
Mucin Multilayers Assembled through Sugar–Lectin Interactions
8
Mucin Multilayers Assembled through Sugar−Lectin Interactions Thomas Crouzier, Colin H. Beckwitt, and Katharina Ribbeck* Department of Biological Engineering, 77 Massachusetts Avenue, Building 56-341c, Massachusetts Institute of Technology, Cambridge, Massachusetts, United States * S Supporting Information ABSTRACT: Multilayer films of biopolymers are attractive tools to exploit the extraordinary properties of certain biomacromolecules and introduce new functionalities to surfaces. Mucins, the gel-forming constituents of mucus, are versatile glycoproteins that have potential as new building blocks for biomaterial surface coatings. Multilayer films have mostly been assembled through the electrostatic pairing of polyelectrolytes, which results in limited pH and salt stability and screens charges otherwise available for useful payload binding. Here, we aim at assembling mucin multilayer films that differ from conventional paired polyelectrolyte assemblies to obtain highly stable and functional surface modifications. Using the lectin wheat germ agglutinin (WGA) to cross-link mucin- bound sugar residues, we show that (Mucin/WGA) films can grow into hydrated films and sustain exceptional resistance to extreme salt conditions and a large range of pH. Furthermore, we show that the addition of soluble N-acetyl-D-glucosamine can induce the controlled release of WGA from (Mucin/WGA) films. Last, we show that (Mucin/WGA) films can repeatedly incorporate and release a positively charged model cargo. The lubricating, hydration, barrier, and antimicrobial properties of mucins open multiple applicative perspectives for these highly stable mucin-based multilayer films. ■ INTRODUCTION Natural hydrogels such as the extracellular matrix or mucus have the capacity to accommodate and regulate a range of biological properties of macromolecules and cells. Accordingly, their constituents have been exploited to create biomimetic materials that recapitulate selected functions. For example, extracellular matrix proteins such as collagen and fibronectin can interact with specific cell surface receptors and accordingly have been used to create coatings or 3D matrices that promote cell adhesion. 1 Polysaccharides also find abundant use in biomedical applications. For instance, hyaluronic acid is injected into joints for lubrication in cases of osteoarthritis because its rheological properties are close to those of natural synovial fluids. 2 Heparin is an effective anticoagulant, but it can also interact with certain growth factors, and has been used to build scaffolds for the controlled delivery of growth factors. 3 Hence, biopolymers have gained increasing attention due to their desirable features for the engineering of biomimetic materials. 4,5 One important, yet underexplored, family of biopolymers are mucins, the main gel-forming constituents of mucus. Mucins are glycoproteins that exist in secreted and membrane-bound form and coat all wet surfaces in the human body, including the eyes, and the epithelia of the respiratory, digestive, and female genital tracts. Secreted mucins are densely glycosylated and assume a largely extended conformation, with molecular weights up to several tens of megadaltons. 6 Mucins have several exquisite features that make them interesting building blocks for synthetic materials. For example, they can interact with a broad spectrum of particles of different sizes and biochemical properties, ranging from protons 7 to certain pathogenic viruses, bacteria, and neutrophils. 8,9 At a functional level, mucins provide a protective barrier toward pathogens and toxins and also serve to hydrate and lubricate the epithelial surfaces. 10 Mucins can be isolated from native tissues or from mucus-secreting cell lines, and they can be reconstituted to form hydrogels. Mucins have been used to generate relatively thin monolayer coatings on a variety of surfaces including glass, polystyrene, and implantable materials such as polyurethane. 11 Such thin mucin coatings have the capacity to limit bacterial surface adhesion 12,13 and appear to trigger low immune responses, 14,15 making them potentially biocompatible. Thicker multilayer coatings are also of interest because they have several critical advantages over monolayers: they offer more volume to incorporate molecules of interest such as drugs, 16 antibiotics, 17 or growth factors, 18,19 they provide more structural flexibility to the polymers, thus potentially mimicking more closely their physiological conformation, and they are tunable in thickness, structure, and morphology. In addition, multilayer films generated with mucins could be of specific interest to generate biocompatible surface modifications or to reconstitute native functions of the mucosal barrier on synthetic surfaces. Mucin-based multilayer films can be formed by complexing mucins electrostatically with positively charged molecules such as chitosan, 20,21 polyallylamine hydrochloride 22 polymers, or Received: August 1, 2012 Revised: August 22, 2012 Published: August 24, 2012 Article pubs.acs.org/Biomac © 2012 American Chemical Society 3401 dx.doi.org/10.1021/bm301222f | Biomacromolecules 2012, 13, 3401−3408
Transcript of Mucin Multilayers Assembled through Sugar–Lectin Interactions

Mucin Multilayers Assembled through Sugar−Lectin InteractionsThomas Crouzier, Colin H. Beckwitt, and Katharina Ribbeck*
Department of Biological Engineering, 77 Massachusetts Avenue, Building 56-341c, Massachusetts Institute of Technology,Cambridge, Massachusetts, United States
*S Supporting Information
ABSTRACT: Multilayer films of biopolymers are attractivetools to exploit the extraordinary properties of certainbiomacromolecules and introduce new functionalities tosurfaces. Mucins, the gel-forming constituents of mucus, areversatile glycoproteins that have potential as new buildingblocks for biomaterial surface coatings. Multilayer films havemostly been assembled through the electrostatic pairing ofpolyelectrolytes, which results in limited pH and salt stabilityand screens charges otherwise available for useful payloadbinding. Here, we aim at assembling mucin multilayer films that differ from conventional paired polyelectrolyte assemblies toobtain highly stable and functional surface modifications. Using the lectin wheat germ agglutinin (WGA) to cross-link mucin-bound sugar residues, we show that (Mucin/WGA) films can grow into hydrated films and sustain exceptional resistance toextreme salt conditions and a large range of pH. Furthermore, we show that the addition of soluble N-acetyl-D-glucosamine caninduce the controlled release of WGA from (Mucin/WGA) films. Last, we show that (Mucin/WGA) films can repeatedlyincorporate and release a positively charged model cargo. The lubricating, hydration, barrier, and antimicrobial properties ofmucins open multiple applicative perspectives for these highly stable mucin-based multilayer films.
■ INTRODUCTION
Natural hydrogels such as the extracellular matrix or mucushave the capacity to accommodate and regulate a range ofbiological properties of macromolecules and cells. Accordingly,their constituents have been exploited to create biomimeticmaterials that recapitulate selected functions. For example,extracellular matrix proteins such as collagen and fibronectincan interact with specific cell surface receptors and accordinglyhave been used to create coatings or 3D matrices that promotecell adhesion.1 Polysaccharides also find abundant use inbiomedical applications. For instance, hyaluronic acid isinjected into joints for lubrication in cases of osteoarthritisbecause its rheological properties are close to those of naturalsynovial fluids.2 Heparin is an effective anticoagulant, but it canalso interact with certain growth factors, and has been used tobuild scaffolds for the controlled delivery of growth factors.3
Hence, biopolymers have gained increasing attention due totheir desirable features for the engineering of biomimeticmaterials.4,5
One important, yet underexplored, family of biopolymers aremucins, the main gel-forming constituents of mucus. Mucinsare glycoproteins that exist in secreted and membrane-boundform and coat all wet surfaces in the human body, including theeyes, and the epithelia of the respiratory, digestive, and femalegenital tracts. Secreted mucins are densely glycosylated andassume a largely extended conformation, with molecularweights up to several tens of megadaltons.6 Mucins haveseveral exquisite features that make them interesting buildingblocks for synthetic materials. For example, they can interactwith a broad spectrum of particles of different sizes and
biochemical properties, ranging from protons7 to certainpathogenic viruses, bacteria, and neutrophils.8,9 At a functionallevel, mucins provide a protective barrier toward pathogens andtoxins and also serve to hydrate and lubricate the epithelialsurfaces.10 Mucins can be isolated from native tissues or frommucus-secreting cell lines, and they can be reconstituted toform hydrogels.Mucins have been used to generate relatively thin monolayer
coatings on a variety of surfaces including glass, polystyrene,and implantable materials such as polyurethane.11 Such thinmucin coatings have the capacity to limit bacterial surfaceadhesion12,13 and appear to trigger low immune responses,14,15
making them potentially biocompatible. Thicker multilayercoatings are also of interest because they have several criticaladvantages over monolayers: they offer more volume toincorporate molecules of interest such as drugs,16 antibiotics,17
or growth factors,18,19 they provide more structural flexibility tothe polymers, thus potentially mimicking more closely theirphysiological conformation, and they are tunable in thickness,structure, and morphology. In addition, multilayer filmsgenerated with mucins could be of specific interest to generatebiocompatible surface modifications or to reconstitute nativefunctions of the mucosal barrier on synthetic surfaces.Mucin-based multilayer films can be formed by complexing
mucins electrostatically with positively charged molecules suchas chitosan,20,21 polyallylamine hydrochloride22 polymers, or
Received: August 1, 2012Revised: August 22, 2012Published: August 24, 2012
Article
pubs.acs.org/Biomac
© 2012 American Chemical Society 3401 dx.doi.org/10.1021/bm301222f | Biomacromolecules 2012, 13, 3401−3408

lactoperoxidase.23,24 In general, multilayer films with oppositelycharged polyelectrolytes has enabled the engineering ofinteresting functional coatings for a wide range of applications,from sensors and solar cells to biomaterial and tissueengineering.25 However, they also have certain shortcomings.First, these films are sensitive to salt and the ionization state ofthe polymers, making them relatively unstable unless covalentlycross-linked.26,27 Second, the bulk of charges of thepolyelectrolyte are complexed by the partner polymer, thuspotentially screening charges that could serve to sequesterparticles or drugs into the multilayer films. Alternativetechniques that bypass the need for electrostatic pairing havealso been presented and include hydrogen bonding28 and clickchemistry,29 but these techniques are best achieved withsynthetic polymers of well-controlled chemistry. Otherstrategies use biological interactions such as biotin/strepati-vin,30 antibody−antigen pairs,31 and lectin/sugar interac-tions,32−35 all representing relatively strong interactions thatdrive the autoassembly of biopolymers and other biomacro-molecules in vivo. Lectin/sugar interactions could be well-suited to generate robust mucin films that are independent ofelectrostatic pairing of polyelectrolytes, as sugars that arecommonly found in mucins; namely, N-acetyl-D-glucosamine(GlcNAc) and sialic acid are ligands to the lectin wheat germagglutinin (WGA). WGA forms dimers and can bind up toeight sugar residues simultaneously.36
We here describe the use of the WGA lectin to cross-linkmucin layers. We study the physicochemical properties of thesefilms such as thickness, level of hydration, morphology,robustness toward ionic strength, and their capacity to bind amodel cargo.
■ MATERIAL AND METHODSMaterials and Reagents. Pig gastric mucin was purified from pig
stomachs and lyophilized as reported in ref 37, omitting the cesiumchloride density gradient. This preparation is enriched in the secretedmucin MUC5AC. Lyophilized mucin samples were dissolved over-night at 4 °C in deionized water at 2 mg/mL. Purified pig gastricmucin was labeled with the fluorescence dye Alexa488-carboxylic acidsuccinimidyl ester (Invitrogen). The dye was dissolved and stored at10 mg/mL (15.5 mM) in DMSO. For the labeling reaction, 4 mg ofmucin was dissolved in 0.1 M bicarbonate buffer (pH 9) andcombined with 0.1 mg of the dye. The mixture was incubated for 1 h atroom temperature; then, the pH was lowered to 7, and the unbounddye was removed by centrifuge filtration (Pall, MWCO 50 kDa). WGAlectin, poly L-lysine (PLL), fluorescein-labeled PLL (PLL-FITC), andN-acetyl-D-glucosamine were purchased from Sigma (St. Louis, MO).(Mucin/WGA) Film Buildup. Films were built by alternating
depositions of mucin and the lectin WGA from dilute solutions. Mucinwas prepared at a concentration of 0.2 mg/mL and lectin was preparedat 0.1 mg/mL in buffer containing 20 mM Hepes (pH 7.4) and 0, 200,or 400 mM NaCl. Mucin was left to adsorb for 15 min, and lectin wasleft for 5 min. Between the layering steps, two washes of 5 min wereperformed with the same buffer as used for buildup. For 96-well plates,50 μL of mucin and WGA solution and 150 μL of washing solutionwere used. For QCM-D experiments, 300 μL of WGA and mucinsolution and 1 mL of washing solution were used. The resulting filmsare termed (Mucin/WGA)n, with n being the number of layer pairs.The (Mucin/WGA)12 films were assembled on both the gold surfaceused for AFM measurement and the polystyrene surface for all otheranalyses. No differences in thickness or growth curve were measuredon these two surfaces by QCM-D (Figure SI 2 of the SupportingInformation).(Mucin/WGA) Film Composition. To determine the composition
of the (Mucin/WGA) multilayer films, we spiked the mucin and WGAsolutions with 40% (w/w) fluorescent mucin and 10% (w/w)
fluorescent WGA, respectively. The films were assembled in untreatedpolystyrene 96-well plates (Falcon, 351172) with 0 mM NaCl in 20mM Hepes buffer (pH 7.4). The fluorescence of each well wasmeasured after the addition of each new layer using a fluorescenceplate reader (Spectramax M3, Molecular Devices). Mucin and lectincontents were quantified by calibrating fluorescence in solution withknown amounts of mucin and lectin.
Dry and Hydrated Mass and Thickness of the (Mucin/WGA)Films. The dry thickness was determined by spectroscopicellipsometry using a XLS-100 ellipsometer (J.A. Woollam, Lincoln,NE). This surface-sensitive optical technique records changes of lightpolarization upon reflection on the dried film. The optical thicknessand the refractive index of the films were calculated from suchmeasurements performed at an angle of 70° and from wavelength 190to 993 nm. The films assembled on QCM-D crystals were rinsed withwater and dried under nitrogen flow. We performed 70 spectroscopicscans per measurement and 4 measurements per sample. The datawere modeled using the WVASE32 software (version 3.768) andassuming a multilayer model composed of a known Si substrate (0.2mm), a gold layer (75 nm), and a Cauchy layer of unknown thicknessand optical properties corresponding to the (Mucin/WGA) film. Themass of the dry film was estimated by assuming a density of 1200 kg/m3, as previously measured for other similar systems.38 The refractiveindices were 1.62 ± 0.06 for films built in 0 mM NaCl, 1.59 ± 0.05 for200 mM NaCl, 1.59 ± 0.1 for 400 mM NaCl, and 1.59 ± 0.05 for 0mM NaCl after WGA was released. A refractive index of ∼1.6 is in theupper regime of what has been reported before for biopolymer-basedmultilayer films.39
The hydrated mass and thickness were measured by quartz crystalmicrobalance with dissipation monitoring (QCM-D, E4 system, Q-Sense, Sweden). The films were grown on a gold-covered quartzcrystal, cleaned with warm 2% SDS and 0.1 M HCl, rinsed withdeionized water, and further cleaned by ozone treatment for 15 min.Film growth on polystyrene-coated crystals was similar to that on gold(Figure SI 2 of the Supporting Information). The crystal vibration wasfollowed at its fundamental frequency (∼5 MHz) and the sixovertones (15, 25, 35, 45, 55, and 65 MHz). Changes in theresonance frequencies, and in dissipation of the vibration once theexcitation is stopped, were followed at the seven frequencies. Assuggested by the high dissipation values (Mucin/WGA), films arehighly hydrated and possess viscoelastic properties, requiring themeasurement data to be modeled. The Voigt-based model40 (i.e., aspring and dashpot in parallel under no slip conditions) was used tocalculate the hydrated thickness, assuming a density of 1050 kg/m3 41
and that the film is homogeneous in thickness over the crystal’ssurface. The multilayer films were built up to 12 layer pairs.
The level of hydration of the film was deduced from the dry andhydrated mass by the relationship:
=−
hydrationhydrated mass dry mass
hydrated mass
(Mucin/WGA) Film Imaging. For fluorescence microscopy, thefilms were built using the fluorescent mucin-Alexa488 conjugate in 96-well plates with thin untreated polystyrene bottoms, optimized foroptical observation (Costar 3615 Corning, Corning, NY). The filmswere scratched with a pipet tip and imaged using an Observer Z1inverted fluorescent microscope (Zeiss, Oberkochen, Germany) and a10× 0.3 NA or 100× 1.4 NA lens (Zeiss, Germany). For AFMimaging, the films were built on QCM-D gold-covered crystals, kept in20 mM Hepes buffer (pH 7.4), and observed in tapping mode usingpyramidal cantilevers (NP-S10, Veeco, Santa Barbara, CA) and anAsylum MFP-3D-BIO AFM (Asylum Research, Santa Barbara, CA).
WGA Release and Film Resistance to pH, Salt, and Media.WGA was released from the mucin-capped films by adding 200 μL of a100 mM N-acetyl-D-glucosamine solution to each well. Resistance teststo various chemicals were done on films built in 20 mM Hepes bufferwithout NaCl. For low-pH treatment, 200 μL of acetate buffer (0.1 M,pH 3) was used. The high-pH treatment consisted of 200 μL ofcarbonate buffer (0.1 M sodium carbonate and 0.1 M sodium
Biomacromolecules Article
dx.doi.org/10.1021/bm301222f | Biomacromolecules 2012, 13, 3401−34083402

bicarbonate, adjusted to pH 9) or KCl/NaOH buffer (0.1 M, pH 12).Of note, the labeled mucin retained its fluorescence intensity afterexposure to pH 12 solutions, suggesting that the fluorophore and itslinkage to mucin are stable under these conditions (Figure SI 5 of theSupporting Information). To test the resistance to salts, the films weresubjected to 200 μL of a 5 M NaCl, 75 mM MgCl2, or 75 mM CaCl2solution buffered with 20 mM Hepes to pH 7.4. Resistance to cellculture media was followed over 14 days by subjecting the films to 200μL of DMEM supplemented with 10% fetal bovine serum(Invitrogen), 25 U/mL penicillin, and 25 μg/mL streptomycin(Invitrogen). The medium was exchanged after each measurement.Films were washed three times with 150 μL of 20 mM Hepes buffer(pH 7.4) after treatment. Compositional changes in (Mucin/WGA))12films were measured by comparing the total fluorescence before andafter treatment, relative to the fluorescence of nontreated wells.PLL Incorporation and Release. To investigate the capacity of
(Mucin/WGA) films to incorporate positively charged molecules, 40μL of a 0.5 mg/mL solution of PLL-FITC was deposited on (Mucin/WGA)12 or (Mucin/WGA)11.5 films built in 0 mM NaCl buffered with20 mM Hepes at pH 7.4. The films were either untreated or treatedwith GlcNAc to release WGA. After 30 min, the PLL-FITC solutionwas removed, and the wells were washed five times with 150 μL of 20mM Hepes solution (pH 7.4) before fluorescence was quantified.Fluorescence was converted to mass of PLL by calibration in solution.The PLL incorporation relative to the mucin content in the film wascalculated based on the mucin mass obtained by fluorescencemeasurements in separate experiments. (See the (Mucin/WGA)Film Composition section.) PLL was released from the film by adding150 μL of 20 mM Hepes (pH 7.4) supplemented with either or 0.15or 5 M NaCl. At each time point, the film’s fluorescence was measuredin 150 μL of a 20 mM Hepes solution at pH 7.4. For repeated loading/unloading, the PLL was released for 30 min using a 5 M NaClsolution.(Mucin/WGA) Film Cytotoxicity. HeLa cells were grown to 70%
confluency in T25 flasks with DMEM media supplemented with 10%fetal bovine serum (Invitrogen), 25 U/mL penicillin, and 25 μg/mLstreptomycin (Invitrogen). The cells were detached using trypsin-EDTA (Invitrogen). The detached cells were washed to remove thetrypsin before being plated at a density of 27 000 cells/cm2 on thefilms constructed in the wells of 96-well plates. The cells wereincubated at 37 °C, 5% CO2 under a humidified atmosphere for 1, 3,or 7 days before being stained with the live-dead stain (2 μM calceinand 2 μM Ethidium homodimer-1 in DMED media, Invitrogen) for 20min. Images were acquired on an Axio Observer Z1 microscope (Zeiss,Oberkochen, Germany) using a EC-Plan Neofluar 10× 0.3 NA lens(Zeiss) in phase contrast or fluorescence mode. Live (green) and dead(red) cells were counted with the image analysis software ImageJ usingthe cell count plug-in.
■ RESULTS AND DISCUSSION
Mucin Can Be Cross-Linked via Lectins to Form(Mucin/WGA) Films. When a dilute solution of mucins issubjected to the substrate of a quartz crystal microbalance withdissipation monitoring (QCM-D), a 40−60 nm thick andhydrated mucin coating forms.11 It is not trivial to increase thecoating thickness by depositing further mucin layers, possiblybecause adsorbed mucins prevent further binding of mucinsfrom solution by steric and electrostatic effects (Figure SI 1 ofthe Supporting Information).42,43 We tested if this limitationcould be overcome by cross-linking individual mucin layerswith lectins. In this experiment, the deposition of mucins wasalternated with the deposition of the lectin WGA. The QCM-Dmeasurements were modeled to obtain the hydrated thicknessof the films as the mucin and lectin layers were deposited.Figure 1A shows that the initial mucin layer decreased inthickness on deposition of the first lectin layer, possibly due toa collapse of the glycan chains protruding from the mucins.
Then, for the 12 subsequent mucin−WGA layer pairs, the filmgrew almost linearly, suggesting that mucins and WGA canindeed autoassemble into multilayer films. It appears that thelectins are able to cross-link soluble mucins onto alreadyexisting mucin layers.To analyze the robustness of assembly of (Mucin/WGA)
films in further detail, we performed the assembly reaction atthree different ionic strengths: 0, 200, and 400 mM NaCl.Figure 1A shows that the final hydrated thickness of the(Mucin/WGA) films was comparable under the threeconditions. For comparison, the growth of films composed ofmucins and the positively charged PLL polymer was sensitive tosalt (Figure SI 3 of the Supporting Information), resulting inabout a two-fold difference in the final thickness with no saltand 200 mM NaCl.High ionic strength can influence the growth of polyelec-
trolyte-based assemblies as it affects charge shielding andchanges the number of charged groups available for ionicparing.44 The relative insensitivity of (Mucin/WGA) filmgrowth toward ambient ionic strength when measured byQCM-D suggests that specific lectin/sugar interactions aredriving the film’s assembly by mechanisms that differ from thesalt-sensitive ionic pairing occurring in (Mucin/PLL) films.Also of note is that the growth of the (Mucin/WGA) films was
Figure 1. (Mucin/WGA) film growth. Hydrated thickness obtained byQCM-D measurements, as a function of layer number for (Mucin/WGA) films built in the presence of 0, 200, or 400 mM NaCl (A). Thegrowth of the films was also followed using fluorescently labeled mucin(B) and WGA (B′). The corresponding masses were calculated basedon calibration with mucin and WGA in solution.
Figure 2. (Mucin/WGA) film hydration. The hydrated and drythicknesses (scale on the left) and the corresponding hydration (scaleon the right) for (Mucin/WGA)12 were determined for films built inbuffer containing 0, 200, or 400 mM NaCl.
Biomacromolecules Article
dx.doi.org/10.1021/bm301222f | Biomacromolecules 2012, 13, 3401−34083403

roughly linear. This profile is indicative for relatively stronginteractions that generate tightly stacked polymers, resulting ina linear increase in thickness at each deposition step.45,46
To measure the interaction of WGA and mucin with the filmindependently, the components were labeled with Alexa488,and their incorporation was followed by fluorescence imaging.(See the Materials and Methods.) WGA adsorbed to the filmmore efficiently at high salt concentrations, whereas nodifference in incorporation was measured for mucin at low orhigh ionic strength (Figure 1B,B′). This experiment alsorevealed that 10−20% of WGA was released from the film oneach mucin deposition step, and conversely, 5−10% of mucinswere released on WGA deposition. This may suggest that acertain fraction of newly formed Mucin−WGA complexesdissociated from the surface of the film in each deposition step.(Mucin/WGA) Films Are Highly Hydrated. Hydration
corresponds to the fraction of the film’s mass that is composedof water and is an important parameter that relates to
properties such as thickness, porosity, diffusion, and function-alities such as lubrication. The hydration of the (Mucin/WGA)films was calculated based on the hydrated mass obtained byQCM-D and the dry mass obtained by ellipsometry. (See theMaterial and Methods.) Figure 2 shows that ionic strength hadno significant effect on the hydrated thickness of the film.However, the dry thickness increased from 26 ± 3 (0 mMNaCl) to 46 ± 11 nm (200 mM NaCl) (Figure 2). Thisincrease may be attributed to the more effective incorporationof WGA at higher ionic strength (Figure 1B′). Combining thechanges in hydrated and dry mass implies that the level ofhydration decreased from 84 ± 0.6 (0 mM NaCl) to 78 ± 0.6%(400 mM NaCl), suggesting that the films are mildly sensitiveto ionic strength.In the three buildup conditions tested here, (Mucin/WGA)
films consisted of ∼80% water, which is comparable to previousmeasurements on mucin- and other biopolymer-based multi-layer films.20,45 Although this level of hydration is lower thanwould be provided by a single mucin coating (estimated at∼90% water24,47), it appears that the mucin molecules maintaintheir water binding capacity to a certain degree when arrangedas multilayer films.
Morphology of (Mucin/WGA) Films. The morphology ofthe films was characterized both by fluorescence microscopyusing fluorescently labeled mucins and by atomic forcemicroscopy (AFM). For this experiment, films were assembledin Hepes buffer without NaCl. At low (10×) magnification, thefilms appeared relatively homogeneous (Figure 3A). However,at 100× magnification, aggregates in the micrometer range werevisualized (Figure 3B). AFM measurements on hydrated filmsrevealed these ∼1 μm aggregates in more detail (Figure 3C).The film’s root mean-square-average roughness was 6.5 (±3.7)nm, which is comparable to the roughness in cross-linked(PLL/hyaluronic acid) multilayer films.48,49
Figure 3. (Mucin/WGA) film morphology. (Mucin-Alexa488/WGA)12 films observed by epifluorescence microscopy at different magnifications:10× objective; scale bar is 100 μm (A) and 100× objective; scale bar is 20 μm (B); and by AFM, the image is 10 × 10 μm (C). All observations weremade with films in the hydrated state.
Figure 4. (Mucin/WGA) film resistance to degradation. (Mucin-Alexa488/WGA)12 and (Mucin/WGA-Alexa488)12 films were testedfor their resistance to a range of pH values and ionic strengths. Thereported values are the percentage of remaining mucin and WGAinside the films after the indicated treatments. The films remainedlargely intact under these tested conditions, except at pH 12.
Figure 5. GlcNAc-induced WGA release. (Mucin/WGA)12 films were built in 96-well plates and subjected to three consecutive treatments with 100mM N-acetyl-D-glucosamine (GlcNAc). The percentage of mucin and WGA remaining in the film in response to each release treatment is plotted(A). The GlcNAc-mediated selective release of WGA results in an enrichment of the film in mucin (B).
Biomacromolecules Article
dx.doi.org/10.1021/bm301222f | Biomacromolecules 2012, 13, 3401−34083404
(Mucin/WGA) Films Are Exceptionally Resistant toHigh Salt Concentrations. Because the hydrated thickness ofmucin multilayer films is independent of the ionic strength usedfor assembly (Figure 2), it is also possible that established filmsremain resistant to high ionic strength and possibly extreme pHconditions. This feature would be desirable because it maysignificantly prolong the lifetime of the film if put in contactwith harsh biological fluids, such as the gastric acid of thestomach. (Mucin/WGA)12 films were built in 0 mM NaCl atpH 7.4 and then subjected to pH 3, 9, or 12 or 5 M NaCl, 75
mM MgCl2 or CaCl2. Indeed, already established filmsremained largely intact even under extreme conditions, sinceneither the mucin nor WGA content decreased by more than20% when exposed to pH 3 and 9 or 5 M NaCl. The films werealso resistant toward divalent salts at a supraphysiologicalconcentration of 75 mM. Only a pH of 12 dismantled thestructure of the film (Figure 4). By fluorescence microscopy, nochanges could be observed after the permissive treatments(Figure SI 4 of the Supporting Information).These results suggest that (Mucin/WGA) films are more
robust than mucin multilayer films that are based on electro-static interactions. For example (Mucin/Chitosan) tend todismantle when exposed to a different pH or ionic strengththan used for the buildup.20 Several other polyelectrolytemultilayer systems and, in particular, synthetic polymer systemssuch as poly(allylamine hydrochloride)/poly(acrylic acid) films,can resist pH changes within the range tested here.50,51
However, resistance to 5 M NaCl is unusual. This effect may bebrought about by the relatively high concentrations of WGAand WGA-binding sites within the film or the specific nature ofhydrogen bonding that largely contributes to the WGA−carbohydrate interactions.52
Controlled Release of WGA from (Mucin/WGA) Filmsby N-Acetyl-D-Glucosamine. Lectins have successfully beenused to modulate the delivery and targeting of certaindrugs.53,54 Hence, if it is possible to release the mucin-boundlectins from the films, for example, by introducing smallmolecules that specifically compete for lectin binding, then thissystem may provide an interesting carrier for lectin-coupleddrugs. To test this possibility, we exposed (Mucin/WGA)12films to N-acetyl-D-glucosamine, a known ligand for WGA. Thefilms were treated three consecutive times with a 100 mMGlcNAc solution at pH 7.4 for 30 min. Figure 5A shows thatthe WGA content of the film decreased by ∼80% after GlcNActreatment. For comparison, the amount of mucin dropped byonly 20% under the same conditions, suggesting that mucinremained largely associated with the films even after a
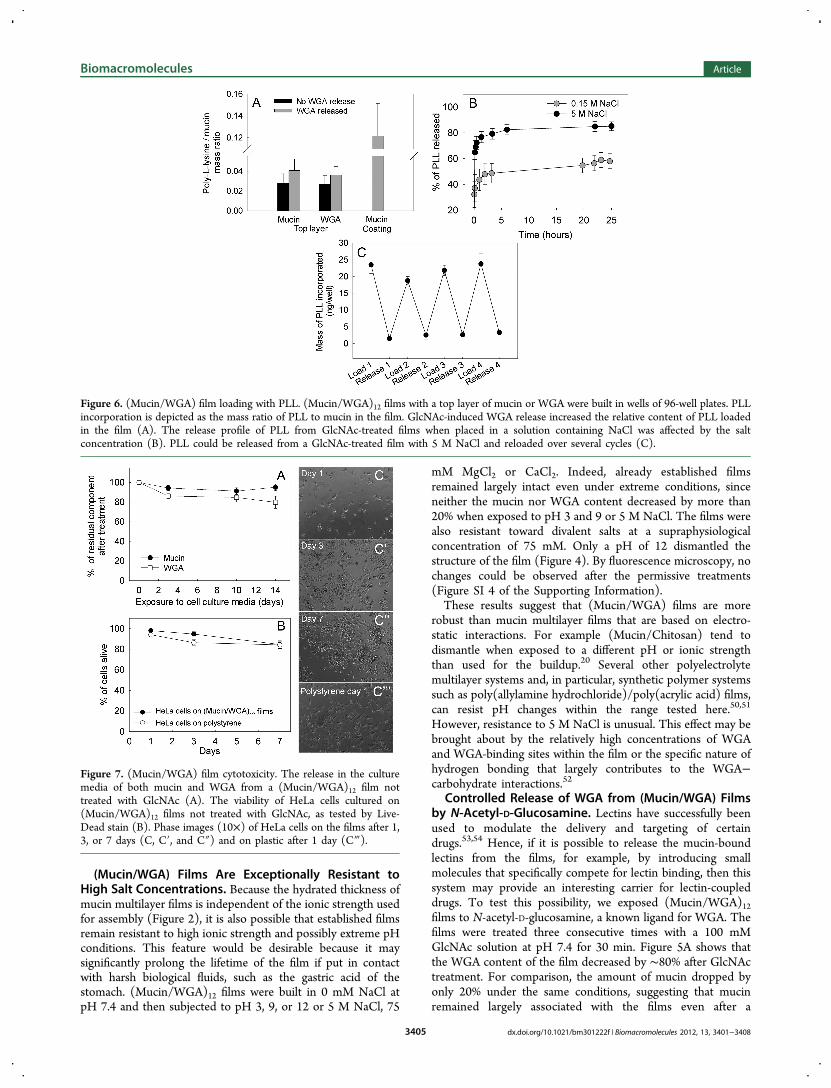
Figure 6. (Mucin/WGA) film loading with PLL. (Mucin/WGA)12 films with a top layer of mucin or WGA were built in wells of 96-well plates. PLLincorporation is depicted as the mass ratio of PLL to mucin in the film. GlcNAc-induced WGA release increased the relative content of PLL loadedin the film (A). The release profile of PLL from GlcNAc-treated films when placed in a solution containing NaCl was affected by the saltconcentration (B). PLL could be released from a GlcNAc-treated film with 5 M NaCl and reloaded over several cycles (C).
Figure 7. (Mucin/WGA) film cytotoxicity. The release in the culturemedia of both mucin and WGA from a (Mucin/WGA)12 film nottreated with GlcNAc (A). The viability of HeLa cells cultured on(Mucin/WGA)12 films not treated with GlcNAc, as tested by Live-Dead stain (B). Phase images (10×) of HeLa cells on the films after 1,3, or 7 days (C, C′, and C″) and on plastic after 1 day (C‴).
Biomacromolecules Article
dx.doi.org/10.1021/bm301222f | Biomacromolecules 2012, 13, 3401−34083405

substantial fraction of WGA had been released. One directconsequence of WGA dissociation from the film is a relativeenrichment of the film in mucin, increasing the Mucin/WGAratio from 2 to nearly 6 after three GlcNAc treatments (Figure5B).A competitor-induced release of lectins has been demon-
strated on several occasions in 3D hydrogels and multilayerfilms, mostly in the context of autoregulated insulin deliverysystems.55 The related characteristics of (Mucin/WGA) filmsbear potential for the design of novel sugar-specific inducibledelivery systems, in particular if combined with the film’sresilience toward a wide range of ionic strengths and pH values.(Mucin/WGA) Films Can Repeatedly Incorporate and
Release the Positively Charged Polymer PLL. Thepotential to release mucin-bound WGA is of interest in thecontext of drug delivery, where WGA-tethered molecules couldbe specifically dissociated in the presence of sugars. However,secreted mucins are acidic proteins that contain abundantnegative charge at neutral pH. Accordingly, they may interactwith positively charged molecules such as polycationicpolymers, growth factors, antimicrobial peptides, and certainsmall drugs. To test if (Mucin/WGA) films can be loaded withpositively charged molecules, we analyzed their ability toincorporate PLL. Figure 6A depicts that mucin multilayers canindeed incorporate PLL. However, the ratio of mucin to PLLwas lower than that on simple mucin coatings, possibly due tothe occupation of binding sites on the mucins by WGA and thepresence of repulsive positive charges introduced by WGA (Ip= 9).The release of WGA should uncover charged groups like
sialic acid, which could serve as further binding sites forpositively charged molecules. Therefore, the controlled releaseof WGA from the mucins could potentially be used to adjustthe binding capacity for positively charged molecules within thefilm. Indeed, the PLL-to-mucin ratio increased slightly afterpartial release of WGA (Figure 6A).If PLL adsorbs to mucins based on electrostatic interactions,
then it should be possible to release it with salt. Figure 6Bshows the release of PLL over time at relatively low (0.15 MNaCl) or high ionic strength (5 M NaCl). At 0.15 M NaCl, asmaller fraction of the loaded PLL was released than at 5 MNaCl, suggesting that the extent, and to some degree the rate,of the release can be tuned by varying the salt concentration.One further feature of the mucin-multilayer films that can beexploited for drug delivery applications is their resistance tohigh ionic strength: because the mucin multilayer films remains
intact even at 5 M NaCl, the same structure can be dischargedand recharged with PLL at least four times with no loss of PLLbinding capacity (Figure 6C).While we studied the loading of mucin multilayer films with
the model cargo PLL, we note that the mucus barrieraccommodates a range of bioactive molecules such as growthfactors and antimicrobial peptides in vivo.56 Hence, one mightenvision mucin-based coatings with complexed bioactivemolecules for biomedical applications such as drug delivery,wound healing, and antimicrobial surfaces. In addition, sialicacid is recognized as a ligand with high biological significance57
because it can interact with a range of different viruses andbacteria. We foresee that mucin-multilayers may be used todetect or inactivate such biological functional entities.
(Mucin/WGA) Films Are Noncytotoxic. In the context ofbiomedical applications or as a model substrate to study cell−mucin interactions, mucin multilayer films would be placed incontact with cells and must thus prove to be noncytotoxic.Previous studies report a certain toxicity of lectins towardcancer cell lines, for example.58,59 Components of multilayerscan potentially be released during cell culture procedures andinfluence cell behavior.60 We found that over 14 days, (Mucin/WGA)12 films released ∼20% of the lectin and less than ∼5% ofmucin (Figure 7A). Next, we directly tested if the (Mucin/WGA)12 films and the released film components were cytotoxictoward epithelial HeLa cells. Our data revealed that HeLa cellsgrown on (Mucin/WGA)12 films for 1, 3, and 7 days remainedalmost as viable as those on standard polystyrene surfaces asjudged by a fluorescence-based live-dead assay (Figure 7B).While the cells adhered to the films as depicted in Figure7C,C′,C″, they were less spread than on standard tissue culturesurface (Figure 7C‴).Lectins represent an integral part of our diet, and one may
thus argue that these molecules should be, to some degree,biocompatible. Nevertheless, few studies directly test thetoxicity of recombinant or purified lectins in vivo. Our systemconsisting of HeLa cells clearly is a simplification and cannot beextrapolated to live organisms; however, it may provide anapproximation to mucin-associated lectin behavior in context ofliving cells. WGA appears to be cytotoxic when uptaken by thecell,58 hence its relatively stable sequestration to the mucinsmay limit toxicity by reducing its ability to penetrate into cells.
■ CONCLUSIONS
We here report a new type of biopolymer-based multilayer filmmade from mucins and the lectin WGA. These films are
Figure 8. (Mucin/WGA) films as a multilfuntional delivery vehicle. Mucins (protein core in black and attached glycans in blue) interact with lectins(orange cross) to form nanothick mutlilayer films. The loading and release of molecules of interest inside the (Mucin/WGA) films is possiblethrough multiple strategies. The cargo can electrostatically interact with the mucin molecules and be released with salt. This does not alter the film’sstructure, and hence loading−unloading cycles can be repeated. In addition, the specific release of lectins from the films can be induced by solublesugars. If drugs are conjugated to the lectins, then the films could act as a sugar-triggered drug delivery system.
Biomacromolecules Article
dx.doi.org/10.1021/bm301222f | Biomacromolecules 2012, 13, 3401−34083406

resistant to extreme NaCl and divalent ion concentrations aswell as a broad range of pH values. In addition, they canundergo multiple rounds of loading and release of a modelsubstrate, PLL, without disintegrating (Figure 8). Because of itsunique properties, this system may be used to generate mucin-based materials for a range of biomedical applications. Forexample, the high stability could enable the generation ofpermanent drug-eluting coatings for materials that areotherwise poorly biocompatible and which should remaincoated after delivery of the drug cargo. The uptake of certaindrugs by the body can be improved by coupling them tolectins.53,54 Hence, mucin multilayers in which lectins can bereleased in a controlled way may be suitable vectors for suchlectin-bound drugs (Figure 8). Last, the creation of thin filmswith mucins could be a starting point for exploiting the nativeability of the biomolecules to trap specific pathogens, such ascertain viruses. Mucin multilayers may be used to deplete suchviruses from, for example, blood, and potentially be the startingpoint for diagnostic tools.
■ ASSOCIATED CONTENT
*S Supporting InformationQCM-D data showing five experiments: (1) that mucins cannotspontaneously form multilayer films in the conditions used; (2)that the (Mucin/WGA) films grow on both gold andpolystyrene covered surfaces, (3) the growth curves of theelectrostatically paired (Mucin/PLL) films, fluorescencemicroscopy images of (Mucin/WGA) films after treatmentwith salts or sugar (4); we also show a control experimentverifying the stability of the Alea488 dye and its linkage tomucin when exposed to pH 12 solutions (5). This material isavailable free of charge via the Internet at http://pubs.acs.org.
■ AUTHOR INFORMATION
Corresponding Author*E-mail: [email protected]. Tel: +617-715-4575. Fax: 617-324-7554.
NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTSWe thank the Johnson & Johnson Corporate Office of Scienceand Technology for the postdoctoral fellowship provided toT.C, and MIT startup funds to K.R. This work made use of theMRSEC Shared Experiment Facilities supported by theNational Science Foundation under award number DMR −819762. We are also grateful to Ronn Friedlander for hisassistance with the AFM experiments.
■ REFERENCES(1) Van den Dolder, J.; Bancroft, G. N.; Sikavitsas, V. I.; Spauwen, P.H. M.; Mikos, A. G.; Jansen, J. A. Tissue Eng. 2003, 9, 505−515.(2) Burdick, J. A.; Prestwich, G. D. Adv. Mater. (Weinheim, Ger.)2011, 23, H41−56.(3) Calarco, A.; Petillo, O.; Bosetti, M.; Torpedine, A.; Cannas, M.;Perrone, L.; Galderisi, U.; Melone, M. A. B.; Peluso, G. J. Cell. Biochem.2010, 110, 903−909.(4) Crouzier, T.; Boudou, T.; Picart, C. Curr. Opin. Colloid InterfaceSci. 2010, 15, 417−426.(5) Rinaudo, M. Polym. Int. 2008, 57, 397−430.(6) Bansil, R.; Stanley, E.; LaMont, J. T. Annu. Rev. Physiol. 1995, 57,635−657.
(7) Tanaka, S.; Podolsky, D. K.; Engel, E.; Guth, P. H.; Kaunitz, J. D.Am. J. Physiol. 1997, 272, G1473−1480.(8) Aknin, M.-L. R.; Berry, M.; Dick, A. D.; Khan-Lim, D. Cell TissueRes. 2004, 318, 545−551.(9) Linden, S. K.; Sutton, P.; Karlsson, N. G.; Korolik, V.; McGuckin,M. A. Mucosal Immunol. 2008, 1, 183−197.(10) Bansil, R.; Turner, B. S. Curr. Opin. Colloid Interface Sci. 2006,11, 164−170.(11) Svensson, O.; Arnebrant, T. Curr. Opin. Colloid Interface Sci.2010, 15, 395−405.(12) Bushnak, I.; Labeed, F.; Sear, R.; Keddie, J. Biofouling 2010, 26,387−397.(13) Shi, L.; Ardehali, R.; Caldwell, K. D.; Valint, P. Colloids Surf., B2000, 17, 229−239.(14) Sandberg, T.; Carlsson, J.; Karlsson Ott, M. J. Mater. Sci.: Mater.Med. 2008, 20, 621−631.(15) Sandberg, T.; Karlsson Ott, M.; Carlsson, J.; Feiler, A.; Caldwell,K. D. J. Biomed. Mater. Res. 2009, 91A, 773−785.(16) Berg, M. C.; Zhai, L.; Cohen, R. E.; Rubner, M. F.Biomacromolecules 2006, 7, 357−364.(17) Chuang, H. F.; Smith, R. C.; Hammond, P. T. Biomacromole-cuules 2008, 9, 1660−1668.(18) Crouzier, T.; Ren, K.; Nicolas, C.; Roy, C.; Picart, C. Small2009, 5, 598−608.(19) Shah, N. J.; Macdonald, M. L.; Beben, Y. M.; Padera, R. F.;Samuel, R. E.; Hammond, P. T. Biomaterials 2011, 32, 6183−6193.(20) Svensson, O.; Lindh, L.; Cardenas, M.; Arnebrant, T. J. ColloidInterface Sci. 2006, 299, 608−616.(21) Dedinaite, A.; Lundin, M.; Macakova, L.; Auletta, T. Langmuir2011, 21, 9502−9509.(22) Vreuls, C.; Zocchi, G.; Garitte, G.; Archambeau, C.; Martial, J.;Van de Weerdt, C. Biofouling 2010, 26, 645−656.(23) Lindh, L.; Svendsen, I. E.; Svensson, O.; Cardenas, M.;Arnebrant, T. J. Colloid Interface Sci. 2007, 310, 74−82.(24) Halthur, T. J.; Arnebrant, T.; Macakova, L.; Feiler, A. Langmuir2010, 26, 4901−4908.(25) Boudou, T.; Crouzier, T.; Ren, K.; Blin, G.; Picart, C. Adv.Mater. 2010, 22, 441−467.(26) Hoogeveen, N. G.; Cohen Stuart, M. A.; Fleer, G. J.; Bohmer,M. R. Langmuir 2011, 12, 3675−3681.(27) Richert, L.; Boulmedais, F.; Lavalle, P.; Mutterer, J.; Ferreux, E.;Decher, G.; Schaaf, P.; Voegel, J.-C.; Picart, C. Biomacromolecules2011, 5, 284−294.(28) Stockton, W.; Rubner, M. F. Macromolecules 1997, 30, 2717−2725.(29) Such, G. K.; Quinn, J. F.; Quinn, A.; Tjipto, E.; Caruso, F. J. Am.Chem. Soc. 2011, 128, 9318−9319.(30) Cassier, T.; Lowack, K.; Decher, G. Supramol. Sci. 1998, 5, 309−315.(31) Bourdillon, C.; Demaille, C.; Moiroux, J.; Saveant, J.-M. J. Am.Chem. Soc. 1994, 116, 10328−10329.(32) Sato, K.; Imoto, Y.; Sugama, J.; Seki, S.; Inoue, H.; Odagiri, T.;Hoshi, T.; Anzai, J. Langmuir 2005, 21, 797−799.(33) Sato, K.; Kodama, D.; Endo, Y.; Anzai, J. J. Nanosci. Nanotechnol.2009, 9, 386−390.(34) Anzai, J.; Kobayashi, Y. Langmuir 2000, 16, 2851−2856.(35) Sato, K.; Imoto, Y.; Sugama, J.; Seki, S.; Inoue, H.; Odagiri, T.;Anzai, J. Anal. Sci. 2004, 20, 1247−1248.(36) Schwefel, D.; Maierhofer, C.; Beck, J. G.; Seeberger, S.;Diederichs, K.; Moller, H. M.; Welte, W.; Wittmann, V. J. Am. Chem.Soc. 2010, 132, 8704−8719.(37) Celli, J.; Gregor, B.; Turner, B.; Afdhal, N. H.; Bansil, R.;Erramilli, S. Biomacromolecules 2005, 6, 1329−1333.(38) Caruso, F.; Furlong, D. N.; Ariga, K.; Ichinose, I.; Kunitake, T.Langmuir 1998, 14, 4559−4565.(39) Lavalle, P.; Gergely, C.; Cuisinier, F. J. G.; Decher, G.; Schaaf,P.; Voegel, J. C.; Picart, C. Macromolecules 2002, 35, 4458−4465.(40) Voinova, M. V.; Rodahl, M.; Jonson, M.; Kasemo, B. Phys. Scr.1999, 59, 391.
Biomacromolecules Article
dx.doi.org/10.1021/bm301222f | Biomacromolecules 2012, 13, 3401−34083407

(41) Weber, N.; Wendel, H. P.; Kohn, J. J. Biomed. Mater. Res., Part A2005, 72A, 420−427.(42) Lundin, M.; Sandberg, T.; Caldwell, K. D.; Blomberg, E. J.Colloid Interface Sci. 2009, 336, 30−39.(43) Iijima, M.; Yoshimura, M.; Tsuchiya, T.; Tsukada, M.; Ichikawa,H.; Fukumori, Y.; Kamiya, H. Langmuir 2008, 24, 3987−3992.(44) McAloney, R. A.; Sinyor, M.; Dudnik, V.; Goh, M. C. Langmuir2001, 17, 6655−6663.(45) Laugel, N.; Betscha, C.; Winterhalter, M.; Voegel, J.-C.; Schaaf,P.; Ball, V. J. Phys. Chem. B 2006, 110, 19433−19449.(46) Picart, C.; Mutterer, J.; Richert, L.; Luo, Y.; Prestwich, G. D.;Schaaf, P.; Voegel, J.-C.; Lavalle, P. Proc. Natl. Acad. Sci. U. S. A. 2002,99, 12531−12535.(47) Haberska, K.; Svensson, O.; Shleev, S.; Lindh, L.; Arnebrant, T.;Ruzgas, T. Talanta 2008, 76, 1159−1164.(48) Crouzier, T.; Fourel, L.; Boudou, T.; Albiges-Rizo, C.; Picart, C.Adv. Mater. 2011, 23, H111−H118.(49) Zahn, R.; Thomasson, E.; Guillaume-Gentil, O.; Voros, J.;Zambelli, T. Biomaterials 2012, 33, 3421−3427.(50) Cho, C.; Zacharia, N. S. Langmuir 2011, 841−848.(51) Kharlampieva, E.; Sukhishvili, S. A. Langmuir 2003, 19, 1235−1243.(52) Wright, C. S.; Kellogg, G. E. Protein Sci. 1996, 5, 1466−1476.(53) Bies, C.; Lehr, C.-M.; Woodley, J. F. Adv. Drug Delivery Rev.2004, 56, 425−435.(54) Lehr, C. M. J. Controlled Release 2000, 65, 19−29.(55) Ravaine, V.; Ancla, C.; Catargi, B. J. Controlled Release 2008,132, 2−11.(56) Murphy, M. S. Nutrition (N. Y., NY, U. S.) 1998, 14, 771−774.(57) Roland, S. Trends Biochem. Sci. 1985, 10, 357−360.(58) Schwarz, R. E.; Wojciechowicz, D. C.; Picon, A. I.; Schwarz, M.A.; Paty, P. B. Br. J. Cancer 1999, 80, 1754−1762.(59) Dalla Pellegrina, C.; Matucci, A.; Zoccatelli, G.; Rizzi, C.;Vincenzi, S.; Veneri, G.; Andrighetto, G.; Peruffo, A. D. B.; Chignola,R. Toxicol. In Vitro 2004, 18, 821−827.(60) Blin, G.; Lablack, N.; Louis-Tisserand, M.; Nicolas, C.; Picart,C.; Puceat, M. Biomaterials 2010, 31, 1742−1750.
Biomacromolecules Article
dx.doi.org/10.1021/bm301222f | Biomacromolecules 2012, 13, 3401−34083408




















