
Roles of Tumor Markers in Central Nervous System Germ Cell ...
Upload
khangminh22Category
view
0download
0
Neuroembryology and Brain Malformations
Myriam Srour MDCM, FRCP(C)
Pediatric Neurology
Academic Half Day
January 16th, 2013

Embryology
► 3% neonates have major systemic or CNS malformations
► 75% fetal deaths & 40% deaths within first year of life are associated with CNS malformations
► Most common CNS malformation involves neural tube closure and this happens by 28 days gestation, often before the woman knows she is pregnant

Major Stages of Nervous System formation
Primary neurulation 3-4 weeks
Prosencephalic development 2-3 months
Neuronal proliferation 3-4 months
Neural migration 3-5 months
Organization 5 mo – years
Myelination birth-years

NEURULATION

Neurulation
►Definition:
Formation of the neural tube which will give rise to brain and spinal cord
►The notochord (d. 16-21)
Defines longitudinal axis of embryo
Induces overlying ectoderm to form neural plate
►Occurs in dorsal aspect of embryo

Neurulation

Neurulation

► Neuropores: openings at either end
► Cranial neuropore closes before caudal
Anterior closes at ~ d 24
Posterior closes at ~ d 26 at lumbosacral level
► Caudal cord formed from a different process: secondary neurulation

►Neural tube brain and spinal cord
►Neural crest cells peripheral nervous
system (sensory ganglion cells, schwann cells)+ other ( pia, autonomic cells, melanocytes)
Neurulation

►Neural tube has 3 layers: ►ependymal
►mantle
►marginal
► Alar plate: (dorsal) sensory and coordinating
neurons
► Basal plate: (ventral) motor control neurons
Neurulation Alar plate

►Craniorachischisis totalis
►Anencephaly
►Myeloschisis
►Encephalocele
►Myelomeningocele, Arnold-Chiari malformation
Abnormalities in neurulation
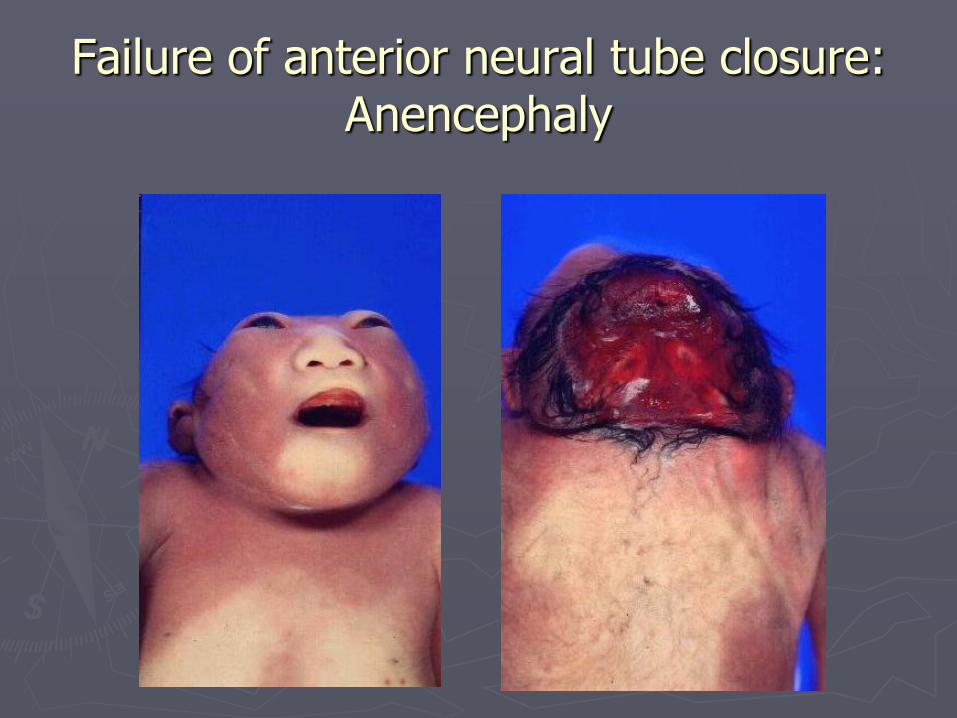
Failure of anterior neural tube closure: Anencephaly
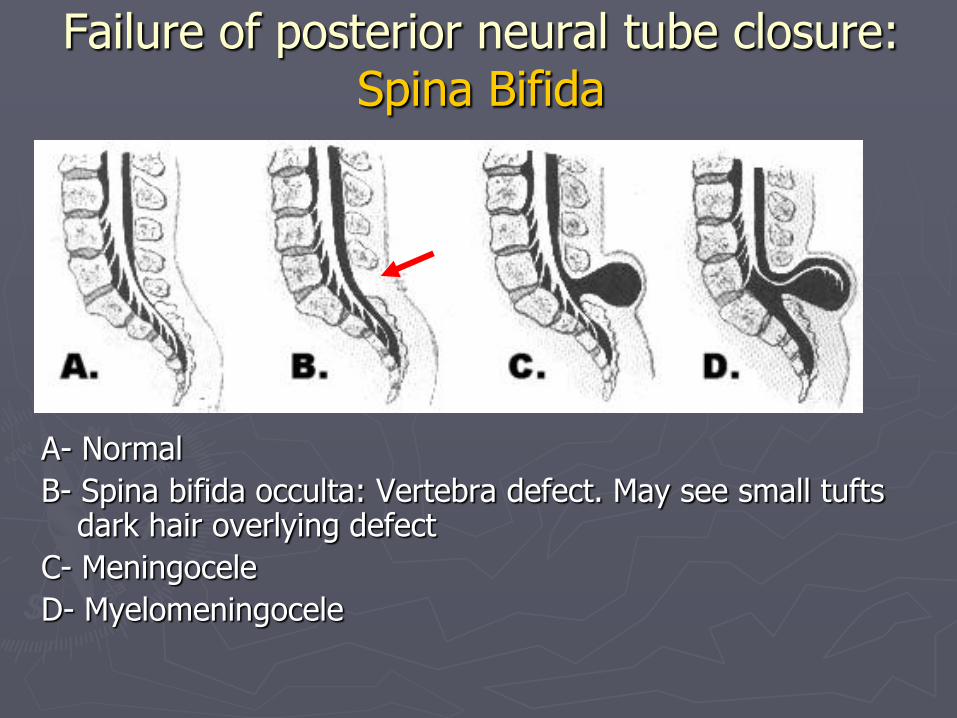
A- Normal
B- Spina bifida occulta: Vertebra defect. May see small tufts dark hair overlying defect
C- Meningocele
D- Myelomeningocele
Failure of posterior neural tube closure: Spina Bifida

Meningocele
►Sac contains meninges, NO nervous tissue

Prosencephalic Development

Prosencephalic development and Early brain structures (2-3 months)
► Following closure of anterior neuropore, there is rapid growth of neural tissue in the cranial region
► This occurs along with formation of the face Severe disorders of formation of brain development at
this time result in facial anomalies
► Inductive role of Sonic Hedgehog and Retinoic acid
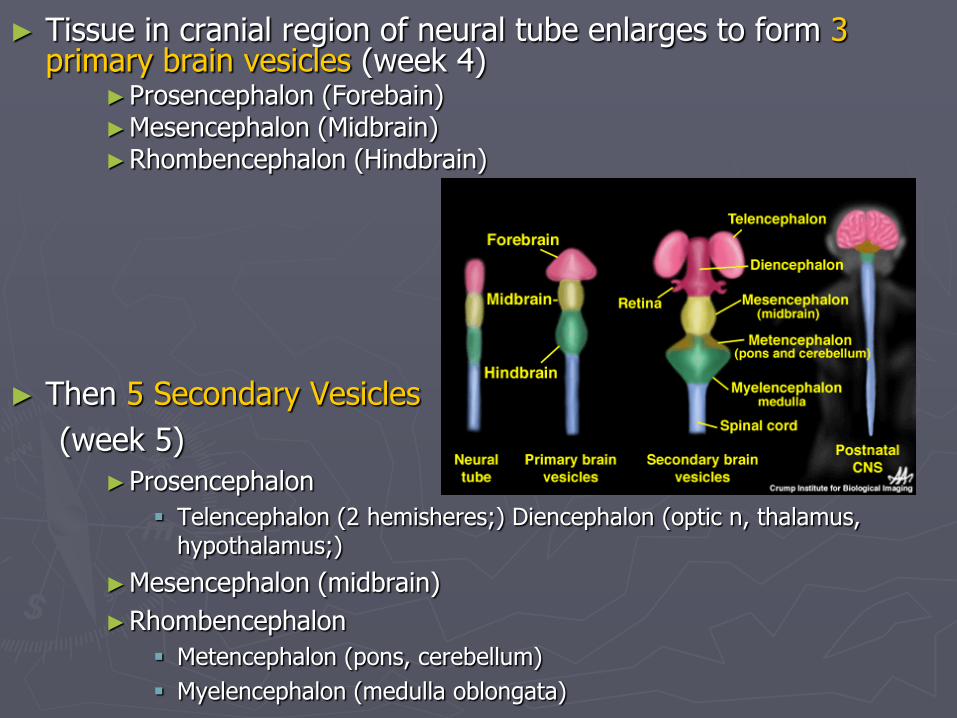
► Tissue in cranial region of neural tube enlarges to form 3 primary brain vesicles (week 4)
►Prosencephalon (Forebain) ►Mesencephalon (Midbrain) ►Rhombencephalon (Hindbrain)
► Then 5 Secondary Vesicles
(week 5)
►Prosencephalon
Telencephalon (2 hemisheres;) Diencephalon (optic n, thalamus, hypothalamus;)
►Mesencephalon (midbrain)
►Rhombencephalon
Metencephalon (pons, cerebellum)
Myelencephalon (medulla oblongata)

Prosencephalic development and Early brain structures
► Two flexures develop in the neural tube:
Cervical flexure – at rhonbencephalon/spinal cord junction
Cephalic flexure – at level of mesencephalon

Disorders of Prosencephalic development
► Prosencephalic formation Aprosencephaly Atelencephaly
► Prosencephalic Cleavage Holoprosencephaly Holotelencephaly
► Midline Prosencephalic Development Agenesis of CC Agenesis of septum pelucidum Septo-Optic dysplasia Septo-optic-hypothalamic dysplasia

Holoprosencephaly
►Failure of cleavage of telencephalon
18 weeks fetus 13 weeks
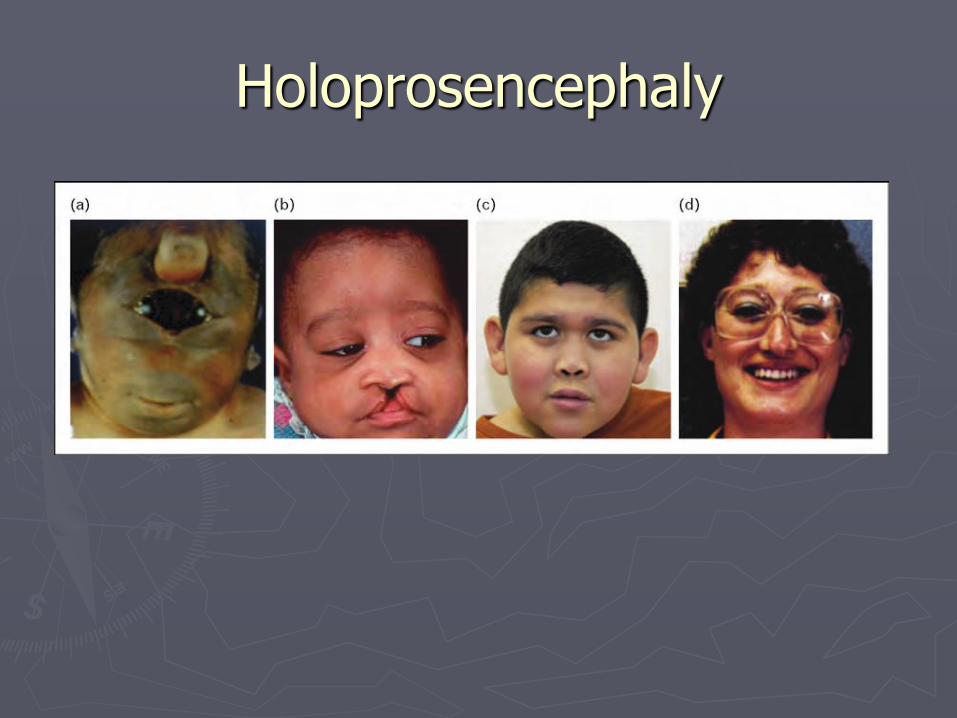
Holoprosencephaly

Holoprosencephaly (HPE)
► Continuity of the right and left hemispheres across all or part of the midline
► Associated with malformation of face and eyes
► 3 subtypes based on continuity of the hemispheres
Alobar
semilobar
lobar

► Alobar Complete failure of division of forebrain
Absent interhemispheric fissure
Single horseshoe-shaped ventricle
Undivided thalamus and basal ganglia
► Semilobar Presence of posterior interhemispheric fissure
Continuity of L and R frontal and parietal lobes
Continuity of thalamus and basal ganglia
► Lobar Most of hemispheres separate
Continuity of posterior frontal region, sometimes thalamus and basal ganglia
Continuity of the rostral corpus callosum, giving an appearance on MRI of absence of the anterior CC

Alobar Semi-lobar
Lobar

Holoprosencephaly
► Facial anomalies: Hypotelorism Cyclopsia Cebocephaly Midline cleft lip and palate Single central incisor
► “face predicts the brain” in 70-90%
► Severe handicap and death within the first months of life in
alobar HPE ► Semilobar HPE: also severe handicap ► Lobar HPE:
moderate to severe MR. Typically learn to walk and limited language
“forme fruste” normal intelligence or mild to moderate MR

Holoprosencephaly
► All patients with HPE should have an endocrine evaluation as associated with pituitary defects
► In presence of hydrocephalus, prognosis should be differed until shunt has been placed because of difficulty with identifying between alobar, semi and lobar
► Associated with maternal diabetes, retinoic acid exposure, CMV, and
rubella
Chromosmal abnormalities (trisomies 13 and 18)

Holoprosencephaly
► Genetics:
Mostly sporadic
Familial cases
►AD with incomplete penetrance and variable expressivity
►Also AR and X-linked forms
Molecular basis understood in 10% of patients
►Most common HPE genes: SHH, ZIC2, SIX3, TGIF
►Expressed inventral portion of rostral neural tube
►Role in ventral neural tube induction
Empiric recurrence to future sibs in sporadic HPE is 6%

De Morsier syndrome

Septo-optic dysplasia (de Morsier syndome)
► Characterized by 1. Absence of septum pellucidum 2. optic nerve hypoplasia 3. hypothalamic dysfunction
► For diagnosis, need 2 of the above ► Whenever one of triad discovered, look for others ► Two distinct syndromes:
1. Isolated SOD 1. Usually present with pituitary insufficiency 2. Usually mild development delay, or normal 3. Seizures uncommon
2. SOD associated with schizencephaly 1. Usually present with visual loss and neurologic abnormalities 2. Dev delay, MR, hemi or quadriplegia
► Equal frequency ► Sporadic

Neuronal Proliferation

Neuronal Proliferation
►All neurons and glia and derived from the ventricular zones
►“To-and-fro” migration
Cells from periphery of ventricular zone migrate to luminal surface and divide
The two daughter cells then migrate back to the periphery of the ventricular zone

“to and fro” migration and division

Disorders of Neuronal Proliferation
►Microcephaly Micrencephaly vera
►Macrocephaly Isolated macrocephaly
Hemimegalencephaly

Hemimegalencephaly
Hemimegalencephaly

Hemimegalencephaly ► Enlargement and dysplasia of one cerebral hemisphere
► Associated with other anatomic abnormalities: Thick cortex
Abnormal signal of white matter on T2
Heterotopias
Enlarged ventricles
► Clinical presentation:
Variable developmental delay and MR
Unilateral neurologic signs
Seizures severe and intractable
► Partial with 2y generalization
► Isolated vs. associated with neurocutaneous disorders:
Klippel-Trenaunay, epidermal nevus syndrome, hypomelanosis of Ito, Proteus syndrome…

Hemimegalencephaly
► Management
Aggressive management of epilepsy
Should consider surgical options
►Hemispherectomy
► Genetics
Somatic mosaicism with mutations in AKT gene (mTOR pathway)
No examples of familial recurrences
Low recurrence risk in siblings
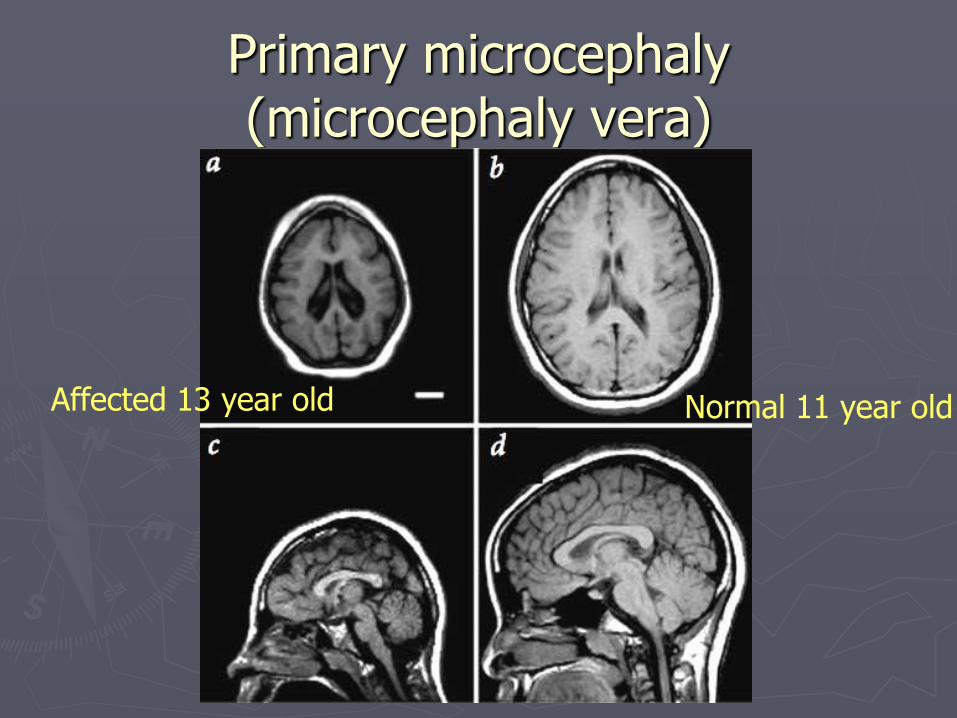
Primary microcephaly (microcephaly vera)
Affected 13 year old Normal 11 year old

Primary microcephaly (microcephaly vera)
► Defined as congenital microcephaly (HC < 2 SD) and otherwise normal brain structure Usually HC <4SD
► Clinical course and prognosis: Variable Some children only have moderate developmental delay
and moderate to severe MR
Others profound MR and spastic quadriparesis and epilepsy
► Genetics AR
X-linked

Neuronal Migration

Migration to cerebral cortex and deep nuclei
►Neuron migrate by following radial glia guides
►Early-arriving neurons take deep positions in cortex, and later arriving neurons take superficial positions
“inside-out” pattern
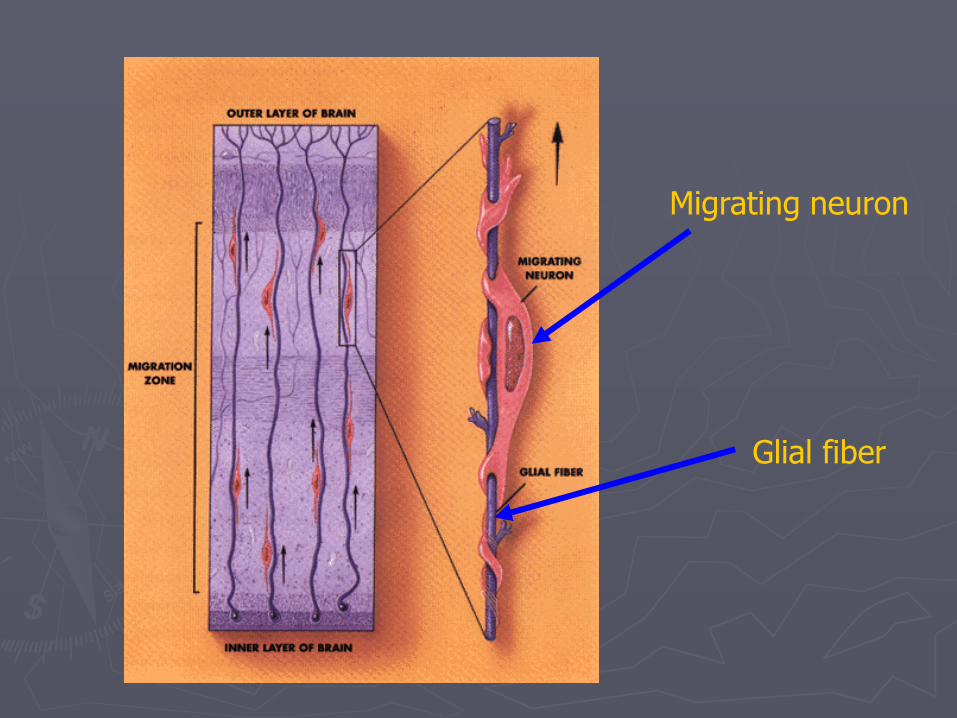
Glial fiber
Migrating neuron
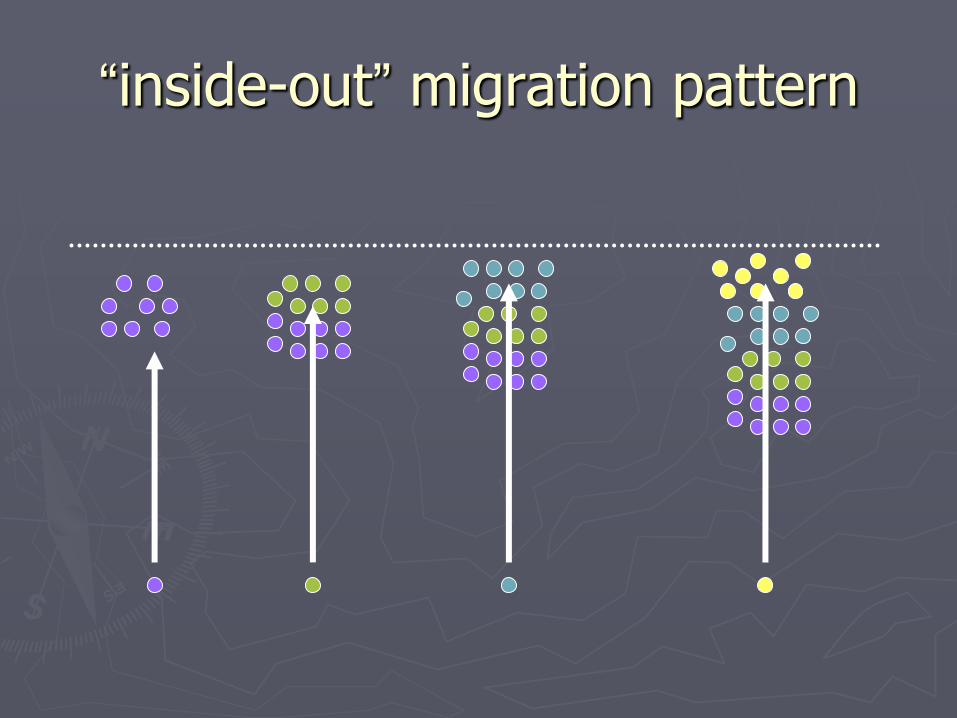
“inside-out” migration pattern

Identified key proteins
► Filamin-1 gene encodes actin-cross-linking phosphoprotein ► Key function in growth-cone extension of the migrating
neuron along radial glial cells
• LIS1 has a role in stabilizing microtubules
• DCX encodes a microtubules-associate protein expressed in migrating neuroblasts
• TUBA1A (tubulin A1A) assemble to form microtubules essentual for neuronal migration, neurite outgrowth…
► Termination of neuronal migration linked to reelin protein and mDab1 protein

Disorders of Neuronal Migration (Volpe)
►Schizencephaly
►Lissencephaly-Pachygyria
►Polymicrogyria
►Heterotopia
►Focal dysgenesis

Lissencephaly smooth brain
Brain of 18 week fetus

Lissencephaly (LIS) or agyria-pachygyria
► Lissencephaly= smooth brain ► Should be considered as an agyria-pachygyria
spectrum Subcortical band heterotopia/double cortex part of same
spectrum
► Two types: Classic lissencephaly (type 1) Cobblestone lissencephaly (type 2)
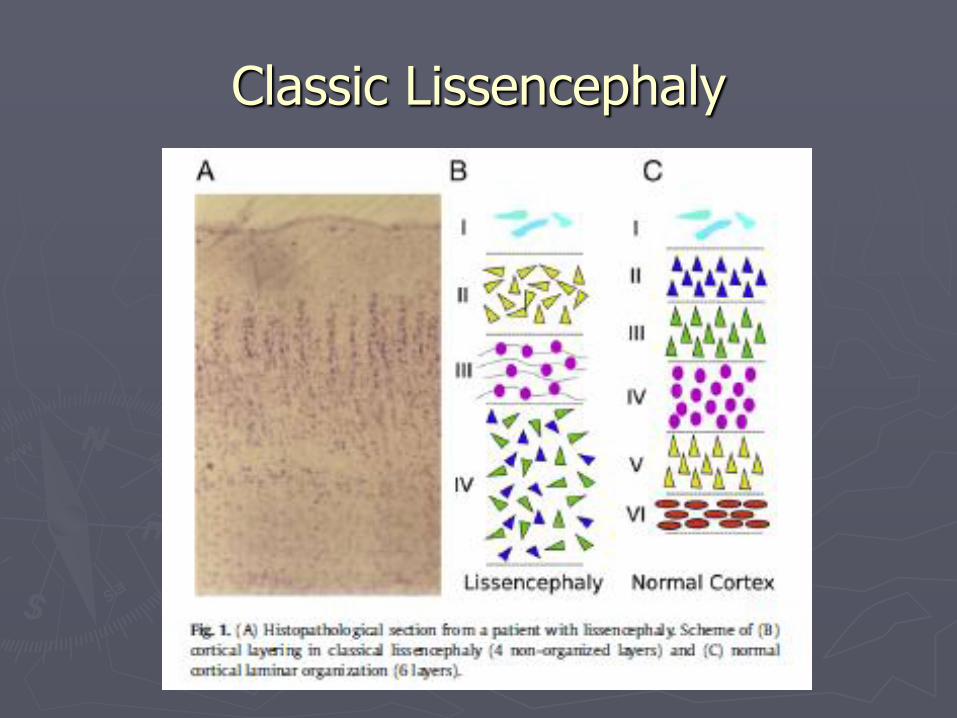
Classic Lissencephaly

Lissencephaly (LIS) or agyria-pachygyria
► Most have normal facial appearance Some with cranioafacial abnormalities Miller-Dieker syndrome
► Prominent forehead, bitemporal hallowing ► Short nose with upturned nares, prominent upper lip and small jaw
► Clinical Course of Lissencephaly Intractable seizures
► Infantile spasms early on ► Multiple seizure types
Profound MR Early hypotonia then spastic quadriparesis Gastrostomy Repeated aspiration pneumonia

Miller-Dieker syndrome

Genetics of LIS-SBH ► 6 genes account for 80%
► LIS1, DCX, ARX, RELN, TUBA1A, VLDLR
► Isolated Lissencephaly: LIS1, DCX and TUBA1A
► Can be associated with cerebellar hypoplasia (esp. TUBA1A)
► LIS1: posterior> anterior gradient
► DCX: anterior>posterior gradient
► All patients with Miller-Dieker have large deletions of 17p13.3, which includes LIS1 70% cytogenetically visible
► In patients with classical LIS but no other anomalies: 40% microdeletions of 17p13.3
24% intragenic mutations of LIS1
12% intragenic mutation of DCX
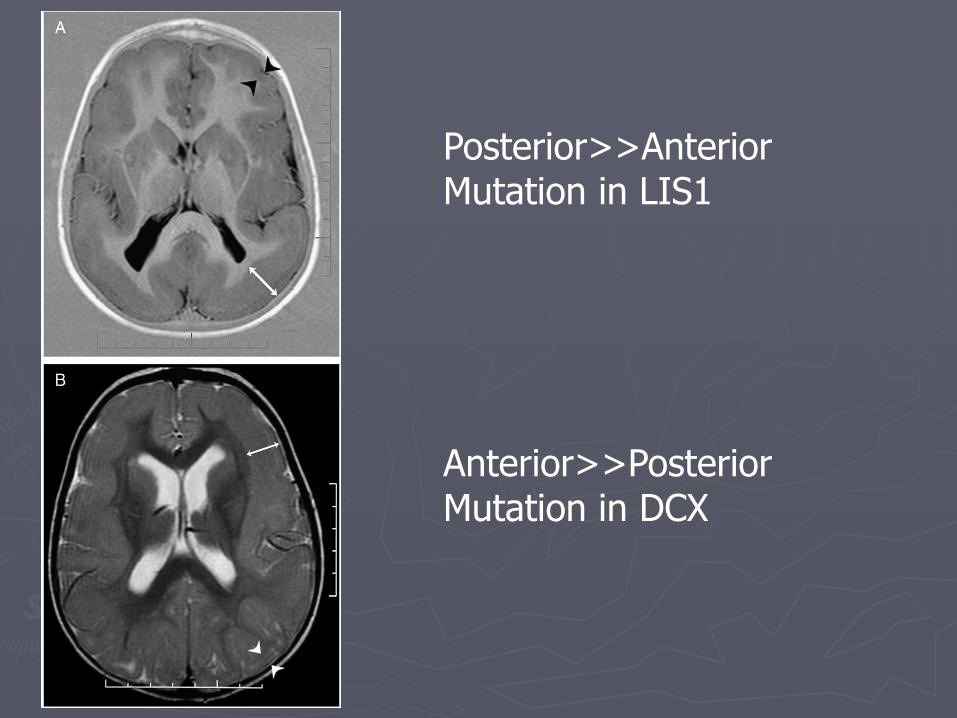
Posterior>>Anterior Mutation in LIS1
Anterior>>Posterior Mutation in DCX

►RELN: Lissencephaly with cerebellar hypoplasia
►ARX: X-linked lissencephaly with abnormal genitalia

Subcortical band heterotopia

Subcortical Band Heterotopia
►Course and prognosis
Variable
Severe MR to normal
►Most mild to moderate MR
Variable seizure frequency and severity
Neurologic outcome depends in part on thickness of the heterotopic band
►Genetics
Mutations in DCX in 100% of girls with family history, 80% sporadic females and 25% males
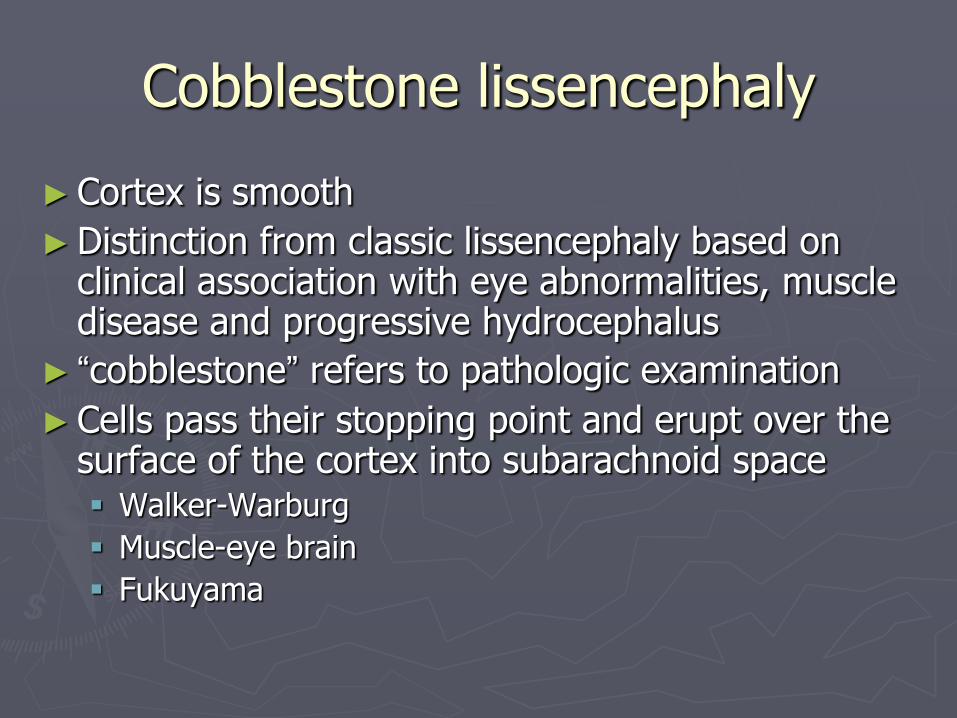
Cobblestone lissencephaly
► Cortex is smooth
► Distinction from classic lissencephaly based on clinical association with eye abnormalities, muscle disease and progressive hydrocephalus
► “cobblestone” refers to pathologic examination
► Cells pass their stopping point and erupt over the surface of the cortex into subarachnoid space Walker-Warburg
Muscle-eye brain
Fukuyama

Walker Warburg syndrome

Periventricular heterotopias
+ corpus callosum agenesis
+ bilateral periventricular heterotopia

Periventricular Heterotopia ► Nodular or diffuse ► Contiguous bilateral periventricular nodular heterotopia
(BPNH) FLNA: Filamin A Large actin-binding phosphoprotein that stablizes the cytoskeleton Failure of the neuron to attach to radial glia X-linked Explain almost all females with + family history and 25% of
sporadic females Explain approx 7% males Other associated abnormalities: coagulopathy and cardiovascular
abnormalities
► Most patients with nodular heterotopias are of normal intelligence Subcortical heterotopias tend to have lower IQ
► Seizures ► PNH with severe congenital microcephaly: ARGEF2

Disorders associated with Neuronal Heterotopias
► X-linked disorders- BPNH and X-linked double cortex ► Metabolic disorders- neonatal ALD, glutaric aciduria type II,
non-ketotic hyperglycinemia, Leigh disease, Menkes, GM2-gangliosidosis, Hurler disease
► Myotonic dystrophy ► Neurocutaneous syndromes- NF, TS, incontinentia
pigmenti, Ito’s hypomelanosis, linear nevus sebaceus
► Multiple congenital anomaly syndromes- Smith-Lemli-Opitz, de Lange, Potter
► Chromosomal syndromes- trisomy 18, trisomy 13, deletion 4p
► Fetal toxic exposures- carbon monoxide, isotretinoic acid, ethanol, organic mercurial

Schizencephaly

Schizencephaly-Polymicrogyria complex
► Disorder of migration “Malformations due to abnormal cortical organization (including late
neuronal migration)”, under Barkovich classification
► Deep clefts lined by polymicrogyria from the pial surface to the ependymal surface of the lateral ventricle
► Open-lip and closed-lip ► Unilateral in 63% ► There is NO reported case of schizencephaly WITHOUT
polymicrogyria Grey matter lining clefts help differentiate from porencephalic cysts
► Other causes: TORCH infections, especially CMV Early vascular event

Schizencephaly
Clinical Developmental delay and MR
►In 2 series, cognitive disturbances only in 24% of unilateral lesions
Spastic hemi or quadriparesis
Epilepsy
Distribution and severity related to size and location of clefts
Open-lipped and bilateral clefts are more severe

Schizencephaly
Genetics Sporadic
Some familial forms
EMX2 homeobox-containing gene ►Expressed in neuroblasts of the ventricular zone
Disorder of segmentation vs migration?

Polymicrogyria
► Cerebral cortex with multiple excessive small convolutions
► Cortex appears thickened – but microscopically, is not truly thickened
► May be misdiagnosed as pachygyria
► Classified based on distribution and bilaterality
Bilateral perisylvian –> pseudobulbar, speech delay, ID
Unilateral perisylvian
Generalized
Frontal
Parasagital parieto-occipital

Unilateral perisylvian syndrome Bilateral perisylvian dysplasia

Focal cortical dysplasias (FCD)
►Key feature is presence of abnormal cortical lamination
►Classified based on absence (type I) or presence (type II) of balloon cells
►FCDI: may not be detectable on MRI
►FCDII: increased cortical thickness, blurring of grey-white junction, abnormal sulcatiom, high T2 or FLAIR signal at base of the lesion

Organization

Organization
►Lamination: alignment, orientation and layering of cortical neurons
►Neurite outgrowth: dendritic and axonal ramification
►Synaptogenesis
►Cell death and selective elimination of neural processes and synapses

Disorders of Organization
►Primary disturbance Mental Retardation
Down Syndrome
Angelman syndrome
Infantile Autism
►Potential disturbance Premature infants
Nutrition
Other perinatal and postnatal insults

2012 Developmental and genetic classification of malformations of cortical debelopment-
Barkovich et al, Brain 2012
► Group 1: due to abnormal neuronal and glial proliferation or apoptosis
Microcephalies
Megalencephalies
Cortical dysgeneses with abnormal cell proliferation
► Group 2: due to abnormal neuronal migration
Heterotopias
Lissencephaly
Subcortical heterotopias
Cobblestone malformations
► Group 3: due to abnormal post-migrational development
Polymicrogyria and schizencephaly
Focal cortical dysplasias

Questions?

►http://laxmi.nuc.ucla.edu:8888/Teachers/pphelps//Published_Trays/PS107-Fall98-Lec2/Pindex.html
Copyright © 2022 FDOKUMEN




















