
Synthesis of SAPO-35 molecular sieve and its catalytic properties in the methanol-to-olefins...
7
Chinese Journal of Catalysis 34 (2013) 798–807 available at www.sciencedirect.com journal homepage: www.elsevier.com/locate/chnjc Article Synthesis of SAPO‐35 molecular sieve and its catalytic properties in the methanol‐to‐olefins reaction LI Bing a , TIAN Peng a , LI Jinzhe a , CHEN Jingrun a,b , YUAN Yangyang a,b , SU Xiong a,b , FAN Dong a,b , WEI Yingxu a , QI Yue a , LIU Zhongmin a, * a Dalian National Laboratory for Clean Energy, National Engineering Laboratory for Methanol‐to‐Olefins, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, Liaoning, China b University of Chinese Academy of Sciences, Beijing 100049, China ARTICLE INFO ABSTRACT Article history: Received 30 December 2012 Accepted 30 January 2013 Published 20 April 2013 SAPO‐35 molecular sieve samples with different Si contents were hydrothermally synthesized using hexamethyleneimine as the template and characterized by XRD, XRF, SEM, MAS NMR, XPS and N2 physisorption. Three SAPO‐35 samples were tested as methanol‐to‐olefins catalysts. After the reac‐ tion, the evolution of coke species was investigated over SAPO‐35 and SAPO‐34 catalysts with simi‐ lar Si concentrations. A correlation between the cage size of the molecular sieves and the coke spe‐ cies was obtained. © 2013, Dalian Institute of Chemical Physics, Chinese Academy of Sciences. Published by Elsevier B.V. All rights reserved. Keywords: SAPO‐35 Methanol to olefins SAPO‐34 Coke species Deactivation 1. Introduction Silicoaluminophosphate molecular sieves (SAPO‐n) were first synthesized and reported by Union Carbide Corporation (UCC) in 1984 [1]. Due to their mild acidity and special pore structures, SAPO molecular sieves have widespread applica‐ tions in the chemical industry [2]. SAPO‐35, as a member of the family with the levyne‐like crystal structure (LEV), is a small pore molecular sieve with a pore diameter of 3.6 nm × 4.8 nm. The framework of SAPO‐35 comprises levyne cages connected by single six‐membered rings and double six‐membered rings. There are two distinct T sites in the framework: one is in the double six‐membered ring and the other is in the single six‐membered ring. The distribution of these two sites is in the ratio of 2:1 [3]. Due to the special structure of SAPO‐35, it has been employed as the catalyst in methanol conversion and aminomethylation [4–7]. Moreover, SAPO‐35 is also explored as an adsorbent for CO2/CH4 separation [8,9]. The methanol‐to‐olefins (MTO) process is the key process to produce light olefins from coal or natural gas. The molecular sieves ZSM‐5 and SAPO‐34 with eight‐membered ring and CHA cages exhibit excellent catalytic performance in the reaction [6,7,10–12]. Other small pore molecular sieves with the eight‐membered pore size such as SAPO‐17, SAPO‐18, and SAPO‐35 have also been explored as catalysts for the MTO re‐ action [7,13,14]. The SAPO‐35 molecular sieve showed a faster coking deactivation rate compared with SAPO‐34 [4,6]. At pre‐ sent, the correlations between the deactivation process (de‐ posited coke species) and the reaction conditions (such as time and temperature) during the MTO reaction over the SAPO‐34 catalyst are relatively clear [15,16]. However, there is no report so far on the deactivation process during methanol conversion * Corresponding author. Tel/Fax: +86‐411‐84379335; E‐mail: [email protected] DOI: 10.1016/S1872‐2067(12)60557‐9 | http://www.sciencedirect.com/science/journal/18722067 | Chin. J. Catal.,Vol. 34, No. 4, April 2013
-
Upload
independent -
Category
Documents
-
view
4 -
download
0
Transcript of Synthesis of SAPO-35 molecular sieve and its catalytic properties in the methanol-to-olefins...

ChineseJournalofCatalysis34(2013)798–807
a v a i l a b l e a t www. s c i e n c ed i r e c t . c om
j o u r n a l h omep a g e : www. e l s e v i e r. c om / l o c a t e / c h n j c
Article
SynthesisofSAPO‐35molecularsieveanditscatalyticpropertiesinthemethanol‐to‐olefinsreaction
LIBinga,TIANPenga,LIJinzhea,CHENJingruna,b,YUANYangyanga,b,SUXionga,b,FANDonga,b,WEIYingxua,QIYuea,LIUZhongmina,*aDalianNationalLaboratoryforCleanEnergy,NationalEngineeringLaboratoryforMethanol‐to‐Olefins,DalianInstituteofChemicalPhysics,ChineseAcademyofSciences,Dalian116023,Liaoning,China
bUniversityofChineseAcademyofSciences,Beijing100049,China
A R T I C L E I N F O
A B S T R A C T
Articlehistory:Received30December2012Accepted30January2013Published20April2013
SAPO‐35molecularsievesampleswithdifferentSicontentswerehydrothermallysynthesizedusinghexamethyleneimineasthetemplateandcharacterizedbyXRD,XRF,SEM,MASNMR,XPSandN2
physisorption.ThreeSAPO‐35samplesweretestedasmethanol‐to‐olefinscatalysts.Afterthereac‐tion,theevolutionofcokespecieswasinvestigatedoverSAPO‐35andSAPO‐34catalystswithsimi‐larSiconcentrations.Acorrelationbetweenthecagesizeofthemolecularsievesandthecokespe‐cieswasobtained.
©2013,DalianInstituteofChemicalPhysics,ChineseAcademyofSciences.PublishedbyElsevierB.V.Allrightsreserved.
Keywords:SAPO‐35MethanoltoolefinsSAPO‐34CokespeciesDeactivation
1. Introduction
Silicoaluminophosphate molecular sieves (SAPO‐n) werefirst synthesized and reported by Union Carbide Corporation(UCC) in 1984 [1]. Due to theirmild acidity and special porestructures, SAPO molecular sieves have widespread applica‐tionsinthechemicalindustry[2].SAPO‐35,asamemberofthefamilywith the levyne‐like crystal structure (LEV), is a smallporemolecularsievewithaporediameterof3.6nm×4.8nm.TheframeworkofSAPO‐35compriseslevynecagesconnectedbysinglesix‐memberedringsanddoublesix‐memberedrings.Thereare twodistinctTsites in the framework:one is in thedouble six‐membered ring and the other is in the singlesix‐memberedring.Thedistributionofthesetwositesisintheratioof2:1[3].DuetothespecialstructureofSAPO‐35,ithasbeen employed as the catalyst in methanol conversion and
aminomethylation [4–7].Moreover, SAPO‐35 is also exploredasanadsorbentforCO2/CH4separation[8,9].
Themethanol‐to‐olefins(MTO)processisthekeyprocesstoproduce light olefins from coal or natural gas. ThemolecularsievesZSM‐5andSAPO‐34witheight‐memberedringandCHAcages exhibit excellent catalytic performance in the reaction[6,7,10–12]. Other small pore molecular sieves with theeight‐membered pore size such as SAPO‐17, SAPO‐18, andSAPO‐35havealsobeenexploredascatalystsfortheMTOre‐action[7,13,14].TheSAPO‐35molecularsieveshowedafastercokingdeactivationratecomparedwithSAPO‐34[4,6].Atpre‐sent, the correlations between the deactivation process (de‐positedcokespecies)andthereactionconditions(suchastimeand temperature)during theMTOreactionover theSAPO‐34catalystarerelativelyclear[15,16].However,thereisnoreportsofaronthedeactivationprocessduringmethanolconversion
*Correspondingauthor.Tel/Fax:+86‐411‐84379335;E‐mail:[email protected] DOI:10.1016/S1872‐2067(12)60557‐9|http://www.sciencedirect.com/science/journal/18722067|Chin.J.Catal.,Vol.34,No.4,April2013

LIBingetal./ChineseJournalofCatalysis34(2013)798–807
catalyzedbySAPO‐35. It is speculated that theactivitydiffer‐encesbetweenSAPO‐34andSAPO‐35areduetotheirdifferentstructures.ResearchonthedeactivationduringtheMTOreac‐tionoverSAPO‐35willhelpunderstand theeffectof thecagestructuresofthemolecularsievesandthedepositedcokespic‐es.
In this work, SAPO‐35 molecular sieves with different Sicontentswerehydrothermallysynthesizedbyusinghexameth‐yleneimine as the template. The effect of Si content on thephysical and chemical properties of themolecular sieveswasstudied indetail. In addition, three SAPO‐35molecular sieveswithdifferentSicontentswerechosenasMTOcatalyststoin‐vestigatetheeffectsoftheacidityonthereaction.Theevolutionofthecokespecies inthereactionwasinvestigatedoverbothSAPO‐35andSAPO‐34catalystswithsimilarSiconcentrations.Thecorrelationbetweenthecagesizeof themolecularsievesanddeactivationduetothecokespeciesisdiscussed.
2. Experimental
2.1. Synthesisofmolecularsieves
TheSAPO‐35molecularsievesweresynthesizedasreport‐ed elsewhere [17]. Pseudoboehmite powder (w(Al2O3) = 70wt%),sol(w(SiO2)=31.1wt%)andphosphoricacid(w(H3PO4)= 85 wt%) were the aluminum source, silicon source, andphosphorus source, respectively. Hexamethyleneimine (HMI,chemicalpure)wasusedasthetemplateagent.Themolarratioof reactants was 0.96P2O5:1.0Al2O3:nSiO2:1.51HMT:55.47H2O(n=0.1,0.2,0.3,0.5,0.8,1.0).Thedetailedprocedurewasasfollows.Intoabeaker,deionizedwater,aluminumsource,sili‐con source, phosphorus source, and HMI were sequentiallyaddedwithvigorousstirringtomaketheinitialgelhomogene‐ous. This was then transferred into a 100 ml stainless steelreactor,whichwasthensealedandheatedat200oCfor24h.After crystallization, the productwas collected by centrifugalseparation, rinsedwithwater until the pH levelwas neutral,andthendriedat120oC.ThesampleswithdifferentSicontentsweredenotedas0.1Si,0.2Si,0.3Si,0.5Si,0.8Si,and1.0Si.
2.2. Characterization
X‐ray powder diffraction (XRD) measurements were per‐formed using a PANalytical X’Pert PRO X‐ray diffractometerwithCuanode,Kαradiation(λ=0.15418nm),voltageof40kVandcurrentof40mA.TheelementalanalysiswascarriedoutusingaPhilipsMagix2424XX‐rayFluorescenceSpectrometer(XRF).MorphologyimageswereacquiredonaHitachiS‐3400Nscanning electron microscopy (SEM). X‐ray photoelectronspectroscopy (XPS) was determined employing a ThermoESCALAB 250Xi X‐ray photoelectron spectrometerwith AlKαradiation.ThepeakofAl2pat74.7eVfromAl2O3onthesur‐faceof the sampleswasusedas the internal standard.N2ad‐sorption measurement was performed on a MicromeriticsASAP2010volumetricadsorptionanalyzer.
1H MAS NMR experiments were performed on a BrukerAvance III‐600 solid phase NMR spectrometer with a proton
resonance frequency of 600.13MHz by using a 4mm probehead. One pulse programwas used and the π/2 pulse lengthwas4.4µs.Therecycledelaytimeis10s.Thespinratewas12kHzwith32timessamplingfrequency.Beforethe1HMASNMRexperiments, all samples underwent vacuum dehydration at400oCand<10–3Pafor20htoremovewaterandimpuritiesinthemolecular sieve.The sampleswere transferred inanitro‐gen‐filledgloveboxtotheNMRrotatorfortesting.Thequanti‐tative processing of the 1HMASNMR datawas as follows. Acalibration curve for the correlation between peak areas andmass of samplewere firstmade by using adamantine as theexternalstandard.Thenthe1HMASNMRspectraofthesamplewith known mass were acquired under identical acquisitionconditions.ThepeakareasofBrönstedacidsiteswascalculat‐ed by Gauss‐Lowrance linear fitting to get the density ofBrönstedacidsitesofthesamplesfromthecalibrationcurve.
2.3. Reactionevaluation
Thecatalyticpropertiesofthemolecularsieveswereevalu‐atedusinganatmosphericfixedbedequipment.1.2gcalcinedsample (40–60mesh) was packed in the reactor, which wasthenpurgedwithnitrogenat520oCfor30min.Afterpurging,methanolsolution(40wt%)wasinjectedintothereactorbyapump. The products were analyzed online using an Agilent7890Agaschromatograph(GC)withPoraPLOTQ‐HTcapillarycolumnandFIDdetector.
2.4. Collectionsofcokedepositsandanalysisofcokespecies
Thedepositedcokespecieswerecollectedbystoppingthefeedafterapre‐determinedreactiontime,unloadingthecata‐lystsquicklyandthenquenchinginliquidnitrogen.
TheGuisnetmethodwasusedforthequalitativeanalysisofthe coke species [18,19]. In aTeflonbottle, 50mgof catalystwasaddedto1mlHFwatersolution(20wt%).Afterthemix‐ture was shaken well and allowed to sit for 1 h, 0.5 ml di‐chloromethanewasaddedintothesolution.5minlater,NaOHwasaddedandmixedwell.Themixturewasthentransferredtoaseparatoryfunnelandshakenvigorously.ThelowerlayerliquidwascollectedintoasamplevialforGCtestingbyanAg‐ilent7890‐5975CMSDgaschromatograph‐massspectrometerwith aHP‐5 capillary column. Each compoundwas identifiedusingtheNIST08library.
3. Resultsanddiscussion
3.1. SynthesisandcharacterizationofSAPO‐35withdifferentSicontents
TheXRDresultsofthesamplesareshowninFig.1.AlltheSAPO‐35 molecular sieves with different Si contents had theLEV framework structure, as shown by comparing to thestandardpattern[7,20].Therelativecrystallinity,relativeyield,andchemicalcompositionoftheSAPO‐35sampleswithdiffer‐entSicontentsarelistedinTable1.Thesolidyieldofthesam‐plesincreasedwithincreasingamountofSiO2intheinitialgels.

LIBingetal./ChineseJournalofCatalysis34(2013)798–807
However, the crystallinity exhibited first an increase then adecrease trend.0.3Si sample showed thehighest crystallinity.InourpreviousresearchonthesynthesisofSAPO‐34,wealsofound that samples with 0.2–0.3 Si contents had the highestcrystallinity[21].ThissuggestedthattheSicontentintheini‐tialgelnotonlyaffectedthechemicalcompositionofthesam‐ples but also the crystallinity. In addition, the XRF elementalanalysis results indicated that the Si content of SAPO‐35 in‐creasedwiththeincreaseofSicontentintheinitialgel.Tobet‐tercorrelatetheSicontentsofthestartingmaterialsandprod‐ucts,weintroduceaconceptof“siliconincorporation”definedas [Si/(Si+Al+P)]product/[Si/(Si+Al+P)]gel (shown in Table 1). Adescending tendency was found for silicon incorporation inSAPO‐35withtheincreaseofSicontentinthestartingmateri‐als.The0.1Sisampleexhibitedthehighestsiliconincorporationof 1.82. The silicon incorporation decreased to less than 1.0whentheSicontentoftheinitialgelwasmorethan0.3.Siliconincorporationwasonly0.7whentheSicontentoftheinitialgelwas1.0.Therefore,webelievedthatitisthehighsiliconincor‐porationassociatedwiththesampleswithlowSicontentsthatresulted in their relatively low solidyields.Catlowet al. [22].employedlatticesimulationtocalculatetheenergychangesof
silicon incorporation in the framework of SAPO‐5 molecularsieves and found thathighly dispersedand singly substitutedsiliconwasthemoststable,followed5Siand8Siislands.Theseresults suggested that the energy of Si incorporation in theframework increased with increasing content of substitutedsilicon, leading to thedecreasing ability for silicon incorpora‐tionintheframework.Theirconclusionisconsistentwithourexperimentalresults.
The SEM images of the sampleswith different Si contentsareshowninFig.2.TheSAPO‐35grainspresentedthetypicalrhombohedral morphology, but the change of silicon contenthadasignificanteffectonthesurfaceroughnessofthemolecu‐lar sieve crystals. With a low silicon content, the molecularsievesurfacewasrelativelysmooth.Thecrystalsurfacebecamerough and irregular small holes appeared when the siliconcontentwasincreasedto0.8.Whenthesiliconcontentwas1.0,thesurfaceofthecrystalwaswrappedbysmallparticlesanditshowed a ‘core‐shell’ structure, which presumably resultedfrom a secondary crystal growth on the rough surface of thecrystalortheenrichmentofexcessiveamorphoussilicainthegelonthecrystalsurface.Thesurfacecompositionsofthe0.5Siand1.0SisampleswereanalyzedbyXPS(Table2).Itwasfoundthatthecrystalsurfaceofthesetwosamplescomprisedsilica,phosphorusoxide,andalumina.Therelativesiliconcontentofthesurfacewashigherthanthatofthebulkphase,andtheen‐richmentof siliconon thehighsiliconcontentsamplesurfacewas higher. It was thus speculated that the shell of the 1.0Si
10 20 30 40 50
Inte
nsit
y
2/( o )
0.1Si
0.2Si
0.3Si
0.5Si
0.8Si
1.0Si
Fig.1.XRDpatternsoftheSAPO‐35sampleswithdifferentSicontents.
Table1 RelativecrystallinityandchemicalcompositionoftheSAPO‐35sampleswithdifferentSicontents.
SampleRelative
crystallinity(%)Relativeyield(%)
Molarcomposition
Siincorporation*
0.1Si 84 62 Si0.060Al0.490P0.450 1.820.2Si 94 74 Si0.079Al0.497P0.424 1.250.3Si 100 86 Si0.092Al0.485P0.423 1.010.5Si 94 Si0.122Al0.487P0.391 0.890.8Si 91 95 Si0.169Al0.465P0.367 0.801.0Si 85 100 Si0.175Al0.457P0.368 0.70*Definedasthemolarratioof[Si/(Si+Al+P)]product/[Si/(Si+Al+P)]gel.
Fig.2.SEMimagesoftheSAPO‐35sampleswithdifferentSicontents.

LIBingetal./ChineseJournalofCatalysis34(2013)798–807
sample grains was formed by a secondary crystal growth onthe rough surface of the crystal. Meanwhile, from the siliconenrichmentontheSAPO‐35crystalsurfacerevealedbyXPS,wecan suppose that the distribution of silicon in the SAPO‐35crystalswasnon‐uniformandtherewasagradual increaseofthesiliconcontent fromtheinsideoutwards.Wealso foundasimilar situation in the crystallization of SAPO‐34 with di‐ethylamineasatemplateagent[23].ThemainreasonwouldbethegradualincreaseofthegelpHduringthesynthesisofSAPOmolecularsieves.TheincreaseofpHpromoteddepolymeriza‐tionofthesiliconsourceinthegelsystem,thustheability forsilicon incorporation into themolecular sieve frameworkwasenhanced(thepHvalueofthegel inoursynthesissystembe‐foreandafterthecrystallizationof0.5Sisamplewere5.56and8.20,respectively).
Figure 3 and Table 3 show the N2 adsorption‐desorptionisotherms,specificsurfaceareas,andporevolumesofthesam‐ples with different Si contents. All three samples displayed ahighmicroporesurfaceareaandmicroporevolume.Moreover,the outer specific surface area and mesopore volume of thesamplesincreasedwiththeincreaseofsiliconcontents,whichwould be due to the rough grain surface of the sampleswithhighsiliconcontents,asrevealedbytheSEMimages.
3.2. MethanolconversionbySAPO‐35molecularsievewithdifferentSicontents
0.1Si,0.3Si,and0.5Sisampleswereselectedforthestudyofmethanolconversion.TheresultsareshowninFig.4andTable4. The results of the MTO reaction on SAPO‐34 (elementalcomposition is Si0.086Al0.493P0.421) are also listed inTable4 forcomparison.Duringaninitialperiod,theconversionofmetha‐nol on SAPO‐35with different Si contentswasmaintained atabove 99%. The conversion decreased as the reaction timeincreasedandthehighertheSicontent,thefastertheconver‐sionofmethanoldropped.Theselectivityforethyleneofthesemolecularsievesgraduallyroseasthereactiontimeincreased,while the selectivity forpropylenedecreased.ComparedwithSAPO‐34,SAPO‐35exhibitedthecharacteristicoffastdeactiva‐tion.
TheMTO reaction is a typical acid catalyzed reaction, andtheacidityofthemolecularsieveplaysanimportantroleonthelifetimeandtheselectivitytolightolefins. We determined theamountofBrönstedacidof the three SAPO‐35 samplesusing1H MAS NMR (shown in Table 5). Generally, the density ofBrönstedacidsitesincreasedinthesampleswiththeincreaseoftheSicontents.Botharealmostinalinearrelationship.Thehigher acid density in the samples with higher Si contentcausedserioussidereactions,suchascokedepositionandthehydrogentransferreaction,whichshortenedthelifetimeofthecatalystandgeneratedmoremethaneandpropane.Moreover,thedeposited coke species gradually formedduring the reac‐tionwilldecreasethecagesizeofthemolecularsieves,reducethe generation and diffusion rates of molecules with largerdiameters, and consequently increase the selectivity for eth‐yleneanddecreasetheselectivityforpropylene.
Table2 BulkandsurfacecompositionsoftheSAPO‐35sampleswithdifferentSicontents.
SampleComposition
Sisurface/SibulkBulk Surface
0.5Si Si0.122Al0.487P0.391 Si0.178Al0.478P0.335 1.461.0Si Si0.175Al0.457P0.368 Si0.275Al0.405P0.320 1.58
0.0 0.2 0.4 0.6 0.8 1.00
20
40
60
80
100
120
140
160
180
200
Vol
ume
(ml/
g)
Relative pressure (p/p0)
0.8Si 1.0Si 0.2Si
Fig. 3. N2 adsorption‐desorption isotherms of the SAPO‐35 sampleswithdifferentSicontents.
Table3 Pore structure parameters of the SAPO‐35 samples with different Sicontents.
SampleSurfacearea(m2/g) Porevolume(cm3/g)
Micropore External Total Micropore Mesopore Total0.2Si 443.5 26.3 469.7 0.22 0.01 0.210.8Si 497.4 41.0 538.4 0.23 0.05 0.281.0Si 475.4 45.8 521.2 0.22 0.07 0.29
0 20 40 60 80 100 120 140 160 18060
65
70
75
80
85
(4)
(3)
(2)(1)
C2H
4+C
3H6 s
elec
tivi
ty (
%)
MeO
H c
onve
rsio
n (%
)
Time on stream (min)
(b)
30
45
60
75
90
105
(4)
(3)
(2)
(1)
(a)
Fig.4.CH3OHconversion(a)andC2H4+C3H6selectivity(b)intheMTOreactionoverSAPO‐35andSAPO‐34.(1)0.1Si;(2)0.3Si;(3)0.5Si;(4)SAPO‐34.Reaction condition:450 °C, 40%CH3OH solution (SAPO‐35,WHSV=2h–1;SAPO‐34,WHSV=4h–1).
LIBingetal./ChineseJournalofCatalysis34(2013)798–807
3.3. AnalysisofthedepositedcokespeciesformedintheMTOreactionoverSAPO‐35andSAPO‐34
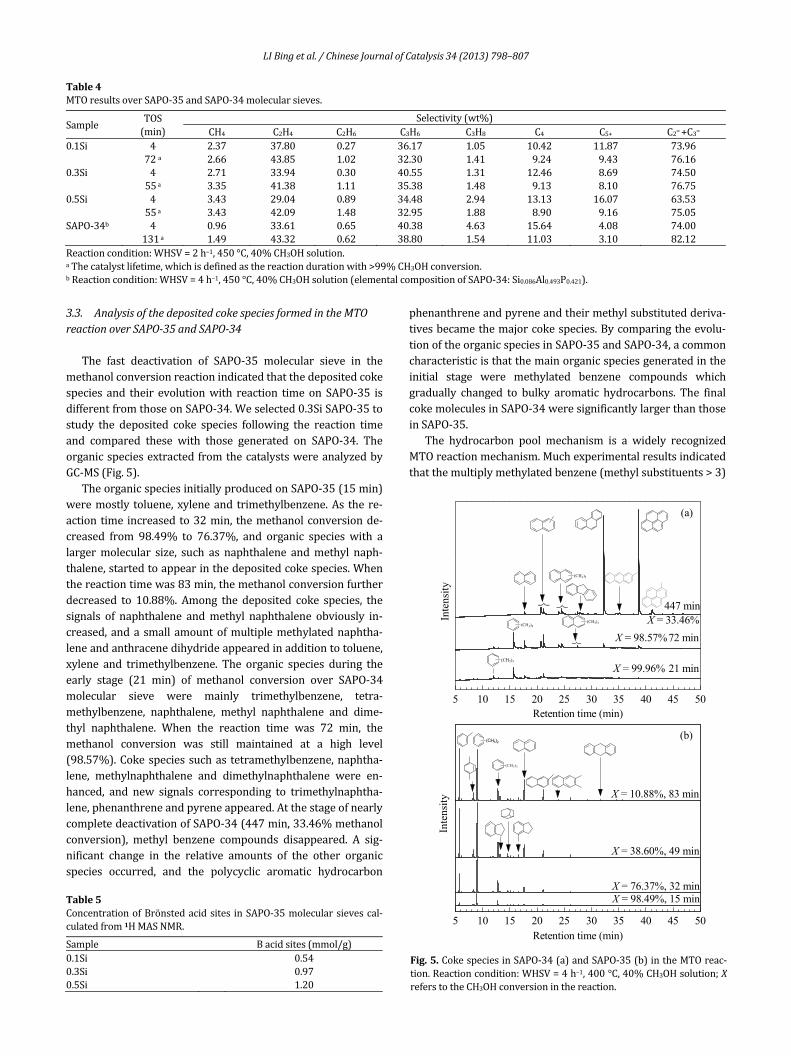
The fast deactivation of SAPO‐35 molecular sieve in themethanolconversionreactionindicatedthatthedepositedcokespecies and their evolutionwith reaction timeon SAPO‐35 isdifferentfromthoseonSAPO‐34.Weselected0.3SiSAPO‐35tostudy the deposited coke species following the reaction timeand compared these with those generated on SAPO‐34. TheorganicspeciesextractedfromthecatalystswereanalyzedbyGC‐MS(Fig.5).
TheorganicspeciesinitiallyproducedonSAPO‐35(15min)weremostly toluene,xyleneand trimethylbenzene.As there‐actiontime increasedto32min, themethanolconversionde‐creased from 98.49% to 76.37%, and organic species with alargermolecular size, such as naphthalene andmethyl naph‐thalene,startedtoappearinthedepositedcokespecies.Whenthereactiontimewas83min,themethanolconversionfurtherdecreased to 10.88%. Among the deposited coke species, thesignals of naphthalene andmethyl naphthalene obviously in‐creased, anda small amountofmultiplemethylatednaphtha‐leneandanthracenedihydrideappearedinadditiontotoluene,xylene and trimethylbenzene. The organic species during theearly stage (21 min) of methanol conversion over SAPO‐34molecular sieve were mainly trimethylbenzene, tetra‐methylbenzene, naphthalene, methyl naphthalene and dime‐thyl naphthalene. When the reaction time was 72 min, themethanol conversion was still maintained at a high level(98.57%).Cokespeciessuchas tetramethylbenzene,naphtha‐lene, methylnaphthalene and dimethylnaphthalene were en‐hanced, and new signals corresponding to trimethylnaphtha‐lene,phenanthreneandpyreneappeared.AtthestageofnearlycompletedeactivationofSAPO‐34(447min,33.46%methanolconversion), methyl benzene compounds disappeared. A sig‐nificant change in the relative amounts of the other organicspecies occurred, and the polycyclic aromatic hydrocarbon
phenanthreneandpyreneandtheirmethylsubstitutedderiva‐tivesbecamethemajorcokespecies.Bycomparingtheevolu‐tionoftheorganicspeciesinSAPO‐35andSAPO‐34,acommoncharacteristicisthatthemainorganicspeciesgeneratedintheinitial stage were methylated benzene compounds whichgradually changed to bulky aromatic hydrocarbons. The finalcokemoleculesinSAPO‐34weresignificantlylargerthanthoseinSAPO‐35.
The hydrocarbon pool mechanism is a widely recognizedMTOreactionmechanism.Muchexperimentalresultsindicatedthatthemultiplymethylatedbenzene(methylsubstituents>3)
Table4 MTOresultsoverSAPO‐35andSAPO‐34molecularsieves.
SampleTOS(min)
Selectivity(wt%)CH4 C2H4 C2H6 C3H6 C3H8 C4 C5+ C2=+C3=
0.1Si 4 2.37 37.80 0.27 36.17 1.05 10.42 11.87 73.96 72a 2.66 43.85 1.02 32.30 1.41 9.24 9.43 76.160.3Si 4 2.71 33.94 0.30 40.55 1.31 12.46 8.69 74.50 55a 3.35 41.38 1.11 35.38 1.48 9.13 8.10 76.750.5Si 4 3.43 29.04 0.89 34.48 2.94 13.13 16.07 63.53 55a 3.43 42.09 1.48 32.95 1.88 8.90 9.16 75.05SAPO‐34b 4 0.96 33.61 0.65 40.38 4.63 15.64 4.08 74.00 131a 1.49 43.32 0.62 38.80 1.54 11.03 3.10 82.12Reactioncondition:WHSV=2h–1,450°C,40%CH3OHsolution.aThecatalystlifetime,whichisdefinedasthereactiondurationwith>99%CH3OHconversion.bReactioncondition:WHSV=4h–1,450°C,40%CH3OHsolution(elementalcompositionofSAPO‐34:Si0.086Al0.493P0.421).
Table5 ConcentrationofBrönstedacidsites inSAPO‐35molecularsievescal‐culatedfrom1HMASNMR.
Sample Bacidsites(mmol/g)0.1Si 0.540.3Si 0.970.5Si 1.20
5 10 15 20 25 30 35 40 45 50
Int
ensi
ty
(CH3)3
(CH3)3(CH3)4
(CH3)2
Retention time (min)
21 min
72 min
447 min
(a)
X = 99.96%
X = 98.57%
X = 33.46%
5 10 15 20 25 30 35 40 45 50
(C H3)3
(CH3)2
Int
ensi
ty
Retention time (min)
(b)
X = 10.88%, 83 min
X = 38.60%, 49 min
X = 76.37%, 32 minX = 98.49%, 15 min
Fig.5.CokespeciesinSAPO‐34(a)andSAPO‐35(b)intheMTOreac‐tion.Reactioncondition:WHSV=4h–1,400°C,40%CH3OHsolution;XreferstotheCH3OHconversioninthereaction.

LIBingetal./ChineseJournalofCatalysis34(2013)798–807
isahydrocarbonpoolactivecenter,onwhichthemethylationofmethanol/diethyletheroccurredtoformethyleneandpro‐pylene, while the resulting less methylated benzene isre‐methylated to start a new catalytic cycle [15,16,24–27]. Inthepresentexperiments,intheinitialstageofreactiontheex‐istence of some hydrocarbon pool active species (multiplymethylatedbenzenes)wereobservedinSAPO‐34,whereasthecoke species was dominated by less methylated benzenes inSAPO‐35.With increased reaction time, at the stageofnearlycompletedeactivation,themaincokespeciesinSAPO‐34werebulky aromatic hydrocarbons such as phenanthrene and py‐rene(generated fromringcondensationbyhydrogentransferfrommethylbenzene andmethylnaphthalene),while the cokespecies in SAPO‐35 were methylbenzene, naphthalene andmethylnaphthalene. The difference in the coke species ofSAPO‐35andSAPO‐34 isprobably related to their structures.The CHA cage of SAPO‐34 (0.67 nm × 0.67 nm × 1.0 nm) islargerthantheLEVcageofSAPO‐35(0.63nm×0.63nm×0.73nm).The smaller cageof SAPO‐35restricted the formationofthehydrocarbonpoolactivespecies(multiplymethylatedben‐zene)anddeactivatedcokespecies(macromolecularfused‐ringaromatichydrocarbon).Also, it has a smaller accommodationcapacity for deactivated coke species, which thus resulted inthe rapid deactivation. The lack of hydrocarbon pool activespecies also led to the relatively low ethylene and propyleneselectivity with SAPO‐35. Moreover, it is worth noting thatduring the reaction, the organic compounds deposited inSAPO‐35alwayscontainedasmallamountofsaturatedcyclo‐alkaneadamantanecompounds.InourrecentresearchonthecokespeciesinSAPO‐34intheMTOreaction[28],wereportedthat adamantane compounds are the coke species in the lowtemperaturereaction(≤350 oC),whichweregradually trans‐formed intonaphthaleneandsubstitutednaphthalenespeciesat elevated temperatures. In this experiment, the reactiontemperaturewas400oC,sotheexistenceofadamantanecom‐poundsinSAPO‐35onceagaindemonstratedthatthedifferentcage size of themolecular sieve has a large influence on thegenerationandstabilityofthecokespecies.
4. Conclusions
SAPO‐35molecular sieveswith different Si contents were
hydrothermally synthesized. The yield of solid sample in‐creasedwiththeincreaseofSicontentinthesynthesisgel.Thecrystallinityofthesampleincreasedfirstandthendecreasedasthe Si content increased. 0.3Si sample exhibited the highestcrystallinity. The Si incorporation degree showed a droppingtrend when the silica content rose. The surface of SAPO‐35crystalswithahighsilicacontentwasrough.Inparticular,1.0Sishowedthecharacteristicofacore‐shellstructure,whichcouldbe due to secondary crystallization on the rough surface.SAPO‐35 showed fast deactivation than SAPO‐34 in theMTOreaction,andahighersilicacontentwillcausefasterdeactiva‐tion.ThemainreasonisthatthehigherBrönstedacidconcen‐trationinthesamplewithhighersilicacontentledtomoresidereactions such as coke deposition and hydrogen transfer. BycomparingandanalyzingthedepositedcokespeciesformedinSAPO‐35andSAPO‐34duringtheMTOreaction,weconcludedthatthesmallercageofSAPO‐35limitedthegenerationofhy‐drocarbon pool active species (polymethylbenzens) andmac‐romolecularcokespecies(polyaromatichydrocarbons).More‐over, thesmallercageofSAPO‐35hada lowercapacitytoac‐commodatedepositedcokespecies.Thesetworeasonsresult‐edinthefasterdeactivationofSAPO‐35.
References
[1] LokBM,MessinaCA,PattonRL,GajekRT,CannanTR,FlanigenEM.USPatent4440871.1984
[2] FuY,WangL F, TanYX, JiHB.ChemReactEngTechnol, 2000,16(1):55
[3] http://www.iza‐structure.org/databases/[4] ZhuZhD,HartmannM,KevanL.ChemMater,2000,12:2781[5] JeonHY,ShinCH,JungHJ,HongSB.ApplCatalA,2006,305:70[6] DjieugoueMA,PrakashAM,KevanL.JPhysChemB,2000,104:
6452
[7] VenkatathriN,YooJW.ApplCatalA,2008,340:265
[8] Cheung O, Liu Q L, Bacsik Z, Hedin N.MicroporousMesoporous
Mater,2012,156:90
[9] FalconerJL,LiShG,NobleRD.USPatent20050204916A1.2005
[10] LiuGY,TianP,LiuZhM.ProgrChem,2010,22:1531
[11] Zhang JCh, ZhangHB, YangXY,HuangZh,CaoWL. JNatGas
Chem,2011,20:266
[12] LiuGY,TianP,XiaQH,LiuZhM.JNatGasChem,2012,21:431[13] NawazS,KolboeS,StöckerM. StudSurfSciCatal,1994, 81:393
GraphicalAbstract
Chin.J.Catal.,2013,34:798–807 doi:10.1016/S1872‐2067(12)60557‐9
SynthesisofSAPO‐35molecularsieveanditscatalyticpropertiesinthemethanol‐to‐olefinsreaction
LIBing,TIANPeng,LIJinzhe,CHENJingrun,YUANYangyang,SUXiong,FANDong,WEIYingxu,QIYue,LIUZhongmin*DalianInstituteofChemicalPhysics,ChineseAcademyofSciences; UniversityofChineseAcademyofSciences
SAPO‐35 was hydrothermally synthesized using hexamethyleneimine asthetemplate.ThecokespeciesintheMTOreactionoverbothSAPO‐35andSAPO‐34wereinvestigatedandcorrelatedwiththeircagesize.
SAPO‐35(LEV)
5 10 15 20 25 30 35 40 45 50
(CH3)3
(CH3)2
(CH3)3
(CH3)2
Retention time (min)
Coke species
SAPO‐34(CHA)

LIBingetal./ChineseJournalofCatalysis34(2013)798–807
[14] Chen J Sh, Wright P A, Thomas J M, Natarajan S, Marchese L,BradleySM,SankarG,CatlowCRA,Gai‐BoyesPL.JPhysChem,1994,98:10216
[15] YuanCY,WeiYX,LiJZh,XuShT,ChenJR,ZhouY,WangQY,XuL,LiuZhM.ChinJCatal,2012,33:367
[16] Hereijgers B P C, Bleken F, Nilsen M H, Svelle S, Lillerud K P,BjørgenM,WeckhuysenBM,OlsbyeU.JCatal,2009,264:77
[17] ZhangFM,KuangXH,LiLSh,ShuXT,WangWD,QinFM,CNPatent101397143.2009
[18] GuisnetM.JMolCatalA,2002,182‐183:367[19] GuisnetM,CostaL,RibeiroFR.JMolCatalA,2009,305:69[20] PrakashAM,HartmannM,KevanL.ChemMater,1998,10:932
[21] ZhangDZh.[PhDDissertation].Dalian:DalianInstituteofChemi‐calPhysics,CAS, 2007
[22] SastreG,LewisDW,CatlowCRA.JPhysChem,1996,100:6722[23] Liu G Y, Tian P, Zhang Y, Li J Zh, Xu L, Meng Sh H, Liu Zh M.
MicroporousMesoporousMater,2008,114:416[24] WangJD,YuXB,LiuY,YangYR.ProgrChem,2009,21:1757[25] ArstadB,KolboeS.JAmChemSoc,2001,123:8137[26] ArstadB,KolboeS.CatalLett,2001,71:209[27] Lesthaeghe D, De Sterck B, Van Speybroeck V, Marin G B,
WaroquierM.AngewChem,IntEd,2007,46:1311[28] WeiYX,Li JZh,YuanCY,XuShT,ZhouY,ChenJR,WangQY,
ZhangQ,LiuZhM.ChemCommun,2012,48:3082




















