ions from cis-CA+. The C−C and C−O bond
lengths are mentioned in Å units.
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp403178x | J. Phys. Chem. A 2013, 117, 8710−87178716

(22) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.;Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci,B.; Petersson, G. A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.:Wallingford, CT, 2010.(23) Baba, M.; Hanazaki, I.; Nagashima, U. The S1 (nπ*) States ofAcetaldehyde and Acetone in Supersonic Nozzle Beam: MethylInternal Rotation and CO out-of-Plane Wagging. J. Chem. Phys.1985, 82, 3938−3947.(24) Watanabe, K. Ionization Potentials of Some Molecules. J. Chem.Phys. 1957, 26, 542−547.(25) Morita, H.; Freitas, J. E.; El-Sayed, M. A. Laser MultiphotonDissociation Ionization of Acrolein Clusters. J. Phys. Chem. A 1997,101, 3699−3701.
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp403178x | J. Phys. Chem. A 2013, 117, 8710−87178717




















