
ChemInform Abstract: Self-Assembled Discrete Metal-Organic Complexes: Recent Advances
30
Review Self-assembled discrete metal–organic complexes: Recent advances Rana A. Bilbeisi a , John-Carl Olsen b , Loïc J. Charbonnière c , Ali Trabolsi a,⇑ a Centre for Science and Engineering, New York University Abu Dhabi (NYUAD), Abu Dhabi, United Arab Emirates b School of Sciences, Indiana University Kokomo, Kokomo, IN 46904, USA c Laboratoire d’Ingénierie Moléculaire Appliquée à l’Analyse, IPHC, UMR 7178 CNRS/UdS, ECPM, 25 rue Becquerel, 67200 Strasbourg, France article info Article history: Available online 28 December 2013 Metals In Supramolecular Chemistry Special Issue Keywords: Chemical topology Dynamic structural interconversions Metal-organic complexes Molecular capsules Non-trivial structures Template directed self-assembly abstract Novelty, aesthetic appeal and the promise of a wide range of applications drive the current surge of inter- est in discrete metal–organic coordination complexes. This review covers achievements in the design, synthesis, characterization, and application of these multinuclear complexes in the years 2011–2013. Examples of their structural interconversion within dynamic combinatorial libraries are also presented. Ó 2014 Elsevier B.V. All rights reserved. Rana A. Bilbeisi was born and raised in Amman, Jordan. She received a BSc (Hons) degree in biochemistry from Concordia University, Canada in 2005. As part of her co-op undergraduate degree she worked in the pharmaceutical industry at MDS Pharma Services and at Merck Frosst. Rana completed her MSc degree in medicinal chemistry at McGill University, Canada in 2008. She taught chemistry at Kings Academy in Madaba, Jordan for a year before beginning her doctoral studies in science and chemistry in 2009 at Cambridge University, UK. Rana completed her PhD work on guest-binding and magnetic properties of metal-organic capsules under the supervision of Dr. Jonathan R. Nitschke in 2013. In September 2013, Rana joined the Trabolsi group as a post doctoral researcher at NYUAD in the UAE. J.-C. Olsen received a BA in English from Pomona College and a MS degree in chemistry from California State University, Northridge. He spent 2 years synthesizing MRI contrast agents at MetaProbe LLC, a small pharmaceutical company in Pasadena, California, before entering the chemistry PhD program at UCLA. His work there and as a visiting pre-doctoral scholar at Northwestern University earned him a PhD in 2010. After 2 years of postdoctoral research and teaching at the W.M. Keck Science Department in Claremont, California, he began his independent career at Indiana University Kokomo as an assistant professor. http://dx.doi.org/10.1016/j.ica.2013.12.015 0020-1693/Ó 2014 Elsevier B.V. All rights reserved. ⇑ Corresponding author. Tel.: +971 26284575. E-mail address: [email protected] (A. Trabolsi). Inorganica Chimica Acta 417 (2014) 79–108 Contents lists available at ScienceDirect Inorganica Chimica Acta journal homepage: www.elsevier.com/locate/ica
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of ChemInform Abstract: Self-Assembled Discrete Metal-Organic Complexes: Recent Advances

Inorganica Chimica Acta 417 (2014) 79–108
Contents lists available at ScienceDirect
Inorganica Chimica Acta
journal homepage: www.elsevier .com/locate / ica
Review
Self-assembled discrete metal–organic complexes: Recent advances
Rana A. Bilbeisi was born and raised in Amman, Jordan. She received a BSc (Hons) degree in biochemistry from Concordia University, Cin 2005. As part of her co-op undergraduate degree she worked in the pharmaceutical industry at MDS Pharma Services and at MerckRana completed her MSc degree in medicinal chemistry at McGill University, Canada in 2008. She taught chemistry at Kings AcadMadaba, Jordan for a year before beginning her doctoral studies in science and chemistry in 2009 at Cambridge University, UK. Rana comher PhD work on guest-binding and magnetic properties of metal-organic capsules under the supervision of Dr. Jonathan R. Nitschke inIn September 2013, Rana joined the Trabolsi group as a post doctoral researcher at NYUAD in the UAE.
J.-C. Olsen received a BA in English from Pomona College and a MS degree in chemistry from California State University, Northridge. H2 years synthesizing MRI contrast agents at MetaProbe LLC, a small pharmaceutical company in Pasadena, California, before enterichemistry PhD program at UCLA. His work there and as a visiting pre-doctoral scholar at Northwestern University earned him a PhD inAfter 2 years of postdoctoral research and teaching at the W.M. Keck Science Department in Claremont, California, he began his indepcareer at Indiana University Kokomo as an assistant professor.
http://dx.doi.org/10.1016/j.ica.2013.12.0150020-1693/� 2014 Elsevier B.V. All rights reserved.
⇑ Corresponding author. Tel.: +971 26284575.E-mail address: [email protected] (A. Trabolsi).
Rana A. Bilbeisi a, John-Carl Olsen b, Loïc J. Charbonnière c, Ali Trabolsi a,⇑a Centre for Science and Engineering, New York University Abu Dhabi (NYUAD), Abu Dhabi, United Arab Emiratesb School of Sciences, Indiana University Kokomo, Kokomo, IN 46904, USAc Laboratoire d’Ingénierie Moléculaire Appliquée à l’Analyse, IPHC, UMR 7178 CNRS/UdS, ECPM, 25 rue Becquerel, 67200 Strasbourg, France
a r t i c l e i n f o
Article history:Available online 28 December 2013Metals In Supramolecular Chemistry SpecialIssue
Keywords:Chemical topologyDynamic structural interconversionsMetal-organic complexesMolecular capsulesNon-trivial structuresTemplate directed self-assembly
a b s t r a c t
Novelty, aesthetic appeal and the promise of a wide range of applications drive the current surge of inter-est in discrete metal–organic coordination complexes. This review covers achievements in the design,synthesis, characterization, and application of these multinuclear complexes in the years 2011–2013.Examples of their structural interconversion within dynamic combinatorial libraries are also presented.
� 2014 Elsevier B.V. All rights reserved.
anadaFrosst.
emy inpleted2013.
e spentng the
2010.endent

Loïc J. Charbonnière received a BS degree in chemical engineering in 1991, followed by a MS in organic chemistry in 1993 (University ofStrasbourg, France). He obtained his PhD from the University of Geneva (Switzerland) in 1996 and joined the research groups of Pr. Jean-Claude Bünzli’s (Lausanne, Switzerland) and Dr. Françoise Arnaud’s (Strasbourg) as a postdoctoral scholar. In 1998, he was appointed assistantresearcher at the French National Centre for Scientific Research (CNRS). Since 2011, he has been Director of Research at the CNRS and head ofthe Laboratory of Molecular Engineering for Analytical Applications in Strasbourg. His main scientific interests are organic synthesis andcoordination chemistry applied to imaging and diagnostic technologies.
Ali Trabolsi received his BSc degree in chemistry from the Lebanese University in Beirut. In 2002, he moved to Strasbourg, France, where heobtained a MSc (2003) degree in analytical chemistry and a PhD (2006) degree in physical chemistry. He then joined the group of ProfessorFraser Stoddart as a postdoctoral scholar at UCLA where he spent 1 year before relocating with the group to Northwestern University. From2009 to 2011, he worked at KAUST in Saudi Arabia as a research scientist in the membrane center. Now an assistant professor at New YorkUniversity Abu Dhabi, Dr. Trabolsi pursues research in the areas of molecular topology, supramolecular chemistry, and materials chemistry.
80 R.A. Bilbeisi et al. / Inorganica Chimica Acta 417 (2014) 79–108
Contents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 802. Container molecules. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 81
2.1. Polyhedral structures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 81
2.1.1. Tetrahedral structures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 812.1.2. Cubic structures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 832.1.3. Octahedral structures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 832.1.4. Icosahedral structures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 842.1.5. Archimedean structures/container molecules. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 852.2. Larger spherical structures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 85
3. Topologically non-trivial structures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 873.1. Catenanes. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 873.2. Knots . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 913.3. Ravels . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 933.4. Interlocked cages. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 94
4. Miscellaneous structures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 95
4.1. Metallomacrocycles. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 954.2. Helicates . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 984.3. Cyclic helicates . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 994.4. Grids. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1005. Structural Interconversions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1026. Conclusions. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 107
Acknowledgments . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 107References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 107
1. Introduction
In nature, supramolecular self-assembly is ubiquitous. Numer-ous examples exist whereby weak interactions, such as hydro-gen-bonding, van der Waals, p–p stacking, dipole–dipole, andmetal coordination, direct the organization of multiple compo-nents into discrete, complex architectures that, in most cases, havevital functions. Representative examples of biological assembliesinclude: (i) viral capsids, which are assembled from identical pro-tein subunits [1], and (ii) the octahedral iron storage protein apo-
ferritin, which is composed of 24 non-covalently linked proteinsubunits [2].
Owing to the weakness of the forces involved, supramolecularself-assembly presents significant synthetic challenges. However,by following nature’s lead [3–5], chemists, over the past 50 yearsor thereabouts, have begun to develop effective strategies for cre-ating complex yet stable supramolecular assemblies with diversepotential applications [6,7]. Metal cations, in particular, have pro-ven to be effective synthetic tools. Metal-coordination is, more of-ten than not, the strongest of the weak intermolecular forces, and,

R.A. Bilbeisi et al. / Inorganica Chimica Acta 417 (2014) 79–108 81
with the transition metals, provides highly directional bonding andtherefore structural control. Metals, then, are excellent templatesfor the self-assembly of discrete 2D and 3D supramolecular con-structs including topologically non-trivial ones such as knots andlinks, and various coordination-based synthetic approaches havebeen developed [8–19].
In this review we highlight synthetic developments, structuralachievements, and practical applications in the field of discretemetallosupramolecular self-assembly that have been publishedduring the past 3 years, 2011–2013. The review is divided into fourmain sections; the first three describe, respectively, (i) metal–or-ganic capsules, (ii) topologically non-trivial structures and (iii) anassortment of grids, helicates, metallomacrocycles and novel struc-tures. The last section (iv) discusses the dynamic nature of com-plexes that enables structural transformation and interconversionupon the addition of an external stimulus.
2. Container molecules
As a consequence of their well defined inner cavities which arecapable of binding guest molecules, metal–organic container com-plexes [20–28] have applications in the areas of separations tech-nology (involving anions [29,30], fullerenes [31–34] and gases[35]), catalysis [10–15], photochemistry [36–38], and sensing[39–41]. In this section we present examples of highly symmetricalcapsules with Platonic (in which the faces of containers are definedby a regular polygon) and Archimedean (where the faces are de-fined by two or more polygons) structures, as well as examples oflarger more spherical assemblies prepared through self-assembly.
2.1. Polyhedral structures
2.1.1. Tetrahedral structuresTetrahedral metal–organic capsules are usually prepared by two
main methods that are characterized by the geometry of the multi-dentate ligand used. Edge-directed self-assembly [30,42–48,21] incor-porates octahedral metal ions and linear C2-symmetric ligands havingone bidentate coordination site on each end. Each ligand bridges two
N12
4 FeII
CH3CN
ON
N
NN
NH2
NH2H2N
4
Fe2+
Fe2+
Fig. 1. Subcomponent self-assembly of FeII4L4 tetrahedral capsules: mixing triamines 1–4
capsules. Crystal structure of capsules 5–7 and the MM2 model capsule 8.
vertices and defines an edge of an M4L6 tetrahedron. Alternatively,face-directed synthesis [19,49,50] employs C3-symmetric trisbiden-tate ligands and octahedrally coordinated metal ions. Each ligandbridges three vertices and defines one face of an M4L4 tetrahedron.Numerous tetrahedral capsules have been synthesized in the past3 years. Here, we discuss three representative examples.
Nitschke and coworkers have developed an elegant face-direc-ted synthetic protocol for FeII
4L4 capsules [51]. The capsules self-assemble in solution from three subcomponents: C3-symmetric tri-amines, 2-formylpyridine and iron(II) salts as depicted in Fig. 1. Thecondensation of a triamine with three 2-formylpyridine units yieldsa C3-symmetric ligand that coordinates iron(II) and defines the tetra-hedral faces. Four triamine building blocks (1–4, with 1, 3 and 4 beingflat and 2 pyramidal) were tested. Reaction of all four with 2-formyl-pyridine and iron(II) yielded FeII
4L4 cages of various sizes.Guest binding properties of the capsules were investigated and itwas found that only capsule 7 has a sufficiently large and well-en-closed cavity to bind a wide variety of organic guests in a size andshape-selective fashion.The wide scope of this method was demonstrated in subsequentstudies involving different metal ions [52], aldehydes [39,53],and triamines [52].
Custelcean and coworkers have reported the design and synthe-sis of a novel class of urea based tetrahedral M4L6 capsules [54].Their assembly is achieved through the coordination of ligand 9(Scheme 1) to octahedral metal ions (either ZnII or NiII) in the pres-ence of a templating tetrahedral anion. The capsules’ cavities arefunctionalized with six anion-binding urea groups [55], resultingin size and charge selective encapsulation of hydrophilic oxoanionsEO4
n� (where E = S, Cr, Se, Mo or W when n = 2 or P when n = 3).The tetrahedral anions form hydrogen bonds with the centrallydirected urea groups and template the formation of the capsulesin aqueous media. Encapsulation of such strongly hydrophilic an-ions is unusual, but can be explained by the stabilization of theEO4
n� �M4L6 complex provided by twelve hydrogen bonds. In theabsence of a templating multi-charged tetrahedral anion, theM4L6 capsules do not form. The binding affinities and selectivitiesfor EO4
n� oxoanions were investigated through anion exchange
5
6
7
8
N
HO
NNFe2+
≡
Fe2+
4
3
2
1
, 2-formylpyridine, and iron(II) salts resulted in the formation of the corresponding

9 10
NN
NHO
NH
NN
4 Zn(NO3)2Na2MoO4
MeOH/H2O6
Scheme 1. Self-assembly of anion templated ZnII4L6 and X-ray crystal structure of the capsule 10.
82 R.A. Bilbeisi et al. / Inorganica Chimica Acta 417 (2014) 79–108
experiments using 77SeO42� as an anionic probe monitored by 77Se
NMR spectroscopy. The selectivity trend among the tetrahedtralanions is: PO4
3�� CrO42� > SO4
2� > SeO42� > MoO4
2� > WO42�. Factors
that contribute to the observed selectivity include: anion size,charge, hydration, hydrogen-bond acceptor ability and basicity.
Chiral molecular capsules possess chirotopic internal cavities[46,56–60] that allow for the enatioselective recognition andseparation of chiral guests. Chiral capsules also provide an asym-metric environment for stereo-selective reactions to take place[7,57,61,62].Nitschke and coworkers have described the diastereoselective sub-component self-assembly of FeII
4L6 capsules (Scheme 2) through the
*H2N
H
N
N
O
O+
12
6
N
N
N
N
Fe2+
Fe2+
Fe2+
FeII
NH2
NH2
O*
OHOH
O*HOOH
6
N
O+
12
FeII
ΔΔΔΔΔΔΔΔ - 13
15
11
S-12
(a)
(b)
Scheme 2. Diastereoselective formation of tet
incorporation of enantiopure building blocks [60,63]. Capsule 13 [6],with twelve stereogenic centers arrayed on its periphery, wasprepared from (S)-1-phenylethylamine (S-12), 6,6-diformyl-3,3-bipyridine (11) and iron(II) triflate (Scheme 2a). NMR and ESI-MSmeasurements in solution and single-crystal X-ray data confirmedits structure. Diastereoselective formation of the same DDDD-isomer,S-12 was further confirmed by circular dichroism spectroscopy (CD).CD analysis of the KKKK-enantiomer, R-12, prepared from(R)-1-phenylethylamine, resulted in the expected mirror image CDspectrum. The authors also investigated the conversion of a racemiccapsule, 14, to the optically active capsule 13 through the additionof S-12 to capsule 14. CD measurements indicated that two S-12 sub-
Fe2+
N
N
N
N
Fe2+
Fe2+Fe2+
Fe2+
*H2N
H
NH2
N
N
N
N
OO
*OH
OH*
HO
HO
Fe2+
Fe2+
Fe2+
Fe2+
ΔΔΔΔΔΔΔΔ - 16
Racemic- 14
rahedral FeII4L6 capsules (a) 13 and (b) 16.
R.A. Bilbeisi et al. / Inorganica Chimica Acta 417 (2014) 79–108 83
components per capsule are sufficient to induce all of the capsule’s FeII
stereocentres to adopt the same D configurational twist, a resultwhich reflects cooperative communication between metal centers.
In a follow-up investigation, Nitschke et al. prepared a watersoluble enantiopure capsule FeII
4L6 with a chiral core rather than achiral periphery. Capsule 16 (Scheme 2b) was assembled from anenantiopure diaminoterphenylene (bearing chiral glyceryl groups),2-formylpyridine and iron(II) sulfate. CD analysis indicated thatthe chirality of the glyceryl groups dictates the stereochemistry ofthe iron(II) centers. Capsule 16 possesses a large cavity volume thatwas observed to bind a wide range of organic guest molecules.
2.1.2. Cubic structuresLike the tethrahedrons, metal–organic cubic capsules can be
prepared by either the edge-directed or face-directed methods toform M8L12 or face-capped M8L6 complexes, depending on thegeometry of the ligand used. Two representative examples of awide range of recently reported cubic capsules [64–67] are high-lighted below.
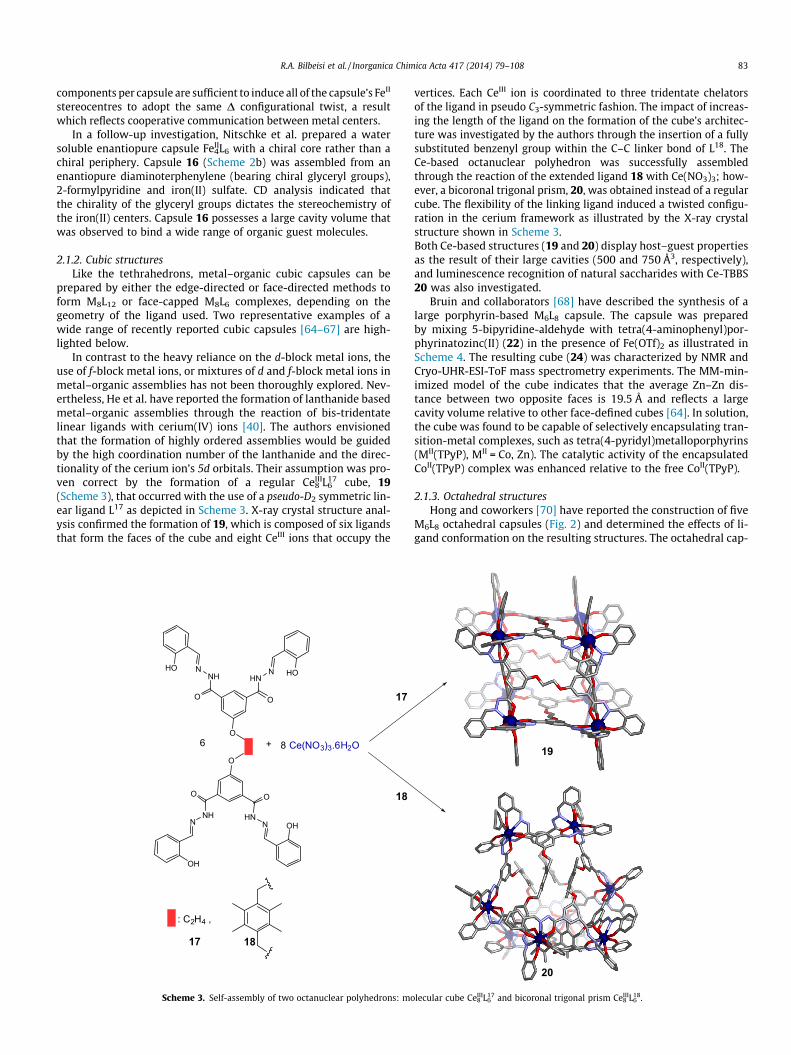
In contrast to the heavy reliance on the d-block metal ions, theuse of f-block metal ions, or mixtures of d and f-block metal ions inmetal–organic assemblies has not been thoroughly explored. Nev-ertheless, He et al. have reported the formation of lanthanide basedmetal–organic assemblies through the reaction of bis-tridentatelinear ligands with cerium(IV) ions [40]. The authors envisionedthat the formation of highly ordered assemblies would be guidedby the high coordination number of the lanthanide and the direc-tionality of the cerium ion’s 5d orbitals. Their assumption was pro-ven correct by the formation of a regular CeIII
8 L176 cube, 19
(Scheme 3), that occurred with the use of a pseudo-D2 symmetric lin-ear ligand L17 as depicted in Scheme 3. X-ray crystal structure anal-ysis confirmed the formation of 19, which is composed of six ligandsthat form the faces of the cube and eight CeIII ions that occupy the
NH
O O
HNN OHN
OH
NH
O O
HNNNHO
O
O
HO
6 8 Ce(NO3)3.6H2O
: C2H4 ,
+
17 18
17
18
Scheme 3. Self-assembly of two octanuclear polyhedrons: mo
vertices. Each CeIII ion is coordinated to three tridentate chelatorsof the ligand in pseudo C3-symmetric fashion. The impact of increas-ing the length of the ligand on the formation of the cube’s architec-ture was investigated by the authors through the insertion of a fullysubstituted benzenyl group within the C–C linker bond of L18. TheCe-based octanuclear polyhedron was successfully assembledthrough the reaction of the extended ligand 18 with Ce(NO3)3; how-ever, a bicoronal trigonal prism, 20, was obtained instead of a regularcube. The flexibility of the linking ligand induced a twisted configu-ration in the cerium framework as illustrated by the X-ray crystalstructure shown in Scheme 3.Both Ce-based structures (19 and 20) display host–guest propertiesas the result of their large cavities (500 and 750 Å3, respectively),and luminescence recognition of natural saccharides with Ce-TBBS20 was also investigated.
Bruin and collaborators [68] have described the synthesis of alarge porphyrin-based M6L8 capsule. The capsule was preparedby mixing 5-bipyridine-aldehyde with tetra(4-aminophenyl)por-phyrinatozinc(II) (22) in the presence of Fe(OTf)2 as illustrated inScheme 4. The resulting cube (24) was characterized by NMR andCryo-UHR-ESI-ToF mass spectrometry experiments. The MM-min-imized model of the cube indicates that the average Zn–Zn dis-tance between two opposite faces is 19.5 Å and reflects a largecavity volume relative to other face-defined cubes [64]. In solution,the cube was found to be capable of selectively encapsulating tran-sition-metal complexes, such as tetra(4-pyridyl)metalloporphyrins(MII(TPyP), MII = Co, Zn). The catalytic activity of the encapsulatedCoII(TPyP) complex was enhanced relative to the free CoII(TPyP).
2.1.3. Octahedral structuresHong and coworkers [70] have reported the construction of five
M6L8 octahedral capsules (Fig. 2) and determined the effects of li-gand conformation on the resulting structures. The octahedral cap-
19
20
lecular cube CeIII8 L17
6 and bicoronal trigonal prism CeIII8 L18
6 .

Scheme 4. Subcomponent self-assembly of capsule 24 through two synthetic routes. (Adapted with permission from Ref. [68]).
PO
HNHN NH
N
N
N
PO
HNHN NH
N
N
N
Syn-25
Anti-25
CoII-26PdII-27
Fig. 2. Two conformations of 25 observed in capsules PdII-27 and CoII-26; X-ray structures of capsules 27 and 26 constructed from the syn and anti-forms of the ligand,respectively.
84 R.A. Bilbeisi et al. / Inorganica Chimica Acta 417 (2014) 79–108
sules were prepared through the reaction of a tripodal ligand,N,N0,N00-tris(3-pyridinyl)phosphoric triamide (TPPA), and any offive transition-metal ions [M = NiII, CoII, ZnII, CdII and PdII]. Six me-tal ions occupy vertices and form an octahedral framework, andthe eight ligands span the framework’s faces. Single crystal X-rayanalysis in addition to NMR and MS data confirmed the existenceof the capsules in the solid state and in solution. Solid state analy-sis of the capsules revealed that they possess large internal cavitiesand flexible windows. The ligand itself is flexible and can adopteither a syn or an anti conformation, and this flexibility affectsthe internal cavity volume of the capsules. With NiII, CoII, ZnII orCdII the ligand has a syn conformation, whereas with PdII, the li-gand assumes the anti conformation. The switch form syn to antiresults in an increase in the size of the portals and the internal cav-ity of the capsule.
2.1.4. Icosahedral structuresPasquale et al. have described giant [71], highly symmetrical
polyhedral capsules that self-assemble from calixarene carboxyl-
OHOH
OH HO
HO
HOOC
COOH
COOH
COOHHOOC
20 UO22+
pyr12
28
Scheme 5. Synthesis and X-ray crystal structure of 29 of an icoasa
ates and the uranyl cation, UO22+, under mild conditions (pyridine
base in DMF). Calix[4]arene and calix[5]arene yield an octahedralcomplex and an icosahedral complex 29 (Scheme 5), respectively.The ligands display a wide-open cone conformation at the verticesof the structure and bridge the metal centers which reside on thefaces [72]. The structures are anionic, with each metal contributingonly one negative charge. Few counter cations are required forneutrality, which leaves considerable space for guest encapsula-tion. X-ray crystal data show a radius of 1.4 Å for the larger, icoasa-hedral capsule. Considering their large sizes, they are composed ofrelatively few components. The icosahedral capsule 29, for exam-ple, consists of 32 subcomponents and 20 counterions (as com-pared to Fujita’s slightly larger capsule [73] which is made of 72subcomponents and 24 counterions). Studies of the photolumines-cent and photocatalytic properties of these assemblies are poten-tial avenues for further research.
Nitschke and coworkers have reported the synthesis of an FeII12-
L12 capsule 30 with an icosahedral framework (Scheme 6) from theC3-symmetric triamine 1,3,5-tri(4-aminophenyl)benzene and 2-
29
hedral coordination capsule from a calix[5]arene and uranyl.

Fe(OTf)2
NH2H2N
NH2
NH
O
12
4
4
CH3OH:CH3CN (1:1)70 oC
3
30
Scheme 6. Synthesis and X-ray crystal structure of icoasahedral 30.
R.A. Bilbeisi et al. / Inorganica Chimica Acta 417 (2014) 79–108 85
formylpyridine in the presence of iron(II) triflate in a polar solvent.Formation of the capsule was confirmed by NMR, MS and X-ray crys-tallographic analyses. The twelve iron(II) centers occupy the vertices,and the twelve tris-bidentate ligands cap twelve of the twenty facespresent in the regular icosahedron as depicted by the X-ray crystalstructure presented in Scheme 6. All iron(II) centers in 30 display alower symmetry meridional (mer) coordination geometry, and over-all, the structure possesses T-symmetry. The icosahedron encloses alarge cavity with a volume of approximately 2768 ÅA
03, allowing for
the encapsulation of large guests. The capsule was found to binddodecafluoro-closo-dodecaborate ([B12F12]2�) selectively in solutionover other screened anionic guests.
2.1.5. Archimedean structures/container moleculesSeveral metal–organic capsules with Archimedean structures
were published over the past 3 years as highlighted by the workof Ward [74], Fujita [75], Hong [76], Zhang [77].
Fujita and coworkers [75] reported the self-assembly of a stel-lated cuboctahedron through post-stellation of a cuboctahedronby metal coordination. The M12L24 cubotahedron was assembledthrough the coordination of tris(4-pyridyl) ligands to Pd(II) metalions as presented in Scheme 7. The assembly of the M12L24 takesplace through the selective complexation of two coordination sitesof tris(4-pyridyl) to Pd(II) leaving an uncoordinated site. This freecoordination site complexes to additionally added Pd(II) ions toform a stellated M18L24 cuboctahedron as shown in Scheme 7. For-mation of the stellated cuboctahedron was confirmed in solutionby NMR measurements and in the solid state as demonstrated by
NN
O
N
12 Pd(II)24
Scheme 7. Self-assembly of M12L24 cubotahedron and the subsequent reversible converswith permission from [75]).
the X-ray analysis. The formation of the stellated structure isreversible upon removal of the six metal ions from the vertices.
2.2. Larger spherical structures
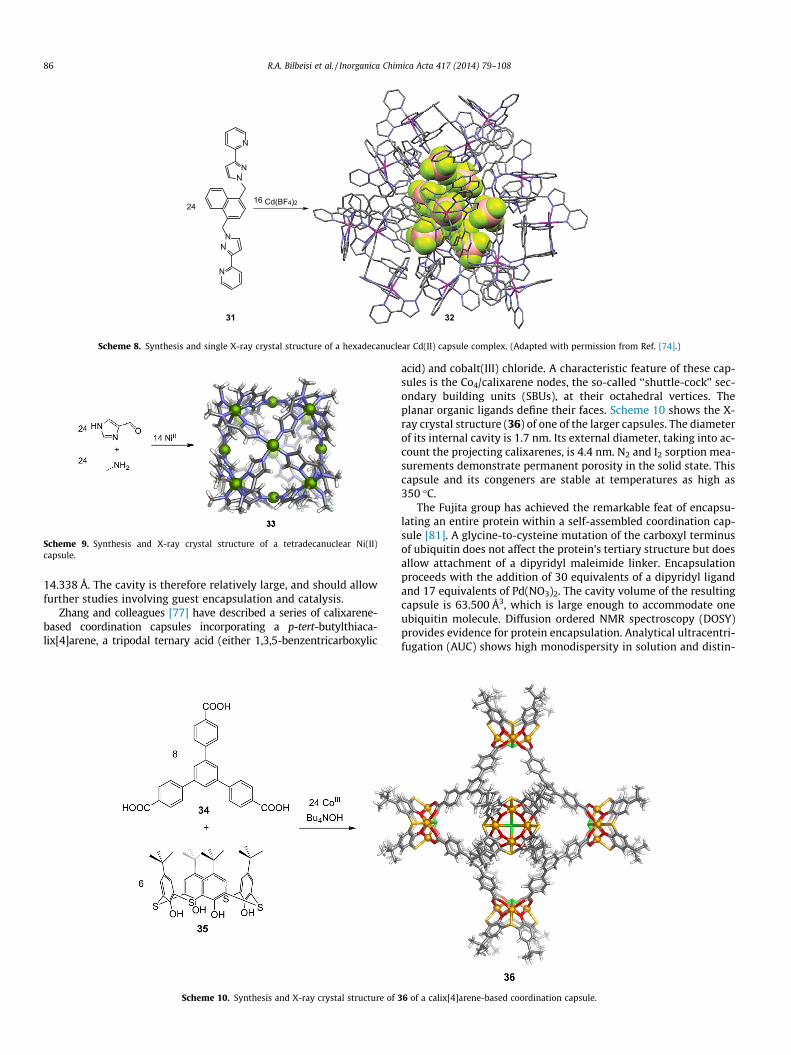
Ward and coworkers have reported that a large spherical Cd16
coordination capsule (Scheme 8) forms when the flexible ditopicnaphthyl ligand 31 is mixed with Cd(II) tetrafluoroborate in organ-ic solvents [74]. The naphthyl moieties of the ligands provide, viainter-ligand p-stacking, a major element of stability, as well as adiagnostic fluorescent marker for capsule formation—a red-shiftedexciplex-like emission. The capsule consists of cyclic, helical sub-units of formula CdII
3L3. Provided that the subunits can be pre-formed, this feature might be adapted to allow for a more rational,stepwise approach to the synthesis of larger capsules by subcompo-nent self-assembly [78,79]. The X-ray crystal structure of 32 is pre-sented in Scheme 8. Its central cavity has an approximate volumeof 1300 Å3 and was found to contain nine tetrafluoroborate counte-rions. The capsule’s relative stability in solution and large cavity vol-ume should permit it to function as a supramolecular host.
Zhou et al. [80] have reported the self-assembly of 62 compo-nents including 4-formylimidazole, methyl amine, and Ni(II) intoa highly symmetrical tetrahedral structure (Scheme 9). Alterna-tively, combination of the pre-formed imine ligand and Ni(II) re-sults in the same structure, in which, notably, both octahedraland square planar Ni(II) complexes are present. The Ni–Ni distancebetween two opposite square planar Ni(II) sites is 11.339 Å, andthe diagonal distance between two octahedral Ni(II) sites is
6 Pd(II)
ion between the cuboctahedron and an M18L24 stellated cuboctahedron (reproduced
N
N
N
N
N
N
16 Cd(BF4)224
31 32
Scheme 8. Synthesis and single X-ray crystal structure of a hexadecanuclear Cd(II) capsule complex. (Adapted with permission from Ref. [74].)
Scheme 9. Synthesis and X-ray crystal structure of a tetradecanuclear Ni(II)capsule.
86 R.A. Bilbeisi et al. / Inorganica Chimica Acta 417 (2014) 79–108
14.338 Å. The cavity is therefore relatively large, and should allowfurther studies involving guest encapsulation and catalysis.
Zhang and colleagues [77] have described a series of calixarene-based coordination capsules incorporating a p-tert-butylthiaca-lix[4]arene, a tripodal ternary acid (either 1,3,5-benzentricarboxylic
Scheme 10. Synthesis and X-ray crystal structure of 3
acid) and cobalt(III) chloride. A characteristic feature of these cap-sules is the Co4/calixarene nodes, the so-called ‘‘shuttle-cock’’ sec-ondary building units (SBUs), at their octahedral vertices. Theplanar organic ligands define their faces. Scheme 10 shows the X-ray crystal structure (36) of one of the larger capsules. The diameterof its internal cavity is 1.7 nm. Its external diameter, taking into ac-count the projecting calixarenes, is 4.4 nm. N2 and I2 sorption mea-surements demonstrate permanent porosity in the solid state. Thiscapsule and its congeners are stable at temperatures as high as350 �C.
The Fujita group has achieved the remarkable feat of encapsu-lating an entire protein within a self-assembled coordination cap-sule [81]. A glycine-to-cysteine mutation of the carboxyl terminusof ubiquitin does not affect the protein’s tertiary structure but doesallow attachment of a dipyridyl maleimide linker. Encapsulationproceeds with the addition of 30 equivalents of a dipyridyl ligandand 17 equivalents of Pd(NO3)2. The cavity volume of the resultingcapsule is 63.500 Å3, which is large enough to accommodate oneubiquitin molecule. Diffusion ordered NMR spectroscopy (DOSY)provides evidence for protein encapsulation. Analytical ultracentri-fugation (AUC) shows high monodispersity in solution and distin-
6 of a calix[4]arene-based coordination capsule.

N N
N+(CH3)3NO3-
CH3
N N
O
NO O
S
ubiquitin
23
12 PdII
37
38 39
Scheme 11. Encapsulation of ubiquitin in a metal–organic capsule complex. (Adapted with permission from Ref. [81].)
R.A. Bilbeisi et al. / Inorganica Chimica Acta 417 (2014) 79–108 87
guishes empty from ubiquitin-containing spheres. Gel electropho-retic analysis (SDS–PAGE) confirms encapsulation, as does X-raycrystallography coupled with maximum entropy measurement(MEM) analysis. Scheme 11 shows an illustration of the encapsu-lated protein. The ultimate goal of this line of work is to achieveconformational and functional control of encapsulated proteins[81].
3. Topologically non-trivial structures
This section is devoted to topologically non-trivial metal–or-ganic architectures. Thorough descriptions of such molecules andthe mathematical approach of chemical topology, in general, canbe found in excellent reviews on the subject [82]. Here, we focuson recently described structures.
Topological isomers are defined as molecules that have the samemolecular formulas and bond connectivities but that cannot beinterconverted by any deformative action in the three-dimensionalspace [83]. Such molecules can be classified according to the num-ber of components and nodes that they contain (where a node is acrossing point in the two-dimensional graph-representation of themolecule). Following this approach, this section is organized bystructure, with simpler links at the beginning followed by thosethat have greater numbers of components.
N
N
N
N
N
N
N
N
NN
N
N
CuI
CuI
40 41
Scheme 12. Quantitative formation of a [2] c
3.1. Catenanes
The simplest topologically non-trivial structure, the [2]catenaneor ‘‘Hopf link’’, played a pivotal historical role, as it was the firstmechanically interlocked structure to be synthesized in high yield.The landmark achievement in the synthesis of a metal templated[2]catenane was accomplished by Sauvage and coworkers who re-lied on Cu(I) assisted templation to produce a metallo-catenane[84].
In a recent variation on the original theme, the ring closingsteps were accomplished by coordinative bond formation betweenpyridines and Zn-porphyrins rather than by double alkylationwhich produced a final catenated structure (42) in quantitativeyield (Scheme 12) [85].In a first step, a Cu(I) complex was obtained by the strongcoordination of two phenantroline units functionalized at the 2and 9 positions by para-phenyl-4-pyridines. The rings were thenclosed quantitatively by the introduction of two strands eachcomposed of two Zn porphyrins judiciously spaced by an aromaticlinker. The apical coordination of the terminal pyridines to theZn-porphyrins afforded the [2]catenane 42. The formation of thecatenane was monitored by UV–Vis absoption spectroscopytitration experiments. Formation of the ‘‘free’’ macrocycle wasmonitored in the absence of Cu(I). The association constants for
N NNN Zn
N NNN Zn
R
RR
RR
R
NN
N NZn
NN
N NZn
R
R
R
R
R
R
N
N
N
N
NN
N
N
CuI
42
atenane by coordination chemistry [85].

88 R.A. Bilbeisi et al. / Inorganica Chimica Acta 417 (2014) 79–108
both processes (4 � 1010 M�1, and 2 � 105 M�1, respectively)indicate that no cooperative effects are present during catenaneformation.
Taking advantage of the Cu(I) templation, Schuster and cowork-ers have optimized protocols for the preparation of catenanes androtaxanes that incorporate fullerene and Zn-porphyrin moieties asphotoactive components for charge separation (Scheme 13) [86].In these cases, the final linking steps were achieved byCu(I)-catalyzed-1,3-dipolar ‘‘click’’ cycloaddition of organic azides(on the phenantroline strands) to terminal alkynes (on the porphy-rin-containing components). With this tactic, the authors wereable to isolate a [3]catenane (45) that incorporates two fullerenerings. Interestingly, the Cu(I) bis-phenantroline core was not onlyused for its templating effect, but also for its intrinsic photoactivitywhich allows it to act as a relay in electron transfer processes. Afollow-up article [87], describes a comparative study of a seriesof these catenanes. Based on steady-state emission and transientabsorption spectroscopy, the photoexcitation of the compoundsled to the formation of a long lived ZnPþ=C�60 charge separated rad-ical pair. Catenation was shown to increase the distance betweenthe donor and acceptor moieties and resulted in a stabilized andlonger lived excited state. In addition, the removal of the pivotalCu(I) templating cations resulted in an increased motility of thestrands that ultimately led to quenching of the charge separatedstate.
At the nanoscopic scale, the Cu(I) templating strategy was em-ployed by Telfer and coworkers [88] to join gold and silver nano-particles. In this case, the phenanthroline ligands were
46
N
N
N
O
OO
O
FeII
Scheme 14. Catenane formation using a tridentate dpiq-py ligand around oct
43
44
45
Scheme 13. Cu(I)-templated synthesis of catenanes containing photoactive probesfor charge separation.
terminated by thiol groups and the final closing step consisted inthe formation of thiol-metal bonds at the surfaces of the nanopar-ticles. The interlocked nanoparticles where studied by dynamiclight scattering, transmission electron microscopy and surface-en-hanced Raman scattering, and as a function of the Cu(I) precaten-ane/nanoparticle ratio.
Although the tetrahedral coordination of Cu(I) was historicallythe first templation method to be developed, an increasing numberof catenanes that incorporate octahedral complexes are being de-scribed. Sauvage and Sour recently reported the design of a newtridentate ligand based on (dpiq-py, Scheme 14) [89].
This new ligand, 46 (Scheme 14), was developed to avoid a ste-rically unfavorable pinching effect that is present when two triden-tate terpyridines coordinate to octahedral cations. With dpiq-py,the authors were able to link ligand strands by using ring closingmetathesis. The product alkenes were subsequently reduced toavoid isolation of mixtures of cis and trans stereoisomers. Thelinking/reduction sequence could be applied to the Fe(II) andCo(III) complexes to give overall chemical yields of 75% and 42%,respectively.
Octahedral coordination was also used by Trabolsi, Char-bonnière and coworkers who isolated a Zn(II)-containing [2]caten-ane by dynamic coordination chemistry [90]. [The self-assemblyprocess produced a library a topologically non-trivial structureswhose major component was a trefoil knot (see Section 4.2)].
Leigh et al. took advantage of the different stereochemical pref-erences of Pd(II) and Co(III) to achieve a half rotation in a [2]caten-ane [91]. Starting from (i) a macrocycle that featured a monodentatepyridine site and a tridentate picoline diamide and (ii) a PdL(CH3CN)complex in which L was a tridentate ligand with appended terminalvinyl groups, a square planar Pd complex was formed. Closure of thesecond macrocycle was achieved by ring-closing metathesis fol-lowed by alkene reduction. Potassium cyanide displaced the Pd(II)to furnish the metal-free [2]catenane. Addition of Co(II) in basicmedia, followed by oxidation to Co(III) produced a new metallocatenane in which the Co(III) was coordinated by two tridentate pic-oline diamindes to achieve a coordination number of 6. The metalexchange phenomenon led to a net half rotation of one of themacrocycles.
Piguet and coworkers have taken advantage of octahedral coor-dination around Fe(II) to clip together two ligands, one containinga central tridentate coordination site and two terminal bidentatecoordination sites. Once coordinated, the addition of two Ag(I) cat-ions resulted in the exclusive formation of a trimetallic [2]caten-ane, and the thermodynamic parameters governing the assemblywere studied to understand the reason for the catenane’s beingfavored over the expected helicate [92].
An interesting way of using octahedral coordination complexesto form catenanes was developed by Liu and coworkers [93]. Theirlink-forming reaction involved coordination of two terminal phe-
47
ahedral complexes and the X-ray crystal structure of the Fe(II) catenane.
Sn
O
O O O O
+H2N
O
O O O O
NNN N
Ph
Ph
Ph
PhO
O OOO
OOO
O
O O O O
+H2N
O
O O O O
NNN N
Ph
Ph
Ph SnO
O OOO
OOO
48 49
Scheme 15. Tin based [2]catenanes obtained by the axial coordination of phenolate on a metal porphyrine [93].
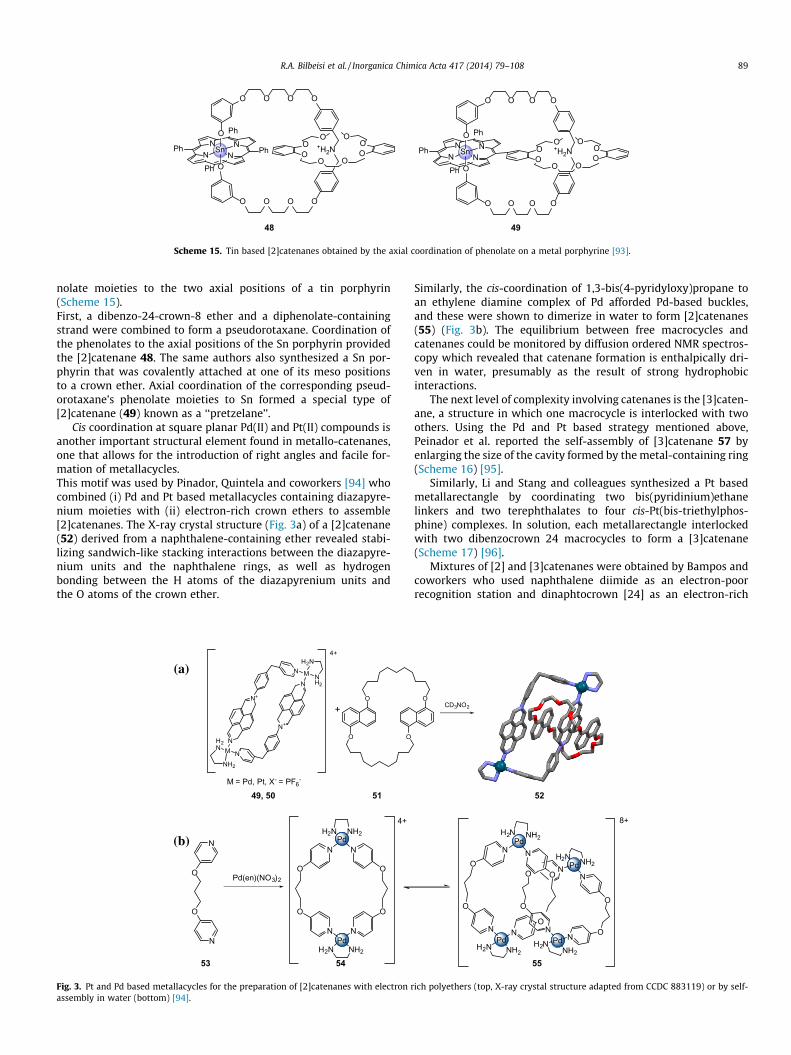
R.A. Bilbeisi et al. / Inorganica Chimica Acta 417 (2014) 79–108 89
nolate moieties to the two axial positions of a tin porphyrin(Scheme 15).First, a dibenzo-24-crown-8 ether and a diphenolate-containingstrand were combined to form a pseudorotaxane. Coordination ofthe phenolates to the axial positions of the Sn porphyrin providedthe [2]catenane 48. The same authors also synthesized a Sn por-phyrin that was covalently attached at one of its meso positionsto a crown ether. Axial coordination of the corresponding pseud-orotaxane’s phenolate moieties to Sn formed a special type of[2]catenane (49) known as a ‘‘pretzelane’’.
Cis coordination at square planar Pd(II) and Pt(II) compounds isanother important structural element found in metallo-catenanes,one that allows for the introduction of right angles and facile for-mation of metallacycles.This motif was used by Pinador, Quintela and coworkers [94] whocombined (i) Pd and Pt based metallacycles containing diazapyre-nium moieties with (ii) electron-rich crown ethers to assemble[2]catenanes. The X-ray crystal structure (Fig. 3a) of a [2]catenane(52) derived from a naphthalene-containing ether revealed stabi-lizing sandwich-like stacking interactions between the diazapyre-nium units and the naphthalene rings, as well as hydrogenbonding between the H atoms of the diazapyrenium units andthe O atoms of the crown ether.
N
O
O
NN
O
O
N N
O
O
N
Pd
PdH2N NH2
H2N NH2
Pd(en)(NO3)2
4+
(a)
(b)
49, 50 51
53 54
N+
N
N
N+
N
NM
M
H2N
NH2
H2N
NH2
M = Pd, Pt, X- = PF6-
O
O O
4+
+
Fig. 3. Pt and Pd based metallacycles for the preparation of [2]catenanes with electron rassembly in water (bottom) [94].
Similarly, the cis-coordination of 1,3-bis(4-pyridyloxy)propane toan ethylene diamine complex of Pd afforded Pd-based buckles,and these were shown to dimerize in water to form [2]catenanes(55) (Fig. 3b). The equilibrium between free macrocycles andcatenanes could be monitored by diffusion ordered NMR spectros-copy which revealed that catenane formation is enthalpically dri-ven in water, presumably as the result of strong hydrophobicinteractions.
The next level of complexity involving catenanes is the [3]caten-ane, a structure in which one macrocycle is interlocked with twoothers. Using the Pd and Pt based strategy mentioned above,Peinador et al. reported the self-assembly of [3]catenane 57 byenlarging the size of the cavity formed by the metal-containing ring(Scheme 16) [95].
Similarly, Li and Stang and colleagues synthesized a Pt basedmetallarectangle by coordinating two bis(pyridinium)ethanelinkers and two terephthalates to four cis-Pt(bis-triethylphos-phine) complexes. In solution, each metallarectangle interlockedwith two dibenzocrown 24 macrocycles to form a [3]catenane(Scheme 17) [96].
Mixtures of [2] and [3]catenanes were obtained by Bampos andcoworkers who used naphthalene diimide as an electron-poorrecognition station and dinaphtocrown [24] as an electron-rich
N
O
O
N N
O
N
Pd
PdH2N NH2
H2N NH2
NO
O
NN O
O
N
Pd
PdH2N NH2
H2N NH2
O
8+
52
55
OCD3NO2
ich polyethers (top, X-ray crystal structure adapted from CCDC 883119) or by self-

N+
N+
N
N
+ 4 Pt
PEt3OTf
OTf
Et3P
+ 2
O ONa
ONaO
N+
N+
N
N
Pt
PEt3Et3P
Pt
PEt3
Et3P
O
O O
O
N+
N+
N
N
Pt
PEt3
PEt3
Pt
PEt3PEt3
O
OO
O
DB24C8N+
N+
N
N
Pt
PEt3Et3P
Pt
PEt3
Et3P
O
O O
O
N+
N+
N
N
Pt
PEt3
PEt3
Pt
PEt3PEt3
O
OO
O
O OO
OO
O
O
O OO
OO
O
O
58 60
61
62 63
Scheme 17. Formation of a [3]catenane from the self-assembly of dibenzocrown 24 ethers and a Pt based metallarectangle and the corresponding X-ray crystal structure.
N+
N+
N+
N+
M
H2N
NH2
MII = Pd, Pt
O
O O
O
N+
N+
N+
N+
MNH2
H2N
8+
+ 2
5156 57
Scheme 16. Strategy for the assembly of a [3]catenane with Pt and Pd based units and the X-ray crystal structure of the [3]catenane [95].
90 R.A. Bilbeisi et al. / Inorganica Chimica Acta 417 (2014) 79–108
macrocycle [97]. A component strand containing the diimide wasterminated at one end by a pyridyl ring and at the other by aterpyridyl group. Catenation was ultimately achieved by the intro-duction of (i) a second strand composed of a polyethylene glycolchain terminated by a Ru porphyrin on one end and a terpyridylgroup on the other and (ii) one equivalent of Fe(II). The two linearpieces were linked by coordination of the terminal pyridine of thefirst strand to the axial position of the porphyrin of the secondstrand, as well as by coordination of the two terminal terpyridines
to Fe(II). The [2] and [3]catenane’s were identified on the basis ofelectrospray mass spectrometry and characterized by 1H NMR.
A further step in the development of complex catenanes wastaken by Anderson and colleagues who synthesized a [4]catenane(65) in 62% yield [98]. By coupling two Zn porphyrins in the pres-ence of a phenanthroline-containing macrocyle, a bisporphyrin[2]rotaxane was first obtained. A radial hexapyridine template(Scheme 18, blue strand) then facilitated (via coordination of itspyridyl groups to Zn) palladium-catalyzed Glasser coupling of

Scheme 18. Radial hexapyridine templated formation of a [4]catenane (left) and the MM + force field calculated structure (reproduced with permission from [98]).
R.A. Bilbeisi et al. / Inorganica Chimica Acta 417 (2014) 79–108 91
three [2]rotaxane subcomponents. Addition of an excess of base(DABCO) removed the hexapyridine template from the product[4]catenane. Furthermore, product analysis indicated the concom-itant formation of a [7]catenane in 6% yield.
The Anderson group’s approach is thus a kinetic covalent link-ing strategy. An alternative method was used by Sanders, Nitschkeand coworkers who were able to identify the presence of a[7]catenane in a dynamic covalent library (i.e. under thermody-namic conditions) [99]. The first step of their strategy was thedevelopment of a tetrahedral [FeII
4L6]8+ complex in which the fourFe ions occupy the corners of a tetrahedron and the six ligandsconstitute the edges. Each ligand is linear and composed of twobidentate pyridylimine termini separated by a rigid spacer that is it-self composed of an electron-poor naphthalene dimimide core andfour phenyl rings. When the electron-rich dinaphthocrown [24]macrocycle is in solution with the tetrahedral complex, the forma-tion of catenated structures was observed in which the crown ethersare slipped onto the linear ligands. This catenation process wasstudied by NMR spectroscopy and mass spectrometry which re-vealed the equilibration to be slow (requiring hours) and the forma-tion of the [n]catenated structures (n = 2–7) to result from a subtleinterplay between coordination chemistry, supramolecular assemblyand p-stacking interactions.
Finally, coordination chemistry can lead to the formation of infi-nite catenated structures of discrete components. Loots andBarbour demonstrated this possibility by the coordination of CuCl2
to a pyrenyl ligand (Scheme 19) [100].Their ligand (66) is composed of a pyrene core functionalized withtwo terminal imidazole rings. The self-assembly process is stronglydriven by the formation of p–p stacking interactions between thepyrene units. Dangling imidazol rings coordinate tetrahedral Cu(II)ions which buckle pairs of ligands together to form metallacycles.The space between each pair of pyrenes within a metallacycle per-
66
N
N N
N
CuCl2.H2O
CH2Cl2
Scheme 19. Formation of an infinite catenane (67) from dis
fectly accommodates two intercalated pyrenes from two othermetallacycles.
A second example of an infinite catenane was reported by Luand coworkers who obtained discrete tetrahedral complexes bycombining a tri(4-carboxybenzyl)amine pod and a 1,4,8,11-tetr-azaundecane nickel complex. Interpenetration of the tetrahedralcomplexes gave rise to the polycatenane [101].
3.2. Knots
In contrast to catenanes, knots are obtained by the entangle-ment of a single strand. The simplest example of a knot is the trefoilknot in which the strand forms three crossings. Charbonnière andTrabolsi have recently reported the synthesis of a metal-templatedtrefoil knot 70 that used dynamic covalent chemistry of iminebonds [90]. Inspired by the work of Stoddart on the synthesis of Bor-romean rings [102], and with the aid of DFT calculation, the authorsanticipated that the combination of a 5,50-dimethyl-2,20-bipyridinefunctionalized with para-aminomethyl-phenyl ethers (Scheme 20)would provide an adequate geometry for the bidentate coordina-tion of zinc(II).
The presence of the trefoil knot in the reaction mixture wasdemonstrated by a combination of high resolution mass spectrom-etry and NMR experiments. Interestingly, the synthesis of the tre-foil knot was accomplished in 56% yield but was accompanied bythe formation of a [2]catenane specie in 22% yield and a largerSolomon link structure, which was only detected in minor amountsby mass spectrometry during the course of the reaction. From theX-ray crystal structure of the [2]catenane and the DFT-calculatedstructure of the trefoil knot, it can be observed that the formationof the knotted structures is a result of strong zinc-templated che-lation combined with the formation of dynamic covalent iminebonds that are catalyzed by (i) Lewis acidic Zn(II) and (ii) strong
67
crete loops assembled by coordination to copper [100].
N N
O O
H2N NH2
NO O
Zn(OAc)2
i-PrOH+
O
N N
N
N NZn
N
N N
N
N
ZnN
N
NN
NZn
OO
O
OO
68
69 70
Scheme 20. Preparation and semiempirical PM6 model of the trefoil knot [90].
92 R.A. Bilbeisi et al. / Inorganica Chimica Acta 417 (2014) 79–108
p-stacking interactions between bipyridines and phenoxy ringsthat place the amine functions in close proximity to the formylgroups.
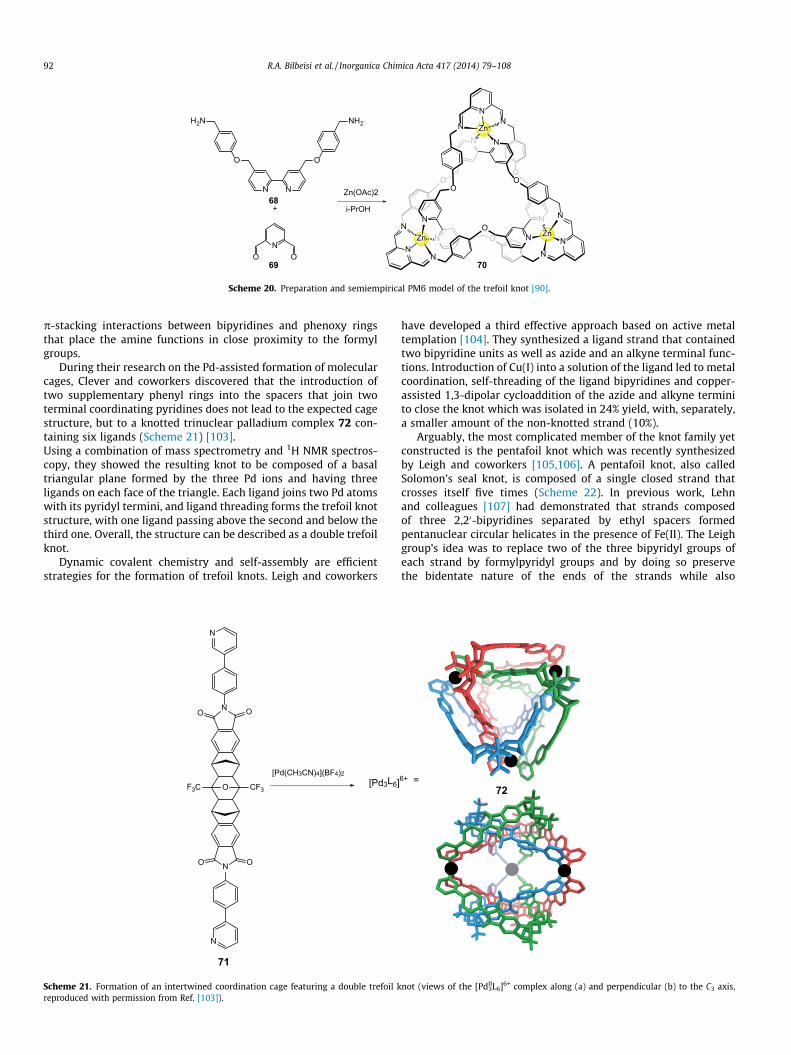
During their research on the Pd-assisted formation of molecularcages, Clever and coworkers discovered that the introduction oftwo supplementary phenyl rings into the spacers that join twoterminal coordinating pyridines does not lead to the expected cagestructure, but to a knotted trinuclear palladium complex 72 con-taining six ligands (Scheme 21) [103].Using a combination of mass spectrometry and 1H NMR spectros-copy, they showed the resulting knot to be composed of a basaltriangular plane formed by the three Pd ions and having threeligands on each face of the triangle. Each ligand joins two Pd atomswith its pyridyl termini, and ligand threading forms the trefoil knotstructure, with one ligand passing above the second and below thethird one. Overall, the structure can be described as a double trefoilknot.
Dynamic covalent chemistry and self-assembly are efficientstrategies for the formation of trefoil knots. Leigh and coworkers
N
N
OO
O O
CF3OF3C
N
N
[Pd(CH3CN)4](BF4)2
[Pd3L6]
71
Scheme 21. Formation of an intertwined coordination cage featuring a double trefoil kreproduced with permission from Ref. [103]).
have developed a third effective approach based on active metaltemplation [104]. They synthesized a ligand strand that containedtwo bipyridine units as well as azide and an alkyne terminal func-tions. Introduction of Cu(I) into a solution of the ligand led to metalcoordination, self-threading of the ligand bipyridines and copper-assisted 1,3-dipolar cycloaddition of the azide and alkyne terminito close the knot which was isolated in 24% yield, with, separately,a smaller amount of the non-knotted strand (10%).
Arguably, the most complicated member of the knot family yetconstructed is the pentafoil knot which was recently synthesizedby Leigh and coworkers [105,106]. A pentafoil knot, also calledSolomon’s seal knot, is composed of a single closed strand thatcrosses itself five times (Scheme 22). In previous work, Lehnand colleagues [107] had demonstrated that strands composedof three 2,20-bipyridines separated by ethyl spacers formedpentanuclear circular helicates in the presence of Fe(II). The Leighgroup’s idea was to replace two of the three bipyridyl groups ofeach strand by formylpyridyl groups and by doing so preservethe bidentate nature of the ends of the strands while also
6+ =72
not (views of the [PdII3L6]6+ complex along (a) and perpendicular (b) to the C3 axis,

N
N
N
O
N
O
H2N O ONH2
FeCl2, d6-DMSOKPF6
73
74
75
Scheme 22. Synthesis of a pentafoil knot (schematized on the right) by clipping a circular pentanuclear helicate [25].
R.A. Bilbeisi et al. / Inorganica Chimica Acta 417 (2014) 79–108 93
allowing for strand linkage via Schiff base formation. This modifi-cation allowed for strand closure and hence the formation of theknot.
Another complex non-trivial structure is the Solomon link whichis composed of two interwoven rings that cross each other four times(Scheme 22). The strategy developed by Leigh and coworkers to syn-thesize Solomon links involves the clipping of the strands of a circu-lar tetrahelicate [108], as was done with the pentanuclear circularhelicate to generate the pentafoil knot presented in Scheme 22[25]. To improve the chances of forming a tetranuclear rather thana pentanuclear helicate, a supplementary oxygen atom was intro-duced in the spacer linking the bipyridine units. This allowed formore flexibility of the strands, as would be necessary to form a smal-ler, more constrained ring. In the presence of FeCl2 salts, the tris-bidentate strands did form circular helicates of [FeII
4L4] composition.The introduction of the judiciously chosen 2,2-(ethylenedioxy)bis(ethylamine) linker led to the formation of imine bonds betweenthe diamine and the terminal formyl groups of the ligand strands,thereby joining separate strands and affording the expected Solomonlink.
Trace amounts of a Solomon link were also observed in the li-brary of compounds obtained by the dynamic coordination chem-istry approach of Trabolsi and Charbonnière [90].
One of the most complex types of link, the Borromean rings, wasrecently synthesized by Jin and coworkers [109]. By mixing
NNCu
O
OO
O
O O
2-
Rh Cl
NCl
S
RhCl
NCl
RhN
S
RhN
NNCu
OOO
OO O
RhN
S
RhN
N NCu
OO O
OOO
S = ;NH
O
O
HN Metallarectangles
Ag(OTf)
78 8079
Fig. 4. Strategy for the formation of metallarectangles that entangle to f
[RhCp⁄Cl2]2, and bismonodentate pyridine ligands that incorporatedvarious spacers, dincluear Rh precursor complexes were isolated.These complexes were then reacted with an o-phenylenebis(oxamato) Cu(II) complex to form metallarectangles that had theRhCp⁄ complexes at their corners (Fig. 4). When the spacer lengthwas long enough, metallarectangles self-assembled to formBorromean-like structures.
A comparison of the properties of the metallarectangles versusthe Borromean rings structures revealed the former to be catalyti-cally active in acyl transfer reactions between N-acetylimidazoleand 3-pyridylcarbinol. The Borromean structures had no catalyticactivity.
3.3. Ravels
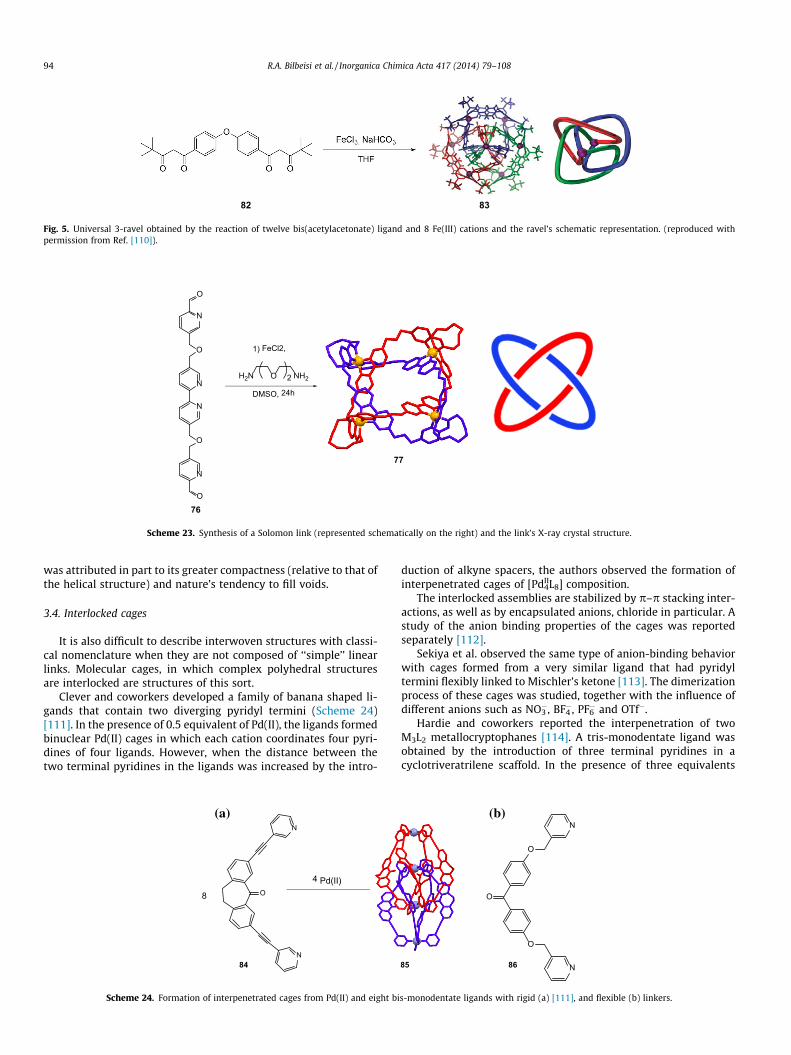
Although the mathematical description of knots and links isprecise [82], there are some entanglements that cannot be definedwith this nomenclature. Such is the case with the universal 3-ravel(Fig. 5) obtained by Lindoy and coworkers (Scheme 23) [110].
When a mixture of a bis-dione ligand and Fe(III) was allowed tocrystallize from a dichloromethane/methanol solution, a dinucleartriple helix was isolated. In contrast, when diethylether was veryslowly diffused into a THF solution over 3 months, crystals of the3-ravel were isolated, and X-ray analysis revealed their unprece-dented, interwoven molecular structure. Formation of the ravel
81
orm a Borromean rings structure (schematized on the right) [109].
82 83
Fig. 5. Universal 3-ravel obtained by the reaction of twelve bis(acetylacetonate) ligand and 8 Fe(III) cations and the ravel’s schematic representation. (reproduced withpermission from Ref. [110]).
N
O
O
N
N
O
N
O
1) FeCl2,
DMSO, 24h
H2N O NH22
76
77
Scheme 23. Synthesis of a Solomon link (represented schematically on the right) and the link’s X-ray crystal structure.
94 R.A. Bilbeisi et al. / Inorganica Chimica Acta 417 (2014) 79–108
was attributed in part to its greater compactness (relative to that ofthe helical structure) and nature’s tendency to fill voids.
3.4. Interlocked cages
It is also difficult to describe interwoven structures with classi-cal nomenclature when they are not composed of ‘‘simple’’ linearlinks. Molecular cages, in which complex polyhedral structuresare interlocked are structures of this sort.
Clever and coworkers developed a family of banana shaped li-gands that contain two diverging pyridyl termini (Scheme 24)[111]. In the presence of 0.5 equivalent of Pd(II), the ligands formedbinuclear Pd(II) cages in which each cation coordinates four pyri-dines of four ligands. However, when the distance between thetwo terminal pyridines in the ligands was increased by the intro-
O
N
N
4 Pd(II)
8
(a)
84
Scheme 24. Formation of interpenetrated cages from Pd(II) and eight bi
duction of alkyne spacers, the authors observed the formation ofinterpenetrated cages of [PdII
4L8] composition.The interlocked assemblies are stabilized by p–p stacking inter-
actions, as well as by encapsulated anions, chloride in particular. Astudy of the anion binding properties of the cages was reportedseparately [112].
Sekiya et al. observed the same type of anion-binding behaviorwith cages formed from a very similar ligand that had pyridyltermini flexibly linked to Mischler’s ketone [113]. The dimerizationprocess of these cages was studied, together with the influence ofdifferent anions such as NO3
�, BF4�, PF6
� and OTf�.Hardie and coworkers reported the interpenetration of two
M3L2 metallocryptophanes [114]. A tris-monodentate ligand wasobtained by the introduction of three terminal pyridines in acyclotriveratrilene scaffold. In the presence of three equivalents
O
O
O
N
N
(b)
8685
s-monodentate ligands with rigid (a) [111], and flexible (b) linkers.

OMeOMeO O
OMeO
NNN
AgX[Ag6L4]X6
87 87 88
Scheme 25. Interpenetration of two M3L2 metallocryptophanes [114].
R.A. Bilbeisi et al. / Inorganica Chimica Acta 417 (2014) 79–108 95
of Ag(I), this ligand assembled to form non-interlocked Ag3L2.However, when the spacers between the scaffold and the coordi-nating pyridines are short and flexible, the ligands and metalsself-assemble into interpenetrated cages (Scheme 25).Although their two-dimensional graphs have no fewer than fourcrossing points, these interpenetrated cages were loosely desig-nated‘‘[2]catenanes’’ (Scheme 24). (The two-dimensional graphsof conventional [2]catenanes, i.e. Hopf links, have two crossingpoints.)
Mukherjee and coworkers have reported the self-assembly ofhexanuclear prism-like complexes of Pd, some of which havinginterpenetrated structures [115]. The prisms were obtained byassembly of two benzene-1,3,5-tricarboxylic acids, six[Pd(en)(NO3)2] complexes and three 4,40-bipyridines (Scheme 26).The interpenetration was shown to be disfavored in the presence ofan excess of the tricarboxylic acid, which entered into the cavitiesof the prisms and blocked the self-assembly of interpenetratedstructures.
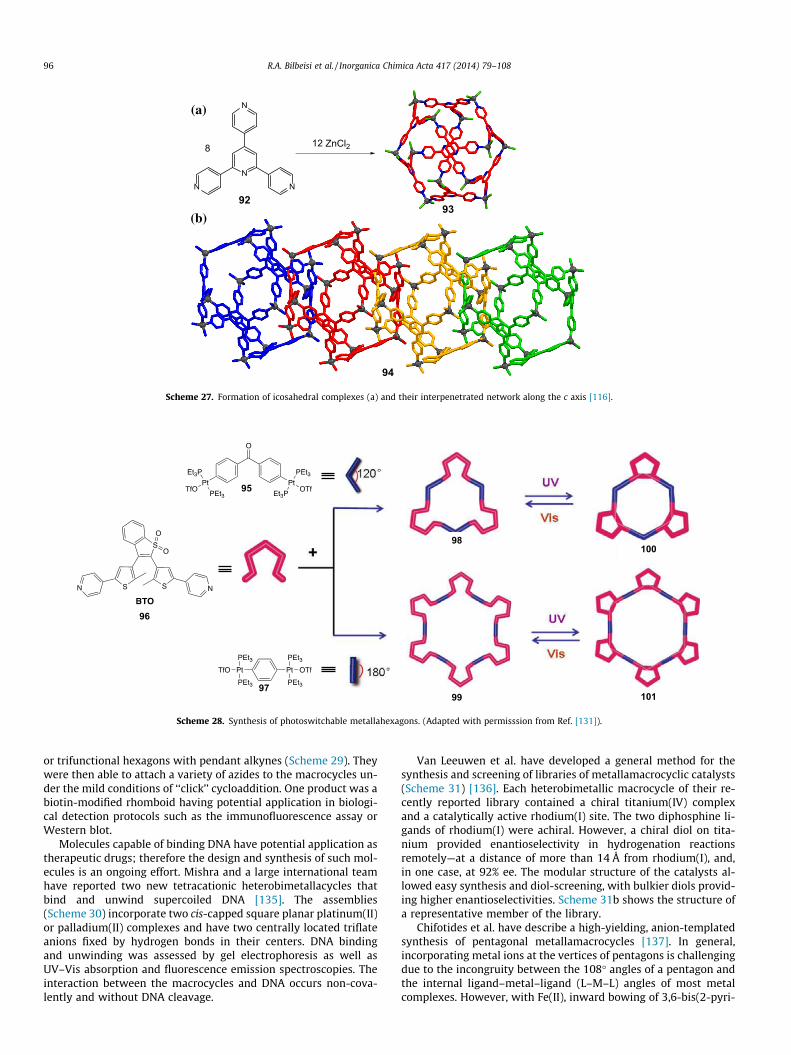
Finally, a very elegant and simple example of interpenatratedcages was synthesized by Dehnen and coworkers [116], who, bysimply combining 2,4,6-tris(4-pyridyl)pyridine (92) with zincchloride in methanol, isolated colorless crystals of a compoundhaving the formula [(ZnCl2)12L8], 93, and a molecular structureconsisting of interpenetrated icosahedral cages 94. The twelveZnCl2 units were found at the vertices of the icosahedra whilethe ligands constituted the icosadheras’ 20 triangular facial planes(Scheme 27). In the solid state, each icosahedron was interpene-trated by two neighbors along the c crystallographic axis, therebyforming an infinite interpenetrated chain. Owing to their largeinner volumes (2700 Å3) the metallocages were investigated bysolid-state NMR spectroscopy for their ability to encapsulate guestmolecules.
4. Miscellaneous structures
In this section we highlight recent advances in the fields of grids[117–120], helicates [121–124], and metallomacrocycles
89 90
2 +3 OKO
O
OK O
KO
2
N
N
6 [Pd(en)(NO3)2]
Scheme 26. Formation of interpenetra
[12,72,125–128]. We have also included two novel structures thatresist easy categorization. The large number of articles that havebeen published during the last 3 years preclude a comprehensivesurvey, so, in place of complete coverage, we have tried to selectrepresentative examples that demonstrate progress in the areasof design, synthesis, and applications. Metallofoldamers, whichgenerally contain only one organic component, have been excludedhere, but have been reviewed extensively elsewhere [129,130].
4.1. Metallomacrocycles
Chen et al. have designed and synthesized two robust, switchablehexagons (Scheme 28) incorporating multiple 2,3-bis(2-methyl-5-(pyridin-4-yl)thiophen-3-yl)benzo[b]thiophene-1,1-dioxide (BTO)donor units and complementary Pt(II) acceptor moieties [131].Ultraviolet light irradiation induces quantitative ring closure of allBTO units within the hexagons, which is remarkable consideringthat ring-closure yields for the free BTO monomer is 88%. Theauthors attribute the higher yield of ring closure within the metalla-cycles to the presence of a higher proportion of BTOs in the photore-active antiparallel conformation. (In solution, the ratio ofantiparallel to parallel conformers in the monomer is 1:1.) Molecu-lar modeling suggests that, within the hexagons, the antiparallelconformation is more stable and is therefore preferentially incorpo-rated during the self-assembly process. Visible light irradiation ofthe macrocycle with cyclized BTOs results in quantitative cyclore-version. The complete (ring opening/closing) cycle is reversibleand exhibits good fatigue resistance over ten iterations.
Functional groups, especially polar ones, can interfere in theself-assembly process of supramolecular coordination complexes.Within the context of metallamacrocycle synthesis, Chakrabartyand Stang have overcome this problem by developing an efficientmethod for post-assembly functionalization [132]. The key tacticof their approach involves strain-promoted [3+2] Huisgen cycload-dition [133,134]. The authors found that the self-assembly of Pt(II)-based acceptors and cyclooctyne-modified donors goes smoothly,and they were able to synthesize either difunctional rhomboids
91
ted hexanuclear Pd prisms [115].
N
N
N
N
12 ZnCl28
(a)
(b)92
93
94
Scheme 27. Formation of icosahedral complexes (a) and their interpenetrated network along the c axis [116].
95
96
97
98
99
100
101
Scheme 28. Synthesis of photoswitchable metallahexagons. (Adapted with permisssion from Ref. [131]).
96 R.A. Bilbeisi et al. / Inorganica Chimica Acta 417 (2014) 79–108
or trifunctional hexagons with pendant alkynes (Scheme 29). Theywere then able to attach a variety of azides to the macrocycles un-der the mild conditions of ‘‘click’’ cycloaddition. One product was abiotin-modified rhomboid having potential application in biologi-cal detection protocols such as the immunofluorescence assay orWestern blot.
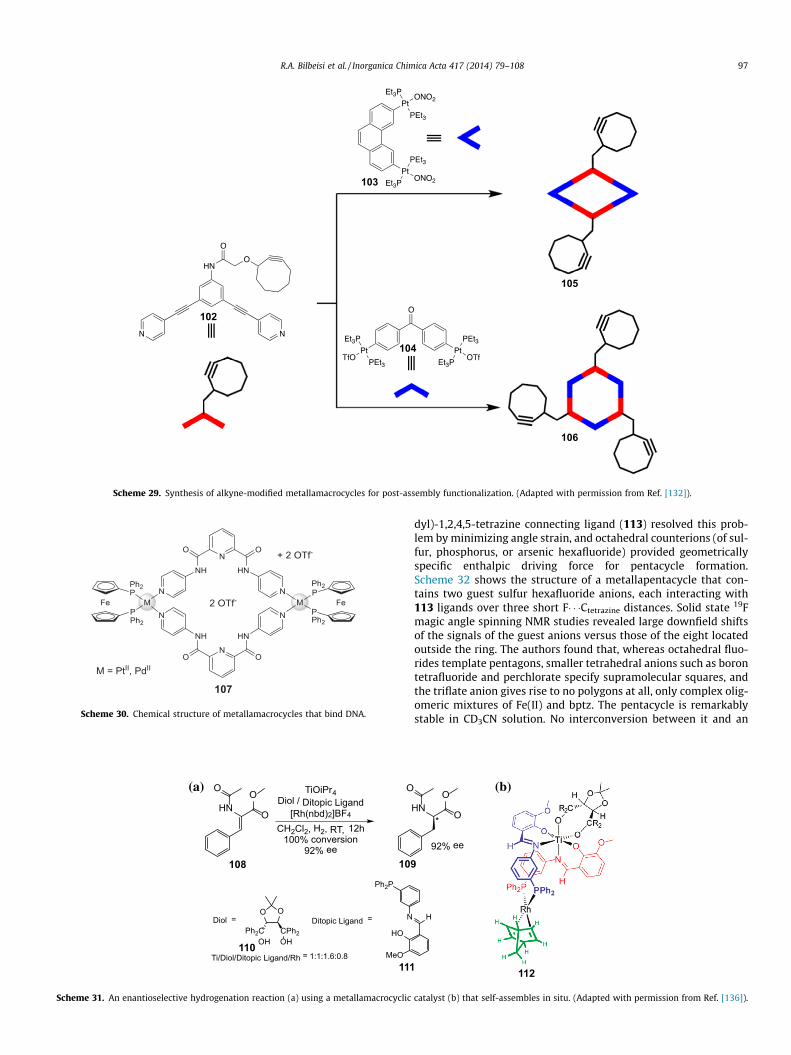
Molecules capable of binding DNA have potential application astherapeutic drugs; therefore the design and synthesis of such mol-ecules is an ongoing effort. Mishra and a large international teamhave reported two new tetracationic heterobimetallacycles thatbind and unwind supercoiled DNA [135]. The assemblies(Scheme 30) incorporate two cis-capped square planar platinum(II)or palladium(II) complexes and have two centrally located triflateanions fixed by hydrogen bonds in their centers. DNA bindingand unwinding was assessed by gel electrophoresis as well asUV–Vis absorption and fluorescence emission spectroscopies. Theinteraction between the macrocycles and DNA occurs non-cova-lently and without DNA cleavage.
Van Leeuwen et al. have developed a general method for thesynthesis and screening of libraries of metallamacrocyclic catalysts(Scheme 31) [136]. Each heterobimetallic macrocycle of their re-cently reported library contained a chiral titanium(IV) complexand a catalytically active rhodium(I) site. The two diphosphine li-gands of rhodium(I) were achiral. However, a chiral diol on tita-nium provided enantioselectivity in hydrogenation reactionsremotely—at a distance of more than 14 Å from rhodium(I), and,in one case, at 92% ee. The modular structure of the catalysts al-lowed easy synthesis and diol-screening, with bulkier diols provid-ing higher enantioselectivities. Scheme 31b shows the structure ofa representative member of the library.
Chifotides et al. have describe a high-yielding, anion-templatedsynthesis of pentagonal metallamacrocycles [137]. In general,incorporating metal ions at the vertices of pentagons is challengingdue to the incongruity between the 108� angles of a pentagon andthe internal ligand–metal–ligand (L–M–L) angles of most metalcomplexes. However, with Fe(II), inward bowing of 3,6-bis(2-pyri-
107
Scheme 30. Chemical structure of metallamacrocycles that bind DNA.
Scheme 29. Synthesis of alkyne-modified metallamacrocycles for post-assembly functionalization. (Adapted with permission from Ref. [132]).
HN
OO
O
OTiOiPr4Diol / Ditopic Ligand
[Rh(nbd)2]BF4
CH2Cl2, H2, RT, 12h100% conversion
92% ee
Ditopic Ligand =Diol =OO
CPh2Ph2COH OH
Ph2P
N
HO
MeOTi/Diol/Ditopic Ligand/Rh = 1:1:1.6:0.8
(a)
108 109
111
110
Scheme 31. An enantioselective hydrogenation reaction (a) using a metallamacrocyclic
R.A. Bilbeisi et al. / Inorganica Chimica Acta 417 (2014) 79–108 97
dyl)-1,2,4,5-tetrazine connecting ligand (113) resolved this prob-lem by minimizing angle strain, and octahedral counterions (of sul-fur, phosphorus, or arsenic hexafluoride) provided geometricallyspecific enthalpic driving force for pentacycle formation.Scheme 32 shows the structure of a metallapentacycle that con-tains two guest sulfur hexafluoride anions, each interacting with113 ligands over three short F� � �Ctetrazine distances. Solid state 19Fmagic angle spinning NMR studies revealed large downfield shiftsof the signals of the guest anions versus those of the eight locatedoutside the ring. The authors found that, whereas octahedral fluo-rides template pentagons, smaller tetrahedral anions such as borontetrafluoride and perchlorate specify supramolecular squares, andthe triflate anion gives rise to no polygons at all, only complex olig-omeric mixtures of Fe(II) and bptz. The pentacycle is remarkablystable in CD3CN solution. No interconversion between it and an
HNO
O*
H
92% ee
(b)
112
catalyst (b) that self-assembles in situ. (Adapted with permission from Ref. [136]).

N
NN N
N
N
4 Fe(BF4)24
CH3CN
113
114
Scheme 32. Anion-templated synthesis of a metallapentacycle.
NN
N
FeIINN
N
N
116116
117
Scheme 34. Synthesis of a square metallacycle that has switchable magnetic states.
98 R.A. Bilbeisi et al. / Inorganica Chimica Acta 417 (2014) 79–108
analogous square macrocycle was detected by NMR after overnightstirring at room temperature, despite the presence of a 15-fold ex-cess of [nBu4N][BF4].
Vajpayee et al. have recently reported the synthesis of severalheterometallic squares and found one of them (115, Scheme 33)to be a sensitive and selective host for nitroaromatic guests[138]. The squares were composed of linear platinum(II)-baseddonors as the edge components, and cis-blocked platinum(II) orpalladium(II) acceptors as corners. The blocking ligands wereferrocenyldiphosphine ligands. NMR and UV–Vis spectroscopiesprovided minimal evidence for guest binding. However, due tothe presence of the Pt-ethynyl moieties, the squares are fluores-cent. Quenching of fluorescence occurred in the presence of1,3,5-trinitrobezene and especially picric acid and provided strongevidence for sensitive and selective binding. Thus, these squaresmay have potential application as sensors for picric acid.
Spin-crossover (SCO) compounds have stable high spin (HS) andlow spin (LS) magnetic states that can be interconverted by theapplication of heat, pressure or light [139]. Such compounds havepotential application as components of display devices or data stor-age elements. Wei et al. [140] recently synthesized and tested amolecular square (Scheme 34) that has SCO properties. The com-pound 117 contains four iron(II) sites that are each capped by atris(2-pyridylmethyl)amine (tpa) ligand (116) and linked to twoother iron(II) cations by two dicyanamide ligands. In the solid state,the square exists in water-solvated and unsolvated forms. Solid-state magnetic measurements of both forms were carried out inthe temperature range of 300 to 5 K and both complexes were foundto undergo a two-step spin transition from a high-spin state (FeII
HS)4
above 300 K, to an intermediate state (HSFe1-LSFe2-HSFe1-LSFe2) be-tween 260 and 220 K, and abruptly to a low spin state (FeII
LS)4 below
N Pt N MN N
Pt Pt
N NN NPt
Et3P PEt3 Et3P PEt3
PEt3
PEt3
PEt3
PEt3
PPh
Ph
PPhPh
Fe
MPPh
Ph
PPh
Ph
Fe
MPPh
Ph
PPhPh
Fe
MPPh
Ph
PPhPh
Fe8 OTf-
M = PtII, PdII
115
Scheme 33. Chemical structure of square metallacycles that act as receptors fornitroaromatic compounds.
156 K. A complete LS to HS spin transition could be induced in thesolvated form by light irradiation (457 nm) at 5 K. Such studies willaid in the design of multi-stable materials.
4.2. Helicates
Like metallacycles, helicates have potential medical and diag-nostic applications. In order to fulfill this potential, helicates willneed to be water-soluble, accessible in large quantities, opticallypure, and non-racemizing. Howson et al. [141] have synthesizeda series of flexible helicates (dubbed ‘‘flexicates’’) satisfying all ofthese criteria. The ligands of these new complexes incorporate (i)flexible linkers between metal-chelating groups and (ii) chiral cen-ters that induce helical chirality. The flexibility of the ligandsmechanically uncouples the two metal centers, yet enantiomericpurity is enforced at each metal center independently, and in thehelicate as a whole, by the point chirality of the ligands. Also, themodular ‘‘subcomponent self-assembly’’ strategy employed allowsfor easy synthesis in relatively few steps as well as access to a vari-ety of similar structures.
Scheme 35 shows the well-resolved structure of a zinc(II) triple-stranded flexicate (119). An iron(II) version of the complex bindsstrongly to the major groove of DNA, as measured in ethidium bro-mide displacement assays and melting point (Tm) experiments.The iron(II) flexicate also exhibits antimicrobial activity (MIC = 4and 8 lg/ml for Staphylococcus aureus and Escherichia coli,respectively), but comparatively little toxicity toward Caenorhabdi-tis elegans (IC50 = 408 lg/ml).
The self-assembly of the two helicates (121 and 122 shown inScheme 36) is notable for the incorporation of lanthanide cationswhich are kinetically labile and lack strong directional bondingpreferences. Anion-controlled self-assembly of lanthanide heli-cates is rarer still. Nevertheless, Wang et al. [142] have reportedthat, in the presence of lanthanum(III), the flexible phenolic ligand(120) shown below forms two elaborate structures, either a hexa-nuclear cyclic helicate (121) or a tetranuclear quadruple-strandedhelicate (122) depending on whether the counterion is perchlorate
ONH2
OH2N
PhPh
3
+
NH
O
6
2 ZnII118
119
Scheme 35. Synthesis and X-ray crystal structure of an RNA-binding ‘‘flexicate’’.

O
HON
NHO
OHN
NOH
O
6 La(ClO4)3
4 La(NO3)3
6
4
120
121
122
Scheme 36. Synthesis of a lanthanum-containing cyclic helicate (top) and quadruple stranded helicate (bottom). (Adapted with permission from Ref. [142].)
R.A. Bilbeisi et al. / Inorganica Chimica Acta 417 (2014) 79–108 99
or nitrate, respectively. The authors designed the ligand to haveamide groups in order to facilitate anion templation. In the cyclichelicate, these amide hydrogens donate two hydrogen bonds totwo central, encapsulated perchlorate ions. In the linear helicate,however, the oxygens of one central nitrate anion are directlybound to La(III) in a l4-g1:g1:g2 bridging fashion. In methanol,addition of nitrate salts such as sodium, ammonium, or tetrabutyl-ammonium nitrate converts the cyclic helicate to the linear heli-cate, suggesting that nitrate has a strong templating effect.
Freisinger and collaborators [143] have reported a first-of-its-kind study that involves the complexation of a triple-stranded heli-cate within the central cavity of an RNA three-way junction (3WJ).A high-resolution X-ray crystal structure of the helicate within the3WJ (Scheme 37) reveals p-stacking interactions between the
N
N
N
N
2 FeII3
M P
(a)
(c)
123 124
(b)
Scheme 37. Synthesis of a pair of enantiomeric helicates for RNA-binding (a), aswell as top view (b) and side view (c) of the X-ray crystal structure of the Menantiomer bound to an RNA three-way junction. (Adapted with permission fromRef. [143]).
bases of RNA and the central phenylene rings of the helicate. Gelelectrophoresis, circular dichroism (CD) and UV–Vis absorptionexperiments all confirmed the formation of the complex betweenthe helicate and the 3WJ. Interestingly, RNA binding and stabiliza-tion assays showed that the RNA had no preference for either heli-cal enantiomer, yet the M enantiomer was the only one toco-crystallize with the RNA. Further studies and development ofRNA-binding helicates such as these may produce effective anti-cancer therapeutics.
Few examples of functionalized homochiral coordination cageshave been reported. Xuan et al. [59] have described a quadruple-stranded helicate cage for enantioselective recognition andseparation. Scheme 38 shows a crystal structure of the cage whichis composed of four pyridyl-functionalized metallosalen ligands125 and eight zinc(II) metal centers. The overall structure displaysP helicity. In solution, the molar optical rotation of the cage is >45times that of the free salen ligand (31208.7 and 674.9 deg cm3 -dm�1 mol�1, respectively). In the solid state, the cage contains achiral central cavity of �13.7 Å � 6.4 Å that is occupied by a singleTHF molecule. One-dimensional channels of �4.0 Å � 4.0 Å arepresent between stacked cages. The chiral coordinated aminesare oriented toward the outside of the cages and are exposed tothe interstitial pores. Fluorescence emission spectroscopy providedevidence that solid samples of the cage enantioselectively adsorbsmall chiral molecules including 1-phenylethanol, 1-phenylethyl-amine, and alanine. The cage’s modular construction based on met-allosalan, should allow for the synthesis of other chiral assemblieswith potential enantioselective functions.
4.3. Cyclic helicates
Rice and collaborators [144] have reported the self-assembly ofa series of cyclic helicates from thiazole containing ligand 127.When heated in acetonitrile with zinc(II) triflate, an achiral versionof the ligand produces a helicate 128 in 50% yield (Scheme 39). Theauthors postulate that steric interactions between the phenylspacer units of the ligands destabilize linear assemblies and leadto the formation of the cyclic structures. Incorporation of chiralenantiopure amino ester moieties within the ligand results in thediastereoselective formation of circular helicates with d.e. of upto 80%.

(a) (b)
HNNH
N N
HOOH 8 ZnCl2
125
126
124
Scheme 38. Synthesis of a helical cage complex (a) and its X-ray crystal structure (b).
SN
N S
N
N
N
N
5 ZnII
NO
NO
5
127 128
Scheme 39. Synthesis and X-ray crystal structure of a cyclic pentanuclear zinc helicate.
100 R.A. Bilbeisi et al. / Inorganica Chimica Acta 417 (2014) 79–108
High net positive charge (NPC) of supramolecular coordinationcomplexes can hinder the formation of high-nuclearity structuresand complicate their isolation. Wu et al. [145] designed a dianionicdithiolate ligand (129) that assembles into a decanuclear zinc(II)helicate of NPC = 0 when mixed with Zn(II) in solvent (Scheme 40).No solvent molecules or counterions other than the ligand appearto have a templating role in the assembly process. The ten octahe-dral zinc(II) centers display an alternating K and D chirality, lead-ing to an overall meso helical structure. An interesting feature ofthe complex is that it can be disassembled by an excess (5–10equivalents) of H2PO4
�, but is stable in the presence of other anionssuch as Br�, I�, HSO4
�, and NO3�, as well as in acetic acid. It is the
highest nuclearity circular helicate yet reported.The Hamacek group [146] has combined structural motifs to
create a series of hybrid pentanuclear lanthanide complexes(Scheme 41) that have a tetrahedral and a linear domain, each witha helical twist. Mixing two carefully designed ligands with Nd(III),Eu(III), Er(III), or Eu(III) perchlorate produced Eiffel tower-likestructures (132), as verified by NMR and electrospray mass spec-trometry (ESI-MS) and corroborated by molecular modeling (SPAR-KLE/AM1). The key to the design was the choice of the largerdiaminodiphenyl linker [131] which allowed extension of one ofthe arms of the tetrahedral domain. Further development will in-volve the synthesis of heteronuclear towers.
Nitschke and colleagues [52] have reported the self-assembly ofa novel triangular triple helicate 135, in acetonitrile at 343 Kfrom a C3 symmetric aldehyde 133, Zn(II) triflimide, and 4-methoxyaniline 134. Addition of quinolin-8-amine to this novelstructure triggers its disassembly and the subsequent self-assemblyof another triangular helicate, one that has a central cavity capableof accommodating a range of planar, aromatic guests such asanthracene, perylene, and pyrene, the binding constant for thelatter being 1240 ± 80 M�1. Non-planar guests such as triptyceneand C60 do not bind, nor does cyclohexane or cyclodecane.Scheme 42 shows an X-ray crystal structure of the non-guestbinding helicate.
4.4. Grids
The reaction of an iminopyridly ligand with copper(II) triflateproduces a [2 � 2] grid complex, [Cu4L4]8+, which was character-ized by NMR and X-ray crystallography (Scheme 43) [69]. Theaddition of trifilc acid, ascorbic acid and triethylamine convertsthe grid to a double helicate of formula [Cu2L2]2+. The helicatecan be transformed back to the grid by air oxidation or the additionof copper(II) triflate. This oxidation state-dependent interconver-sion is the first reported transformation between a grid and ahelicate.
N
NHO
ONH
NH
N
ON
O
NHO
N
ON
NNO O
HN
3
1. LnIII
Ln = Eu, Lu
2.
HN
ONN
O
2
131
132
Scheme 41. Synthesis of pentanulcear lanthanide tower-complexes. (Adapted with permission from Ref. [146]).
NH HN
SH HS
10 ZnII
Et3N
10
129
130
Scheme 40. Synthesis and X-ray crystal structure of a decanuclear zinc(II) helicate incorporating a dithiolphenolic ligand.
N
N
N
N
N
N
O
O
O
O
NH2
ZnII+
3
9
133
134
135
Scheme 42. Synthesis and X-ray crystal structure of a triangular zinc(II) helicate.
R.A. Bilbeisi et al. / Inorganica Chimica Acta 417 (2014) 79–108 101
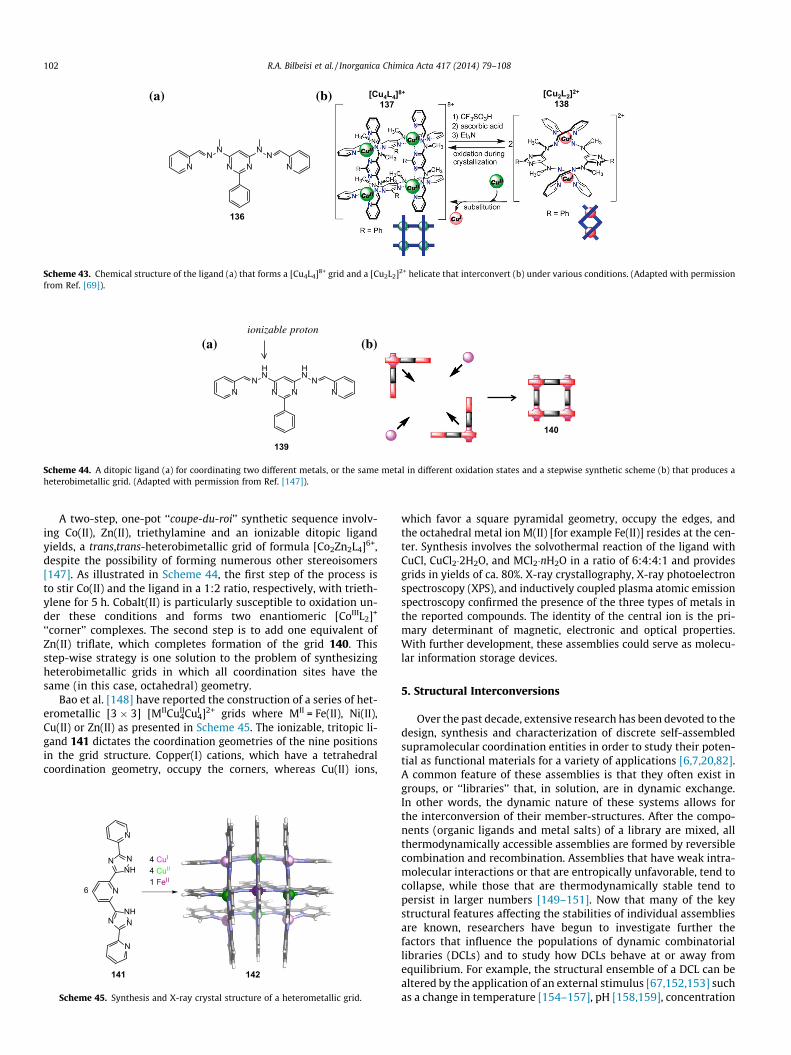
N N
N NN N
N N
(b) [Cu4L4]8+
137[Cu2L2]2+
138(a)
136
Scheme 43. Chemical structure of the ligand (a) that forms a [Cu4L4]8+ grid and a [Cu2L2]2+ helicate that interconvert (b) under various conditions. (Adapted with permissionfrom Ref. [69]).
(a) (b)ionizable proton
N N
HN
HN
N NN N
139
140
Scheme 44. A ditopic ligand (a) for coordinating two different metals, or the same metal in different oxidation states and a stepwise synthetic scheme (b) that produces aheterobimetallic grid. (Adapted with permission from Ref. [147]).
102 R.A. Bilbeisi et al. / Inorganica Chimica Acta 417 (2014) 79–108
A two-step, one-pot ‘‘coupe-du-roi’’ synthetic sequence involv-ing Co(II), Zn(II), triethylamine and an ionizable ditopic ligandyields, a trans,trans-heterobimetallic grid of formula [Co2Zn2L4]6+,despite the possibility of forming numerous other stereoisomers[147]. As illustrated in Scheme 44, the first step of the process isto stir Co(II) and the ligand in a 1:2 ratio, respectively, with trieth-ylene for 5 h. Cobalt(II) is particularly susceptible to oxidation un-der these conditions and forms two enantiomeric [CoIIIL2]+
‘‘corner’’ complexes. The second step is to add one equivalent ofZn(II) triflate, which completes formation of the grid 140. Thisstep-wise strategy is one solution to the problem of synthesizingheterobimetallic grids in which all coordination sites have thesame (in this case, octahedral) geometry.
Bao et al. [148] have reported the construction of a series of het-erometallic [3 � 3] [MIICuII
4CuI4]2+ grids where MII = Fe(II), Ni(II),
Cu(II) or Zn(II) as presented in Scheme 45. The ionizable, tritopic li-gand 141 dictates the coordination geometries of the nine positionsin the grid structure. Copper(I) cations, which have a tetrahedralcoordination geometry, occupy the corners, whereas Cu(II) ions,
N
NHNN
N NNH
N
N
4 CuI
4 CuII
1 FeII
6
141 142
Scheme 45. Synthesis and X-ray crystal structure of a heterometallic grid.
which favor a square pyramidal geometry, occupy the edges, andthe octahedral metal ion M(II) [for example Fe(II)] resides at the cen-ter. Synthesis involves the solvothermal reaction of the ligand withCuCl, CuCl2�2H2O, and MCl2�nH2O in a ratio of 6:4:4:1 and providesgrids in yields of ca. 80%. X-ray crystallography, X-ray photoelectronspectroscopy (XPS), and inductively coupled plasma atomic emissionspectroscopy confirmed the presence of the three types of metals inthe reported compounds. The identity of the central ion is the pri-mary determinant of magnetic, electronic and optical properties.With further development, these assemblies could serve as molecu-lar information storage devices.
5. Structural Interconversions
Over the past decade, extensive research has been devoted to thedesign, synthesis and characterization of discrete self-assembledsupramolecular coordination entities in order to study their poten-tial as functional materials for a variety of applications [6,7,20,82].A common feature of these assemblies is that they often exist ingroups, or ‘‘libraries’’ that, in solution, are in dynamic exchange.In other words, the dynamic nature of these systems allows forthe interconversion of their member-structures. After the compo-nents (organic ligands and metal salts) of a library are mixed, allthermodynamically accessible assemblies are formed by reversiblecombination and recombination. Assemblies that have weak intra-molecular interactions or that are entropically unfavorable, tend tocollapse, while those that are thermodynamically stable tend topersist in larger numbers [149–151]. Now that many of the keystructural features affecting the stabilities of individual assembliesare known, researchers have begun to investigate further thefactors that influence the populations of dynamic combinatoriallibraries (DCLs) and to study how DCLs behave at or away fromequilibrium. For example, the structural ensemble of a DCL can bealtered by the application of an external stimulus [67,152,153] suchas a change in temperature [154–157], pH [158,159], concentration

R.A. Bilbeisi et al. / Inorganica Chimica Acta 417 (2014) 79–108 103
[157,160], solvent [157,161], light intensity [162,163] or theaddition of a chemical template or other chemical trigger[52,164–169]. This area of research is often inspired by the abilityof biochemical systems to adapt and reconfigure.
Although structurally complex molecular links are beingsynthesized individually more frequently, full characterization ofmixtures (i.e. dynamic libraries) of such structures is still rare.Recently, Trabolsi and coworkers [90] designed a simple pair ofchelating ligands that, in the presence of transition metal cationsand via reversible imine and metal–ligand bond formation, self-assembles into three links. The design of the ligands was inspiredby work on the Borromean rings (BRs) previously published bythe Stoddart group [102]. Symmetric extension of the diaminobi-pyridine used for BRs synthesis was found to maintain structurallyimportant p–p stacking interactions but also provide more rota-tional freedom and flexibility. Thus, when the new diaminopyri-dine ligand (DAB) and diformylpyridine (DFP) were combinedwith Zn(II) ions, a dynamic library of three links was formed(Scheme 46): a [2]catenane ([2]C), a trefoil knot (TK), and aSolomon knot (SK). The [2]C and TK were isolated in 22% and56% yields, respectively, while SK was detected by mass spectrom-etry. The structures of both, TK and [2]C were confirmed by 1HNMR spectroscopy and mass spectrometry, and by X-ray crystal-lography in the case of [2]C. Mass spectrometric data obtained overthe course of the reaction suggested a plausible mechanism for theformation of each link. The results highlighted in this work clearlydemonstrate the synthetic efficiency of combining dynamic cova-
Fe2+
N
NN
N
NH2
N
NN
N
N
H2N NH2
N
N
N
N
N
+
Fe2+
Fe2+
H2N
NH2
NO
Fe(NTf2)2
12
4
4
+
CD3CN
NH2
148 FeII
2L3
3
Scheme 47. Subcomponent self-assembly of three ir
N
N
O
O
NH2
NH2
NOO
ZnII
NN
N
NN
Zn
N N
N
N NZn
OO
OO
O
N N
N
N Zn
N
N
NN
N
Zn
O
OO
145 146
143
144
Scheme 46. One pot synthesis of a topological triptych consisting of a [2]catenane, a t
lent imine bond formation and metal templation for creatinglibraries of links.
The Nitschke group has recently studied how very differentproduct structures form from the same starting materials but un-der different reaction conditions [170,171] and/or with the addi-tion of a template [165,171,172]. A recent publication [171]describes the self-assembly and characterization of a library of me-tal–organic complexes, and how particular conditions lead to thepreferential formation of each member of the library.A library of three iron(II) based complexes was formed in solutionfrom a mixture of tritopic triamine 1,3,5-tri(4-aminophenyl)ben-zene (4 equiv), 2-formylpyridine (12 equiv), and iron(II) triflimide(4 equiv) as demonstrated in Scheme 47. The assembled com-plexes; triple helicate FeII
2L3 148, face-capped tetrahedronFeII
4L4 7 and icosahedron capsules FeII12L12 149 were characterized
in solution, the gas phase and in the case of 7 and 149 the solidstate. The complexes vary in the number of subcomponents andthe geometry of the metal centers incorporated. Complex 148 (atriple helicate) is composed of fewer subcomponents and has a dif-ferent empirical formula than complexes 7 and 149, whereas bothcapsules (discussed in Section 2 of this review) have the sameempirical formula but different metal coordination geometries(mer versus fac, respectively) which results in them havingdifferent polyhedral structures. Also, the two capsules possessradically different cavity volumes and shapes which allows forthe encapsulation of a range of guests that have different charges,shapes and sizes.
Fe2+
Fe2+
Fe2+
Fe2+
Fe2+
N
N
N
N
Fe2+
Fe2+
Fe2+ Fe2+
Fe2+
Fe2+ Fe2+
Fe2+
N
N
N
N
Fe2+
N
NFe2+
+
7FeII
4L4
149FeII
12L12
on(II) based complexes in acetonitrile at 323 K.
O
N
N
N
N
N
Zn
N
NN
N
N
ZnN
N
NN
NZn
O
O
O
OO
N
N
NN
NZn
O
ON
N
N N
N
N
Zn
OO
147
refoil knot, and a Solomon link from a simple pair of chelating ligands and Zn(II).

104 R.A. Bilbeisi et al. / Inorganica Chimica Acta 417 (2014) 79–108
The dynamic system shown in Scheme 47 could be induced toexpress one of the complexes predominantly in solution by chang-ing the reaction conditions. The triple helicate (148) was preferen-tially formed from Fe(NTF2)2 in acetonitrile at room temperature.Its formation could be reduced by increasing the temperatureand the concentration of the starting material. Alternatively, thetetrahedral complex was formed selectively in acetonitrile at323 K from Fe(NTF2)2 and in the presence of an organic template,such as cyclohexane. Finally, increasing the polarity of the solventto 50:50 (v/v) methanol:acetonitrile led to an increase in the yieldof the icosahedron (�88%) when employing Fe(OTF)2 at 343 K.
A more elaborate study of templation, in this case with anions,was also reported by Nitschke and coworkers [165]. The teaminvestigated the formation of diverse anion-templated multinu-clear metal–organic assemblies starting from the same subcompo-nents. The self-assembled discrete structures included tetrahedra(M4L6), extended circular helicates (M6L8), distorted cuboids(M8L12) and pentagonal prisms (M10L15).The self-assembly of a series of NiII-based metal–organic structuresfrom p-toluidine, 6,60-diformyl-3,3-bipyridine and nickel(II) salts ispresented in Scheme 48. An anion with a suitable size and shape isnecessary for the formation of one of the five discrete structures. Inthe case of using nickel(II) with the relatively large counter aniontriflimide, a collection of uncharacterized products was obtainedas indicated by ESI mass spectroscopy.On the other hand, the ligand was observed to react with nickel(II)perchlorate to form the pentagonal prism (NiII
10L15) as indicated bythe single crystal X-ray structure. The pentagonal prism is composedof two parallel NiII
5L5 pentagonal rings, each composed of equatorialligands, and the two rings are joined by axial ligands. The overallstructure has idealized D5 point symmetry, with the metal centersdisplaying meridional (mer) stereochemistry. This structure pos-sesses six distinct anion binding pockets, one central and five whichlie along the twofold symmetry axis. This unique architecture is sta-
NiIIX
+
N
N
O
O
6
3
2+
X = NTf2-
X = ClO4-
X = NO3-
X = OTf-
X = BF4-
D
Ni
Ni
NiII4L6
NiII4L6 +
NH2
11
150
Scheme 48. Preparation of a series of NiII-based discrete structures: Ni-DCL, pentagonal phelicates (Ni6L8).
bilized through the templation of a chloride anion occupying thecentral pocket and five perchlorate anions occupying the rest ofthe pockets.A distorted cuboid (NiII
8L12) with an overall idealized S4 point sym-metry was obtained when reacting the ligand with nickel(II) nitrate.Unlike the pentagonal prism, the metal centers in the cuboid alter-nate between mer and fac conformation, resulting in four mer andfour fac metal centers per structure. The cuboid possesses two dis-tinct binding pockets, each of which able to bind anions of a sizesimilar to NO3
� or BF4�.
The reaction of nickel(II) triflate with the ligand yielded a mixtureof a tetrahedron (NiII
4L6) and an extended circular helicate (NiII6L8) as
indicated by X-ray diffraction analysis. The tetrahedron has an ideal-ized T symmetry with all metal centers adopting the fac conforma-tion. The tetrahedron has one central binding pocket. The isolatedstructure was filled with solvent molecules rather than counterions.The extended circular helicate, on the other hand, has two types ofmetal coordination sites. The four central metal complexes that in-volve three pyridyl imine groups and have mer configuration con-stitute the first, whereas the two peripheral complexes involvingtwo pyridyl imine groups and two triflate anions constitute thesecond. The overall structure is composed of a [2 � 2] helicate unit,consisting of four ligands that adopt anti conformations to bridgethe four metal centers. This architecture has an idealized D2 sym-metry. Two distinct types of metal binding site are present. Thetwo peripheral sites that encapsulate the nickel-bound triflateare of one type. The central site, which is occupied by a distortedtriflate anion, is of the other.The same mixture of NiII
4L6 and NiII6L8 was obtained when the ligand
components were reacted with nickel(II) tetrafluoroborate at 323 K.Heating the reaction at 363 K for 2 months, however, resulted in theformation of the NiII
10L15 pentagonal prism exclusively, as indicatedby ESI-MS. This observation indicates that NiII
10L15 is the thermody-namically favored product in the presence of BF4
�.
CL
II10L15
II8L12
NiII6L8
+
NiII6L8 NiII10L15
risms (Ni10L15), distorted cuboids (Ni8L12), tetrahedron (Ni4L6) and extended circular

R.A. Bilbeisi et al. / Inorganica Chimica Acta 417 (2014) 79–108 105
The formation of the five discrete structures was investigated usingother metal ions (FeII, ZnII and CoII) with different ionic radii. It wasobserved that the architectural diversity increased with increasingionic radius of the metal.The authors also systematically probed the synergic and competingeffects of anion and metal templation on the formation of each ofthese structures. This study complements the use of geometric de-sign principles by highlighting the effects that secondary interac-tions have on the metal–organic self-assembly process.
While gaining the necessary understanding of the effects ofdifferent templates (including metal ions, anions and organic tem-plates) on the formation of multinuclear metal–organic assembliesfrom complex dynamic systems, several groups also conductedresearch into the effects of chemical triggers on the transformationof architectures in dynamic systems [153,173–175]. Two represen-tative studies of this work are presented below.
Early in 2010, Zhou and Li described [176] a ground-breakingsynthetic strategy for the preparation and interconversion of sev-eral metal–organic-polyhedra (MOPs) as shown in Scheme 49.Their approach relies on ‘‘bridging-ligand-substitution’’ which in-volves the introduction, to an initial MOP structure, of linkers withdifferent properties. This type of perturbation can lead to MOPswith new structures including mixed ligand species that are other-wise difficult to obtain.
More specifically, three initial MOPs, [Cu24(A)24S24]�xS, [Cu24
(B)24S24]�xS and Na6H8[Cu24(C)24S24]�xS where S represents a solventmolecule, were prepared by direct reaction of Cu2(OAC)�H2O andthree angular diacid ligands A, B, and C (Scheme 49). As a represen-tative example of their strategy, reaction of (i) the initial cuboctahedralMOP 1 constructed from 5-t-butyl-1,3-benzenedicarboxilic acid (A)which has a bend angle of 120� with (ii) 3,30-(ethyne-1,2-diyl)-diben-zoic acid (D) which has a bend angle of 60�, in N,N-diethylforma-mide, resulted in a mixed-ligand MOP 4 [Cu24(A)6(D)6S12]�xS witha distorted octahedral geometry. Alternatively, the reaction of 1 with2,7-naphthalenedicarboxylic, (E), a ligand that possesses a 120�bend angle, resulted in a quasi-spherical structure, MOP 5, consist-ing of 12 A ligands, 12 E ligands and 12 Cu2 paddlewheel units. Forthe sake of comparison, Zhou and Li also tried ‘‘one-pot’’ preparation
MOP 1 MOP 5MOP 4
MOP 3 MOP 2
MOP 8 MOP 6MOP 9
MOP 7
Scheme 49. Schematic representation of the bridging-ligand-substitution strategyemployed by Zhou and Li to synthesize their MOPs library. (Adapted withpermission from Ref. [176]).
of mixed ligand MOPs from their corresponding ligands demonstrat-ing that the formation of such polyhedral cages is favored eitherthermodynamically or kinetically in the bridging-ligand-substitu-tion reactions. These MOP materials presented some potential appli-cation in gas separation.
Nitschke and coworkers also investigated a network of architec-tures induced by chemical triggers [177]. The system involved thereaction 6,6-diformyl-3,3-bipyridine 11, cadmium(II) triflate and avariety of different amines to produce assemblies of formulaCdII
xLy. The effects of different amines on the reaction outcome isillustrated in Scheme 50. The assembled structures, each formedwith a different amine, included triple helicate CdII
2L3, triangularcircular helicate CdII
3L3, tetrahedral cage CdII4L and hexagonal prism
CdII12L18.Subsequent to the self-assembly and characterization of the di-
verse architectures, the authors investigated structural intercon-versions that occurred with addition of new subcomponents (i.e.a chemical stimulus) to a pre-assembled structure. A network ofarchitectural transformations was achieved through imine ex-change upon addition of an amine subcomponent with differentelectronic and steric properties [79,178–180]. The network ofselective transformations is summarized in Scheme 51, which de-picts the energetic hierarchy between structures.
Ward and coworkers [67] reported a different class of transforma-tion triggered by crystallization and involving a simple pyrazole-based bis-bidentate bridging ligand that, when combined with differ-ent six-coordinate dicationic transition metals such as Ni(II), Cu(II),Zn(II) and Cd(II) yields three different types of polyhedral cage. Morespecifically, reaction of the a,a0-bis-[3-(2-pyridyl)-pyrazol-1-yl]-1,4-dimethylbenzene, with ZnII(BF4)2 or CdII(ClO4)2 in MeCN in a 3:2ratio resulted in the formation of [M16L24]X32 cages (where M isthe metal and X represents the counterion, Fig. 6). The structure ofthese cages was confirmed by X-ray crystallography. Each of thesetwo polyhedra has 24 edges and one bridging ligand along each edgethat spans two metal ions at the vertices of the polyhedron. Furtherstructural analysis revealed the importance of the aromaticpi-stacking interactions in guiding the assembly of such a large
N
N
O
O
3
2Cd(OTf)2
+
H2NN
NH26
6
NH2N
H2N
NH2
6
O
NH2
6
CdII3L3
CdII4L
CdII12L18
CdII2L3
11
151
152
153
154
Scheme 50. Preparation of diverse architectures from the self-assembly of 11 withamines 151–154 and cadmium(II).

Scheme 51. Transformations of cadmium(II) architectures. (Adapted with permission from Ref. [177].)
155M16L24
156M6L9
Fig. 6. Dynamic behavior representation of the interconversion of the largest[M16L24]32+ cages formed on crystallization to the [M6L9]12+ trigonal prismatic cagein solution. (Adapted with permission from Ref. [67]).
106 R.A. Bilbeisi et al. / Inorganica Chimica Acta 417 (2014) 79–108
and—as compared to a set of smaller structures—entropically disfa-vored structure.Solution studies such as 1H, 13C NMR and ESI-MS were in agree-ment with the solid-state structure of the CdII
16L24(BF4)32 complex.However, a slow rearrangement occured when the sample was leftin solution for an extended period (over 2 months) to give theCdII
6L6 trigonal prism, as deduced from NMR analysis.Alternatively, reacting the ligand with Cu(BF4)2 under the sameconditions of solvent and ligand-to-metal ratio afforded a hexanu-clear cage [Cu6L9](BF4)12 with an approximately trigonal prismatictopology where the two triangular faces were formed by 3 CuII ionseach and there were nine bridging ligands along the edges.
Fig. 7. Temperature-controlled interconversion between the hexamer (kinetic propyrogallol[4]arene.
Finally, when the same ligand was mixed with Ni(II) in the sameM:L ratio, a classic octanuclear cubic cage NiII
8L12 was formed.Building on their previously reported work dealing with the
conversion of pyrogallol[4]arene monomers into hexameric nano-capsucles, Atwood and coworkers [181] have recently shown thepreferential formation of one specie over the other (dimer versushexamer), upon carrying out the reaction in methanol solution athigher temperatures and when employing nickel(II) salts (Fig. 7).Moreover, conditions under which the nickel-seamed hexamerconverts to the dimer were identified. Small-angle neutron scatter-ing was employed to help elucidate the role of temperature, solventand metal in controlling the formation of the corresponding metalorganic nanocapsules. Dimers were found to be thermodynamicallystable at all temperatures whereas hexamers were kineticallyfavored at low temperatures.
In addition to transformations involving reagents and reactionconditions, those of light-responsive metal self-assembled struc-tures have also been reported. Zhu and coworkers recently reportedthe design and self-assembly of a family of multi-bisthienylethenehexagons where the reversible size/shape transformation can betriggered by light. Mixing 2,3-bis(2-methyl-5-(pyridine-4-yl)thiophen-3-yl)benzo[b]thiophene-1,1-dioxide, a photoresponsiveligand that possesses a 120� angle, with suitable 120� and 180�diplatinum(II) substrates, in a 1:1 ratio yielded [3+3] and [6+6]hexagons, respectively. Upon irradiation at 365 nm in CH2Cl2 solu-
duct) and the dimer (thermodynamic product) nickel/copper capsules from

R.A. Bilbeisi et al. / Inorganica Chimica Acta 417 (2014) 79–108 107
tion of both macrocycles, a color change was observed for bothstructures as a result of the ring closing of the bisthienyletheneunits.
6. Conclusions
Established design principles based on thermodynamics andsymmetry are reliable starting points for the construction of com-plex metal–organic assemblies. In the past 3 years, the relativelynew subcomponent self-assembly method, which allows for theintegration of multiple simple building blocks, has been frequentlyemployed. New step-wise syntheses have also been devised andhave been proven useful for building heterometallic assembliesthat have identical metal coordination sites or for attaching polarfunctional groups in post-assembly fashion. Chemists continue todetermine and exploit the influences that weak interactions exerton the self-assembly process. Subtle factors such as anionic tem-plation, pi-stacking, hydrogen bonding, and the metal cation’sradius often foil expectations based on idealized geometric guide-lines but are now under increasing scrutiny. Methods for synthe-sizing optically pure assemblies and assemblies that are stable inaqueous solvents are active areas of endeavor, as is the analysisof structural interconversions within dynamic systems. With re-gard to functions, examples are diverse. Helicates and metallamac-rocycles are being designed for therapeutic purposes; links forsensing; and container molecules for the encapsulation and analy-sis of biomolecules. Thus, the pace of progress is brisk and warrantsperiodic assessment and summary.
Acknowledgments
R.A.B. and A.T. thank NYUAD for their generous support. L.C.thanks the Centre National de la Recherche Scientifique (CNRS),the University of Strasbourg, and the Region Alsace in France forfinancial support.
References
[1] R.W. Horne, Virus Structure, Academic Press, New York, 1974.[2] P.M. Proulx-Curry, N.D. Chasteen, Coord. Chem. Rev. 144 (1995) 347.[3] J.-M. Lehn, Angew. Chem., Int. Ed. 27 (1988) 89.[4] C.J. Pedersen, Angew. Chem., Int. Ed. Engl. 27 (1988) 1021.[5] C.J. Pedersen, Angew. Chem. 100 (1988) 1053.[6] M.D. Ward, P.R. Raithby, Chem. Soc. Rev. 42 (2013) 1619.[7] M. Yoshizawa, J.K. Klosterman, M. Fujita, Angew. Chem., Int. Ed. 48 (2009)
3418.[8] D.L. Caulder, K.N. Raymond, Acc. Chem. Res. 32 (1999) 975.[9] D.L. Caulder, K.N. Raymond, J. Chem. Soc., Dalton Trans. (1999) 1185.
[10] D.L. Caulder, C. Bruckner, R.E. Powers, S. Konig, T.N. Parac, J.A. Leary, K.N.Raymond, J. Am. Chem. Soc. 123 (2001) 8923.
[11] M. Fujita, M. Tominaga, A. Hori, B. Therrien, Acc. Chem. Res. 38 (2005) 371.[12] M. Fujita, Chem. Soc. Rev. 27 (1998) 417.[13] C.G. Oliveri, P.A. Ulmann, M.J. Wiester, C.A. Mirkin, Acc. Chem. Res. 41 (2008)
1618.[14] N.C. Gianneschi, M.S. Masar, C.A. Mirkin, Acc. Chem. Res. 38 (2005) 825.[15] F.A. Cotton, C. Lin, C.A. Murillo, Proc. Natl. Acad. Sci. 99 (2002) 4810.[16] S. De, K. Mahata, M. Schmittel, Chem. Soc. Rev. 39 (2010) 1555.[17] J.R. Nitschke, Acc. Chem. Res. 40 (2007) 103.[18] J.P. Collin, V. Heitz, J.P. Sauvage, Mol. Mach. 262 (2005) 29.[19] R.W. Saalfrank, H. Maid, A. Scheurer, Angew. Chem., Int. Ed. 47 (2008) 8794.[20] R. Chakrabarty, P.S. Mukherjee, P.J. Stang, Chem. Rev. 111 (2011) 6810.[21] M.D. Ward, Chem. Commun. (2009) 4487.[22] S.J. Dalgarno, N.P. Power, J.L. Atwood, Coord. Chem. Rev. 252 (2008) 825.[23] M. Fujita, M. Tominaga, A. Hori, B. Therrien, Acc. Chem. Res. 38 (2005) 369.[24] D.J. Tranchemontagne, Z. Ni, M. O’Keeffe, O.M. Yaghi, Angew. Chem., Int. Ed.
47 (2008) 5136.[25] S.R. Seidel, P.J. Stang, Acc. Chem. Res. 35 (2002) 972.[26] K. Ghosh, J. Hu, H.S. White, P.J. Stang, J. Am. Chem. Soc. 131 (2009) 6695.[27] Y.-R. Zheng, Z. Zhao, H. Kim, M. Wang, K. Ghosh, J.B. Pollock, K.-W. Chi, P.J.
Stang, Inorg. Chem. 49 (2010) 10238.[28] P. Jin, S.J. Dalgarno, J.L. Atwood, Coord. Chem. Rev. 254 (2010) 1760.[29] R. Custelcean, P. Remy, P.V. Bonnesen, D.-E. Jiang, B.A. Moyer, Angew. Chem.,
Int. Ed. 47 (2008) 1866.
[30] C.R.K. Glasson, J.K. Clegg, J.C. McMurtrie, G.V. Meehan, L.F. Lindoy, C.A. Motti,B. Moubaraki, K.S. Murray, J.D. Cashion, Chem. Sci. 2 (2011) 540.
[31] E. Huerta, G.A. Metselaar, A. Fragoso, E. Santos, C. Bo, J. de Mendoza, Angew.Chem., Int. Ed. 46 (2007) 202.
[32] N. Kishi, Z. Li, K. Yoza, M. Akita, M. Yoshizawa, J. Am. Chem. Soc. 133 (2011)11438.
[33] W. Meng, B. Breiner, K. Rissanen, J.D. Thoburn, J.K. Clegg, J.R. Nitschke, Angew.Chem., Int. Ed. 50 (2011) 3479.
[34] K. Mahata, P.D. Frischmann, F. Wurthner, J. Am. Chem. Soc. 135 (2013) 15656.[35] I.A. Riddell, M.M.J. Smulders, J.K. Clegg, J.R. Nitschke, Chem. Commun. 47
(2011) 457.[36] M. Yoshizawa, S. Miyagi, M. Kawano, K. Ishiguro, M. Fujita, J. Am. Chem. Soc.
126 (2004) 9172.[37] M. Yoshizawa, Y. Takeyama, T. Kusukawa, M. Fujita, Angew. Chem., Int. Ed. 41
(2002) 1347.[38] M. Yoshizawa, Y. Takeyama, T. Okano, M. Fujita, J. Am. Chem. Soc. 125 (2003)
3243.[39] R.A. Bilbeisi, S. Zarra, H.L. Feltham, G.N. Jameson, J.K. Clegg, S. Brooker, J.R.
Nitschke, Chem. Eur. J. 19 (2013) 8058.[40] L. Zhao, S. Qu, C. He, R. Zhang, C. Duan, Chem. Commun. 47 (2011) 9387.[41] J. Wang, C. He, P. Wu, J. Wang, C. Duan, J. Am. Chem. Soc. 133 (2011) 12402.[42] D.L. Caulder, R.E. Powers, T.N. Parac, K.N. Raymond, Angew. Chem., Int. Ed. 37
(1998) 1840.[43] D.L. Caulder, C. Brückner, R.E. Powers, S. Koenig, T.N. Parac, J.A. Leary, K.A.
Raymond, J. Am. Chem. Soc. 123 (2001) 8923.[44] R.W. Saalfrank, B. Demleitner, H. Glaser, H. Maid, D. Bathelt, F. Hampel, W.
Bauer, M. Teichert, Chem. Eur. J. 8 (2002) 2679.[45] R.L. Paul, S.P. Argent, J.C. Jeffery, L.P. Harding, J.M. Lynam, M.D. Ward, Dalton
Trans. (2004) 3453.[46] S.P. Argent, T. Riis-Johannessen, J.C. Jeffery, L.P. Harding, M.D. Ward, Chem.
Commun. (2005) 4647.[47] C.R.K. Glasson, G.V. Meehan, J.K. Clegg, L.F. Lindoy, P. Turner, M.B. Duriska, R.
Willis, Chem. Commun. (2008) 1190.[48] I.S. Tidmarsh, B.F. Taylor, M.J. Hardie, L. Russo, W. Clegg, M.D. Ward, New J.
Chem. 33 (2009) 366.[49] A.J. Amoroso, J.C. Jeffery, P.L. Jones, J.A. McCleverty, P. Thornton, M.D. Ward,
Angew. Chem., Int. Ed. Engl. 34 (1995) 1443.[50] C. Brückner, R.E. Powers, K.N. Raymond, Angew. Chem., Int. Ed. 37 (1998)
1837.[51] R.A. Bilbeisi, J.K. Clegg, N. Elgrishi, X.d. Hatten, M. Devillard, B. Breiner, P. Mal,
J.R. Nitschke, J. Am. Chem. Soc. 134 (2012) 5110.[52] A. Sorensen, A.M. Castilla, T.K. Ronson, M. Pittelkow, J.R. Nitschke, Angew.
Chem., Int. Ed. Engl. 52 (2013) 11273.[53] A. Ferguson, M.A. Squire, D. Siretanu, D. Mitcov, C. Mathoniere, R. Clerac, P.E.
Kruger, Chem. Commun. 49 (2013) 1597.[54] R. Custelcean, P.V. Bonnesen, N.C. Duncan, X. Zhang, L.A. Watson, G. Van
Berkel, W.B. Parson, B.P. Hay, J. Am. Chem. Soc. 134 (2012) 8525.[55] R. Custelcean, Chem. Commun. 49 (2013) 2173.[56] O. Chepelin, J. Ujma, X. Wu, A.M. Slawin, M.B. Pitak, S.J. Coles, J. Michel, A.C.
Jones, P.E. Barran, P.J. Lusby, J. Am. Chem. Soc. 134 (2012) 19334.[57] D. Fiedler, D.H. Leung, R.G. Bergman, K.N. Raymond, Acc. Chem. Res. 38 (2005)
349.[58] A.V. Davis, D. Fiedler, M. Ziegler, A. Terpin, K.N. Raymond, J. Am. Chem. Soc.
129 (2007) 15354.[59] W. Xuan, M. Zhang, Y. Liu, Z. Chen, Y. Cui, J. Am. Chem. Soc. 134 (2012) 6904.[60] N. Ousaka, J.K. Clegg, J.R. Nitschke, Angew. Chem., Int. Ed. 51 (2012) 1464.[61] C.J. Brown, R.G. Bergman, K.N. Raymond, J. Am. Chem. Soc. 131 (2009) 17530.[62] J.M. Rivera, T. Martin, J. Rebek, Science 279 (1998) 1021.[63] J.L. Bolliger, A.M. Belenguer, J.R. Nitschke, Angew. Chem., Int. Ed. Engl. 52
(2013) 7958.[64] W. Meng, B. Breiner, K. Rissanen, J.D. Thoburn, J.K. Clegg, J.R. Nitschke, Angew.
Chem., Int. Ed. 50 (2011) 3479.[65] M.M. Smulders, A. Jimenez, J.R. Nitschke, Angew. Chem., Int. Ed. Engl. 51
(2012) 6681.[66] M.H. Alkordi, J.L. Belof, E. Rivera, L. Wojtas, M. Eddaoudi, Chem. Sci. 2 (2011)
1695.[67] A. Stephenson, S.P. Argent, T. Riis-Johannessen, I.S. Tidmarsh, M.D. Ward, J.
Am. Chem. Soc. 133 (2011) 858.[68] M. Otte, P.F. Kuijpers, O. Troeppner, I. Ivanovic-Burmazovic, J.N.H. Reek, B. de
Bruin, Chem. Eur. J. 19 (2013) 10170.[69] A.M. Stadler, C. Burg, J. Ramirez, J.M. Lehn, Chem. Commun. 49 (2013) 5733.[70] X.-J. Li, F.-L. Jiang, M.-Y. Wu, S.-Q. Zhang, Y.-F. Zhou, M.-C. Hong, Inorg. Chem.
51 (2012) 4116.[71] S. Pasquale, S. Sattin, E.C. Escudero-Adan, M. Martinez-Belmonte, J. de
Mendoza, Nat. Commun. 3 (2012) 1.[72] T.R. Cook, Y.R. Zheng, P.J. Stang, Chem. Rev. 113 (2013) 734.[73] Q.-F. Sun, J. Iwasa, D. Ogawa, Y. Ishido, S. Sato, T. Ozeki, Y. Sei, K. Yamaguchi,
M. Fujita, Science 328 (2010) 1144.[74] A. Stephenson, D. Sykes, M.D. Ward, Dalton Trans. 42 (2013) 6756.[75] Q.F. Sun, S. Sato, M. Fujita, Nat. Chem. 4 (2012) 330.[76] K. Xiong, F. Jiang, Y. Gai, D. Yuan, L. Chen, M. Wu, K. Su, M. Hong, Chem. Sci. 3
(2012) 2321.[77] M. Liu, W. Liao, C. Hu, S. Du, H. Zhang, Angew. Chem., Int. Ed. Engl. 51 (2012)
1585.[78] T.K. Ronson, S. Zarra, S.P. Black, J.R. Nitschke, Chem. Commun. 49 (2013)
2476.

108 R.A. Bilbeisi et al. / Inorganica Chimica Acta 417 (2014) 79–108
[79] V.E. Campbell, J.R. Nitschke, Synlett (2008) 3077.[80] X.P. Zhou, J. Liu, S.Z. Zhan, J.R. Yang, D. Li, K.M. Ng, R.W. Sun, C.M. Che, J. Am.
Chem. Soc. 134 (2012) 8042.[81] D. Fujita, K. Suzuki, S. Sato, M. Yagi-Utsumi, Y. Yamaguchi, N. Mizuno, T.
Kumasaka, M. Takata, M. Noda, S. Uchiyama, K. Kato, M. Fujita, Nat. Commun.3 (2012) 1093.
[82] R.S. Forgan, J.-P. Sauvage, J.F. Stoddart, Chem. Rev. 111 (2011) 5434.[83] H.R. Frisch, E. Wasserman, J. Am. Chem. Soc. 83 (1961) 3789.[84] C.O. Dietrich-Buchecker, J.P. Sauvage, J.P. Kintzinger, Tetrahedron Lett. 24
(1983) 5095.[85] M. Beyler, V.r. Heitz, J.-P. Sauvage, J. Am. Chem. Soc. 132 (2010) 4409.[86] J.D. Megiatto Jr., D.I. Schuster, Org. Lett. 13 (2011) 1808.[87] J.D. Megiatto Jr., D.I. Schuster, G. de Miguel, S. Wolfrum, D.M. Guldi, Chem.
Mater. 24 (2012) 2472.[88] C.A. Otter, P.J. Patty, M.A. Williams, M.R. Waterland, S.G. Telfer, Nanoscale 3
(2011) 941.[89] J.F. Ayme, J. Lux, J.P. Sauvage, A. Sour, Chemistry 18 (2012) 5565.[90] T. Prakasam, M. Lusi, M. Elhabiri, C. Platas-Iglesias, J.C. Olsen, Z. Asfari, S.
Cianferani-Sanglier, F. Debaene, L.J. Charbonniere, A. Trabolsi, Angew. Chem.,Int. Ed. Engl. 52 (2013) 9956.
[91] D.A. Leigh, P.J. Lusby, A.M. Slawin, D.B. Walker, Chem. Commun. 48 (2012)5826.
[92] L. Aboshyan-Sorgho, M. Cantuel, G. Bernardinelli, C. Piguet, Dalton Trans. 41(2012) 7218.
[93] M. Han, H.-Y. Zhang, L.-X. Yang, Z.-J. Ding, R.-J. Zhuang, Y. Liu, Eur. J. Org.Chem. 2011 (2011) 7271.
[94] C. Alvarino, A. Terenzi, V. Blanco, M.D. Garcia, C. Peinador, J.M. Quintela,Dalton Trans. 41 (2012) (1998) 11992.
[95] V. Blanco, M.D. Garcia, C. Peinador, J.M. Quintela, Chem. Sci. 2 (2011) 2407.[96] S. Li, J. Huang, T.R. Cook, J.B. Pollock, H. Kim, K.W. Chi, P.J. Stang, J. Am. Chem.
Soc. 135 (2013) 2084.[97] M.A. Aleman Garcia, N. Bampos, Org. Biomol. Chem. 11 (2013) 27.[98] M.J. Langton, J.D. Matichak, A.L. Thompson, H.L. Anderson, Chem. Sci. 2 (2011)
1897.[99] S.P. Black, A.R. Stefankiewicz, M.M. Smulders, D. Sattler, C.A. Schalley, J.R.
Nitschke, J.K. Sanders, Angew. Chem., Int. Ed. Engl. 52 (2013) 5749.[100] L. Loots, L.J. Barbour, Chem. Commun. 49 (2013) 671.[101] L. Jiang, P. Ju, X.R. Meng, X.J. Kuang, T.B. Lu, Sci. Rep. 2 (2012) 668.[102] K.S. Chichak, S.J. Cantrill, A.R. Pease, S.-H. Chiu, G.W.V. Cave, J.L. Atwood, J.F.
Stoddart, Science 304 (2004) 1308.[103] D.M. Engelhard, S. Freye, K. Grohe, M. John, G.H. Clever, Angew. Chem., Int.
Ed. 51 (2012) 4747.[104] P.E. Barran, H.L. Cole, S.M. Goldup, D.A. Leigh, P.R. McGonigal, M.D. Symes, J.
Wu, M. Zengerle, Angew. Chem., Int. Ed. 50 (2011) 12280.[105] J.-F. Ayme, J.E. Beves, D.A. Leigh, R.T. McBurney, K. Rissanen, D. Schultz, Nat.
Chem. 4 (2012) 15.[106] J.F. Ayme, J.E. Beves, D.A. Leigh, R.T. McBurney, K. Rissanen, D. Schultz, J. Am.
Chem. Soc. 134 (2012) 9488.[107] B. Hasenknopf, J.M. Lehn, N. Boumediene, A. Dupont Gervais, A. Van
Dorsselaer, B. Kneisel, D. Fenske, J. Am. Chem. Soc. 119 (1997) 10956.[108] J.E. Beves, C.J. Campbell, D.A. Leigh, R.G. Pritchard, Angew. Chem., Int. Ed.
Engl. 52 (2013) 6464.[109] S.-L. Huang, Y.-J. Lin, T.S.A. Hor, G.-X. Jin, J. Am. Chem. Soc. 135 (2013) 8125.[110] F. Li, J.K. Clegg, L.F. Lindoy, R.B. MacQuart, G.V. Meehan, Nat. Commun. 2
(2011) 205.[111] S. Freye, J. Hey, A. Torras-Galan, D. Stalke, R. Herbst-Irmer, M. John, G.H.
Clever, Angew. Chem., Int. Ed. Engl. 51 (2012) 2191.[112] S. Freye, D.M. Engelhard, M. John, G.H. Clever, Chemistry 19 (2013) 2114.[113] R. Sekiya, M. Fukuda, R. Kuroda, J. Am. Chem. Soc. 134 (2012) 10987.[114] J.J. Henkelis, T.K. Ronson, L.P. Harding, M.J. Hardie, Chem. Commun. 47 (2011)
6560.[115] A.K. Bar, S. Raghothama, D. Moon, P.S. Mukherjee, Chemistry 18 (2012) 3199.[116] J. Heine, J. Schmedt auf der Gunne, S. Dehnen, J. Am. Chem. Soc. 133 (2011)
10018.[117] M. Ruben, J. Rojo, F.J. Romero-Salguero, L.H. Uppadine, J.M. Lehn, Angew.
Chem., Int. Ed. 43 (2004) 3644.[118] L.N. Dawe, K.V. Shuvaev, L.K. Thompson, Chem. Soc. Rev. 38 (2009) 2334.[119] A.R. Stefankiewicz, G. Rogez, J. Harrowfield, M. Drillon, J.M. Lehn, Dalton
Trans. (2009) 5787.[120] J.G. Hardy, Chem. Soc. Rev. 42 (2013) 7881.[121] C. Piguet, G. Bernardinelli, G. Hopfgartner, Chem. Rev. 97 (1997) 2005.[122] M.J. Hannon, L.J. Childs, Supramol. Chem. 16 (2004) 7.[123] S.E. Howson, P. Scott, Dalton Trans. 40 (2011) 4332.[124] H. Miyake, H. Tsukube, Chem. Soc. Rev. 41 (2012) 6977.[125] S. Leininger, B. Olenyuk, P.J. Stang, Chem. Rev. 100 (2000) 853.[126] F.C.J.M. van Veggel, W. Verboom, D.N. Reinhoudt, Chem. Rev. 94 (1994) 279.[127] C.J. Jones, Chem. Soc. Rev. 27 (1998) 289.[128] B.H. Northrop, H.B. Yang, P.J. Stang, Chem. Commun. (2008) 5896.[129] G. Maayan, M.D. Ward, K. Kirshenbaum, Proc. Natl. Acad. Sci. 106 (2009)
13679.[130] H.A. Scheidt, A. Sickert, T. Meier, N. Castellucci, C. Tomasini, D. Huster, Org.
Biomol. Chem. 9 (2011) 6998.[131] S. Chen, L.J. Chen, H.B. Yang, H. Tian, W. Zhu, J. Am. Chem. Soc. 134 (2012)
13596.[132] R. Chakrabarty, P.J. Stang, J. Am. Chem. Soc. 134 (2012) 14738.
[133] E.M. Sletten, C.R. Bertozzi, Angew. Chem., Int. Ed. Engl. 48 (2009) 6974.[134] M.F. Debets, S.S. van Berkel, J. Dommerholt, A.T. Dirks, F.P. Rutjes, F.L. van
Delft, Acc. Chem. Res. 44 (2011) 805.[135] A. Mishra, S. Ravikumar, S.H. Hong, H. Kim, V. Vajpayee, H. Lee, B. Ahn, M.
Wang, P.J. Stang, K.W. Chi, Organometallics 30 (2011) 6343.[136] P.W. van Leeuwen, D. Rivillo, M. Raynal, Z. Freixa, J. Am. Chem. Soc. 133
(2011) 18562.[137] H.T. Chifotides, I.D. Giles, K.R. Dunbar, J. Am. Chem. Soc. 135 (2013) 3039.[138] V. Vajpayee, H. Kim, A. Mishra, P.S. Mukherjee, P.J. Stang, M.H. Lee, H.K. Kim,
K.W. Chi, Dalton Trans. 40 (2011) 3112.[139] P. Gütlich, H.A. Goodwin, Top. Curr. Chem. 233 (2004) 1.[140] R.-J. Wei, Q. Huo, J. Tao, R.-B. Huang, L.-S. Zheng, Angew. Chem., Int. Ed.
(2011) 8940.[141] S.E. Howson, A. Bolhuis, V. Brabec, G.J. Clarkson, J. Malina, A. Rodger, P. Scott,
Nat. Chem. 4 (2012) 31.[142] B. Wang, Z. Zang, H. Wang, W. Dou, X. Tang, W. Liu, Y. Shao, J. Ma, Y. Li, J.
Zhou, Angew. Chem., Int. Ed. 52 (2013) 3756.[143] S. Phongtongpasuk, S. Paulus, J. Schnabl, R.K. Sigel, B. Spingler, M.J. Hannon, E.
Freisinger, Angew. Chem., Int. Ed. Engl. 52 (2013) 11513.[144] O.R. Clegg, R.V. Fennessy, L.P. Harding, C.R. Rice, T. Riis-Johannessen, N.C.
Fletcher, Dalton Trans. 40 (2011) 12381.[145] Z.S. Wu, J.T. Hsu, C.C. Hsieh, Y.C. Horng, Chem. Commun. 48 (2012) 3436.[146] B. El Aroussi, S. Zebret, C. Besnard, P. Perrottet, J. Hamacek, J. Am. Chem. Soc.
133 (2011) 10764.[147] A.R. Stefankiewicz, J. Harrowfield, A. Madalan, K. Rissanen, A.N. Sobolev, J.M.
Lehn, Dalton Trans. 40 (2011) 12320.[148] X. Bao, W. Liu, L.L. Mao, S.D. Jiang, J.L. Liu, Y.C. Chen, M.L. Tong, Inorg. Chem.
52 (2013) 6233.[149] J.M. Lehn, Chem. Eur. J. 5 (1999) 2455.[150] C.D. Meyer, C.S. Joiner, J.F. Stoddart, Chem. Soc. Rev. 36 (2007) 1705.[151] S.J. Rowan, S.J. Cantrill, G.R.L. Cousins, J.K.M. Sanders, J.F. Stoddart, Angew.
Chem., Int. Ed. 41 (2002) 899.[152] W.J. Meng, T.K. Ronson, J.K. Clegg, J.R. Nitschke, Angew. Chem., Int. Ed. 52
(2013) 1017.[153] Y.-R. Zheng, W.-J. Lan, M. Wang, T.R. Cook, P.J. Stang, J. Am. Chem. Soc. 133
(2011) 17045.[154] D.H. Qu, B.L. Feringa, Angew. Chem., Int. Ed. Engl. 49 (2010) 1107.[155] T. Malah, S. Rolf, S.M. Weidner, A.F. Thunemann, S. Hecht, Chem. Eur. J. 18
(2012) 5837.[156] T. Bark, M. Duggeli, H. Stoeckli-Evans, A. Von Zelewsky, Angew. Chem., Int.
Ed. 40 (2001) 2848.[157] R.A. Bilbeisi, T.K. Ronson, J.R. Nitschke, Angew. Chem., Int. Ed. Engl. 52 (2013)
9027.[158] M. Barboiu, G. Vaughan, N. Kyritsakas, J.-M. Lehn, Chem. Eur. J. 9 (2003) 763.[159] S.J. Pike, P.J. Lusby, Chem. Commun. 46 (2010) 8338.[160] M. Fujita, O. Sasaki, T. Mitsuhashi, T. Fujita, J. Yazaki, K. Yamaguchi, K. Ogura,
Chem. Commun. (1996) 1535.[161] C. Browne, S. Brenet, J.K. Clegg, J.R. Nitschke, Angew. Chem., Int. Ed. Engl. 52
(2013) 1944.[162] N. Koumura, R.W.J. Zijistra, R.A. Van Delden, N. Harada, B.L. Feringa, Nature
401 (1999) 152.[163] T. Muraoka, K. Kinbara, T. Aida, Nature 440 (2006) 512.[164] I.A. Riddell, M.M.J. Smulders, J.K. Clegg, Y.R. Hristova, B. Breiner, J.D. Thoburn,
J.R. Nitschke, Nat. Chem. 4 (2012) 860.[165] I.A. Riddell, Y.R. Hristova, J.K. Clegg, C.S. Wood, B. Breiner, J.R. Nitschke, J. Am.
Chem. Soc. 135 (2013) 2723.[166] M.C. O’Sullivan, J.K. Sprafke, D.V. Kondratuk, C. Rinfray, T.D.W. Claridge, A.
Saywell, M.O. Blunt, J.N. O’Shea, P.H. Beton, M. Malfois, H.L. Anderson, Nature469 (2011) 72.
[167] E. Stulz, S.M. Scott, A.D. Bond, S.J. Teat, J.K.M. Sanders, Chem. Eur. J. 9 (2003)6039.
[168] E. Stulz, Y.-F. Ng, S.M. Scott, J.K.M. Sanders, Chem. Commun. (2002) 524.[169] S. Hiraoka, K. Harano, M. Shiro, M. Shionoya, Angew. Chem., Int. Ed. Engl. 44
(2005) 2727.[170] S. Zarra, J.K. Clegg, J.R. Nitschke, Angew. Chem., Int. Ed. 52 (2013) 4837.[171] R.A. Bilbeisi, T.K. Ronson, J.R. Nitschke, Angew. Chem., Int. Ed. 125 (2013)
9197.[172] I.A. Riddell, M.M. Smulders, J.K. Clegg, Y.R. Hristova, B. Breiner, J.D. Thoburn,
J.R. Nitschke, Nat. Chem. 4 (2012) 751.[173] A. Granzhan, C. Schouwey, T. Riis-Johannessen, R. Scopelliti, K. Severin, J. Am.
Chem. Soc. 133 (2011) 7106.[174] Y.R. Zheng, Z. Zhao, M. Wang, K. Ghosh, J.B. Pollock, T.R. Cook, P.J. Stang, J. Am.
Chem. Soc. 132 (2010) 16873.[175] I.A. Riddell, M.M.J. Smulders, J.K. Clegg, Y.R. Hristova, B. Breiner, J.D. Thoburn,
J.R. Nitschke, Nat. Chem. 4 (2012) 751.[176] J.R. Li, H.C. Zhou, Nat. Chem. 2 (2010) 893.[177] W. Meng, T.K. Ronson, J.K. Clegg, J.R. Nitschke, Angew. Chem., Int. Ed. Engl. 52
(2013) 1017.[178] M.E. Belowich, J.F. Stoddart, Chem. Soc. Rev. 41 (2012) 5881.[179] Y.R. Hristova, M.M.J. Smulders, J.K. Clegg, B. Breiner, J.R. Nitschke, Chem. Sci. 2
(2011) 638.[180] D. Schultz, J.R. Nitschke, J. Am. Chem. Soc. 128 (2006) 9887.[181] H. Kumari, A.V. Mossine, S.R. Kline, C.L. Dennis, D.A. Fowler, S.J. Teat, C.L.
Barnes, C.A. Deakyne, J.L. Atwood, Angew. Chem., Int. Ed. Engl. 51 (2012)1452.




















