
ChemInform Abstract: Exchange Bias Effect of Ferro-/Antiferromagnetic Heterostructures
Upload
independentCategory
view
2download
0
www.rsc.org/pccpRegistered Charity Number 207890
As featured in:
See Lee et al., Phys. Chem. Chem.
Phys., 2012, 14, 4333.
www.rsc.org/pccpRegistered Charity Number 207890
Theoretical Chemistry Research Laboratory of
Professor Pratim Kumar Chattaraj at the Indian
Institute of Technology Kharagpur
Title: Some novel molecular frameworks involving
representative elements
Professor Pratim Kumar Chattaraj and his research group have been
actively engaged in research work comprising density functional
theory, ab initio calculations, chemical reactivity, nonlinear
dynamics, aromaticity in metal clusters, hydrogen storage, etc. for
the last quarter of a century. The stability of diff erent types of novel
helical molecular assemblies as well as a series of fi ve membered
and six membered star-like clusters has been explored.
As featured in:
See Chattaraj et al., Phys. Chem. Chem.
Phys., 2012, 14, 14784.
Publ
ishe
d on
09
July
201
2. D
ownl
oade
d by
Ind
ian
Inst
itute
of
Tec
hnol
ogy
Kha
ragp
ur o
n 25
/01/
2015
17:
26:4
4.
View Article Online / Journal Homepage / Table of Contents for this issue

14784 Phys. Chem. Chem. Phys., 2012, 14, 14784–14802 This journal is c the Owner Societies 2012
Cite this: Phys. Chem. Chem. Phys., 2012, 14, 14784–14802
Some novel molecular frameworks involving representative elementsw
Arindam Chakraborty,aSateesh Bandaru,
aRanjita Das,
aSoma Duley,
a
Santanab Giri,bKoushik Goswami,
aSukanta Mondal,
aSudip Pan,
a
Soumya Senaand Pratim K. Chattaraj*
a
Received 3rd May 2012, Accepted 6th June 2012
DOI: 10.1039/c2cp41424d
Several new molecular frameworks with interesting structures, based on clusters of main group
elements have been studied at different levels of theory with various basis sets. Conceptual density
functional theory based reactivity descriptors and nucleus independent chemical shift provide
important insights into their bonding, reactivity, stability and aromaticity.
1. Introduction
The first hint of the application of quantum chemistry1 in
molecular modeling dates back to the earlier half of the last
century when Heitler and London2 discussed the bonding
pattern of the simple H2 molecule. The development of quantum
chemistry from the self-consistent field method to more sophisti-
cated techniques like the complete active space-SCF (CASSCF)
and CASPT2 is well known3–17 and has been broadly applied.
High-level computations surely yield more accurate results, but,
at the same time are more expensive than the SCF methods.
Density functional theory (DFT) developed by Hohenberg
and Kohn18,19 is another unique approach to elucidate the
bonding and reactivity in molecules. The foundation of conceptual
density functional theory (CDFT)20–24 by Robert G. Parr20
proved to be a very powerful algorithm towards modeling
molecular structures and predicting their subsequent chemical
reactivity trends.
The idea of modeling new molecular motifs, over the past
few decades has become a wide arena of fundamental research
for both the experimentalists and theoreticians. While an
experimentalist exploits the best instrumental techniques and
plausible reactions to discover new molecules, a theoretician
applies sophisticated mathematical algorithms and computer
software to design and predict the stability of novel molecular
assemblies. The theoretical design of a new molecular frag-
ment along with an idea about its stability and reactivity does
serve as a useful pathfinder for the experimental chemists
to synthesize and hence utilize them to serve nature. Compu-
tational chemistry in this regard has progressed a lot and a
plethora of molecular motifs has been designed, which can
potentially serve as medicinal drugs,25,26 building blocks for
multi-decker bulk cluster assemblies27–31 useful for various
applications and hydrogen storage templates32–40 required for
the trapping of H2 gas and its further use as an alternative
energy and fuel source. Conceptual DFT as a useful mathe-
matical tool has been successfully implemented in this regard
to model various types of novel molecular clusters suitable for
practical usage. The various CDFT based global reactivity
descriptors like electronegativity41,42 (w), hardness43,44 (Z) andelectrophilicity45–48 (o) as well as local indices like atomic
charges49 (Qk), Fukui functions50 (f(r)) and their condensed-to-
atom variant51 (fk), which, in turn, are a set of mathematical
response functions, provide invaluable insights into predicting
the bonding, stability and reactivity patterns of the molecules.
Additional inputs explaining the intricate bonding patterns and
stability criteria of the molecular clusters can be gained, which
in turn may be utilized for the further refinement of associated
experimental data. A rigorous benchmarking of the choices of
the level of theory and the type of basis set for a particular row
of elements also provides more accurate results that are closely
on a par with experimental findings. The stability of these
cluster molecules can also be assessed by virtue of the existence
of an ‘‘aromaticity criterion’’ which, in turn, can be computed
and compared through the various aromaticity indices.52–58
In this article, we have made an attempt to design different
types of novel molecular assemblies under a conceptual DFT
paradigm. These include boron–carbon linked compounds,
representing a family of cage-like carboranes. Boron–carbon–
hydrogen based 1,7-C2B5H7, 1,6-C2B4H6 and 1,5-C2B3H5
closo-carborane units have been further chosen as the building
blocks towards modeling straight chain-like, planar sheet-like,
helical and twisted carborane structures. Half-cage templates
comprising a continuous C–N based saturated and/or unsatu-
rated network have also been designed for possible practical
applications. Some innovative ‘‘star-shaped’’ molecules con-
sisting of the different second and third row elements in the
basal plane with a Li-atom occupying the vertices of the
geometric star have been designed for their utility as possible
aDepartment of Chemistry and Center for Theoretical Studies,Indian Institute of Technology, Kharagpur – 721 302, India.E-mail: [email protected]
bCIMAT, Universidad de Chile & QTC,Pontificia Universidad Catolica de Chile, Santiago, Chile
w Electronic supplementary information (ESI) available. See DOI:10.1039/c2cp41424d
PCCP Dynamic Article Links
www.rsc.org/pccp PAPER
Publ
ishe
d on
09
July
201
2. D
ownl
oade
d by
Ind
ian
Inst
itute
of
Tec
hnol
ogy
Kha
ragp
ur o
n 25
/01/
2015
17:
26:4
4.
View Article Online

This journal is c the Owner Societies 2012 Phys. Chem. Chem. Phys., 2012, 14, 14784–14802 14785
hydrogen storage materials. The Li-center has been supposed
to serve as the active site towards binding with a hydrogen
atom/molecule. The stability of these molecular ‘‘stars’’ in
terms of an aromaticity criterion has been assessed from the
nucleus independent chemical shift (NICS)52,53 values com-
puted at the ring-center (NICS(0)) on the basal plane or n A
(NICS(n)) vertically away from the ring. A series of clusters
with planar pentacoordinated boron centers have also been
proposed in silico. The stability and reactivity of some metal-ion
doped cationic complexes, comprising an alkali or an alkaline
earth metal ion bound to neutral molecules (like HF, NH3 and
H2O) have been reported. The ability of these cationic complexes
as possible hydrogen binding templates has also been proposed.
The bonding and stability of a range of isomeric [Be8]2� clusters
and a variety of double-decker sandwich complexes have further
been analyzed. A natural population analysis (NPA) of the
different atomic centers of the various molecular motifs modeled
in this study further invokes the proneness of the given sites
towards plausible chemical attack.
2. Theoretical background
The stability of a molecular system is often dictated by a subtle
interplay of molecular properties that, in accordance with
theoretical chemistry practice, are conventionally referred to
using global reactivity descriptors like hardness (Z)43,44 and
electrophilicity (o).45–48 Deeper insights into the stability of a
molecular species and subsequent reactivity upon chemical
response can be obtained from a careful scrutiny of the valida-
tion of various electronic structure principles like the Principle of
Maximum Hardness59–61 (PMH), Minimum Polarizability
Principle62,63 (MPP) and Minimum Electrophilicity Principle64,65
(MEP). For an N-electron system, the electronegativity41,42 (w)and hardness43,44 (Z) can be defined as follow:
w ¼ �m ¼ � @E
@N
� �vð~rÞ
ð1Þ
Z ¼ @2E
@N2
� �vð~rÞ: ð2Þ
Here E is the total energy of the N-electron system and m and
v(-r) are its chemical potential and external potential respec-
tively. The electrophilicity45–48 (o) is defined as:
o ¼ m2
2Z¼ w2
2Z: ð3Þ
A finite difference approximation to eqn (1) and (2) can be
expressed as:
w ¼ I þ A
2ð4Þ
and
Z = I – A (5)
where I and A represent the ionization potential and electron
affinity of the system respectively.
The local reactivity descriptor, Fukui function50 (FF) measures
the change in electron density at a given point when an
electron is added to or removed from a system at constant v(-r).
It may be written as:
f ð r*Þ ¼ @rð r*Þ@N
!vð r*Þ
¼ dm
dvðr*Þ
!N
ð6Þ
Condensation of this Fukui function, f(-r) to an individual
atomic site k in a molecule gives rise to the following expres-
sions in terms of electron population51 qk
f+k = qk(N+1) � qk(N) for nucleophilic attack (7a)
f �k = qk(N) � qk(N � 1) for electrophilic attack (7b)
f 0k = [qk(N+1) � qk(N � 1)]/2 for radical attack (7c)
3. Computational details
The molecular geometries of the different types of cluster motifs
have been optimized at various levels of theory which include
the standard DFT-based B3LYP procedure as well as other
methods, viz. MP2 and M052X. The computations have been
performed with several basis sets by using the GAUSSIAN 03
program package.66 Their existence at a minimum on the
potential energy surface (PES) has been confirmed by computing
the harmonic vibrational frequency at the same level and by
ensuring that no imaginary frequency is present. Single point
calculations have been further performed to evaluate the
energies of theN� 1 electron systems by adopting the geometries
of the corresponding N-electron systems optimized at the given
level of theory. The I and A values have been calculated using the
DSCF technique. The CDFT based global reactivity descriptors
have been computed by utilizing eqn (3)–(5).
The small closo-carboranes 1,5-C2B3H5 and 1,6-C2B4H6
units have been modeled according to the structures reported
by Astheimer et al.67 and Shapir et al.68 The structure of
1,7-C2B5H7 has been obtained by placing a boron atom at the
B4 square plane of square bi-pyramid 1,6-C2B4H6 resulting in
a pentagonal bi-pyramidal structure. Dimers of 1,5-C2B3H5,
1,6-C2B4H6 and 1,7-C2B5H7 have been obtained by joining
axial –CH groups with removal of a H2 molecule. Single-stranded
chain structures of 1,5-C2B3H5, 1,6-C2B4H6 and 1,7-C2B5H7 units
have been generated similarly by joining six dimers of each
through C–C linkages. These optimized (at the same level of
theory) single stranded structures have been joined through
the B atom of a –BH unit with the simultaneous replacement
of twelve hydrogen molecules, which thereby results in a
double stranded structure in each case. The optimization and
subsequent frequency calculation of all the above-mentioned
structures have been carried out at the M052X/6-31G(d) level.
For the sake of simplicity, all the structures throughout the text
C2B3, C2B4, and C2B5 have been written in place of 1,5-C2B3H5,
1,6-C2B4H6 and 1,7-C2B5H7 respectively.
The boron–carbon based carborane cages and the C–N
based saturated and/or unsaturated half-cage templates have
also been optimized at the M052X level of theory using
6-311+G(d,p) and 6-31G(d,p) as basis sets respectively.
The geometry optimizations followed by subsequent frequency
calculations for all the star-like motifs have been performed at
three different levels of theory, viz., B3LYP/6-311++G(d,p),
Publ
ishe
d on
09
July
201
2. D
ownl
oade
d by
Ind
ian
Inst
itute
of
Tec
hnol
ogy
Kha
ragp
ur o
n 25
/01/
2015
17:
26:4
4.
View Article Online
14786 Phys. Chem. Chem. Phys., 2012, 14, 14784–14802 This journal is c the Owner Societies 2012
MP2/6-311++G(d,p) and M052X/6-311++G(d,p). The NICS(0)
values at the ring plane of the star moieties and the subsequent
NICS(n) (n 4 0) values have been computed through the
Schleyer’s52,53 method at B3LYP/6-311++G(d,p) level. The
conceptual density functional theory (CDFT) based descrip-
tors for all star-like clusters have been computed at the
B3LYP/6-311++G(d,p) level. In case of planar pentacoordinated
boron clusters, the related studies have been made at the
B3LYP/6-311+G(d) level.
For the metal-ion doped cationic complexes (with neutral
molecules like HF, NH3 and H2O), single point calculations at
the MP2 and M052X levels have been performed at even higher
basis sets by adopting the molecular geometry optimized at the
respective levels of theory. The CCSD(T) single point energies
have however been computed by adopting the corresponding
geometries optimized at the B3LYP level of theory.
All the isomeric analogues of the [Be8]2� system have been
optimized at the B3LYP level of theory using 6-311+G(d) as
basis set whereas the stabilities of double-decker sandwich
complexes have been scrutinized at HF/6-311+G(d), B3LYP/
6-311+G(d) and M052X/6-311+G(d) levels of theory.
The atomic charges (Qk) on the possible active sites of the
different molecular clusters have been computed by applying
the natural population analysis (NPA) scheme. The frontier
molecular orbitals (FMOs) have been generated through the
Gaussview 03 program package.66
4. Results and discussion
A. Polymeric single stranded and double stranded
closo-carboranes
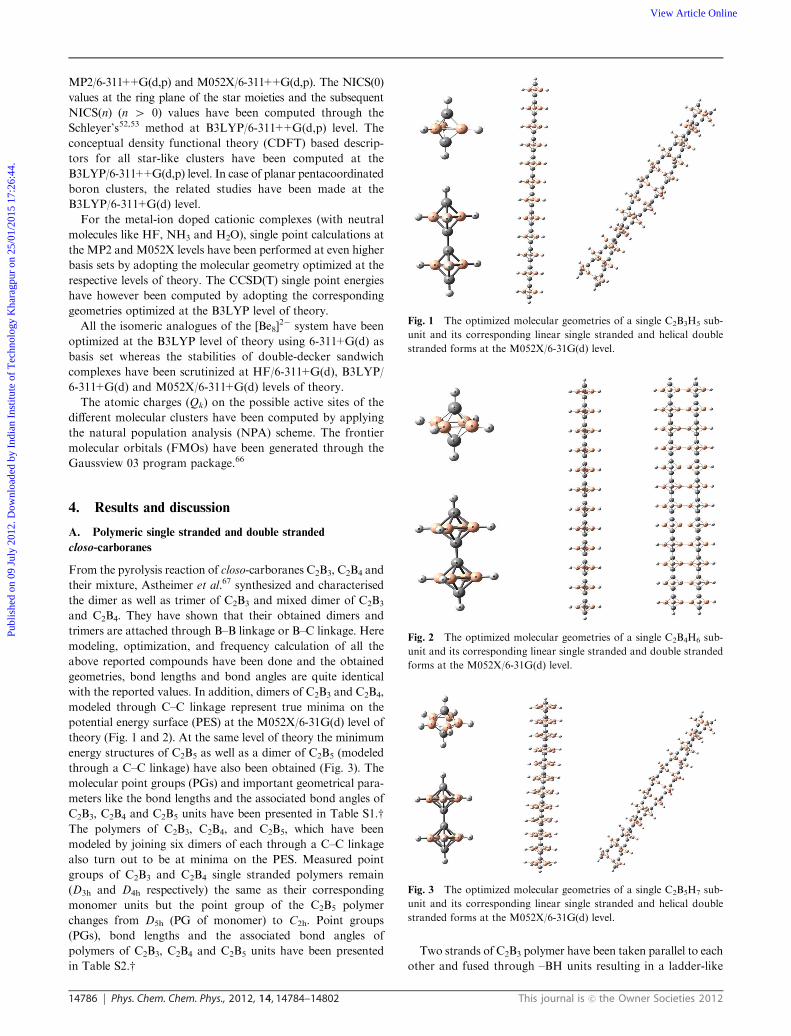
From the pyrolysis reaction of closo-carboranes C2B3, C2B4 and
their mixture, Astheimer et al.67 synthesized and characterised
the dimer as well as trimer of C2B3 and mixed dimer of C2B3
and C2B4. They have shown that their obtained dimers and
trimers are attached through B–B linkage or B–C linkage. Here
modeling, optimization, and frequency calculation of all the
above reported compounds have been done and the obtained
geometries, bond lengths and bond angles are quite identical
with the reported values. In addition, dimers of C2B3 and C2B4,
modeled through C–C linkage represent true minima on the
potential energy surface (PES) at the M052X/6-31G(d) level of
theory (Fig. 1 and 2). At the same level of theory the minimum
energy structures of C2B5 as well as a dimer of C2B5 (modeled
through a C–C linkage) have also been obtained (Fig. 3). The
molecular point groups (PGs) and important geometrical para-
meters like the bond lengths and the associated bond angles of
C2B3, C2B4 and C2B5 units have been presented in Table S1.wThe polymers of C2B3, C2B4, and C2B5, which have been
modeled by joining six dimers of each through a C–C linkage
also turn out to be at minima on the PES. Measured point
groups of C2B3 and C2B4 single stranded polymers remain
(D3h and D4h respectively) the same as their corresponding
monomer units but the point group of the C2B5 polymer
changes from D5h (PG of monomer) to C2h. Point groups
(PGs), bond lengths and the associated bond angles of
polymers of C2B3, C2B4 and C2B5 units have been presented
in Table S2.wTwo strands of C2B3 polymer have been taken parallel to each
other and fused through –BH units resulting in a ladder-like
Fig. 1 The optimized molecular geometries of a single C2B3H5 sub-
unit and its corresponding linear single stranded and helical double
stranded forms at the M052X/6-31G(d) level.
Fig. 2 The optimized molecular geometries of a single C2B4H6 sub-
unit and its corresponding linear single stranded and double stranded
forms at the M052X/6-31G(d) level.
Fig. 3 The optimized molecular geometries of a single C2B5H7 sub-
unit and its corresponding linear single stranded and helical double
stranded forms at the M052X/6-31G(d) level.
Publ
ishe
d on
09
July
201
2. D
ownl
oade
d by
Ind
ian
Inst
itute
of
Tec
hnol
ogy
Kha
ragp
ur o
n 25
/01/
2015
17:
26:4
4.
View Article Online

This journal is c the Owner Societies 2012 Phys. Chem. Chem. Phys., 2012, 14, 14784–14802 14787
double strand structure. Similar ladder-like structures of C2B4
and C2B5 have also been modeled. In case of polymeric double
stranded C2B3 and C2B5 structures, two B-DNA (a long, thin
form of deoxyribonucleic acid in which the helix is right-
handed) like helical geometries with D2 point groups have
been obtained at minima on the PES whereas in case of
polymeric double stranded C2B4, a perfect ladder-like struc-
ture has been identified. Table S3w elaborates the geometrical
parameters for the single and double stranded (ladder-like and
helical) analogues of the monomeric C2B3, C2B4 and C2B5
units. Fig. 1 depicts the molecular geometry of a single C2B3
sub-unit, its dimer and corresponding single stranded and
helical double stranded forms. An analogous portrayal of
the molecular structure of the monomeric C2B4 and C2B5
units and its associated single and double stranded conforma-
tions have also been provided in Fig. 2 and 3 respectively.
Geometrical constraints of the double stranded ladder-like
and helical structures have been measured by considering the
middle units. The total energy (E, au), electronegativity
(w, eV), hardness (Z, eV) and electrophilicity (o, eV) of the
monomeric C2B3, C2B4 and C2B5 systems and their associated,
polymeric single stranded and double stranded (linear and
helical) structures have been given in Table S4.wFig. 4 illustrates a ball and stick depiction of a DNA model
containing 10 A–T base pairs adopted from elsewhere69 as well
as the helical double stranded C2B3 and C2B5 systems. While
the C2B3 system consists of eight pairs of C2B3 sub-units in a
single loop, the C2B5 system comprises seven pairs of C2B5
sub-units in the same. Both the helical forms of the polymeric,
three and five-membered C2B3 and C2B5 systems, respectively,
resemble with DNA double helix structure in many respects.
A detailed scrutiny of the Tables S1–S3w and Fig. 1–3 reveal
some interesting facts on the structural patterns of the B–C–H
based stranded molecules. The bond lengths and the allied
bond angles of the monomeric C2B3, C2B4 and C2B5 units
show slight alterations upon conversion to the polymeric
single and double stranded structures. However the molecular
point groups of the given moieties do change upon trans-
formation from the single to double stranded forms. Fig. 1–3
clearly show that the double stranded structures containing
the C2B3, and C2B5 units attain a helical alignment while the
double stranded C2B4 unit is parallel. The double stranded
moiety containing the C2B4 unit attains a non-helical, parallel
configuration due to a symmetric, orthogonal alignment of the
hydrogen atoms around the B4 rings of the two adjoining
strands. Such an arrangement renders two H-atoms from the
two strands to be spaced in a ‘‘face-to-face’’ manner which
hinders the possibility of any twisting amongst the two
strands. Thus the C2B4 units assume a ‘‘ladder-like’’ geometry.
In spite of a symmetrical alignment, the hydrogen atoms
around the Bn (n = 3, 5) rings of the double stranded C2B3,
and C2B5 systems are not held eclipsed with the repeated units
and thus, unlike the C2B4 polymer, do avoid a ‘‘head-to-head’’
disposition in between two adjoining strands, which might
have attributed to the helical nature of the same. In practice,
however, it has also been observed that for the double
stranded DNA helix, both the base-pairs connecting the
adjoining strands contain a five-membered (odd) ring moiety.
Such an outcome seems to be quite relevant to this present study
as the helical nature of the newly designed C2B3, and C2B5 closo-
carborane based polymeric structures closely mimic a double-
stranded DNA molecule. Deeper insights into the structure of
a DNA molecule from Fig. 4 shows a 61 tilt in the double helix
(B-DNA).70 A similar depiction of the double stranded C2B3
and C2B5 systems in Fig. 4, however, shows an upright helix
with no tilting. A tilted helix in the double stranded DNA
molecule may be due to the fact that the chemical environment
around the adjacently connected five-membered rings is not
similar. The rings are heterocyclic in nature and contain
different types of atoms. On the other hand, for the double
stranded C2B3 and C2B5 helices, the adjoining all-boron three
and five-membered rings offer a homogeneous chemical
environment around the helical structure. This seems to minimize
the possibility of any tilting. All the other parameters corre-
sponding to the DNA helix and the same computed for the
two helical forms of the C2B5H7 system are quite comparable,
excluding the helix radius, as given in Table 1.
The stability of a double-helical, polymeric C2B5 system
along with its close correspondence to the DNA molecule
offers new ideas and even better possibilities for the experi-
mentalists to design such purely inorganic helical structures
for further practical applications.
Fig. 4 Ball and stick depiction of a DNA model containing 10 A–T
base pairs adopted from J. Phys. Chem. B, 2006, 110, 15742–15748 as
well as the helical double stranded C2B3H5 and C2B5H7 systems
optimized at M052X/6-31G(d) level.
Table 1 Calculated structural features of double stranded helicalpolymers of C2B5 and C2B3 closo-carboranes and their comparisonwith B-DNA (ref. 69)
Structural properties C2B5 C2B3 B-DNA
No. of stacked pairs in a loop 8 7 10Stacking height 3.446 A 3.643 A 3.380 ATwist angle 25.6531 25.0631 361Helix radius 3.46 A 2.625 A 10 ATilt/roll angle 01 01 61
Publ
ishe
d on
09
July
201
2. D
ownl
oade
d by
Ind
ian
Inst
itute
of
Tec
hnol
ogy
Kha
ragp
ur o
n 25
/01/
2015
17:
26:4
4.
View Article Online

14788 Phys. Chem. Chem. Phys., 2012, 14, 14784–14802 This journal is c the Owner Societies 2012
B. Hydrogen-bridged carboranes
Two innovative cage-like hydrogen-bridged carborane struc-
tures (Fig. 5) have been proposed here which may serve as
plausible reaction templates particularly for the binding and
storage of hydrogen. A closer look at the nature of chemical
bonding in the B–C–H based carborane cages in Fig. 5 clearly
shows the existence of an electron-deficient three-centered–
two-electron (3c–2e) ‘banana’ bonding among the B–H–B
centers. While the (C12B6H6)2H6 molecule contains single
3c–2e type linkages in between the B–H–B centers, the given
cage upon further hydrogenation yields (C12B6H6)2H12, which
shows the existence of two such electron-deficient bonds
amongst B–H–B centers, thereby, mimicking the bonding
pattern of the stable diborane molecule. Table S5w presents
the molecular point groups and the important geometrical
parameters (bond lengths and bond angles) of the hydrogen-
bonded carborane cages. The bridged B–H bond lengths in
(C12B6H6)2H6 carborane (1.34 A) and (C12B6H6)2H12 carborane
(1.35 A) are very much comparable with those of other
molecules containing 3c–2e bonds e.g., diborane (B–H bond
length = 1.31 A).71 The higher stability of (C12B6H6)2H12 than
its former analogue is also reflected through the increasing
hardness and decreasing electrophilicity values from Table S6.wBoth the hydrogen-loaded carborane cages are shown to be
considerably stable and therefore deserve to be synthesized
experimentally.
C. C–N based half-cages
In this part, an attempt has been made to design some novel
C–N based half cages. In this regard, three new half-cage
frameworks namely C12N6, C12N6H12 and C12N6H18 have
been proposed (Fig. 6). Table S7w outlines the molecular point
groups and the important geometrical parameters (bond
lengths and bond angles) of the bare C–N based unsaturated
framework as well as its associated hydrogen-bonded clusters.
The total energy (E, au) and the various conceptual DFT
based global reactivity descriptors for the C–N based clusters
have been given in Table S8.w A careful scrutiny of Fig. 6
shows that gradual hydrogen loading on to the unsaturated,
hemispherical C12N6 molecule induces saturation. The resulting
C12N6H12 cluster contains only one unsaturated benzenoid
ring which upon further hydrogenation becomes unsaturated
and non-planar to yield C12N6H18. Associated hardness and
electrophilicity values of the C12N6 cluster and its hydrogen-
bound complexes in Table S8w presupposes an increase in
molecular stability with gradually increasing Z values followed
by a hand-in-hand decrease in the respective magnitudes of o.Thus, all the studied hydrogenated C–N based hemispherical
clusters obey the principles of maximum hardness59–61 and
minimum electrophilicity64,65 which often serve as useful
pathfinders towards molecular stability, reaction spontaneity
and the theoretical modeling of molecular motifs. Hydrogena-
tion of the C–N based half-cage structures, therefore, seems to
be favorable and endows an increasing molecular stability. So
these cluster frameworks can act as useful reaction vessels for
hydrogen binding and storage, important for further industrial
applications.
D. Star-shaped molecules
The design of molecular clusters resembling popular geome-
trical configurations has always catapulted both the physicists
and chemists to introduce new ideas and methods. In this
regard, an arrangement of atoms in a molecule producing a
‘‘star-like’’ geometry is quite innovative and attractive at the
same time. Modeling of three-dimensional (3D) ‘‘molecular
stars’’ has been detailed in a few relevant articles.72,73 The
designing of 2D-molecular stars and their plausible usage as
effective hydrogen trapping materials have also been recently
reported.74 In this study we have chosen a variety of well-
known five-membered and six-membered aromatic organic
molecules and substitute the H-centers with an Li-atom to
create a ‘‘star-like’’ moiety. Computations at three different
levels namely B3LYP/6-311++G(d,p), MP2/6-311++G(d,p)
andM052X/6-311++G(d,p) have been performed to assess their
stability. The electronic states, molecular point groups (PGs),
total energy (E, au) of the optimized geometrical structures
Fig. 5 The optimized molecular geometries of the carboranes at the
M052X/6-311+G(d,p) level.
Fig. 6 The optimized molecular geometries of C–N based half-cage molecular networks at the M052X/6-31G(d,p) level.
Publ
ishe
d on
09
July
201
2. D
ownl
oade
d by
Ind
ian
Inst
itute
of
Tec
hnol
ogy
Kha
ragp
ur o
n 25
/01/
2015
17:
26:4
4.
View Article Online

This journal is c the Owner Societies 2012 Phys. Chem. Chem. Phys., 2012, 14, 14784–14802 14789
containing the five-membered and six-membered rings
(computed at different levels of theory) and their allied con-
ceptual DFT based global reactivity descriptors have been
presented in Tables S9 and S10 respectively.w The NICS values
of the five and six-membered planar rings constituting a
molecular star have been computed at the basal plane
(NICS(0)) and up to 3 A vertically away from the ring-center
at regular intervals of 0.5 A, the corresponding values being
outlined in Tables S11 and S12 respectively.w Fig. 7(a–h) and
Fig. 8(a–c) depict the optimized geometries and the important
occupied molecular orbitals of the given ‘‘molecular stars’’
containing the five and six-membered rings respectively.
A comparative variation of the NICS(n) (n = 0, 1, 2) values
for the five-membered and six-membered ‘‘molecular stars’’ can
be envisaged from Fig. 9 and Fig. 10 respectively. We segregate
this discussion into a few sub-sections depending on the nature
of the ring involved in creating the ‘‘star-like’’ moiety.
Stars containing five-membered rings
Pentalithio furan star. Furan (C4H4O) is a very well-known
aromatic heterocyclic organic compound and has been first
prepared by Heinrich Limpricht75 in 1870, initially named as
tetraphenol. Here an attempt has been made to explore the
viability of beautiful star-like models based on furan as the
mother motif. For that purpose, five Li atoms have been
introduced by replacing four H atoms and it has been found
that the Li centers prefer bridging positions as a result of ionic
character in bonding.72,73,76,77 Now starting with a C4OLi5molecular formula, the obvious choice of a structural arrange-
ment corresponds to a C2V point group and 2A1 electronic
state (Fig. 7(a)) which has been found to have NIMAG = 0
structure (at the minimum on the potential energy surface) at
both B3LYP and M052X levels whereas at MP2 level it turns
out to be a saddle point having nmin = 115.6i. An effort to
bring it to the minimum on the PES at MP2 level, a structure
having a C1 point group with a 2A electronic state is obtained
which is 33.4 kcal mol�1 more stable than the corresponding
C2V analogue (Table S9w) although the structure with a C1
point group differs marginally from that of C2V. To assess
whether the structure with a C1 point group is energetically
more stable than that of the C2V structure at the B3LYP and
M052X levels, a free optimization taking the C1 symmetrized
structure at the above mentioned two levels has been per-
formed which, however, brings it back almost to the same
structure as that of the C2V point group (almost the same
energy values and same values of global reactivity descriptors).
The aromaticity of the pentalithio furan star (C4OLi5) has
been assessed in terms of the nucleus independent chemical shift
(NICS) criterion which has turned out to be quite negative,
affirming the existence of a favorable aromaticity phenomenon –
a useful benchmark towards assessing molecular stability. The
values further reveal a steady decreasing trend up to 1 A
(NICS(1)) from the ring-center, which goes on increasing
further with distance (Table S11w). This observation becomes
pictorially quite relevant from Fig. 9 where the NICS(1) value
corresponding to C4OLi5 is lower than the corresponding
NICS(0) value. These facts trigger the existence of a favorable
aromatic p-ring current of the pentalithio furan star which
remains quite dominant up to a certain distance from the base
and eventually ceases with increasing vertical height. A molecular
orbital analysis tells the origin of aromaticity in pentalithio furan
star and it is due to the presence of three delocalized p-MOs
namely HOMO-1, HOMO-5 and HOMO-7 (Fig. 7(a)).
Pentalithio pyrrole star. Pyrrole is another aromatic five-
membered heterocyclic organic compound, with the molecular
formula C4H4NH and can be prepared industrially by the
exposure of NH3 upon furan in presence of solid acid catalysts.78
Here a replacement of five H atoms by five Li atoms (at bridging
positions) gives a beautiful star-like structure with a C2v point
group and a 1A1 electronic state with a molecular formula
of C4NLi5 (Fig. 7(b)). The C2v symmetrized structure of
the pentalithio pyrrole star (C4NLi5) turns out to be with
NIMAG= 0, thereby confirming its existence at the minimum
on the potential energy surface (PES) at all the studied levels
(Table S9w). The high hardness value (4.607 eV) of C4NLi5and the existence at a minimum on the PES at all the studied
levels imply the stability of this configuration (Table S9w). Thepentalithio pyrrole star, being analogous to the earlier furan
system, also sustains an aromaticity criterion with a high
negative NICS value at the ring center as well as different
distances perpendicular to the ring plane, as evident from
Table S11w and Fig. 9. The presence of aromaticity in a
pentalithio pyrrole star can be properly justified by molecular
orbital analysis in which it has been found that three delocalized
p-MOs, namely HOMO, HOMO-2 and HOMO-6 (Fig. 7(b)),
are present satisfying Huckel’s (4n + 2; n = 1) p electron rule.
Here an assessment of the stability of the C4NLi5 cluster in
terms of atomization energy has also been performed. In this
present case atomization energy (AE) has been defined as
AE = [EC4NLi5� (4EC + EN + 5ELi)]. The atomization energy
for the C4NLi5 cluster has been found to be �1068.3 kcal mol�1,
implying the high stability of the studied cluster in the given
configuration.
Pentalithio oxazole star. Oxazole is a member of azole
family having an oxygen and a nitrogen atom separated by
one carbon atom. This aromatic heterocyclic oxazole
(C3H3NO) molecule is taken as mother template to design
star-like motif. In order to design star-like clusters, five Li
atoms replacing three H atoms have been incorporated into
the system offering Cs symmetry and 1A0 electronic state. In
both B3LYP and M052X levels, the Cs symmetrized geometry
turns out to be minimum on the PES (NIMAG = 0) but at
MP2 level it turns out to be a second order saddle point
(NIMAG = 2) as given in Table S9.w At B3LYP level, the Cs
symmetrized geometry has all the Li centers in a bridging
position, whereas at M052X level the optimized structure (Cs)
differs from that of the B3LYP level with one Li center in an
open position (terminal position), as displayed in Fig. 7(c).
Now the presence of two imaginary frequencies at MP2 level
hints at the probable existence of a lower energy structure. A
structure having C1 point group and 1A has been obtained at
minimum on the PES at MP2 level which is 48.9 kcal mol�1
lower in energy than the corresponding Cs analogue. In the C1
structure, the Li centers bend slightly from the C3NO ring.
Again, to evaluate the stability of this C1 geometry at the
B3LYP and M052X levels, a free optimization taking this
Publ
ishe
d on
09
July
201
2. D
ownl
oade
d by
Ind
ian
Inst
itute
of
Tec
hnol
ogy
Kha
ragp
ur o
n 25
/01/
2015
17:
26:4
4.
View Article Online

14790 Phys. Chem. Chem. Phys., 2012, 14, 14784–14802 This journal is c the Owner Societies 2012
Fig. 7 (a) The optimized geometries of pentalithio furan star (C4OLi5) and their occupied delocalized pi molecular orbitals at the B3LYP/
6-311++G(d,p) level (other levels are mentioned below the structures). (b) The optimized geometries of pentalithio pyrrole Star (C4NLi5) and their
occupied delocalized pi molecular orbitals at the B3LYP/6-311++G(d,p) level. (c) The optimized geometries of pentalithio oxazole Star (C3NOLi5)
and their occupied delocalized pi molecular orbitals at the B3LYP/6-311++G(d,p) level (other levels are mentioned below the structures). (d) The
optimized geometries of pentalithio pyrazole Star and pentalithio imidazole star (C4NLi5) and their occupied delocalized pi molecular orbitals at
the B3LYP/6-311++G(d,p) level (other levels are mentioned below the structures). (e) The optimized geometries of pentalithio-1,2,3-triazole star
and pentalithio-1,2,4-triazole star (C2N3Li5) and their occupied delocalized pi molecular orbitals at the B3LYP/6-311++G(d,p) level (other levels
are mentioned below the structures). (f) The optimized geometries of pentalithio phosphole star (C4PLi5), phosphole and their occupied delocalized
pi molecular orbitals at the B3LYP/6-311++G(d,p) level (other levels are mentioned below the structures). (g) The optimized geometries of
Pentalithio-1,3-aza phosphole, Pentalithio-1,4-aza phosphole (C3PNLi5 and their occupied delocalized pi molecular orbitals at the B3LYP/
6-311++G(d,p) level. (h) The optimized geometries of Pentalithio thiophene (C4SLi5) and their occupied delocalized pi molecular orbitals at the
B3LYP/6-311++G(d,p) level.
Publ
ishe
d on
09
July
201
2. D
ownl
oade
d by
Ind
ian
Inst
itute
of
Tec
hnol
ogy
Kha
ragp
ur o
n 25
/01/
2015
17:
26:4
4.
View Article Online
This journal is c the Owner Societies 2012 Phys. Chem. Chem. Phys., 2012, 14, 14784–14802 14791
C1 geometry at both levels has been carried out and found to
be slightly at a higher energy than that of Cs (Table S9w). Thearomaticity of the pentalithio oxazole star has been confirmed
by both negative NICS values as evident from Table S11wand Fig. 9, as well as from the presence of three delocalized
p-MOs, viz. HOMO-1, HOMO-5 and HOMO-7 (Fig. 7(c)).
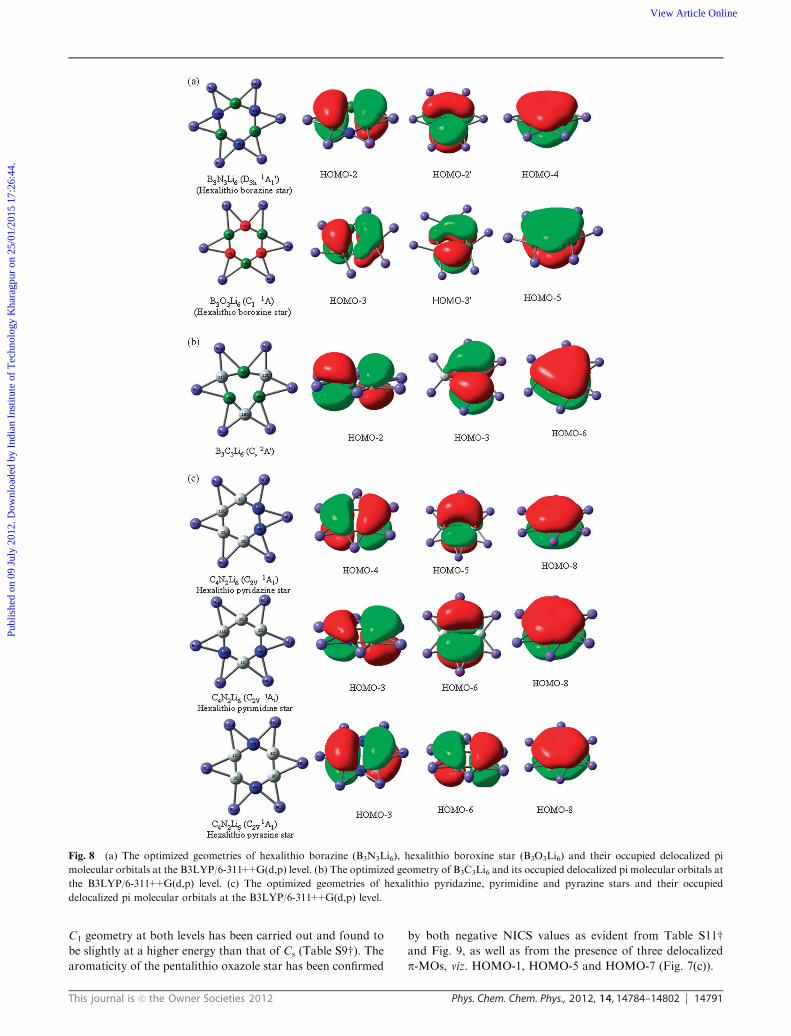
Fig. 8 (a) The optimized geometries of hexalithio borazine (B3N3Li6), hexalithio boroxine star (B3O3Li6) and their occupied delocalized pi
molecular orbitals at the B3LYP/6-311++G(d,p) level. (b) The optimized geometry of B3C3Li6 and its occupied delocalized pi molecular orbitals at
the B3LYP/6-311++G(d,p) level. (c) The optimized geometries of hexalithio pyridazine, pyrimidine and pyrazine stars and their occupied
delocalized pi molecular orbitals at the B3LYP/6-311++G(d,p) level.
Publ
ishe
d on
09
July
201
2. D
ownl
oade
d by
Ind
ian
Inst
itute
of
Tec
hnol
ogy
Kha
ragp
ur o
n 25
/01/
2015
17:
26:4
4.
View Article Online

14792 Phys. Chem. Chem. Phys., 2012, 14, 14784–14802 This journal is c the Owner Societies 2012
Pentalithio pyrazole and imidazole stars. Pyrazole and
imidazole (common molecular formula of C3H4N2) are two
other candidates in the azole family, having two nitrogen atoms
adjacent to each other in pyrazole, whereas in imidazole two
nitrogen atoms are separated by a carbon atom. A substitution
of four H atoms by five Li atoms provides a star-like shape to
the given system (Fig. 7(d)). Pentalithio pyrazole star (C3N2Li5)
with a C2v point group and 2A1 electronic state turns out to be
at minimum on the PES at each studied level (Table S9w),thereby, confirming the viability of this configuration. But
in the case of the pentalithio imidazole star (C3N2Li5), the C2v
symmetrized geometry is a minimum on the PES at the B3LYP
and M052X levels but is a first order saddle point at MP2 level
(Table 9). The pentalithio imidazole star having Cs point
group is a minimum on the PES at MP2 level (27.1 kcal more
stable than C2v) but a re-optimization taking Cs geometry at
B3LYP and M052X levels drives back to the C2V configu-
ration (same energy for C2v and Cs at both B3LYP and
M052X levels and same value of global reactivity descriptors)
(Table S9w). The NICS data in Table S11w and Fig. 9 imply the
presence of a favorable aromaticity phenomenon in penta-
lithio pyrazole and imidazole stars. In pentalithio pyrazole
star, the p-aromaticity arises due to the presence of HOMO-2,
HOMO-3 and HOMO-7 delocalized p-MOs whereas in
pentalithio imidazole star, HOMO-1, HOMO-5 and HOMO-7
are responsible for the p-aromaticity as displayed in Fig. 7(d).
Pentalithio triazole stars. Triazole implies either one of a
pair of isomeric chemical compounds namely 1,2,3-triazole and
1,2,4-triazole having general formula C2H3N3. 1,2,3-Triazole,
a basic aromatic heterocycle has a five membered ring having
two carbon atoms and three nitrogen atoms. At first, by
placing five Li atoms in the bridging positions by replacing
three H atoms, a star-like structure (pentalithio-1,2,3-triazole
star) having a C2V point group and a 1A1 electronic state
(Fig. 7(e)) has been generated which has been found to be at a
minimum on the PES at the B3LYP and MP2 levels but at
M052X level, it has a small imaginary frequency (nmin = 33i)
(Table S9w). This imaginary frequency corresponds to an out-
of-plane bending mode of all Li centers. Therefore, an effort
has been made to search for lower energy structures by moving
all the Li centers slightly out of plane at M052X level and has
been obtained as a minimum energy structure having a C1
point group which is 0.483 kcal mol�1 more stable than the
C2V geometry at the studied level (Table S9w). Then, a free
optimization taking the C1 structure as an initial input has
been carried out at both the B3LYP and MP2 levels. At the
B3LYP level, all the Li centers come to the plane of the ring
and give a structure which is very close to that of C2V geometry,
but at the MP2 level a new lower energy (58.7 kcal mol�1
lower than C2V geometry) non-planar structure has been
attained at a minimum on the PES, as displayed in Fig. 7(e).
Now, to design star-like structures based on the 1,2,4-triazole
mother motif, another basic aromatic heterocycle, the placing
of five Li atoms at the bridging position by replacing three
H atoms has been done to generate a structure with a Cs
symmetry (pentalithio-1,2,4-triazole star) (Fig. 7(e)) which has
been found to be a minimum energy structure at both the
B3LYP and MP2 levels but has an imaginary frequency
(nmin = 74.6i) at the M052X level (Table S9w). Following
the mode of imaginary frequency, a new non-planar minimum
energy structure (Fig. 9(e)) has been obtained at the M052X
level which is 6.9 kcal mol�1 more stable than the corre-
sponding Cs geometry (Table S9w). Now unlike pentalithio-
1,2,3-triazole, this C1 geometry turns out to be at a minimum
on the PES at both the B3LYP and MP2 levels which are
1.2 kcal mol�1 and 3.6 kcal mol�1 higher and a lower-energy
structure than the corresponding Cs geometry, respectively
(Table S9w). Therefore different levels of theory show different
pictures regarding the stability of a given configuration.
Fig. 9 The plot of NICS (0), NICS (1) and NICS (2), in ppm units, of
various five membered star-like clusters at the B3LYP/6-311++G(d,p)
level of theory. [where 1 = C4OLi5 (C2V), 2 = C4OLi5 (C1), 3 =
C4NLi5 (C2V), 4 = C3NOLi5 (Cs), 5 = C3NOLi5 (C1), 6 = C3N2Li5(1,2) (C2V), 7 = C3N2Li5 (1,3) (C2V), 8 = C2N3Li5 (1,2,3) (C2V), 9 =
C2N3Li5 (1,2,3) (C1), 10 = C2N3Li5 (1,2,4) (Cs), 11 = C2N3Li5 (1,2,4)
(C1), 12 = C4PLi5(C2V), 13 = C3PNLi5 (1,2) (Cs), 14 = C3PNLi5 (1,3)
(Cs), 15 = C4SLi5 (C2V), 16 = C4SLi5 (C1)].
Fig. 10 The plot of NICS (0), NICS (1) and NICS (2), in ppm
units, of various six membered star-like clusters at the B3LYP/
6-311++G(d,p) level of theory. [1 = B3C3Li6 (Cs), 2 = B3N3Li6(D3h), 3 = B3O3Li6 (C1), 4 = C4N2Li6 (1,2) (C2V), 5 = C4N2Li6(1,3) (C2V), 6 = C4N2Li6 (1,4) (C2V)].
Publ
ishe
d on
09
July
201
2. D
ownl
oade
d by
Ind
ian
Inst
itute
of
Tec
hnol
ogy
Kha
ragp
ur o
n 25
/01/
2015
17:
26:4
4.
View Article Online

This journal is c the Owner Societies 2012 Phys. Chem. Chem. Phys., 2012, 14, 14784–14802 14793
The aromatic nature of both the pentalithio-1,2,3-triazole star
and pentalithio-1,2,4-triazole star has been justified by highly
negative NICS values (Table S11w and Fig. 9). The delocalized
HOMO-1, HOMO-4 and HOMO-7 p-MOs in pentalithio-
1,2,3-triazole and delocalized HOMO-2, HOMO-4 and
HOMO-7 p-MOs in pentalithio-1,2,4-triazole are responsible
for their p-aromaticity (Fig. 7(e)).
Pentalithio phosphole star. Phosphole is an organic com-
pound with the chemical formula C4H4PH, formed by a fully
unsaturated five-membered ring containing a phosphorus
atom. The most debated issue regarding the phosphole has
been for a long time its aromaticity.79–81 Now it is well
established that phosphole is basically non-aromatic, since
the stabilization of the planar configuration due to electronic
delocalization is not sufficient to compensate the barrier of
inversion around phosphorus. Hence, it prefers a pyramidal
configuration, reducing the overlap between the phosphorus
lone pair and the cis-1,3-butadiene p-orbitals significantly.
It has been proved that a planar configuration of phosphole
has a greatly enhanced aromaticity with respect to its non-
planar analogue.82,83 Here to design a star-like structure based
on phosphole, two significant outcomes have been attained.
Firstly, a replacement of H atoms by Li atoms (bridging
position) tends to give a nice planar star-like structure (penta-
lithio phosphole star) (Fig. 7(f)). Then the stability of a planar
C2V symmetrized pentalithio phosphole star has been studied,
which is found to be a minimum energy structure at both the
B3LYP and M052X levels, but at the MP2 level a small
imaginary frequency (31.9i) appears (Table S9w). This imaginary
frequency corresponds to an out of plane bending mode of two
Li centers connected to the P atom. Following this mode, a
slightly lower energy structure (0.006 kcal mol�1 more stable
than the C2V analogue) having a Cs point group has been
attained in which two Li centers connected to the P atom are
bent slightly away from the ring plane. But a re-optimization at
both the B3LYP and M052X levels taking this Cs symmetrized
structure gives back the planar C2V structure. Therefore,
phosphole which has a non-planar minimum energy structure,
(Fig. 7(f)) can be planarized by substituting H atoms with Li
atoms. Secondly, another beauty of this planar pentalithio
phosphole star is its high aromaticity as indicated by quite
high negative NICS values given in Table S11w and Fig. 9. So a
nonaromatic phosphole molecule is converted to a highly
aromatic one by substituting H with Li. A molecular orbital
analysis reveals that p-aromaticity arises due to the presence of
three p-MOs namely HOMO, HOMO-3 and HOMO-6. The
HOMO corresponds to the strong overlap between the phos-
phorus lone pair and the cis-1,3-butadiene p-orbitals, whichwas absent in the case of phosphole (Fig. 7(f)).
Pentalithio-1,2- azaphosphole and pentalithio-1,3-azaphosphole.
Now an investigation has been made regarding the potential of
aza phosphole template to be used in designing star-like motifs.
Both 1,2-azaphosphole and 1,3-azaphosphole have been tested in
this regard, but an aromatic Cs-imposed configuration of penta-
lithio-1,2-azaphosphole and pentalithio-1,3-azaphosphole having
the same molecular formula (C3NPLi5) (Fig. 7(g)) turn out as
minimum energy structures only at the B3LYP level, whereas at
the MP2 and M052X levels these structures correspond to a
saddle point with the mode of imaginary frequency towards
out of plane bending of the Li centers, suggesting the presence
of a lower energy structure in a non-planar configuration at
the studied levels (Table S9w). In a planar configuration of both
pentalithio-1,2-azaphosphole and pentalithio-1,3-azaphosphole,
the aromaticity, as indicated by Table S11w and Fig. 9, arises due
to the presence of three delocalized p-MOs namely HOMO-1,
HOMO-4 and HOMO-7(Fig. 7(g)).
Pentalithio thiophene star. Thiophene, commonly known as
thiofuran, is an aromatic heterocyclic compound with molecular
formula of C4H4S, which is now chosen to give a star-like shape.
For this purpose, five Li atoms have been incorporated by
replacing four H atoms resulting in pentalithio thiophene
(C4SLi5). At first, the stability of a planar C2v imposed star-like
geometry (Fig. 7(h)) has been scrutinized and found to be a
saddle point (first order at B3LYP level and higher order atMP2
and M052X levels) at the studied levels of theory (Table S9w).Carefully following the mode with imaginary frequency, a non-
planar geometry of C4SLi5 with a C1 point group has been
obtained at the minimum on the PES at each studied level of
theory, in which one Li atom resides vertically above the five-
membered C4S plane (Fig. 7(h)). The resulting conformation
cannot, therefore, be considered as a star. The non-planar C1
analogue of C4SLi5 is energetically more stable, as well as harder
than its C2v configuration, suggesting its greater stability over
the C2V structure. From the NICS values (Table S11w and
Fig. 9), it has been found that C4SLi5 in its non-planar C1
configuration has almost the same degree of aromaticity as that of
its planar C2V geometry, although a molecular orbital analysis
shows less delocalization of the p-electron cloud in itsC1 structure
compared to that of C2V analogue, as displayed in Fig. 7(h).
Stars containing six-membered rings
Hexalithio borazine and boroxine stars. Borazine (B3N3H6) is
supposed to be the brainchild of inorganic chemists who have
been searching for an inorganic substitute of the typical prototype
of the planar aromatic organic benzene molecule. The molecule
bears some resemblance to benzene and therefore is called
‘‘inorganic benzene’’. But, unlike benzene, the six-membered
B3N3 ring in borazine does not allow a complete p-electron drift
across the heteronuclear plane. Several views on the aromaticity
of borazine have already been reported. Islas et al.84 have
attempted to separate the s and p contributions to the resultant
ring current in borazine. Calculations reveal that the s-electronsare much more localized than the p-electrons, thereby rendering
the borazine molecule as a p-aromatic species but not globally
aromatic like benzene, where electron delocalization is even more
profuse. Some earlier studies85–89 also establish the ‘‘lower’’
aromaticity of borazine with regard to benzene. Assessments of
the aromatic measure based on various aromaticity indices have
also proposed that borazine and boroxine molecules are
‘‘non-aromatic’’ in nature.90,91 A recent study92 further shows
that the aromaticity of the heteronuclear ring of boroxine may
be increased upon substitution with electron-withdrawing
groups. Electron donating groups subsequently reduce the
aromaticity of boroxine. Here the viability of six-membered
star-like clusters based on borazine and boroxine mother motifs
Publ
ishe
d on
09
July
201
2. D
ownl
oade
d by
Ind
ian
Inst
itute
of
Tec
hnol
ogy
Kha
ragp
ur o
n 25
/01/
2015
17:
26:4
4.
View Article Online

14794 Phys. Chem. Chem. Phys., 2012, 14, 14784–14802 This journal is c the Owner Societies 2012
has been investigated. In case of borazine, substituting six H
atoms with six Li atoms, a beautiful planar star-like structure
(hexalithio borazine star) having a D3h point group and a 1A10
electronic state (Fig. 8(a)) has been obtained at a minimum on
the PES at each studied level of theory, thereby indicating the
stability of this configuration (Table S10w). In case of boroxine,
a star-like geometry of the hexalithio boroxine star, which is
slightly distorted from the D3h configuration, (Fig. 8(a)) has
turned out as the minimum energy structure at both the B3LYP
and MP2 levels, but it does not meet the convergence criterion
at the M052X level. The NICS values provided in Table S12wand Fig. 10 reveal that the hexalithio borazine and boroxine
star are almost nonaromatic, like borazine and boroxine,
although they possess six p electrons as given in Fig. 8(a).
B3C3Li6. An analogous carbon counterpart of borazine,
B3C3Li6 has also been modelled, where the six-membered B3C3
ring is deformed, unlike the benzene-like geometry (Fig. 8(b)). The
structure having a Cs point group and a 2A0 electronic state is of
minimum energy at both the B3LYP and M052X levels of theory,
but at the MP2 level, the calculation does not converge. Another
interesting aspect regarding B3C3Li6 is its high aromaticity, unlike
B3N3Li6 and B3O3Li6, as given in Table S12w and Fig. 10 origi-
nating mainly from the presence of three delocalized p-molecular
orbitals, viz., HOMO-2, HOMO-3 and HOMO-6 (Fig. 8(b)).
Hexalithio pyridazine, pyrimidine and pyrazine stars. Pyridazine,
pyrimidine and pyrazine are three isomeric heterocyclic aromatic
organic compounds with the same molecular formula, C4H4N2
and only differing in the positions of two N atoms in the
hexagonal ring. Now in order to design star-like motifs based
on these three mother heterocycles, incorporation of six Li atoms
by replacing four H atoms has been done. The geometry optimi-
zation followed by subsequent frequency calculation give three
perfect star-like isomeric structures of general molecular formula,
C4N2Li6 having a C2V point group at the minima on the PES at
each studied level of theory (Fig. 8(c) and Table S10w). Sincepyrazine has a D2h structure in its ground state, an investigation
has also been made to assess the fate of D2h symmetrized
hexalithio pyrazine on the PES, but it turns out to be a higher
order saddle point (Table S10w). The NICS values (Table S12wand Fig. 10) indicate that these isomeric C4N2Li6 stars are
aromatic, like their mother motifs. The aromaticity arises due
to the presence of three delocalized p-MOs (six p e�s) satisfying
the (4n + 2)p electrons rule, as displayed in Fig. 8(c).
Although here a proposal has been made regarding the stability
of a series of star-like clusters based on well-known molecules,
they may be only local minima on the PES, like the beautiful star-
like perlithio annulenes CnLin (n = 3–6) proposed by Schleyer93
(except C3Li3+) and Minkin.94 Since only the global minimum
energy structure is experimentally obtainable, therefore it is
necessary to survey in detail the potential energy surface of these
clusters taking into account all probable configurations. Here it is
worth mentioning that many Li decorated clusters are stable
and they have been successfully synthesized and characterized
experimentally.95–97 Hence, the proposed Li-decorated star-like
clusters may also be achievable experimentally.
Schemes. Here an effort has been made to provide probable
schemes (Fig. 11) to synthesize these star-like systems taking
pentalithio pyrrole and hexalithio borazine star as model
systems. Tiznado et al.73 prescribed a scheme to derive Si5Li7+
from Si5H5� in which they considered the first step as the loss
of five protons generating Si56� and then seven Li+ ions
interacting with Si56� forming an Si5Li7
+ cluster. Following
the same path, we have considered the first step as the loss of
five and six protons from pyrrole (C4NH5) and borazine
(B3N3H6) generating C4N5� and B3N3
6� respectively. Both
C4N5� and B3N3
6� have been obtained at a minima on the
PES with C2V and D3h point groups, respectively, at B3LYP/
6-311++G(d,p) level (Fig. 11). We guess that a very strong base
would be able to eliminate protons from the mother moiety. So
the challenge to the experimentalists is to derive C4N5� and
B3N36� species by adopting a suitable experimental method.
Then addition of Li+ as counter-ions will produce the desired
star-like molecule. The very high interaction energies provide
Fig. 11 The schemes for the plausible syntheses of C4NLi5 and B3N3Li6 from pyrrole (C4NH5) and borazine (B3N3H6) studied at B3LYP/
6-311++G(d,p) level.
Publ
ishe
d on
09
July
201
2. D
ownl
oade
d by
Ind
ian
Inst
itute
of
Tec
hnol
ogy
Kha
ragp
ur o
n 25
/01/
2015
17:
26:4
4.
View Article Online

This journal is c the Owner Societies 2012 Phys. Chem. Chem. Phys., 2012, 14, 14784–14802 14795
sufficient justification regarding the feasibility of the subsequent
steps (Fig. 11).
E. Clusters having planar pentacoordinated boron
The molecules having unusual bonding pattern, such as hyper-
coordination, have received considerable attention from scientists
and so far, many such systems are well-known and saturate the
scientific literature.98–101 However, most of the systems are just
local mimima. The first global minimum structure with planar
pentacoordinated carbon (ppC) reported in the literature is
CAl5+ (D5h) with its 18 valence electrons.102 In 1991, Schleyer
and Boldyrev103 first demonstrated the importance of 18 valence
electrons (magic number) in order to provide stability of such
of planar hypercoordinated systems. Based on CAl5+ (D5h)
structure,102 a series of global minima structures of ppCs104–106
have been reported by replacing Al with Be, maintaining
18 valence electrons which, indeed, further justifies the role
of 18 valence electrons towards assessing the stability of such
type of systems. Very recently, Castro et al.107 have shown that
the systems having the formula CBe5E� (E =Al, Ga) are global
minima in which the C atom is in a ppC environment and one Be
atom is in a capping position of the Be–Be bond in-plane. They
satisfy the 18 valence electrons count by considering CBe5E� to
be like CBe4E3� interacting with Be2+. Now, since BAl5 (D5h)
(iso-electronic with CAl5+)108 is already a known system with
planar pentacoordinated boron (ppB), one should be able to
design a series of systems with ppB by replacing Al with Be in
the same spirit of maintaining 18 valence electrons as is the case
for ppCs. A series of systems with ppBs have been successfully
modeled by replacing the Al of BAl5 (D5h) with Be and adjusting
the charge in such a way that 18 valence electrons remain
engaged in bonding around the ppB moiety. In this regard, a
scheme has been provided depicting all the lower energy
structures in Fig. 12. All the computations have been made at
the B3LYP/6-311+G(d) level. It should be mentioned that here
we have tried to explore the probable existence of structures
having ppBs similar to that of ppCs, considering only some
probable planar isomers of a given system for discussion.
BAl5 system. The optimized geometry of BAl5 having a D5h
point group and a 1A10 electronic state has been given in
Fig. S1.w The NPA charge analysis (Fig. S15w) reveals that anet negative charge of �2.71|e| resides on the central B atom,
whereas the same for Al atoms is +0.54|e| indicating signifi-
cant charge transfer from the peripheral Al to the central B.
Here B acts as a s-acceptor, like C in ppCs. However, it is
relevant to mention that the NPA charge on B atom is some-
what smaller than that of C in CAl5+ (�2.9|e|),102 CAl4Be
(�2.87|e|), and CAl3Be2� (�2.96|e|).105 This is presumably due
to the lesser electronegativity of B than C. The Wiberg bond
indices (WBIs)109 for individual B–Al bonds vary from 0.640
to 0.648 (Table S13w) giving a total WBI of 3.218 to the B atom.
It should be noted that the total WBI of B in BAl5 is consider-
ably larger than that of C in CAl5+ (WBIC(Tot) = 1.99).102 The
valence orbital population at B in BAl5 is (2s1.39 2px
1.20 2py1.55
2pz1.55). The higher 2py and 2pz occupancies in comparison to
those in 2px is an outcome of back-donation from the
perpendicular 2px orbital to the p-orbital (HOMO-3) (Fig. S1w)(in this present case, 2px orbital is perpendicular to the plane),
similar to the electronic stabilization mechanism offered by
Hoffmann et al.110 To understand the nature of electron
delocalization in BAl5, a detailed MO analysis has also been
performed (Fig. S1w) in which the presence of two p-electronsin HOMO-3 suggests it to be a p-aromatic system. However,
a NICS study calculated on a triangular plane of BAl5reveals that it is doubly aromatic (both s- and p-aromatic)
(Table S15w).
BAl4Be� and its Li+/Be2+ doped systems. A substitution of
Al of BAl5 with Be� (since Al is iso-electronic with Be�)
produces BAl4Be� (18 valence electrons) having C2V point
group as given in Fig. S2.w In BAl4Be�, the central B atom
gets an NPA charge of �2.51|e| (Fig. S15w) which is smaller
than that of B in BAl5, and this is because of the greater
electropositivity of Al than Be. Incorporation of Be� into BAl5system, replacing Al, results in the removal of degeneracy of
the MO levels (Fig. S2w), as well as improvement of the total
WBI at the B atom (3.386) and the HOMO–LUMO gap
(Tables S13 and S14w). The vertical electron detachment
energy (VEDE) calculated for the HOMO electron using the
outer-valence Green’s functional (OVGF), in conjunction with
the 6-311+G(d) basis set indicates the bound nature of the
HOMO electron (+2.801eV), therefore, it is a stable anionic
species. Since we know that all systems having ppCs or ppBs
can only be detected and characterized experimentally as
anions, in this spirit BAl4Be� should attract attention from
the experimentalists. The effect of counter ions (Li+/Be2+) on
the electronic structure of BAl4Be� has also been tested. For
Li+ doped BAl4Be�, two different isomers have been identi-
fied in which LiBAl4Be (Cs1A0) is energetically more stable
than its C2V analogue (Fig. S3 and Table S14w) whereas in case
of Be2+ doped BAl4Be�, three different isomers have been
obtained at a minima on the PES (Fig. S4w): among them
BAl4Be2+ (Cs
1A0) [1] is the most stable (Table S14w). As
shown in Fig. S15,w the NPA analysis reveals the obvious
charge redistribution within the molecule when Li+/Be2+ has
been doped. The total WBI value at B (Table S13w) and the
HOMO–LUMO gap (Table S14w) are very similar for
BAl4Be� (WBIB = 3.386, gap = 1.941 eV) and LiBAl4Be
(WBIB = 3.384, gap = 1.939 eV), whereas, it improves to
some extent in BAl4Be2+ (WBIB = 3.511, gap = 2.524 eV).
Therefore, BAl4Be2+ seems to be more stable than BAl4Be
�
and LiBAl4Be by virtue of its larger total WBI and larger
HOMO–LUMO gap. The MO analysis (Fig. S2–S4w) showsthat the incorporation of Li+ and Be2+ into the system only
alters the shape of the MO, and in a few cases the associated
energy order (HOMO-4 is of p-type for BAl4Be2+ but
HOMO-5 is of p-type for BAl4Be� and LiBAl4Be). The
valence orbital populations at B are (2s1.33 2px1.43 2py
1.55
2pz1.18) for BAl4Be
�, (2s1.31 2px1.46 2py
1.54 2pz1.16) for
LiBAl4Be, and (2s1.33 2px1.43 2py
1.55 2pz1.18) for BAl4Be2
+.
The lower occupancies at the 2pz orbital, compared to the 2pxand 2py orbitals, are due to the p-back-donation (here the 2pzorbital is perpendicular to plane). The NICS study on a three
membered ring of BAl4Be�, LiBAl4Be and BAl4Be2
+ systems
shows that these systems are both s- and p-aromatic
(Table S15w). p-Aromaticity arises due to the presence of
two p-electrons in these systems as shown in Fig. S2–S4.w
Publ
ishe
d on
09
July
201
2. D
ownl
oade
d by
Ind
ian
Inst
itute
of
Tec
hnol
ogy
Kha
ragp
ur o
n 25
/01/
2015
17:
26:4
4.
View Article Online

14796 Phys. Chem. Chem. Phys., 2012, 14, 14784–14802 This journal is c the Owner Societies 2012
BAl3Be22� and its Li+/Be2+ doped systems. Upon the
introduction of another Be� replacing one Al of BAl4Be�,
two isomers having C2V point group have been obtained in
which BAl3Be22� (C2v
1A1) [1] is more stable than the corre-
sponding C2V analogue (Fig. S5 and Table S14w). The total
WBI at B in BAl3Be22� (3.477) is larger than that in BAl4Be
�
(3.386) (Table S13w) but VEDE calculation shows that the
HOMO electron is not in a bound state (VEDE=�0.984 eV),therefore, it is not a stable dianionic species. Upon Li+
doping, three different isomers have been attained at a minima
on the PES in which LiBAl3Be2� (C2v
1A1) is energetically
more stable than the two others (Fig. S6 and Table S14w).However, upon Be2+ doping, two different isomers have been
obtained in which BAl3Be3 (C2v1A1) is more stable than its Cs
analogue (Fig. S7 and Table S14w). Here it is relevant to
mention that for the electron count, to effectively engage in
bonding within the ppB moiety, LiBAl3Be2� and BAl3Be3
should be considered like BAl3Be22� interacting with Li+
and Be2+, as prescribed by Castro et al.,107 thereby satisfying
the 18 valence electrons criterion. Introduction of Li+ and Be2+
on to BAl3Be22� alters the charge distribution (Fig. S15w) as
well as the nature of LUMO and the associated MO energy
order (Fig. S5–S7w). Moreover, these counter ions have a
significant role in providing molecular stability. Due to
their presence, the energies of all occupied molecular
orbitals become negative and, correspondingly, the asso-
ciated VEDE becomes positive suggesting the bound nature
of the electrons (Table S14w). Additionally, the presence
of Li+ and Be2+ improves the total WBI at B (Table S13w)but the HOMO–LUMO gap is reduced somewhat in
the presence of Li+ but it enhances with the incorporation
of Be2+ (Table S14w), indicating greater stability of BAl3Be3over the others. The valence orbital populations at the
central B are: (2s1.29 2px1.18 2py
1.54 2pz1.54) for BAl3Be2
2�;
(2s1.26 2px1.16 2py
1.48 2pz1.39) for LiBAl3Be2
�; and (2s1.29
2px1.18 2py
1.54 2pz1.54) for BAl3Be3. Here also the p-back-
donation from 2px orbital to the p-MO is responsible for the
smaller occupancies at the 2px orbital with respect to 2py and
2pz orbitals (here the 2px orbital is perpendicular to the plane).
Both the MO analysis and NICS study reveal that these
systems are both s- and p-aromatic (Table S15w).
BAl2Be33� and its Be2+ doped systems. Upon further
incorporation of Be�, replacing one Al of BAl3Be22�, two
isomers with a C2V point group have been obtained in which
BAl2Be33� (C2v
1A1) [1] is more stable than the other C2V
analogue (Fig. S8 and Table S14w). Here it should be stated
that this trianionic species will surely be unstable with respect to
spontaneous emission of electrons, but still we have analyzed it
in detail to enhance our understanding regarding the impor-
tance of the 18 valence electrons rule to design such planar
hyper-coordinated species and, of course, one can then under-
stand its electronic stability in the presence of counter-ions.
Now, in the presence of one Be2+ ion, three isomers have been
identified at a minima on the PES: among them BAl2Be4�
(Cs1A0) [1] is energetically more stable than the others (Fig. S9
and Table S14w). Again in presence of two Be2+ ions, a total
of five isomers have been obtained, but only two of them
possess a minima on the PES (Fig. S10 and Table S14w).A comparison of the total energy reveals that BAl2Be5
+ (Cs1A0)
[1] is a lower energy structure with respect to others (Table S14w).All the three systems possess two p-electrons, indicating their
p-aromaticity (Fig. S8–S10w), however, the NICS study
implies that the systems are not only p-aromatic but also
s-aromatic (Table S15w). The positive value of VEDE (+2.879 eV)
for BAl2Be4� suggests its stability towards spontaneous emission
Fig. 12 The scheme depicting the optimized geometries of lower energy structures of the studied clusters having ppBs at B3LYP/6-311+G(d)
level.
Publ
ishe
d on
09
July
201
2. D
ownl
oade
d by
Ind
ian
Inst
itute
of
Tec
hnol
ogy
Kha
ragp
ur o
n 25
/01/
2015
17:
26:4
4.
View Article Online

This journal is c the Owner Societies 2012 Phys. Chem. Chem. Phys., 2012, 14, 14784–14802 14797
of an electron. The valence orbital populations at the central B
are: (2s1.31 2px1.21 2py
1.37 2pz1.53) for BAl2Be3
3�; (2s1.21 2px1.34
2py1.45 2pz
1.12) for BAl2Be4�; and (2s1.18 2px
1.38 2py1.43 2pz
1.09)
for BAl2Be5+. In case of BAl2Be3
3�, some p-back-donationoccurs from the 2px orbital to the p-MO (HOMO-5) (here the
2px orbital is perpendicular to the plane) whereas in case of
BAl2Be4� and BAl2Be5
+, 2pz orbitals have been engaged in
back-donation to the p-MOs (HOMO-4 and HOMO-3 respec-
tively) (in these cases, 2pz orbitals are perpendicular to plane).
The study of total WBI at the B atom and the HOMO–LUMO
gap reveals that, upon Be2+ doping, both the total WBI at B
atom and the HOMO–LUMO gap improve (Tables S13
and S14w). However, the BAl2Be4� system attains a maximum
HOMO–LUMO gap indicating its greater stability over the
others.
BAlBe44�
and its Be2+
doped systems. The next product as a
result of Be� introduction replacing one Al atom of BAl2Be33�
is BAlBe44� (C2v
1A1) (Fig. S11w). Now, to provide electronic
stability to this tetraanionic species, two Be2+ ions have been
incorporated into the system, producing three isomers, however,
only BAlBe6 (Cs1A0) [1] turns out to be at a minimum on the PES
(Fig. S12 and Table S14w). The higher stability of BAlBe6 than
BAlBe44� can be properly justified by the HOMO–LUMO gap,
which has been found to increase upon Be2+ doping (Table S14w),however, the total WBI at B remains the same (Table S13w).Similar to the earlier cases, the Be2+ doping alters the natural
charge distribution (Fig. S15w) as well as the nature of the LUMO
and energy levels of the occupied MOs (Fig. S11 and S12w). Thevalence orbital populations at the central B have been found to be
(2s1.32 2px1.25 2py
1.48 2pz1.43) for BAlBe4
4� and (2s1.15 2px1.44 2py
1.31
2pz1.08) for BAlBe6, consistent with earlier cases, and it has been
found that the occupancy of one 2p orbital is lower than the other
due to the p-back donation. Here the 2px orbital for BAlBe44� and
the pz orbital for BAlBe6 are perpendicular to the molecular plane,
therefore, they are engaged in the p-back donation. The NICS
study shows that they are doubly aromatic, similar to systems in
the earlier cases (Table S15w).
BBe55�
and its Be2+
doped systems. The last system derived
by substituting last Al atom of BAlBe44� with Be� is BBe5
5�
(D5h1A1
0) (Fig. S13w). Excepting monoanionic systems, any
multianionic species studied here are unstable due to unbound
nature of the HOMO electron, but the total WBI at the B
center reveals that WBIB(Tot) gradually improves with succes-
sive Be� doping into the BAl5 system, replacing Al and finally
it is maximized for BBe55� (WBIB(Tot) = 4.602) (Table S13w).
Now, upon two Be2+ ions doping in BBe55�, two isomers with a
C2V point group have been obtained, however, BBe7� (C2v
1A1)
[1] is a minimum energy structure whereas the other one is just a
saddle point (Fig. S14 and Table S14w). The introduction of two
Be2+ ions to BBe55� increases the HOMO–LUMO gap, indi-
cating increased stability upon Be2+ doping as well as making
the VEDE positive, suggesting its stability towards spontaneous
emission of an electron (Table S14w). The variation of NPA
charge upon the introduction of Be2+ has been depicted in
Fig. S15.w The valence orbital populations at B are (2s1.36
2px1.48 2py
1.48 2pz1.29) for BBe5
5� and (2s1.12 2px1.06 2py
1.36
2pz1.43) for BBe7
� indicating p-back donation from the 2pz
orbital in case of BBe55� (in this case the 2pz orbital is
perpendicular to the plane) and from the 2px orbital in BBe7�
(in this case the 2px orbital is perpendicular to plane). These
two systems are also both s- and p-aromatic as understood
from the NICS calculations (Table S15w).Thus, we have successfully designed a series of clusters with
ppBs by carefully monitoring the 18 valence electrons rule. All
the systems are doubly aromatic (both s- and p-aromatic). In
all cases, the central B atom acts as a s-acceptor and p-back-donor, similar to C in ppCs.
F. Metal-ion doped complexes
The chemical reactivity of a neutral molecule can be enhanced
upon doping with a charged species, such as a metal ion. The
alkali and alkaline earth metal ions, owing to a higher ionic
potential, have the ability to bind smaller molecules or groups to
produce complex motifs. These metal-doped cationic complexes
can therefore serve as suitable templates for the trapping of
small molecules. Some simple neutral molecules like HF, NH3
and H2O doped with alkali metal ions like Li+, Na+ and
alkaline earths like Be2+, Mg2+ have been chosen for this study.
The total energy (E, au) of the optimized structures of the metal-
ion doped complexes computed at various levels of theory has
been given in Table S16.w Single point energy calculations at
higher levels or using higher basis sets at a given level of theory
have also been performed and the values have been shown in
Table S17.w Tables S18–S20w present the values of the important
global reactivity parameters and the atomic charges on the metal
centers computed at different levels of theory. The optimized
molecular geometries of the various metal-ion doped molecular
complexes have been pictorially depicted in Fig. 13. The energy
(E, au) trends of the metal-ion bound complexes in Table S16wreveal that the corresponding values of the Na+ and Mg2+
bound systems are lowest, irrespective of the choices of the
parent moiety or the level of theory. A glance at the atomic
charges on the metal centers in Tables S18–S20w clearly shows
that the Na and the Mg centers (for a given parent moiety or at
any level of theory) bear a slightly high positive charge than the
respective Li or Be counterparts. This observation highlights
that the Na+ and the Mg2+ ions have the tendency to bind
more ‘‘strongly’’ with the neutral moieties through a favorable
ion–dipole type of interaction to yield a stable complex.
G. Be82– isomers
Molecular clusters serve as a useful link between smaller assemblies
and bulky multi-dimensional materials. The modeling of small to
medium all-metal 2D as well as large 3D multidecker clusters has
been a widely cultivated topic in the past few years. Conceptual
DFT20–24 in conjunction with the novel concept of all-metal
aromaticity111 has been successfully executed towards deciphering
the structure, bonding, aromaticity and reactivity of a plethora of
metal clusters.112–115 This study reports the modeling of fifteen
plausible isomers of a dianionic all-beryllium [Be82–] system
(Fig. 14). Table S21w describes the optimized molecular geo-
metries along with the respective total energy (E, au) values
and the various conceptual DFT based global reactivity descrip-
tors of all the generated isomeric forms of the [Be82–] molecule.
The atomic charges on the Be-centers and the associated
Publ
ishe
d on
09
July
201
2. D
ownl
oade
d by
Ind
ian
Inst
itute
of
Tec
hnol
ogy
Kha
ragp
ur o
n 25
/01/
2015
17:
26:4
4.
View Article Online

14798 Phys. Chem. Chem. Phys., 2012, 14, 14784–14802 This journal is c the Owner Societies 2012
geometrical parameters (bond lengths and bond angles) of all
the [Be82–] isomers have been given in Table S22.w The
important frontier molecular orbitals (FMOs) of some repre-
sentative [Be82–] isomers have been depicted in Fig. S16.w The
hardness values of almost all the generated isomers are compar-
able except for structure 14, a linear conformation where Z turnsout to be the lowest among all. The global electrophilicity value
for the linear [Be82–] isomer also shows the highest magnitude,
thereby rendering the given conformation to be a highly reactive
species. Structure 12 of the [Be82–] system has already been
reported28 and has the lowest o value among all the generated
conformers. This may be ascribed to the presence of a favorable
aromaticity criterion in the two trigonal Be32– rings stabilizing
the Be–Be bond in the [Be82–] cluster.
H. Double-decker sandwich-type molecules
The design of all-metal and non-metal complexes assuming a
double-decker or a multi-decker form is already well known.27–31
The various 2D and 3D clusters show the existence of an
aromaticity/antiaromaticity criterion along the various all-metal
and non-metal rings, which have been further stabilized by
appropriate molecular groups above and below the plane. This
study reports the design of some sandwich structures involving a
trigonal [B3(BO)3] unit to bind with an all-metal or non-metal
moiety through a metallic linkage. The optimized molecular
geometries of the given complexes and their corresponding
frontier molecular orbitals (FMOs) have been portrayed in
Fig. 15. The total energy (E, au), molecular point groups (PG)
and the important global reactivity descriptors of the sandwich-
type clusters studied at the HF/6-311+G(d), B3LYP/6-311+G(d)
and M052X/6-311+G(d) levels have been presented in
Tables S23–S25 respectively.w Table S26 illustrates the NICS(0)
values computed at the ring centers of the different complexes.
The values of the relevant parameters in Tables S23–S25 reveal
that for most of the di-anionic complexes, the electronegativity
(w) values are negative which presupposes the reluctance of the
given molecules towards further acceptance of electrons. The
magnitudes of the chemical hardness (Z) of the given complexes
are quite high as compared to their associated electrophilicity (o)values. The molecules are thus supposed to be less reactive and
hence stable. An assessment of the stability norms of these clusters
in terms of an aromaticity criterion can be rationalized from the
corresponding NICS values of the various rings of the respective
complexes in Table S26.w The NICS(0) values for the central
all-metal/non-metal ring of the complexes are negative in most
cases (Be3, Mg3, Al4, C5H5, N5, P4) excepting the all-nitrogen N4
and N6 systems, which show positive NICS(0) values. The
trigonal B3 ring unit positioned above and below the all-metal/
non-metal plane shows a varied trend in the NICS(0) values.
Thus, the central rings mostly retain their aromatic nature upon
complexation, thereby rendering stability to the given multi-
decker systems. Moreover, as is evident from Fig. 15, the Al4,
N4 and N6 bound complexes are not perfect sandwiches, as the
central all-metal/non-metal ring tends to remain more or less
perpendicular with the trigonal B3 unit placed above and below
the same. The important frontier molecular orbitals of the various
sandwich-type clusters show prominent electron delocalization
above and below the different ring planes, which probably justify
the sustenance of a diatropic/paratropic current to establish an
aromaticity/antiaromaticity phenomenon.
Potential utility of molecular clusters in hydrogen storage
applications. The modeling of molecular materials as useful
templates for the storage of hydrogen in its atomic and
Fig. 13 Structures of Mn+@ (–H2O, –NH3, –HF): (M = Li, Na, Be, Mg: n = 1,2).
Publ
ishe
d on
09
July
201
2. D
ownl
oade
d by
Ind
ian
Inst
itute
of
Tec
hnol
ogy
Kha
ragp
ur o
n 25
/01/
2015
17:
26:4
4.
View Article Online

This journal is c the Owner Societies 2012 Phys. Chem. Chem. Phys., 2012, 14, 14784–14802 14799
molecular forms has become a topic of top priority in chemical
research. Hydrogen has been conceived in the past decade as a
fuel source, as well as an energy carrier for applications in
industry and the automobile sector. An uncontrolled use of
fossil fuels over the past few decades has ultimately met up
with the dual curses of global environmental pollution and a
dearth in the former. So with an aim to mend these crises, the
use of hydrogen as an alternative fuel has become necessary.
Hydrogen, being a clean fuel alternative (generating water as
a harmless by-product) is quite abundant in the universe and
is also available from rocks, soil, air and of course, water.
For practical purposes, to be used as a fuel, hydrogen needs to
be stored in manageable quantities under proper volumetric
and gravimetric standards. In this regard, other than lique-
faction and storage in high-pressure conditions, hydrogen
can be adsorbed in considerable amounts upon various
molecular motifs which can further lead to its prompt release
during use. A plethora of different classes of molecular
clusters have been designed so far which include graphene-
based sheet like materials,116,117 nanomaterials118,119 and cage-
like clusters,120,121 metal hydrides and metal clusters122,123 and
metal–organic frameworks (MOFs).124,125 A recent review126
Fig. 14 Optimized geometries of all the possible isomers of Be82� molecule at the B3LYP/6-311+G(d) level.
Publ
ishe
d on
09
July
201
2. D
ownl
oade
d by
Ind
ian
Inst
itute
of
Tec
hnol
ogy
Kha
ragp
ur o
n 25
/01/
2015
17:
26:4
4.
View Article Online

14800 Phys. Chem. Chem. Phys., 2012, 14, 14784–14802 This journal is c the Owner Societies 2012
and a few other relevant articles32–40 delineate the modeling of
various molecular assemblies useful for hydrogen storage
applications.
In this present backdrop, with an aim towards further enrich-
ment of the library of hydrogen storage materials, we propose
the plausible usage of the newly designed molecular frameworks
as effective templates for the same. The potency of ‘‘star-like’’
molecules as useful hydrogen trapping materials are already
known.74 Thus the new five and six-membered molecular stars
should aptly perform for the same purpose. Moreover, based on
the efficacy of a metal site towards hydrogen binding,32–34,36,38–40
metal-ion doped cationic complexes can be utilized as effective
storage materials for molecular hydrogen. The ability of the
carborane cages, C–N based half-cage frameworks, as suitable
templates for hydrogen storage is also studied.
5. Conclusion
The application of sophisticated theoretical methods towards
molecular modeling has now become a well advanced topic of
fundamental research. Scientists as diverse as astrophysicists,
biologists, chemists, materials scientists and zoologists can
reach for a handful of utility software programs to design
molecular frameworks of unanticipated complexity. The new
molecules are supposed to possess unique structural features
and can be used for many potential applications. The present
work makes an attempt to design a variety of different forms of
such molecular assemblies which are supposed to have some
fruitful practical usage. Apart from that, they make a very good
case for promoting the use of theoretical methods in order to
avoid following unproductive lines of inquiry in synthesizing
rather exotic, and sometimes very toxic, chemicals. Theoretical
approaches such as this allow the researcher to ‘‘zoom in’’ to
more productive lines of enquiry. The attractive ‘‘star-shaped’’
molecules, carborane cages, C–N based half-cage frameworks
along with the small metal-ion bound cationic complexes have
the ability to act as effective media for the trapping and storage
of hydrogen. The clusters with ppBs will stimulate the curiosity
of scientists for further work. The all-beryllium clusters and the
double-decker, complexed sandwich materials can be used for
further molecular modeling towards building 3D frameworks,
useful for bulk applications. Further work is in progress.
Acknowledgements
P. K. C. would like to thank Professors Gabriel Merino and
Thomas Heine for kindly inviting him to present an article in
this Special Issue of Physical Chemistry Chemical Physics on
‘‘Predicting New Molecules by Quantum Chemical Methods’’.
He also thanks the Indo-EU HYPOMAP project for financial
assistance and DST, New Delhi for the Sir J. C. Bose National
Fellowship. S. M. and R. D. thank UGC and S. P. thanks
CSIR for their research fellowships.
References
1 P. Atkins and J. De Paula, Atkins’ Physical Chemistry, OxfordUniversity Press, 8th edn, 2006.
2 W. Heitler and F. London, Z. Phys., 1927, 44, 455–472.3 H. Nakano and K. Hirao, Chem. Phys. Lett., 2000, 317, 90–96.
Fig. 15 The optimized structures of several double-decker sandwich
complexes studied at the B3LYP/6-311+G(d) level.
Publ
ishe
d on
09
July
201
2. D
ownl
oade
d by
Ind
ian
Inst
itute
of
Tec
hnol
ogy
Kha
ragp
ur o
n 25
/01/
2015
17:
26:4
4.
View Article Online

This journal is c the Owner Societies 2012 Phys. Chem. Chem. Phys., 2012, 14, 14784–14802 14801
4 R. S. Mulliken, Rev. Mod. Phys., 1960, 32, 232.5 J. C. Slater, Phys. Rev., 1951, 81, 385–390.6 D. R. Hartree, The Calculation of Atomic Structures, Chapmanand Hall, New York, J. Wiley and Sons, London, 1957.
7 V. Fock, Z. Phys., 1930, 61, 126–148.8 S. F. Boys, Proc. R. Soc. London, Ser. A, 1950, 201, 125–137.9 C. D. Sherrill and H. F. Schaefer III, The Configuration Inter-action Method: Advances in Highly Correlated Approaches, Adv.Quantum Chem., 1999, 34, 143–269.
10 C. J. Cramer, Essentials of Computational Chemistry, John Wiley& Sons, Ltd, Chichester, 2002, pp. 191–232.
11 A. C. Wahl and G. Das, in Methods of Electronic StructureTheory, ed. Schaefer HF III, Plenum Press, New York, 1977.
12 C. Møller and M. S. Plesset, Phys. Rev., 1934, 46, 618–622.13 G. D. Purvis and R. J. Bartlett, J. Chem. Phys., 1982, 76,
1910–1918.14 P. O. Lowdin, Phys. Rev., 1955, 97, 1474–1489.15 B. O. Roos, P. R. Taylor and P. E. M. Siegbahn, Chem. Phys.,
1980, 48, 157–173.16 K. Andersson, P.-A. Malmqvist, B. O. Roos, A. J. Sadlej and
K. Wolinski, J. Phys. Chem., 1990, 94, 5483–5488.17 K. Andersson, P.-A. Malmqvist and B. O. Roos, J. Chem. Phys.,
1992, 96, 1218–1226.18 P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871.19 P. Hohenberg and W. Kohn, Phys. Rev. A, 1965, 140, 1133–1138.20 R. G. Parr and W. Yang, Density Functional Theory of Atoms and
Molecules, Oxford University Press, New York, 1989.21 Chemical Reactivity Theory: A Density Functional View, ed. P. K.
Chattaraj, Taylor & Francis/CRC Press, Florida, 2009.22 P. Geerlings, F. De Proft and W. Langenaeker, Chem. Rev., 2003,
103, 1793–1873.23 P. K. Chattaraj and S. Giri, Annu. Rep. Prog. Chem., Sect. C,
2009, 105, 13–39.24 A. Chakraborty, S. Duley, S. Giri and P. K. Chattaraj, An
Understanding of the Origin of Chemical Reactivity from aConceptual DFT approach, in A Matter of Density: Exploringthe Electron Density Concept in the Chemical, Biological, andMaterials Sciences, ed. N. Sukumar, John Wiley, 2010.
25 M. Sulpizi, G. Folkers, U. Rothlisberger, P. Carloni andL. Scapozza, Quant. Struct.–Act. Relat., 2002, 21, 173–181.
26 A. Chakraborty, S. Pan and P. K. Chattaraj, Biological Activityand Toxicity: A Conceptual DFT Approach, in Structure andBonding, ed. M. V. Putz, 2012.
27 D. R. Roy, S. Duley and P. K. Chattaraj, Proc. Ind. Natl. Sci.Acad., Part A, 2008, 74, 11–18.
28 P. K. Chattaraj, D. R. Roy and S. Duley,Chem. Phys. Lett., 2008,460, 382–385.
29 S. Duley, P. Goyal, S. Giri and P. K. Chattaraj, Croat. Chim.Acta, 2009, 82, 193–205.
30 P. K. Chattaraj and S. Giri, Int. J. Quantum Chem., 2009, 109,2373–2382.
31 S. Giri, R. P. S. A. Kumar, A. Chakraborty, D. R. Roy, S. Duley,R. Parthasarathi, M. Elango, R. Vijayraj, V. Subramanian,G. Merino and P. K. Chattaraj, Bonding, Aromaticity andPossible Bond-Stretch Isomerism in an ‘‘All-Metal’’ cluster –[Be6Zn2]
2�, in Aromaticity and Metal Clusters, ed. P. K. Chattaraj,Taylor & Francis, Florida, 2009.
32 S. Giri, A. Chakraborty and P. K. Chattaraj, J. Mol. Model.,2010, 17, 777–784.
33 A. Chakraborty, S. Giri and P. K. Chattaraj, Struct. Chem., 2011,22, 823–837.
34 S. Duley, S. Giri, N. Sathymurthy, R. Islas, G. Merino andP. K. Chattaraj, Chem. Phys. Lett., 2011, 506, 315–320.
35 S. Giri, A. Chakraborty and P. K. Chattaraj, Nano Rev., 2011, 2,5767–5773.
36 S. Bandaru, A. Chakraborty, S. Giri and P. K. Chattaraj, Int. J.Quantum Chem., 2012, 112, 695–702.
37 P. K. Chattaraj, S. Bandaru and S. Mondal, J. Phys. Chem. A,2011, 115, 187–193.
38 S. Pan, S. Giri and P. K. Chattaraj, J. Comput. Chem., 2012, 33,425–434.
39 R. Das and P. K. Chattaraj, J. Phys. Chem. A, 2012, 116,3259–3266.
40 K. Srinivasu, S. K. Ghosh, R. Das, S. Giri and P. K. Chattaraj,RSC Adv., 2012, 2, 2914–2922.
41 P. K. Chattaraj, J. Ind. Chem. Soc., 1992, 69, 173–183.42 R. G. Parr, R. A. Donnelly, M. Levy and W. E. Palke, J. Chem.
Phys., 1978, 68, 3801–3807.43 R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105,
7512–7516.44 R. G. Pearson, Chemical Hardness: Applications from Molecules
to Solids, Wiley-VCH, Weinheim, 1997.45 R. G. Parr, L. v. Szentpaly and S. Liu, J. Am. Chem. Soc., 1999,
121, 1922–1924.46 P. K. Chattaraj, U. Sarkar and D. R. Roy, Chem. Rev., 2006, 106,
2065–2091.47 P. K. Chattaraj and D. R. Roy, Chem. Rev., 2007, 107,
PR46–PR74.48 P. K. Chattaraj, S. Giri and S. Duley, Chem. Rev., 2011, 111,
PR43–PR75.49 R. S. Mulliken, J. Chem. Phys., 1955, 23, 1833–1840.50 R. G. Parr and W. Yang, J. Am. Chem. Soc., 1984, 106,
4049–4050.51 W. Yang and W. J. Mortier, J. Am. Chem. Soc., 1986, 108,
5708–5711.52 P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and
N. J. R. v. E. Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318.53 Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and
P. v. R. Schleyer, Chem. Rev., 2005, 105, 3842–3888.54 A. Stanger, J. Org. Chem., 2006, 71, 883–893.55 J. C. Jimenez-Halla, E. Matito, L. Blancafort, J. Robles and
M. Sola, J. Comput. Chem., 2009, 30, 2764–2776.56 S. Noorizadeh and M. Dardab, Chem. Phys. Lett., 2010, 493,
376–380.57 J. A. N. F. Gomes and R. B. Mallion, Chem. Rev., 2001, 101,
1349–1384.58 J. Kruszewski and T. M. Krygowski, Tetrahedron Lett., 1972, 13,
3839–3842.59 R. G. Pearson, J. Chem. Educ., 1987, 64, 561–567.60 R. G. Parr and P. K. Chattaraj, J. Am. Chem. Soc., 1991, 113,
1854–1855.61 P. W. Ayers and R. G. Parr, J. Am. Chem. Soc., 2000, 122,
2010–2018.62 P. K. Chattaraj and S. Sengupta, J. Phys. Chem., 1996, 100,
16126–16130.63 P. Fuentealba, Y. Simon-Manso and P. K. Chattaraj, J. Phys.
Chem. A, 2000, 104, 3185–3187.64 E. Chamorro, P. K. Chattaraj and P. Fuentealba, J. Phys. Chem.
A, 2003, 107, 7068–7072.65 R. Parthasarathi, M. Elango, V. Subramanian and
P. K. Chattaraj, Theor. Chem. Acc., 2005, 113, 257–266.66 Gaussian 03, Revision B. 03, Gaussian, Inc., Pittsburgh, PA.67 R. J. Astheimer and L. G. Sneddon, Inorg. Chem., 1983, 22,
1928–1934.68 (a) I. Shapiro, C. David. Good and R. E. Williams, J. Am. Chem.
Soc., 1962, 84, 3837–3840; (b) I. Shapiro, B. Keilin, R. E. Williamsand C. D. Good, J. Am. Chem. Soc., 1963, 85, 3167–3171.
69 J. B. MacNaughton, A. Moewes, J. S. Lee, S. D. Wettig,H.-B. Kraatz, L. Z. Ouyang, W. Y. Ching and E. Z. Kurmaev,J. Phys. Chem. B, 2006, 110, 15742–15748.
70 Biomacromolecules: Introduction to structure, function and infor-matics, ed. C. S. Tsai, Wiley-Liss, 2007, Science.
71 P. Laslo, Angew. Chem., Int. Ed., 2000, 39, 2071–2072.72 N. Perez-Peralta, M. Contreras, W. Tiznado, J. Stewart,
K. J. Donald and G. Merino, Phys. Chem. Chem. Phys., 2011,13, 12975–12980.
73 W. Tiznado, N. Perez-Peralta, R. Islas, A. Toro-Labbe,J. M. Ugalde and G. Merino, J. Am. Chem. Soc., 2009, 131,9426–9431.
74 S. Giri, S. Bandaru, A. Chakraborty and P. K. Chattaraj, Phys.Chem. Chem. Phys., 2011, 13, 20602–20614.
75 H. Limpricht, Ber. Dtsch. Chem. Ges., 1870, 3, 90–91.76 K. Gregory, P. v. R. Schleyer and R. AdV. Snaith, Inorg. Chem.,
1991, 37, 47–142.77 P. v. R. Schleyer, Pure Appl. Chem., 1984, 56, 151–162.78 A. L. Harreus, ‘‘Pyrrole’’, in Ullmann’s Encyclopedia of Industrial
Chemistry, Wiley-VCH, Weinheim, 2002.79 L. Nyulaszi, Chem. Rev., 2001, 101, 1229–1246.80 W. Schaefer, A. Schweig and F. Mathey, J. Am. Chem. Soc., 1976,
98, 407–414.
Publ
ishe
d on
09
July
201
2. D
ownl
oade
d by
Ind
ian
Inst
itute
of
Tec
hnol
ogy
Kha
ragp
ur o
n 25
/01/
2015
17:
26:4
4.
View Article Online

14802 Phys. Chem. Chem. Phys., 2012, 14, 14784–14802 This journal is c the Owner Societies 2012
81 N. D. Epiotis and W. Cherry, J. Am. Chem. Soc., 1976, 98,4365–4370.
82 D. B. Chesnut and L. D. Quin, J. Am. Chem. Soc., 1994, 116,9638–9643.
83 L. Nyulaszi, J. Phys. Chem., 1995, 99, 586–591.84 R. Islas, E. Chamorro, J. Robles, T. Heine, J. C. Santos and
Gabriel Merino, Struct. Chem., 2007, 18, 833–839.85 B. Kiran, A. K. Phukan and E. D. Jemmis, Inorg. Chem., 2001,
40, 3615–3618.86 W. H. Fink and J. C. Richards, J. Am. Chem. Soc., 1991, 113,
3393–3398.87 N. Matsunaga and M. S. Gordon, J. Am. Chem. Soc., 1994, 116,
11407–11419.88 N. Matsunaga, T. R. Cundari, M. W. Schmidt andM. S. Gordon,
Theor. Chim. Acta, 1992, 83, 57–68.89 R. C. Haddon, Pure Appl. Chem., 1982, 54, 1129–1142.90 J. J. Engelberts, R. W. A. Havenith, J. H. v. Lenthe,
L. W. Jenneskens and P. W. Fowler, Inorg. Chem., 2005, 44,5266–5272.
91 P. v. R. Schleyer, H. Jiao, N. J. R. V. E. Hommes, V. G. Malkinand O. L. Malkina, J. Am. Chem. Soc., 1997, 119, 12669–12670.
92 L. Turker, S. Gumus and T. Atalar, Bull. Korean Chem. Soc.,2009, 30, 2233–2239.
93 E. D. Jemmis, G. Subramanian, A. J. Kos and P. v. R. Schleyer,J. Am. Chem. Soc., 1997, 119, 9504–9512.
94 V. I. Minkin, R. M. Minyaev, A. G. Starikov andT. N. Gribanova, Russ. J. Org. Chem., 2005, 41, 1289–1295.
95 A. Kumar, A. L. M. Reddy, A. Mukherjee, M. Dubey, X. Zhan,N. Singh, L. Ci, W. E. Billups, J. Nagurny, G. Mital andP. M. Ajayan, ACS Nano, 2011, 5, 4345–4349.
96 H. Miyaoka, W. Ishida, T. Ichikawa and Y. Kojima, J. AlloysCompd., 2011, 509, 719–723.
97 C. H. Wu, J. Phys. Chem., 1983, 87, 1534–1540.98 R. Keese, Chem. Rev., 2006, 106, 4787–4808.99 G. Merino, M. A. Mendez-Rojas, A. Vela and T. Heine,
J. Comput. Chem., 2007, 28, 362–372.100 E. D. Jemmis, E. G. Jayasree and P. Parameswaran, Chem. Soc.
Rev., 2006, 35, 157–168.101 L. Radom and D. R. Rasmussen, Pure Appl. Chem., 1998, 70,
1977–1984.102 Y. Pei, W. An, K. Ito, P. V. R. Schleyer and X. C. Zeng, J. Am.
Chem. Soc., 2008, 130, 10394–10400.103 P. V. R. Schleyer and A. I. Boldyrev, J. Chem. Soc., Chem.
Commun., 1991, 1536–1538.
104 Y.-B. Wu, H.-G. Lu, S.-D. Li and Z.-X. Wang, J. Phys. Chem. A,2009, 113, 3395–3402.
105 J. O. C. Jimenez-Halla, Y.-B. Wu, Z.-X. Wang, R. Islas, T. Heineand G. Merino, Chem. Commun., 2010, 46, 8776–8778.
106 Y.-B. Wu, Y. Duan, H.-G. Lu and S.-D. Li, J. Phys. Chem. A,2012, 116, 3290–3294.
107 A. C. Castro, G. Martınez-Guajardo, T. Johnson, J. M. Ugalde,Y.-b. Wu, J. M. Mercero, T. Heine, K. J. Donald and G. Merino,Phys. Chem. Chem. Phys., 2012, DOI: 10.1039/c2cp40839b.
108 Z.-Y. Jiang, C.-J. Yang and S.-T. Li, J. Chem. Phys., 2005,123, 204315.
109 K. B. Wiberg, Tetrahedron, 1968, 24, 1083–1096.110 R. Hoffmann, R. W. Alder and C. F. Wilcox Jr, J. Am. Chem.
Soc., 1970, 92, 4992–4993.111 A. I. Boldyrev, Chem. Rev., 2005, 105, 3716–3757.112 S. Duley, S. Giri, A. Chakraborty and P. K. Chattaraj, J. Chem.
Sci., 2009, 121, 849–858.113 S. Duley, A. Chakraborty, S. Giri and P. K. Chattaraj, J. Sulfur
Chem., 2010, 31, 231–246.114 S. Giri, D. R. Roy, S. Duley, A. Chakraborty, R. Parthasarathi,
M. Elango, R. Vijayaraj, V. Subramanian, R. Islas, G. Merinoand P. K. Chattaraj, J. Comp. Chem., 2010, 31, 1815–1821.
115 S. Giri, A. Chakraborty, S. Duley, G. Merino, V. Subramanianand P. K. Chattaraj, ICCMSE, AIP Issue (2009).
116 K. Srinivasu, K. R. S. Chandrakumar and S. K. Ghosh, Phys.Chem. Chem. Phys., 2008, 10, 5832–5839.
117 C. Ataca, E. Akturk, S. Ciraci and H. Ustunel, Appl. Phys. Lett.,2008, 93, 043123.
118 H. W. Kroto, J. R. Heath, S. C. O’Brien, R. F. Curl andR. E. Smalley, Nature, 1985, 318, 162–163.
119 S. Iijima, Nature, 1991, 354, 56–58.120 Q. Sun, P. Jena, Q. Wang and M. Marquez, J. Am. Chem. Soc.,
2006, 128, 9741–9745.121 G. Wu, J. Wang, X. Zhang and L. Zhu, J. Phys. Chem. C, 2009,
113, 7052–7057.122 S. V. Alapati, J. K. Johnson and D. S. Sholl, J. Phys. Chem. B,
2006, 110, 8769–8776.123 B. Sakintuna, F. L-Darkrimb and M. Hirscherc, Int. J. Hydrogen
Energy, 2007, 32, 1121–1140.124 S. L. James, Chem. Soc. Rev., 2003, 32, 276–288.125 M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O’.
Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319–330.126 A. Chakraborty, S. Duley and P. K. Chattaraj, Ind. J. Chem.,
2012, 51A, 226–244.
Publ
ishe
d on
09
July
201
2. D
ownl
oade
d by
Ind
ian
Inst
itute
of
Tec
hnol
ogy
Kha
ragp
ur o
n 25
/01/
2015
17:
26:4
4.
View Article Online
Copyright © 2022 FDOKUMEN


![Aromaticity of the planar hetero[8]circulenes and their doubly charged ions: NICS and GIMIC characterization](https://static.fdokumen.com/doc/165x107/6335e0f002a8c1a4ec01f590/aromaticity-of-the-planar-hetero8circulenes-and-their-doubly-charged-ions-nics.jpg)








![ChemInform Abstract: Recent Trends in the Chemistry of Aminobenzo[b]thiophenes](https://static.fdokumen.com/doc/165x107/6323ed8c3c19cb2bd106befc/cheminform-abstract-recent-trends-in-the-chemistry-of-aminobenzobthiophenes.jpg)








