
Chronic exposure to imidacloprid induces inflammation and oxidative stress in the liver & central...
7
Chronic exposure to imidacloprid induces inflammation and oxidative stress in the liver & central nervous system of rats Vesile Duzguner b,⇑ , Suat Erdogan a a Mustafa Kemal University, Veterinary Faculty, Biochemistry Department, 31040 Antakya, Hatay, Turkey b Ardahan University, Health Services Vocational School, 75000 Ardahan, Turkey article info Article history: Received 13 October 2011 Accepted 27 June 2012 Available online 27 July 2012 Keywords: Imidacloprid Inflammation Insecticide Nitric oxide Oxidative stress abstract Imidacloprid is the most important example of the neonicotinoid insecticides known to target the nico- tinic acetylcholine receptor (nAChR) in insects, and potentially in mammals. In the present study, oxidant and inflammatory responses to chronic exposure of imidacloprid was studied in rats. Wistar rats were randomly allocated into two groups as control and imidacloprid-exposed group (n = 10 rat/each group). 1 mg/kg/BW/day imidacloprid was administrated orally by gavage for 30 days. After exposure, rats were euthanized and liver and brain samples were surgically removed for analyses. Imidacloprid application caused a significant increase in nitric oxide production in brain (p < 0.05) and liver (p < 0.001). The quan- titative analyses of mRNA confirmed the finding that imidacloprid induced the mRNA transcriptions of the three isoforms of nitric oxide synthases (iNOS, eNOS, nNOS) in brain and two isoforms (iNOS, eNOS) in the liver. Exposure to imidacloprid caused significant lipid peroxidation in plasma, brain (p < 0.001) and liver (p < 0.003). While the superoxide-generating enzyme xanthine oxidase activity was elevated in both tissues (p < 0.001), myeloperoxidase activity was increased only in the liver (p < 0.001). Antioxi- dant enzyme activities showed various alterations following exposure, but a significantly depleted anti- oxidant glutathione level was detected in brain (p < 0.008). Evidence of chronic inflammation by imidacloprid was observed as induction of pro-inflammatory cytokines such as TNF-a, IL-1b, IL-6, IL- 12 and IFN-c in the liver and brain. In conclusion, chronic imidacloprid exposure causes oxidative stress and inflammation by altering antioxidant systems and inducing pro-inflammatory cytokine production in the liver and central nervous system of non-target organisms. Crown Copyright Ó 2012 Published by Elsevier Inc. All rights reserved. 1. Introduction The neonicotinoids are a new major class of highly potent insec- ticides that are used for crop protection against piercing–sucking insects of cereals, vegetables, tea and cotton, and for flea control in cats and dogs. Currently the best known neonicotinoid is imida- cloprid [IMI, 1-(6-chloro-3-pyridylmethyl)-N-nitroimidazolidin-2- ylideneamine] which is an active substance in such commercial insecticide preparations as Confidor, Gaucho, Prestige, Admire, and Premier which are increasingly used in agriculture [1,2]. Imi- dacloprid and its analogs are remarkably potent neurotoxic insec- ticides, which act as nicotinic acetylcholine receptor agonists (nAChRs) [3]. nAChRs play a central role in rapid cholinergic synap- tic transmission and are important targets of insecticides [3,4]. Most neonicotinoids are partial agonists of native and recombinant nAChRs in both mammals and insects with differential selectivity conferred by only minor structural changes [5,6]. Tomizawa [7] suggests that nAChRs may be up-regulated in mammals by chronic exposure to imidacloprid or its metabolites. It is also known that free radicals play an important role in the toxicity of pesticides and environmental chemicals [8]. Pesticide chemicals such as insecticides may induce oxidative stress leading to generation of free radicals and alterations in antioxidants or free radical scavenging enzyme systems [9,10]. The data on experimen- tal animals either in vivo or in vitro [11–13] indicate that the en- zymes associated with antioxidant defense mechanisms are altered under the influence of pesticides. Moreover, oxidative stress and DNA damage have been proposed as mechanisms link- ing pesticide exposure to health effects such as cancer and neuro- logical diseases. During metabolism of the insecticides reactive oxygen species (ROS) and reactive nitrogen species (RNS) such as nitric oxide (NO) can be generated. Nitric oxide is a diatomic free radical which plays critical roles in the homeostatic regulation of cardiovascular, neuronal and immune systems. Despite its impor- tant physiological functions, NO is also a well-known toxic agent [14]. Subsequently there is onset of an oxidative stress in central nervous system (CNS) structures. These structures include the hip- pocampus, cortex, striatum and cerebellum where mitochondrial 0048-3575/$ - see front matter Crown Copyright Ó 2012 Published by Elsevier Inc. All rights reserved. http://dx.doi.org/10.1016/j.pestbp.2012.06.011 ⇑ Corresponding author. Tel.: +90 478 211 37 50. E-mail addresses: [email protected] (V. Duzguner), serdogan1967@ hotmail.com (S. Erdogan). Pesticide Biochemistry and Physiology 104 (2012) 58–64 Contents lists available at SciVerse ScienceDirect Pesticide Biochemistry and Physiology journal homepage: www.elsevier.com/locate/pest
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Chronic exposure to imidacloprid induces inflammation and oxidative stress in the liver & central...

Pesticide Biochemistry and Physiology 104 (2012) 58–64
Contents lists available at SciVerse ScienceDirect
Pesticide Biochemistry and Physiology
journal homepage: www.elsevier .com/locate /pest
Chronic exposure to imidacloprid induces inflammation and oxidative stressin the liver & central nervous system of rats
Vesile Duzguner b,⇑, Suat Erdogan a
a Mustafa Kemal University, Veterinary Faculty, Biochemistry Department, 31040 Antakya, Hatay, Turkeyb Ardahan University, Health Services Vocational School, 75000 Ardahan, Turkey
a r t i c l e i n f o a b s t r a c t
Article history:Received 13 October 2011Accepted 27 June 2012Available online 27 July 2012
Keywords:ImidaclopridInflammationInsecticideNitric oxideOxidative stress
0048-3575/$ - see front matter Crown Copyright � 2http://dx.doi.org/10.1016/j.pestbp.2012.06.011
⇑ Corresponding author. Tel.: +90 478 211 37 50.E-mail addresses: [email protected] (V.
hotmail.com (S. Erdogan).
Imidacloprid is the most important example of the neonicotinoid insecticides known to target the nico-tinic acetylcholine receptor (nAChR) in insects, and potentially in mammals. In the present study, oxidantand inflammatory responses to chronic exposure of imidacloprid was studied in rats. Wistar rats wererandomly allocated into two groups as control and imidacloprid-exposed group (n = 10 rat/each group).1 mg/kg/BW/day imidacloprid was administrated orally by gavage for 30 days. After exposure, rats wereeuthanized and liver and brain samples were surgically removed for analyses. Imidacloprid applicationcaused a significant increase in nitric oxide production in brain (p < 0.05) and liver (p < 0.001). The quan-titative analyses of mRNA confirmed the finding that imidacloprid induced the mRNA transcriptions ofthe three isoforms of nitric oxide synthases (iNOS, eNOS, nNOS) in brain and two isoforms (iNOS, eNOS)in the liver. Exposure to imidacloprid caused significant lipid peroxidation in plasma, brain (p < 0.001)and liver (p < 0.003). While the superoxide-generating enzyme xanthine oxidase activity was elevatedin both tissues (p < 0.001), myeloperoxidase activity was increased only in the liver (p < 0.001). Antioxi-dant enzyme activities showed various alterations following exposure, but a significantly depleted anti-oxidant glutathione level was detected in brain (p < 0.008). Evidence of chronic inflammation byimidacloprid was observed as induction of pro-inflammatory cytokines such as TNF-a, IL-1b, IL-6, IL-12 and IFN-c in the liver and brain. In conclusion, chronic imidacloprid exposure causes oxidative stressand inflammation by altering antioxidant systems and inducing pro-inflammatory cytokine production inthe liver and central nervous system of non-target organisms.
Crown Copyright � 2012 Published by Elsevier Inc. All rights reserved.
1. Introduction
The neonicotinoids are a new major class of highly potent insec-ticides that are used for crop protection against piercing–suckinginsects of cereals, vegetables, tea and cotton, and for flea controlin cats and dogs. Currently the best known neonicotinoid is imida-cloprid [IMI, 1-(6-chloro-3-pyridylmethyl)-N-nitroimidazolidin-2-ylideneamine] which is an active substance in such commercialinsecticide preparations as Confidor, Gaucho, Prestige, Admire,and Premier which are increasingly used in agriculture [1,2]. Imi-dacloprid and its analogs are remarkably potent neurotoxic insec-ticides, which act as nicotinic acetylcholine receptor agonists(nAChRs) [3]. nAChRs play a central role in rapid cholinergic synap-tic transmission and are important targets of insecticides [3,4].Most neonicotinoids are partial agonists of native and recombinantnAChRs in both mammals and insects with differential selectivityconferred by only minor structural changes [5,6]. Tomizawa [7]
012 Published by Elsevier Inc. All r
Duzguner), serdogan1967@
suggests that nAChRs may be up-regulated in mammals by chronicexposure to imidacloprid or its metabolites.
It is also known that free radicals play an important role in thetoxicity of pesticides and environmental chemicals [8]. Pesticidechemicals such as insecticides may induce oxidative stress leadingto generation of free radicals and alterations in antioxidants or freeradical scavenging enzyme systems [9,10]. The data on experimen-tal animals either in vivo or in vitro [11–13] indicate that the en-zymes associated with antioxidant defense mechanisms arealtered under the influence of pesticides. Moreover, oxidativestress and DNA damage have been proposed as mechanisms link-ing pesticide exposure to health effects such as cancer and neuro-logical diseases. During metabolism of the insecticides reactiveoxygen species (ROS) and reactive nitrogen species (RNS) such asnitric oxide (NO) can be generated. Nitric oxide is a diatomic freeradical which plays critical roles in the homeostatic regulation ofcardiovascular, neuronal and immune systems. Despite its impor-tant physiological functions, NO is also a well-known toxic agent[14]. Subsequently there is onset of an oxidative stress in centralnervous system (CNS) structures. These structures include the hip-pocampus, cortex, striatum and cerebellum where mitochondrial
ights reserved.

V. Duzguner, S. Erdogan / Pesticide Biochemistry and Physiology 104 (2012) 58–64 59
respiratory chain dysfunctions have been noted. Oxidative mecha-nisms play an important role in insecticide-induced tissue damagenot only by balancing oxidant-antioxidant status but also by inhib-iting neutrophil infiltration and regulating inflammatory media-tors [15,16].
Insecticides are associated with direct or indirect modulation ofmajor and vital immune mechanisms and inflammatory activationmight be an important mechanism underlying neurotoxicity ofpesticides [17]. Its known that exposure to different insecticidescould accelerate the synthesis of cytokines such as interferon gam-ma-c (IFN) and tumor necrosis factor-a (TNF), reduce anti-inflam-matory cytokine interleukin-10 (IL) and also inhibit signaltransduction correlated with their toxicity [10,18,19].
Few studies have been performed in mammals with neonicoti-noid insecticides and their relationship with oxidative and inflam-matory events. Tomizawa [7] suggested that stimulation of thenervous system by acute or sustained exposure to these chemicalsmay lead to synaptic plasticity or attenuated neuronal functions.We have previously demonstrated that acute exposure to imida-cloprid leads to oxidative and inflammatory effects in rats [10].Although imidacloprid is the most frequently used insecticideamongst the pesticides, it is possible that its chronic effects innon-target organisms such as mammals remain to be elucidated.In this study, we investigated the potential oxidative and chronicinflammatory effects of imidacloprid on the central nervous sys-tem and liver in rats.
2. Materials and methods
2.1. Animals and study design
Ten-week-old female Wistar rats (150 ± 25 g) were acclima-tized for 10 days before starting the experimental procedure. Therats were assigned randomly to either control (n = 10) or imidaclo-prid (n = 10) group and housed in a temperature controlled room(21–22 �C) with a 12 h dark–light cycle during the study.
Concentration of imidacloprid was calculated based on 1/15 ofits LD50 values and on body weight data [20]. Imidacloprid (Riedel,Sigma–Aldrich) was suspended in corn oil and administered to ratsat the dose of 1 mg/kg/bw-day by gavage during a 30 day period,whilst controls were treated with corn oil as vehicle. At the endof the exposure, the rats were anesthetized by intramuscular injec-tion of 50 mg/kg ketamine hydrochloride and blood was taken bypuncturing the heart ventricle. The brain and liver were taken fromeach rat, washed with ice-cold physiological saline and used forbiochemical studies. The protocols were approved by the AnimalsExperiments Ethics Committee of Firat University, Elazig, Turkey.
2.2. Biochemical assays
Tissue samples were homogenized in ice-cold homogenizationbuffer (10 mM Tris, 1 mM EDTA, 25 mM MgCl2, 0.1 mM dithiothre-itol, 0.25 M sucrose, pH 7.4) containing complete protease inhibitormixture (aprotinin, phenylmethylsulphonyl fluoride, leupeptin, so-dium floride) (Sigma, Germany). Homogenates were centrifuged at4 �C, 15 000 rpm for 10 min and the soluble fraction was retained.Protein concentrations of supernatants were measured by themethod of Bradford [21] using bovine serum albumin as a stan-dard. The degree of lipid peroxidation was assessed by measuringmalondialdehyde (MDA) levels in plasma and tissue samples [22].Total superoxide dismutase (SOD) activity in the homogenates andplasma samples was determined according to the method of Sunet al. [23]. Nitric oxide concentration in plasma and tissue sampleswas analyzed indirectly by measuring the nitrite levels based onGriess reaction [24].
Xanthine oxidase (XO) activity was analyzed spectrophotomet-rically by the formation of uric acid from xanthine through in-creased absorbency at 293 nm, according to the method of Prajdaand Weber [25]. Myeloperoxidase (MPO) activity was measuredby the method of Andrews and Krinski [26]. Catalase activity wasmeasured according to the method of Luck [27]. Glutathione per-oxidase (GSH-Px) activity was determined by a kinetic methodusing a commercial kit (RANSEL, Randox Laboratorius). The meth-od is based on Paglia and Valentine’s study [28].
Reduced glutathione (GSH) concentrations found in liver andbrain homogenates were determined according to Sedlak and Lind-say [29]. GSH was reacted with 5,5-dithiobis-2-nitrobenzoic acidresulting in the formation of a product with a maximal absorbanceat 410 nm. The results were expressed as lmol/mg protein.
To reveal the relationship between activation of nicotinic recep-tors on neurons and intracellular calcium levels, plasma calciumconcentrations were determined using a commercial kit (TecoDiagnostics, USA). Finally, the activities of liver enzymes such asalanine aminotransferase (ALT), aspartate aminotransferase (AST)and lactate dehydrogenase (LDH) were measured using commer-cial kits (Thermo Infinity, USA).
2.3. RNA preparation and reverse transcriptase polymerase chainreaction (RT–PCR)
For the evaluation of mRNA expression, real-time PCR was per-formed in a Strategene Mx 3005P QPCR system. Total RNA was ex-tracted from brain and liver samples using TRIZOL reagent (Sigma)according to the manufacturer’s instructions. Two micrograms oftotal RNA was reverse transcribed in a reaction volume of 20 llusing a reverse transcriptase kit (Fermentase). One microgram ofeach cDNA was used as templates for amplification using SYBERGreen PCR amplification reagent and gene-specific primers. Theprimer sets used were from Thermo Electron Corporation (Ger-many). The specific primers for TNF-a, IFN-c, IL-1b, IL-6, IL-12,neuronal- (nNOS), inducible- (iNOS), and endothelial nitric oxidesynthase (eNOS) and the PCR reaction conditions are given in Ta-ble 1. The threshold cycle (Ct) number for the transcripts is nor-malized to b-actin by subtracting the average Ct number for eachtreatment. Expression of this housekeeping gene should not alterin response to imidacloprid exposure. Each PCR reaction was per-formed in triplicate. The mean relative expressions of the targetinggenes were calculated and the differences were determined usingthe 2�DDCt method [30].
3. Results
3.1. The effect of imidacloprid on oxidant and antioxidant systems
Nitric oxide is produced from the amino acid L-arginine by theenzymatic action of NOS. Whilst it has physiological actions,abnormal production of NO can damage numerous molecules(including lipids, proteins and DNA) causing alterations in thefunctioning of target cells potentially leading to cell death. In thepresent study, we found that long term exposure to imidaclopridsignificantly increased NO production by approximately 23% inbrain (p < 0.05) and 50% in liver (p < 0.001) (Table 2). The level oflipid peroxidation, as indicated by malondialdehyde (MDA) forma-tion, was markedly increased by 1.24-, 1.45-, 2.35-fold respectivelyin liver (p < 0.003), brain and plasma (p < 0.001) when imidaclopridexposed animals were compared to control (Table 2). Similar to NOproduction and MDA formation, imidacloprid significantly stimu-lated the activity of oxidant generating enzymes such as xanthineoxidase in the brain and liver (p < 0.001), and myeloperoxidase inthe liver (p < 0.001) (Table 3). Catalase activity was also elevated
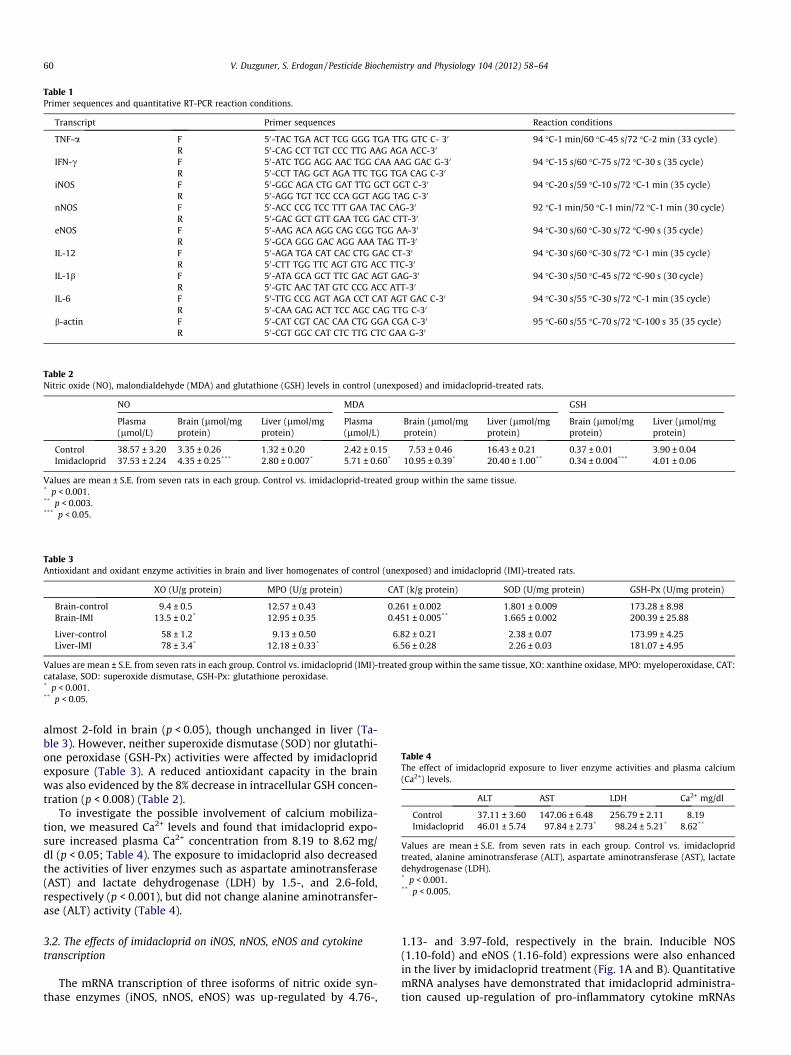
Table 1Primer sequences and quantitative RT-PCR reaction conditions.
Transcript Primer sequences Reaction conditions
TNF-a FR
50-TAC TGA ACT TCG GGG TGA TTG GTC C- 30
50-CAG CCT TGT CCC TTG AAG AGA ACC-3094 �C-1 min/60 �C-45 s/72 �C-2 min (33 cycle)
IFN-c FR
50-ATC TGG AGG AAC TGG CAA AAG GAC G-30
50-CCT TAG GCT AGA TTC TGG TGA CAG C-3094 �C-15 s/60 �C-75 s/72 �C-30 s (35 cycle)
iNOS FR
50-GGC AGA CTG GAT TTG GCT GGT C-30
50-AGG TGT TCC CCA GGT AGG TAG C-3094 �C-20 s/59 �C-10 s/72 �C-1 min (35 cycle)
nNOS FR
50-ACC CCG TCC TTT GAA TAC CAG-30
50-GAC GCT GTT GAA TCG GAC CTT-3092 �C-1 min/50 �C-1 min/72 �C-1 min (30 cycle)
eNOS FR
50-AAG ACA AGG CAG CGG TGG AA-30
50-GCA GGG GAC AGG AAA TAG TT-3094 �C-30 s/60 �C-30 s/72 �C-90 s (35 cycle)
IL-12 FR
50-AGA TGA CAT CAC CTG GAC CT-30
50-CTT TGG TTC AGT GTG ACC TTC-3094 �C-30 s/60 �C-30 s/72 �C-1 min (35 cycle)
IL-1b FR
50-ATA GCA GCT TTC GAC AGT GAG-30
50-GTC AAC TAT GTC CCG ACC ATT-3094 �C-30 s/50 �C-45 s/72 �C-90 s (30 cycle)
IL-6 FR
50-TTG CCG AGT AGA CCT CAT AGT GAC C-30
50-CAA GAG ACT TCC AGC CAG TTG C-3094 �C-30 s/55 �C-30 s/72 �C-1 min (35 cycle)
b-actin FR
50-CAT CGT CAC CAA CTG GGA CGA C-30
50-CGT GGC CAT CTC TTG CTC GAA G-3095 �C-60 s/55 �C-70 s/72 �C-100 s 35 (35 cycle)
Table 2Nitric oxide (NO), malondialdehyde (MDA) and glutathione (GSH) levels in control (unexposed) and imidacloprid-treated rats.
NO MDA GSH
Plasma(lmol/L)
Brain (lmol/mgprotein)
Liver (lmol/mgprotein)
Plasma(lmol/L)
Brain (lmol/mgprotein)
Liver (lmol/mgprotein)
Brain (lmol/mgprotein)
Liver (lmol/mgprotein)
Control 38.57 ± 3.20 3.35 ± 0.26 1.32 ± 0.20 2.42 ± 0.15 7.53 ± 0.46 16.43 ± 0.21 0.37 ± 0.01 3.90 ± 0.04Imidacloprid 37.53 ± 2.24 4.35 ± 0.25*** 2.80 ± 0.007* 5.71 ± 0.60* 10.95 ± 0.39* 20.40 ± 1.00** 0.34 ± 0.004*** 4.01 ± 0.06
Values are mean ± S.E. from seven rats in each group. Control vs. imidacloprid-treated group within the same tissue.* p < 0.001.** p < 0.003.*** p < 0.05.
Table 3Antioxidant and oxidant enzyme activities in brain and liver homogenates of control (unexposed) and imidacloprid (IMI)-treated rats.
XO (U/g protein) MPO (U/g protein) CAT (k/g protein) SOD (U/mg protein) GSH-Px (U/mg protein)
Brain-control 9.4 ± 0.5 12.57 ± 0.43 0.261 ± 0.002 1.801 ± 0.009 173.28 ± 8.98Brain-IMI 13.5 ± 0.2* 12.95 ± 0.35 0.451 ± 0.005** 1.665 ± 0.002 200.39 ± 25.88
Liver-control 58 ± 1.2 9.13 ± 0.50 6.82 ± 0.21 2.38 ± 0.07 173.99 ± 4.25Liver-IMI 78 ± 3.4* 12.18 ± 0.33* 6.56 ± 0.28 2.26 ± 0.03 181.07 ± 4.95
Values are mean ± S.E. from seven rats in each group. Control vs. imidacloprid (IMI)-treated group within the same tissue, XO: xanthine oxidase, MPO: myeloperoxidase, CAT:catalase, SOD: superoxide dismutase, GSH-Px: glutathione peroxidase.* p < 0.001.** p < 0.05.
Table 4The effect of imidacloprid exposure to liver enzyme activities and plasma calcium(Ca2+) levels.
ALT AST LDH Ca2+ mg/dl
Control 37.11 ± 3.60 147.06 ± 6.48 256.79 ± 2.11 8.19Imidacloprid 46.01 ± 5.74 97.84 ± 2.73* 98.24 ± 5.21* 8.62**
Values are mean ± S.E. from seven rats in each group. Control vs. imidaclopridtreated, alanine aminotransferase (ALT), aspartate aminotransferase (AST), lactatedehydrogenase (LDH).* p < 0.001.** p < 0.005.
60 V. Duzguner, S. Erdogan / Pesticide Biochemistry and Physiology 104 (2012) 58–64
almost 2-fold in brain (p < 0.05), though unchanged in liver (Ta-ble 3). However, neither superoxide dismutase (SOD) nor glutathi-one peroxidase (GSH-Px) activities were affected by imidaclopridexposure (Table 3). A reduced antioxidant capacity in the brainwas also evidenced by the 8% decrease in intracellular GSH concen-tration (p < 0.008) (Table 2).
To investigate the possible involvement of calcium mobiliza-tion, we measured Ca2+ levels and found that imidacloprid expo-sure increased plasma Ca2+ concentration from 8.19 to 8.62 mg/dl (p < 0.05; Table 4). The exposure to imidacloprid also decreasedthe activities of liver enzymes such as aspartate aminotransferase(AST) and lactate dehydrogenase (LDH) by 1.5-, and 2.6-fold,respectively (p < 0.001), but did not change alanine aminotransfer-ase (ALT) activity (Table 4).
3.2. The effects of imidacloprid on iNOS, nNOS, eNOS and cytokinetranscription
The mRNA transcription of three isoforms of nitric oxide syn-thase enzymes (iNOS, nNOS, eNOS) was up-regulated by 4.76-,
1.13- and 3.97-fold, respectively in the brain. Inducible NOS(1.10-fold) and eNOS (1.16-fold) expressions were also enhancedin the liver by imidacloprid treatment (Fig. 1A and B). QuantitativemRNA analyses have demonstrated that imidacloprid administra-tion caused up-regulation of pro-inflammatory cytokine mRNAs
0
1
2
3
4
5
6
iNOS nNOS eNOS
rela
tive
mRN
A in
bra
in (
fold
cha
nge)
0
0,2
0,4
0,6
0,8
1
1,2
1,4
eNOSiNOS
rela
tive
mRN
A in
live
r (fo
ld c
hang
e)
Fig. 1. RT–PCR analyses of NOS in imidacloprid treated groups compared withcontrol in brain (4.76 ± 0.07; 1.13 ± 0.01; 3.97 ± 0.07, respectively in (A) and iNOS1.1 ± 0.06 and eNOS 1.16 ± 0.03 in liver in (B)).
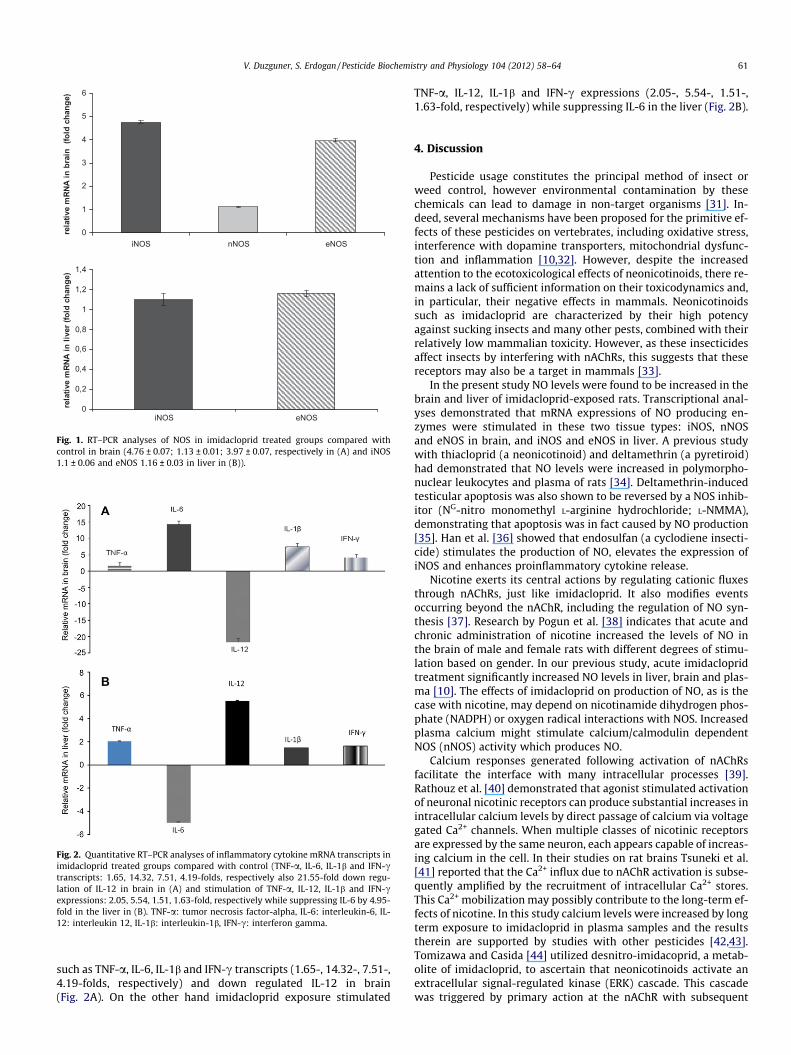
Fig. 2. Quantitative RT–PCR analyses of inflammatory cytokine mRNA transcripts inimidacloprid treated groups compared with control (TNF-a, IL-6, IL-1b and IFN-ctranscripts: 1.65, 14.32, 7.51, 4.19-folds, respectively also 21.55-fold down regu-lation of IL-12 in brain in (A) and stimulation of TNF-a, IL-12, IL-1b and IFN-cexpressions: 2.05, 5.54, 1.51, 1.63-fold, respectively while suppressing IL-6 by 4.95-fold in the liver in (B). TNF-a: tumor necrosis factor-alpha, IL-6: interleukin-6, IL-12: interleukin 12, IL-1b: interleukin-1b, IFN-c: interferon gamma.
V. Duzguner, S. Erdogan / Pesticide Biochemistry and Physiology 104 (2012) 58–64 61
such as TNF-a, IL-6, IL-1b and IFN-c transcripts (1.65-, 14.32-, 7.51-,4.19-folds, respectively) and down regulated IL-12 in brain(Fig. 2A). On the other hand imidacloprid exposure stimulated
TNF-a, IL-12, IL-1b and IFN-c expressions (2.05-, 5.54-, 1.51-,1.63-fold, respectively) while suppressing IL-6 in the liver (Fig. 2B).
4. Discussion
Pesticide usage constitutes the principal method of insect orweed control, however environmental contamination by thesechemicals can lead to damage in non-target organisms [31]. In-deed, several mechanisms have been proposed for the primitive ef-fects of these pesticides on vertebrates, including oxidative stress,interference with dopamine transporters, mitochondrial dysfunc-tion and inflammation [10,32]. However, despite the increasedattention to the ecotoxicological effects of neonicotinoids, there re-mains a lack of sufficient information on their toxicodynamics and,in particular, their negative effects in mammals. Neonicotinoidssuch as imidacloprid are characterized by their high potencyagainst sucking insects and many other pests, combined with theirrelatively low mammalian toxicity. However, as these insecticidesaffect insects by interfering with nAChRs, this suggests that thesereceptors may also be a target in mammals [33].
In the present study NO levels were found to be increased in thebrain and liver of imidacloprid-exposed rats. Transcriptional anal-yses demonstrated that mRNA expressions of NO producing en-zymes were stimulated in these two tissue types: iNOS, nNOSand eNOS in brain, and iNOS and eNOS in liver. A previous studywith thiacloprid (a neonicotinoid) and deltamethrin (a pyretiroid)had demonstrated that NO levels were increased in polymorpho-nuclear leukocytes and plasma of rats [34]. Deltamethrin-inducedtesticular apoptosis was also shown to be reversed by a NOS inhib-itor (NG-nitro monomethyl L-arginine hydrochloride; L-NMMA),demonstrating that apoptosis was in fact caused by NO production[35]. Han et al. [36] showed that endosulfan (a cyclodiene insecti-cide) stimulates the production of NO, elevates the expression ofiNOS and enhances proinflammatory cytokine release.
Nicotine exerts its central actions by regulating cationic fluxesthrough nAChRs, just like imidacloprid. It also modifies eventsoccurring beyond the nAChR, including the regulation of NO syn-thesis [37]. Research by Pogun et al. [38] indicates that acute andchronic administration of nicotine increased the levels of NO inthe brain of male and female rats with different degrees of stimu-lation based on gender. In our previous study, acute imidaclopridtreatment significantly increased NO levels in liver, brain and plas-ma [10]. The effects of imidacloprid on production of NO, as is thecase with nicotine, may depend on nicotinamide dihydrogen phos-phate (NADPH) or oxygen radical interactions with NOS. Increasedplasma calcium might stimulate calcium/calmodulin dependentNOS (nNOS) activity which produces NO.
Calcium responses generated following activation of nAChRsfacilitate the interface with many intracellular processes [39].Rathouz et al. [40] demonstrated that agonist stimulated activationof neuronal nicotinic receptors can produce substantial increases inintracellular calcium levels by direct passage of calcium via voltagegated Ca2+ channels. When multiple classes of nicotinic receptorsare expressed by the same neuron, each appears capable of increas-ing calcium in the cell. In their studies on rat brains Tsuneki et al.[41] reported that the Ca2+ influx due to nAChR activation is subse-quently amplified by the recruitment of intracellular Ca2+ stores.This Ca2+ mobilization may possibly contribute to the long-term ef-fects of nicotine. In this study calcium levels were increased by longterm exposure to imidacloprid in plasma samples and the resultstherein are supported by studies with other pesticides [42,43].Tomizawa and Casida [44] utilized desnitro-imidacoprid, a metab-olite of imidacloprid, to ascertain that neonicotinoids activate anextracellular signal-regulated kinase (ERK) cascade. This cascadewas triggered by primary action at the nAChR with subsequent

62 V. Duzguner, S. Erdogan / Pesticide Biochemistry and Physiology 104 (2012) 58–64
involvement of intracellular Ca2+ mobilization, possibly mediatedby inositol-3-phosphate. The same group has also suggested thatintracellular Ca2+ activates a sequential pathway from protein ki-nase C (PKC) to ERK. Studies indicate that stimulation of this path-way through nAChR may activate Ca2+-dependent eNOS [43] andconsequently increase production of NO [45].
Exposure to pesticides has been associated with many harmfuleffects, including: acute and chronic toxicities in human and ani-mals, liver and heart diseases, hormonal disorders, mutagenicand carcinogenic events and effects on lipid peroxidation [46].Oxygen free radicals generated due to exposure to pesticides cancause tissue damage by triggering several oxidative mechanismsand lipid peroxidation [9]. Indeed, Atessahin et al. [47] reportedthat cypermetrin leads to lipid peroxidation in brain, kidney andblood, whilst Fortunato et al. [48] showed that lipid peroxidationis increased in cerebrospinal fluid and brain samples in Malathionexposed rats. The significant increase of MDA, the index of lipidperoxidation, in brain, liver and plasma following imidaclopridtreatment is important evidence of oxidative stress in the presentstudy.
Xanthine oxidase catalyses the last steps in purine catabolism,namely the conversion of hypoxanthine to xanthine and that ofxanthine to uric acid; the byproduct of these processes is a toxicsuperoxide radical [49]. Xanthine oxidase activity and superoxideanion generation by XO have been shown to increase in the pres-ence of nicotine, an agonist of nAChR like imidacloprid [50,51]. Inaddition, various insecticides such as organophosphate poisonsare known to stimulate the generation of superoxide anions byincreasing XO activity [49,52,53].
Imidacloprid treatment increased the activity of catalase but nostatistical changes were observed in SOD and GSH-Px activity. Eventhough some studies show an increase in intracellular antioxidantsto compensate for the generation of free radicals induced by pesti-cide exposure [54], others have reported that the elevation in oxi-dant molecule production causes an inhibition of antioxidantenzyme activity [9,55]. Different studies have demonstrated thatlong term pesticide exposure leads to a significant decrease inGSH levels [56,57]. The decreased brain GSH content observed inour current study may reflect, at least partially, GSH conjugationor oxidation of GSH to glutathione disulfide (GSSG) due to the pes-ticide-induced generation of oxygen free radicals and their by-products.
Myeloperoxidase, present in the granules of neutrophils, catal-yses the production of hypochlorous acid which has bacteriocidalproperties [58]. It is reported that chronic exposure to endosul-phane causes an increase in MPO activity as a result of neutrophilactivation [59]. Gabbianelli et al. [60] have suggested that superox-ide anion levels and activation of the hydrogen peroxide-myelo-peroxidase system were increased 33- and 67-fold, respectivelyin long term permethrin-exposed rats. Based on these observationsit is suggested that the increased activity of MPO following chronicimidacloprid exposure may evidence the intensity of the oxidativeprocess and the presence of inflammation.
In the present study, the mRNA transcriptions of TNF-a, IL-6, IL-1b and IFN-c were up regulated and that of IL-12 was down regu-lated in brain. Additionally, although TNF-a, IL-1b, IL-12 and IFN-cmRNA transcriptions were enhanced in liver, IL-6 transcriptionwas inhibited. A raft of evidence exist linking exposure to pesti-cides to changes in cytokine activity. For example, Omurtag et al.[61] reported that endosulfan administration resulted in hepatox-icity related to proinflammatory cytokine expression (TNF-a andIFN-c) which in turn was increased by oxidative stress. It has alsobeen demonstrated that subcutaneous injection of dieldrin (10 mg/kg/day for 7 days), an organochloride, inhibited IL-6 and IL-12 pro-duction and c-JUN activation in a dose dependent manner [62].Diazinon treatment is reported to deplete IL-2, IL -4, IL-12 and
IFN-c synthesis in mice [63], whilst DDT metabolites trigger ROSproduction, leading to oxidative stress and enhanced activationof the NF-jB pathway [64]. Additionally, DDT induces significantproduction of TNF-a and NO in macrophages and thus contributesto inflammatory reactions, cytokine imbalance and immune-dysregulation [65]. Videla et al. [66] have proposed that lindane-induced oxidative stress in liver triggers DNA binding activity ofNF-jB, with a consequent increase in the expression of NF-jB-dependent genes for TNF-a and IL-1a, therein identifyingfactors that may mediate the hepatotoxic effect of insecticides. Inour study the observed increase of NO levels in liver and brain,and the stimulation of proinflammmatory cytokine expressionsuggests that, as with other insecticides, imidacloprid may mediateits effect through the NF-jB pathway in the chronic phase ofinflammation.
The liver plays a major role in metabolism and carries out vitalfunctions such as detoxification, so in order to evaluate hepaticdamage serum enzymes such as ALT, AST and LDH were moni-tored. It was noted that chronic imidacloprid exposure markedlyinhibited AST and LDH activities whilst ALT activity was un-changed. Supporting our findings, chlorpyrifos treatment has beenshown to decrease the activity of ALT and AST [67,68], whilst Ribe-iro et al. [69] have shown parathion treatment to decrease theactivities of LDH and acetylcholinesterase: parameters that maybe used as criteria for toxicity tests. Our results may therefore betaken as evidence of altered liver function due to imidaclopridexposure.
In conclusion, the findings obtained in the present study showthat chronic exposure to imidacloprid, which is generally acceptedas being a less toxic compound in comparison with other insecti-cides, may indeed induce oxidative stress and trigger chronicinflammation in non-target organisms.
Acknowledgments
This work was financially supported by Mustafa Kemal Univer-sity, Scientific Research Projects Committee (Project No. 08L0201).The authors thank Altug Kucukgul (Antakya, Turkey) for his techni-cal assistance. Authors wish to thank Dr. Sandra Spence for herhelpful reading of the manuscript.
References
[1] A. Elbert, R. Nauen, W. Leicht, Imidacloprid a Novel Chloronicotinyl Insecticide:Biological Activity and Agricultural Importance, in: I. Ishaaya, D. Degheele(Eds.), Insecticides with Novel Modes of Action, Mechanism, and Application,Springer, Verlag, 1998, pp. 50–73.
[2] J.N. Tasei, J. Lerin, G. Ripault, Sub-lethal effects of imidacloprid on bumblebees,Bombus terrestris (Hymenoptera: Apidae), during a laboratory feeding test, PestManage. Sci. 56 (2000) 784–788.
[3] K. Matsuda, M. Shimomura, M. Ihara, M. Akamatsu, D.B. Sattelle,Neonicotinoids show selective and diverse actions on their nicotinic receptortargets: electrophysiology, molecular biology, and receptor modeling studies,Biosci. Biotechnol. Biochem. 69 (2005) 1442–1452.
[4] D.B. Sattelle, H. Breer, Cholinergic nerve terminals in the central nervoussystem of insects: molecular aspects of structure, function and regulation, J.Neuroendocrinol. 2 (1990). 241–156.
[5] M. Shimomura, M. Yokota, M. Ihara, M. Akamatsu, D.B. Sattelle, K. Matsuda,Role in the selectivity of neonicotinoids of insect-specific basic residues in loopD of the nicotinic acetylcholine receptor agonist binding site, Mol. Pharmacol.70 (2006) 1255–1263.
[6] M. Ihara, L.A. Brown, C. Ishida, H. Okuda, D.B. Sattelle, K. Matsuda, Actions ofimidacloprid, clothianidin and related neonicotinoids on nicotinicacetylcholine receptors of American cockroach neurons and theirrelationships with insecticidal potency, J. Pestic. Sci. 31 (1) (2006) 35–40.
[7] M. Tomizawa, Neonicotinoids and derivatives: effects in mammalian cells andmice, J. Pestic. Sci. 29 (3) (2004) 177–183.
[8] V. D’Almeida, D.C. Hipólide, L.A. Azzalis, L.L. Lobo, V.B.C. Junqueira, S. Tufik,Absence of oxidative stress following paradoxical sleep deprivation in rats,Neurosci. Lett. 235 (1997) 25–28.
[9] M. Kanbur, B.C. Liman, G. Eraslan, S. Altinordulu, Effects of cypermethrin,propetamphos, and combination involving cypermethrin and propetamphoson lipid peroxidation in mice, Environ. Toxicol. 23 (4) (2008) 473–479.

V. Duzguner, S. Erdogan / Pesticide Biochemistry and Physiology 104 (2012) 58–64 63
[10] V. Duzguner, S. Erdogan, Acute oxidant and inflammatory effects ofimidacloprid on the mammalian central nervous system and liver in rats,Pest. Biochem. Physiol. 97 (2010) 13–18.
[11] M. Singh, R. Sandhir, R. Kiran, Erythrocyte antioxidant enzymes in toxilogicalevaluation of commonly used organophasphate pesticides, Indian J. Exp. Biol.44 (2006) 580–583.
[12] S. John, M. Kale, N. Rathore, D. Bhatnagar, Protective effect of vitamin E indimethoate and malathion induced oxidative stress in rat erythrocytes, J. Nutr.Biochem. 12 (2001) 500–504.
[13] A. Thapar, R. Sandhir, R. Kiran, Acephate induced oxidative stress inerythrocytes, Indian J. Exp. Biol. 40 (2002) 963–996.
[14] M.B. Grisham, D. Jourd’Heuil, D.A. Wink, Nitric Oxide 431 I. Physiologicalchemistry of nitric oxide and its metabolites: implications in inflammation,Am. J. Physiol. 433 Gastrointest. Liver Physiol. 276 (1999) G315–G321.
[15] J.F. Muniz, L. McCauley, J. Scherer, M. Lasarev, M. Koshy, Y.W. Kow, V. Nazar-Stewart, G.E. Kisby, Biomarkers of oxidative stress and DNA damage inagricultural workers: a pilot study, Toxicol. Appl. Pharmacol. 227 (2008) 97–107.
[16] E.H.B. Delgado, E.L. Streck, J.L. Quevedo, F. Dal-Pizzol, Mitochondrialrespiratory dysfunction and oxidative stress after chronic malathionexposure, Neurochem. Res. 31 (2006) 1021–1025.
[17] R.L. Miller, M. James-Kracke, G.Y. Sun, A.Y. Sun, Oxidative and inflammatorypathways in Parkinson’s disease, Neurochemistry 34 (2009) 55–65.
[18] A.M. Alluwaimi, Y. Hussein, Diazinon immunotoxicity in mice. Modulation ofcytokines level and their gene expression, Toxicology 236 (2007) 123–131.
[19] I. Ikizceli, Y. Yurumez, L. Avsarogullari, C. Kucuk, E.M. Sozuer, I. Soyuer, Y.Yavuz, S. Muhtaroglu, Effect of interleukin-10 on pancreatic damage caused byorganophosphate poisoning, Regul. Toxicol. Pharmacol. 42 (2005) 260–264.
[20] OECD Guidelines for the Testing of Chemicals/Section 4: Health Effects, TestNo. 408: Repeated Dose 90-Day Oral Toxicity Study in Rodents (1998), OECDPublishing.
[21] M. Bradford, A rapid and sensitive method for the quantification of microgramquantities of protein utilizing the principle of protein–dye binding, Anal.Biochem. 72 (1976) 248–254.
[22] T. Yoshioka, K. Kawada, T. Shimada, M. Mori, Lipid peroxidation in maternaland cord blood and protective mechanism against activated-oxygens toxicityin the blood, Am. J. Obstet. Gynecol. 135 (1979) 372–376.
[23] Y. Sun, L.W. Oberley, L.A. Ying, Simple method for clinical assay of superoxidedismutase, Clin. Chem. 34 (1988) 497–500.
[24] N.K. Cortas, N.W. Wakid, Determination of inorganic nitrate in serum andurine by a kinetic cadmium-reduction method, Clin. Chem. 36 (1990) 1440–1443.
[25] N. Prajda, G. Weber, Malignant transformation-linked imbalance: decreasedXO activity in hepatomas, FEBS Lett. 59 (1975) 245–249.
[26] P.C. Andrews, N.I. Krinski, Quantitative determination of myeloperoxidaseusing tetramethylbenzidine as substrate, Anal. Biochem. 127 (1982) 346–350.
[27] H. Luck, Catalase, in: H.U. Bergmeyer (Ed.), Methods in Enzyme Analysis,Verlag-Chemic, Weinheim/Bergstrasse, Germany, 1965.
[28] D.E. Paglia, W.N. Valentine, Studies on the quantitative and qualitativecharacterization of erythrocyte GSH-Px, J. Lab. Clin. Med. 70 (1) (1967) 158–169.
[29] J. Sedlak, R.H. Lindsay, Estimation of total, protein-bound, and nonproteinsulfhydryl groups in tissue with Ellman’s reagent, Anal. Biochem. 25 (1) (1968)192–205.
[30] M.W. Pfaffl, A new mathematical model for relative quantification in real-timeRT–PCR. Nucleic Acids Res. 1, 29(9) (2001), e45.
[31] H. Qian, W. Chen, L. Sun, Y. Jin, W. Liu, Z. Fu, Inhibitory effects of paraquat onphotosynthesis and the responseto oxidative stress in Chlorella vulgaris,Ecotoxicology 18 (2009) 537–543.
[32] A. Ascherio, H. Chen, M.G. Weisskopf, E. O’Reilly, M.L. McCullough, E.E. Calle,M.A. Schwarzschild, M.J. Thun, Pesticide exposure and risk for Parkinson’sdisease, Ann. Neurol. 60 (2006) 197–203.
[33] S.L. Chao, J.E. Casida, Interaction of imidacloprid metabolites and analogs withthe nicotinic acetylcholine receptor of mouse brain in relation to toxicity, Pest.Biochem. Physiol. 58 (1997) 77–88.
[34] B. Aydin, Effects of thiacloprid, deltamethrin and their combination onoxidative stress in lymphoid organs, polymorphonuclear leukocytes andplasma of rats, Pest. Biochem. Physiol. 100 (2011) 165–171.
[35] M. El-Gohary, W.M. Awara, S. Nassar, S. Hawas, Deltamethrin-inducedtesticular apoptosis in rats: the protective effect of nitric oxide synthaseinhibitor, Toxicology 132 (1999) 1–8.
[36] E.H. Han, Y.P. Hwang, H.G. Kim, H.G. Jeong, Inflammatory effect of endosulfanvia NF-jB activation in macrophages, Biochem. Biophys. Res. Commun. 355(2007) 860–886.
[37] B.H. Tonnessen, S.R. Severson, R.D. Hurt, V.M. Miller, Modulation of nitric-oxide synthase by nicotine, JPET 295 (2000) 601–606.
[38] S. Pogun, S. Demirgoren, D. Taskiran, L. Kanit, O. Yilmaz, E.O. Koylu, B. Balkan,E.D. London, Nicotine modulates nitric oxide in rat brain, Eur.Neuropsychopharmacol. 10 (2000) 463–472.
[39] F. Dajas-Bailador, S. Wonnacott, Nicotinic acetylcholine receptors and theregulation of neuronal signalling, Trends in Pharmacol. Sci. 25 (2004) 317–324.
[40] M.M. Rathouz, S. Vijayaraghavan, D.K. Berg, Elevation of intracellular calciumlevels in neurons by nicotinic acetylcholine receptors, Mol. Neurobiol. 12(1996) 117–131.
[41] H. Tsuneki, R. Klink, C. Léna, H. Korn, J.P. Changeux, Calcium mobilizationelicited by two types of nicotinic acetylcholine receptors in mouse substantianigra pars compacta, Eur. J. Neurosci. 12 (2000) 2475–2485.
[42] L. Imamura, M. Yasuda, K. Kuramitsu, D. Hara, A. Tabuchi, M. Tsuda,Deltamethrin, a pyrethroid insecticide, is a potent inducer for the activity-dependent gene expression of brain-derived neurotrophic factor in neurons,JPET 316 (2006) 136–143.
[43] N. Pourkhalilii, S. Pournourmohammadi, F. Rahimi, S. Vosough-Ghanbari, M.B.Seyed, Comparatıve effects of calcium channel blockers, autonomic nervoussystem blockers, and free radical scavengers on diazinon-inducedhyposecretion of insulin from isolated islets of langerhans in rats, Arh. Hig.Rada. Toksikol. 60 (2009) 157–164.
[44] M. Tomizawa, J.E. Casida, Desnitro-imidacloprid activates the extracellularsignal-regulated kinase cascade via the nicotinic receptor and intracellularcalcium mobilization in N1E–115 cells, Toxicol. Appl. Pharmacol. 184 (3)(2002) 180–186.
[45] C. Heeschen, M. Weis, A. Aicher, S. Dimmeler, J.P. Cooke, A novel angiogenicpathway mediated by non-neuronal nicotinic acetylcholine receptors, J. Clin.Invest. 110 (2002) 527–536.
[46] T.S.S. Dikshith, Toxicology of pesticides in animals, CRC Press (1991) BocaRaton, Boston.
[47] A. Atessahin, S. Yilmaz, I. Karahan, I. Pirinci, The effects of vitamin E andselenium on cypermethrin induced oxidative stress in rats, Turk. J. Vet. Anim.Sci. 29 (2005) 385–391.
[48] J.J. Fortunato, F.R. Agostinho, G.Z. Réus, F.C. Petronilho, F.D. Pizzol, J. Quevedo,Lipid peroxidative damage on malathion exposure in rats, Neurotox. Res. 9(2006) 23–28.
[49] C.E. Berry, J.M. Hare, Xanthine oxidoreductase and cardiovascular disease:molecular mechanisms and pathophysiological implications, J. Physiol. 555 (3)(2004) 589–606.
[50] A. Cormier, C. Morin, R. Zini, J.P. Tillement, G. Lagrue, Nicotine protects ratbrain mitochondria against experimental injuries, Neuropharmacology 44(2003) 642–652.
[51] U.S. Kayyali, R. Budhiraja, C.M. Pennella, S. Cooray, J.J. Lanzillo, R. Chalkley,P.M. Hassoun, Upregulation of xanthine oxidase by tobacco smokecondensate in pulmonary endothelial cells, Toxicol. Appl. Pharmacol. 188(2003) 59–68.
[52] A. Terzi, M. Iraz, S. Sahin, A. Ilhan, N Idiz, E. Fadillioglu, Protective effects oferdosteine on rotenone-induced oxidant injury in liver tissue, Toxicol. Indust.Health. 20 (6–10) (2004) 141–147.
[53] L.M. Domico, K.R. Cooper, L.P. Bernard, G.D. Zeevalk, Reactive oxygen speciesgeneration by the ethylene-bis-dithiocarbamate (EBDC) fungicide mancozeband its contribution to neuronal toxicity in mesencephalic cells, Neuro.Toxicol. 28 (2007) 1079–1091.
[54] A.E. Bayoumi, A.J. Garcia-Fernandez, C. Ordonez, Y. Perez-Pertejo, J.C. Cubria,R.M. Reguera, R. Balana-Fouce, D. Ordonez, Cyclodiene organochlorineinsecticide induced alterations in the sulfur-redox cycle in CHO-K1 cells,Comp. Biochem. Physiol. Part C. 130 (2001) 315–323.
[55] M. Panemangalore, F.N. Bebe, Dermal exposure to pesticides modifiesantioxidant enzymes in tissues of rats, J. Environ. Sci. Health B 35 (4) (2000)399–416.
[56] B.D. Banerjee, V. Seth, A. Bhattacharya, S.T. Pasha, A.K. Chakraborty,Biochemical effects of some pesticides on lipid peroxidation and free-radicalscavengers, Toxicol. Lett. 107 (1999) 33–47.
[57] R.S. Ahmed, V. Seth, S.T. Pasha, B.D. Banerjee, Influence of dietary ginger(Zingiber officinales Rosc) on oxidative stress induced by malathion in rats,Food Chem. Toxicol. 38 (2000) 443–450.
[58] M. Tokarska-Rodak, S. Tos-Luty, A. Haratym-Maj, Selected parameters ofimmunological response in hop growers during the period of intensiveapplication of pesticides, Ann. Agric. Environ. Med. 11 (2004) 227–231.
[59] F. Mor, O. Ozmen, Endosulfan-induced neurotoxicity and serumacetylcholinesterase inhibition in rabbits: The protective effect of Vit C, Pest.Biochem. Physiol. 96 (2010) 108–112.
[60] R. Gabbianelli, M.L. Falcioni, C. Nasuti, F. Cantalamessa, I. Imada, M. Inoue.Effect of permethrin insecticide on rat polymorphonuclear neutrophils, Chem.Biologic. Int. 182 (2009) 245–252.
[61] G.Z. Omurtag, A. Tozan, A.O. Sehirli, G. Sener, Melatonin protects againstsendosulfan induced oxidative tissues damage, J. Pineal Res. 44 (2008) 432–438.
[62] S.B. Pruett, R. Fan, S. Oppenheimer, Greater than additive suppression of TLR3-induced IL-6 responses by administration of dieldrin and atrazine, J.Immunotoxicol. 3 (4) (2006) 253–262.
[63] F. Diel, B. Horr, H. Borck, T. Irman-Florjanc, Pyrethroid insecticides influencethe signal transduction in T helper lymphocytes from atopic and nonatopicsubjects, Inflamm. Res. 52 (2003) 154–163.
[64] Y. Shi, Y. Song, Y. Wang, X. Liang, Y. Hu, X. Guan, J. Cheng, K. Yang, p, p-DDEInduces Apoptosis of Rat Sertoli Cells via a FasL-Dependent Pathway, J.Biomed. Biotechn. 2009 (2009) 1–11.
[65] R. Dutta, A.M. Mondal, V. Arora, T.C. Nag, N. Das, Immunomodulatory effect ofDDT (bis[4-chlorophenyl]-1,1,1-trichloroethane) on complement system andmacrophages, Toxicology 252 (2008) 78–85.
[66] L.A. Videla, G. Tapia, P. Varela, P. Cornejo, J. Guerrero, Y. Israel, V. Fernández,Effects of acute c-hexachlorocyclohexane intoxication in relation to the redoxregulation of nuclear factor-jB, cytokine gene expression, and liver injury inthe rat, Antiox. Redox Signal. 6 (2) (2004) 471–480.
[67] T. Barna-Lloyd, J.R. Szabo, N.L. Davis, Chlorpyrifos-methyl rat subchronicdietary toxicity and recovery study Unpublished report TXT: K-046193-0,31from Dow chemical, Texas, USA. Submitted to WHO by Dow Elenco, (1990),Indianapolis, USA.

64 V. Duzguner, S. Erdogan / Pesticide Biochemistry and Physiology 104 (2012) 58–64
[68] S. Ambali, D. Akanbi, N. Igbokwe, M. Shittu, M. Kawu, J. Ayo, Evaluation ofsubchronic chlorpyrifos poisoning on hematological and serum biochemicalchanges in mice and protective effect of vitamin C, J. Toxicol. Sci. 32 (2) (2007)111–120.
[69] S. Ribeiro, L. Guilhermino, J.P. Sousa, A.M.V.M. Soares, Novel bioassay based onacetylcholinesterase and lactate dehydrogenase activities to evaluate thetoxicity of chemicals to soil isopods, Ecotoxicol. Environ. Saf. 44 (3) (1999)287–293.




















