
Role of metallothionein-III following central nervous system damage
15
Role of metallothionein-III following central nervous system damage Javier Carrasco, a Milena Penkowa, b Mercedes Giralt, a Jordi Camats, a Amalia Molinero, a Iain L. Campbell, c Richard D. Palmiter, d and Juan Hidalgo a, * a Institute of Neurosciences and Department of Cellular Biology, Physiology and Immunology, Animal Physiology Unit, Faculty of Sciences, Autonomous University of Barcelona, Bellaterra, Barcelona, Spain 08193 b Department of Medical Anatomy, The Panum Institute, University of Copenhagen, DK-2200 Copenhagen, Denmark c Department of Neuropharmacology, The Scripps Research Institute, La Jolla, CA 92037, USA d Howard Hughes Medical Institute, Department of Biochemistry, University of Washington, Seattle, WA 98195, USA Received 18 April 2002; revised 29 November 2002; accepted 6 January 2003 Abstract We evaluated the physiological relevance of metallothionein-III (MT-III) in the central nervous system following damage caused by a focal cryolesion onto the cortex by studying Mt3-null mice. In normal mice, dramatic astrogliosis and microgliosis and T-cell infiltration were observed in the area surrounding the lesioned tissue, along with signs of increased oxidative stress and apoptosis. There was also significant upregulation of cytokines/growth factors such as tumor necrosis factor-, interleukin (IL)-1 /, and IL-6 as measured by ribonuclease protection assay. Mt3-null mice did not differ from control mice in these responses, in sharp contrast to results obtained in Mt1- Mt2-null mice. In contrast, Mt3-null mice showed increased expression of several neurotrophins as well as of the neuronal sprouting factor GAP-43. Thus, unlike MT-I and MT-II, MT-III does not affect the inflammatory response elicited in the central nervous system by a cryoinjury, nor does it serve an important antioxidant role, but it may influence neuronal regeneration during the recovery process. © 2003 Elsevier Science (USA). All rights reserved. Introduction Traumatic injuries of the central nervous system (CNS) initiate a complex sequence of pathophysiological responses at the lesion site. In an early phase the disruption of the blood vessels causes an extravasation of blood into the lesion site, filling it with cells that initiate a characteristic inflammatory response. Local ischemia and hypoxia along with release of excitatory amino acids causes necrosis of local neurons and glial cells and degeneration of fibers passing through the area. Following these early responses, resident microglia and infiltrating monocytes/macrophages are activated and transform into ameboid brain macro- phages. Lymphocytes massively infiltrate the injured area, and local endothelial cells migrate and proliferate into the injured area. Astrocytes are also activated after CNS injury. The recruitment and activation of all these cells limits neu- ronal degeneration and spreading of the lesion and ulti- mately results in a glial scar surrounding the injured area. It is generally accepted that this inflammatory response is orchestrated by the secretion of several cytokines and che- mokines by microglia and macrophages, which stimulate glia and neurons their own cytokines, growth factors, and neurotrophins (for reviews, see Giulian et al., 1989; Ed- dleston and Mucke, 1993; Mattson and Scheff, 1994; Hop- kins and Rothwell, 1995; Perry et al., 1995; Rothwell and Hopkins, 1995; Merrill and Benveniste, 1996; Ridet et al., 1997; McIntosh et al., 1998; Mun ˜oz-Ferna ´ndez and Fresno, 1998; Stichel and Verner Mu ¨ ller, 1998; Hughes et al., 1999; Horner and Gage, 2000; Allan and Rothwell, 2001; Aloisi, 2001; Dong and Benveniste, 2001). Evidence supporting a role for oxidative stress in the brain following tissue injury is compelling. The brain is especially prone to damage by oxygen radicals because its membranes are rich in polyunsaturated fatty acids, it pro- duces significant amounts on nitric oxide, has poor catalase * Corresponding author. Departamento de Biologia Celular, Fisiologia e Inmunologia, Unidad de Fisiologia Animal, Facultad de Ciencias, Uni- versidad Auto ´ noma de Barcelona, Bellaterra, Barcelona, Spain 08193. Fax: 34-93-581-23-90. E-mail address: [email protected] (J. Hidalgo). R Available online at www.sciencedirect.com Neurobiology of Disease 13 (2003) 22–36 www.elsevier.com/locate/ynbdi 0969-9961/03/$ – see front matter © 2003 Elsevier Science (USA). All rights reserved. doi:10.1016/S0969-9961(03)00015-9
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Role of metallothionein-III following central nervous system damage

Role of metallothionein-III following central nervous system damage
Javier Carrasco,a Milena Penkowa,b Mercedes Giralt,a Jordi Camats,a Amalia Molinero,a
Iain L. Campbell,c Richard D. Palmiter,d and Juan Hidalgoa,*a Institute of Neurosciences and Department of Cellular Biology, Physiology and Immunology, Animal Physiology Unit, Faculty of Sciences,
Autonomous University of Barcelona, Bellaterra, Barcelona, Spain 08193b Department of Medical Anatomy, The Panum Institute, University of Copenhagen, DK-2200 Copenhagen, Denmark
c Department of Neuropharmacology, The Scripps Research Institute, La Jolla, CA 92037, USAd Howard Hughes Medical Institute, Department of Biochemistry, University of Washington, Seattle, WA 98195, USA
Received 18 April 2002; revised 29 November 2002; accepted 6 January 2003
Abstract
We evaluated the physiological relevance of metallothionein-III (MT-III) in the central nervous system following damage caused by afocal cryolesion onto the cortex by studying Mt3-null mice. In normal mice, dramatic astrogliosis and microgliosis and T-cell infiltrationwere observed in the area surrounding the lesioned tissue, along with signs of increased oxidative stress and apoptosis. There was alsosignificant upregulation of cytokines/growth factors such as tumor necrosis factor-�, interleukin (IL)-1 �/�, and IL-6 as measured byribonuclease protection assay. Mt3-null mice did not differ from control mice in these responses, in sharp contrast to results obtained in Mt1-Mt2-null mice. In contrast, Mt3-null mice showed increased expression of several neurotrophins as well as of the neuronal sprouting factorGAP-43. Thus, unlike MT-I and MT-II, MT-III does not affect the inflammatory response elicited in the central nervous system by acryoinjury, nor does it serve an important antioxidant role, but it may influence neuronal regeneration during the recovery process.© 2003 Elsevier Science (USA). All rights reserved.
Introduction
Traumatic injuries of the central nervous system (CNS)initiate a complex sequence of pathophysiological responsesat the lesion site. In an early phase the disruption of theblood vessels causes an extravasation of blood into thelesion site, filling it with cells that initiate a characteristicinflammatory response. Local ischemia and hypoxia alongwith release of excitatory amino acids causes necrosis oflocal neurons and glial cells and degeneration of fiberspassing through the area. Following these early responses,resident microglia and infiltrating monocytes/macrophagesare activated and transform into ameboid brain macro-phages. Lymphocytes massively infiltrate the injured area,and local endothelial cells migrate and proliferate into the
injured area. Astrocytes are also activated after CNS injury.The recruitment and activation of all these cells limits neu-ronal degeneration and spreading of the lesion and ulti-mately results in a glial scar surrounding the injured area. Itis generally accepted that this inflammatory response isorchestrated by the secretion of several cytokines and che-mokines by microglia and macrophages, which stimulateglia and neurons their own cytokines, growth factors, andneurotrophins (for reviews, see Giulian et al., 1989; Ed-dleston and Mucke, 1993; Mattson and Scheff, 1994; Hop-kins and Rothwell, 1995; Perry et al., 1995; Rothwell andHopkins, 1995; Merrill and Benveniste, 1996; Ridet et al.,1997; McIntosh et al., 1998; Munoz-Fernandez and Fresno,1998; Stichel and Verner Muller, 1998; Hughes et al., 1999;Horner and Gage, 2000; Allan and Rothwell, 2001; Aloisi,2001; Dong and Benveniste, 2001).
Evidence supporting a role for oxidative stress in thebrain following tissue injury is compelling. The brain isespecially prone to damage by oxygen radicals because itsmembranes are rich in polyunsaturated fatty acids, it pro-duces significant amounts on nitric oxide, has poor catalase
* Corresponding author. Departamento de Biologia Celular, Fisiologiae Inmunologia, Unidad de Fisiologia Animal, Facultad de Ciencias, Uni-versidad Autonoma de Barcelona, Bellaterra, Barcelona, Spain 08193. Fax:�34-93-581-23-90.
E-mail address: [email protected] (J. Hidalgo).
R
Available online at www.sciencedirect.com
Neurobiology of Disease 13 (2003) 22–36 www.elsevier.com/locate/ynbdi
0969-9961/03/$ – see front matter © 2003 Elsevier Science (USA). All rights reserved.doi:10.1016/S0969-9961(03)00015-9

and only moderate superoxide dismutase and glutathioneperoxidase activities, and it is rich in iron, which can bereleased from dying cells and generate the highly reactivehydroxyl radical by the Fenton reaction. When perfusion isreestablished, there is an imbalance between antioxidantsystems and free radical formation and thus oxidative stress.Cells protect themselves from oxidative stress by producingthe antioxidant enzymes mentioned above, free radicalscavengers such as glutathione, vitamin E, and �-carotene,as well as proteins that bind transition metals such as ironand copper (for reviews, see Halliwell, 1992; Coyle andPuttfarcken, 1993; Olanow, 1993; Mattson and Scheff,1994; Merrill and Benveniste, 1996; Heales et al., 1999;Lipton, 1999; Pentreath and Slamon, 2000).
The metallothionein (MT) family of proteins may helpprotect cells from oxidative damage. MTs are small, cys-teine-rich proteins that bind heavy metals such as zinc andcopper with high affinity. Four, closely linked Mt genes arepresent in rodents (Palmiter et al., 1992; Quaife et al., 1994),but multiple Mt1 gene variants are present in ungulates andprimates. Mt1 and Mt2 (Mt1 & 2) are expressed coordi-nately in most tissues including the CNS (Yagle andPalmiter, 1985; van Lookeren Campagne et al., 2000); theyare transcriptionally activated by inflammatory stimuli, glu-cocorticoid hormones, and heavy metals. Mt1&2 are pref-erentially expressed in astrocytes and activated microglia(Young et al., 1991; Blaauwgeers et al., 1993; Nakajima andSuzuki, 1995; Holloway et al., 1997; Vela et al., 1997;Acarin et al., 1999b; Penkowa et al., 1999a). In contrast,Mt3 is expressed predominantly in neurons and this gene isrelatively unresponsive to cytokines, hormones, and metals(Masters et al., 1994b; Carrasco et al., 1998a,b, 2000a;Uchida, 1999; Giralt et al., 2001). However, Mt3 is alsoexpressed in astrocytes under some conditions (Uchida etal., 1991; Tsuji et al., 1992; Kobayashi et al., 1993; Masterset al., 1994b). Mt4 is not expressed in the brain (Quaife etal., 1994). Our previous work has demonstrated that Mt1 &2 genes protect the CNS in response to injury. We havecharacterized in considerable detail the role of MT-I&IIfollowing a cryolesion of the parietal cortex, an establishedexperimental neurotrauma model that features a standard-ized and highly reproducible focal disruption of the blood-brain barrier and massive brain edema, gliosis, recruitmentof inflammatory cells, and upregulation of a number ofgenes attempting to increase neuronal survival and promot-ing the regeneration of brain parenchyma (Chan et al., 1991;Cook et al., 1998; Knerlich et al., 1999; Murakami et al.,1999). Mt1&2 mRNAs are induced in response to cryoin-jury and Mt1&2-null mice manifest poor recovery followinginjury. The inflammatory response is prolonged in theknockout (KO) mice and oxidative stress and apoptosis areenhanced (Penkowa et al., 1999a, 2000, 2001b; Giralt et al.,2002b). These effects are reversed in transgenic mice over-expressing Mt1, and in mice injected with MT-II protein(Giralt et al., 2002b). Mt1&2 genes also provide protectionin other models of CNS injury such as experimental auto-
immune encephalomyelitis (Penkowa and Hidalgo, 2000,2001; Penkowa et al., 2001a), kainic acid-induced seizures(Carrasco et al., 2000b), 6-aminonicotinamide administra-tion (Penkowa et al., 1999b, 2002), transgenic mice with afamilial amyotrophic lateral sclerosis-linked mutation of theSODI gene (Nagano et al., 2001) and in the transgenicinterleukin (IL)-6-induced neuropathology (Giralt et al.,2002a). The function of MT-III, which is preferentiallyexpressed in neurons, is less clear. Mt3 mRNA levelschange in response to CNS injury, including stab wounds,cortical ablation, intracerebral kainic acid and NMDA ad-ministration, facial nerve transection or middle cerebralartery occlusion-induced ischemia (Anezaki et al., 1995;Hozumi et al., 1995, 1996; Yuguchi et al., 1995a, b; Inuzukaet al., 1996; Yamada et al., 1996; Acarin et al., 1999a;Penkowa et al., 2000). Depending on the injury model, Mt3mRNA levels increase, decrease or do both a different timesafter injury. However, it is not clear from these studieswhether MT-III protein is directly involved in protectingneurons from injury. The aim of the present study was tosubject Mt3-null mice to a cortical cryolesion and comparetheir responses to that of wild-type (WT) and Mt1&2-nullmice.
Materials and methods
Animals
Homozygous Mt1&2-null and Mt3-null mice and con-trol, wild-type (WT) mice were all maintained on inbred129/Sv genetic background (Masters et al., 1994a; Ericksonet al., 1997). All mice were kept under constant temperatureand had free access to food and water. All experiments werecarried out in a humane manner and were approved by theComissio d’Experimentacio Animal del Departamentd’Agricultura, Ramaderia i Pesca de la Generalitat de Cata-lunya.
Cryolesion procedure
Adult mice in the different experiments were lesionedunder tribromethanol anesthesia. The skull over the rightfrontoparietal cortex was exposed, and a focal cryoinjurywas produced on the surface of the brain by applying dry icefor 60 s (Penkowa and Moos, 1995). The animals werereturned to the animal room, and were killed from 1 to 14days after the cryolesion. Although the results will be pre-sented together, several separate experiments were per-formed. The brains were prepared for analysis followingdifferent procedures depending on whether they were for insitu hybridization and northern blot or for histochemistry-immunohistochemistry studies (see below).
23J. Carrasco et al. / Neurobiology of Disease 13 (2003) 22–36
In situ hybridization and northern blot
The mice were killed and their brains removed, frozen inliquid nitrogen, and stored at �80°C. Later, serial coronalsections (20 �m in thickness) were obtained with a cryostatand mounted on slides coated with poly(L)lysine. The in situhybridization analysis of the Mt1 and Mt3 isoforms wascarried out as previously described (Carrasco et al., 1998b).All sections to be compared were prepared simultaneouslyand exposed to the same autoradiographic film. The north-ern blot procedure for neurotrophins and GAP-43 was asdescribed previously (Hernandez et al., 2000). The GAP-43cDNA was kindly provided by Dr. Juan Carlos de la Torre,The Scripps Research Institute, La Jolla, CA.
Histochemistry and immunohistochemistry
Tissue processingMice were deeply anesthetized with 100 mg/kg of body
weight of Brietal (Methohexital 10 mg/ml, Lilly ResearchLaboratories, Lyngby, Denmark) and were transcardially per-fused with 0.9% saline with 0.3% heparin (15000 IU/liter) for3–5 min followed by perfusion with 0.1% Na2S in 0.1 Mphosphate-buffered saline (PBS), pH 7.4, for 5 min, followedby perfusion with Zamboni’s fixative for 8–10 min, pH 7.4.Afterwards, all the brains were fixed by immersion in Zam-boni’s fixative for 4 h, pH 7.4, at room temperature. Brainswere dehydrated according to standard procedures, embeddedin paraffin, and cut in serial, coronal 3-�m-thick sections to be
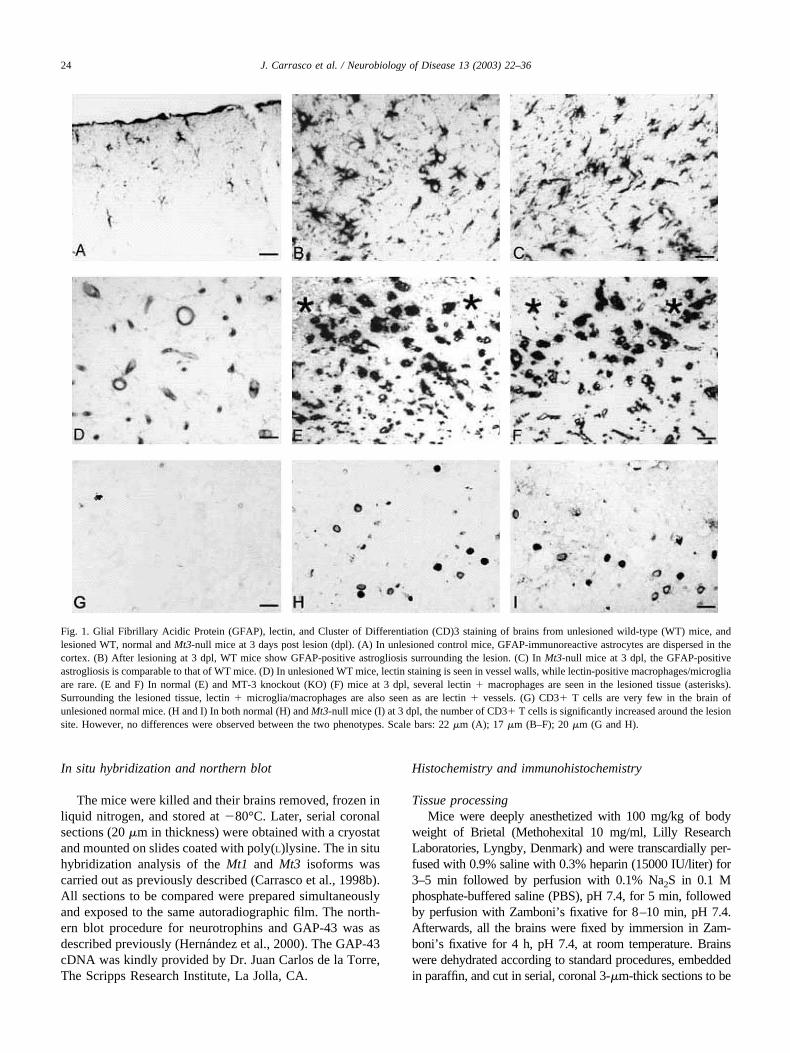
Fig. 1. Glial Fibrillary Acidic Protein (GFAP), lectin, and Cluster of Differentiation (CD)3 staining of brains from unlesioned wild-type (WT) mice, andlesioned WT, normal and Mt3-null mice at 3 days post lesion (dpl). (A) In unlesioned control mice, GFAP-immunoreactive astrocytes are dispersed in thecortex. (B) After lesioning at 3 dpl, WT mice show GFAP-positive astrogliosis surrounding the lesion. (C) In Mt3-null mice at 3 dpl, the GFAP-positiveastrogliosis is comparable to that of WT mice. (D) In unlesioned WT mice, lectin staining is seen in vessel walls, while lectin-positive macrophages/microgliaare rare. (E and F) In normal (E) and MT-3 knockout (KO) (F) mice at 3 dpl, several lectin � macrophages are seen in the lesioned tissue (asterisks).Surrounding the lesioned tissue, lectin � microglia/macrophages are also seen as are lectin � vessels. (G) CD3� T cells are very few in the brain ofunlesioned normal mice. (H and I) In both normal (H) and Mt3-null mice (I) at 3 dpl, the number of CD3� T cells is significantly increased around the lesionsite. However, no differences were observed between the two phenotypes. Scale bars: 22 �m (A); 17 �m (B–F); 20 �m (G and H).
24 J. Carrasco et al. / Neurobiology of Disease 13 (2003) 22–36
used for hematoxylin-eosin (H&E), lectin histochemistry, im-munohistochemistry, or in situ detection of DNA fragmenta-tion/TUNEL labeling. Brains of unlesioned mice were pro-cessed along with those of lesioned animals.
HistochemistryH&E staining was performed according to standard proce-
dures. Other sections were incubated overnight at 4°C withbiotinylated tomato lectin from Lycopersicon esculentum (Sig-ma, St. Louis, MO, USA; code L9389), which was diluted1:500 in 10% goat serum and used as a marker for all cells ofthe myelomonocytic cell lineage, such as ramified and plumpmicroglial cells and macrophages. Labeling by lectin was ver-ified using streptavidin-biotin-peroxidase complex (StreptAB-Complex/HRP, DakoCytomation, Glostrup, Denmark; codeK377) prepared at manufacturer’s recommended dilution andincubated for 30 min at room temperature. The immunoreac-tion was visualized using 0.015% H2O2 in 3,3-diaminobenzi-dine-tetrahydrochloride (DAB)/Tris-buffered saline, (TBS: 0.5M Tris, pH 7.4, 0.15 M NaCl; with 0.01% Nonidet p-40)(TBS) (Sigma-Aldrich, St. Louis, MO, USA, code N-6507) for10 min at room temperature.
ImmunohistochemistrySections were incubated overnight at 4°C with one of the
following primary antibodies: rabbit anti-cow Glial FabrillaryAcidic Protein (GFAP) (as a marker for astrocytes), 1:250(DakoCytomation, DK; code Z 334); mouse anti-rat CD-3 (asa marker for all T lymphocytes), 1:50 (Serotec, Oxford, OX5,UK; code KD MCA 772); rabbit anti-MT-I�II, 1:500 (Gasullet al., 1993, 1994); rabbit anti-NITT (a marker for peroxyni-trite-induced nitration of tyrosine residues, which monitorsoxidative stress), 1:100 (Alpha Diagnostic Int., San Antonio,TX, USA; code NITT 12-A); rabbit anti-MDA (a marker formalondialdehyde produced as a byproduct of fatty acid per-oxidation, which also monitors oxidative stress), 1:100 (AlphaDiagnostic Int., USA; code MDA 11-S); goat anti-humanNT-3, 1:20 (RD Systems, Minneapolis, MN, USA; codeAF-267-NA); rabbit anti-human NT-4/5, 1:50 (Santa Cruz,Santa Cruz, CA, USA; code sc-545); mouse anti-rat phosphor-ylated GAP-43, 1:100 (Calbiochem, San Diego, CA, USA;code CP09).
The methods for detecting primary and secondary antibod-ies as well as for examining nonspecific binding have beenpublished previously (Carrasco et al., 2000b; Penkowa et al.,1999a, 2000, 2001a; Giralt et al., 2002a, b). Additionally, somecontrol sections were incubated with isotypic IgG instead ofthe primary antibody. For this purpose, sections were incu-bated overnight with normal mouse IgG (Santa Cruz, USA;code sc-2025) or with normal rabbit IgG (Santa Cruz, USA;code sc-2027) followed by the steps mentioned above.
Triple immunostainingsTo determine which cells contain MT-I�II, sections were
incubated overnight at 4°C with mouse anti-porcine vimentin
1:50 (Dakopatts, DK; code M0725) (as a marker for reactiveastrocytes and macrophages), and goat anti-human NeuroFila-ment (NF) 1:50 (Santa Cruz, USA; code sc-12980) (as aneuronal marker), and rabbit anti-rat MT-I�II simultaneously.The anti-vimentin antibodies were detected by using donkeyanti-mouse IgG linked with fluorescein (FITC) 1:20 (JacksonImmunoResearch Lab. Inc., West Grove, PA, USA; code 715-095-151), while anti-NF antibodies were detected by usingdonkey anti-goat IgG linked with aminomethylcoumarin(AMCA) 1:30 (Jackson ImmunoResearch Lab. Inc.; code 713-155-147), while anti-MT-I�II antibodies were detected byusing Texas Red (TXRD)-linked donkey anti-rabbit IgG 1:40(Jackson ImmunoResearch Lab. Inc.; code 711-075-152).
To determine which cells suffer from oxidative stress, sec-tions were incubated overnight at 4°C with mouse anti-porcinevimentin, and goat anti-human NF, and rabbit anti-NITT orrabbit anti-MDA. The anti-vimentin antibodies were detectedas above, while anti-NF antibodies were detected by usingdonkey anti-goat IgG linked with TXRD 1:30 (Jackson Immu-noResearch Lab. Inc.; code 713-075-147), and the anti-NITTand anti-MDA antibodies were detected by using AMCA-linked donkey anti-rabbit IgG 1:40 (Jackson ImmunoResearchLab. Inc.; code 711-155-152).
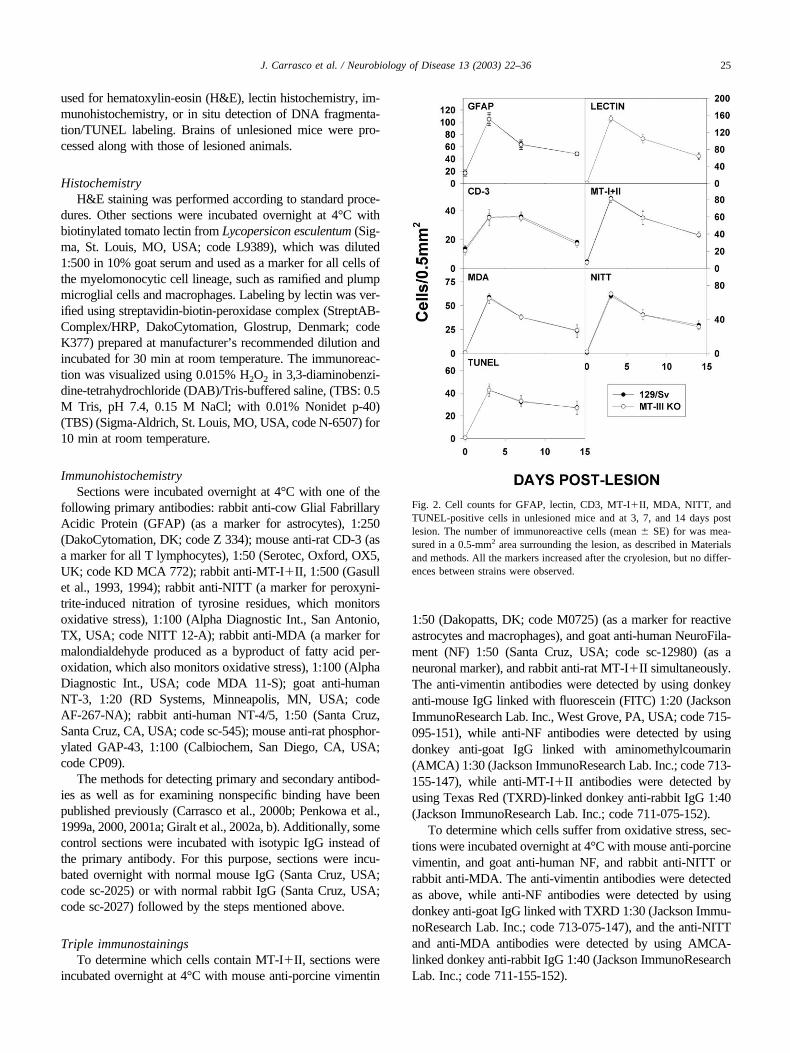
Fig. 2. Cell counts for GFAP, lectin, CD3, MT-I�II, MDA, NITT, andTUNEL-positive cells in unlesioned mice and at 3, 7, and 14 days postlesion. The number of immunoreactive cells (mean � SE) for was mea-sured in a 0.5-mm2 area surrounding the lesion, as described in Materialsand methods. All the markers increased after the cryolesion, but no differ-ences between strains were observed.
25J. Carrasco et al. / Neurobiology of Disease 13 (2003) 22–36

To determine which cells suffer from apoptosis, triplestainings for Terminal deoxynucleotidyl transferase (TdT)-mediated deoxyuridine triphosphate (dUTP)-digoxigeninnick end labeling (TUNEL) and cellular markers were per-formed. Hence, sections were incubated with rhodamine-linked TUNEL (Oncor, Gaithersburg, MD, USA; codeS7165-KIT) according to manufacturer’s protocol, and af-terwards incubated overnight at 4°C with rabbit anti-humanNeuron-Specific-Enolase NSE 1:1000 (Calbiochem, USA;code D05059) and mouse anti-porcine vimentin simulta-neously. The anti-NSE antibodies were detected by usingdonkey anti-rabbit IgG linked with FITC 1:50 (JacksonImmunoResearch Lab. Inc.; code 711-095-152), while theanti-vimentin antibodies were detected by using donkeyanti-mouse IgG linked with AMCA 1:30 (Jackson Im-munoResearch Lab. Inc.; code 715-155-151).
To determine which cells contain NT-4/5, sectionswere incubated overnight at 4°C with mouse anti-porcinevimentin and goat anti-human NF, and rabbit anti-humanNT-4/5. The anti-vimentin and the anti-NF antibodieswere detected by using donkey anti-mouse IgG linkedwith AMCA and donkey anti-goat IgG linked withTXRD, respectively, while the anti-NT-4/5 antibodieswere detected by using donkey anti-rabbit IgG linkedwith FITC.
The sections were embedded in 20 �l of fluorescentmounting (Dakopatts, DK; code S3023) and kept in dark-ness at 4°C. To evaluate the extent of nonspecific binding ofthe antisera in the fluorescence stainings, control sectionswere incubated in the absence of primary antibody. Resultswere considered only if these controls were negative. Forthe simultaneous examination and recording of the three
Fig. 3. Effects of cryolesion on mRNA abundance of host-response genes. Ipsilateral mRNA levels were analyzed by RNase protection assay (RPA) at 0,8, 24, 48, and 144 h post lesion, h PL). Wild-type (C), Mt1&2-null (1 � 2) and Mt3-null (3) mice were studied. The markers on the right column migratedifferently because the specific sequences have some extra nucleotides because of how the plasmids were built, so they are bigger than the samples. In theleft we state the names and locations of the main mRNAs of interest. The cryolesion upregulated the expression of the Glial Fabrillary Acidic Protein (GFAP),the acute-phase protein EB22/5.3 (EB22; the murine homologue for alpha1-antichymotrypsin), the macrophage activation marker protein Mac-1, and of theintracellular adhesion Molecule-I (ICAM), but no dramatic differences between strains were observed.
26 J. Carrasco et al. / Neurobiology of Disease 13 (2003) 22–36

stains, a Zeiss Axioplan2 light microscope equipped with atripleband (FITC/TXRD/AMCA) filter was used.
Detection of DNA fragmentation (TUNEL) in situ
Terminal deoxynucleotidyl transferase (TdT)-mediateddeoxyuridine triphosphate (dUTP)-digoxigenin nick end-labeling (TUNEL) staining was performed after tissue pro-cessing as described above according to manufacturer’sprotocol. Sections were incubated with 20 mg/ml proteinaseK (Sigma, USA; code P2308) for 5 min to strip off nuclearproteins. TUNEL was accomplished using the ApoptagPlus, In Situ Apoptosis Detection Kit (Oncor, USA; codeS7101-KIT). After immersion in equilibration buffer for 10min, sections were incubated with TdT and dUTP-digoxi-genin in a humified chamber at 37°C for 1 h and thenincubated in the stop/wash buffer at 37°C for 30 min to stopthe reaction. Afterwards, the sections were incubated inanti-digoxigenin-peroxidase solution for 30 min. DAB wasused as chromogen (as mentioned above), and the sectionswere counterstained with methyl green. Negative controlsections were treated similarly but incubated in the absenceof TdT enzyme, dUTP-digoxigenin, or anti-digoxigenin an-tibody. Sections were compared with positive control slidesfrom (Oncor, USA; code S7115). Furthermore, morpho-
logic criteria for apoptosis were evaluated too, since theTUNEL may stain necrotic cells.
RNase protection assay
RNase protection assay for the detection of host-re-sponse (ICAM, iNOS, the apoptosis inhibitor A20, Macl,the acute phase gene EB22, and GFAP) and cytokine geneswas performed as described previously (Hobbs et al., 1993;Campbell et al., 1994; Chiang et al., 1994). The mRNAfrom the ribosomal protein L32 was used to normalize thedata of each sample.
Cell counts
In addition to morphological analysis, the number ofstained cells was quantified in a blinded manner in matched0.5-mm2 areas of 3-�m-thick sections of the cortex. Inlesioned brains, the counts were done in the deepest borderof the lesion. To this end, positively stained cells, defined ascells with staining of the soma, or in the case of TUNELcells with nuclear staining, were counted. Cell counts wereperformed from three sections per mouse for three mice pergroup. Results were confirmed in separate experiments.
Fig. 4. Analysis of cytokine mRNA expression. Several cytokines were analyzed by RNase protection assay as in Fig. 2. Again, the markers on the rightcolumn migrate differently because the specific sequences have some extra nucleotides because of how the plasmids were built, so they are bigger than thesamples. The cryolesion dramatically upregulated tumor necrosis factor (TNF)-�, interleukin (IL)-1�, IL-6, and IL-1� in all strains peaking at 24 hours postlesion. Mt3-knockout (KO) mice did not differ significantly from wild-type (WT) controls, whereas Mt1&2-null mice showed greater induction. Quantitationis shown in Fig. 4.
27J. Carrasco et al. / Neurobiology of Disease 13 (2003) 22–36
Statistical analysis
Results were evaluated by two-way analysis of variance(ANOVA), with strain and lesion as main factors. When theinteraction was significant, it was interpreted to be theconsequence of a specific effect of the genetic Mt1&2 orMt3 deficiency during the inflammation. When only twogroups were compared, the Student t test was used.
Results
Astrogliosis, microgliosis, and T-cell infiltration followingcryolesion are unaffected by MT-III deficiency
In all unlesioned mice, some GFAP-positive astro-cytes and lectin-staining in ramified microglia were seen
in the cortex; most lectin staining, however, was ob-served in blood vessels, as expected. Mt3-null mice didnot alter the morphological appearance of these cells, ortheir anatomical distribution throughout the brain (Fig.1A and D and Fig. 2). In agreement with these data,GFAP (reflecting astrocyte reactivity) and Mac-1 (re-flecting microglia/macrophage activation) gene expres-sion measured by RNase protection assay (RPA) weresimilar in unlesioned animals of all three strains (Fig. 3).Brains of all the unlesioned mice were basically devoidof CD3-positive T cells (Fig. 1G).
Following the cryolesion, WT and Mt3 KO mice dis-played a comparable extent of injury to the cortex andcomparable responses to the injury. Astrocytes becamehypertrophic (Fig. 1A–C), while resident microglia wereactivated, and peripheral monocytes/macrophages wererecruited to the site of the lesion (Fig. 1D–F). GFAP andlectin stainings were maximal 3–5 days post lesion (dpl),and declined progressively thereafter, but at 14 dpl theywere still significantly higher than those of unlesionedmice (Fig. 2). The responses of Mt3 KO mice wereessentially the same as those of 129/Sv mice, both interms of morphological appearance of these cells but alsoin the number of cells positive for either GFAP or lectin,both within and surrounding the lesioned area. In accor-dance with GFAP and lectin stainings, there was a sig-nificant upregulation of both GFAP and Mac-1 mRNA at2– 6 dpl, reflecting the prominent gliosis in all threegroups of mice (Fig. 3). The acute phase protein EB22also showed a significant upregulation. Densitometricanalysis of these RPA revealed no statistical differencesbetween the strains (data not shown). There was a dra-matic increase of the number of CD3� cells in theinjured area following cryolesion, but Mt3 deficiency didnot alter number of infiltrating cells (Fig. 1G–I andFig. 2).
Cytokine expression is unaltered in Mt3-null mice
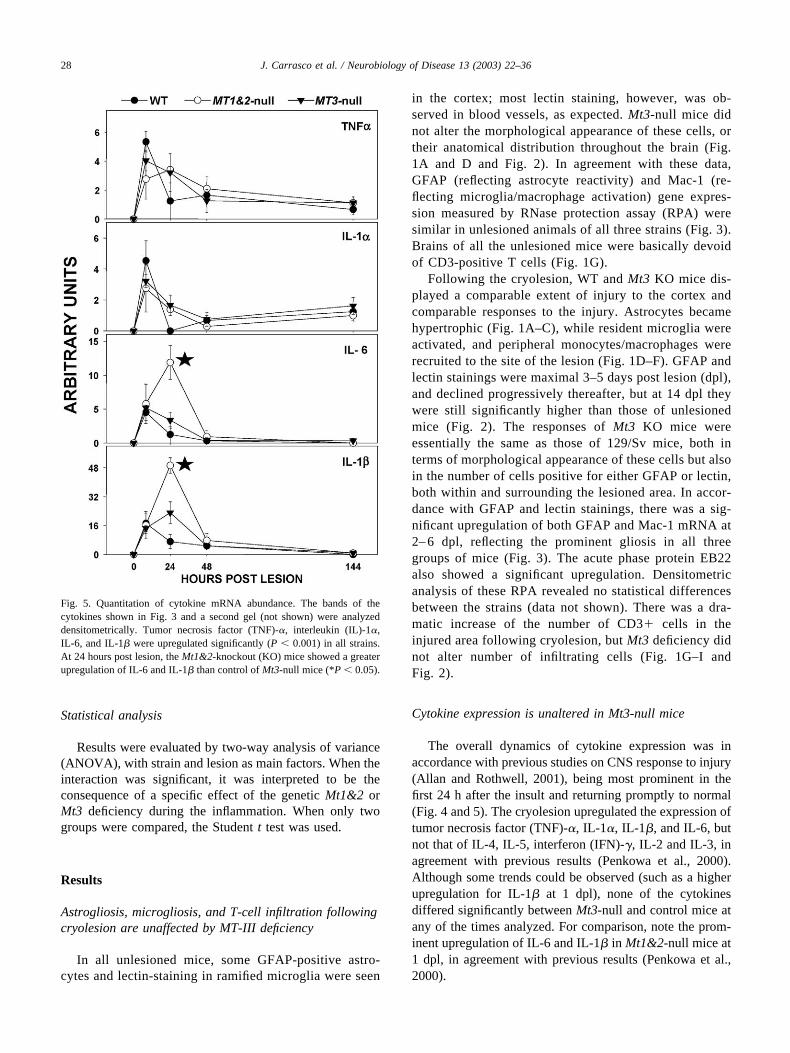
The overall dynamics of cytokine expression was inaccordance with previous studies on CNS response to injury(Allan and Rothwell, 2001), being most prominent in thefirst 24 h after the insult and returning promptly to normal(Fig. 4 and 5). The cryolesion upregulated the expression oftumor necrosis factor (TNF)-�, IL-1�, IL-1�, and IL-6, butnot that of IL-4, IL-5, interferon (IFN)-�, IL-2 and IL-3, inagreement with previous results (Penkowa et al., 2000).Although some trends could be observed (such as a higherupregulation for IL-1� at 1 dpl), none of the cytokinesdiffered significantly between Mt3-null and control mice atany of the times analyzed. For comparison, note the prom-inent upregulation of IL-6 and IL-1� in Mt1&2-null mice at1 dpl, in agreement with previous results (Penkowa et al.,2000).
Fig. 5. Quantitation of cytokine mRNA abundance. The bands of thecytokines shown in Fig. 3 and a second gel (not shown) were analyzeddensitometrically. Tumor necrosis factor (TNF)-�, interleukin (IL)-1�,IL-6, and IL-1� were upregulated significantly (P � 0.001) in all strains.At 24 hours post lesion, the Mt1&2-knockout (KO) mice showed a greaterupregulation of IL-6 and IL-1� than control of Mt3-null mice (*P � 0.05).
28 J. Carrasco et al. / Neurobiology of Disease 13 (2003) 22–36
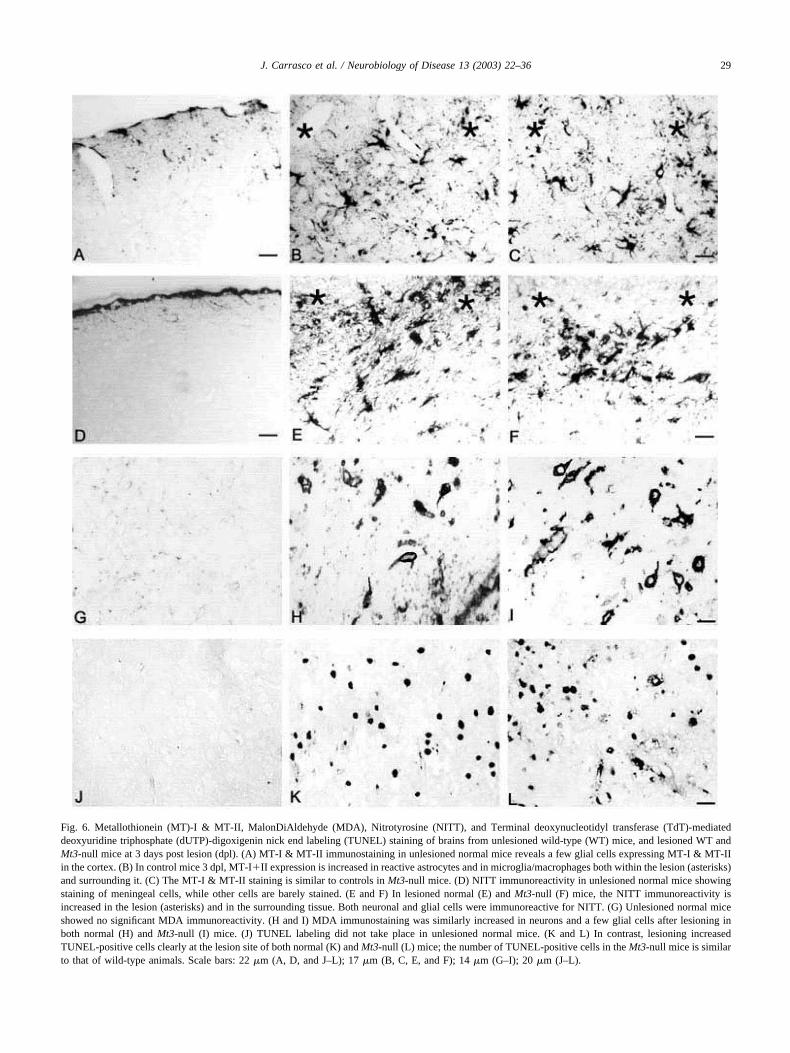
Fig. 6. Metallothionein (MT)-I & MT-II, MalonDiAldehyde (MDA), Nitrotyrosine (NITT), and Terminal deoxynucleotidyl transferase (TdT)-mediateddeoxyuridine triphosphate (dUTP)-digoxigenin nick end labeling (TUNEL) staining of brains from unlesioned wild-type (WT) mice, and lesioned WT andMt3-null mice at 3 days post lesion (dpl). (A) MT-I & MT-II immunostaining in unlesioned normal mice reveals a few glial cells expressing MT-I & MT-IIin the cortex. (B) In control mice 3 dpl, MT-I�II expression is increased in reactive astrocytes and in microglia/macrophages both within the lesion (asterisks)and surrounding it. (C) The MT-I & MT-II staining is similar to controls in Mt3-null mice. (D) NITT immunoreactivity in unlesioned normal mice showingstaining of meningeal cells, while other cells are barely stained. (E and F) In lesioned normal (E) and Mt3-null (F) mice, the NITT immunoreactivity isincreased in the lesion (asterisks) and in the surrounding tissue. Both neuronal and glial cells were immunoreactive for NITT. (G) Unlesioned normal miceshowed no significant MDA immunoreactivity. (H and I) MDA immunostaining was similarly increased in neurons and a few glial cells after lesioning inboth normal (H) and Mt3-null (I) mice. (J) TUNEL labeling did not take place in unlesioned normal mice. (K and L) In contrast, lesioning increasedTUNEL-positive cells clearly at the lesion site of both normal (K) and Mt3-null (L) mice; the number of TUNEL-positive cells in the Mt3-null mice is similarto that of wild-type animals. Scale bars: 22 �m (A, D, and J–L); 17 �m (B, C, E, and F); 14 �m (G–I); 20 �m (J–L).
29J. Carrasco et al. / Neurobiology of Disease 13 (2003) 22–36

Oxidative stress and apoptosis are unaltered aftercryolesion of Mt3-null mice
In all unlesioned animals, MDA and NITT staining wereonly detected in a few meningeal and glial cells, while noTUNEL-positive cells were observed (Fig. 6). As expected(Penkowa et al., 1999a, 2001b; Giralt et al., 2002b), afterthe cryolesion the number of cells positive for MDA, NITT(markers of lipid oxidation and nitrotyrosine, respectively),and TUNEL (a marker of apoptosis) increased significantly,peaking at 3 dpl and then slowly returning to that of unle-sioned mice (but not completely) over the next 2 weeks(Figs. 2 and 6). Triple staining revealed that MDA andNITT stainings were predominantly in neurons; however,some reactive astrocytes and macrophages/microglia in thearea surrounding the necrotic tissue were also stained (Fig.7). Most of the cells undergoing apoptosis were neurons(Fig. 7). There were no differences in the number of cellsstaining for MDA, NITT, or TUNEL when comparing WTand Mt3-null mice (Figs. 2 and 6).
Neurotrophin and GAP-43 expression were increased inMt3-null mice after cryolesion
In control, unlesioned mice, a faint NT-3 and NT-4/5expression was observed in a few ramified microglia,perivascular macrophages, grey matter astrocytes, and neu-rons (Fig. 8A and D); a similar pattern of expression wasobserved in Mt3-null mice. After the lesion, both neurotro-phins were upregulated in brain macrophages and in reac-tive astrocytes as well as in surviving neurons (Fig. 8B andE). Mt3-null mice showed a greater staining intensity andnumber of NT-3- and NT-4/5-positive cells compared toWT mice (Fig. 8C and F and 9). Mostly neurons but alsosome astrocytes and macrophage/microglia were NT-4/5positive (Fig. 7). Unlesioned WT or Mt3-null mice showedno significant staining of phosphorylated GAP-43 through-out the parenchyma (Fig. 8G). After the lesion, however,phosphorylated GAP-43 immunoreactivity increased dra-matically in the areas surrounding the lesion (Fig. 8H), andthis increase was greater in Mt3-null mice (Figs. 8H and Iand 9). Specificity of the immunostaining procedure wasassured by results obtained with isotype IgG (Fig. 10).
In an attempt to extend these immunohistochemistryresults, a preliminary experiment with pooled hemispheres
Fig. 7. Representative triple immunostainings at 3 days post lesion (dpl).(A) Immunostainings for vimentin-positive reactive astrocytes and macro-phages (green), NF-positive neurons (blue), and MT-I�II (red). As shown,MT-I�II are seen in vimentin-positive reactive astrocytes and macro-phages (yellow), while not in the neurons. (B) Immunostainings for vi-mentin-positive reactive astrocytes and macrophages (green), NF-positive
neurons (red), and NITT (blue). As shown, mainly neurons contain NITT(pink), while also a few vimentin-positive cells show NITT (turquoise). (C)Triple stainings of vimentin-positive reactive astrocytes and macrophages(blue), NSE-positive neurons (green), and TUNEL (red), showing thatprimarily neurons are undergoing apoptosis. (D) Vimentin-positive reac-tive astrocytes and macrophages (blue), NF-positive neurons (red), andNeurotrophin (NT)-4/5 (green). As shown, both neurons and some vimen-tin-positive cells contain NT-4/5. Besides, a vimentin-negative astrocyte aswell as a macrophage show NT-4/5 expression. Scale bars: 40 �m (A, B,and D); 55 �m (C).
30 J. Carrasco et al. / Neurobiology of Disease 13 (2003) 22–36
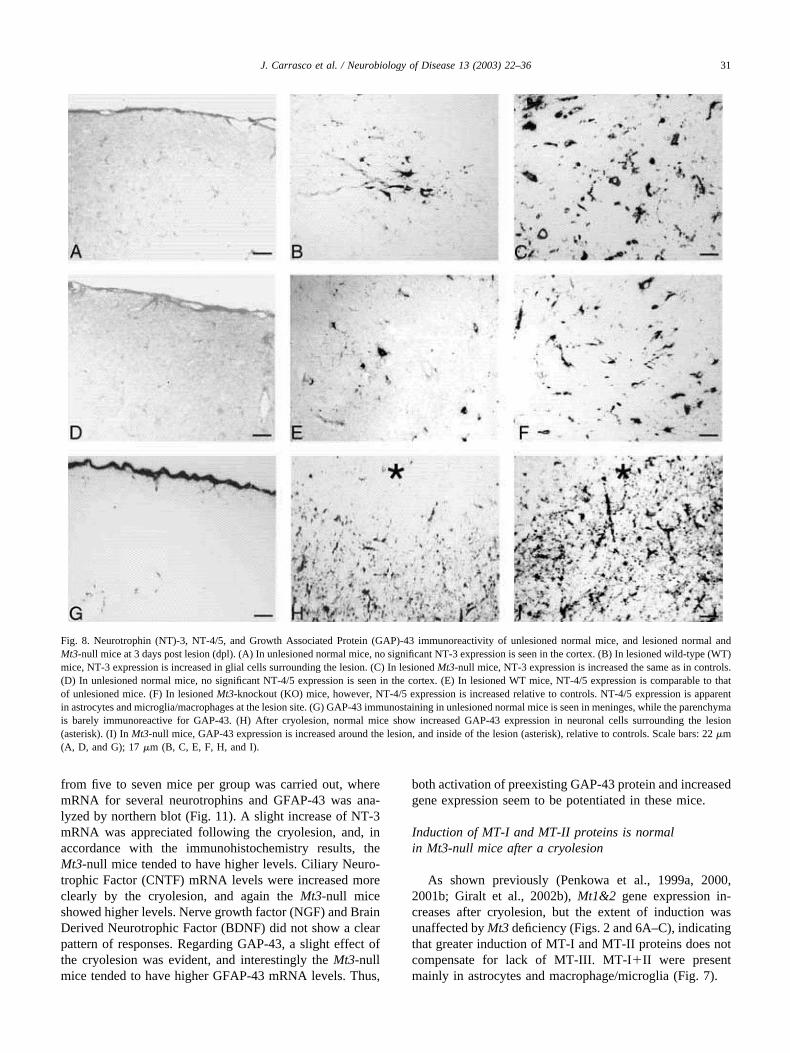
from five to seven mice per group was carried out, wheremRNA for several neurotrophins and GFAP-43 was ana-lyzed by northern blot (Fig. 11). A slight increase of NT-3mRNA was appreciated following the cryolesion, and, inaccordance with the immunohistochemistry results, theMt3-null mice tended to have higher levels. Ciliary Neuro-trophic Factor (CNTF) mRNA levels were increased moreclearly by the cryolesion, and again the Mt3-null miceshowed higher levels. Nerve growth factor (NGF) and BrainDerived Neurotrophic Factor (BDNF) did not show a clearpattern of responses. Regarding GAP-43, a slight effect ofthe cryolesion was evident, and interestingly the Mt3-nullmice tended to have higher GFAP-43 mRNA levels. Thus,
both activation of preexisting GAP-43 protein and increasedgene expression seem to be potentiated in these mice.
Induction of MT-I and MT-II proteins is normalin Mt3-null mice after a cryolesion
As shown previously (Penkowa et al., 1999a, 2000,2001b; Giralt et al., 2002b), Mt1&2 gene expression in-creases after cryolesion, but the extent of induction wasunaffected by Mt3 deficiency (Figs. 2 and 6A–C), indicatingthat greater induction of MT-I and MT-II proteins does notcompensate for lack of MT-III. MT-I�II were presentmainly in astrocytes and macrophage/microglia (Fig. 7).
Fig. 8. Neurotrophin (NT)-3, NT-4/5, and Growth Associated Protein (GAP)-43 immunoreactivity of unlesioned normal mice, and lesioned normal andMt3-null mice at 3 days post lesion (dpl). (A) In unlesioned normal mice, no significant NT-3 expression is seen in the cortex. (B) In lesioned wild-type (WT)mice, NT-3 expression is increased in glial cells surrounding the lesion. (C) In lesioned Mt3-null mice, NT-3 expression is increased the same as in controls.(D) In unlesioned normal mice, no significant NT-4/5 expression is seen in the cortex. (E) In lesioned WT mice, NT-4/5 expression is comparable to thatof unlesioned mice. (F) In lesioned Mt3-knockout (KO) mice, however, NT-4/5 expression is increased relative to controls. NT-4/5 expression is apparentin astrocytes and microglia/macrophages at the lesion site. (G) GAP-43 immunostaining in unlesioned normal mice is seen in meninges, while the parenchymais barely immunoreactive for GAP-43. (H) After cryolesion, normal mice show increased GAP-43 expression in neuronal cells surrounding the lesion(asterisk). (I) In Mt3-null mice, GAP-43 expression is increased around the lesion, and inside of the lesion (asterisk), relative to controls. Scale bars: 22 �m(A, D, and G); 17 �m (B, C, E, F, H, and I).
31J. Carrasco et al. / Neurobiology of Disease 13 (2003) 22–36

Discussion
We have compared the CNS responses of Mt3-null micewith WT mice and mice lacking expression of Mt1 and Mt2after cryoinjury of the cortex. We showed previously thatMT-I (and presumably MT-II) are dramatically inducedduring the first 2 dpl in microglia and reactive astrocytesand then levels of these proteins gradually return to baselineduring the next 2–3 weeks (for 30–60 s of cryolesion,respectively) as the lesions are repaired (Penkowa et al.,1999a, 2000, 2001b; Giralt et al., 2002b). In the absence ofMT-I and II, microglial and reactive astrocytes markersremain elevated even 3 months after the cryolesion. Anincreased astrogliosis caused by MT-I&II deficiency hasalso been found in the G93A SODI mice (Puttaparthi et al.,2002). Likewise, evidence of oxidative stress and apoptosispersists for 20 dpl in the Mt1&2-null mice, whereas theyreturn to normal, nearly undetectable levels within 10 dpl inWT mice. Thus, wound repair is dramatically slowed inmice lacking MT-I and MT-II (reviewed in Hidalgo et al.,2001, 2002). Mt3 expression declines during the first dpl,but is elevated between 7 and 13 dpl (Penkowa et al., 2000).Based on the above findings, it seemed plausible that MT-IIImight also contribute to the recovery from traumatic injury.However, the results obtained in this study do not supportthat conclusion. We herewith show that none of the MT-
I&II-related effects detailed above are shared by MT-III invivo. Thus, the inflammatory response of Mt3-null mice, aswell as the oxidative stress and apoptosis elicited by thecryolesion, were all essentially identical to what was ob-served in wild-type mice. A compensatory response of theother brain MT isoforms (MT-I&II) is ruled out, since, ifanything, their expression was either normal or even de-creased in the Mt3-null mice compared to WT mice.
In planning these experiments, we favored the hypothesisthat MT-III might protect neurons against neurotoxic insultsin a manner analogous to ways that MT-I and MT-II protectglia and neurons. Our observations that the Mt3-null miceshow a normal progression of wound responses, which arepredominantly mediated by astrocytes and microglia, areconsistent with this observation, but we also observed nor-mal degree of neuronal apoptosis, rather than more exten-sive cell death as might be expected if MT-III normallyprotects against neuronal demise. The data available fordiscussing these results are scarce. MT-III was originallycalled growth inhibitory factor (GIF) because it inhibitssurvival of cultured neurons (Uchida et al., 1991). Thisinhibitory activity was more abundant in control brain ex-tracts than in extracts from people with Alzheimer’s diseaseand it correlated with a depletion of Mt3 mRNA and MT-IIIprotein in Alzheimer brains (Uchida et al., 1991; Tsuji et al.,1992). The inhibitory activity is a property of the � domainof MT-III and is not observed with other MT isoforms(Erickson et al., 1994; Sewell et al., 1995; Uchida and Ihara,1995). While the difference in inhibitory activity of controland Alzheimer extracts has been confirmed, the depletion ofMT-III has not been confirmed in other populations (Erick-son et al., 1994; Amoureux et al., 1997; Carrasco et al.,1999; Yu et al., 2001). Furthermore, Mt3-null mice do notreveal morphological signs or behavioral symptoms sugges-tive of Alzheimer’s disease (Erickson et al., 1997). In con-trast to the so-called neuronal growth inhibitory activity ofMT-III (Uchida et al., 1991; Tsuji et al., 1992), a neuropro-tective role of MT-III appears likely judging the response ofMt3-null mice to kainate-induced seizures. Thus, Mt3-nullmice manifest more neuronal death in response to kainate-induced seizures, and mice expressing excess MT-III pro-tein are resistant to this damage, suggesting that MT-IIIplays a protective role in this seizure model (Erickson et al.,1997). We demonstrated that MT-III protects cerebellarneurons in vitro against glutamate and nitric oxide neuro-toxicity (Montoliu et al., 2000), despite the control of theexperimental conditions has been underestimated by certainauthors (Uchida et al., 2002), and other laboratories havealso demonstrated a neuroprotective role of MT-III in invitro studies following injuries caused by �-amyloid 25–35peptide (Ren et al., 2001), by S-nitroso-thiols and H2O2
(Chen et al., 2002) and by high oxygen conditions (Uchidaet al., 2002). MT-III is also effective protecting humanfibroblasts against H2O2 (You et al., 2002). In this regard, itis noteworthy to mention that MT-III has actually a neuro-protective role in the absence of brain extracts in the same
Fig. 9. Cell counts for Neurotrophin (NT)-3-, NT-4/5-, and Growth Asso-ciated Protein (GAP)-43-positive cells in unlesioned mice and at 3, 7, and14 days post lesion (dpl). The number of immunoreactive cells (mean �SE) for NT-3, NT-4/5, and GAP-43 was measured in a 0.5-mm2 areasurrounding the lesion, as described in Materials and Methods. All themarkers increased after the cryolesion, but to a greater extent in Mt3-nullmice (P � 0.001).
32 J. Carrasco et al. / Neurobiology of Disease 13 (2003) 22–36
bioassay originally described by Uchida et al. (Uchida et al.,1991; Erickson et al., 1994; Sewell et al., 1995). The abovestudies suggest that MT-III would be acting as an antioxi-dant. MT-III could help protect against oxidative damage byreleasing zinc as its sulfhydryl residues become oxidized, orby directly interacting with oxygen and/or nitrogen radicals.However, despite all these in vitro evidences of a potentantioxidant role(s) of MT-III, we could not observe signif-icant oxidative stress differences between Mt3-null miceand wild-type mice in the cryolesion model. It is possiblethat MT-III could afford only a limited protection. In thisregard, the protection afforded by MT-III in the kainicacid-induced seizures model was evident only followinglow intensity seizures (Erickson et al., 1997). The cryole-sion model of brain injury causes a large damage, and itseems reasonable that the lack of evidence of MT-III actingas antioxidant in this model resembles the lack of effect ofMT-III during high intensity kainic acid-induced seizuresmodel (Erickson et al., 1997). Since MT-I&II are highlyinducible in contrast to MT-III, it is possible that such
induction could compensate for MT-III deficiency, but it isobvious that this is speculative and that the generation of atriple KO mouse could greatly help to ascertain such pos-sibility.
While this manuscript was under revision, a study ap-peared describing the phenotypes of transgenic G93ASOD1 mice crossed with either Mt1&2-null or Mt3-nullmice (Puttaparthi et al., 2002). These mice showed accel-erated motor dysfunction with regard to onset or progres-sion, respectively, and decreased cumulative survival inboth cases (dose-dependently only for MT-III deficiency).Mutated superoxide dismutase (mSOD1) shows abnormal-ities in zinc binding and has enhanced nitration activity(Puttaparthi et al., 2002, and references therein), and thusoxidative damage could contribute to the progressive motorneurons impairment and death. The deficiency of MT-I&IIor MT-III potentiated motor neuron impairment in theG93A SOD1 mice as revealed by stride length and gripstrength, but the mechanisms underlying such effects appearto be different. Thus, MT-I&II deficiency causes motorneuron impairment that is not matched by loss of cellnumber, while MT-III deficiency does potentiate motor neu-ron death. Such effects is in accordance with the neuropro-tective roles discussed above. However, MT-III deficiencypotentiated motor neuron death in early symptomatic butnot in end-stage G93A SOD1 animals. As for the kainicacid-induced seizures model and likely for the cryolesionmodel, the neuroprotection afforded by MT-III is limited innature and thus in more severe diseased G93A SOD1 miceMT-III deficiency does not make a difference. A mechanismthat might be involved in the phenotype of these mice is adisturbance of zinc homeostasis, since MT-III deficiencydecreases overall brain levels in areas where it is normallyexpressed (e.g., cortex and hippocampus) and zinc-depletedSOD1 shows an increased ability to react with peroxynitriteand catalyze the addition of nitro groups to tyrosine residues(Erickson et al., 1997; Puttaparthi et al., 2002). However,such mechanism would not operate prominently in the cry-olesion model since NITT and MDA immunostainings did
Fig. 10. Control sections for the immunohistochemistry. (A and B) Isotypic normal mouse IgG (A) or isotypic normal rabbit IgG (B) was used instead ofthe primary antibody. As shown, in both cases, no significant or specific staining can be seen. Scale bars: 22 �m (A and B).
Fig. 11. Northern blot for neurotrophins and GAP-43. A northern blot wascarried out in pooled samples (n � 5–7) from control and cryolesionedwild-type (WT) (a) and Mt3-null (b) mice. Neurotrophin (NT-3); CiliaryNeurotrophic Factor (CNTF); nerve growth factor (NGF); Brain DerivedNeurotrophic Factor (BDNF); dpl, days post lesion.
33J. Carrasco et al. / Neurobiology of Disease 13 (2003) 22–36

not differ between Mt3-null mice and WT mice. Anotherpossible mechanism(s) through which MT-III might affectneuronal functions and/or survival is through the alterationof neurotrophins and other factors such as GAP-43 (Beno-witz and Routtenberg, 1997; Bibel and Barde, 2000), whichwere significantly affected by MT-III deficiency. Althoughin our studies we did not observe differences between Mt3-null mice and WT mice regarding neuronal survival, weneed to keep in mind that the cryolesion model causessevere injury against which MT-III might not afford signif-icant protection (in contrast to MT-I&II). In contrast, suchalteration of neurotrophins and GAP-43 might be underly-ing the phenotype of the early symptomatic G93A SOD1mice (Puttaparthi et al., 2002), which warranties furtherstudies. The mechanisms by which MT-III may normallysuppress the expression of GAP-43 or neurotrophins areunknown.
In summary, the present results suggest that, in contrastto MT-I�II, MT-III is unlikely to be a significant factor forcontrolling the inflammatory response, oxidative stress, andapoptosis after significant brain damage. They are consis-tent with the idea that MT-III may have a unique functioncompared to MT-I and MT-II, perhaps related to neuralrecovery as suggests the results for neurotrophins and GAP-43, but additional experiments will be required to under-stand what that unique function may be.
Acknowledgments
The authors are grateful to Dr. Joaquın Hernandez and toNatalia Lago, H. Hadberg, P.S. Thomsen, M. Søberg, G.Hahn, and K. Stub for valuable help. These studies weresupported by PSPGC PM98-0170, Ministerio de Ciencia yTecnologıa and Feder SAF2002-01268, and Direccio Gen-eral de Recerca 2001SGR 00203 (J.H.); by Statens Sund-hedsvidenskabelige Forskningsråad (SSVF), Novo NordiskFonden, Nordisk Forsknings Komite, University of Copen-hagen/SVF Fond, The Danish Medical Association Re-search Fund, Schrøders Fond, Holger Rabitz Mindelegat,Dir. Ib Henriksens Fond, Dir. Jacob Madsen’s Fond,Fonden af 17.12.1981, Kong Christian IX og DronningLouises Jubilæumslegat, Fonden til LægevidenskabensFremme, Gerda og Aage Haensch’s Fond, Eva og HenryFrænkels Mindelegat, Marshalls Fond, Lily Benthine LundsFond, Kathrine og Vigo Skovgaards Fond, Øster-JørgensensFond, Karen A Tolstrups Fond, Eva og Robert Voss Han-sens Fond, Warwara Larsens Fond, Dansk Parkinsonforen-ing, Bernhard og Marie Kleins Legat til Sukkersygeforskn-ing, Ragnhild Ibsens Legat for Medicinsk Forskning,Hestehandler Ole Jacobsens Mindelegat, Øjenfonden, LægeEilif Trier-Hansen og Hustru Ane Trier-Hansens Legat(M.P.); and by NIH grant MH 50426 and DA 12444(I.L.C.).
References
Acarin, L., Carrasco, J., Gonzalez, B., Hidalgo, J., Castellano, B., 1999a.Expression of growth inhibitory factor (metallothionein-III) mRNAand protein following excitotoxic immature brain injury. J. Neuro-pathol. Exp. Neurol. 58, 389–397.
Acarin, L., Gonzalez, B., Hidalgo, J., Castro, A.J., Castellano, B., 1999b.Primary cortical glial reaction versus secondary thalamic glial responsein the excitotoxically injured young brain: astroglial response andmetallothionein expression. Neuroscience 92, 827–839.
Allan, S.M., Rothwell, N.J., 2001. Cytokines and acute neurodegeneration.Natl. Rev. Neurosci. 2, 734–744.
Aloisi, F., 2001. Immune function of microglia. Glia 36, 165–179.Amoureux, M.C., Van Gool, D., Herrero, M.T., Dom, R., Colpaert, F.C.,
Pauwels, P.J., 1997. Regulation of metallothionein-III (GIF) mRNA inthe brain of patients with Alzheimer disease is not impaired. Mol.Chem. Neuropathol. 32, 101–121.
Anezaki, T., Ishiguro, H., Hozumi, I., Inuzuka, T., Hiraiwa, M., Kobayashi,H., Yuguchi, T., Wanaka, A., Uda, Y., Miyatake, T., Yamada, K.,Tohyama, M., Tsuji, S., 1995. Expression of growth inhibitory factor(GIF) in normal and injured rat brains. Neurochem. Int. 27, 89–94.
Benowitz, L.I., Routtenberg, A., 1997. GAP-43: an intrinsec determinantof neuronal development and plasticity. Trends Neurosci. 20, 84–91.
Bibel, M., Barde, Y.-A., 2000. Neurotrophins: key regulators of cell fateand cell shape in the vertebrate nervous system. Genes Dev. 14,2919–2937.
Blaauwgeers, H.G., Sillevis Smitt, P.A., De Jong, J.M., Troost, D., 1993.Distribution of metallothionein in the human central nervous system.Glia 8, 62–70.
Campbell, I.L., Hobbs, M.V., Kemper, P., Oldstone, M.B.A., 1994. Cere-bral expression of multiple cytokine genes in mice with lymphocyticchoriomeningitis. J. Immunol. 152, 716–723.
Carrasco, J., Giralt, M., Molinero, A., Penkowa, M., Moos, T., Hidalgo, J.,1999. Metallothionein (MT)-III: generation of polyclonal antibodies,comparison with MT-I�II in the freeze lesioned rat brain and in abioassay with astrocytes, and analysis of Alzheimer’s disease brains.J. Neurotrauma 16, 1115–1129.
Carrasco, J., Giralt, M., Penkowa, M., Stalder, A.K., Campbell, I.L.,Hidalgo, J., 2000a. Metallothioneins are upregulated in symptomaticmice with astrocyte-targeted expression of tumor necrosis factor-�.Exp. Neurol. 163, 46–54.
Carrasco, J., Hernandez, J., Bluethmann, H., Hidalgo, J., 1998a. Interleu-kin-6 and tumor necrosis factor-alpha type 1 receptor deficient micereveal a role of IL-6 and TNF-alpha on brain metallothionein-I and -IIIregulation. Mol. Brain Res. 57, 221–234.
Carrasco, J., Hernandez, J., Gonzalez, B., Campbell, I.L., Hidalgo, J.,1998b. Localization of metallothionein-I and -III expression in the CNSof transgenic mice with astrocyte-targeted expression of interleukin 6.Exp. Neurol. 153, 184–194.
Carrasco, J., Penkowa, M., Hadberg, H., Molinero, A., Hidalgo, J., 2000b.Enhanced seizures and hippocampal neurodegeneration followingkainic acid induced seizures in metallothionein-I�II deficient mice.Eur. J. Neurosci. 12, 2311–2322.
Chan, P.H., Yang, G.Y., Chen, S.F., Carlson, E., Epstein, C.J., 1991.Cold-induced brain edema and infarction are reduced in transgenicmice overexpressing CuZn-superoxide dismutase. Ann. Neurol. 29,482–486.
Chen, Y., Irie, Y., Keung, W.M., Maret, W., 2002. S-Nitrosothiols reactpreferentially with zinc thiolate clusters of metallothionein III throughtransnitrosation. Biochemistry 41, 8360–8367.
Chiang, C.-S., Stalder, A., Samimi, A., Campbell, I.L., 1994. Reactivegliosis as a consequence of interleukin 6 expression in the brain: studiesin transgenic mice. Dev. Neurosci. 16, 212–221.
Cook, J.L., Marcheselli, V., Alam, J., Deininger, P.L., Bazan, N.G., 1998.Temporal changes in gene expression following cryogenic rat braininjury. Mol. Brain Res. 55, 9–19.
34 J. Carrasco et al. / Neurobiology of Disease 13 (2003) 22–36

Coyle, J., Puttfarcken, P., 1993. Oxidative stress, glutamate, and neurode-generative disorders. Science 262, 689–695.
Dong, Y., Benveniste, E.N., 2001. Immune function of astrocytes. Glia 36,180–190.
Eddleston, M., Mucke, L., 1993. Molecular profile of reactive astrocytes-Implications for their role in neurological disease. Neuroscience 1,15–36.
Erickson, J.C., Hollopeter, G., Thomas, S.A., Froelick, G.J., Palmiter,R.D., 1997. Disruption of the metallothionein-III gene in mice: analysisof brain zinc, behavior, and neuron vulnerability to metals, aging, andseizures. J. Neurosci. 17, 1271–1281.
Erickson, J.C., Sewell, A.K., Jensen, L.T., Winge, D.R., Palmiter, R.D.,1994. Enhanced neurotrophic activity in Alzheimer’s disease cortex isnot associated with down-regulation of metallothionein-III (GIF). BrainRes. 649, 297–304.
Gasull, T., Giralt, M., Hernandez, J., Martinez, P., Bremner, I., Hidalgo, J.,1994. Regulation of metallothionein concentrations in rat brain: effectof glucocorticoids, zinc, copper, and endotoxin. Am. J. Physiol. 266,E760–E767.
Gasull, T., Rebollo, D.V., Romero, B., Hidalgo, J., 1993. Development ofa competitive double antibody radioimmunoassay for rat metallothio-nein. J. Immunoassay 14, 209–225.
Giralt, M., Carrasco, J., Penkowa, M., Morcillo, M.A., Santamaria, J.,Campbell, I.L., Hidalgo, J., 2001. Astrocyte-targeted expression ofinterleukin-3 and interferon-� causes specific changes in metallothio-nein expression in the brain. Exp. Neurol. 168, 334–346.
Giralt, M., Penkowa, M., Hernandez, J., Molinero, A., Carrasco, J., Lago,N., Camats, J., Campbell, I.L., Hidalgo, J., 2002a. Metallothionein-1�2 deficiency increases brain pathology in transgenic mice withastrocyte-targeted expression of interleukin 6. Neurobiol. Dis. 9, 319–338.
Giralt, M., Penkowa, M., Lago, N., Molinero, A., Hidalgo, J., 2002b.Metallothionein-1�2 protect the CNS after a focal brain injury. Exp.Neurol. 173, 114–128.
Giulian, D., Chen, J., Ingemann, J., George, J., Noponen, M., 1989. Therole of mononuclear phagocytes in wound healing after traumaticinjury to adult mammalian brain. J. Neurosci. 12, 4416–4429.
Halliwell, B., 1992. Reactive oxygen species and the central nervoussystem. J. Neurochem. 59, 1609–1623.
Heales, S.J., Bolanos, J.P., Stewart, V.C., Brookes, P.S., Land, J.M., Clark,J.B., 1999. Nitric oxide, mitochondria and neurological disease. Bio-chim. Biophys. Acta 1410, 215–228.
Hernandez, J., Carrasco, J., Belloso, E., Giralt, M., Bluethmann, H., Lee,D.K., Andrews, G.K., Hidalgo, J., 2000. Metallothionein induction byrestraint stress: role of glucocorticoids and IL-6. Cytokine 12, 791–796.
Hidalgo, J., Aschner, M., Zatta, P., Vasak, M., 2001. Roles of the metal-lothionein family of proteins in the central nervous system. Brain Res.Bull. 55, 133–145.
Hidalgo, J., Penkowa, M., Giralt, M., Carrasco, J., Molinero, A., 2002.Metallothionein expression and oxidative stress in the brain. MethodsEnzymol. 348, 238–249.
Hobbs, M.V., Weigle, W.O., Noonan, D.J., Torbett, B.E., McEvilly, R.J.,Koch, R.J., Cardenas, G.J., Ernst, D.N., 1993. Patterns of cytokine geneexpression by CD4� T cells from young and old mice. J. Immunol.150, 3602–3608.
Holloway, A.F., Stennard, F.A., Dziegielewska, K.M., Weller, L., West,A.K., 1997. Localisation and expression of metallothionein immuno-reactivity in the developing sheep brain. Int. J. Dev. Neurosci. 15,195–203.
Hopkins, S., Rothwell, N., 1995. Cytokines and the nervous system. I:Expression and recognition. Trends Neurosci. 18, 83–88.
Horner, P.J., Gage, F.H., 2000. Regenerating the damaged central nervoussystem. Nature 407, 963–970.
Hozumi, I., Inuzuka, T., Hiraiwa, M., Uchida, Y., Anezaki, T., Ishiguro,H., Kobayashi, H., Uda, Y., Miyatake, T., Tsuji, S., 1995. Changes ofgrowth inhibitory factor after stab wounds in rat brain. Brain Res. 688,143–148.
Hozumi, I., Inuzuka, T., Ishiguro, H., Hiraiwa, M., Uchida, Y., Tsuji, S.,1996. Immunoreactivity of growth inhibitory factor in normal rat brainand after stab wounds—an immunocytochemical study using confocallaser scan microscope. Brain Res. 741, 197–204.
Hughes, P.E., Alexi, T., Walton, M., Williams, C.E., Dragunow, M., Clark,R.G., Gluckman, P.D., 1999. Activity and injury-dependent expressionof inducible transcription factors, growth factors and apoptosis-relatedgenes within the central nervous system. Prog. Neurobiol. 57, 421–450.
Inuzuka, T., Hozumi, I., Tamura, A., Hiraiwa, M., Tsuji, S., 1996. Patternsof growth inhibitory factor (GIF) and glial fibrillary acidic proteinrelative level changes differ following left middle cerebral artery oc-clusion in rats. Brain Res. 709, 151–153.
Knerlich, F., Schilling, L., Gorlach, C., Wahl, M., Ehrenreich, H., Siren,A.L., 1999. Temporal profile of expression and cellular localization ofinducible nitric oxide synthase, interleukin-1 beta and interleukin con-verting enzyme after cryogenic lesion of the rat parietal cortex. Mol.Brain Res. 68, 73–87.
Kobayashi, H., Uchida, Y., Ihara, Y., Nakajima, K., Kohsaka, S.,Miyatake, T., Tsuji, S., 1993. Molecular cloning of rat growth inhibi-tory factor cDNA and the expression in the central nervous system.Mol. Brain Res. 19, 188–194.
Lipton, P., 1999. Ischemic cell death in brain neurons. Physiol. Rev. 79,1431–1568.
Masters, B.A., Kelly, E.J., Quaife, C.J., Brinster, R.L., Palmiter, R.D.,1994a. Targeted disruption of metallothionein I and II genes increasessensitivity to cadmium. Proc. Natl. Acad. Sci. USA 91, 584–588.
Masters, B.A., Quaife, C.J., Erickson, J.C., Kelly, E.J., Froelick, G.J.,Zambrowicz, B.P., Brinster, R.L., Palmiter, R.D., 1994b. Metallothio-nein III is expressed in neurons that sequester zinc in synaptic vesicles.J. Neurosci. 14, 5844–5857.
Mattson, M., Scheff, S., 1994. Endogenous neuroprotection factors andtraumatic brain injury: mechanisms of action and implications fortherapy. J. Neurotrauma 11, 3–33.
McIntosh, T., Juhler, M., Wieloch, T., 1998. Novel pharmacologic strate-gies in the treatment of experimental traumatic brain injury. J. Neuro-trauma 15, 731–769.
Merrill, J.E., Benveniste, E.N., 1996. Cytokines in inflammatory brainlesions: helpful and harmful. Trends Neurosci. 19, 331–338.
Montoliu, C., Monfort, P., Carrasco, J., Palacios, O., Capdevila, M.,Hidalgo, J., Felipo, V., 2000. Metallothionein-III prevents glutamateand nitric oxide neurotoxicity in primary cultures of cerebellar neurons.J. Neurochem. 75, 266–273.
Munoz-Fernandez, M.A., Fresno, M., 1998. The role of tumour necrosisfactor, interleukin 6, interferon-gamma and inducible nitric oxide syn-thase in the development and pathology of the nervous system. Prog.Neurobiol. 56, 307–340.
Murakami, K., Kondo, T., Yang, G., Chen, S.F., Morita-Fujimura, Y.,Chan, P.H., 1999. Cold injury in mice: a model to study mechanisms ofbrain edema and neuronal apoptosis. Prog. Neurobiol. 57, 289–299.
Nagano, S., Satoh, M., Sumi, H., Fujimura, H., Tohyama, C., Yanagihara,T., Sakoda, S., 2001. Reduction of metallothioneins promotes thedisease expression of familial amyotrophic lateral sclerosis mice in adose-dependent manner. Eur. J. Neurosci. 13, 1363–1370.
Nakajima, K., Suzuki, K., 1995. Immunochemical detection of metallo-thionein in brain. Neurochem. Int. 27, 73–87.
Olanow, C., 1993. A radical hypothesis for neurodegeneration. TrendsNeurosci. 16, 439–444.
Palmiter, R.D., Findley, S.D., Whitmore, T.E., Durnam, D.M., 1992. MT-III, a brain-specific member of the metallothionein gene family. Proc.Natl. Acad. Sci. USA 89, 6333–6337.
Penkowa, M., Carrasco, J., Giralt, M., Molinero, A., Hernandez, J., Camp-bell, I.L., Hidalgo, J., 2000. Altered central nervous system cytokine-growth factor expression profiles and angiogenesis in metallothionein-I�II deficient mice. J. Cereb. Blood Flow Metab. 20, 1174–1189.
35J. Carrasco et al. / Neurobiology of Disease 13 (2003) 22–36

Penkowa, M., Carrasco, J., Giralt, M., Moos, T., Hidalgo, J., 1999a. CNSwound healing is severely depressed in metallothionein I- and II-deficient mice. J. Neurosci. 19, 2535–2545.
Penkowa, M., Espejo, C., Martinez-Caceres, E.M., Poulsen, C.B., Mont-alban, X., Hidalgo, J., 2001a. Altered inflammatory response and in-creased neurodegeneration in metallothionein I�II deficient mice dur-ing experimental autoimmune encephalomyelitis. J. Neuroimmunol.119, 248–260.
Penkowa, M., Giralt, M., Camats, J., Hidalgo, J., 2002. Metallothionein 1� 2 protect the CNS during neuroglial degeneration induced by 6-ami-nonicotinamide. J. Comp. Neurol. 444, 174–189.
Penkowa, M., Giralt, M., Moos, T., Thomsen, P.S., Hernandez, J., Hidalgo,J., 1999b. Impaired inflammatory response to glial cell death in genet-ically metallothionein-I- and -II-deficient mice. Exp. Neurol. 156, 149–164.
Penkowa, M., Giralt, M., Thomsen, P., Carrasco, J., Hidalgo, J., 2001b.Zinc or copper deficiency-induced impaired inflammatory response tobrain trauma may be caused by the concomitant metallothioneinchanges. J. Neurotrauma 18, 447–463.
Penkowa, M., Hidalgo, J., 2000. Metallothionein I�II expression and theirrole in experimental autoimmune encephalomyelitis. Glia 32, 247–263.
Penkowa, M., Hidalgo, J., 2001. Metallothionein treatment reduces proin-flammatory cytokines IL-6 and TNF-a and apoptotic cell death duringexperimental autoimmune encephalomyelitis. Exp. Neurol. 170, 1–14.
Penkowa, M., Moos, T., 1995. Disruption of the blood-brain interface inneonatal rat neocortex induces a transient expression of metallothioneinin reactive astrocytes. Glia 13, 217–227.
Pentreath, V.W., Slamon, N.D., 2000. Astrocyte phenotype and preventionagainst oxidative damage in neurotoxicity. Human Exp. Toxicol. 19,641–649.
Perry, V., Bell, M., Brown, H., Matyszak, M., 1995. Inflammation in thenervous system. Curr. Opin. Neurobiol. 5, 636–641.
Puttaparthi, K., Gitomer, W.L., Krishnan, U., Son, M., Rajendran, B.,Elliott, J.L., 2002. Disease progression in a transgenic model of familialamyotrophic lateral sclerosis is dependent on both neuronal and non-neuronal zinc binding proteins. J. Neurosci. 22, 8790–8796.
Quaife, C.J., Findley, S.D., Erickson, J.C., Froelick, G.J., Kelly, E.J.,Zambrowicz, B.P., Palmiter, R.D., 1994. Induction of a new metallo-thionein isoform (MT-IV) occurs during differentiation of stratifiedsquamous epithelia. Biochemistry 33, 7250–7259.
Ren, H., Ji, Q., Liu, Y., Ru, B., 2001. Different protective roles in vitro ofalpha- and beta-domains of growth inhibitory factor (GIF) on neuroninjuries caused by oxygen free radicals. Biochim. Biophys. Acta. 1568,129–134.
Ridet, J.L., Malhotra, A., Gage, F., 1997. Reactive astrocytes: cellular andmolecular cues to biological function. Trends Neurosci. 20, 570–577.
Rothwell, N.J., Hopkins, S.J., 1995. Cytokines and the nervous system II:actions and mechanisms of action. Trends Neurol. Sci. 18, 130–136.
Sewell, A.K., Jensen, L.T., Erickson, J.C., Palmiter, R.D., Winge, D.R.,1995. Bioactivity of metallothionein-3 correlates with its novel betadomain sequence rather than metal binding properties. Biochemistry34, 4740–4747.
Stichel, C., Verner Muller, H., 1998. Experimental strategies to promoteaxonal regeneration after traumatic central nervous system injury. Prog.Neurobiol. 56, 119–148.
Tsuji, S., Kobayashi, H., Uchida, Y., Ihara, Y., Miyatake, T., 1992. Mo-lecular cloning of human growth inhibitory factor cDNA and its down-regulation in Alzheimer’s disease. EMBO J. 11, 4843–4850.
Uchida, Y., 1999. Regulation of growth inhibitory factor expression byepidermal growth factor and interleukin-1� in cultured rat astrocytes.J. Neurochem. 73, 1945–1953.
Uchida, Y., Gomi, F., Masumizu, T., Miura, Y., 2002. Growth inhibitoryfactor prevents neurite extension and death of cortical neurons causedby high oxygen exposure through hydroxyl radical scavenging. J. Biol.Chem. 277, 32353–32359.
Uchida, Y., Ihara, Y., 1995. The N-terminal portion of growth inhibitoryfactor is sufficient for biological activity. J. Biol. Chem. 270, 3365–3369.
Uchida, Y., Takio, K., Titani, K., Ihara, Y., Tomonaga, M., 1991. Thegrowth inhibitory factor that is deficient in the Alzheimer’s diseasebrain is a 68 amino acid metallothionein-like protein. Neuron 7, 337–347.
van Lookeren Campagne, M., Thiobodeaux, H., van Bruggen, N., Cairns,B., Lowe, D.G., 2000. Increased binding activity at an antioxidant-responsive element in the metallothionein-1 promoter and rapid induc-tion of metallothionein-1 and -2 in response to cerebral ischemia andreperfusion. J. Neurosci. 20, 5200–5207.
Vela, J.M., Hidalgo, J., Gonzalez, B., Castellano, B., 1997. Induction ofmetallothionein in astrocytes and microglia in the spinal cord from themyelin-deficient jimpy mouse. Brain Res. 767, 345–355.
Yagle, M.K., Palmiter, R.D., 1985. Coordinate regulation of mouse me-tallothionein I and II genes by heavy metals and glucocorticoids. Mol.Cell. Biol. 5, 291–294.
Yamada, M., Hayashi, S., Hozumi, I., Inuzuka, T., Tsuji, S., Takahashi, H.,1996. Subcellular localization of growth inhibitory factor in rat brain:light and electron microscopic immunohistochemical studies. BrainRes. 735, 257–264.
You, H.J., Lee, K.J., Jeong, H.G., 2002. Overexpression of human metal-lothionein-III prevents hydrogen peroxide-induced oxidative stress inhuman fibroblasts. FEBS Lett. 521, 175–179.
Young, J.K., Garvey, J.S., Huang, P.C., 1991. Glial immunoreactivity formetallothionein in the rat brain. Glia 4, 602–610.
Yu, W.H., Lukiw, W.J., Bergeron, C., Niznik, H.B., Fraser, P.E., 2001.Metallothionein III is reduced in Alzheimer’s disease. Brain Res. 894,37–45.
Yuguchi, T., Kohmura, E., Yamada, K., Sakaki, T., Yamashita, T., Otsuki,H., Kataoka, K., Tsuji, S., Hayakawa, T., 1995a. Expression of growthinhibitory factor mRNA following cortical injury. J. Neurotrauma 12,299–306.
Yuguchi, T., Kohmura, E., Yamada, K., Sakaki, T., Yamashita, T., Otsuki,H., Wanaka, A., Tohyama, M., Tsuji, S., Hayakawa, T., 1995b.Changes in growth inhibitory factor mRNA expression compared withthose in c-jun mRNA expression following facial nerve transection.Mol. Brain Res. 28, 181–185.
36 J. Carrasco et al. / Neurobiology of Disease 13 (2003) 22–36




















