Targeted Exon Skipping to Address “Leaky” Mutations in the Dystrophin Gene
Upload
independentCategory
view
7download
0
Cardiomyopathy in dystrophin-deficient heartsis prevented by expression of a neuronal nitricoxide synthase transgene in the myocardium
Michelle Wehling-Henricks1, Maria C. Jordan3, Kenneth P. Roos3, Bo Deng4
and James G. Tidball1,2,4,*
1Department of Physiological Science, 2Department of Pathology and Laboratory Medicine, 3Department
of Physiology and 4Molecular, Cellular and Integrative Physiology Program, David Geffen School of Medicine,
5833 Life Science Building, University of California, Los Angeles, CA 90095, USA
Received January 26, 2005; Revised April 18, 2005; Accepted May 17, 2005
Null mutation of dystrophin causes the lethal pathology of Duchenne muscular dystrophy (DMD) in whichthere is progressive pathology of skeletal and cardiac muscles. A large proportion of DMD patient deathsare attributable to cardiac dysfunction associated with ventricular fibrosis, arrhythmias and conductionabnormalities, although the relationships between the dystrophin mutation and the cardiac defects areunknown. Here, we tested whether cardiac pathology in dystrophin-deficient mdx mice can be correctedby the elevated production of nitric oxide (NO) by the myocardium. Dystrophin-deficient mdx mice were pro-duced in which there was myocardial expression of a neuronal nitric oxide synthase (nNOS) transgene.Expression of the transgene prevented the progressive ventricular fibrosis of mdx mice and greatly reducedmyocarditis. Electrocardiographs (ECG) attained by radiotelemetry of freely ambulatory mice showed thatmdx mice displayed cardiac abnormalities that are characteristic of DMD patients, including deepQ-waves, diminished S:R ratios, polyphasic R-waves and frequent premature ventricular contractions. Allof these ECG abnormalities in mdx mice were improved or corrected by nNOS transgene expression. Inaddition, defects in mdx cardiac autonomic function, which were reflected by decreased heart rate variability,were significantly reduced by nNOS transgene expression. These findings indicate that increasing NO pro-duction by dystrophic hearts may have therapeutic value.
INTRODUCTION
Cardiomyopathy frequently causes mortality in Duchennemuscular dystrophy (DMD) patients. More than 30% ofDMD deaths reported between 1985 and 1989 resultedfrom cardiac failure (1). This figure is surely increasing aspreviously fatal respiratory deficits are treated with ventilatoryassist devices (2) permitting cardiac involvement to emerge asa more prevalent feature of the disease. Current managementoptions for patients with dystrophin-deficient cardiomyopathyare typically limited to the use of drugs prescribed to relievesymptoms of congestive heart failure (3,4). Unfortunately,DMD patients are largely inactive due to loss of ambulationin their early teens and the increasing severity of the cardio-myopathy can undergo unnoticed, untreated and result in
sudden death. The dystrophic cardiomyopathy also affectsfemale carriers of dystrophin mutations who are generallyspared from the severe skeletal muscle pathology. More than50% of female carriers experience clinically relevant cardiacinvolvement often severe enough to merit heart transplantation(5,6,7).
The DMD cardiomyopathy presents clinically and histologi-cally with characteristic features. Electrocardiography (ECG)of DMD patients shows tall right precordial R-waves,decreased S:R ratios, deep Q-waves in the limb and left pre-cordial leads, and often, polyphasic R-waves (8–12). As thecardiomyopathy progresses, arrhythmias, conduction abnorm-alities, regional left ventricular wall motion abnormalities, leftventricular dilation and cardiac autonomic dysfunction occur(8–10,13–20). Preclinical abnormalities are detectable by
# The Author 2005. Published by Oxford University Press. All rights reserved.
For Permissions, please email: [email protected]
*To whom correspondence should be addressed. Tel: þ1 3102063395; Fax: þ1 3108258489; Email: [email protected]
Human Molecular Genetics, 2005, Vol. 14, No. 14 1921–1933doi:10.1093/hmg/ddi197Advance Access published on May 25, 2005
by guest on Decem
ber 6, 2014http://hm
g.oxfordjournals.org/D
ownloaded from

ECG in 59% of patients between 6 and 10 years of age andprogressively develop into clinically apparent cardiomyopathyin 100% of patients over 18 years of age (21). Postmortemexamination of cardiac tissue from DMD patients shows thatextreme fibrosis, generally localized to the posterobasal wallof the left ventricle, is the most prevalent histologicalfeature of the pathology, while fibrosis of the peripheral con-duction system has also been observed (1,8–10,13,15). Fur-thermore, remaining viable cardiomyocytes can becomeencased in connective tissue, thereby compromising theirintercellular connections and ability to conduct signals (22).The electrical and functional cardiac abnormalities in DMDpatients have been attributed to cardiac fibrosis, whichsuggests that fibrosis is an important component of cardiomyo-pathy in DMD (8).
Hearts of dystrophin-deficient mdx mice share many fea-tures of the DMD cardiomyopathy. Similar to DMD patients,mdx mice experience a progressive development of cardiacdefects, although the pathology is less severe. Young, adultmice show little pathology at 2–3 months of age, while func-tion and morphology markedly deteriorate by 12 months ofage (23–25). Like DMD patients, mdx mice display ECGabnormalities, autonomic dysfunction, impaired conduction,arrhythmias, deteriorated left ventricular function and dilatedcardiomyopathy (23,24,26,27). Mdx hearts also experience asevere, progressive accumulation of connective tissue(27,28), suggesting that fibrosis may also be responsible forsome features of the mdx cardiomyopathy.
Pathological fibrosis is also a prominent feature in dystrophin-deficient skeletal muscle (28–30) and may indicate thatcommon mechanisms underlie fibrosis of dystrophin-deficienthearts and muscles. Because dystrophin-deficient skeletalmuscles and hearts experience inflammation (27,31–36), andinflammatory cells are known to promote fibrosis in numerouspathologies (37–39), it is possible that fibrosis of dystrophictissues results, in part, from inflammatory processes. Thispossibility has been supported by the finding that mdx micerendered T-cell deficient by breeding onto the nu/nubackground showed less muscle and cardiac fibrosis thanmdx mice with T-cells (28). Inflammatory cells may inducefibrosis by secreting cytokines such as transforming growthfactor-b (TGF-b), which stimulate connective tissueproduction by fibroblasts (40,41). Previous investigatorshave shown that TGF-b is elevated in the skeletal musclesof dystrophin-deficient mice, dogs and humans (42–44)during the stages of muscle pathology in which fibrosisoccurs and that TGF-b blockade can reduce collagenexpression in mdx diaphragms (40).
If cardiac inflammation contributes significantly to myocar-dial fibrosis and subsequent functional defects in dystrophin-deficient hearts, then interventions that affect the inflammatoryprocess could be valuable in reducing cardiomyopathy inDMD and mdx dystrophy. In our previous work, we showedthat inflammation of dystrophin-deficient skeletal muscle isexacerbated by the loss of nNOS from muscle (33), whichoccurs as a secondary consequence of dystrophin-deficiency(45,46). Expression of a nNOS transgene in mdx mice skeletalmuscle to normalize the levels of muscle (nitric oxide) NOproduction caused great reductions in muscle inflammationand in muscle membrane lysis (33), which indicated that
muscle-derived NO can serve anti-inflammatory and cytopro-tective roles in dystrophic skeletal muscle.
Previous investigators have shown that the hearts of mdxmice lose �80% of normal nNOS activity (23), which issimilar to the loss of NOS activity that occurs in mdx skeletalmuscle (46). This similarity suggests that nNOS defects mayalso contribute to cardiomyopathy in dystrophin-deficiency.However, there are important distinctions between the distri-bution and the molecular associations of nNOS in cardiacand skeletal muscles, which can affect the functions of NOin the two muscle types. For example, nNOS in skeletalmuscle appears to be localized primarily at the sarcolemmathrough its association with the dystrophin-complex (45). Incardiac muscle, nNOS is located at the sarcolemma (47) buthas also been reported to be localized at the sarcoplasmicreticulum (48) and mitochondria (49,50), where it is not co-distributed with dystrophin. Thus, the deficiency in nNOSactivity in cardiac muscle may primarily reflect defects in itsregulation rather than loss of binding to the dystrophin-complex. Predicting the effects of perturbed NOS activityin the heart is more complex because the specific site ofNO production in cardiac muscle may have antagonisticeffects on the functions mediated by NO. Although NOgenerated by endothelial NOS at the sarcolemma can have anegative inotrophic effect in cardiac muscle, NO generatedby nNOS at the sarcoplasmic reticulum membrane can havea positive effect on contractility (51). Competing functionsof NO-derived from different NOS isoforms are not knownto occur in skeletal muscle.
In the present investigation, we test whether modificationsin NO production by nNOS in cardiomyocytes can affectthe pathology of mdx hearts. In our hypothetical model,experimental elevation of myocardial NO production in dys-trophin-deficient hearts would reduce cardiac inflammation,decrease fibrosis and, as a consequence, normalize those fea-tures of cardiac function that have been attributed to fibrosisof the mdx or DMD myocardium. We test our hypothesisby generating a dystrophin-deficient mice line in which thereis myocardial expression of a nNOS transgene. The effect ofelevated myocardial NO production by nNOS in mdx heartsis assessed by telemetric monitoring of cardiac function infreely ambulatory mice and by testing whether increases incardiac inflammation and fibrosis are affected.
RESULTS
nNOS transgene insertion increases nNOS expressionand activity in cardiac tissue
Western blot analysis of hearts from wild-type C57 mice andmdx mice at 3, 5, 9 or 18 months of age showed that there isno difference in nNOS concentration between wild-type andmdx hearts at any age that was assayed (Fig. 1A shows datafrom 5-month-old mice). The concentration of nNOS in thehearts of nNOS transgenic, dystrophin-expressing mice(nNOS Tgþ mice) was 9.4 times the concentration in thehearts of non-transgenic littermates (nNOS Tg2 mice)(Fig. 1B) and 2.0-fold greater than the expression levelsin nNOS transgenic, dystrophin-deficient mice (nNOSTgþ/mdx mice) (Fig. 1C). NOS activity assays showed that
1922 Human Molecular Genetics, 2005, Vol. 14, No. 14
by guest on Decem
ber 6, 2014http://hm
g.oxfordjournals.org/D
ownloaded from

relative NO production levels generally exhibited the sametrends as NOS concentration in each line assayed, althoughthe magnitudes of the differences were smaller. Activity datawere expressed relative to nNOS Tg2 hearts, for which themean value was set at 100% (SEM ¼ 5.2; n ¼ 9). Hearts ofnNOS Tgþ mice had 51% more NOS activity (SEM ¼ 16.2;n ¼ 5) than nNOS Tg2 hearts and nNOS Tgþ/mdx hearts
had 18% more activity (SEM ¼ 2.99; n ¼ 6) (Fig. 1D).However, NOS activity in nNOS Tg2 hearts did not differsignificantly from activity in nNOS Tg2/mdx hearts (101%;SEM ¼ 3.7; n ¼ 5).
Dystrophin-deficient mice experience myocarditis that isreduced with increased NO production
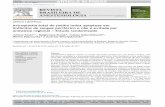
Histological analysis showed no evidence of inflammationof nNOS Tgþ or nNOS Tg2 hearts at any age sampled inmice from 3 months to 18 months of age. However, nNOSTg2/mdx hearts contained increased numbers of inflammatorycells when compared with nNOS Tg2 hearts at all agesexamined. In nNOS Tg2/mdx hearts from mice �12months of age, macrophages were increased by 497%(nNOS Tg2/mdx ¼ 1110 cells/mm3, SEM ¼ 154; nNOSTg2 ¼ 223 cells/mm3, SEM ¼ 41), eosinophils wereincreased by 342% (nNOS Tg2/mdx ¼ 222 cells/mm3,SEM ¼ 27; nNOS Tg2 ¼ 65 cells/mm3, SEM ¼ 14), CD4þT-cells were increased by 1046% (nNOS Tg2/mdx ¼ 251cells/mm3, SEM ¼ 50; nNOS Tg2 ¼ 24 cells/mm3,SEM ¼ 10) and CD8þ T-cells were increased by 438%(nNOS Tg2/mdx ¼ 464 cells/mm3, SEM ¼ 14; nNOSTg2 ¼ 106 cells/mm3, SEM ¼ 27) (Fig. 2). Macrophages,the predominant inflammatory cell type present in mdx hearts,were significantly reduced in concentration by cardiac expressionof the nNOS transgene. nNOS Tgþ/mdx mice exhibited an 88%reduction in macrophage concentration (128 cells/mm3,SEM ¼ 26) that lowered the concentration of macrophagesto levels not statistically different from dystrophin-expressingcontrols. The concentrations of other inflammatory cell typesanalyzed were not affected by the nNOS transgene, suggestinga specific effect of NO upon macrophages.
Macrophages lyse cardiomyocytes in vitro, althoughthere is little membrane lysis in vivo
Macrophage-mediated lysis of dystrophin-deficient, skeletalmuscle fibers plays a major role in promoting the pathologyof mdx skeletal muscle (33). We tested whether cardiacmacrophages could also contribute to lysis of mdx cardio-myocytes by performing co-culture assays using primarycardiomyocytes and freshly isolated macrophages and to testwhether the concentration of macrophages present in mdxhearts is sufficient to cause cardiomyocyte lysis. We observeda dose-dependent cytotoxic-response in cardiomyocyte lysiswith increasing concentrations of macrophages (Fig. 3A). Ata macrophage concentration comparable to that observed innNOS Tg2/mdx hearts (1000 cells/mm3), we observed lysisof 19.4% of cardiomyocytes, showing that cardiomyocytesare susceptible to lysis by activated macrophages.
Then, we tested whether the reduction of macrophageconcentrations that resulted from nNOS transgene expressionin the mdx myocardium decreased cardiomyocyte membranelysis in vivo. Cardiomyocytes with damaged cell membranesin vivo were identified immunohistochemically by the pre-sence of IgG in the muscle cell cytoplasm (Fig. 3B). No myo-cytes containing IgG were observed in dystrophin-expressinghearts. Areas occupied by IgG-containing myocytes werepresent in nNOS Tg2/mdx hearts, but were only 1.1% ofthe total tissue area (SEM ¼ 0.23) (Fig. 3C). Areas containing
Figure 1. Expression of an active nNOS transgene in cardiac tissue. (A)Whole heart homogenates show that endogenous nNOS expression does notvary between adult mdx and C57 mice. Left three lanes are mdx mice; rightthree lanes are C57 mice. (B) Whole heart homogenates from nNOS Tgþmice show a 9.4-fold increase in nNOS expression. Left three lanes arenNOS Tg2 hearts; right three lanes are nNOS Tgþ hearts. (C). nNOS trans-gene expression in hearts of nNOS Tgþ mice is twice the expression level innNOS Tgþ/mdx hearts. Left three lanes are nNOS Tgþ/mdx hearts; rightthree lanes are nNOS Tgþ hearts. (D). Whole heart homogenates fromnNOS Tgþ mice (n ¼ 5) show a 51% increase in total NOS activity,whereas nNOS Tgþ/mdx hearts (n ¼ 6) show an 18% increase in total NOSactivity when compared with nNOS Tg2 control hearts (n ¼ 9). Total NOSactivity did not differ between nNOS Tg2 hearts and nNOS Tg2/mdxhearts (n ¼ 5). Asterisk indicates significant difference from nNOS Tg2 con-trols at P, 0.05. Error bars represent SEM.
Human Molecular Genetics, 2005, Vol. 14, No. 14 1923
by guest on Decem
ber 6, 2014http://hm
g.oxfordjournals.org/D
ownloaded from
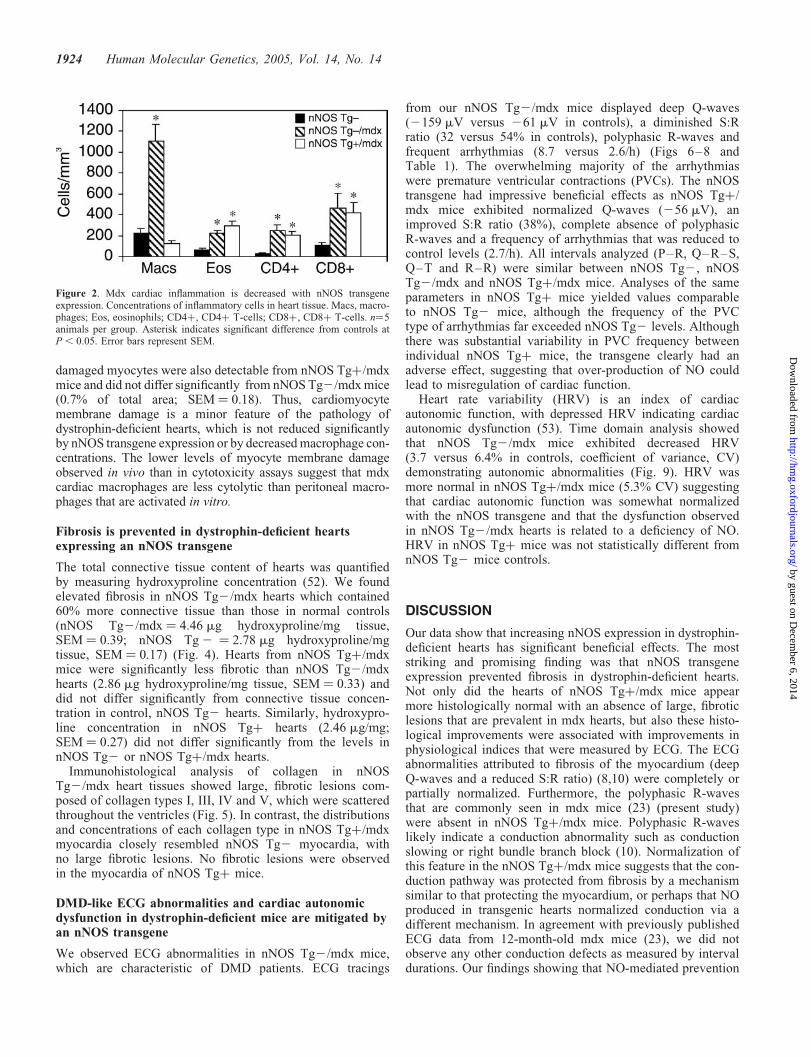
damaged myocytes were also detectable from nNOS Tgþ/mdxmice and did not differ significantly from nNOS Tg2/mdxmice(0.7% of total area; SEM ¼ 0.18). Thus, cardiomyocytemembrane damage is a minor feature of the pathology ofdystrophin-deficient hearts, which is not reduced significantlyby nNOS transgene expression or by decreasedmacrophage con-centrations. The lower levels of myocyte membrane damageobserved in vivo than in cytotoxicity assays suggest that mdxcardiac macrophages are less cytolytic than peritoneal macro-phages that are activated in vitro.
Fibrosis is prevented in dystrophin-deficient heartsexpressing an nNOS transgene
The total connective tissue content of hearts was quantifiedby measuring hydroxyproline concentration (52). We foundelevated fibrosis in nNOS Tg2/mdx hearts which contained60% more connective tissue than those in normal controls(nNOS Tg2/mdx ¼ 4.46 mg hydroxyproline/mg tissue,SEM ¼ 0.39; nNOS Tg2 ¼ 2.78 mg hydroxyproline/mgtissue, SEM ¼ 0.17) (Fig. 4). Hearts from nNOS Tgþ/mdxmice were significantly less fibrotic than nNOS Tg2/mdxhearts (2.86 mg hydroxyproline/mg tissue, SEM ¼ 0.33) anddid not differ significantly from connective tissue concen-tration in control, nNOS Tg2 hearts. Similarly, hydroxypro-line concentration in nNOS Tgþ hearts (2.46 mg/mg;SEM ¼ 0.27) did not differ significantly from the levels innNOS Tg2 or nNOS Tgþ/mdx hearts.
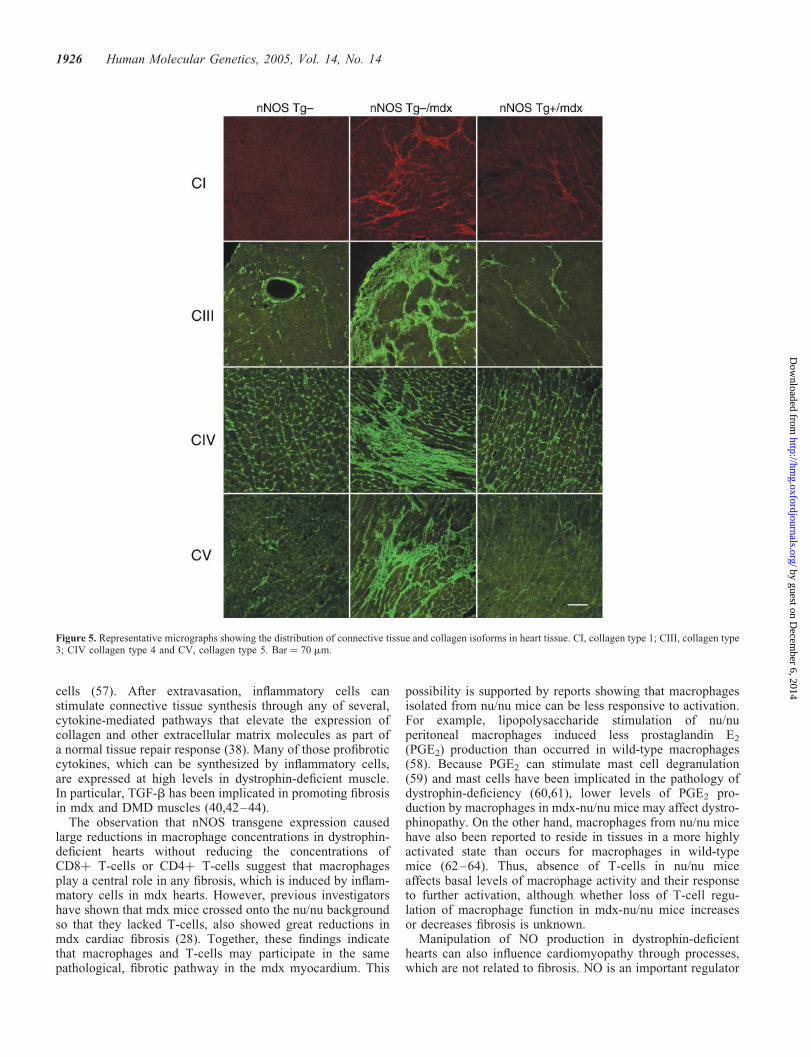
Immunohistological analysis of collagen in nNOSTg2/mdx heart tissues showed large, fibrotic lesions com-posed of collagen types I, III, IV and V, which were scatteredthroughout the ventricles (Fig. 5). In contrast, the distributionsand concentrations of each collagen type in nNOS Tgþ/mdxmyocardia closely resembled nNOS Tg2 myocardia, withno large fibrotic lesions. No fibrotic lesions were observedin the myocardia of nNOS Tgþ mice.
DMD-like ECG abnormalities and cardiac autonomicdysfunction in dystrophin-deficient mice are mitigated byan nNOS transgene
We observed ECG abnormalities in nNOS Tg2/mdx mice,which are characteristic of DMD patients. ECG tracings
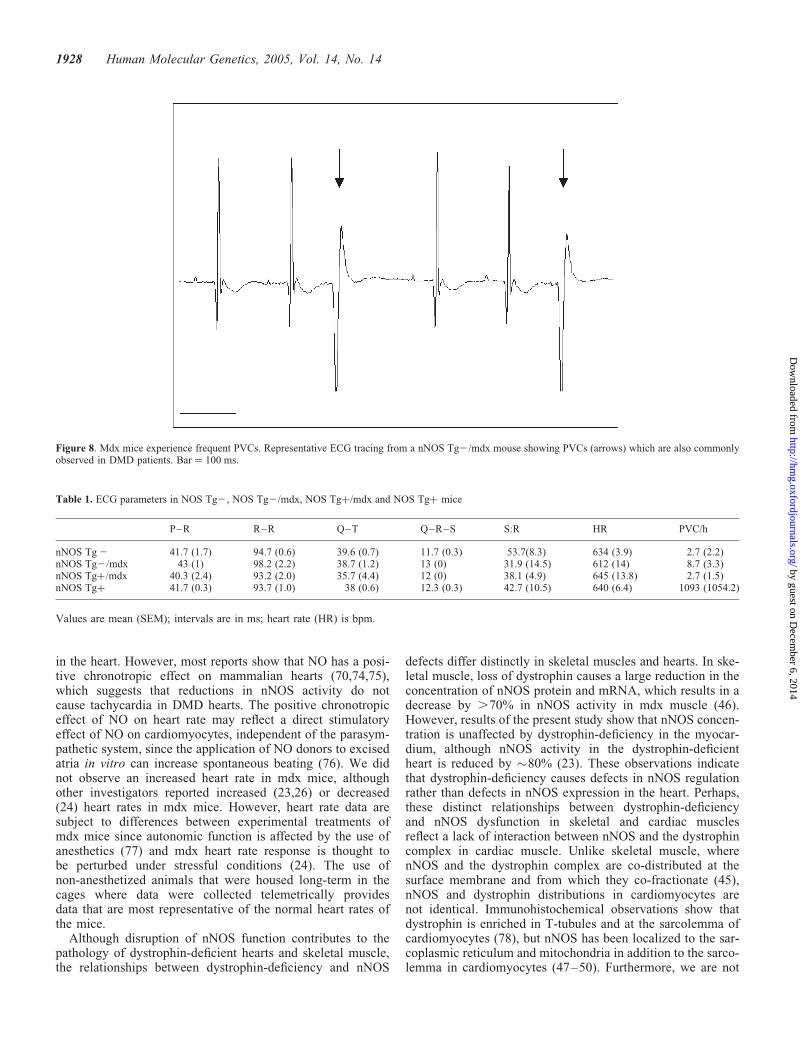
from our nNOS Tg2/mdx mice displayed deep Q-waves(2159 mV versus 261 mV in controls), a diminished S:Rratio (32 versus 54% in controls), polyphasic R-waves andfrequent arrhythmias (8.7 versus 2.6/h) (Figs 6–8 andTable 1). The overwhelming majority of the arrhythmiaswere premature ventricular contractions (PVCs). The nNOStransgene had impressive beneficial effects as nNOS Tgþ/mdx mice exhibited normalized Q-waves (256 mV), animproved S:R ratio (38%), complete absence of polyphasicR-waves and a frequency of arrhythmias that was reduced tocontrol levels (2.7/h). All intervals analyzed (P–R, Q–R–S,Q–T and R–R) were similar between nNOS Tg2, nNOSTg2/mdx and nNOS Tgþ/mdx mice. Analyses of the sameparameters in nNOS Tgþ mice yielded values comparableto nNOS Tg2 mice, although the frequency of the PVCtype of arrhythmias far exceeded nNOS Tg2 levels. Althoughthere was substantial variability in PVC frequency betweenindividual nNOS Tgþ mice, the transgene clearly had anadverse effect, suggesting that over-production of NO couldlead to misregulation of cardiac function.
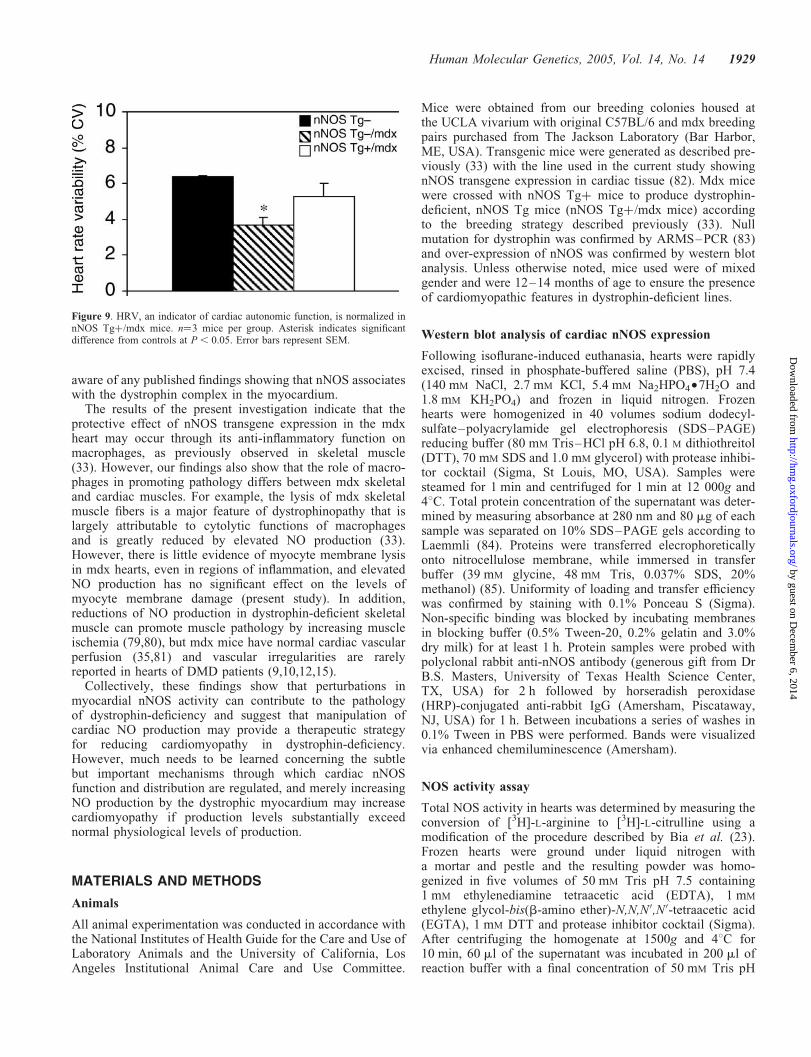
Heart rate variability (HRV) is an index of cardiacautonomic function, with depressed HRV indicating cardiacautonomic dysfunction (53). Time domain analysis showedthat nNOS Tg2/mdx mice exhibited decreased HRV(3.7 versus 6.4% in controls, coefficient of variance, CV)demonstrating autonomic abnormalities (Fig. 9). HRV wasmore normal in nNOS Tgþ/mdx mice (5.3% CV) suggestingthat cardiac autonomic function was somewhat normalizedwith the nNOS transgene and that the dysfunction observedin nNOS Tg2/mdx hearts is related to a deficiency of NO.HRV in nNOS Tgþ mice was not statistically different fromnNOS Tg2 mice controls.
DISCUSSION
Our data show that increasing nNOS expression in dystrophin-deficient hearts has significant beneficial effects. The moststriking and promising finding was that nNOS transgeneexpression prevented fibrosis in dystrophin-deficient hearts.Not only did the hearts of nNOS Tgþ/mdx mice appearmore histologically normal with an absence of large, fibroticlesions that are prevalent in mdx hearts, but also these histo-logical improvements were associated with improvements inphysiological indices that were measured by ECG. The ECGabnormalities attributed to fibrosis of the myocardium (deepQ-waves and a reduced S:R ratio) (8,10) were completely orpartially normalized. Furthermore, the polyphasic R-wavesthat are commonly seen in mdx mice (23) (present study)were absent in nNOS Tgþ/mdx mice. Polyphasic R-waveslikely indicate a conduction abnormality such as conductionslowing or right bundle branch block (10). Normalization ofthis feature in the nNOS Tgþ/mdx mice suggests that the con-duction pathway was protected from fibrosis by a mechanismsimilar to that protecting the myocardium, or perhaps that NOproduced in transgenic hearts normalized conduction via adifferent mechanism. In agreement with previously publishedECG data from 12-month-old mdx mice (23), we did notobserve any other conduction defects as measured by intervaldurations. Our findings showing that NO-mediated prevention
Figure 2. Mdx cardiac inflammation is decreased with nNOS transgeneexpression. Concentrations of inflammatory cells in heart tissue. Macs, macro-phages; Eos, eosinophils; CD4þ, CD4þ T-cells; CD8þ, CD8þ T-cells. n¼5animals per group. Asterisk indicates significant difference from controls atP, 0.05. Error bars represent SEM.
1924 Human Molecular Genetics, 2005, Vol. 14, No. 14
by guest on Decem
ber 6, 2014http://hm
g.oxfordjournals.org/D
ownloaded from
of fibrosis in mdx hearts results in clinically detectableimprovements in cardiac function emphasizes the importantrole of fibrosis in the disease and suggests the possibility fornew therapeutic strategies.
The reduction in myocardial fibrosis in nNOS Tgþ/mdxhearts corresponds with the reduction in myocardial macro-phage concentrations, although we cannot definitively con-clude that mdx myocardial fibrosis is a consequence ofinflammation. However, that interpretation of the data is con-sistent with the results of numerous investigations, which havedemonstrated that NO can inhibit inflammation and thatinflammatory cells can promote tissue fibrosis. NO canreduce the expression of adhesion molecules, which mediateleukocyte interactions with the vascular endothelia thatprecede extravasation (54–56). Conversely, NOS inhibitionincreases adhesion of leukocytes to vascular endothelial cellsand can thereby promote extravasation of inflammatory
Figure 3. Macrophages in mdx myocardium are not potently cytolytic. (A) Activated macrophages can lyse cultured myocardial cells. Striped bar representsconcentration of macrophages found in nNOS Tg2/mdx hearts. Results are the mean value of three experiments. Error bars represent SEM. (B). Representativemicrograph of a nNOS Tg2/mdx heart section stained with anti-mouse IgG showing an area of cells with membrane damage (brackets). Bar ¼ 70 mm.(C). IgG-positive areas from heart sections expressed per total section area. Black bar for nNOS Tg2 mice is too small to appear in histogram. n¼5 miceper group.
Figure 4. Cardiac fibrosis is prevented in mdx mice with a nNOS transgene.Concentration of connective tissue in hearts. n¼5 mice per group. Asteriskindicates significant difference from controls at P , 0.05. Error bars representSEM.
Human Molecular Genetics, 2005, Vol. 14, No. 14 1925
by guest on Decem
ber 6, 2014http://hm
g.oxfordjournals.org/D
ownloaded from
cells (57). After extravasation, inflammatory cells canstimulate connective tissue synthesis through any of several,cytokine-mediated pathways that elevate the expression ofcollagen and other extracellular matrix molecules as part ofa normal tissue repair response (38). Many of those profibroticcytokines, which can be synthesized by inflammatory cells,are expressed at high levels in dystrophin-deficient muscle.In particular, TGF-b has been implicated in promoting fibrosisin mdx and DMD muscles (40,42–44).
The observation that nNOS transgene expression causedlarge reductions in macrophage concentrations in dystrophin-deficient hearts without reducing the concentrations ofCD8þ T-cells or CD4þ T-cells suggest that macrophagesplay a central role in any fibrosis, which is induced by inflam-matory cells in mdx hearts. However, previous investigatorshave shown that mdx mice crossed onto the nu/nu backgroundso that they lacked T-cells, also showed great reductions inmdx cardiac fibrosis (28). Together, these findings indicatethat macrophages and T-cells may participate in the samepathological, fibrotic pathway in the mdx myocardium. This
possibility is supported by reports showing that macrophagesisolated from nu/nu mice can be less responsive to activation.For example, lipopolysaccharide stimulation of nu/nuperitoneal macrophages induced less prostaglandin E2
(PGE2) production than occurred in wild-type macrophages(58). Because PGE2 can stimulate mast cell degranulation(59) and mast cells have been implicated in the pathology ofdystrophin-deficiency (60,61), lower levels of PGE2 pro-duction by macrophages in mdx-nu/nu mice may affect dystro-phinopathy. On the other hand, macrophages from nu/nu micehave also been reported to reside in tissues in a more highlyactivated state than occurs for macrophages in wild-typemice (62–64). Thus, absence of T-cells in nu/nu miceaffects basal levels of macrophage activity and their responseto further activation, although whether loss of T-cell regu-lation of macrophage function in mdx-nu/nu mice increasesor decreases fibrosis is unknown.
Manipulation of NO production in dystrophin-deficienthearts can also influence cardiomyopathy through processes,which are not related to fibrosis. NO is an important regulator
Figure 5. Representative micrographs showing the distribution of connective tissue and collagen isoforms in heart tissue. CI, collagen type 1; CIII, collagen type3; CIV collagen type 4 and CV, collagen type 5. Bar ¼ 70 mm.
1926 Human Molecular Genetics, 2005, Vol. 14, No. 14
by guest on Decem
ber 6, 2014http://hm
g.oxfordjournals.org/D
ownloaded from

of cardiac autonomic function through direct, agonistic effectsupon the parasympathetic pathway and through indirect,inhibitory effects upon the sympathetic pathway, which col-lectively serves to protect against ventricular arrhythmias(65). The deleterious effects of disrupted NO production inthe heart are evident in animal models where NOS inhibitiondecreased parasympathetic function (66–68), decreased HRV(69) and increased ventricular arrhythmias (70). Furthermore,modest overexpression of eNOS in cardiac tissue increasedvagal drive and appeared to be anti-arrhythmogenic (71).Similarly, DMD patients experience decreased parasympa-thetic drive, increased sympathetic drive, decreased HRV,sinus tachycardia and an increased frequency of ventriculararrhythmias (most prominently, PVCs), all of whichmay reflect disruptions in cardiac NO production(14,15,17,19,20,72,73). We observed that features consistent
with DMD-associated cardiac autonomic dysfunction such asdecreased HRV and an increased frequency of PVCs in dystro-phin-deficient mice and found that these abnormalities werepalliated in mdx mice expressing the nNOS transgene. Thesedata further support the hypothesis that the cardiac autonomicdysfunction associated with dystrophin-deficiency is attribu-table, at least in part, to disruptions in NO production.However, autonomic dysfunctions that occur in hearts ofnNOS2/2mice (26) are not identical to those that occur inmdx hearts, which indicates that the impaired autonomic func-tion in mdx hearts is not simply attributable to a reduction ofnNOS-derived NO.
The beneficial effects of increased nNOS activity in mdxmice that express the nNOS transgene may be highly depen-dent on the level of nNOS-derived NO production. Expressionof the transgene in mdx hearts in which endogenous levels ofnNOS activity have been shown to be reduced by �80% (23),yielded levels of total NO production that were 18% higherthan levels measured in healthy, nNOS Tg2 hearts. Our find-ings show that this 18% increase was associated with norma-lization of cardiac function. However, we also observed thatnNOS transgene expression in dystrophin-expressing myocar-dia, in which endogenous nNOS-derived NO and transgenicnNOS-derived NO were both produced, yielded an increasein total NO production of 51%. This greater elevation of NOproduction was associated with a tremendous increase in therate of PVCs, so that they occurred hundreds of times morefrequently than in nNOS Tg2 hearts. We observed noincrease in myocardial inflammation or fibrosis in nNOSTgþ hearts, suggesting that the elevation of PVCs resultedfrom disruption of autonomic function.
Sinus tachycardia, which may be caused by impaired para-sympathetic function, is frequently observed in DMD patients(9,10,13,14,72,73) and is potentially influenced by NO levels
Figure 6. ECG tracings from mdx mice show DMD-like abnormalities that are ameliorated with cardiac expression of a nNOS transgene. Representative ECGtracings collected using implantable telemetry devices. (A) Normal tracing from a nNOS Tg2 mouse. (B) nNOS Tg2/mdx tracing shows deep Q-waves, peakedR-waves and diminished S-waves. (C) nNOS Tg2/mdx tracing showing polyphasic R-waves. (D) Tracing from nNOS Tgþ/mdx mouse showing normalizedQ-waves, R-waves and S-waves. Bar ¼ 100 ms.
Figure 7. Q-wave amplitude is normalized in mdx mice with cardiacexpression of a nNOS transgene. n¼3 mice per group. Asterisk indicates sig-nificant difference from controls at P, 0.05. Error bars represent SEM.
Human Molecular Genetics, 2005, Vol. 14, No. 14 1927
by guest on Decem
ber 6, 2014http://hm
g.oxfordjournals.org/D
ownloaded from
in the heart. However, most reports show that NO has a posi-tive chronotropic effect on mammalian hearts (70,74,75),which suggests that reductions in nNOS activity do notcause tachycardia in DMD hearts. The positive chronotropiceffect of NO on heart rate may reflect a direct stimulatoryeffect of NO on cardiomyocytes, independent of the parasym-pathetic system, since the application of NO donors to excisedatria in vitro can increase spontaneous beating (76). We didnot observe an increased heart rate in mdx mice, althoughother investigators reported increased (23,26) or decreased(24) heart rates in mdx mice. However, heart rate data aresubject to differences between experimental treatments ofmdx mice since autonomic function is affected by the use ofanesthetics (77) and mdx heart rate response is thought tobe perturbed under stressful conditions (24). The use ofnon-anesthetized animals that were housed long-term in thecages where data were collected telemetrically providesdata that are most representative of the normal heart rates ofthe mice.
Although disruption of nNOS function contributes to thepathology of dystrophin-deficient hearts and skeletal muscle,the relationships between dystrophin-deficiency and nNOS
defects differ distinctly in skeletal muscles and hearts. In ske-letal muscle, loss of dystrophin causes a large reduction in theconcentration of nNOS protein and mRNA, which results in adecrease by .70% in nNOS activity in mdx muscle (46).However, results of the present study show that nNOS concen-tration is unaffected by dystrophin-deficiency in the myocar-dium, although nNOS activity in the dystrophin-deficientheart is reduced by �80% (23). These observations indicatethat dystrophin-deficiency causes defects in nNOS regulationrather than defects in nNOS expression in the heart. Perhaps,these distinct relationships between dystrophin-deficiencyand nNOS dysfunction in skeletal and cardiac musclesreflect a lack of interaction between nNOS and the dystrophincomplex in cardiac muscle. Unlike skeletal muscle, wherenNOS and the dystrophin complex are co-distributed at thesurface membrane and from which they co-fractionate (45),nNOS and dystrophin distributions in cardiomyocytes arenot identical. Immunohistochemical observations show thatdystrophin is enriched in T-tubules and at the sarcolemma ofcardiomyocytes (78), but nNOS has been localized to the sar-coplasmic reticulum and mitochondria in addition to the sarco-lemma in cardiomyocytes (47–50). Furthermore, we are not
Figure 8. Mdx mice experience frequent PVCs. Representative ECG tracing from a nNOS Tg2/mdx mouse showing PVCs (arrows) which are also commonlyobserved in DMD patients. Bar ¼ 100 ms.
Table 1. ECG parameters in NOS Tg2, NOS Tg2/mdx, NOS Tgþ/mdx and NOS Tgþ mice
P–R R–R Q–T Q–R–S S:R HR PVC/h
nNOS Tg2 41.7 (1.7) 94.7 (0.6) 39.6 (0.7) 11.7 (0.3) 53.7(8.3) 634 (3.9) 2.7 (2.2)nNOS Tg2/mdx 43 (1) 98.2 (2.2) 38.7 (1.2) 13 (0) 31.9 (14.5) 612 (14) 8.7 (3.3)nNOS Tgþ/mdx 40.3 (2.4) 93.2 (2.0) 35.7 (4.4) 12 (0) 38.1 (4.9) 645 (13.8) 2.7 (1.5)nNOS Tgþ 41.7 (0.3) 93.7 (1.0) 38 (0.6) 12.3 (0.3) 42.7 (10.5) 640 (6.4) 1093 (1054.2)
Values are mean (SEM); intervals are in ms; heart rate (HR) is bpm.
1928 Human Molecular Genetics, 2005, Vol. 14, No. 14
by guest on Decem
ber 6, 2014http://hm
g.oxfordjournals.org/D
ownloaded from
aware of any published findings showing that nNOS associateswith the dystrophin complex in the myocardium.
The results of the present investigation indicate that theprotective effect of nNOS transgene expression in the mdxheart may occur through its anti-inflammatory function onmacrophages, as previously observed in skeletal muscle(33). However, our findings also show that the role of macro-phages in promoting pathology differs between mdx skeletaland cardiac muscles. For example, the lysis of mdx skeletalmuscle fibers is a major feature of dystrophinopathy that islargely attributable to cytolytic functions of macrophagesand is greatly reduced by elevated NO production (33).However, there is little evidence of myocyte membrane lysisin mdx hearts, even in regions of inflammation, and elevatedNO production has no significant effect on the levels ofmyocyte membrane damage (present study). In addition,reductions of NO production in dystrophin-deficient skeletalmuscle can promote muscle pathology by increasing muscleischemia (79,80), but mdx mice have normal cardiac vascularperfusion (35,81) and vascular irregularities are rarelyreported in hearts of DMD patients (9,10,12,15).
Collectively, these findings show that perturbations inmyocardial nNOS activity can contribute to the pathologyof dystrophin-deficiency and suggest that manipulation ofcardiac NO production may provide a therapeutic strategyfor reducing cardiomyopathy in dystrophin-deficiency.However, much needs to be learned concerning the subtlebut important mechanisms through which cardiac nNOSfunction and distribution are regulated, and merely increasingNO production by the dystrophic myocardium may increasecardiomyopathy if production levels substantially exceednormal physiological levels of production.
MATERIALS AND METHODS
Animals
All animal experimentation was conducted in accordance withthe National Institutes of Health Guide for the Care and Use ofLaboratory Animals and the University of California, LosAngeles Institutional Animal Care and Use Committee.
Mice were obtained from our breeding colonies housed atthe UCLA vivarium with original C57BL/6 and mdx breedingpairs purchased from The Jackson Laboratory (Bar Harbor,ME, USA). Transgenic mice were generated as described pre-viously (33) with the line used in the current study showingnNOS transgene expression in cardiac tissue (82). Mdx micewere crossed with nNOS Tgþ mice to produce dystrophin-deficient, nNOS Tg mice (nNOS Tgþ/mdx mice) accordingto the breeding strategy described previously (33). Nullmutation for dystrophin was confirmed by ARMS–PCR (83)and over-expression of nNOS was confirmed by western blotanalysis. Unless otherwise noted, mice used were of mixedgender and were 12–14 months of age to ensure the presenceof cardiomyopathic features in dystrophin-deficient lines.
Western blot analysis of cardiac nNOS expression
Following isoflurane-induced euthanasia, hearts were rapidlyexcised, rinsed in phosphate-buffered saline (PBS), pH 7.4(140 mM NaCl, 2.7 mM KCl, 5.4 mM Na2HPO4†7H2O and1.8 mM KH2PO4) and frozen in liquid nitrogen. Frozenhearts were homogenized in 40 volumes sodium dodecyl-sulfate–polyacrylamide gel electrophoresis (SDS–PAGE)reducing buffer (80 mM Tris–HCl pH 6.8, 0.1 M dithiothreitol(DTT), 70 mM SDS and 1.0 mM glycerol) with protease inhibi-tor cocktail (Sigma, St Louis, MO, USA). Samples weresteamed for 1 min and centrifuged for 1 min at 12 000g and48C. Total protein concentration of the supernatant was deter-mined by measuring absorbance at 280 nm and 80 mg of eachsample was separated on 10% SDS–PAGE gels according toLaemmli (84). Proteins were transferred elecrophoreticallyonto nitrocellulose membrane, while immersed in transferbuffer (39 mM glycine, 48 mM Tris, 0.037% SDS, 20%methanol) (85). Uniformity of loading and transfer efficiencywas confirmed by staining with 0.1% Ponceau S (Sigma).Non-specific binding was blocked by incubating membranesin blocking buffer (0.5% Tween-20, 0.2% gelatin and 3.0%dry milk) for at least 1 h. Protein samples were probed withpolyclonal rabbit anti-nNOS antibody (generous gift from DrB.S. Masters, University of Texas Health Science Center,TX, USA) for 2 h followed by horseradish peroxidase(HRP)-conjugated anti-rabbit IgG (Amersham, Piscataway,NJ, USA) for 1 h. Between incubations a series of washes in0.1% Tween in PBS were performed. Bands were visualizedvia enhanced chemiluminescence (Amersham).
NOS activity assay
Total NOS activity in hearts was determined by measuring theconversion of [3H]-L-arginine to [3H]-L-citrulline using amodification of the procedure described by Bia et al. (23).Frozen hearts were ground under liquid nitrogen witha mortar and pestle and the resulting powder was homo-genized in five volumes of 50 mM Tris pH 7.5 containing1 mM ethylenediamine tetraacetic acid (EDTA), 1 mM
ethylene glycol-bis(b-amino ether)-N,N,N 0,N0-tetraacetic acid(EGTA), 1 mM DTT and protease inhibitor cocktail (Sigma).After centrifuging the homogenate at 1500g and 48C for10 min, 60 ml of the supernatant was incubated in 200 ml ofreaction buffer with a final concentration of 50 mM Tris pH
Figure 9. HRV, an indicator of cardiac autonomic function, is normalized innNOS Tgþ/mdx mice. n¼3 mice per group. Asterisk indicates significantdifference from controls at P, 0.05. Error bars represent SEM.
Human Molecular Genetics, 2005, Vol. 14, No. 14 1929
by guest on Decem
ber 6, 2014http://hm
g.oxfordjournals.org/D
ownloaded from

7.5, 5 mM CaCl2, 1 mM MgCl2, 14 mM tetrahydrobiopterin,10 mg/ml calmodulin, 4 mM flavin adenine dinucleotide,4 mM flavin adenine mononucleotide, 1 mM reduced nicotina-mide adenine dinucleotide phosphate (NADPH) and 1 ml of1 mCi/ml [3H]-L-arginine. After a 30 min incubation at378C, the reactions were stopped with 20 mM sodium acetatepH 5.5, 0.2 mM EGTA, 1 mM L-citrulline and 2 mM EDTAand poured over Dowex-50W columns (Biorad, Hercules,CA, USA) previously converted to Naþ form to separate the[3H]-L-arginine from the [3H]-L-citrulline. The eluate wasmixed with scintillation cocktail (Packard Bioscience,Meriden, CT, USA) and the samples counted in a LS-3133Tscintillation counter (Beckman, Fullerton, CA, USA). Scintil-lation counts were normalized to total protein content of thehomogenate as determined by measuring the absorbance at280 nm and expressed as a percent of control values.
Histology
Following euthanasia with isoflurane, hearts were rapidlyexcised, transferred to liquid nitrogen-cooled isopentane-filled vials and stored at 2808C until use. Frozen heartswere sectioned through the short axis of the ventricles at athickness of 10 mm. For immunostaining, sections were air-dried and fixed in acetone, and endogenous peroxidase activitywas quenched with 0.03% hydrogen peroxide. Sections wereincubated in PBS with 2% gelatin and 3% bovine serumalbumin to prevent non-specific binding of antibodies.Tissues were incubated with primary antibodies overnight at48C, host-appropriate biotinylated secondary antibodies(Vector Laboratories, Burlingame, CA, USA) for 30 min atroom temperature, and HRP-avidin D for 30 min. Sectionswere washed with PBS between antibody incubations. Stain-ing was visualized using 3-amino-9-ethyl carbazole (AEC,red) (Vector) as substrate. Antibodies used for inflammatorycell staining were rat monoclonal anti-mouse F4/80 (formacrophages), anti-mouse CD4 and anti-mouse CD8, allobtained from supernatants of hybridoma cultures (hybrido-mas obtained from American Type Culture Collection,Bethesda, MD, USA) and polyclonal rabbit anti-murineeosinophil granule major basic protein (a generous gift fromDr J.J Lee, Mayo Clinic Scottsdale, AZ, USA) (86). Theconcentrations of inflammatory cells were measured byhistomorphometry as previously described (87).
Collagen staining was performed as described earlier with afew exceptions. Sections were incubated with primary anti-bodies for 3 h at room temperature and with host-appropriatefluorescent-conjugated secondary antibodies (Vector) for 1 h.Primary antibodies used were rabbit anti-rat collagen type I(Chemicon, Temecula, CA, USA), rabbit anti-rat collagentype III (Chemicon), goat anti-human collagen type IV(Southern Biotech, Birmingham, AL, USA) and goat anti-human collage type V (Southern Biotech). Sections stainedfor collagen were qualitatively analyzed for the presence offoci of fibrosis.
IgG staining
Cardiomyocytes with damaged membranes were identified byIgG staining as performed previously by Hainsey et al. (27).
Transverse sections through the ventricles, 10 mm in thick-ness, were incubated for 30 min with a 1% gelatin solutionin PBS to block non-specific binding. After rinsing withPBS, the sections were incubated with FITC-conjugatedmouse anti-IgG (Vector Laboratories) for 1 h. A series ofthree PBS washes were performed and the sections wereviewed and analyzed on a microscope equipped withfluorescent optics. Areas of damaged cells were quantifiedaccording to the previous technique (88); a sampling gridwas superimposed on the tissue and the number of interceptsthat overlie cell IgG-positive cells were counted and expressedas a percentage of the total number of intercepts.
Cytotoxicity assays
Primary cardiac cultures were generated from neonatal C57mice according to the modified protocol of Sen et al. (89).Hearts were removed from 2-day-old C57 mice and trimmedof connective tissue and atria. Cardiomyocytes were dispersedfrom the ventricles by digestion with 0.2% collagenase II(Invitrogen, Carlsbad, CA, USA) and 0.6% pancreatin(Sigma) in a buffer containing 0.8 mM MgSO4, 116 mM
NaCl, 0.5 mM KCl, 11 mM NaH2PO4, 5.5 mM glucose and20 mM HEPES. The cell suspension was preplated twice for30 min each to enrich for cardiomyocytes. Cells werecounted and the enriched cardiomyocytes were plated in 4:1DMEM/medium 199 (Sigma) containing penicillin and strep-tomyocin and supplemented with 10% FBS (Omega Scientific,Tarzana, CA, USA) at 1800 cells/mm2 which produced aconfluent layer in gelatin-coated 96-well plates. The cultureswere maintained at 378C with 5% CO2 and began beatingsynchronously 2–3 days following plating.
Macrophages for co-culturing were obtained by modifi-cation of our previously described technique (90). Peritonealexudate was collected from adult C57 mice 3 days followingintraperitoneal injection of 12% sodium caseinate. Theexudate was filtered through 70 mm nylon mesh, cells pelletedat 500g and resuspended in 0.85% NH4Cl to lyse erythrocytes.The cells were re-pelleted, resuspended in HBSS and overlaidonto Histopaque 1077 (Sigma). After centrifuging at 500g for45 min the purified macrophages were collected from theinterface and purity of the population was determined histo-logically to be .90%.
Approximately 5 days after plating the cardiomyocyte cul-tures, the cells were loaded with 51Cr (as previously described)(33) and co-cultured for 16 h with varying concentrations offreshly isolated macrophages activated with 0.6 mM PMA.The medium was then assayed for 51Cr release by scintillationcounting. Cytotoxicity was expressed as a percentage of totallysis by setting 0% as 51Cr released spontaneously by cardio-myocytes incubated without macrophages. The 51Cr releaseinto the medium by cardiomyocytes lysed with 0.1% TritonX-100 (Sigma) was determined to establish 100% cytotoxicity.
Hydroxyproline assay
Total connective tissue content of hearts was quantified bymeasuring the amount of hydroxyproline concentration inthe tissues according to the technique of Kivirikko et al.(52) that we have used previously (91).
1930 Human Molecular Genetics, 2005, Vol. 14, No. 14
by guest on Decem
ber 6, 2014http://hm
g.oxfordjournals.org/D
ownloaded from

Electrocardiography
ECG data was collected in awake, freely moving mice byimplanting radio telemetry devices (TA10ETA-F20, Tran-soma-Data Science Intl. (DSI), St Paul, MN, USA). MeasuringECG activity in conscious mice is advantageous since the con-founding effects of anesthesia upon cardiac function areobviated. Transmitter units were implanted in the peritonealcavity of anesthetized mice and the two electrical leads weresecured near the apex of the heart and the right acromion ina lead II orientation. Mice were housed singly in cages overantenna receivers connected to a computer system for datarecording. Unfiltered ECG data was collected for 10 s eachhour for 35 days. The first 7 days of data were discarded toallow for recovery from the surgical procedure andensure any effects of anesthesia had subsided. Datawaveforms and parameters were analyzed with the DSIanalysis packages (ART 3.01 and Physiostat 4.01) andmeasurements were compiled and averaged to determineheart rates, ECG wave heights and interval durations. RawECG waveforms were scanned for arrhythmias by two inde-pendent observers.
Time-domain measures of HRV
The CV (%), an index of HRV, was determined fromsequential R–R intervals as previously described byGehrmann et al. (53). Briefly, the standard deviation ofall normal R–R intervals was divided by mean of all R–Rintervals: SDNN/RRmean � 100.
Statistics
Statistical significance of differences between groups wasdetermined using the two-tailed Student’s t-test. The P-valuewas set at 0.05. ANOVA was used for four-way analysis ofcardiac telemetry data.
ACKNOWLEDGEMENTS
The authors thank Dr Joshua Goldhaber for helpfuldiscussions on the ECG data analysis and Helen C. Changfor assistance in ECG data analysis. This work was supportedby grants from the American Heart Association 0325146Y(M.W.H.), the National Institutes of Health AR40343(J.G.T.), AR47721 (J.G.T.), the Muscular Dystrophy Associ-ation (J.G.T.) and the Laubisch Endowment (K.P.R.).
Conflict of Interest statement. None declared.
REFERENCES
1. Moriuchi, T., Kagawa, N., Mukoyama, M. and Hizawa, K. (1993)Autopsy analysis of the muscular dystrophies. Tokushima J. Exp. Med.,40, 83–93.
2. Eagle, M., Baudouin, S.V., Chandler, C., Giddings, D.R., Bullock, R.and Bushby, K. (2002) Survival in Duchenne muscular dystrophy:improvements in life expectancy since 1967 and the impact of homenocturnal ventilation. Neuromuscul. Disord., 12, 926–929.
3. Ishikawa, Y., Bach, J.R. and Minami, R. (1999) Cardioprotection forDuchenne’s muscular dystrophy. Am. Heart J., 137, 895–902.
4. Bushby, K., Muntoni, F. and Bourke, J.P. (2003) 107th ENMCinternational workshop: the management of cardiac involvement inmuscular dystrophy and myotonic dystrophy, 7th–9th June 2002,Naarden, The Netherlands. Neuromuscul. Disord., 13, 166–172.
5. Hoogerwaard, E.M., van der Wouw, P.A., Wilde, A.A.M., Bakker, E.,Ippel, P.F., Oosterwijk, J.C., Majoor-Krakauer, D.F., van Essen, A.J.,Leschot, N.J. and de Visser, M. (1999) Cardiac involvement in carriers ofDuchenne and Becker muscular dystrophy. Neuromuscul. Disord., 9,347–351.
6. Politano, L., Nigro, V., Nigro, G., Petretta, V.R., Passamano, L.,Papparella, S., Di Somma, S. and Comi, L.I. (1996) Development ofcardiomyopathy in female carriers of Duchenne and Becker musculardystrophies. JAMA, 275, 1335–1338.
7. Mirabella, M., Servidei, S., Manfredi, G., Ricci, E., Frustaci, A., Bertini, E.,Rana, M. and Tonali, P. (1993) Cardiomyopathy may be the only clinicalmanifestation in female carriers of Duchenne muscular dystrophy.Neurology, 43, 2342–2345.
8. Perloff, J.K., Roberts, W.C., De Leon, A.C., Jr and O’Doherty, D. (1967)The distinctive electrocardiogram of Duchenne’s progressive musculardystrophy. Am. J. Med., 42, 179–188.
9. Perloff, J.K., Henze, E. and Schelbert, H. (1984) Alterations in regionalmyocardial metabolism, perfusion, and wall motion in Duchennemuscular dystrophy studied by radionuclide imaging. Circulation, 69,33–42.
10. Slucka, C. (1968) The electrocardiogram in Duchenne progressivemuscular dystrophy. Circulation, 38, 933–940.
11. Manning, G.W. and Cropp, G.J. (1958) The electrocardiogram inprogressive muscular dystrophy. Br. Heart J., 20, 416–420.
12. Gnecchi-Ruscone, T., Taylor, J., Mercuri, E., Paternostro, G., Pogue, R.,Bushby, K., Sewry, C., Muntoni, F. and Camici, P.G. (1999)Cardiomyopathy in Duchenne, Becker and sarcoglycanopathies: a role forcoronary dysfunction? Muscle Nerve, 22, 1549–1556.
13. Perloff, J.K., De Leon, A.C., Jr and O’Doherty, D. (1966) Thecardiomyopathy of progressive muscular dystrophy. Circulation, 33,625–648.
14. Akita, H., Matsuoka, S. and Kuroda, Y. (1993) Predictiveelectrocardiographic score for evaluating prognosis in patients withDuchenne’s muscular dystrophy. Tokushima J. Exp. Med., 40, 55–60.
15. Nomura, H. and Hizawa, K. (1982) Histopathological study of theconduction system of the heart in Duchenne progressive musculardystrophy. Acta Pathol. Jpn., 32, 1027–1033.
16. de Kermadec, J.-M., Becane, H.-M., Chenard, A., Tertrain, F. andWeiss, Y. (1994) Prevalence of left ventricular systolic dysfunction inDuchenne muscular dystrophy: an echocardiographic study. Am. Heart J.,127, 618–623.
17. Yanagisawa, A., Miyagawa, M., Yotsukura, M., Tsuya, T., Shirato, C.,Ishihara, T., Aoyagi, T. and Ishikawa, K. (1992) The prevalence andprognostic significance of arrhythmias in Duchenne type musculardystrophy. Am. Heart J., 124, 1244–1250.
18. Sanyal, S.K. and Johnson, W.W. (1982) Cardiac conduction abnormalitiesin children with Duchenne’s progressive muscular dystrophy:electrocardiographic features and morphologic correlates. Circulation, 4,853–863.
19. Lanza, G.A., Russo, A.D., Giglio, V., De Luca, L., Messano, L., Santini, C.,Ricci, E., Damiani, A., Fumagalli, G., De Martino, G., Mangiola, F. andBellocci, F. (2001) Impairment of cardiac autonomic function in patientswith Duchenne muscular dystrophy: relationship to myocardial andrespiratory function. Am. Heart J., 141, 808–812.
20. Chenard, A.A., Becane, H.M., Tertrain, F., de Kermadec, J.M. andWeiss, Y.A. (1993) Ventricular arrhythmia in Duchenne musculardystrophy: prevalence, significance and prognosis. Neuromuscul. Disord.,3, 201–206.
21. Nigro, G., Comi, L.I., Politano, L. and Bain, R.J.I. (1990) The incidenceand evolution of cardiomyopathy in Duchenne muscular dystrophy.Int. J. Cardiol., 26, 271–277.
22. Fenoglio, J.J., Jr, Pham, T.D., Harken, A.H., Horowitz, L.N., Josephson,M.E. and Wit, A.L. (1983) Recurrent sustained ventricular tachycardia:structure and ultrastructure of subendocardial regions in whichtachycardia originates. Circulation, 68, 518–533.
23. Bia, B.L., Caddisy, P.J., Young, M.E., Rafael, J.A., Leighton, B.,Davies, K.E., Radda, G.K. and Clarke, K. (1999) Decreased myocardialnNOS, increased iNOS and abnormal ECGs in mouse models ofDuchenne muscular dystrophy. J. Mol. Cell. Cardiol., 31, 1857–1862.
Human Molecular Genetics, 2005, Vol. 14, No. 14 1931
by guest on Decem
ber 6, 2014http://hm
g.oxfordjournals.org/D
ownloaded from

24. Quinlan, J.G., Hahn, H.S., Wong, B.L., Lorenz, J.N., Wenisch, A.S. and
Levin, L.S. (2004) Evolution of the mdx mouse cardiomyopathy:physiological and morphological findings. Neuromuscul. Disord., 14,491–496.
25. Wilding, J.R., Schneider, J.E., Sang, A.E., Davies, K.E., Neubauer, S. andClarke, K. (2005) Dystrophin- and MLP-deficient mouse hearts: marked
differences in morphology and function, but similar accumulation ofcytoskeletal proteins. FASEB J., 19, 79–81.
26. Chu, V., Otero, J.M., Lopez, O., Sullivan, M.F., Morgan, J.P., Amende, I.and Hampton, T.G. (2002) Electrocardiographic findings in mdx mice: acardiac phenotype of Duchenne muscular dystrophy. Muscle Nerve, 26,
513–519.
27. Hainsey, T.A., Senapati, S., Kuhn, D.E. and Rafael, J.A. (2003)Cardiomyopathic features associated with muscular dystrophy areindependent of dystrophin absence in cardiovasculature. Neuromuscul.Disord., 13, 294–302.
28. Morrison, J., Lu, W.L., Pastoret, C., Partridge, T. and Bou-Gharios, G.(2000) T-cell-dependent fibrosis in the mdx dystrophic mouse. Lab.Invest., 80, 881–891.
29. Laws, N. and Hoey, A. (2004) Progression of kyphosis in mdx mice.J. Appl. Physiol., 97, 1970–1977.
30. Roig, M., Roma, J., Fargas, A. and Munell, F. (2004) Longitudinalpathologic study of the gastrocnemius muscle group in mdx mice. ActaNeuropathol., 107, 27–34.
31. Cai, B., Spencer, M.J., Nakamura, G., Tseng-Ong, L. and Tidball, J.G.(2000) Eosinophilia of dystrophin-deficient muscle is promoted by
perforin-mediated cytotoxicity by T cell effectors. Am. J. Pathol., 156,1789–1796.
32. Spencer, M.J., Montecino-Rodriguez, E., Dorshkind, K. and Tidball, J.G.(2001) Helper (CD4þ) and cytotoxic (CD8þ) T cells promote thepathology of dystrophin-deficient muscle. Clin. Immunol., 98, 235–243.
33. Wehling, M., Spencer, M.J. and Tidball, J.G. (2001) A nitric oxidesynthase transgene ameliorates muscular dystrophy in mdx mice. J. Cell.Biol., 155, 123–131.
34. Cullen, M.J. and Fulthorpe, J.J. (1975) Stages in fibre breakdown inDuchenne muscular dystrophy. An electron-microscopic study. J. Neurol.
Sci., 24, 179–200.
35. Bridges, L.R. (1986) The association of cardiac muscle necrosis andinflammation with the degenerative and persistent myopathy of mdx mice.J. Neurol. Sci., 72, 147–157.
36. Adams, R.D. (1971) Diseases of Muscle—A Study in Pathology. Harper
and Row, Hagerstown, MD, p. 298.
37. Strutz, F. and Neilson, E.G. (2003) New insights into mechanisms offibrosis in immune renal injury. Springer Semin. Immunopathol., 24,459–476.
38. Nicoletti, A. and Michel, J.B. (1999) Cardiac fibrosis and inflammation:
interaction with hemodynamic and hormonal factors. Cardiovasc. Res.,41, 532–543.
39. Koehler, D.R., Downey, G.P., Sweezey, N.B., Tanswell, A.K. andHu, J. (2004) Lung inflammation as a therapeutic target in cysticfibrosis. Am. J. Respir. Cell Mol. Biol., 31, 377–381.
40. Gosselin, L.E., Williams, J.E., Deering, M., Brazeau, D., Koury, S. andMartinez, D.A. (2004) Localization and early time course of TGF-b1mRNA expression in dystrophic muscle. Muscle Nerve, 30, 645–653.
41. Straino, S., Germani, A., Di Cardok, A., Porcelli, D., De Mori, R.,Mangoni, A., Napolitano, M., Martelli, F., Biglioli, P. and Capogrossi,
M.C. (2004) Enhanced arteriogenesis and wound repair in dystrophin-deficient mdx mice. Circulation, 110, 3341–3348.
42. Iannaccone, S., Quattrini, A., Smirne, S., Sessa, M., de Rino, F.,Ferini-Strambi, L. and Nemni, R. (1995) Connective tissue proliferationand growth factors in animal models of Duchenne muscular dystrophy.
J. Neurol. Sci., 128, 36–44.
43. Bernasconi, P., Torchiana, E., Confalonieri, P., Brugnoni, R., Barresi, R.,Mora, M., Cornelio, F., Morandi, L. and Mantegazza, R. (1995)Expression of transforming growth factor-beta 1 in dystrophic patientmuscles correlates with fibrosis. Pathogenetic role of a fibrogenic
cytokine. J. Clin. Invest., 96, 1137–1144.
44. Passerini, L., Bernasconi, P., Baggi, F., Confalonieri, P., Cozzi, F.,Cornelio, F. and Mantegazza, R. (2002) Fibrogenic cytokines and extentof fibrosis in muscle of dogs with X-linked golden retriever musculardystrophy. Neuromuscul. Disord., 12, 828–835.
45. Brenman, J.E., Chao, D.S., Xia, H., Aldape, K. and Bredt, D.S. (1995)Nitric oxide synthase complexed with dystrophin and absent from skeletalmuscle sarcolemma in Duchenne muscular dystrophy. Cell, 82, 743–752.
46. Chang, W.-J., Iannaccone, S.T., Lau, K.S., Masters, B.S.S., McCabe, T.J.,McMillan, K., Padre, R.C., Spencer, M.J., Tidball, J.G. and Stull, J.T.(1996) Neuronal nitric oxide synthase and dystrophin-deficient musculardystrophy. Proc. Natl Acad. Sci. USA, 93, 9142–9147.
47. Xu, K.Y., Kuppusamy, S.P., Wang, J.Q., Li, H., Cui, H., Dawson, T.M.,Huang, P.L., Burnett, A.L., Kuppusamy, P. and Becker, L.C. (2003) Nitricoxide protects cardiac sarcolemmal membrane enzyme function and ionactive transport against ischemia-induced inactivation. J. Biol. Chem.,278, 41798–41803.
48. Xu, K.Y., Huso, D.L., Dawson, T.M., Bredt, D.S. and Becker, L.C. (1999)Nitric oxide synthase in cardiac sarcoplasmic reticulum. Proc. Natl Acad.Sci. USA, 96, 657–662.
49. Kanai, A.J., Pearce, L.L., Clemens, P.R., Birder, L.A., Van Bibber, M.M.,Choi, S.Y., de Groat, W.C. and Peterson, J. (2001) Identification of aneuronal nitric oxide synthase in isolated cardiac mitochondria usingelectrochemical detection. Proc. Natl Acad. Sci. USA, 98, 14126–14131.
50. Elfering, S.L., Sarkela, T.M. and Giulivi, C. (2002) Biochemistry ofmitochondrial nitric-oxide synthase. J. Biol. Chem., 277, 38079–38086.
51. Barouch, L.A., Harrison, R.W., Skaf, M.W., Rosas, G.O., Cappola, T.P.,Kobeissi, Z.A., Hobai, I.A., Lemmon, C.A., Burnett, A.L., O’Rourke,B. et al. (2002) Nitric oxide regulates the heart by spatial confinement ofnitric oxide synthase isoforms. Nature, 416, 337–339.
52. Kivirikko, K.I., Laitinen, O. and Prockop, D.J. (1967) Modifications of aspecific assay for hydroxyproline in urine. Anal. Biochem., 19, 249–255.
53. Gehrmann, J., Hammer, P.E., Maguire, C.T., Wakimoto, H., Triedman, J.K.and Berul, C.I. (2000) Phenotypic screening for heart rate variability inthe mouse. Am. J. Physiol. Heart Circ. Physiol., 279, H733–H740.
54. Kosonen, O., Kankaanranta, H., Uotila, J. and Moilanen, E. (2000)Inhibition by nitric oxide-releasing compounds of E-selectin expression inand neutrophil adhesion to human endothelial cells. Eur. J. Pharmacol.,394, 149–156.
55. Liu, P., Xu, B., Hock, C.E., Nagele, R., Sun, F.F. and Wong PY. (1998)NO modulates P-selectin and ICAM-1 mRNA expression andhemodynamic alterations in hepatic I/R. Am. J. Physiol., 275,H2191–H2198.
56. Aljada, A., Saadeh, R., Assian, E., Ghanim, H. and Dandona, P. (2000)Insulin inhibits the expression of intercellular adhesion molecule-1 byhuman aortic endothelial cells through stimulation of nitric oxide. J. Clin.Endocrinol. Metab., 85, 2572–2575.
57. Akimitsu, T., Gute, D.C. and Korthuis, R.J. (1995) Leukocyte adhesioninduced by inhibition of nitric oxide production in skeletal muscle.J. Appl. Physiol., 78, 1725–1732.
58. Vogel, S.N., Hansen, C.T. and Rosenstreich, D.L. (1979) Characterizationof a congenitally LPS-resistant, athymic mouse strain. J. Immunol., 122,619–622.
59. Nguyen, M., Solle, M., Audoly, L.P., Tilley, S.L., Stock, J.L., McNeish,J.D., Coffman, T.M., Dombrowicz, D. and Koller, B.H. (2002) Receptorsand signaling mechanisms required for prostaglandin E2-mediatedregulation of mast cell degranulation and IL-6 production. J. Immunol.,169, 4586–4593.
60. Gorospe, J.R., Tharp, M., Demitsu, T. and Hoffman, E.P. (1994)Dystrophin-deficient myofibers are vulnerable to mast cell granule-induced necrosis. Neuromuscul. Disord., 4, 325–333.
61. Granchelli, J.A., Pollina, C. and Hudecki, M.S. (1995) Duchenne-likemyopathy in double-mutant mdx mice expressing exaggerated mast cellactivity. J. Neurol. Sci., 131, 1–7.
62. Cheers, C. and Waller, R. (1975) Activated macrophages in congenitallyathymic ‘nude mice’ and in lethally irradiate mice. J. Immunol., 115,844–847.
63. Nickol, A.D. and Bonventre, P.F. (1977) Anomalous high nativeresistance to athymic mice to bacterial pathogens. Infect. Immun., 18,636–645.
64. Sharp, A.K. and Colston, M.J. (1984) The regulation of macrophageactivity in congenitally athymic mice. Eur. J. Immunol., 14, 102–105.
65. Chowdhary, S. and Townend, J.N. (1999) Role of nitric oxide in theregulation of cardiovascular control. Clin. Sci., 97, 5–17.
66. Choate, J.K., Danson, E.J.F., Morris, J.F. and Paterson, D.J. (2001)Peripheral vagal control of heart rate is impaired in neuronal NOSknockout mice. Am. J. Physiol. Heart Circ. Physiol., 281, H2310–H2317.
1932 Human Molecular Genetics, 2005, Vol. 14, No. 14
by guest on Decem
ber 6, 2014http://hm
g.oxfordjournals.org/D
ownloaded from

67. Mohan, R.M., Heaton, D.A., Danson, E.J.F., Krishnan, S.P.R., Cai, S.,Channon, K.M. and Paterson, D.J. (2002) Neuronal nitric oxide synthasegene transfer promotes cardiac vagal gain of function. Circ. Res., 91,1089–1091.
68. Markos, F., Snow, H.M., Kidd, C. and Conlon, K. (2002) Nitric oxidefacilitates vagal control of heart rate via actions in the cardiacparasympathetic ganglia of the anaesthetised dog. Exp. Physiol., 87,49–52.
69. Markos, F., Snow, H.M., Kidd, C. and Conlon, K. (2001) Inhibition ofneuronal nitric oxide reduces heart rate variability in the anesthetized dog.Exp. Physiol., 86, 539–541.
70. Pabla, R. and Curtis, M.J. (1995) Effects of NO modulation on cardiacarrhythmias in the rat isolated heart. Circ. Res., 77, 984–992.
71. Massion, P.B., Dessy, C., Desjardins, F., Pelat, M., Havaux, X., Belge, C.,Moulin, P., Guiot, Y., Feron, O., Janssens, S. and Ballingand, J.L. (2004)Cardiomyocyte-restricted overexpression of endothelial nitric oxidesynthase (NOS3) attenuates beta-adrenergic stimulation and reinforcesvagal inhibition of cardiac contraction. Circulation, 110, 2666–2672.
72. Yotsukura, M., Sasaki, K., Kachi, E., Sasaki, A., Ishihara, T. andIshikawa, K. (1995) Circadian rhythm and variability of heart rate inDuchenne-type progressive muscular dystrophy. Am. J. Cardiol., 76,947–951.
73. Yotsukura, M., Fujii, K., Katayama, A., Tomono, Y., Ando, H., Sakata, K.,Ishihara, T. and Ishikawa, K. (1998) Nine-year follow-up study of heartrate variability in patients with Duchenne-type progressive musculardystrophy. Am. Heart J., 136, 289–296.
74. Kojda, G., Kottenberg, K. and Noack, E. (1997) Inhibition of nitric oxidesynthase and soluble guanylate cyclase induces cardiodepressive effects innormal rat hearts. Eur. J. Pharmacol., 334, 181–190.
75. Musialek, P., Paterson, D.J. and Casadei, B. (1999) Changes inextracellular pH mediate the chronotropic responses to L-arginine.Cardiovasc. Res., 43, 712–720.
76. Musialek, P., Lei, M., Brown, H.F., Paterson, D.J. and Casadei, B. (1997)Nitric oxide can increase heart rate by stimulating the hyperpolarization-activated inward current, I(f). Circ. Res., 81, 60–68.
77. Chaves, A.A., Dech, S.J., Nakayama, T., Hamlin, R.L., Bauer, J.A. andCarnes, C.A. (2003) Age and anesthetic effects on muringelectrocardiography. Life Sci., 72, 2401–2412.
78. Klietsch, R., Ervasti, J.M., Arnold, W., Cambell, K.P. and Jorgensen, A.O.(1993) Dystrophin-glycoprotein complex and laminin colocalize to thesarcolemma and transverse tubules of cardiac muscle. Circ Res., 72,349–360.
79. Thomas, G.D., Sander, M., Lau, K.S., Huang, P.L., Stull, J.T. and Victor,R.G. (1998) Impaired metabolic modulation of a-adrenergicvasoconstriction in dystrophin-deficient skeletal muscle. Proc. Natl Acad.Sci. USA, 95, 15090–15095.
80. Sander, M., Chavoshan, B., Harris, S.A., Iannaccone, S.T., Stull, J.T.,
Thomas, G.D. and Victor, R.G. (2000) Functional muscle ischemia in
neuronal nitric oxide synthase-deficient skeletal muscle of children withDuchenne muscular dystrophy. Proc. Natl Acad. Sci. USA, 97,
13818–13823.
81. Cohn, R.D., Durbeej, M., Moore, S.A., Coral-Vazquez, R., Prouty, S. andCampbell, K.P. (2001) Prevention of cardiomyopathy in mouse models
lacking the smooth muscle sarcoglycan–sarcospan complex. J. Clin.Invest., 107, R1–R7.
82. Brennan, K.J. and Hardeman, E.C. (1993) Quantitative analysis of the
human a-skeletal actin gene in transgenic mice. J. Biol. Chem., 268,719–725.
83. Amalfitano, A. and Chamberlain, J.S. (1996) The mdx-amplification-
resistant mutation system assay, a simple and rapid polymerasechain reaction-based detection of the mdx allele. Muscle Nerve,
19, 1549–1553.
84. Laemmli, U.K. (1970) Cleavage of structural proteins during assembly ofthe head of bacteriophage T4. Nature (Lond.), 227, 680–685.
85. Burnette, W.N. (1981) ‘Western blotting’: electrophoretic transfer of
proteins from sodium dodecyl sulfate–polyacrylamide gels to unmodifiednitrocellulose and radiographic detection with antibody and
radioindinated protein A. Anal. Biochem., 112, 195–203.
86. Lee, J.J., McGarry, M.P., Farmer, S.C., Denzler, K.L., Larson, K.A.,Carrigan, P.E., Brenneise, I.E., Horton, M.A., Haczku, A., Gelfand, E.W.,
Leikauf, G.D. and Lee, N.A. (1997) Interleukin-5 expression in the lungepitelium of transgenic mice leads to pulmonary changes pathognomonic
of asthma. J. Exp. Med., 185, 2143–2156.
87. Wehling-Henricks, M., Lee, J.J. and Tidball, J.G. (2004) Prednisolone
decreases cellular adhesion molecules required for inflammatory cellinfiltration in dystrophin-deficient skeletal muscle. Neuromuscul. Disord.,
14, 483–490.
88. Spencer, M.J., Marinoa, M.E. and Winckler, W.M. (2000) Alteredpathological progression of diaphragm and quadriceps muscle in
TNF-deficient, dystrophin mice. Neuromuscul. Disord., 10, 612–619.
89. Sen, A., Dunnmon, P., Henderson, S.A., Gerard, R.D. and Chien, K.R.(1988) Terminally differentiated neonatal rat myocardial cells proliferate
and maintain specific differentiated function following expression ofSV40 large T antigen. J. Biol. Chem., 263, 19132–19136.
90. Nguyen, H.X. and Tidball, J.G. (2003) Expression of a muscle-specific,
nitric oxide synthase transgene prevents muscle membrane injury andreduces muscle inflammation during modified muscle use in mice.
J. Physiol., 550, 347–356.
91. Koh, T.J. and Tidball, J.G. (1999) Nitric oxide synthase inhibitors reducesarcomere addition in rat skeletal muscle. J. Physiol., 519, 189–196.
Human Molecular Genetics, 2005, Vol. 14, No. 14 1933
by guest on Decem
ber 6, 2014http://hm
g.oxfordjournals.org/D
ownloaded from
Copyright © 2022 FDOKUMEN