
1Pentene isomerization over SAPO-11, BEA and AlMCM-41 molecular sieves
6
Review 1-Pentene isomerization over SAPO-11, BEA and AlMCM-41 molecular sieves Carmen M. Lo ´pez * , Leyda Ramı ´rez, Virginia Sazo, Vanessa Escobar Universidad Central de Venezuela, Facultad de Ciencias, Escuela de Quı ´mica, Centro de Cata ´lisis, Petro ´leo y Petroquı ´mica, Apartado 47102, Caracas 1020-A, Venezuela Received 14 August 2007; received in revised form 18 January 2008; accepted 8 February 2008 Available online 16 February 2008 Abstract The skeletal isomerization of 1-pentene at atmospheric pressure and 89 mm Hg 1-pentene partial pressure was investigated over SAPO-11, BEA and AlMCM-41 molecular sieves. The effect of the reaction temperature between 250 and 400 8C was studied. Acidity of the catalysts seems to play a more important role than the pore size in the reaction conditions used. # 2008 Elsevier B.V. All rights reserved. Keywords: Skeletal isomerization; 1-Pentene; Molecular sieves; Zeolites Contents 1. Introduction ................................................................................... 1 2. Experimental ................................................................................... 2 3. Results and discussion ............................................................................ 3 4. Conclusions.................................................................................... 5 Acknowledgement ............................................................................... 6 References .................................................................................... 6 1. Introduction The reduction in sulphur and olefin content in gasoline may be one of the best ways to improve automobile emissions. The components of the gasoline pool in a refinery vary from case to case. A post-treating process becomes a desirable schedule for achieving sulphur and olefin reduction in the pool, however, the olefin reduction will lead to octane loss. In this sense, skeletal isomerization can be an alternative reaction to upgrade refinery and petrochemical feed streams. Especially, the isomerization of 1-butene to isobutene has been the subject of many investigations [1–5], because isobutene is the alkene precursor in the synthesis of methyl tertbutyl ether (MTBE). Among the higher alkenes, n-pentenes are an interesting feedstock. The skeletal isomerization of n-pentenes to isopentenes is a topic of increasing interest, because of the demand for isoolefins to ter amyl methyl ether (TAME) synthesis and for alkylation reactions [6–10]. Experimental studies in skeletal isomerization of olefins indicated that this reaction proceeds via either monomolecular or bimolecular (dimerization-cracking) mechanisms. Mono- molecular skeletal isomerization was found to be more common [11–14]. Indeed, n-pentene and higher hydrocarbons can isomerize on zeolites via a monomolecular path, which includes an alkoxy intermediate and a cyclopropane ring [15,16]. Other side reactions such as dimerization–oligomer- ization, cracking, hydride transfer and coking can occur. It is generally accepted that the acid strength required for these reactions decreases in the order: cracking oligomeriza- tion > skeletal isomerization double bond isomerization [17]. According to this, moderate acid sites will be more selective towards skeletal isomerization. www.elsevier.com/locate/apcata Available online at www.sciencedirect.com Applied Catalysis A: General 340 (2008) 1–6 * Corresponding author. Tel.: +58 212 6935981. E-mail address: [email protected] (C.M. Lo ´pez). 0926-860X/$ – see front matter # 2008 Elsevier B.V. All rights reserved. doi:10.1016/j.apcata.2008.02.005
Transcript of 1Pentene isomerization over SAPO-11, BEA and AlMCM-41 molecular sieves

www.elsevier.com/locate/apcata
Available online at www.sciencedirect.com
Applied Catalysis A: General 340 (2008) 1–6
Review
1-Pentene isomerization over SAPO-11, BEA and AlMCM-41
molecular sieves
Carmen M. Lopez *, Leyda Ramırez, Virginia Sazo, Vanessa Escobar
Universidad Central de Venezuela, Facultad de Ciencias, Escuela de Quımica, Centro de Catalisis, Petroleo y Petroquımica,
Apartado 47102, Caracas 1020-A, Venezuela
Received 14 August 2007; received in revised form 18 January 2008; accepted 8 February 2008
Available online 16 February 2008
Abstract
The skeletal isomerization of 1-pentene at atmospheric pressure and 89 mm Hg 1-pentene partial pressure was investigated over SAPO-11,
BEA and AlMCM-41 molecular sieves. The effect of the reaction temperature between 250 and 400 8C was studied. Acidity of the catalysts seems
to play a more important role than the pore size in the reaction conditions used.
# 2008 Elsevier B.V. All rights reserved.
Keywords: Skeletal isomerization; 1-Pentene; Molecular sieves; Zeolites
Contents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1
2. Experimental . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
3. Results and discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3
4. Conclusions. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5
Acknowledgement . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6
1. Introduction
The reduction in sulphur and olefin content in gasoline may
be one of the best ways to improve automobile emissions. The
components of the gasoline pool in a refinery vary from case to
case. A post-treating process becomes a desirable schedule for
achieving sulphur and olefin reduction in the pool, however, the
olefin reduction will lead to octane loss. In this sense, skeletal
isomerization can be an alternative reaction to upgrade refinery
and petrochemical feed streams. Especially, the isomerization
of 1-butene to isobutene has been the subject of many
investigations [1–5], because isobutene is the alkene precursor
in the synthesis of methyl tertbutyl ether (MTBE). Among the
higher alkenes, n-pentenes are an interesting feedstock. The
* Corresponding author. Tel.: +58 212 6935981.
E-mail address: [email protected] (C.M. Lopez).
0926-860X/$ – see front matter # 2008 Elsevier B.V. All rights reserved.
doi:10.1016/j.apcata.2008.02.005
skeletal isomerization of n-pentenes to isopentenes is a topic of
increasing interest, because of the demand for isoolefins to ter
amyl methyl ether (TAME) synthesis and for alkylation
reactions [6–10].
Experimental studies in skeletal isomerization of olefins
indicated that this reaction proceeds via either monomolecular
or bimolecular (dimerization-cracking) mechanisms. Mono-
molecular skeletal isomerization was found to be more
common [11–14]. Indeed, n-pentene and higher hydrocarbons
can isomerize on zeolites via a monomolecular path, which
includes an alkoxy intermediate and a cyclopropane ring
[15,16]. Other side reactions such as dimerization–oligomer-
ization, cracking, hydride transfer and coking can occur. It is
generally accepted that the acid strength required for these
reactions decreases in the order: cracking � oligomeriza-
tion > skeletal isomerization� double bond isomerization
[17]. According to this, moderate acid sites will be more
selective towards skeletal isomerization.

C.M. Lopez et al. / Applied Catalysis A: General 340 (2008) 1–62
Catalyst pore topology, and acid site strength, density and
location are factors that can control the skeletal isomerization
of alkenes. It is often considered that the pore structure plays a
more important role than acidity. The most suitable zeolites are
the 10-membered ring molecular sieves with pore diameters
between 4 and 6 A [18–20]. However, Seo et al. [21] have
reported a high selectivity for isobutene with mesoporous
proton and metal containing MCM-41. On other side,
Trombetta et al. [22] have investigated the catalytic activity
of amorphous silica-alumina, HZSM-5 and H-ferrierite (FER)
zeolites in the skeletal isomerization of 1-butene. The Bronsted
surface acidity correlates well with the catalytic activity in 1-
butene isomerization. The selectivity to isobutene follows
nearly an inverse trend except for FER. According to what has
been mentioned above, it can be considered that the relationship
between acidity and catalyst pore topology with the activity and
selectivity is not clear and is matter of debate.
The main objective of this work is to compare the activity
and selectivity to iso-pentenes of microporous and mesoporous
molecular sieves in the isomerization of 1-pentene. The
influence of acidity and pore size was investigated.
2. Experimental
The acid catalysts used consisted of HDBEA, SAPO-11 and
AlMCM-41 molecular sieves. The starting BEA zeolite was a
commercial sample. First, the starting zeolite was ion-exchanged
with a 3 M ammonium acetate solution for 3 h, under continuous
stirring at 60 8C using 20 ml of solution per gram of zeolite. Then
the exchanged zeolite was calcined in flowing air at 550 8C for
4 h to obtain the hydrogen form (HBEA). HBEA was then
refluxed with 1 M HCl at room temperature for 8 h, using 10 ml
of solution/g of HBEA. After the acid treatment, the sample was
washed free of chloride, dried overnight at 100 8C and calcined
statically under ambient atmosphere at 550 8C, for 15 h. This
product was named HDBEA.
The synthesis procedure for SAPO-11 has been reported
elsewhere [23]. The gel molar composition was Al2O3:P2O5:D-
PA:0.3SiO2:50H2O, the crystallization temperature was 200 8Cand crystallization time was 4 h.
The hydrothermal synthesis of AlMCM-41 mesoporous
material was carried out at 90 8C using a gel with a
molar composition of SiO2:0.26Na2O:0.026 NaAlO2:0.124
CTMABr:135 H2O. Cethyltrimethylammonium bromide
(CTMABr, from Aldrich) was used as template, tetraethylortho-
silicate (TEOS, from Aldrich) as silica source, and sodium
aluminate (made in the laboratory, 49 wt% Al2O3) was added to
the reactant mixture as aluminium source. Sulphuric acid was
added by drops under stirring until the pH became 11.0. The
synthesis mixture was heated at 90 8C for 12 h and then cooled to
room temperature. The precipitated solid was filtered, washed
and dried at 60 8C for 24 h. To remove surfactant, the dried solid
was first heated under nitrogen flow at 250 8C for 3h, and then
calcined under air flow at 550 8C for 8 h. Sodium ions were
exchanged with a 0.5 M ammonium acetate solution at 60 8Cunder agitation for 3 h. H-form AlMCM-41 (HAlMCM-41) was
obtained by calcination in air stream at 500 8C for 8 h.
X-ray diffractograms (XRD) of the as prepared solids were
recorded with a Philips diffractometer PW 1730 Phillips using
Cu Ka radiation operated at 30 kV and 20 mA, and scanning
speed of 28 2u/min. Chemical analysis for Al, P and Si of the
calcined samples were performed using atomic emission
spectroscopy with a source of plasma inductively coupled.
Samples were previously fused with lithium metaborate and
dissolved in dilute nitric acid before analysis. N2-specific
surface areas (SSA) were obtained on Micrometrics 2200
equipment at liquid nitrogen temperature. All the samples were
pre-treated at 350 8C under vacuum conditions overnight.
The total acid site density and the acid strength distribution
of the catalysts were measured by temperature programmed
desorption of ammonia (TPDA), using a Micromeritics TPD/
TPR 2900 analyzer. The total acidity was obtained by
integration of the area under the curve. This curve was fitted
using three peaks, which were classified as weak, moderate and
strong acidity depending on the desorption temperature. It was
a convenient way to categorize the acid strength distribution
obtained by this method.
1-Pentene transformation was carried out under atmospheric
pressure at temperatures ranging from 250 to 400 8C in a
continuous fixed-bed reactor. 1-Pentene was introduced in the
reactor by the gas flow saturation method. The feeding gas
mixture consisted of N2 (15 ml/min) with a partial pressure of
1-pentene of 0.12 atm. To reach this pressure, N2 was passed
through a glass vessel with 1-pentene kept to�19 8C in an ice–
salt mixture. The mass of catalyst was about 0.1 g (weight
hourly space velocity WHSV = 3.5 h�1). Before the reaction
test, the catalyst was activated with a nitrogen flow (30 ml/min)
at 500 8C for 2 h. The product analysis was done after 30 min
on-stream by on-line chromatography using a Hewlett-Packard
5890A. A fused silica KCl/Al2O3 column was used for
separation purposes.
The total conversion (X) of 1-pentene was calculated
according to Eq. (1)
X ¼P
Ai � ðA1-pentene þ Acis-2-pentene þ Atrans-2-penteneÞPAi
� 100
(1)
where Ai is the corrected chromatographic area for a particular
compound, used to express the conversion and selectivity as
molar percentages.
Since the double bond isomerization is much faster than
skeletal isomerization the three isomers of linear pentenes
(1-pentene, cis-2-pentene and trans-2-pentene) were merged to
one component, and the three isomers of branched pentenes (3-
methyl-1-butene, 2-methyl-2-butene and 2-methyl-1-butene)
were also grouped to one component, in agreement with the
reaction network proposed by Sandelin et al. [24].
For a reaction product, or a set of products, the selectivity (S)
was defined according Eq. (2)
Sp ¼ApP
Ai � ðA1-pentene þ Acis-2-pentene þ Atrans-2-penteneÞ
� 100: (2)

C.M. Lopez et al. / Applied Catalysis A: General 340 (2008) 1–6 3
The products were grouped following Hochtl et al. [17] sug-
gestion:
� S
keletal isomers: adding up the corresponding correctedareas of (Ssk) 3-methyl-1-butene (3M1B), 2-methyl-2-butene
(2M2B) and 2-methyl-1-butene (2M1B).
� L
ower C5 (S<C5) products: adding up the correspondingcorrected areas of the C1–C4 products.
� S
aturates C5 (SC5sat): adding up the corresponding areas ofiso-pentane and n-pentane.
� H
Fig. 3. XRD diffractograms for BEA, HBEA and HDBEA zeolites.
igher C5 reaction products (S>C5): sum of the corrected
areas of C6–C8 products.
3. Results and discussion
Figs. 1 and 2 show the XRD patterns of the synthesized
SAPO-11 and calcined AlMCM-41, respectively. The diffrac-
tion patterns, agreed well with those reported previously
[17,21]. Fig. 3 shows the XRD diffractograms of commercial
BEA, HBEA and HDBEA zeolites. It can be observed, that
there are not appreciable changes in the XRD patterns. The
nitrogen adsorption isotherm of calcined AlMCM-41 was type
IV in the IUPAC classification. A narrow pore size distribution
with a mean pore size of 28 Awas determined from the nitrogen
isotherm using the BJH model. When combining these results
with the XRD results (unit cell parameter of 44 A), the pore
wall thickness for AlMCM-41 was 16 A.
Table 1 shows the main characteristics of the solids after
calcination under a flow of air. The SSA of the solids lie within
Fig. 1. XRD diffractogram for SAPO-11.
Fig. 2. XRD diffractogram for AlMCM-41.
the range previously generally reported in the literature [17,21].
The acid treatment of HBEA caused 60% dealumination: Si/Al
molar ratio increases from 15 for HBeta to 41 in HDBEA,
retaining more than 90% crystallinity.
The amount and strength of the acid sites for the different
molecular sieves determined by TPD of ammonia are shown in
Table 2. The ammonia desorption peak temperature was used to
characterize acid strength. Deconvolution of TPDA profiles
resulted in three distinct types of acid sites in the ranges 250–
280, 320–370 and 470–510 8C. These peaks have been
described by Sakthivel et al. [25] as follows: the first and
second peaks referred as type (ii) and (iii), are attributed to
moderate and strong structural Bronsted acid sites, respectively.
The high-temperature peak, designated as type (v) is caused by
ammonia desorbed from strong acid Lewis sites, according to
Kosslick et al. [26]. The order of total acid amounts was
HDBEA > AlMCM-41 � SAPO-11. The order of moderate
Bronsted acid amount was AlMCM-41 � SAPO-11 >HDBEA
whereas the order of strong Bronsted acid amount was
HDBEA > AlMCM-41 � SAPO-11. Finally the order of
high-temperature acid site was AlMCM-41 > SAPO-
11 > HDBEA. According to Sakthivel et al. [25], type (v)
acid sites are not responsible for the catalytic activity in acid
catalyzed reactions.
The skeletal isomerization of 1-pentene over acid catalyst
usually occurs accompanying direct cracking of pentene,
dimerization (and oligomerization) followed by cracking,
hydride transfer and coke deposition. It is generally established
that the linear pentenes act like one reactant while the branched
pentene isomers appear as primary products [17,24]. The main
products were iso-pentenes; small fractions of pentane,
isopentane and C1–C4 products were also obtained.
Table 1
Specific surface area (SSA) and chemical composition of the catalysts
Catalyst Surface area (m2/gcat) Chemical composition
SAPO-11 175 (Al0.50 P0.45 Si0.05)O2
HDBEA 593 H1.5(Al1.5 Si62.5 O128)
AlMCM-41 980 (SiO2)35(AlO2)1

Table 2
Acid distribution and acid strength of different molecular sieves
Catalyst Amount of acid sites (acid strength) (mmol/g) (8C)
Total acidity
(mmol NH3/gcat)
Weak acidity (250–280 8C)
(mmol NH3/gcat)
Moderate acidity (320–370 8C)
(mmol NH3/gcat)
Strong acidity
(470–510 8C) (mmol NH3/gcat)
SAPO-11 0.313 0.087 (252) 0.161 (319) 0.065 (467)
HDBEA 0.470 0.05 (255) 0.400 (365) 0.020 (507)
AlMCM-41 0.370 0.09 (272) 0.190 (351) 0.090 (492)
Fig. 5. Selectivity to skeletal isomers-deactivation curves for HDBEA at the
reaction temperatures studied.
C.M. Lopez et al. / Applied Catalysis A: General 340 (2008) 1–64
A blank experiment was carried out with only glass wood in
the reactor, and it was found that with 0.12 atm of 1-pentene, a
total flow of 15 ml/min, and between 250 and 400 8C, the
maximum level of pentene conversion was 5% at 400 8C. This
result evidences that the contribution of homogeneous reactions
is not appreciable under these reaction conditions.
Selectivity to skeletal isomers-deactivation curves are
shown in Figs. 4–6 for the catalysts, SAPO-11, HDBEA and
AlMCM-41, at 250, 300 and 400 8C. In Fig. 4, for SAPO-11
no deactivation was observed at 300 and 400 8C. The time on
stream (TOS) behaviour at 250 8C was different. Conversion
decreased from 19% to about 5% after 240 min TOS. The
selectivity to skeletal isomerization increased from 50 to
70%, selectivity to other products was very low. For
temperatures of 300 and 400 8C, conversion increased from
16% at 300 8C to 46% at 400 8C. The selectivity to skeletal
isomerization increased from 85% at 300 8C to 95% at
400 8C. As observed when the reaction temperature was
250 8C, selectivity to other products was very low. The higher
deactivation observed at 250 8C, can be attributed to a more
pronounced adsorption and coke formation at lower
temperature. This coke deposits can produce a shape-
selective effect which might prevent formation of C5 saturate
products. At higher temperatures, adsorption of olefins can be
reduced, which might lead to a decrease in the undesired
bimolecular reactions. Since isomerization of n-pentene can
take place via a monomolecular process, bimolecular
reactions that can lead to by-products and consequently
coke are minimized at 300 and 400 8C.
Fig. 4. Selectivity to skeletal isomers-deactivation curves for SAPO-11 at the
reaction temperatures studied.
A similar behaviour in function of reaction temperature was
observed for HDBEA and AlMCM-41, as showed in Figs. 5 and
6, respectively. In these cases, deactivation at 250 8C was lower
compared to SAPO-11; the differences in pore structure of
these solids could explain these results.
The effect of temperature on the catalytic activity and
product selectivity of the catalysts is best depicted in Fig. 7. For
all catalysts evaluated, the conversion increased with an
increase in temperature from 250 to 400 8C. For SAPO-11 and
AlMCM-41 the selectivity to skeletal isomerization increased
with the increase of reaction temperature; whereas for HDBEA,
Fig. 6. Selectivity to skeletal isomers-deactivation curves for AlMCM-41 at the
reaction temperatures studied.
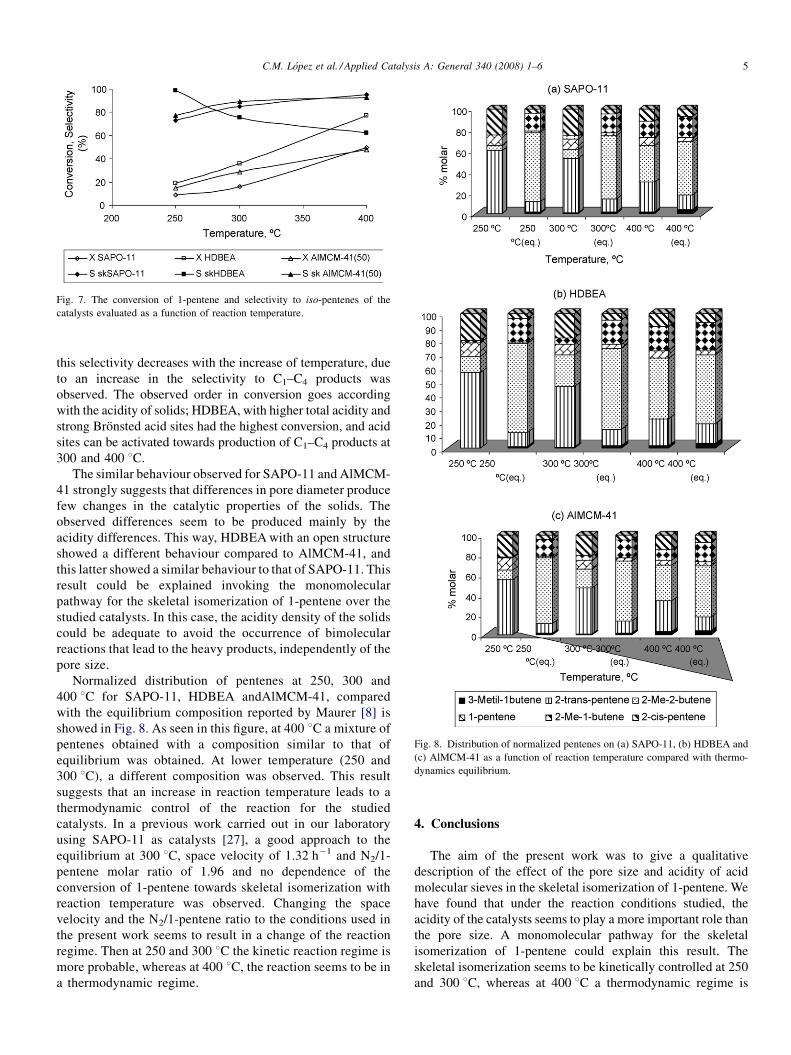
Fig. 7. The conversion of 1-pentene and selectivity to iso-pentenes of the
catalysts evaluated as a function of reaction temperature.
Fig. 8. Distribution of normalized pentenes on (a) SAPO-11, (b) HDBEA and
(c) AlMCM-41 as a function of reaction temperature compared with thermo-
dynamics equilibrium.
C.M. Lopez et al. / Applied Catalysis A: General 340 (2008) 1–6 5
this selectivity decreases with the increase of temperature, due
to an increase in the selectivity to C1–C4 products was
observed. The observed order in conversion goes according
with the acidity of solids; HDBEA, with higher total acidity and
strong Bronsted acid sites had the highest conversion, and acid
sites can be activated towards production of C1–C4 products at
300 and 400 8C.
The similar behaviour observed for SAPO-11 and AlMCM-
41 strongly suggests that differences in pore diameter produce
few changes in the catalytic properties of the solids. The
observed differences seem to be produced mainly by the
acidity differences. This way, HDBEA with an open structure
showed a different behaviour compared to AlMCM-41, and
this latter showed a similar behaviour to that of SAPO-11. This
result could be explained invoking the monomolecular
pathway for the skeletal isomerization of 1-pentene over the
studied catalysts. In this case, the acidity density of the solids
could be adequate to avoid the occurrence of bimolecular
reactions that lead to the heavy products, independently of the
pore size.
Normalized distribution of pentenes at 250, 300 and
400 8C for SAPO-11, HDBEA andAlMCM-41, compared
with the equilibrium composition reported by Maurer [8] is
showed in Fig. 8. As seen in this figure, at 400 8C a mixture of
pentenes obtained with a composition similar to that of
equilibrium was obtained. At lower temperature (250 and
300 8C), a different composition was observed. This result
suggests that an increase in reaction temperature leads to a
thermodynamic control of the reaction for the studied
catalysts. In a previous work carried out in our laboratory
using SAPO-11 as catalysts [27], a good approach to the
equilibrium at 300 8C, space velocity of 1.32 h�1 and N2/1-
pentene molar ratio of 1.96 and no dependence of the
conversion of 1-pentene towards skeletal isomerization with
reaction temperature was observed. Changing the space
velocity and the N2/1-pentene ratio to the conditions used in
the present work seems to result in a change of the reaction
regime. Then at 250 and 300 8C the kinetic reaction regime is
more probable, whereas at 400 8C, the reaction seems to be in
a thermodynamic regime.
4. Conclusions
The aim of the present work was to give a qualitative
description of the effect of the pore size and acidity of acid
molecular sieves in the skeletal isomerization of 1-pentene. We
have found that under the reaction conditions studied, the
acidity of the catalysts seems to play a more important role than
the pore size. A monomolecular pathway for the skeletal
isomerization of 1-pentene could explain this result. The
skeletal isomerization seems to be kinetically controlled at 250
and 300 8C, whereas at 400 8C a thermodynamic regime is

C.M. Lopez et al. / Applied Catalysis A: General 340 (2008) 1–66
possible, however more work will be required in order to make
a complete kinetic analysis.
Acknowledgement
This work was supported by CDCH-UCV Projects 03-12-
5419-2004/2006 and 03-00-5738/2004.
References
[1] B. de Menorval, P. Ayrault, N.S. Gnep, M. Guisnet, Appl. Catal. A: Gen.
304 (2006) 1–13.
[2] S.H. Lee, C.-H. Shin, S. Bong Hong, J. Catal. 223 (2004) 200–211.
[3] Gy. Onyestyak, J. Valyon, G. Pal-Borbely, L.V.C. Rees, Appl. Surf. Sci.
196 (2002) 401–407.
[4] V. Nieminen, N. Kumar, T. Heikkila, E. Laine, J. Villegas, T. Salmi, D.Yu.
Murzin, Appl. Catal. A: Gen. 259 (2000) 227–234.
[5] S.-M. Yang, J.-Y. Lin, D.-H. Guo, S.G. Liaw, Appl. Catal. A: Gen. 181
(1999) 113–122.
[6] J. Walendziewski, B. Pniak, React. Kinet. Catal. Lett. 58 (1996) 359–365.
[7] S.M. Yang, S.Y. Liu, in: Von Ballmoos, et al. (Eds.), Proceedings of the 9th
Int. Zeol. Conf., vol. 1, Butterworth-Heinemann, (1993), pp. 623–630.
[8] T. Maurer, B. Kraushaar-Czarnetzki, J. Catal. 187 (1999) 202–208.
[9] M.R. Apelian, I. Rahmim, A.S. Fung, A. Huss, Jr., US Patent 5,321,194,
1994.
[10] J. Guo, X.W. Cheng, W.-Z. Zhou, Y.-C. Long, Micropor. Mesopor. Mater.
79 (2005) 319–328.
[11] J. Cejka, B. Wichterlova, P. Sarv, Appl. Catal. A: Gen. 179 (1999) 217–
222.
[12] M. Kangas, J. Villegas, N. Kumar, T. Salmi, D.Yu. Murzin, F. Sandelin, E.
Harlin, Catal. Today 100 (2005) 363–366.
[13] P. Ivanov, H. Papp, Appl. Surf. Sci. 179 (2001) 234–239.
[14] B. de Menorval, P. Ayrault, N.S. Gnep, M. Guisnet, J. Catal. 230 (2005)
38–51.
[15] V.B. Kazansky, Catal. Today 51 (1999) 419–434.
[16] T. Demuth, X. Rozanska, L. Benco, J. Hafner, R.A. van Santen, H.
Toulhoat, J. Catal. 214 (2003) 68–77.
[17] M. Hochtl, A. Jentys, H. Vinek, Appl. Catal. A: Gen. 207 (2001) 397–
405.
[18] K. Fottinger, G. Kinger, H. Vinek, Appl. Catal. A: Gen. 249 (2003) 205–
212.
[19] K. Fottinger, G. Kinger, H. Vinek, Catal. Lett. 85 (2003) 117–122.
[20] J. Houzvicka, S. Hansildaar, V. Ponec, J. Catal. 167 (1997) 273–278.
[21] G. Seo, N. Hyeon, Y.-H. Lee, J.-H. Kim, Catal. Lett. 57 (1999) 209–
215.
[22] M. Trombetta, G. Busca, S. Rossini, V. Piccoli, U. Cornaro, A. Guerico, R.
Catani, R.J. Willey, J. Catal. 179 (1998) 581–596.
[23] C.M. Lopez, F.J. Machado, B. Mendez, M. Pinto, V. Sazo, J. Goldwasser,
M.M. Ramırez de Agudelo, Top. Catal. 10 (2000) 65–71.
[24] F. Sandelin, T. Salmi, D.Yu. Murzin, Chem. Eng. Sci. 61 (2006) 1157–
1166.
[25] A. Sakthivel, S.E. Dapurkar, N.M. Gupta, S.K. Kulshreshtha, P. Selvam,
Micropor. Mesopor. Mater. 65 (2003) 177–187.
[26] H. Kosslick, G. Lischke, B. Parlitz, W. Storek, R. Fricke, Appl. Catal. A:
Gen. 184 (1999) 49–60.
[27] E. Gonzalez, Y. Guillen, C.M. Lopez, CIENCIA 14 (2006) 347–356.




















