
Cerium(IV)-mediated oxidation of flavonol with relevance to flavonol 2,4-dioxygenase. Direct...
7
Cerium(IV)-mediated oxidation of flavonol with relevance to flavonol 2,4-dioxygenase. Direct evidence for spin delocalization in the flavonoxy radical Jo ´ zsef Kaizer a , Ildiko ´ Ganszky a , Ga ´bor Speier a, * , Antal Rockenbauer b , La ´szlo ´ Korecz b , Michel Giorgi c , Marius Re ´glier c , Serge Antonczak d a Department of Organic Chemistry, University of Pannonia, 8201 Veszpre ´m, Hungary b Hungarian Academy of Sciences, Chemical Research Center, 1525 Budapest, Hungary c Laboratoire de Cristallochimie et Laboratoire de Bioinorganique Structurale, Universite ´ Paul Cezanne, Aix-Marseille III, Faculte ´ des Sciences et Techniques de Saint Jero ˆme, Service 432, Avenue Escadrille Normandie-Niemen, 13397 Marseille Cedex 20, France d Laboratoire LCMBA, Equipe Chimiome ´trie et Mode ´lisation, Faculte ´ des Sciences, Universite ´ de Nice Sophia-Antipolis, F 06108 Nice Cedex 2, France Received 7 February 2007; accepted 12 February 2007 Available online 21 February 2007 Abstract The cerium(IV)-mediated oxidation of 3-hydroxy-4 0 -methylflavone (1) proceeds by H-atom abstraction forming the flavonoxy radical (7), and the subsequent combination of its resonance forms leads to the 3-hydroxy-4 0 -methylflavone dehydro dimer (9). The above system serves as direct evidence for the intermediacy of the flavonoxy radical, its spin delocalization, and also indirect evidence for valence tau- tomerism as a key step on the substrate activation both in the quercetinase and its biomimic model system. Ó 2007 Elsevier Inc. All rights reserved. Keywords: Valence tautomerism; CAN; Flavonoxy radical; Flavonol 2,4-dioxygenase 1. Introduction Flavonols are widely distributed in vascular plants [1,2], and flavonoids form active constituents of a number of her- bal and traditional medicines [3]. Quercetin which is one of the most abundant natural flavonoids in edible fruits and vegetables has been recognized as antioxidant, scavenging dioxygen-derived radicals [4–10]. The antioxidant activity of flavonoids is well known, although the mechanism(s) by which they act in vivo is not clarified. In a clinical study, the risk of coronary heart disease was shown to correlate inversely with the extent of flavonoid intake [11]. Flavonols are readily degraded by microorganisms. The copper- and iron-containing flavonol 2,4-dioxygenases (quercetinases) catalyze the oxidative degradation of flavo- nol(s) to depside(s) (phenolic carboxylic acid ester(s)) and carbon monoxide by fungi like Aspergillus flavus [12], Aspergillus niger [13], Aspergillus japonicus [14] and the protein YxaG from Bacillus subtilis [15] (Eq. (1)). O O OH O 2 -CO CO 2 H O O 1 2 a) R = H; b) R = Me R R ð1Þ Since the reaction catalyzed by flavonol 2,4-dioxygenase is a spin-forbidden process, it presents the mechanistically intriguing problem of how triplet dioxygen and/or singlet flavonol are activated during the enzymatic process. In pre- vious studies the intermediate formation of binary (ES) and ternary species (ESO 2 ) have been proposed at the oxygen- ation by flavonol 2,4-dioxygenase. One concerns the 0162-0134/$ - see front matter Ó 2007 Elsevier Inc. All rights reserved. doi:10.1016/j.jinorgbio.2007.02.005 * Corresponding author. Tel.: +36 88 624 657; fax: +36 88 624 469. E-mail address: [email protected] (G. Speier). www.elsevier.com/locate/jinorgbio Journal of Inorganic Biochemistry 101 (2007) 893–899 JOURNAL OF Inorganic Biochemistry
-
Upload
uni-pannon -
Category
Documents
-
view
3 -
download
0
Transcript of Cerium(IV)-mediated oxidation of flavonol with relevance to flavonol 2,4-dioxygenase. Direct...

JOURNAL OF
www.elsevier.com/locate/jinorgbio
Journal of Inorganic Biochemistry 101 (2007) 893–899
InorganicBiochemistry
Cerium(IV)-mediated oxidation of flavonol with relevanceto flavonol 2,4-dioxygenase. Direct evidence for spin delocalization
in the flavonoxy radical
Jozsef Kaizer a, Ildiko Ganszky a, Gabor Speier a,*, Antal Rockenbauer b, Laszlo Korecz b,Michel Giorgi c, Marius Reglier c, Serge Antonczak d
a Department of Organic Chemistry, University of Pannonia, 8201 Veszprem, Hungaryb Hungarian Academy of Sciences, Chemical Research Center, 1525 Budapest, Hungary
c Laboratoire de Cristallochimie et Laboratoire de Bioinorganique Structurale, Universite Paul Cezanne, Aix-Marseille III,
Faculte des Sciences et Techniques de Saint Jerome, Service 432, Avenue Escadrille Normandie-Niemen, 13397 Marseille Cedex 20, Franced Laboratoire LCMBA, Equipe Chimiometrie et Modelisation, Faculte des Sciences, Universite de Nice Sophia-Antipolis, F 06108 Nice Cedex 2, France
Received 7 February 2007; accepted 12 February 2007Available online 21 February 2007
Abstract
The cerium(IV)-mediated oxidation of 3-hydroxy-4 0-methylflavone (1) proceeds by H-atom abstraction forming the flavonoxy radical(7), and the subsequent combination of its resonance forms leads to the 3-hydroxy-4 0-methylflavone dehydro dimer (9). The above systemserves as direct evidence for the intermediacy of the flavonoxy radical, its spin delocalization, and also indirect evidence for valence tau-tomerism as a key step on the substrate activation both in the quercetinase and its biomimic model system.� 2007 Elsevier Inc. All rights reserved.
Keywords: Valence tautomerism; CAN; Flavonoxy radical; Flavonol 2,4-dioxygenase
1. Introduction
Flavonols are widely distributed in vascular plants [1,2],and flavonoids form active constituents of a number of her-bal and traditional medicines [3]. Quercetin which is one ofthe most abundant natural flavonoids in edible fruits andvegetables has been recognized as antioxidant, scavengingdioxygen-derived radicals [4–10]. The antioxidant activityof flavonoids is well known, although the mechanism(s)by which they act in vivo is not clarified. In a clinical study,the risk of coronary heart disease was shown to correlateinversely with the extent of flavonoid intake [11].
Flavonols are readily degraded by microorganisms. Thecopper- and iron-containing flavonol 2,4-dioxygenases(quercetinases) catalyze the oxidative degradation of flavo-
0162-0134/$ - see front matter � 2007 Elsevier Inc. All rights reserved.
doi:10.1016/j.jinorgbio.2007.02.005
* Corresponding author. Tel.: +36 88 624 657; fax: +36 88 624 469.E-mail address: [email protected] (G. Speier).
nol(s) to depside(s) (phenolic carboxylic acid ester(s)) andcarbon monoxide by fungi like Aspergillus flavus [12],Aspergillus niger [13], Aspergillus japonicus [14] and theprotein YxaG from Bacillus subtilis [15] (Eq. (1)).
O
OOH
O2
-CO CO2HO
O
1 2a) R = H; b) R = Me
R R
ð1Þ
Since the reaction catalyzed by flavonol 2,4-dioxygenaseis a spin-forbidden process, it presents the mechanisticallyintriguing problem of how triplet dioxygen and/or singletflavonol are activated during the enzymatic process. In pre-vious studies the intermediate formation of binary (ES) andternary species (ESO2) have been proposed at the oxygen-ation by flavonol 2,4-dioxygenase. One concerns the

894 J. Kaizer et al. / Journal of Inorganic Biochemistry 101 (2007) 893–899
change of the character of the flavonolate ligand, i.e. howthe flavonolate ligand is activated for oxygenation: whetherthe flavonolate ligand can be regarded as a radical asshown in (4) due to valence tautomerism (VT) or has ananionic character as in (6) (Fig. 1). The other concern ishow triplet dioxygen is activated to form reactive ESO2
species. Reaction of flavonoxy radical with the biradicaldioxygen, or direct electron transfer from a deprotonatedsubstrate to molecular oxygen to form a caged radical pairmay be discussed as hypothetical mechanisms. The mecha-nism of the above systems is different, however, substrate-derived radicals seem to be the key intermediates in thesereactions. Many reports have appeared for the formationand structures of ES from both enzyme and model studies[16–18], but until now there has been only few mechanisticevidence for the formation of ESO2 species.
Model studies on flavonol 2,4-dioxygenase support amechanism of substrate activation [19–21]. The spin-for-biddance of the reaction can, nonetheless, be circumventedby allowing the existence of a flavonoxy radical-CuI or aflavonoxy radical-FeII valence tautomer, arising from theflow of one electron from the flavonolate to the metal cen-ter, similarly to what was observed in the case of the FeIII-and CuII-dependent intradiol-type catechol dioxygenases(Eq. (2)) [22–24]. Early models for catechol oxidation bythe catechol dioxygenase enzymes contained shifts incharge distribution involving FeIII(Cat) and FeII(SQ) [25],and CuII(Cat) and CuI(SQ) redox isomers too [26].
OO
M(n+1)+LnO
O
Mn+Ln
M(Cat) M(SQ)
VT
ð2Þ
Transition metal complexes containing first row metalions chelated by catecholate (Cat) and semiquinonate(SQ) ligands have been found to have localized electronicstructures with quinone ligands bonded in both Cat andSQ electronic forms. Equilibria occurring betweenMn+(SQ) and M(n+1)+(Cat) redox isomers have beenobserved in solution and in the solid state for complexesof Co, Mn, Fe, and Cu [27,28]. This effect has been consid-
O
OO
R
M(n+1)+Ln
O
OO
Mn+Ln
O
O
R
O
OOO M
4
6
M = Fe, Cu
O2
VT
3
7
Fig. 1. Probable modes of coordination of flavonol in a flavonol–meta
ered as an example of valence tautomerism (VT) [29,30].The radical centers of these complexes can react easily withthe biradical dioxygen.
The pyramidalization of the flavonolate C2 atom,observed and theoretically confirmed in the ES complex,is consistent with the population of the unpaired electronon this atom, and further analysis of the shape of the activesite cavity shows that room is available for a dioxygenmolecule to directly attack the flavonolate C2 atom[14,31]. Dioxygen binding would then produce the depsideand CO via the formation of peroxidic intermediates.Enzyme-like products were obtained by the oxygenationof flavonols initiated by stable free radicals such as2,2,6,6-tetramethyl-1-piperidinyloxyl (TEMPO), 1,1-diphe-nyl-2-picrylhydrazyl (DPPH) or Galvinoxyl also as foundearlier [32,33]. However, there was no direct evidence forthe formation of flavonoxy radical.
In this study we present the oxidative dimerization of 3-hydroxy-4 0-methylflavone mediated by CeIV salts and givedirect evidence for the presence of the flavonoxy radicalas a key intermediate and its resonance forms exhibitingvery similar spin density on the O3 and also on C2 atomsnecessary for the oxygenation process and also for itsdimerization through C,O-coupling.
2. Experimental
2.1. Materials
All manipulations were performed under a pure nitro-gen or argon atmosphere unless otherwise stated, usingstandard Schlenck-type inert gas techniques. Solvents usedfor the reactions were purified by literature methods [34]and stored under argon. All other chemicals were commer-cial products and were used as received without furtherpurification.
2.2. Analytical measurements
Infrared spectra were recorded on a Specord 75 IR (CarlZeiss) spectrophotometer using samples mulled in Nujol
R
R
M
O
OO
R
Mn+Ln
O2O O
O2
O
OO
R
Mn+LnO O
5
8
l binary intermediate and their proposed reactions with dioxygen.

J. Kaizer et al. / Journal of Inorganic Biochemistry 101 (2007) 893–899 895
between KBr plates or in KBr pellets. UV–visible spectrawere recorded on a Shimadzu UV-160 spectrophotometerusing quartz cells. The EPR spectra were recorded at roomtemperature by an X-band Bruker Elexsys 500 spectrome-ter with 100 kHz modulation frequency and 2 G modula-tion amplitude, the microwave power was 25 mW. 1Hand 13C NMR spectra were performed on a Varian Unity300 instrument. Microanalyses were done by the Microan-alytical Service of the University of Pannonia.
2.3. Cerium(IV) mediated oxidation of
3-hydroxy-4 0-methylflavone
A suspension of 3-hydroxy-4 0-methylflavone (0.252 g,1 mmol) and (NH4)2CeIV(NO3)6 (1 mmol) in dry CH2Cl2or toluene (10 mL) was stirred under argon for 3 h. Afterfiltrating the cerium salt the solvent was removed underreduced pressure and the recrystallization of the residuefrom diethylether resulted in the 3-hydroxy-4 0-methylflav-one dehydro dimer (9) as yellow crystals suitable for X-ray analysis [0.138 g, 55% (in CH2Cl2) and 0.15 g, 58%(in toluene), respectively]. mp 144–146 �C. 1H NMR(CDCl3) d 8.09 (dd: doublet of doublets, 2H), 6.95–7.73(m: multiplet, 12H), 6.87 (d: doublet, 2H), 2.36 (s: singlet,3H), 2.29 (s, 3H). 13C NMR (CDCl3) d 178.7, 177.4,172.7, 159.8, 158, 155, 154.6, 141.6, 140, 137.3, 134.8,133.7, 132.1, 129.3, 129.1, 129, 128.9, 128.5, 128.2, 127.5,127.1, 126, 124.8, 122.8, 122.7, 121.4, 118.1, 117.7, 105,23.6, 21.5, 21.1. IR (KBr) 3031, 2958, 2917, 2851, 1736,1699, 1610, 1462, 1385, 1303, 1197, 1152, 1111, 1070,1037, 1013, 915, 817, 747, 665 cm�1. UV–visible (CH2Cl2)kmax 305 (log e 4.21), 247 nm (4.32). Anal. Calcd forC32H22O6: C, 76.48; H, 4.41. Found: C, 75.98; H, 4.38.
The title compound (9) was also prepared by using [(n-Bu)4N]2Ce(NO3)6 in CH2Cl2 under the same conditionsmentioned above (0.18 g, 70%).
Fig. 2. The molecular structure of 9. Atoms are presented as thermalellipsoids at 50% probability.
2.4. X-ray structure determination of (9)
X-ray quality single-crystals were obtained by slow evap-oration of a diethyletheral solution of 9. Measurement wasmade on a Nonius CCD diffractometer with graphite mono-chromatized Mo–Ka radiation (k = 0.71073 A). The struc-ture was solved by direct methods and refinement, based onF2, was made by full-matrix least-squares techniques. Crys-
tal data for 9: CHO, 502.50, triclinic, P�1, a = 7.9320(4),b = 11.8910(10), c = 13.2760(10), a = 93.087(4)�, b =89.997(5)�, c = 96.584(5)�, V = 1242.10(15) A3, Z = 2,qcalcd = 1.344 g cm�3, l = 0.093 mm�1, F(000) = 524,hmax = 26.22� (0 6 h 6 9, �14 6 k 6 14, �16 6 l 6 16).Final residuals (for 343 parameters) were R1 = 0.0632 andwR2 = 0.1473 for 4503 reflections with I > 2r(I), GOF =1.152. Residual electron density was 0.230 and�0.233 e A�3. The CIF file with data for 9 has been depos-ited at the Cambridge Crystallographic Data Centre,CCDC No. 270178.
2.5. Computational methods
Density-functional theory (DFT) calculations have beenperformed at the B3LYP[35–37]/6-31(+)G* [38–41] level oftheory, using GAUSSIAN98 program. Such a level ofcomputation has previously been shown to reproduce fairlywell the structural parameters of similar systems [42].
3. Results and discussion
3.1. Cerium(IV) mediated oxidation of
3-hydroxy-4 0-methylflavone
3-Hydroxy-4 0-methylflavone (1b) reacts with(NH4)2CeIV(NO3)6 (CAN) or ((n-Bu)4N)2CeIV(NO3)6 inCH2Cl2 or toluene solution at room temperature underargon atmosphere to give a yellow crystalline compound(yields: 50–70%) for which elemental analysis and the 1Hand 13C NMR measurements are in agreement with the3-hydroxy-4 0-methylflavone dehydro dimer (9) [32,33].The infrared spectrum of this dehydro dimer exhibits car-bonyl peaks at 1736 and 1699 cm�1, as well as a strongabsorption at 1610 cm�1 indicating the presence of the fla-vone system, since this absorption is also found in flavonol.The X-ray single-crystal structure of 9 was determined, andits computer-generated drawing is shown in Fig. 2.
The EPR spectra of reaction mixture (Fig. 3) were inves-tigated, but only a signal of low intensity was detected afterrepeated scanning, that indicates a low stationary concen-tration. The signal could be decomposed into a broad fea-ture and a resolved triplet, where the shape of triplet linesindicates two slightly different major couplings (8.1 G and6.4 G) according to the best fit obtained by the automaticparameter adjustment process in the computer simulation[43]. The shape of external lines could be well reproduced
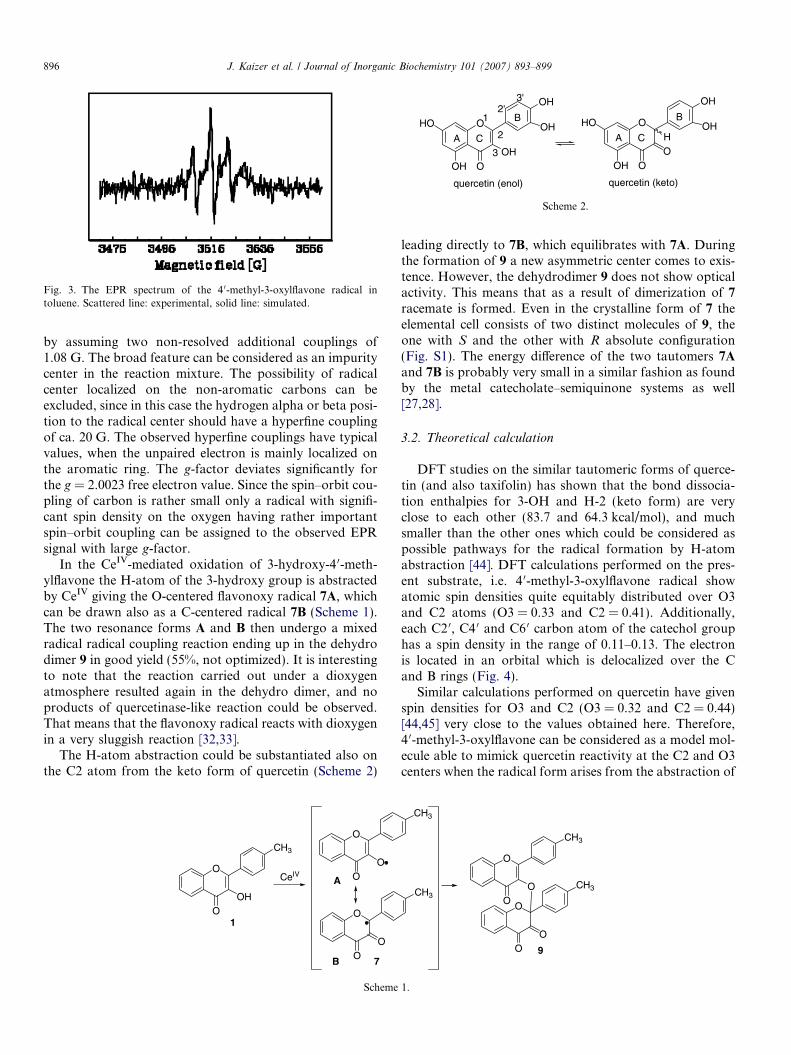
Fig. 3. The EPR spectrum of the 4 0-methyl-3-oxylflavone radical intoluene. Scattered line: experimental, solid line: simulated.
O
OOH
OH
OH
OH
HO
A C
B1
2
3
2'3'
O
OO
OH
OH
OH
HO
A C
B
H
quercetin (enol) quercetin (keto)
Scheme 2.
896 J. Kaizer et al. / Journal of Inorganic Biochemistry 101 (2007) 893–899
by assuming two non-resolved additional couplings of1.08 G. The broad feature can be considered as an impuritycenter in the reaction mixture. The possibility of radicalcenter localized on the non-aromatic carbons can beexcluded, since in this case the hydrogen alpha or beta posi-tion to the radical center should have a hyperfine couplingof ca. 20 G. The observed hyperfine couplings have typicalvalues, when the unpaired electron is mainly localized onthe aromatic ring. The g-factor deviates significantly forthe g = 2.0023 free electron value. Since the spin–orbit cou-pling of carbon is rather small only a radical with signifi-cant spin density on the oxygen having rather importantspin–orbit coupling can be assigned to the observed EPRsignal with large g-factor.
In the CeIV-mediated oxidation of 3-hydroxy-4 0-meth-ylflavone the H-atom of the 3-hydroxy group is abstractedby CeIV giving the O-centered flavonoxy radical 7A, whichcan be drawn also as a C-centered radical 7B (Scheme 1).The two resonance forms A and B then undergo a mixedradical radical coupling reaction ending up in the dehydrodimer 9 in good yield (55%, not optimized). It is interestingto note that the reaction carried out under a dioxygenatmosphere resulted again in the dehydro dimer, and noproducts of quercetinase-like reaction could be observed.That means that the flavonoxy radical reacts with dioxygenin a very sluggish reaction [32,33].
The H-atom abstraction could be substantiated also onthe C2 atom from the keto form of quercetin (Scheme 2)
O
OOH
CH3
O
OO
O
OO
A
B
CeIV
1
7
Scheme
leading directly to 7B, which equilibrates with 7A. Duringthe formation of 9 a new asymmetric center comes to exis-tence. However, the dehydrodimer 9 does not show opticalactivity. This means that as a result of dimerization of 7
racemate is formed. Even in the crystalline form of 7 theelemental cell consists of two distinct molecules of 9, theone with S and the other with R absolute configuration(Fig. S1). The energy difference of the two tautomers 7A
and 7B is probably very small in a similar fashion as foundby the metal catecholate–semiquinone systems as well[27,28].
3.2. Theoretical calculation
DFT studies on the similar tautomeric forms of querce-tin (and also taxifolin) has shown that the bond dissocia-tion enthalpies for 3-OH and H-2 (keto form) are veryclose to each other (83.7 and 64.3 kcal/mol), and muchsmaller than the other ones which could be considered aspossible pathways for the radical formation by H-atomabstraction [44]. DFT calculations performed on the pres-ent substrate, i.e. 4 0-methyl-3-oxylflavone radical showatomic spin densities quite equitably distributed over O3and C2 atoms (O3 = 0.33 and C2 = 0.41). Additionally,each C2 0, C4 0 and C6 0 carbon atom of the catechol grouphas a spin density in the range of 0.11–0.13. The electronis located in an orbital which is delocalized over the Cand B rings (Fig. 4).
Similar calculations performed on quercetin have givenspin densities for O3 and C2 (O3 = 0.32 and C2 = 0.44)[44,45] very close to the values obtained here. Therefore,4 0-methyl-3-oxylflavone can be considered as a model mol-ecule able to mimick quercetin reactivity at the C2 and O3centers when the radical form arises from the abstraction of
CH3
CH3
O
OO
CH3
9
O
OO
CH3
1.

Fig. 4. (a) A highest occupied molecular orbital (HOMO) and (b) distribution of spin density for the 4 0-methyl-3-oxylflavone radical.
Fig. 5. Evolution of O3 and C2 atomic spin densities as well as totalenergy as a function of the angle of pyramidalization at the C2 carbonatom of the 4 0-methyl-3-oxylflavone radical.
J. Kaizer et al. / Journal of Inorganic Biochemistry 101 (2007) 893–899 897
a proton and an electron at the O3 oxygen atom. The sametype of analysis have been carried out on taxifolin (Scheme3) where the C2–C3 double bond is saturated, the corre-sponding spin densities are O3 = 0.81 and C2 = 0.07 indi-cating that p-electron delocalization over the doublebond in quercetin is very high.
The consecutive radical–radical combination of the mes-omeric forms of 7 may result in three products. Why onlyproduct 9 is formed may be reasonable explained that theC2–C2 coupling is unfavored due steric hindrance andthe O3–O3 coupling leads, compared with the C2–O3coupling, to the energetically less stable compound (bondenergies: C–O = 79 kcal/mol; O–O = 34 kcal/mol) [46].Moreover, DFT calculations performed to characterizethese two last forms failed in obtaining an optimized prod-uct arising from the O3–O3 coupling. Indeed, despite deepexploration of the potential energy surface, the O–O dis-tance remains large (about 2.5 A). Furthermore, the forma-tion of adduct through C2–O3 coupling take benefits ofatomic spin density evolution through pyramidalizationat the C2 carbon atom (Fig. 5).
This pyramidalization motion is associated to a very lowfrequency, and thus to a slow deformation of the molecule,and does not cost much energy when a limited angle is con-sidered. Nevertheless, this intrinsic movement of the mole-cule is accompanied by an increase of the atomic spindensity at the C2 carbon atom while the same entitydecreases at the O3 oxygen atom, rendering the additionon C2 more likely to occur.
The results found now give unquestionable firm evi-dence for spin delocalization involving mainly the O3 andC2 atoms in the flavonoxy radical 7, and a high spin den-sity on C2, which probably plays an essential role in thecopper- and iron-containing quercetin 2,3-dioxygenase
O
OO
OH
OH
OH
HO
quercetin
0.44
0.32
O
OOH
HO
taxifolin
0.
Scheme
enzymes as well. Due to valence isomerism in the (flavono-lato)copper(II) mimics (Figs. 1, 3) (flavonoxy)copper(I) (4)is formed which contains two sites to be able to react with3O2. The reaction may lead to a (flavonoxy)(superoxo)cop-per(II) complex (10) or to an organic peroxide moiety (5),from which the former is more favored (Figs. 1, 6). In aconsecutive intramolecular reaction of 10 involving theC2 site with a high spin density leads to a peroxo species(11) and later on to an endoperoxide (12). This breaks thendown to the depside complex (13) (Fig. 6). These resultsseem to suggest that valence tautomerism in the (flavono-lato)copper(II) complexes (and in the enzyme itself) is nec-essary to generate CuI, making the complex able to reactwith triplet dioxygen. While the data obtained suggests thata direct attack of 3O2 on the organic radical part of thecomplex is less probable (slow reaction of 7 with 3O2),the flavonolate ligand is rich enough on energy to be ableto give up one electron to 3O2 yielding 7 and superoxideanion representing an alternative pathway for theenzyme-like reaction. We belive however, that the reactionpath, involving CuI as a result of valence tautomerism, andits reaction with 3O2 is dominant and that is the reason that
O
OH
OH07
0.81
O
OO
CH3
4'-methyl-flavonol
0.41
0.33
3.

O
OO
R
Mn+Ln4
O
OO
R
M(n+1)+(O2)Ln
O2
10
O
O
O
O
R
M(n+1)+Ln13
O
O
O
R
M(n+1)+Ln
O
O
12
O
OO
R
M(n+1)+Ln
O
O
11
Fig. 6. Proposed mechanism for the flavonol 2,4-dioxygenase.
898 J. Kaizer et al. / Journal of Inorganic Biochemistry 101 (2007) 893–899
this type of enzymes all have a metal ion at their active site.The data obtained here may give a better insight into thepossible mechanism of flavonol 2,4-dioxygenases.
Acknowledgements
We thank the Hungarian National Research Fund(OTKA T 043414), COST, Budaconsum Ltd, and UlrichTrade Ltd for financial support of this work.
Appendix A. Supplementary data
Tables giving crystal data and details of the structuredetermination, atomic co-ordinates, anisotropic thermalparameters, hydrogen atom locations, and bond lengthsand angles for 9, and the figure of the elemental cell of 9.Supplementary data associated with this article can befound, in the online version, at doi:10.1016/j.jinorgbio.2007.02.005.
References
[1] E. Wollenweber, in: J.B. Harborne, T.J. Mabry (Eds.), Flavonoids:Advances in Research, Chapman & Hall, London, New York, 1982,pp. 259–335.
[2] J.B. Harborne, C.A. Williams, in: J.B. Harborne, T.J. Mabry (Eds.),Flavonoids: Advances in Research, Chapman & Hall, London,New York, 1982, pp. 337–386.
[3] E. Wollenweber, Prog. Clin. Biol. Res. 2850 (1988) 45–51.[4] Y. Sorata, U. Takaham, M. Kimura, Biochim. Biophys. Acta 799
(1984) 313–317.[5] W.B. Erben-Russ, M. Saran, Int. J. Rad. Biol. 52 (1987) 393–412.[6] S.R. Husain, J. Cillard, P. Cillard, Phytochemistry 26 (1987) 2489–
2491.[7] Y.T. Chen, R.T. Zheng, Z.J. Jia, Y. Ju, Free Rad. Biol. Med. 9 (1990)
19–21.
[8] G. Sichel, C. Corsaro, M. Scalia, A.J. Dibilo, R.P. Bonomo, FreeRad. Biol. Med. 11 (1991) 1–8.
[9] P.-F. Wang, R.-L. Zheng, Chem. Phys. Lipids 63 (1992) 37–40.[10] J. Terao, M. Piskula, Y. Qing, Arch. Biochem. Biophys. 308 (1994)
278–284.[11] M.G.L. Hertog, E.J.M. Feskens, P.C.H. Hollman, M.B. Katan, D.
Kromhout, Lancet 342 (1993) 1007–1011.[12] T. Oka, F.J. Simpson, Biochem. Biophys. Res. Commun. 43 (1971)
1–5.[13] H.-K. Hund, J. Breuer, F. Lingens, J. Huttermann, R. Kappl, S.
Fetzner, Eur. J. Biochem. 263 (1999) 871–878.[14] F. Fusetti, K.H. Schroter, R.A. Steiner, P.I. van Nort, T. Pijning,
H.J. Rozeboom, K.H. Kalk, M.R. Egmond, B.W. Dijkstra, Structure10 (2002) 259–268.
[15] B. Gopal, L.L. Madan, S.F. Betz, A.A. Kossiakoff, Biochemistry 44(2005) 193–201.
[16] E. Makasheva, N.T. Golovkina, Zh. Obsch. Khim. 43 (1973) 1640–1646.
[17] M. Thomson, C.R. Williams, Anal. Chim. Acta 85 (1976) 375–381.[18] K. Takamura, M. Ito, Chem. Pharm. Bull. 25 (1977) 3218–3225.[19] E. Balogh-Hergovich, J. Kaizer, G. Speier, G. Huttner, A. Jacobi,
Inorg. Chem. 39 (2000) 4224–4229.[20] E. Balogh-Hergovich, J. Kaizer, J. Pap, G. Speier, G. Huttner, L.
Zsolnai, Eur. J. Inorg. Chem. (2002) 2287–2295.[21] J. Kaizer, J. Pap, G. Speier, L. Parkanyi, Eur. J. Inorg. Chem. (2004)
2253–2259.[22] M. Costas, M.P. Mehn, M.P. Jensen, L. Que Jr., Chem. Rev. 104
(2004) 939–986.[23] G. Speier, Z. Tyeklar, L. Szabo, P. Toth, C.G. Pierpont, D.N.
Hendrickson, in: D.H.R. Barton, A.E. Martell, D.T. Sawyer (Eds.),The Activation of Dioxygen and Homogeneous Catalytic Oxidation,Plenum Press, New York, 1993, pp. 423–436.
[24] G. Speier, New J. Chem. 18 (1994) 143–147.[25] D.D. Cox, L. Que Jr., J. Am. Chem. Soc. 110 (1988) 8085–8092.[26] G. Speier, Z. Tyeklar, P. Toth, E. Speier, S. Tisza, A.M. Whalen, N.
Alkire, C.G. Pierpont, Inorg. Chem. 40 (2001) 5653–5659.[27] C.G. Pierpont, Coordin. Chem. Rev. 216 (2001) 99–125.[28] C.G. Pierpont, C.W. Lange, Prog. Inorg. Chem. 41 (1994) 331–442.[29] A. Caneschi, A. Dei, Angew. Chem., Int. Ed. Engl. 37 (1998) 3005–
3007.[30] E. Evangelio, D. Ruiz-Molina, Eur. J. Inorg. Chem. (2005) 2957–
2971.[31] P.E.M. Segbahn, Inorg. Chem. 43 (2004) 5944–5953.[32] J. Kaizer, G. Speier, J. Mol. Catal. A: Chem. 171 (2001) 33–36.[33] J. Pap, J. Kaizer, G. Speier, React. Kinet. Catal. Lett. 85 (2005) 115–
121.[34] D.D. Perrin, W.L. Armarego, D.R. Perrin, Purification of Laboratory
Chemicals, second ed., Pergamon, New York, 1990.[35] A.D. Becke, Phys. Rev. A. 38 (1988) 3098–3100.[36] A.D. Becke, J. Chem. Phys. 98 (1993) 5648–5652.[37] C. Lee, W. Yang, R.G. Parr, Phys. Rev. B. 37 (1988) 785–789.[38] R. Ditchfield, W.J. Hehre, J.A. Pople, J. Chem. Phys. 54 (1971) 724–
728.[39] P.C. Hariharan, J.A. Pople, Theor. Chim. Acta 28 (1973) 213–222.[40] T. Clark, J. Chandrasekhar, P.V.R. Schleyer, J. Comp. Chem. 4
(1983) 294–301.[41] R. Krishnan, J.S. Binkley, R. Seeger, J.A. Pople, J. Chem. Phys. 72
(1980) 650–654.[42] M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb,
J.R. Cheeseman, V.G. Zakrzewski, J.A. Montgomery Jr., R.E.Stratmann, J.C. Burant, S. Dapprich, J.M. Millam, A.D. Daniels,K.N. Kudin, M.C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi,
R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J.Ochterski, G.A. Petersson, P.Y. Ayala, Q. Cui, K. Morokuma, D.K.Malick, A.D. Rabuck, K. Raghavachari, J.B. Foresman, J. Cioslow-ski, J.V. Ortiz, A.G. Baboul, B.B. Stefanov, G. Liu, A. Liashenko, P.
Piskorz, I. Komaromi, R. Gomperts, R.L. Martin, D.J. Fox, T.Keith, M.A. Al-Laham, C.Y. Peng, A. Nanayakkara, C. Gonzalez,

J. Kaizer et al. / Journal of Inorganic Biochemistry 101 (2007) 893–899 899
M. Challacombe, P.M.W. Gill, B. Johnson, W. Chen, M.W. Wong,
J.L. Andres, C. Gonzalez, M. Head Gordon, E.S. Replogle, J.A.Pople, Gaussian 98, Revision A.7, Gaussian, Inc, Pittsburgh PA,1998.
[43] A. Rockenbauer, L. Korecz, Appl. Magn. Reson. 10 (1996) 29–43.[44] P. Trouillas, P. Marsal, D. Siri, R. Lazzaroni, J.-L. Duroux, Food
Chem. 97 (2006) 679–688.
[45] S. Fiorucci, J. Golebiowski, D. Cabrol-Bass, S. Antonczak, Chem.Phys. Chem. 5 (2004) 1726–1733.
[46] F.A. Carey, R.J. Sundberg, Advanced Organic Chemistry, third ed.Part A: Structure and Mechanism, Plenum Press, New York, 1977,pp. 12–13.


![Synthesis and inhibition study of monoamine oxidase, indoleamine 2,3-dioxygenase and tryptophan 2,3-dioxygenase by 3,8-substituted 5H-indeno[1,2-c]pyridazin-5-one derivatives](https://static.fdokumen.com/doc/165x107/6343bf46fc30a9d0e204e609/synthesis-and-inhibition-study-of-monoamine-oxidase-indoleamine-23-dioxygenase.jpg)








![Ligand Kedge X-ray Absorption Spectroscopy and DFT Calculations on [Fe 3 S 4 ] 0,+ Clusters: Delocalization, Redox, and Effect of the Protein Environment](https://static.fdokumen.com/doc/165x107/6314290dfc260b71020f6e78/ligand-kedge-x-ray-absorption-spectroscopy-and-dft-calculations-on-fe-3-s-4-0.jpg)








