
ChemInform Abstract: Nanoalloys: From Theory to Applications of Alloy Clusters and Nanoparticles
Upload
independentCategory
view
2download
0
PLEASE SCROLL DOWN FOR ARTICLE
This article was downloaded by: [Max Planck Inst & Research Groups Consortium]On: 3 November 2009Access details: Access Details: [subscription number 909926962]Publisher Taylor & FrancisInforma Ltd Registered in England and Wales Registered Number: 1072954 Registered office: Mortimer House,37-41 Mortimer Street, London W1T 3JH, UK
Synthetic CommunicationsPublication details, including instructions for authors and subscription information:http://www.informaworld.com/smpp/title~content=t713597304
Convenient Methodology for the Synthesis of TrialkylhydrazinesAlvaro J. Vázquez a; Cristian Rodríguez a; N. Sbarbati Nudelman a
a Department of Organic Chemistry, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires,Ciudad Universitaria, Buenos Aires, Argentina
Online Publication Date: 01 January 2009
To cite this Article Vázquez, Alvaro J., Rodríguez, Cristian and Nudelman, N. Sbarbati(2009)'Convenient Methodology for theSynthesis of Trialkylhydrazines',Synthetic Communications,39:22,3958 — 3972
To link to this Article: DOI: 10.1080/00397910902738120
URL: http://dx.doi.org/10.1080/00397910902738120
Full terms and conditions of use: http://www.informaworld.com/terms-and-conditions-of-access.pdf
This article may be used for research, teaching and private study purposes. Any substantial orsystematic reproduction, re-distribution, re-selling, loan or sub-licensing, systematic supply ordistribution in any form to anyone is expressly forbidden.
The publisher does not give any warranty express or implied or make any representation that the contentswill be complete or accurate or up to date. The accuracy of any instructions, formulae and drug dosesshould be independently verified with primary sources. The publisher shall not be liable for any loss,actions, claims, proceedings, demand or costs or damages whatsoever or howsoever caused arising directlyor indirectly in connection with or arising out of the use of this material.

Convenient Methodology for theSynthesis of Trialkylhydrazines
Alvaro J. V�aazquez, Cristian Rodrıguez, and N. Sbarbati NudelmanDepartment of Organic Chemistry, Facultad de Ciencias Exactas yNaturales, Universidad de Buenos Aires, Ciudad Universitaria,
Buenos Aires, Argentina
Abstract: A convenient methodology, based on the addition of organolithiumcompounds to N-nitrosamines, was developed for the synthesis of substitutedhydrazones and trialkyl hydrazines. The reaction is very sensitive to the reactionconditions, particularly the [RLi]–[nitrosamine] ratio. For molar ratios of 1–1.2,an almost quantitative conversion to the N¼O bond addition products (i.e.,alkylhydrazones) can be obtained in very good yields, with any remaining nitro-samine. Using greater molar ratios (in the range of 3–5), a second addition occurs,and branched trialkyl hydrazines are obtained, in good yields, with variableamounts of hydrazone remaining. Substituted hydrazines are known to have use-ful commercial applications and remarkable biological activities. A wide diversityof trialkyl hydrazines could be synthesized by using two different organolithiums.
Keywords: Addition to the N¼O bond, hydrazines, hydrazones, nitrosamines,organolithiums
INTRODUCTION
Hydrazine and its derivatives are of considerable technical andcommercial importance,[1,2] and they are receiving renewed interestbecause of the recent discovery of their remarkable biological activities.
Received September 5, 2008.Address correspondence to N. Sbarbati Nudelman, Department of Organic
Chemistry, Facultad de Ciencias Exactas y Naturales, Universidad de BuenosAires, Pab. II, P. 3, Ciudad Universitaria, 1428 Buenos Aires, Argentina. E-mail:[email protected]
Synthetic Communications1, 39: 3958–3972, 2009
Copyright # Taylor & Francis Group, LLC
ISSN: 0039-7911 print=1532-2432 online
DOI: 10.1080/00397910902738120
3958
Downloaded By: [Max Planck Inst & Research Groups Consortium] At: 20:28 3 November 2009

These derivatives are wellknown as pesticides, drugs, amino acidprecursors, and synthetic building blocks for heterocyclic synthesis.[3]
Substituted hydrazines are key components of azapeptides, a type of pep-tidomimetics, in which one or more of the amino acid residues have beenreplaced by monomers with nitrogen in the position of the chiral carbons.Hydrazine-based peptidomimetics are found to be potent agents againsthepatitis, AIDS, and SARS.[4,5] Isoniacid, a pyiridine hydrazide, is theactive compound in many drugs used against tuberculosis; several hydra-zine derivatives have been shown to be effective for Parkinson’s diseaseand hypertension.[1]
Because of the remarkable interest in substituted hydrazines, a largenumber of synthetic procedures have been developed.[1,6] Many attemptsat alkylation of free hydrazine gave poor results; generally mixtures areproduced, with yields seldom exceeding 40%. To avoid multiple alkyl-ation, some systematic synthetic methods have been developed recentlyusing protective group methodology. Nowadays, a large number ofconvenient protecting groups for amino functions are available,[7] but thisstrategy demands many steps (at least two extra steps) to obtain the endproduct.[8,9] Though the purity and total yield of the products are oftengreater and purification steps are simpler, the protective group methodol-ogy uses several steps, consumes more energy, and produces significantresidues; therefore, development of cleaner synthetic methods aredesired.[10,11] A polystyrene sulfonic acid–catalyzed greener synthesis ofhydrazones was recently reported.[12] Tandem reactions are consideredamong the most efficient and economical synthetic strategies,[13] and theamounts of solvents, reagents, and energy are significant decreased ascompared with stepwise reactions.[14,15] The present article describes aconvenient, direct methodology for the preparation of trialkyl hydrazinesbased on the tandem addition of organolithium compounds to the doublebond of dialkylnitrosamines, producing substituted hydrazones, whichin a one-pot, one-step reaction can be transformed into trialkylhydrazinesby the tandem addition of the same or another organolithium reagent.On the other hand, nitrosamines can be synthesized by the NO insertioninto the N-Li bond of lithium amides.[16]
RESULTS AND DISCUSSION
Synthesis of Hydrazones
New reactions on nitrosamines are being actively studied, especially theirrelationship with carcinogenetic and mutagenic properties[17]; recentstudies showed that they are mainly mutagenic through methylation of
Convenient Synthesis of Trialkylhydrazines 3959
Downloaded By: [Max Planck Inst & Research Groups Consortium] At: 20:28 3 November 2009

DNA.[18] There is special concern about the important roles thatenvironmental nitrosamines play in the etiology of human cancer.[19]
The nitrosamine found in cigarette smoke induces lung tumors in mice[20]
and esophageal and gastroduodenal cancer in animals.[21,22] Nitrosaminesalso have been found in several foods and beverages.[9,23]
The synthetic methodology herewith described is based on thereaction of organolithium compounds, R1CH2Li, with N-dialkyl-nitrosamines. Namely, N-nitrosodicyclohexylamine, 1, in tetrahydrofuran(THF), leads to the production of hydrazones, 2, as indicated in the Fig. 1.
Mechanistic studies on the addition of RLi on a C¼O bond areabundant in the literature, but few involve bonds to nitrogen atoms.[24]
The first adduct between an amine and a carbonyl derivative was isolateda few years ago.[25] By 13C NMR, we characterized the first adductbetween a formamide and R2NLi,[26] and a more complex dilithiatedadduct was later characterized.[27]
The reaction described in this article is extremely sensitive to thereaction conditions. The reaction can be carried out at room temperature;in most cases the reaction time is very short (5min, approx.). One mmolof 1 was dissolved in 2ml of THF, and the organolithium reagent wasadded in a [R1CH2Li]–[1]¼ 1.05:1.2 molar ratio. The concentration oforganolithium was in the range of 0.5–1.5M (approx.) in n-hexane solu-tion. In the case of MeLi (which is less soluble in hexane), a 1M THFsolution was used, and a longer reaction time (2 h) was required. In gen-eral terms, the reaction is very clean, no nitrosamine remains, and goodto high yields (69–93%) are obtained in short reaction times.
Synthesis of Trialkylhydrazines
In the reaction of 1 with RLi, under the conditions described previouslybut using a high excess of RLi (3–5 eq.), an in situ second addition occursat the N¼C bond of intermediate 2, and a trialkyl hydrazine, 3, isformed (Fig. 2).
To the best of our knowledge, few studies have dealt with the addi-tion of RLi on a carbon–nitrogen double bond. We observed that the
Figure 1. R1: (a) H; (b) Et; (c) n-Pr; and (d) n-Bu; 69–93% yield.
3960 A. J. V�aazquez, C. Rodrıguez, and N. S. Nudelman
Downloaded By: [Max Planck Inst & Research Groups Consortium] At: 20:28 3 November 2009

addition to the N¼C bond is slower than to the N¼O. To systematizeresults, in all cases the reactions were carried out for 2 h at room tempera-ture. (Attempts to carry out the reaction at lower temperatures were notsatisfactory.) The results are shown in Table 1. As can be observed, goodyields of 3 were obtained for n-PrLi and n-BuLi (entries b and c). Withn-C5H11Li, instead, lower yields of 3d were obtained, and significantamounts of 2d remained; likely the greater steric hindrance of then-pentyl groups is responsible for the lower reactivity. In the reactionswith MeLi, normal addition to form the hydrazone of formaldehyde(2a) occurred, but when a second molecule of MeLi was added, loss ofthe elements of lithium hydride spontaneously occured and the hydra-zone of acetaldehyde was isolated as the main product instead of theexpected trialkylhydrazine. A similar LiH elimination was observed inthe reaction of diethylnitrosamine with MeLi, for which a tiny amount(<2%) of Et2NN¼CHCH3 was formed.[28]
The formation of 2c (starting from 1, and adding n-BuLi) is very fast(occurs in a few seconds, even at 0�C), The hydrazone 2c formed in situreacts with more n-BuLi, affording 3c (Fig. 3). When some remaininghydrazones are observed, the yields of hydrazines 3 can be increased byadding more n-BuLi after the reaction has advanced (i.e., when someremaining 2 was still present in the reaction mixture).
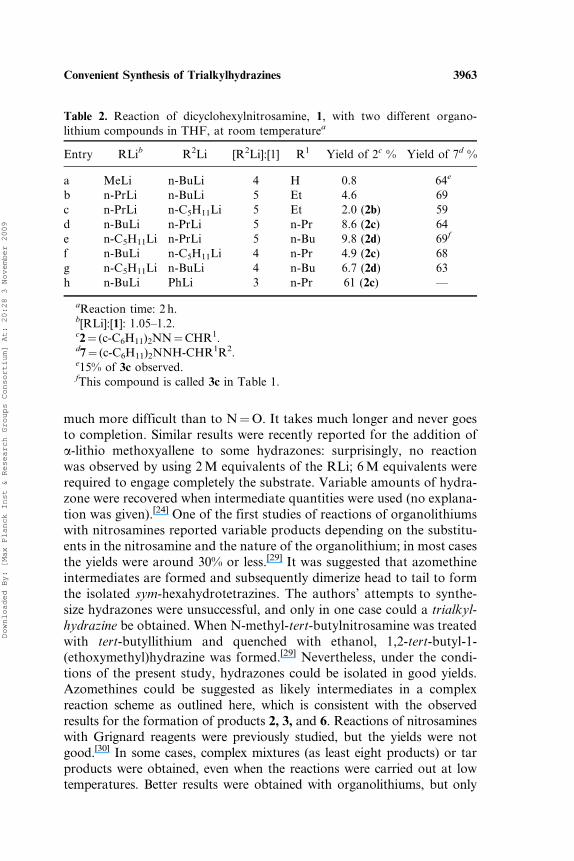
Table 1. Reaction of dicyclohexylnitrosamine, 1, with RLi (in high excess) inTHF, at room temperaturea
Entry RLi [RLi]:[1] R1 Yield of 2b% Yield of 3c%
a MeLi 3 H 28 36d,e
b n-PrLi 4 Et 9.3 77c n-BuLi 5 n-Pr 2.4 75d n-C5H11Li 8 n-Bu 13 61e
aTemp. 20–25�C. Reaction time 2 h.b2¼ (c-C6H11)2NN¼CHR1.c3¼ (c-C6H11)2NNH-CH(R1)R.dMain product¼ (c-C6H11)2NN¼CHCH3.eLateral products in minor yields.
Figure 2. R1: (a) H; (b) Et; (c) n-Pr; and (d) n-Bu.
Convenient Synthesis of Trialkylhydrazines 3961
Downloaded By: [Max Planck Inst & Research Groups Consortium] At: 20:28 3 November 2009

In some cases, by-products are produced in small amounts. Namely,dicyclohexylamine, 4; N-cyclohexylidenecyclohexylamine, 5; and N,N,N0-dicyclohexylalkylhydrazines, 6 were found in poor variable yields (3–5%);they are easily identifiable by gas chromatography.
Synthesis of Several Trialkylhydrazines 7
The reaction of 1was carried out using two different organolithiums with thepurpose of obtaining several branched hydrazines, 7, as shown in Fig. 4.
For the addition of the first organolithium, the procedure described inthe first part for the hydrazone synthesis was followed: addition of a slightexcess of R1CH2Li and short reaction times (a few minutes, except in thecase of MeLi, which required longer times). For the addition of the secondorganolithium, R2Li, a greater excess of the reagent (4–5 equiv.) and longerreaction times (2 h) were needed. As can be observed in Table 2, the yieldsof 7 are good, and little unreacted 2 remained; for the case of n-C5H11Li,the yields are less than in the other cases, as expected. On the other hand,it can be observed that by using PhLi, the hydrazine 7 h is not obtained(even when the reaction was carried out at 50�C), likely as a consequenceof the poor reactivity of PhLi compared to the alkyllithium compounds.
Though the reaction of organolithiums with the N¼O bond is muchless known than the synthetically very useful addition of RLi to C¼Obonds, the efficient formation of hydrazones shown in the present studycan be interpreted by an addition mechanism.
Addition of organolithium to C¼N bond is less frequent thanto C¼O bond, though it is a well-known reaction. Nevertheless, in thepresent conditions, addition of a second molecule of RLi to C¼N is
Figure 3. Reaction carried out with n-BuLi in high excess at r.t.
Figure 4. Sequence of reaction of compound 1 with two different organolithiumsat r.t.
3962 A. J. V�aazquez, C. Rodrıguez, and N. S. Nudelman
Downloaded By: [Max Planck Inst & Research Groups Consortium] At: 20:28 3 November 2009
much more difficult than to N¼O. It takes much longer and never goesto completion. Similar results were recently reported for the addition ofa-lithio methoxyallene to some hydrazones: surprisingly, no reactionwas observed by using 2M equivalents of the RLi; 6M equivalents wererequired to engage completely the substrate. Variable amounts of hydra-zone were recovered when intermediate quantities were used (no explana-tion was given).[24] One of the first studies of reactions of organolithiumswith nitrosamines reported variable products depending on the substitu-ents in the nitrosamine and the nature of the organolithium; in most casesthe yields were around 30% or less.[29] It was suggested that azomethineintermediates are formed and subsequently dimerize head to tail to formthe isolated sym-hexahydrotetrazines. The authors’ attempts to synthe-size hydrazones were unsuccessful, and only in one case could a trialkyl-hydrazine be obtained. When N-methyl-tert-butylnitrosamine was treatedwith tert-butyllithium and quenched with ethanol, 1,2-tert-butyl-1-(ethoxymethyl)hydrazine was formed.[29] Nevertheless, under the condi-tions of the present study, hydrazones could be isolated in good yields.Azomethines could be suggested as likely intermediates in a complexreaction scheme as outlined here, which is consistent with the observedresults for the formation of products 2, 3, and 6. Reactions of nitrosamineswith Grignard reagents were previously studied, but the yields were notgood.[30] In some cases, complex mixtures (as least eight products) or tarproducts were obtained, even when the reactions were carried out at lowtemperatures. Better results were obtained with organolithiums, but only
Table 2. Reaction of dicyclohexylnitrosamine, 1, with two different organo-lithium compounds in THF, at room temperaturea
Entry RLib R2Li [R2Li]:[1] R1 Yield of 2c % Yield of 7d %
a MeLi n-BuLi 4 H 0.8 64e
b n-PrLi n-BuLi 5 Et 4.6 69c n-PrLi n-C5H11Li 5 Et 2.0 (2b) 59d n-BuLi n-PrLi 5 n-Pr 8.6 (2c) 64e n-C5H11Li n-PrLi 5 n-Bu 9.8 (2d) 69f
f n-BuLi n-C5H11Li 4 n-Pr 4.9 (2c) 68g n-C5H11Li n-BuLi 4 n-Bu 6.7 (2d) 63h n-BuLi PhLi 3 n-Pr 61 (2c) ––
aReaction time: 2 h.b[RLi]:[1]: 1.05–1.2.c2¼ (c-C6H11)2NN¼CHR1.d7¼ (c-C6H11)2NNH-CHR1R2.e15% of 3c observed.fThis compound is called 3c in Table 1.
Convenient Synthesis of Trialkylhydrazines 3963
Downloaded By: [Max Planck Inst & Research Groups Consortium] At: 20:28 3 November 2009

in one case was a good yield (80%) of the corresponding hydrazoneobtained.[28] Hydrazones can be converted into hydrazides by a one-potreduction, followed by an in situ reaction with a carboxylic acid.[31]
The first step in Scheme 1 involves attack by the organolithiumreagent on the nitroso moiety to give adduct 8, which, by elimination ofthe elements of HO� Liþ, produces the hydrazone 2c. In smaller amounts,8 could undergo elimination involving the proton of the a-carbon, produ-cing the azomethine 10 (presumably through a diazenium salt 9), whichwould form the hydrazine 6 (which was isolated in small amounts). Byaddition of a second molecule of RLi to the C¼N bond, the adduct 12was formed, which after quenching produces the trialkylhydrazines 3.
Lateral products 4 and 5 can be explained by a denitrosation process,likely occurring through a nitrosiminium ion intermediate stabilized byresonance. Rajamaki and coworkers[32] reported the first evidence ofthe intermediacy of N-nitrosiminium ions in the nonenzymatic decompo-sition of some a-acetoxy-N-nitrosodialkylamines. Denitrosation of theN-nitrosiminium intermediate and simultaneous reduction could be theroute for formation of side products 4 and 5.
CONCLUSIONS
The reaction of dialkylnitrosamines with organolithium reagents providea simple route for the synthesis of substituted hydrazones, and especially
Scheme 1. Overall mechanistic scheme for the reaction of 1 with n-BuLi.
3964 A. J. V�aazquez, C. Rodrıguez, and N. S. Nudelman
Downloaded By: [Max Planck Inst & Research Groups Consortium] At: 20:28 3 November 2009

for the synthesis of multisubstituted hydrazines, which have acquiredrenewed interest for their wide versatile properties. Trialkyl hydrazinescan be formed by a one-pot, one-step reaction using excess of the orga-nolithium reagent; this method circumvents side reactions and avoidslengthy purification of products, problems sometimes associated witholder synthetic methods. Several suitable groups can be introduced byselecting a different RLi, for a further reaction of the initially formedaldehyde hydrazones. This procedure avoids the extra steps requiredby previous methods that use protecting groups and the selective cleav-age of those for the synthesis of multisubstituted hydrazines. Lateralproducts isolated in small amounts suggest some likely mechanisticroutes.
EXPERIMENTAL
Warning: Most N-nitrosamines are powerful, direct-acting carcinogens;they have to be handled and disposed of with special care to avoid skincontact. Manipulations were carried out using frequently changed doublepairs of disposable gloves in a well-ventilated hood. Contaminated mate-rials were disposed of in special containers for further disposition of thestill potentially contaminated materials.
General Remarks
All reactions involving organolithium reagents were carried out bystandard techniques for the manipulation of air- and water-sensitivecompounds, as previously described.[33] The product identification wascarried out by melting point (when available) and spectroscopic data.Product quantification was carried out using a 5890 Series II PlusHewlett-Packard gas chromatograph (equipped with a DB-5 column), at70–280�C programmed temperature. Mass spectra (MS) were recordedon a BG Trio-2 spectrometer. 1H and 13C NMR spectra were recordedin Brucker 200-MHz and Brucker 500-MHz NMR spectrometers.
Solvents and Reagents
Hexane and THF (high-performance liquid-chromatographic grade)were distilled from blue solutions of sodium–benzophenone ketyl imme-diately before use. n-Butyllithium (1M in n-hexane solution) was synthe-sized as previously described, starting from n-butyl-chloride.[20] n-PrLi
Convenient Synthesis of Trialkylhydrazines 3965
Downloaded By: [Max Planck Inst & Research Groups Consortium] At: 20:28 3 November 2009

and n-pentyl Li (0.5M hexane solutions) were prepared similarly ton-BuLi, starting from the respective alkylbromides. Solid MeLi was pre-pared from metal–halogen exchange, starting from n-BuLi (5mmol) andwith the addition of 0.62ml of MeI (5mmol) in several aliquots at 0�C.The precipitated MeLi was centrifuged, the solution was removed, andthe white crystals were washed three times with 3ml of anhydrous hexanefollowed by centrifugation each time. The resulting solid was dried undervacuum at room temperature and dissolved in THF immediately beforeuse. Solid PhLi was prepared from metal–halogen exchange by the sameprocedure described for MeLi, with 1.2ml of PhI (5.25mmol) replacingthe MeI. All organolithium reagents were titrated with the double titra-tion method, previously described.[33] N-Nitrosodicyclohexylamine, 1,was synthesized by NO insertion into the N-Li bond of dicyclohexyllithium amide following the procedure previously described.[34] It waspurified by crystallization from ethanol–water and washed with coldabsolute ethanol (mp 104–105�C).[16] The compound was kept in a desic-cator overnight prior to use.
Synthesis of Hydrazines 3 and 7
N-Nitrosodicyclohexylamine, 1 (210mg, 1mmol), was put in a 10-mlround–bottomed flask equipped with a magnetic stirrer and protectedfrom light by aluminum paper. The flask was tapped with a rubber septum;it was placed in a bath at the desired temperature and then alternatelyevacuated and filled with dry N2 several times. Two ml of anhydrousTHF were added to dissolve the nitrosamine. Then, the organolithiumsolution (4–5mmol in n-hexane) was added by syringe under vigorousstirring and allowed to react for 2 h at room temperature. When two dif-ferent organolithiums are used, the first one was added in a small excess(1.05–1.2mmol), and the second (3–5 equivalents) was added later (after5min). It was allowed to react for 2 h. Workup was carried out by placingthe flask in a water–ice bath, and distilled methanol (ca. 0.65ml) wassyringed. The solvent of the resulting mixture was distilled at reducedpressure, and the liquid residue was purified by silica-gel column chroma-tography (when necessary), obtaining the hydrazines as air-sensitive paleyellow oils, or dissolved in CH2Cl2 prior to GC analysis.
Spectroscopical Data
All the compounds herewith described were fully characterized by 1H and13C NMR and mass spectroscopy.
3966 A. J. V�aazquez, C. Rodrıguez, and N. S. Nudelman
Downloaded By: [Max Planck Inst & Research Groups Consortium] At: 20:28 3 November 2009

N-Nitrosodicyclohexylamine, 1
1H NMR (CDCl3, 200MHz): 4.88 (m, 1H), 3.72 (m, 1H), 1.16–1.96(m, 20H). 13C NMR (CDCl3, 50MHz): 58.6, 52.2, 34.4, 29.4, 26.1,25.5, 25.4, 25.3. MS m=z (rel. ab.): 41 (41), 44 (13), 54 (13), 55 (100),56 (16), 67 (20), 81 (13), 82 (15), 83 (90), 98 (17), 129 (22), 210 (20, Mþ).
2,2-Dicyclohexyl-1-methylenehydrazine, 2a
1H NMR (CDCl3, 200MHz): 6.01 (dd, 2H), 3.29 (tt, 2H), 1.76 (m, 12H),1.35 (m, 8H). 13C NMR (CDCl3, 50MHz): 114.4, 55.5, 31.3, 26.2, 25.9.MS: m=z (rel. int.): 40 (78), 41 (54), 43 (15), 44 (10), 55 (75), 56 (16),69 (44), 70 (16), 83 (100), 84 (10), 125 (14), 165 (27), 208 (19, Mþ).
2,2-Dicyclohexyl-1-propylidenehydrazine, 2b
1H NMR (CDCl3, 200MHz): 6.83 (t, 1H), 3.13 (tt, 2H), 2.24 (m, 2H),1.74 (m, 13H), 1.29 (m, 4H.), 1.06 (t, 3H), 0.87 (m, 3H.). 13C NMR(CDCl3, 50MHz): 139.9, 56.7, 31.1, 26.5, 26.2, 26.1, 11.9. MS m=z (rel.int.): 54 (13), 55 (100), 56 (32), 58 (18), 67 (10), 69 (27), 81 (10), 82(13), 83 (44), 96 (16), 97 (13), 98 (22), 111 (72), 125 (88), 153 (11), 155(17), 193 (48), 207 (65), 236 (39, Mþ).
2,2-Dicyclohexyl-1-butylidenehydrazine, 2c
1H NMR (CDCl3, 200MHz): 6.86 (t, 1H), 3.12 (m, 1H), 2.23 (m, 1H),1.89–1.07 (m, 24H), 0.94 (t, 3H). 13C NMR (CDCl3, 50MHz): 139.9,56.8, 35.2, 31.0, 26.2, 26.1, 20.9, 13.9. MS m=z (rel. int.): 54 (25), 55(97), 56 (65), 57 (30), 67 (24), 68 (17), 69 (39), 70 (21), 81 (19), 82 (31),83 (44), 84 (23), 96 (20), 97 (17), 98 (33), 110 (17), 125 (100), 136 (16),138 (38), 207 (77), 250 (30, Mþ).
2,2-Dicyclohexyl-1-pentylidenehydrazine, 2d
1H NMR (CDCl3, 200MHz): 6.84 (t, 1H), 3.11 (m, 1H), 2.22 (m, 1H),1.75–1.29 (m, 24H), 0.92 (t, 3H). 13C NMR (CDCl3, 50MHz): 140.4,56.8, 32.9, 31.0, 29.8, 26.2, 26.1, 22.4, 14.0. MS m=z (rel. int.): 41 (74),43 (26), 44 (21), 55 (100), 56 (25), 69 (20), 83 (30), 125 (53), 139 (29),207 (25), 264 (8, Mþ).
Convenient Synthesis of Trialkylhydrazines 3967
Downloaded By: [Max Planck Inst & Research Groups Consortium] At: 20:28 3 November 2009

Dicyclohexylethylhydrazone
MS m=z (int. rel.): 54 (12), 55 (100), 56 (21), 69 (27), 82 (13), 83 (60), 84(23), 96 (10), 97 (99), 98 (16), 124 (11), 125 (40), 139 (17), 179 (67), 180(13), 207 (17), 222 (41, Mþ).
N,N-Dicyclohexyl-N’-(1-ethylbutyl)hydrazine, 3b
1H NMR (CDCl3, 200MHz): 2.56 (m, 2H), 1.75 (m, 5H), 1.59 (m, 1H),1.29 (m, 17H), 0.85 (m, 11H). 13C NMR (CDCl3, 50MHz): 60.7, 58.9,34.5, 31.2, 31.0, 26.4, 25.2, 19.0, 14.6, 14.2. MS m=z (rel. int.): 55 (32),83 (24), 96 (20), 113 (79), 195 (100), 196 (15), 197 (28), 280 (19, Mþ).
N,N-Dicyclohexyl-N’-(1-propylpentyl)hydrazine, 3c
1HNMR (CDCl3, 500MHz): 2.66 (m, 1H), 2.56 (m, 1H), 1.87 (m, 1H), 1.76(m, 6H), 1.60 (m, 4H), 1.30 (m, 17H), 1.09 (m, 2H), 0.92 (m, 8H). 13CNMR(CDCl3, 50MHz): 60.7, 57.6, 35.1, 32.5, 31.1, 29.9, 28.0, 26.4, 26.2, 23.2,19.0, 14.6, 14.2. MS m=z (rel. int.): 55 (38), 56 (10), 57 (12), 69 (11), 83(23), 96 (20), 98 (10), 113 (83), 195 (100), 196 (18), 225 (22), 308 (20, Mþ).
N,N-Dicyclohexyl-N’-(1-butylhexyl)hydrazine, 3d
1H NMR (CDCl3, 500MHz): 2.66 (m, 1H), 2.57 (tt, 1H), 1.77 (m, 3H),1.62 (d, 1H), 1.29 (m, 27H), 1.13 (m, 1H), 0.91 (m, 10H). 13C NMR(CDCl3, 50MHz): 60.7, 57.8, 32.5, 31.9, 31.1, 29.7, 29.4, 28.0, 26.4,26.4, 25.4, 23.2, 22.7, 14.2, 14.1. MS m=z (rel. int): 41 (68), 42 (16), 43(60), 44 (40), 55 (100), 56 (22), 57 (30), 69 (19), 71 (11), 79 (12), 81(11), 83 (37), 96 (29), 98 (16), 113 (88), 195 (79), 196 (11), 336 (8, Mþ).
Dicyclohexylamine, 4
1H NMR (CDCl3, 200MHz): 2.60 (m, 2H), 1.73 (m, 10H), 1.17 (m, 10H),0.80 (s, 1H, interchange with D2O). 13C NMR (CDCl3, 50MHz): 53.0,34.4, 26.2, 25.3. MS m=z (rel. int.): 55 (19), 56 (53), 82 (11), 100 (7),138 (100), 139 (11), 181 (12, Mþ).
N-Ciclohexylidenecyclohexylamine, 5
MS m=z (rel. int.): 53 (12), 54 (32), 55 (68), 56 (18), 57 (14), 67 (24), 68(12), 69 (35), 70 (11), 79 (10), 80 (13), 81 (24), 82 (21), 83 (30), 91 (14),
3968 A. J. V�aazquez, C. Rodrıguez, and N. S. Nudelman
Downloaded By: [Max Planck Inst & Research Groups Consortium] At: 20:28 3 November 2009

96 (21), 97 (11), 98 (100), 124 (12), 136 (57), 150 (15), 178 (10), 179(32, Mþ).
N,N-Dicyclohexyl-N’-pentylhydrazine, 7a
1H NMR (CDCl3, 200MHz): 2.64 (m, 1H), 2.55 (m, 1H), 1.84 (m, 1H),1.68 (m, 6H), 1.61 (d, 1H), 1.33 (m, 19H), 1.15 (m, 2H), 0.91 (t, 3H). 13CNMR (CDCl3, 50MHz): 60.3, 57.1, 32.1, 31.7, 26.9, 26.4, 20.9, 18.3, 13.8.MS m=z (rel. int): 41 (79), 42 (18), 43 (38), 44 (21), 45 (17), 54 (12), 55(100), 56 (19), 69 (21), 71 (13), 79 (10), 81 (10), 83 (33), 86 (10), 88(24), 96 (34), 98 (26), 101 (12), 113 (39), 127 (16), 183 (56), 195 (31),266 (14, Mþ).
N,N-Dicyclohexyl-N’-(1-ethylpentyl)hydrazine, 7b
1H NMR (CDCl3, 200MHz): 2.64 (m, 1H), 2.57 (m, 1H), 1.81 (m, 1H),1.75 (m, 6H), 1.60 (m, 2H), 1.32 (m, 17H), 1.12 (m, 2H), 0.90 (m, 8H). 13CNMR (CDCl3, 50MHz): 61.5, 57.4, 32.4, 29.8, 29.7, 27.8, 26.5, 26.5, 24.2,19.2, 13.9. MS m=z (int. rel.): 55 (54), 56 (12), 57 (17), 69 (11), 83 (29),96 (24), 98 (12), 113 (100), 114 (11), 195 (97), 196 (14), 211 (24), 294(15, Mþ).
N,N-Dicyclohexyl-N’-(1-ethylhexyl)hydrazine, 7c
1H NMR (CDCl3, 200MHz): 2.63 (m, 1H), 2.56 (m, 1H), 1.86 (m, 1H),1.74 (m, 6H), 1.62 (m, 2H), 1.33 (m, 19H), 1.11 (m, 2H), 0.92 (m, 8H).13C NMR (CDCl3, 200MHz): 60.6, 57.1, 34.8, 32.0, 30.9, 29.8, 29.7,26.5, 23.8, 17.3, 14.6, 14.1. MS m=z (rel. int.): 41 (58), 42 (11), 43(35), 44 (17), 55 (85), 56 (22), 57 (20), 69 (17), 71 (11), 79 (11), 83(37), 96 (33), 98 (18), 113 (100), 195 (94), 196 (13), 225 (17), 308(10, Mþ).
N,N-Dicyclohexyl-N’-(1-propylbutyl)hydrazine, 7d
1H NMR (CDCl3, 200MHz): 2.63 (m, 1H), 2.55 (m, 1H), 1.84 (m, 1H),1.72 (m, 6H), 1.61 (m, 2H), 1.30 (m, 17H), 1.13 (m, 2H), 0.91 (m, 8H). 13CNMR (CDCl3, 50MHz): 61.3, 57.6, 32.3, 28.7, 26.4, 23.1, 19.8, 13.9. MSm=z (rel. int.): 55 (34), 57 (11), 83 (20), 96 (17), 113 (72), 195 (100), 196(14), 211 (19), 294 (18, Mþ).
Convenient Synthesis of Trialkylhydrazines 3969
Downloaded By: [Max Planck Inst & Research Groups Consortium] At: 20:28 3 November 2009

N,N-Dicyclohexyl-N’-(1-propylhexyl)hydrazine, 7f
1H NMR (CDCl3, 500MHz): 2.65 (m, 1H), 2.57 (tt, 1H), 1.77 (m, 3H),1.61 (d, 1H), 1.30 (m, 25H), 1.12 (m, 1H), 0.91 (m, 10H). 13C NMR(CDCl3, 125MHz): 60.6, 57.4, 32.0, 29.6, 29.3, 26.4, 26.4, 25.3, 22.6,22.6, 18.9, 14.6, 14.0. MS m=z (rel. int.): 41 (32), 43 (23), 55 (68), 56(14), 57 (16), 69 (18), 71 (11), 83 (31), 96 (22), 98 (14), 113 (95), 195(100), 196 (15), 239 (11), 322 (6, Mþ).
N,N-Dicyclohexyl-N’-(1-butylpentyl)hydrazine, 7g
1H NMR (CDCl3, 500MHz): 2.65 (m, 1H), 2.56 (tt, 1H), 1.87 (m, 1H),1.76 (m, 6H), 1.61 (m, 2H), 1.43 (m, 2H), 1.28 (m, 19H), 1.11 (m, 2H),0.92 (m, 8H). 13C NMR (CDCl3, 125MHz): 60.8, 57.8, 32.6, 28.1, 26.4,26.3, 23.2, 22.6, 14.2. MS m=z (rel. int.): 41 (30), 43 (20), 55 (62), 56(15), 57 (17), 69 (17), 71 (10), 83 (28), 96 (22), 98 (12), 113 (96), 195(100), 196 (14), 239 (11), 322 (6, Mþ)
ACKNOWLEDGMENTS
A.V. and C.R. are grateful recipients of the National Research Council(CONICET) fellowships. The authors are indebted to the United NationsEnvironmental Program (Project PNUD=ARG 02=18 Subproj. B-C-53)and to CONICET (PIP no. 6019) for financial support.
REFERENCES
1. Ragnarsson, U. Synthetic methodology for alkyl substituted hydrazines.Chem. Soc. Rev. 2001, 30, 205–213
2. Othmer, D. F.; Kirk, R. E. Hydrazine and its derivatives; In Encyclopedia ofChemical Technology; 4th ed.; Wiley: New York, 1995; vol. 13.
3. Bredihhin, A.; Groth, U. M.; Maeorg, U. Efficient methodology for selectivealkylation of hydrazine derivatives. Org. Lett. 2007, 9, 1097–1099.
4. Zhang, R.; Durkin, J. P.; Windsor, W. T. Azapeptides as inhibitors of thehepatitis C virus NS3 serine protease. Bioorg. Med. Chem. Lett. 2002, 12,1005–1008.
5. Cherney, M. M.; Huitema, C.; Liu, J.; James, K. E.; Powers, J. C.; Eltis, L.D.; James, M. N. Crystal structures of the main peptidase from the SARScoronavirus inhibited by a substrate-like aza-peptide epoxide. J. Mol. Biol.2005, 354, 1137–1151.
6. Huddleston, P. R.; Coutts, I. G. C. Comprehensive Organic Functional GroupTransformations; A. R. Katriztky, O. Meth-Cohn, C. W. Rees (Eds.) Perga-mon=Elsevier: Oxford, 1995; vol. 2, pp. 371–383.
3970 A. J. V�aazquez, C. Rodrıguez, and N. S. Nudelman
Downloaded By: [Max Planck Inst & Research Groups Consortium] At: 20:28 3 November 2009

7. Greene, T. W.; Wuts, P. G. M. Protective Groups in Organic Synthesis; 3rd.ed; Wiley-Interscience: New York, 1999; ch. 7.
8. Brosse, N.; Pinto, M. F.; Jamart-Gregoire, B. New synthesis of 1,1-substituted hydrazines by alkylation of N-acyl- or N-alkyloxycarbonyl-aminophthalimide using the Mitsunobu protocol. J. Org. Chem. 2000, 65,4370–4374.
9. Brosse, N.; Pinto, M. F.; Bodigue, J.; Jamart-Gregoire, B. A new syntheticroute to protected a-hydrazinoesters in high optical purity using the Mitsu-nobu protocol. J. Org. Chem. 2001, 66, 869–2873.
10. Vazquez, A.; Goldberg, R.; Nudelman, N. S. Organolithiums as usefulsynthetic intermediates for tandem reactions; In The Chemistry of FunctionalGroups: Lithium Chemistry; S. Patai, Z. Rappaport (Eds.); Wiley, Chichester,UK, 2005; ch. 2, pp. 63–137.
11. Garcıa, G.V.; Nudelman, N. S. Tandem reactions involving organolithiumreagents; A review. Org. Prep. Proc. Int. 2003, 35, 445–500.
12. Polshettiwar, V.; Varma, R. S. Polystyrene sulfonic acid catalyzed greenersynthesis of hydrazones in aqueous medium using microwaves. TetrahedronLett. 2007, 48, 5649–5652
13. Nicolau, K. C.; Yue, E. W.; Oshima, T. New roads to molecular complexity;In The New Chemistry; N. Hall (Ed.); Cambridge University Press:Cambridge, UK, 2000; p. 178.
14. Waldmann, H. Domino reaction; In Organic Synthesis Highlight II; H.Waldmann (Ed.); VCH: Weinheim, 1995; p. 193.
15. Tietze, L. F. Domino reactions in organic synthesis. Chem. Rev. 1996, 96, 115.16. Nudelman, N. S.; Bonatti, A. E. A highly convenient new methodology
for the synthesis of N-nitrosamines from lithium amides. Synlett 2000,1825–1827.
17. (a) Abnet, C. C. Carcinogenic food contaminants. Cancer Investigat. 2007,25, 189–196. (b) Guza, R.; Rajesh, M.; Fang, Q.; Pegg, A. E.; Tretyakova,N. Kinetics of O6-methyl-20-deoxyguanosine repair by O 6-alkylguanineDNA alkyltransferase within K-ras gene-derived DNA sequences. Chem.Res. Toxicol. 2006, 19, 531–538.
18. Loeber, R.; Rajesh, M.; Fang, Q.; Pegg, A.E.; Tretyakova, N. Cross-linkingof the human DNA repair protein O6-alkylguanine DNA alkyltransferase toDNA in the presence of 1,2,3,4-diepoxybutane. Chem. Res. Toxicol. 2006,19(5), 645–654.
19. Ikeda, M.; Masumura, K.; Sakamoto, Y.; Wang, B.; Nenoi, M.; Sakuma, K.;Hayata, I.; Nohmi, T. Combined genotoxic effects of radiation and atobacco-specific nitrosamine in the lung of gpt delta transgenic mice. Mutat.Res. 2007, 626, 15–25.
20. Loft, S.; Svoboda, P.; Kasai, H.; Tjonneland, A.; Moller, P.; Sorensen, M.;Overbad, K.; Autrup, H.; Raaschou-Nielsen, O. Prospective study of urinaryexcretion of 7-methylguanine and the risk of lung cancer: Effect modificationby mu class glutathione-S-transferases. Int. J. Cancer 2007, 121, 1579–1584.
21. Bonde, P.; Gao, D.; Chen, L.; Miyashita, T.; Montgomery, E.; Harmon,J. W.; Wei, C. Duodenal reflux leads to down regulation of DNA mismatch
Convenient Synthesis of Trialkylhydrazines 3971
Downloaded By: [Max Planck Inst & Research Groups Consortium] At: 20:28 3 November 2009

repair pathway in an animal model of esophageal cancer. Ann. Thorac. Surg.2007, 83, 433–440.
22. Krul, C.; Zeilmaker, M. J.; Schothorst, R. C.; Havenaar, R. Intragastricformation and modulation of N-nitrosodimethylamine in a dynamic in vitrogastrointestinal model under human physiological conditions. Food Chem.Toxicol. 2004, 42, 51–63.
23. (a) Zou, X. N.; Lu, S. H.; Liu, B. Volatile N-nitrosamines and their pre-cursors in Chinese salted fish-–A possible etological factor for NPC in China.Int. J. Cancer 1994, 59, 155–158. (b) Lijinsky, W. N-Nitroso compounds inthe diet. Mutat. Res. 1999, 443, 129–138.
24. Breuil-Desvergnes, V.; Gore, J. Reaction of the lithio-derivative of methox-yallene with hydrazones, part 1: Synthesis and transformation of a-allenylhydrazines. Tetrahedron 2001, 57, 1939–1950.
25. Adler, M.; Marsch, M.; Nudelman, N. S.; Boche, G. [(Ph)2(NMe2)C(O-Li)�THF]2: Crystal structure of the tetrahedral intermediate formed in thereaction of N,N-dimethylbenzamide and phenyllithium. Angew. Chem, Int.Ed. Engl. 1999, 38, 1261–1263.
26. Nudelman, N. S.; Schulz, H.; Garcıa Li~nnares, G.; Bonatti, A. E. Furtherinsights into the chemistry of acyllithium compounds R2NC(O)Li: Character-ization of an amide (R2NLi) adduct (R2NCHNR2(OLi)) to a formamide(R2NC(O)H). Organometallics 1998, 17, 146–150.
27. Nudelman, N. S.; Garcıa Li~nnares, G. New insights into the chemistry oflithium carbamoyls: Characterization of an adduct (R2NC(O)CLi(OLi)NR2).J. Org. Chem. 2000, 65, 1629–1635.
28. Michejda, C. J.; Schluenz, R. W. Reactions of N-nitrosamines with Grignardand lithium reagents. J. Org. Chem. 1973, 38, 2412–2415.
29. Farina, P. R.; Tieckelmann, H. Some reactions of organolithium compoundswith nitrosamines. J. Org. Chem. 1973, 38, 4259–4263.
30. Farina, P. R.; Tieckelmann, H. Reactions of Grignard reagents with nitrosa-mines. J. Org. Chem. 1975, 40, 1070–1074.
31. Perdicchia, D.; Licandro, E.; Mairorana, S.; Baldoli, C. A new ‘‘one-pot’’synthesis of hydrazides by reduction of hydrazones. Tetrahedron 2003, 59,7733.
32. Rajamaki, M.; Vigroux, A.; Chahoua, L.; Fishbein, J. C. N-Nitrosiminiumions are ambident electrophiles. J. Org. Chem. 1995, 60, 2324–2325.
33. V�aazquez, A. J.; Nudelman, N. S. The unexpected effects of added ligands onthe addition of phenyllithium to E-cinnamaldehyde in THF. Arkivoc 2005,332–340.
34. V�aazquez, A. J.; Nudelman, N. S. Complex intermediates in the NO insertionreactions into lithium amides. J. Phys. Org. Chem. 2006, 19, 748–751.
3972 A. J. V�aazquez, C. Rodrıguez, and N. S. Nudelman
Downloaded By: [Max Planck Inst & Research Groups Consortium] At: 20:28 3 November 2009
Copyright © 2022 FDOKUMEN










![ChemInform Abstract: Eco-Friendly Synthesis of 1,4Benzodiazepine2,5-diones in the Ionic Liquid [bmim]Br](https://static.fdokumen.com/doc/165x107/6319c85065e4a6af370ff8b8/cheminform-abstract-eco-friendly-synthesis-of-14benzodiazepine25-diones-in-the.jpg)

![ChemInform Abstract: A Convenient Synthesis of Partially Reduced Benzo[c]phenanthrenes, Its Ketals and Ketones](https://static.fdokumen.com/doc/165x107/6316a0cfc32ab5e46f0dde99/cheminform-abstract-a-convenient-synthesis-of-partially-reduced-benzocphenanthrenes.jpg)







