
Altered functional coupling of coronary K channels in diabetic dyslipidemic pigs is prevented by...
15
Altered functional coupling of coronary K channels in diabetic dyslipidemic pigs is prevented by exercise E. A. Mokelke, 1,3 Q. Hu, 1 M. Song, 4,5 L. Toro, 4,5,6,7 H. K. Reddy, 2 and M. Sturek 1,2,3 1 Departments of Medical Pharmacology and Physiology and 2 Department of Internal Medicine, 3 The Center for Diabetes & Cardiovascular Health, University of Missouri, Columbia, Missouri 65212; and 4 Division of Molecular Medicine, 5 Department of Anesthesiology, 6 Department of Molecular & Medical Pharmacology, and 7 Brain Research Institute, David Geffen School of Medicine, University of California, Los Angeles, California 90095 Submitted 22 October 2002; accepted in final form 30 April 2003 Mokelke, E. A., Q. Hu, M. Song, L. Toro, H. K. Reddy, and M. Sturek. Altered functional coupling of coronary K channels in diabetic dyslipidemic pigs is prevented by exer- cise. J Appl Physiol 95: 1179–1193, 2003. First published May 30, 2003; 10.1152/japplphysiol.00972.2002.—Chronic hyperglycemia and hypercholesterolemia have been shown to alter ionic currents in vascular smooth muscle. We tested the hypothesis that the combined effect of hyperglycemia and hyperlipidemia (diabetic dyslipidemia) would increase the Ca 2 -sensitive K (K Ca ) current as a compensatory response to an increase in intracellular Ca 2 concentration. We also hypothesized that exercise training would prevent this ele- vation in K Ca current. Miniature Yucatan swine were ran- domly assigned to five groups: control, standard pig chow (C, n 6); hyperlipidemic, high-fat pig chow (H, n 5); diabetic, standard pig chow (D, n 7); diabetic, high-fat pig chow (“diabetic dyslipidemic,” DD, n 12); and exercise-trained DD (DDX, n 9). High-fat chow consisted of standard minipig chow supplemented with cholesterol (2%) and coco- nut oil. Increased coronary vasoconstriction assessed in vivo and in vitro in DD was prevented by exercise. Patch-clamp experiments performed on right coronary artery smooth mus- cle cells resulted in greater K current densities in the H, D, and DD groups vs. the DDX group between 10 and 40 mV. In fura 2-loaded cells, current activated by caffeine-induced Ca 2 release was greater in H, D, and DD compared with C and DDX (P 0.05), whereas intracellular Ca 2 concentra- tion was not different across groups. Finally, there were no differences in the K Ca or K v channel protein content between groups. These data indicate that hyperglycemia, hyperlipid- emia, and diabetic dyslipidemia lead to elevated whole cell K current and increased functional coupling of K Ca and Ca 2 release. Endurance exercise prevented increased cou- pling of Ca 2 release to K Ca channel activation in diabetic dyslipidemia. Ca 2 -dependent K channel; sarcoplasmic reticulum; Ca 2 release; dyslipidemia; voltage clamp; porcine; vascular smooth muscle DIABETES IS A MAJOR INDEPENDENT cardiovascular risk factor and causes accelerated atherosclerosis and heart disease (31). The mechanism for this acceleration in atherosclerosis has not been clearly elucidated; how- ever, there is strong evidence that diabetes induces alterations in Ca 2 handling at the level of the isolated arteries and vascular smooth muscle cells (2, 3, 23, 25, 27, 53, 66, 69, 71, 75). The dysregulation of Ca 2 could cause direct significant changes in vascular tone, be- cause Ca 2 is tightly coupled to the contractility of the vessel. Furthermore, because Ca 2 can stimulate po- tassium (K ) channel activity in the vasculature, dia- betes-induced alterations in Ca 2 movement could dis- rupt the balance between contractile agents and K channel-sensitive relaxation, ultimately affecting ves- sel reactivity. Finally, Ca 2 is vital in coordinated and appropriate protein expression; therefore, Ca 2 dys- regulation could activate pathways for pathophysiolog- ical gene transcription (22). The contribution of K channel activity in the regu- lation of vascular smooth muscle tone has been clearly demonstrated (12, 20, 46). The proposed mechanism for K channel-dependent relaxation involves activa- tion of K channels leading to hyperpolarization of the membrane potential (V m ), which then inactivates the voltage-gated Ca 2 channel (VGCC), ultimately lead- ing to a decrease in Ca 2 influx and reduction in cytosolic Ca 2 concentration ([Ca 2 ] i ). The role of the ATP- sensitive K channel (K ATP ) channel in regulation of vascular tone is minor owing to lack of change in the metabolic demands of smooth muscle under normal healthy conditions (28, 59); therefore, the voltage-sen- sitive K channel (K v ) and Ca 2 -sensitive K channel (K Ca ) play the dominant roles in the regulation of vascular tone (12, 46). Because a prominent function of the K channel is to provide a means for smooth muscle relaxation, any alteration in the activity or expression of K channels would have a direct impact on vascular tone. Alterations in the expression and activity of K channels have been shown to occur in several disease states (15, 37, 38, 40, 42). An increase in K Ca channel expression has been interpreted in the particular case of hypertension as a compensatory response to the elevated [Ca 2 ] i (40). Additionally, there is a large body of evidence demonstrating that [Ca 2 ] i is elevated in vascular smooth muscle cells obtained from hypercho- Address for reprint requests and other correspondence: M. Sturek, MA415 Medical Sciences Bldg., Dept. of Medical Pharmacology & Physiology, Univ. of Missouri, Columbia, MO 65212 (E-mail: [email protected]). The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked ‘‘advertisement’’ in accordance with 18 U.S.C. Section 1734 solely to indicate this fact. J Appl Physiol 95: 1179–1193, 2003. First published May 30, 2003; 10.1152/japplphysiol.00972.2002. 8750-7587/03 $5.00 Copyright © 2003 the American Physiological Society http://www.jap.org 1179 by 10.220.33.2 on August 18, 2016 http://jap.physiology.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Altered functional coupling of coronary K channels in diabetic dyslipidemic pigs is prevented by...

Altered functional coupling of coronary K� channels indiabetic dyslipidemic pigs is prevented by exercise
E. A. Mokelke,1,3 Q. Hu,1 M. Song,4,5 L. Toro,4,5,6,7 H. K. Reddy,2 and M. Sturek1,2,3
1Departments of Medical Pharmacology and Physiology and 2Department of Internal Medicine, 3The Centerfor Diabetes & Cardiovascular Health, University of Missouri, Columbia, Missouri 65212; and 4Division ofMolecular Medicine, 5Department of Anesthesiology, 6Department of Molecular & Medical Pharmacology, and7Brain Research Institute, David Geffen School of Medicine, University of California, Los Angeles, California 90095
Submitted 22 October 2002; accepted in final form 30 April 2003
Mokelke, E. A., Q. Hu, M. Song, L. Toro, H. K. Reddy,and M. Sturek. Altered functional coupling of coronary K�
channels in diabetic dyslipidemic pigs is prevented by exer-cise. J Appl Physiol 95: 1179–1193, 2003. First publishedMay 30, 2003; 10.1152/japplphysiol.00972.2002.—Chronichyperglycemia and hypercholesterolemia have been shown toalter ionic currents in vascular smooth muscle. We tested thehypothesis that the combined effect of hyperglycemia andhyperlipidemia (diabetic dyslipidemia) would increase theCa2�-sensitive K� (KCa) current as a compensatory responseto an increase in intracellular Ca2� concentration. We alsohypothesized that exercise training would prevent this ele-vation in KCa current. Miniature Yucatan swine were ran-domly assigned to five groups: control, standard pig chow (C,n � 6); hyperlipidemic, high-fat pig chow (H, n � 5); diabetic,standard pig chow (D, n � 7); diabetic, high-fat pig chow(“diabetic dyslipidemic,” DD, n � 12); and exercise-trainedDD (DDX, n � 9). High-fat chow consisted of standardminipig chow supplemented with cholesterol (2%) and coco-nut oil. Increased coronary vasoconstriction assessed in vivoand in vitro in DD was prevented by exercise. Patch-clampexperiments performed on right coronary artery smooth mus-cle cells resulted in greater K� current densities in the H, D,and DD groups vs. the DDX group between �10 and 40 mV.In fura 2-loaded cells, current activated by caffeine-inducedCa2� release was greater in H, D, and DD compared with Cand DDX (P � 0.05), whereas intracellular Ca2� concentra-tion was not different across groups. Finally, there were nodifferences in the KCa or Kv channel protein content betweengroups. These data indicate that hyperglycemia, hyperlipid-emia, and diabetic dyslipidemia lead to elevated whole cellK� current and increased functional coupling of KCa andCa2� release. Endurance exercise prevented increased cou-pling of Ca2� release to KCa channel activation in diabeticdyslipidemia.
Ca2�-dependent K� channel; sarcoplasmic reticulum; Ca2�
release; dyslipidemia; voltage clamp; porcine; vascularsmooth muscle
DIABETES IS A MAJOR INDEPENDENT cardiovascular riskfactor and causes accelerated atherosclerosis and heartdisease (31). The mechanism for this acceleration inatherosclerosis has not been clearly elucidated; how-ever, there is strong evidence that diabetes induces
alterations in Ca2� handling at the level of the isolatedarteries and vascular smooth muscle cells (2, 3, 23, 25,27, 53, 66, 69, 71, 75). The dysregulation of Ca2� couldcause direct significant changes in vascular tone, be-cause Ca2� is tightly coupled to the contractility of thevessel. Furthermore, because Ca2� can stimulate po-tassium (K�) channel activity in the vasculature, dia-betes-induced alterations in Ca2� movement could dis-rupt the balance between contractile agents and K�
channel-sensitive relaxation, ultimately affecting ves-sel reactivity. Finally, Ca2� is vital in coordinated andappropriate protein expression; therefore, Ca2� dys-regulation could activate pathways for pathophysiolog-ical gene transcription (22).
The contribution of K� channel activity in the regu-lation of vascular smooth muscle tone has been clearlydemonstrated (12, 20, 46). The proposed mechanismfor K� channel-dependent relaxation involves activa-tion of K� channels leading to hyperpolarization of themembrane potential (Vm), which then inactivates thevoltage-gated Ca2� channel (VGCC), ultimately lead-ing to a decrease in Ca2� influx and reduction in cytosolicCa2� concentration ([Ca2�]i). The role of the ATP-sensitive K� channel (KATP) channel in regulation ofvascular tone is minor owing to lack of change in themetabolic demands of smooth muscle under normalhealthy conditions (28, 59); therefore, the voltage-sen-sitive K� channel (Kv) and Ca2�-sensitive K� channel(KCa) play the dominant roles in the regulation ofvascular tone (12, 46). Because a prominent function ofthe K� channel is to provide a means for smoothmuscle relaxation, any alteration in the activity orexpression of K� channels would have a direct impacton vascular tone.
Alterations in the expression and activity of K�
channels have been shown to occur in several diseasestates (15, 37, 38, 40, 42). An increase in KCa channelexpression has been interpreted in the particular caseof hypertension as a compensatory response to theelevated [Ca2�]i (40). Additionally, there is a large bodyof evidence demonstrating that [Ca2�]i is elevated invascular smooth muscle cells obtained from hypercho-
Address for reprint requests and other correspondence: M. Sturek,MA415 Medical Sciences Bldg., Dept. of Medical Pharmacology &Physiology, Univ. of Missouri, Columbia, MO 65212 (E-mail:[email protected]).
The costs of publication of this article were defrayed in part by thepayment of page charges. The article must therefore be herebymarked ‘‘advertisement’’ in accordance with 18 U.S.C. Section 1734solely to indicate this fact.
J Appl Physiol 95: 1179–1193, 2003.First published May 30, 2003; 10.1152/japplphysiol.00972.2002.
8750-7587/03 $5.00 Copyright © 2003 the American Physiological Societyhttp://www.jap.org 1179
by 10.220.33.2 on August 18, 2016
http://jap.physiology.org/D
ownloaded from

lesterolemic and diabetic models, both animal and hu-man (9, 30, 47, 67, 72). Although many investigatorshave shown diabetes-induced increases in vasoreactiv-ity of several different vascular tissue beds (1–3, 53),the effect of diabetes on K� channel activity or expres-sion, Ca2� regulation, and vascular smooth musclereactivity has not been thoroughly studied. A betterunderstanding of this relationship could help explainthe enhanced vascular reactivity to agonists and elu-cidate potential therapeutic targets for the treatmentof altered vascular reactivity.
Endurance exercise training, which is generallythought to provide cardioprotection, has several well-described effects on coronary smooth muscle (CSM)that can be summarized as an enhanced ability to relaxand an attenuated response to vasoconstrictors (9, 16,54). Although the exact mechanism for this protectionis unclear, altered Ca2� regulation is a potential can-didate because of its critical role in the contraction-relaxation cycle of CSM, and, most importantly, Ca2�
homeostasis is affected by chronic endurance exercise(10, 62, 63). The close functional relationship of Ca2�/current and the activity of K� channels make alter-ations in K� channel activity another potential candi-date for the observed effect of exercise-induced cardio-protection. Indeed, Bowles et al. (11) have shown thatCSM cells obtained from exercise-trained miniatureswine exhibit an increase in the contribution of KCachannels to tone, suggesting that K� channel activityis another target for the signal(s) that results fromendurance exercise training that ultimately affects cor-onary blood flow.
The purpose of this study was to determine whetherthe effects of hyperlipidemia (H) and diabetes (D) in-dependently or in combination [diabetic dyslipidemia(DD)] result in alterations in the coupling of Ca2�
regulation and K� channel activity and whether thechanges in diabetic dyslipidemia can be prevented byexercise training. We chose the porcine model of dia-betes because it possesses many qualities similar tohumans, including cardiac and coronary anatomy anddiet-induced atherosclerosis (17, 29, 49, 52, 69), and isalso a well-established model of endurance exercisetraining (62). We found that hyperlipidemia, diabetes,and diabetic dyslipidemia resulted in greater couplingof sarcoplasmic reticulum (SR) Ca2� release to wholecell potassium current (IK) activation but no increasein K� channel protein expression as a compensatorymechanism in an attempt to prevent increased vaso-constriction. All of these events were prevented byendurance exercise training.
METHODS
Porcine model of diabetic dyslipidemia. All procedureswere approved by the University of Missouri Animal Careand Use Committee in accordance with the “Principles for theutilization and care of vertebrate animals used in testing,research and training” and the American Veterinary MedicalAssociation Panel on Euthanasia. Male miniature Yucatanswine (Sinclair Research Center, Columbia, MO) weighingbetween 40 and 55 kg were randomly assigned to five exper-
imental groups: control, standard minipig chow (C, n � 6);hyperlipidemic, high-fat minipig chow (H, n � 5); diabetic,standard minipig chow (D, n � 7); diabetic, high-fat minipigchow (diabetic dyslipidemic, DD, n � 12); and diabetic, high-fat minipig chow, endurance exercise trained (DDX, n � 9).High-fat minipig chow consisted of standard minipig chowsupplemented with cholesterol (2%) and coconut oil, whichincreased the percent of kilocalories provided from fat from 8to 46%. Body weights were monitored weekly, and amount offeed and daily insulin dosage were adjusted as necessary toensure weight gain of 1% of initial body weight per week (8).Animals were fed once at the same time of day and drankwater ad libitum.
Diabetes was induced in the D, DD, and DDX animals byinjecting alloxan (125 mg/kg, Sigma Chemical, St. Louis,MO) into the superior vena cava via a surgically implantedvascular access port (48). Alloxan specifically destroys theinsulin-producing �-cells of the pancreas (69). Animals weremaintained at a fasting blood glucose concentration between300 and 400 mg/dl (6). Additionally, to mimic the dyslipide-mia that is common in the human diabetic population, DDand DDX animals were fed a high-fat feed (see above). Bloodurea nitrogen, creatinine, and liver enzymes all remainedwithin normal limits in all animals as previously reported(17).
Treadmill exercise protocol. DDX animals underwent anacclimatization period to a motorized treadmill (Good Horse-keeping, Ash Grove, MO) over a 2-wk period during whichthe grade and speed were incrementally increased so that, bythe end of the 2 wk, a workload was reached that elicited anexercise heart rate between 65 and 75% of maximal heartrate. The grade was adjusted during the remaining 14-wkexercise training regimen to maintain this target heart rate.Total running time at the target heart rate was 30 min 4days/wk (8).
In vivo assessment of coronary reactivity. The right femoralartery was accessed by arterial cutdown or with an 18-gaugeneedle through which a 0.035-in. J-guidewire was intro-duced. After an introducer and an 8-Fr sheath were insertedover the guidewire, an 8-Fr Amplatz L (sizes 0.75–2.0) guid-ing catheter was advanced into the aortic arch. The guide-wire was removed, and a manifold apparatus was attachedthat allowed direct blood pressure measurements to be ob-tained as well as injection of contrast media or experimentalsolutions. The ostium of the left main artery was engagedwith the guiding catheter, and a 3.2-Fr intravascular ultra-sound catheter (35 MHz, Ultracross, Boston Scientific/SciMed, Maple Grove, MN) was advanced through the guid-ing catheter and positioned in a proximal segment of leftcircumflex artery. A bolus of prostaglandin F2� (PGF2�, 8�g/kg) was injected into the left main artery via the manifold.The contraction to PGF2� was recorded on videotape foroff-line analysis. Peak response to PGF2� across all groupswas obtained by assessment of the luminal area duringdiastole and systole. Distensibility index, which is a measureof vessel stiffness, was calculated by using the equationdistensibility index � [(dA/A)/dP] � 1,000, where dA is thedifference between the smallest and largest luminal areas,dP is the pulse pressure, and A is the diastolic luminal area.Finally, an automated pullback was performed in the circum-flex artery at a rate of 0.5 mm/s to visualize atheroma.Images were captured on videotape for off-line quantitativesegmental analysis. At 1-mm intervals (segments) of thepullback through typically 60–100 mm of circumflex and leftanterior descending arteries, the presence of atheroma wasdefined as any fibrous or soft plaque less echogenic than theadventitia (34). This characteristic was easily resolved as
1180 K� CHANNELS IN DIABETIC DYSLIPIDEMIA
J Appl Physiol • VOL 95 • SEPTEMBER 2003 • www.jap.org
by 10.220.33.2 on August 18, 2016
http://jap.physiology.org/D
ownloaded from

distinct from the nonlayered appearance of all arteries fromcontrol pigs (26). Luminal area was defined by fine scintilla-tions from red blood cells and larger scintillations and/orturbulence from injection of saline. Percent atheroma wasdefined as number of segments having atheroma divided bytotal number of segments � 100. This analysis was per-formed for each pig; the total number of segments studied foreach group is indicated in Fig. 2.
In vitro assessment of coronary reactivity. The right coro-nary artery (RCA) was removed from all animals immedi-ately after exsanguination under isoflurane anesthesia andplaced in ice-cold sterile storage media. Four 3-mm serialsegments were obtained and denuded of endothelium byrubbing a thin dowel along the luminal surface. Denudationwas confirmed by the lack of a response to the endothelium-dependent vasodilator bradykinin after preconstricting with30 � 10�6 M PGF2�. Ring segments were hung on forcetransducers (Grass-Telefactor, West Warwick, RI) and placedin a modified Krebs-Henseleit bicarbonate-based buffer thatcontained (in mM) 2 CaCl2, 118 NaCl, 1 MgCl2, 5 KCl, 24.8NaHCO3, and 10 glucose. The pH was adjusted to 7.4, andthe buffer was continuously gassed with a mixture of 5%CO2-21% O2-75% N2. All data were collected at a samplingfrequency of 0.2 Hz with AxoBASIC software (Axon Instru-ments, Union City, CA) and stored on a personal computerfor off-line analysis.
The rings were stretched to a length that elicited maximalforce and were allowed to equilibrate for 1 h before theexperimental protocol began. A predetermined amount ofstock KCl solution was added to the tissue bath to reach afinal depolarizing concentration of 60 mM KCl. After 5 min,the solution was washed out and replaced with the Krebs-Henseleit. PGF2� (30 � 10�6 M final concentration) or endo-thelin-1 (ET-1, 1 � 10�8 M final concentration) was added toevaluate vessel contractility. Force development to both con-strictors was calculated by determining steady-state forceand subtracting the initial baseline force. The developedforce was then normalized to the response to the 60 mM KClapplication.
Isolation of smooth muscle cells from RCA. Coronarysmooth muscle cells were dispersed from segments of freshlydissected RCA as previously described (64). Briefly, small(0.5–1.0 cm) segments of the proximal RCA were cut longi-tudinally to allow exposure of the luminal side of the RCA.The segment was pinned to the bottom of a Sylgard-coatedspecimen jar into which a solution containing elastase (5U/ml, Worthington Biochemical, Lakewood, NJ), collagenase(CLS II, Worthingon Biochemical), 2 mg/ml bovine serumalbumin (fraction V, Sigma Chemical), 1 mg/ml soybeantrypsin inhibitor (type-I-S, Sigma Chemical), and 0.4 mg/mlDNase I (type IV, Sigma Chemical) was added. The jar wasplaced in a shaking water bath (37°C, 100/min) for an addi-tional 30–60 min. The cell suspension was centrifuged at 150g for 4 min, and the pellet was resuspended in a low-Ca2�-containing storage media.
Whole cell Ca2� measurement. A second cell suspensionwas obtained from the section of the RCA. These CSM cellswere loaded with the acetoxymethyl ester form of the fluo-rescent Ca2� indicator fura 2 (fura 2-AM, 2.5 �M). Fura2-AM-loaded cells were placed in a constant-flow perfusionchamber for the determination of epifluorescence under rest-ing and stimulated conditions. Light was excited with a300-W xenon arc lamp and directed through alternating 340-and 380-nm band-pass filters with a liquid light guide. Theresultant fluorescence from the selected smooth musclecell was reflected to a photomultiplier tube (Products forResearch, Danvers, MA) onto which a photon counter
(Hamamatsu, Bridgewater, NJ) was attached. An opticalprocessor (OP400) received the data, which were stored on apersonal computer for off-line analysis. Ratiometric dataobtained at 340 and 380 nm were used to estimate myoplas-mic Ca2�. Background fluorescence was subtracted for eachcell before the experimental protocol was performed (64).
Perforated patch electrophysiology. The cell suspension (30�l) was pipetted onto a microscope coverslip secured in acustom-made patch-clamp perfusion chamber. The perfo-rated patch-clamp technique was used to preserve the intrin-sic cellular constituents (11). Glass capillary tubes (FisherScientific, Atlanta, GA) were pulled to 4- to 10-M tips, firepolished, and dipped in an intracellular solution that did notcontain amphotericin B (Sigma Chemical). The pipette wasthen back-filled with an amphotericin B-containing solution(240 �g/ml) with the following additional constituents (inmM): 75 K2SO4, 45 KCl, 10 NaCl, 8 MgSO4, and 10 HEPES,pH 7.10. All experiments were performed with the use of aDagan 8900 patch-clamp amplifier, which was interfacedwith a Labmaster analog-to-digital converter and personalcomputer equipped with AxoBASIC 1.0 software (Axon In-struments, Foster City, CA) for data acquisition. Data werefiltered through an eight-pole low-pass filter after digitiza-tion at 495-�s intervals. A gigaohm seal was formed, and theprotocol was begun when series resistance had reached avalue of 25 M or less. A Ca2�-containing solution (2CaNa)was superfused through the chamber by using gravity-as-sisted flow. The constituents of 2CaNa were (in mM) 2 CaCl2,138 NaCl, 0.1 MgCl2, 1.0 KCl, 1.0 HEPES, and 10 glucose,pH 7.4. A current-voltage step protocol was performed bysetting the holding potential (HP) to �80 and applying serialtest pulses in increments of 10 mV (267 ms) from �80 to �60mV. Peak steady-state currents at each step potential weredetermined off-line and normalized to cell surface area by useof cell capacitance. A current density-voltage relationshipwas determined for all groups.
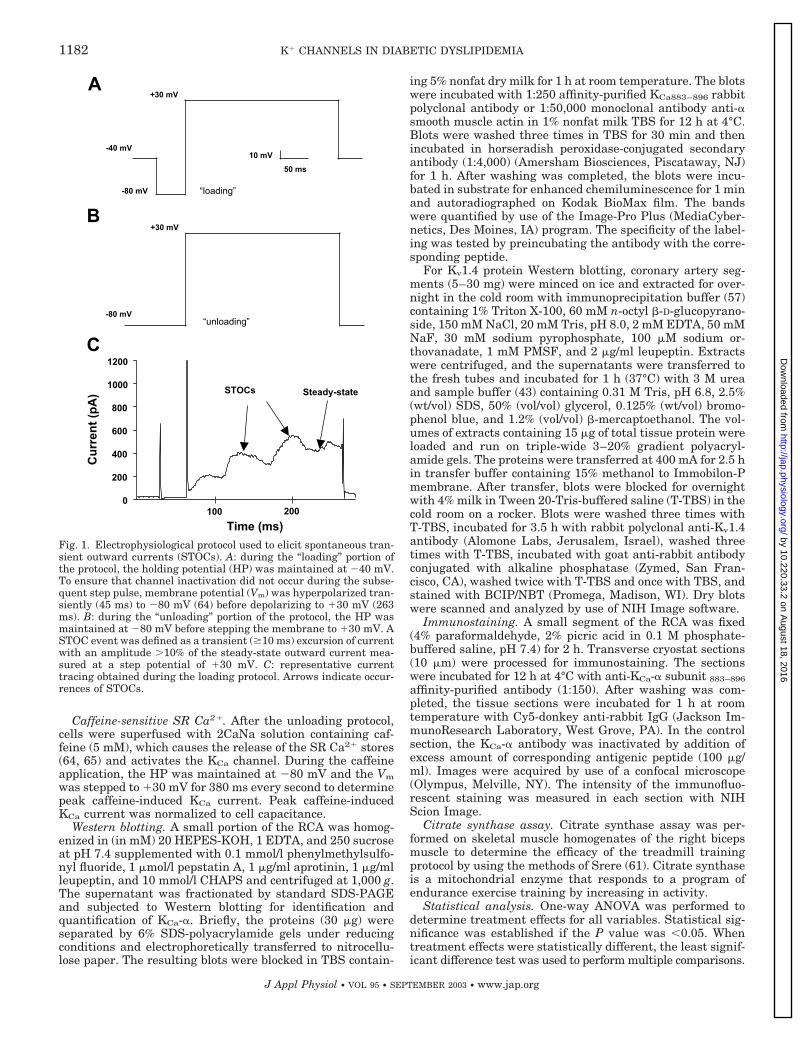
STOC measurement. Spontaneous transient outward cur-rents (STOCs) occur when KCa channels are activated byquantal releases of Ca2� from the SR, which increases thesubsarcolemmal Ca2� concentration (7, 51). STOCs havebeen identified in many types of vascular smooth muscle cellsfrom different animal species (7, 24, 33, 44), including por-cine coronary artery (63, 64). The outward current resultingfrom activation of KCa channels is a useful bioassay fortransient increases in Ca2� (64), quantal releases of Ca2�
from the SR and/or agonist-sensitive SR Ca2� stores (63, 65).In the presence of constant superfusion of 2CaNa solution,we used an electrophysiological protocol designed to optimizethe measurement of STOC events, which is shown in Fig. 1.During the “loading” portion of the protocol (Fig. 1A), theholding potential was maintained at �40 mV for 3 min tofacilitate Ca2� influx via VGCC and loading of the SR Ca2�
store. To prevent channel inactivation, Vm was hyperpolar-ized transiently (45 ms) to �80 mV before depolarizing to�30 mV (263 ms) (64). The depolarizing step to �30 mV waschosen because it has been shown that the sensitivity of theKCa channels to Ca2� is relatively high, approximating Ca2�
sensitivity of fura 2 (64, 65). An 11-min “unloading” protocolfollowed in which the HP was maintained at �80 mV tofacilitate vectoral Ca2� release from the SR toward the sar-colemma (63, 64) (Fig. 1B). Vectoral Ca2� release wouldeither be extruded from the cell via Na�/Ca2� exchanger orplasmalemmal Ca2�-ATPase, or activate KCa channels, thusstimulating STOC events. A STOC event was defined as atransient excursion of current with an amplitude greaterthan 10% of the steady-state outward current at measured at�30 mV with a duration of greater than 10 ms (see Fig. 1C).
1181K� CHANNELS IN DIABETIC DYSLIPIDEMIA
J Appl Physiol • VOL 95 • SEPTEMBER 2003 • www.jap.org
by 10.220.33.2 on August 18, 2016
http://jap.physiology.org/D
ownloaded from
Caffeine-sensitive SR Ca2�. After the unloading protocol,cells were superfused with 2CaNa solution containing caf-feine (5 mM), which causes the release of the SR Ca2� stores(64, 65) and activates the KCa channel. During the caffeineapplication, the HP was maintained at �80 mV and the Vm
was stepped to �30 mV for 380 ms every second to determinepeak caffeine-induced KCa current. Peak caffeine-inducedKCa current was normalized to cell capacitance.
Western blotting. A small portion of the RCA was homog-enized in (in mM) 20 HEPES-KOH, 1 EDTA, and 250 sucroseat pH 7.4 supplemented with 0.1 mmol/l phenylmethylsulfo-nyl fluoride, 1 �mol/l pepstatin A, 1 �g/ml aprotinin, 1 �g/mlleupeptin, and 10 mmol/l CHAPS and centrifuged at 1,000 g.The supernatant was fractionated by standard SDS-PAGEand subjected to Western blotting for identification andquantification of KCa-�. Briefly, the proteins (30 �g) wereseparated by 6% SDS-polyacrylamide gels under reducingconditions and electrophoretically transferred to nitrocellu-lose paper. The resulting blots were blocked in TBS contain-
ing 5% nonfat dry milk for 1 h at room temperature. The blotswere incubated with 1:250 affinity-purified KCa883–896 rabbitpolyclonal antibody or 1:50,000 monoclonal antibody anti-�smooth muscle actin in 1% nonfat milk TBS for 12 h at 4°C.Blots were washed three times in TBS for 30 min and thenincubated in horseradish peroxidase-conjugated secondaryantibody (1:4,000) (Amersham Biosciences, Piscataway, NJ)for 1 h. After washing was completed, the blots were incu-bated in substrate for enhanced chemiluminescence for 1 minand autoradiographed on Kodak BioMax film. The bandswere quantified by use of the Image-Pro Plus (MediaCyber-netics, Des Moines, IA) program. The specificity of the label-ing was tested by preincubating the antibody with the corre-sponding peptide.
For Kv1.4 protein Western blotting, coronary artery seg-ments (5–30 mg) were minced on ice and extracted for over-night in the cold room with immunoprecipitation buffer (57)containing 1% Triton X-100, 60 mM n-octyl �-D-glucopyrano-side, 150 mM NaCl, 20 mM Tris, pH 8.0, 2 mM EDTA, 50 mMNaF, 30 mM sodium pyrophosphate, 100 �M sodium or-thovanadate, 1 mM PMSF, and 2 �g/ml leupeptin. Extractswere centrifuged, and the supernatants were transferred tothe fresh tubes and incubated for 1 h (37°C) with 3 M ureaand sample buffer (43) containing 0.31 M Tris, pH 6.8, 2.5%(wt/vol) SDS, 50% (vol/vol) glycerol, 0.125% (wt/vol) bromo-phenol blue, and 1.2% (vol/vol) �-mercaptoethanol. The vol-umes of extracts containing 15 �g of total tissue protein wereloaded and run on triple-wide 3–20% gradient polyacryl-amide gels. The proteins were transferred at 400 mA for 2.5 hin transfer buffer containing 15% methanol to Immobilon-Pmembrane. After transfer, blots were blocked for overnightwith 4% milk in Tween 20-Tris-buffered saline (T-TBS) in thecold room on a rocker. Blots were washed three times withT-TBS, incubated for 3.5 h with rabbit polyclonal anti-Kv1.4antibody (Alomone Labs, Jerusalem, Israel), washed threetimes with T-TBS, incubated with goat anti-rabbit antibodyconjugated with alkaline phosphatase (Zymed, San Fran-cisco, CA), washed twice with T-TBS and once with TBS, andstained with BCIP/NBT (Promega, Madison, WI). Dry blotswere scanned and analyzed by use of NIH Image software.
Immunostaining. A small segment of the RCA was fixed(4% paraformaldehyde, 2% picric acid in 0.1 M phosphate-buffered saline, pH 7.4) for 2 h. Transverse cryostat sections(10 �m) were processed for immunostaining. The sectionswere incubated for 12 h at 4°C with anti-KCa-� subunit 883–896
affinity-purified antibody (1:150). After washing was com-pleted, the tissue sections were incubated for 1 h at roomtemperature with Cy5-donkey anti-rabbit IgG (Jackson Im-munoResearch Laboratory, West Grove, PA). In the controlsection, the KCa-� antibody was inactivated by addition ofexcess amount of corresponding antigenic peptide (100 �g/ml). Images were acquired by use of a confocal microscope(Olympus, Melville, NY). The intensity of the immunofluo-rescent staining was measured in each section with NIHScion Image.
Citrate synthase assay. Citrate synthase assay was per-formed on skeletal muscle homogenates of the right bicepsmuscle to determine the efficacy of the treadmill trainingprotocol by using the methods of Srere (61). Citrate synthaseis a mitochondrial enzyme that responds to a program ofendurance exercise training by increasing in activity.
Statistical analysis. One-way ANOVA was performed todetermine treatment effects for all variables. Statistical sig-nificance was established if the P value was �0.05. Whentreatment effects were statistically different, the least signif-icant difference test was used to perform multiple comparisons.
Fig. 1. Electrophysiological protocol used to elicit spontaneous tran-sient outward currents (STOCs). A: during the “loading” portion ofthe protocol, the holding potential (HP) was maintained at �40 mV.To ensure that channel inactivation did not occur during the subse-quent step pulse, membrane potential (Vm) was hyperpolarized tran-siently (45 ms) to �80 mV (64) before depolarizing to �30 mV (263ms). B: during the “unloading” portion of the protocol, the HP wasmaintained at �80 mV before stepping the membrane to �30 mV. ASTOC event was defined as a transient (�10 ms) excursion of currentwith an amplitude 10% of the steady-state outward current mea-sured at a step potential of �30 mV. C: representative currenttracing obtained during the loading protocol. Arrows indicate occur-rences of STOCs.
1182 K� CHANNELS IN DIABETIC DYSLIPIDEMIA
J Appl Physiol • VOL 95 • SEPTEMBER 2003 • www.jap.org
by 10.220.33.2 on August 18, 2016
http://jap.physiology.org/D
ownloaded from

RESULTS
Characteristics of animal model. Table 1 containsblood chemistry profiles and selected anatomic charac-teristics from the experimental animals. All animalswere monitored daily to ensure that the overall healthof each animal was not compromised, and animals thatwere febrile or otherwise showing signs of ill healthwere not included in the study. Weekly body weightsfor all animals and weekly blood glucose values for alldiabetic animals were obtained for the purpose ofmaintaining a normal developmental weight gain of1% of initial body weight per week. The mean bodyweights for all groups were not statistically differentwith the exception of D animals having a lower bodyweight compared with the DD animals (P � 0.05). Thisresult serves to highlight the difficulty in maintainingpositive energy balance in many diabetic models (8).Although the mean body weight in the D animals waslower than the DD, it is important to note that theanimals in this group were not in negative energybalance, as indicated by an increase in mean bodyweight during the 20-wk study period. The left ventri-cle-to-right ventricle wall thickness ratio was not dif-ferent between groups. As expected, blood glucose val-ues in D and DD were similar and approximatelyfivefold greater compared with C and H, but were notreduced with exercise (DDX). The effects of the exper-imental treatment on specific lipid levels in this animalmodel are similar to those reported elsewhere for sed-entary pigs (69). Similar to the effect on blood glucose,the diabetic dyslipidemia-induced hypercholesterol-emia and hypertriglyceridemia were not prevented bythe program of endurance exercise training. Theseresults show that exercise has minimal effects on low-ering blood glucose or lipids if a chronic hyperglycemicand/or hyperlipidemic condition is established. Mini-mal effect of exercise on the plasma lipid profile washighly predictable because of the high-fat, high-choles-terol diet. This experimental design enabled us to de-termine more directly the effect of exercise on thevasculature, largely independent of plasma lipids. Theefficacy of the treadmill training program was con-firmed by several well-established indexes of centraland peripheral adaptations to endurance exercisetraining. The animals in the DDX group had a signif-icantly lower resting heart rate (exercise-induced bra-
dycardia). Right hip skeletal muscle obtained from theDDX animals had significantly higher citrate synthaseactivity levels than all other groups; however, only thedifference from the DD group reached statistical sig-nificance. These indexes suggest that the treadmilltraining protocol was adequate to stimulate appropri-ate cardiac and skeletal muscle adaptations in DDXanimals.
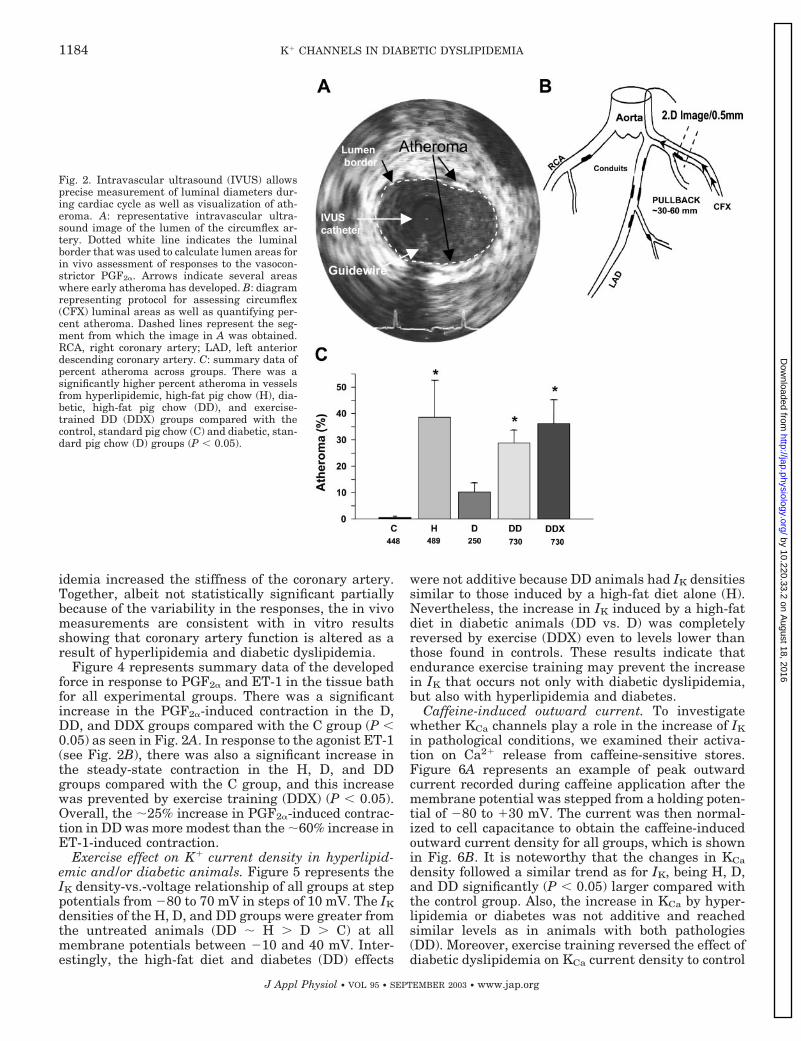
Percent atheroma, in vivo and in vitro responses toagonists. Figure 2 represents the protocol used to as-sess the precise measurement of luminal diametersduring a cardiac cycle as well as visualization of ath-eroma and summary data of percent atheroma. Figure2A contains a representative intravascular ultrasoundimage of the lumen of the circumflex artery. The dottedwhite line marks the luminal border that was used tocalculate lumen areas for in vivo assessment of thedistensibility index as well as responses to the vaso-constrictor PGF2�. Figure 2B represents a schematic ofthe segment of the coronary artery from which thesefunctional data were recorded. The dashed lines repre-sent the segment from which the image in Fig. 2A wasobtained. Figure 2C shows summary data of percentatheroma across groups. There was a significantlyhigher percent atheroma in vessels from H, DD, andDDX compared with the C and D groups (P � 0.05).
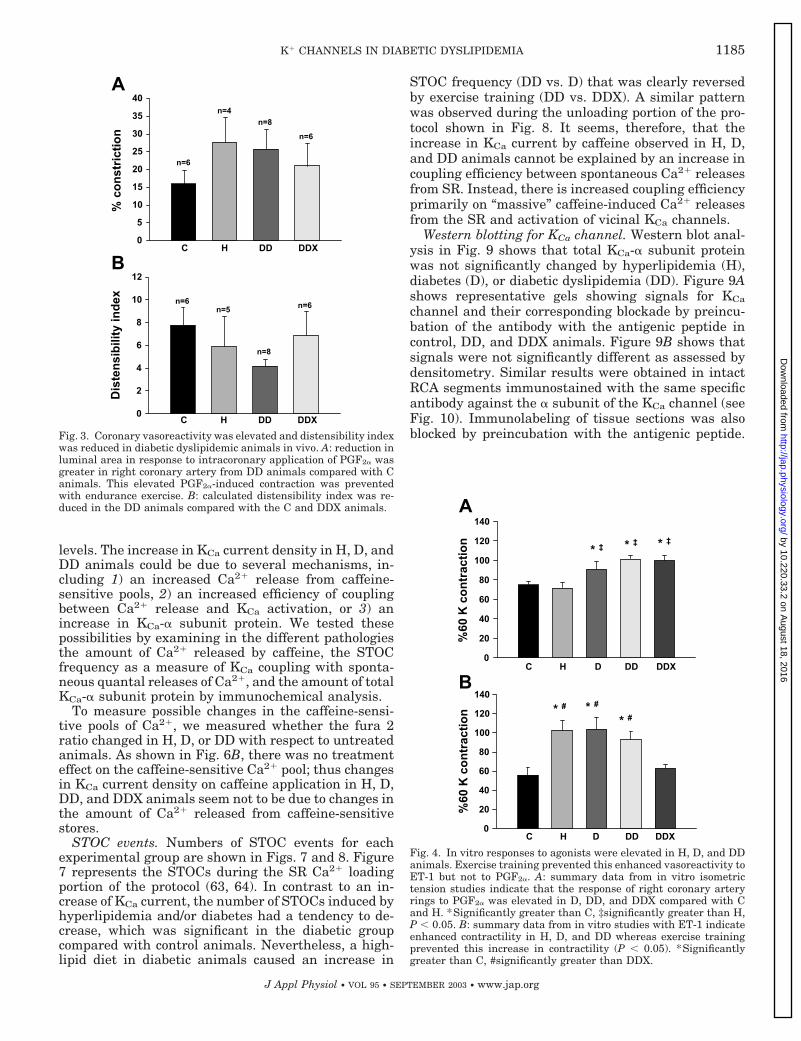
Figure 3 represents summary data for the constric-tion response to the intracoronary application of PGF2�
and distensibility index. The data for the D group werenot included because of lack of sufficient data becausethere were unexpected equipment malfunctions andone incident of premature animal death. As shown inFig. 3A, there was trend toward greater percent con-striction to PGF2� in the H and DD groups comparedwith the C group, but this difference did not reachstatistical significance. Similarly, there was a trend ofa normalization toward the control level of percentconstriction in the DDX. Importantly, the relative mag-nitude of changes in constriction in vivo was greaterthan those noted in vitro in response to PGF2�. Figure3B shows that the baseline distensibility was lower inthe H and DD groups compared with the C and DDXgroups although the differences did not reach statisti-cal significance largely because of the low number ofexperimental animals. In other words, these measuresindicate that both hyperlipidemia and diabetic dyslip-
Table 1. Characteristics of experimental groups
Experimental Groups
C H D DD DDX
Body weight, kg 57.5�1.8# 53.5�2.7 45.0�3.6 56.8�3.0# 53.2�1.0LV/RV 2.9�0.3 3.2�0.3 2.3�0.1 2.6�0.2 2.9�0.3Blood glucose, mg/dl 65.0�2.7 59.0�4.1 320.0�26.2* 344.3�15.8* 344.3�21.9*Triglyceride, mg/dl 35.4�2.5 31.6�3.2 58.6�13.0 64.7�11.3 46.1�13.2Cholesterol, mg/dl 55.2�3.6 366.5�51.1** 68.7�4.6 292.8�38.7** 375.4�71.9**Citrate synthase, �mol �min�1 �g wet wt�1 14.0�3.3 13.9�1.2 10.5�0.8 9.1�1.0 17.8�2.6^Resting HR, beats/min 57�2 57�7 67�4 64�4 52�3&
See text for definition of groups. #Greater than D; *greater than C and H; **greater than C and D; ^greater than H, D, DD; &Less thanD and DD. Triglyceride and cholesterol values were obtained at week 12; all other values were obtained near the end of the 20-wk study.
1183K� CHANNELS IN DIABETIC DYSLIPIDEMIA
J Appl Physiol • VOL 95 • SEPTEMBER 2003 • www.jap.org
by 10.220.33.2 on August 18, 2016
http://jap.physiology.org/D
ownloaded from
idemia increased the stiffness of the coronary artery.Together, albeit not statistically significant partiallybecause of the variability in the responses, the in vivomeasurements are consistent with in vitro resultsshowing that coronary artery function is altered as aresult of hyperlipidemia and diabetic dyslipidemia.
Figure 4 represents summary data of the developedforce in response to PGF2� and ET-1 in the tissue bathfor all experimental groups. There was a significantincrease in the PGF2�-induced contraction in the D,DD, and DDX groups compared with the C group (P �0.05) as seen in Fig. 2A. In response to the agonist ET-1(see Fig. 2B), there was also a significant increase inthe steady-state contraction in the H, D, and DDgroups compared with the C group, and this increasewas prevented by exercise training (DDX) (P � 0.05).Overall, the �25% increase in PGF2�-induced contrac-tion in DD was more modest than the �60% increase inET-1-induced contraction.
Exercise effect on K� current density in hyperlipid-emic and/or diabetic animals. Figure 5 represents theIK density-vs.-voltage relationship of all groups at steppotentials from �80 to 70 mV in steps of 10 mV. The IKdensities of the H, D, and DD groups were greater fromthe untreated animals (DD � H D C) at allmembrane potentials between �10 and 40 mV. Inter-estingly, the high-fat diet and diabetes (DD) effects
were not additive because DD animals had IK densitiessimilar to those induced by a high-fat diet alone (H).Nevertheless, the increase in IK induced by a high-fatdiet in diabetic animals (DD vs. D) was completelyreversed by exercise (DDX) even to levels lower thanthose found in controls. These results indicate thatendurance exercise training may prevent the increasein IK that occurs not only with diabetic dyslipidemia,but also with hyperlipidemia and diabetes.
Caffeine-induced outward current. To investigatewhether KCa channels play a role in the increase of IKin pathological conditions, we examined their activa-tion on Ca2� release from caffeine-sensitive stores.Figure 6A represents an example of peak outwardcurrent recorded during caffeine application after themembrane potential was stepped from a holding poten-tial of �80 to �30 mV. The current was then normal-ized to cell capacitance to obtain the caffeine-inducedoutward current density for all groups, which is shownin Fig. 6B. It is noteworthy that the changes in KCadensity followed a similar trend as for IK, being H, D,and DD significantly (P � 0.05) larger compared withthe control group. Also, the increase in KCa by hyper-lipidemia or diabetes was not additive and reachedsimilar levels as in animals with both pathologies(DD). Moreover, exercise training reversed the effect ofdiabetic dyslipidemia on KCa current density to control
Fig. 2. Intravascular ultrasound (IVUS) allowsprecise measurement of luminal diameters dur-ing cardiac cycle as well as visualization of ath-eroma. A: representative intravascular ultra-sound image of the lumen of the circumflex ar-tery. Dotted white line indicates the luminalborder that was used to calculate lumen areas forin vivo assessment of responses to the vasocon-strictor PGF2�. Arrows indicate several areaswhere early atheroma has developed. B: diagramrepresenting protocol for assessing circumflex(CFX) luminal areas as well as quantifying per-cent atheroma. Dashed lines represent the seg-ment from which the image in A was obtained.RCA, right coronary artery; LAD, left anteriordescending coronary artery. C: summary data ofpercent atheroma across groups. There was asignificantly higher percent atheroma in vesselsfrom hyperlipidemic, high-fat pig chow (H), dia-betic, high-fat pig chow (DD), and exercise-trained DD (DDX) groups compared with thecontrol, standard pig chow (C) and diabetic, stan-dard pig chow (D) groups (P � 0.05).
1184 K� CHANNELS IN DIABETIC DYSLIPIDEMIA
J Appl Physiol • VOL 95 • SEPTEMBER 2003 • www.jap.org
by 10.220.33.2 on August 18, 2016
http://jap.physiology.org/D
ownloaded from
levels. The increase in KCa current density in H, D, andDD animals could be due to several mechanisms, in-cluding 1) an increased Ca2� release from caffeine-sensitive pools, 2) an increased efficiency of couplingbetween Ca2� release and KCa activation, or 3) anincrease in KCa-� subunit protein. We tested thesepossibilities by examining in the different pathologiesthe amount of Ca2� released by caffeine, the STOCfrequency as a measure of KCa coupling with sponta-neous quantal releases of Ca2�, and the amount of totalKCa-� subunit protein by immunochemical analysis.
To measure possible changes in the caffeine-sensi-tive pools of Ca2�, we measured whether the fura 2ratio changed in H, D, or DD with respect to untreatedanimals. As shown in Fig. 6B, there was no treatmenteffect on the caffeine-sensitive Ca2� pool; thus changesin KCa current density on caffeine application in H, D,DD, and DDX animals seem not to be due to changes inthe amount of Ca2� released from caffeine-sensitivestores.
STOC events. Numbers of STOC events for eachexperimental group are shown in Figs. 7 and 8. Figure7 represents the STOCs during the SR Ca2� loadingportion of the protocol (63, 64). In contrast to an in-crease of KCa current, the number of STOCs induced byhyperlipidemia and/or diabetes had a tendency to de-crease, which was significant in the diabetic groupcompared with control animals. Nevertheless, a high-lipid diet in diabetic animals caused an increase in
STOC frequency (DD vs. D) that was clearly reversedby exercise training (DD vs. DDX). A similar patternwas observed during the unloading portion of the pro-tocol shown in Fig. 8. It seems, therefore, that theincrease in KCa current by caffeine observed in H, D,and DD animals cannot be explained by an increase incoupling efficiency between spontaneous Ca2� releasesfrom SR. Instead, there is increased coupling efficiencyprimarily on “massive” caffeine-induced Ca2� releasesfrom the SR and activation of vicinal KCa channels.
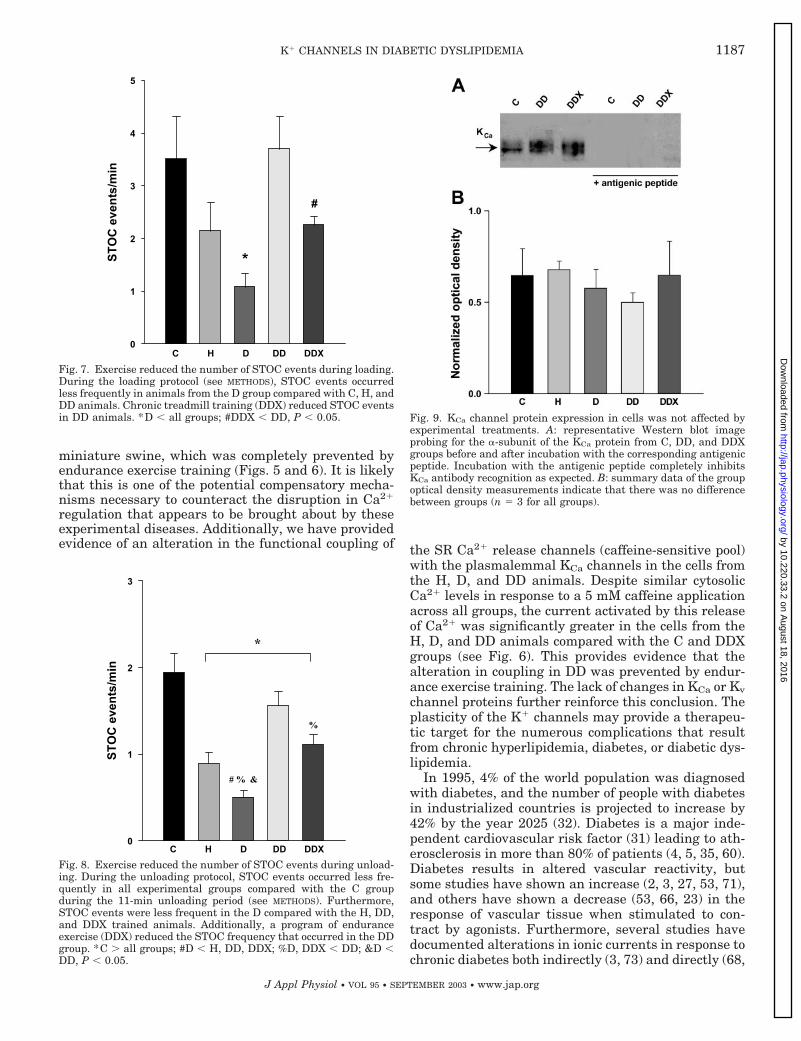
Western blotting for KCa channel. Western blot anal-ysis in Fig. 9 shows that total KCa-� subunit proteinwas not significantly changed by hyperlipidemia (H),diabetes (D), or diabetic dyslipidemia (DD). Figure 9Ashows representative gels showing signals for KCa
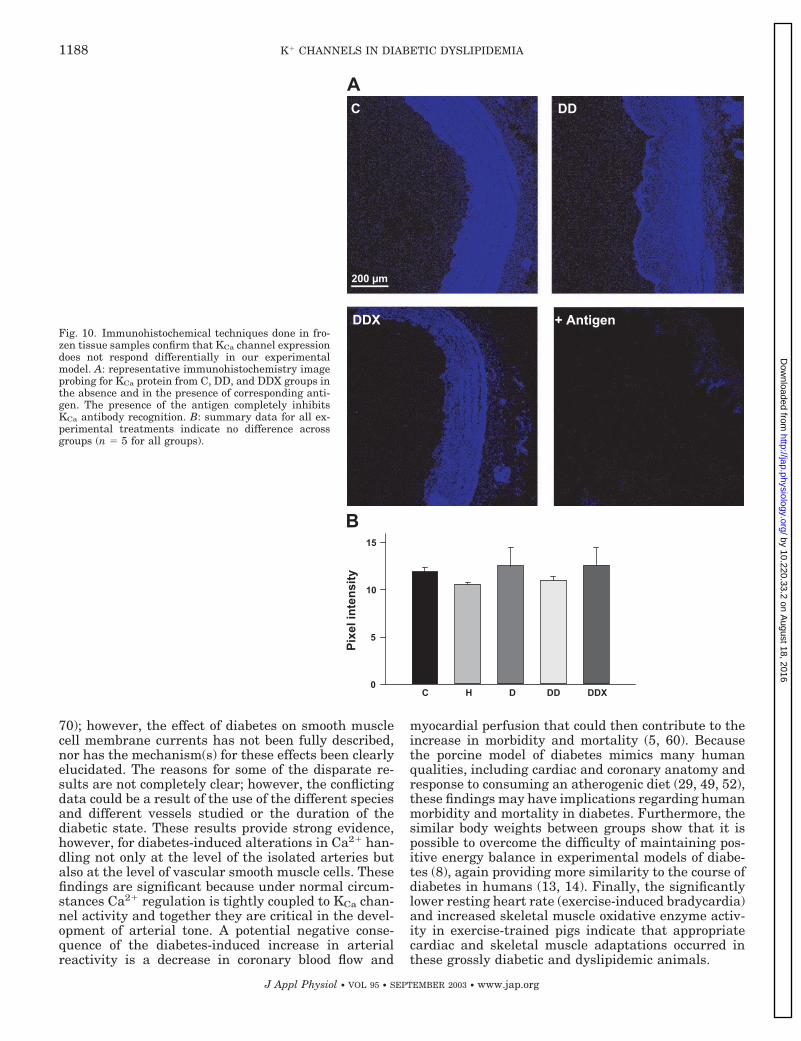
channel and their corresponding blockade by preincu-bation of the antibody with the antigenic peptide incontrol, DD, and DDX animals. Figure 9B shows thatsignals were not significantly different as assessed bydensitometry. Similar results were obtained in intactRCA segments immunostained with the same specificantibody against the � subunit of the KCa channel (seeFig. 10). Immunolabeling of tissue sections was alsoblocked by preincubation with the antigenic peptide.
Fig. 4. In vitro responses to agonists were elevated in H, D, and DDanimals. Exercise training prevented this enhanced vasoreactivity toET-1 but not to PGF2�. A: summary data from in vitro isometrictension studies indicate that the response of right coronary arteryrings to PGF2� was elevated in D, DD, and DDX compared with Cand H. *Significantly greater than C, ‡significantly greater than H,P � 0.05. B: summary data from in vitro studies with ET-1 indicateenhanced contractility in H, D, and DD whereas exercise trainingprevented this increase in contractility (P � 0.05). *Significantlygreater than C, #significantly greater than DDX.
Fig. 3. Coronary vasoreactivity was elevated and distensibility indexwas reduced in diabetic dyslipidemic animals in vivo. A: reduction inluminal area in response to intracoronary application of PGF2� wasgreater in right coronary artery from DD animals compared with Canimals. This elevated PGF2�-induced contraction was preventedwith endurance exercise. B: calculated distensibility index was re-duced in the DD animals compared with the C and DDX animals.
1185K� CHANNELS IN DIABETIC DYSLIPIDEMIA
J Appl Physiol • VOL 95 • SEPTEMBER 2003 • www.jap.org
by 10.220.33.2 on August 18, 2016
http://jap.physiology.org/D
ownloaded from
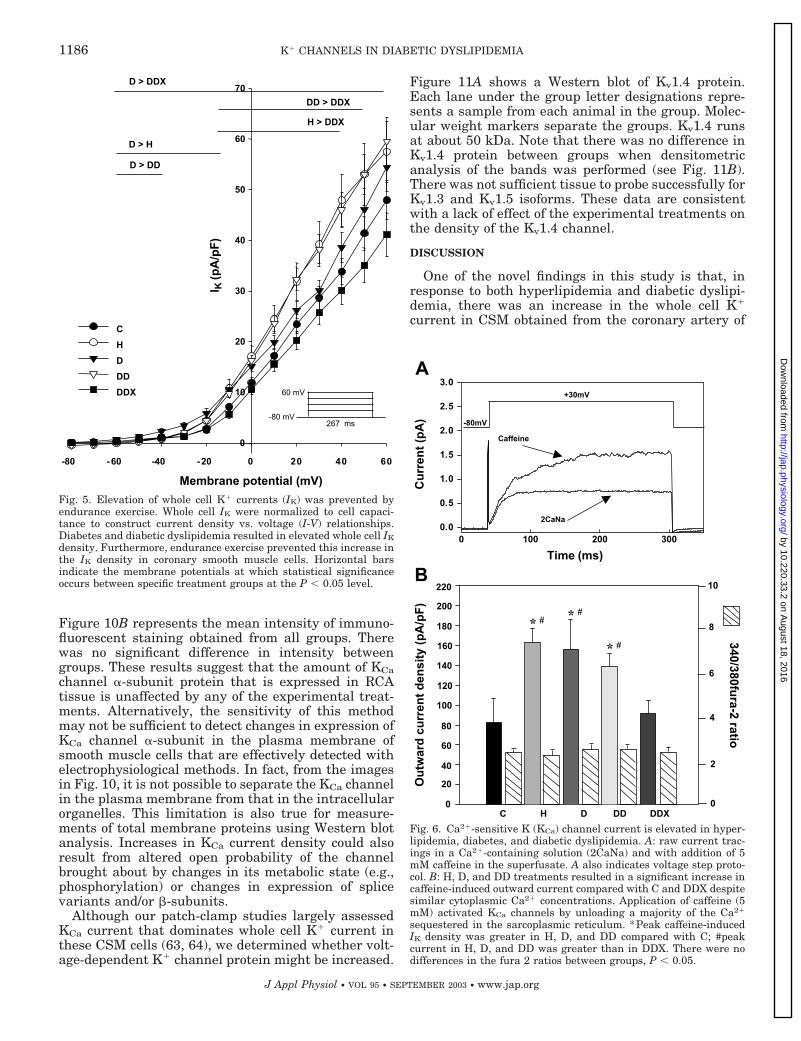
Figure 10B represents the mean intensity of immuno-fluorescent staining obtained from all groups. Therewas no significant difference in intensity betweengroups. These results suggest that the amount of KCachannel �-subunit protein that is expressed in RCAtissue is unaffected by any of the experimental treat-ments. Alternatively, the sensitivity of this methodmay not be sufficient to detect changes in expression ofKCa channel �-subunit in the plasma membrane ofsmooth muscle cells that are effectively detected withelectrophysiological methods. In fact, from the imagesin Fig. 10, it is not possible to separate the KCa channelin the plasma membrane from that in the intracellularorganelles. This limitation is also true for measure-ments of total membrane proteins using Western blotanalysis. Increases in KCa current density could alsoresult from altered open probability of the channelbrought about by changes in its metabolic state (e.g.,phosphorylation) or changes in expression of splicevariants and/or �-subunits.
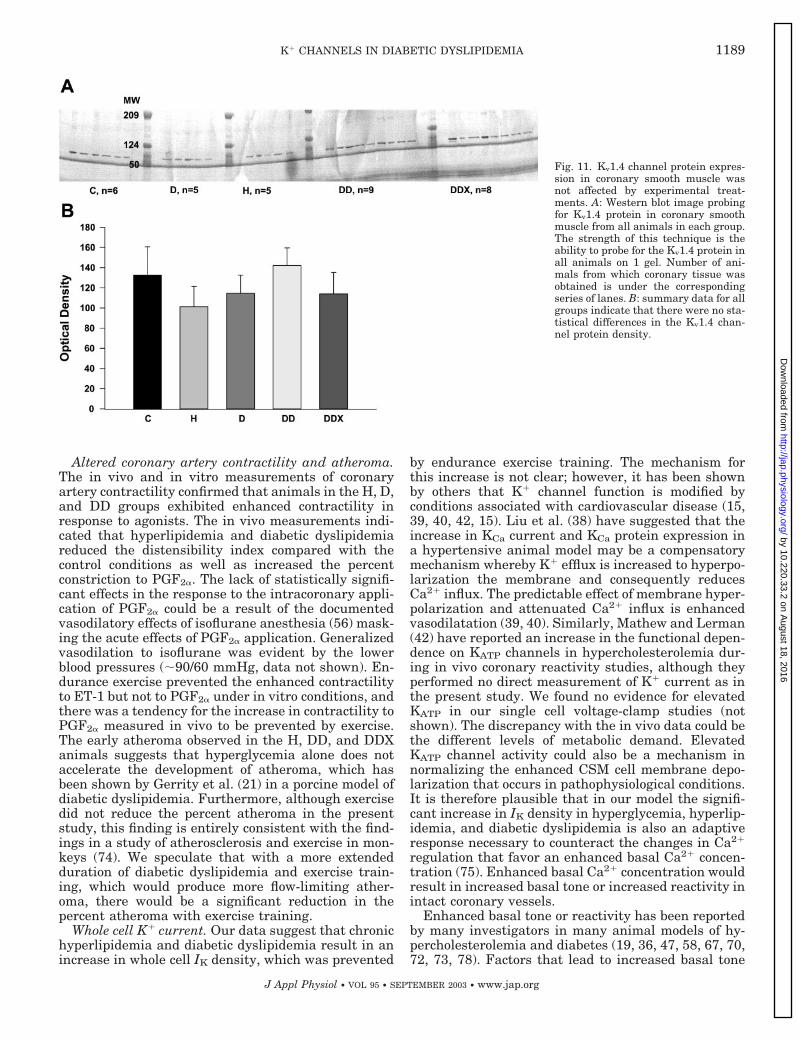
Although our patch-clamp studies largely assessedKCa current that dominates whole cell K� current inthese CSM cells (63, 64), we determined whether volt-age-dependent K� channel protein might be increased.
Figure 11A shows a Western blot of Kv1.4 protein.Each lane under the group letter designations repre-sents a sample from each animal in the group. Molec-ular weight markers separate the groups. Kv1.4 runsat about 50 kDa. Note that there was no difference inKv1.4 protein between groups when densitometricanalysis of the bands was performed (see Fig. 11B).There was not sufficient tissue to probe successfully forKv1.3 and Kv1.5 isoforms. These data are consistentwith a lack of effect of the experimental treatments onthe density of the Kv1.4 channel.
DISCUSSION
One of the novel findings in this study is that, inresponse to both hyperlipidemia and diabetic dyslipi-demia, there was an increase in the whole cell K�
current in CSM obtained from the coronary artery of
Fig. 6. Ca2�-sensitive K (KCa) channel current is elevated in hyper-lipidemia, diabetes, and diabetic dyslipidemia. A: raw current trac-ings in a Ca2�-containing solution (2CaNa) and with addition of 5mM caffeine in the superfusate. A also indicates voltage step proto-col. B: H, D, and DD treatments resulted in a significant increase incaffeine-induced outward current compared with C and DDX despitesimilar cytoplasmic Ca2� concentrations. Application of caffeine (5mM) activated KCa channels by unloading a majority of the Ca2�
sequestered in the sarcoplasmic reticulum. *Peak caffeine-inducedIK density was greater in H, D, and DD compared with C; #peakcurrent in H, D, and DD was greater than in DDX. There were nodifferences in the fura 2 ratios between groups, P � 0.05.
Fig. 5. Elevation of whole cell K� currents (IK) was prevented byendurance exercise. Whole cell IK were normalized to cell capaci-tance to construct current density vs. voltage (I-V) relationships.Diabetes and diabetic dyslipidemia resulted in elevated whole cell IK
density. Furthermore, endurance exercise prevented this increase inthe IK density in coronary smooth muscle cells. Horizontal barsindicate the membrane potentials at which statistical significanceoccurs between specific treatment groups at the P � 0.05 level.
1186 K� CHANNELS IN DIABETIC DYSLIPIDEMIA
J Appl Physiol • VOL 95 • SEPTEMBER 2003 • www.jap.org
by 10.220.33.2 on August 18, 2016
http://jap.physiology.org/D
ownloaded from
miniature swine, which was completely prevented byendurance exercise training (Figs. 5 and 6). It is likelythat this is one of the potential compensatory mecha-nisms necessary to counteract the disruption in Ca2�
regulation that appears to be brought about by theseexperimental diseases. Additionally, we have providedevidence of an alteration in the functional coupling of the SR Ca2� release channels (caffeine-sensitive pool)
with the plasmalemmal KCa channels in the cells fromthe H, D, and DD animals. Despite similar cytosolicCa2� levels in response to a 5 mM caffeine applicationacross all groups, the current activated by this releaseof Ca2� was significantly greater in the cells from theH, D, and DD animals compared with the C and DDXgroups (see Fig. 6). This provides evidence that thealteration in coupling in DD was prevented by endur-ance exercise training. The lack of changes in KCa or Kv
channel proteins further reinforce this conclusion. Theplasticity of the K� channels may provide a therapeu-tic target for the numerous complications that resultfrom chronic hyperlipidemia, diabetes, or diabetic dys-lipidemia.
In 1995, 4% of the world population was diagnosedwith diabetes, and the number of people with diabetesin industrialized countries is projected to increase by42% by the year 2025 (32). Diabetes is a major inde-pendent cardiovascular risk factor (31) leading to ath-erosclerosis in more than 80% of patients (4, 5, 35, 60).Diabetes results in altered vascular reactivity, butsome studies have shown an increase (2, 3, 27, 53, 71),and others have shown a decrease (53, 66, 23) in theresponse of vascular tissue when stimulated to con-tract by agonists. Furthermore, several studies havedocumented alterations in ionic currents in response tochronic diabetes both indirectly (3, 73) and directly (68,
Fig. 7. Exercise reduced the number of STOC events during loading.During the loading protocol (see METHODS), STOC events occurredless frequently in animals from the D group compared with C, H, andDD animals. Chronic treadmill training (DDX) reduced STOC eventsin DD animals. *D � all groups; #DDX � DD, P � 0.05.
Fig. 8. Exercise reduced the number of STOC events during unload-ing. During the unloading protocol, STOC events occurred less fre-quently in all experimental groups compared with the C groupduring the 11-min unloading period (see METHODS). Furthermore,STOC events were less frequent in the D compared with the H, DD,and DDX trained animals. Additionally, a program of enduranceexercise (DDX) reduced the STOC frequency that occurred in the DDgroup. *C all groups; #D � H, DD, DDX; %D, DDX � DD; &D �DD, P � 0.05.
Fig. 9. KCa channel protein expression in cells was not affected byexperimental treatments. A: representative Western blot imageprobing for the �-subunit of the KCa protein from C, DD, and DDXgroups before and after incubation with the corresponding antigenicpeptide. Incubation with the antigenic peptide completely inhibitsKCa antibody recognition as expected. B: summary data of the groupoptical density measurements indicate that there was no differencebetween groups (n � 3 for all groups).
1187K� CHANNELS IN DIABETIC DYSLIPIDEMIA
J Appl Physiol • VOL 95 • SEPTEMBER 2003 • www.jap.org
by 10.220.33.2 on August 18, 2016
http://jap.physiology.org/D
ownloaded from
70); however, the effect of diabetes on smooth musclecell membrane currents has not been fully described,nor has the mechanism(s) for these effects been clearlyelucidated. The reasons for some of the disparate re-sults are not completely clear; however, the conflictingdata could be a result of the use of the different speciesand different vessels studied or the duration of thediabetic state. These results provide strong evidence,however, for diabetes-induced alterations in Ca2� han-dling not only at the level of the isolated arteries butalso at the level of vascular smooth muscle cells. Thesefindings are significant because under normal circum-stances Ca2� regulation is tightly coupled to KCa chan-nel activity and together they are critical in the devel-opment of arterial tone. A potential negative conse-quence of the diabetes-induced increase in arterialreactivity is a decrease in coronary blood flow and
myocardial perfusion that could then contribute to theincrease in morbidity and mortality (5, 60). Becausethe porcine model of diabetes mimics many humanqualities, including cardiac and coronary anatomy andresponse to consuming an atherogenic diet (29, 49, 52),these findings may have implications regarding humanmorbidity and mortality in diabetes. Furthermore, thesimilar body weights between groups show that it ispossible to overcome the difficulty of maintaining pos-itive energy balance in experimental models of diabe-tes (8), again providing more similarity to the course ofdiabetes in humans (13, 14). Finally, the significantlylower resting heart rate (exercise-induced bradycardia)and increased skeletal muscle oxidative enzyme activ-ity in exercise-trained pigs indicate that appropriatecardiac and skeletal muscle adaptations occurred inthese grossly diabetic and dyslipidemic animals.
Fig. 10. Immunohistochemical techniques done in fro-zen tissue samples confirm that KCa channel expressiondoes not respond differentially in our experimentalmodel. A: representative immunohistochemistry imageprobing for KCa protein from C, DD, and DDX groups inthe absence and in the presence of corresponding anti-gen. The presence of the antigen completely inhibitsKCa antibody recognition. B: summary data for all ex-perimental treatments indicate no difference acrossgroups (n � 5 for all groups).
1188 K� CHANNELS IN DIABETIC DYSLIPIDEMIA
J Appl Physiol • VOL 95 • SEPTEMBER 2003 • www.jap.org
by 10.220.33.2 on August 18, 2016
http://jap.physiology.org/D
ownloaded from
Altered coronary artery contractility and atheroma.The in vivo and in vitro measurements of coronaryartery contractility confirmed that animals in the H, D,and DD groups exhibited enhanced contractility inresponse to agonists. The in vivo measurements indi-cated that hyperlipidemia and diabetic dyslipidemiareduced the distensibility index compared with thecontrol conditions as well as increased the percentconstriction to PGF2�. The lack of statistically signifi-cant effects in the response to the intracoronary appli-cation of PGF2� could be a result of the documentedvasodilatory effects of isoflurane anesthesia (56) mask-ing the acute effects of PGF2� application. Generalizedvasodilation to isoflurane was evident by the lowerblood pressures (�90/60 mmHg, data not shown). En-durance exercise prevented the enhanced contractilityto ET-1 but not to PGF2� under in vitro conditions, andthere was a tendency for the increase in contractility toPGF2� measured in vivo to be prevented by exercise.The early atheroma observed in the H, DD, and DDXanimals suggests that hyperglycemia alone does notaccelerate the development of atheroma, which hasbeen shown by Gerrity et al. (21) in a porcine model ofdiabetic dyslipidemia. Furthermore, although exercisedid not reduce the percent atheroma in the presentstudy, this finding is entirely consistent with the find-ings in a study of atherosclerosis and exercise in mon-keys (74). We speculate that with a more extendedduration of diabetic dyslipidemia and exercise train-ing, which would produce more flow-limiting ather-oma, there would be a significant reduction in thepercent atheroma with exercise training.
Whole cell K� current. Our data suggest that chronichyperlipidemia and diabetic dyslipidemia result in anincrease in whole cell IK density, which was prevented
by endurance exercise training. The mechanism forthis increase is not clear; however, it has been shownby others that K� channel function is modified byconditions associated with cardiovascular disease (15,39, 40, 42, 15). Liu et al. (38) have suggested that theincrease in KCa current and KCa protein expression ina hypertensive animal model may be a compensatorymechanism whereby K� efflux is increased to hyperpo-larization the membrane and consequently reducesCa2� influx. The predictable effect of membrane hyper-polarization and attenuated Ca2� influx is enhancedvasodilatation (39, 40). Similarly, Mathew and Lerman(42) have reported an increase in the functional depen-dence on KATP channels in hypercholesterolemia dur-ing in vivo coronary reactivity studies, although theyperformed no direct measurement of K� current as inthe present study. We found no evidence for elevatedKATP in our single cell voltage-clamp studies (notshown). The discrepancy with the in vivo data could bethe different levels of metabolic demand. ElevatedKATP channel activity could also be a mechanism innormalizing the enhanced CSM cell membrane depo-larization that occurs in pathophysiological conditions.It is therefore plausible that in our model the signifi-cant increase in IK density in hyperglycemia, hyperlip-idemia, and diabetic dyslipidemia is also an adaptiveresponse necessary to counteract the changes in Ca2�
regulation that favor an enhanced basal Ca2� concen-tration (75). Enhanced basal Ca2� concentration wouldresult in increased basal tone or increased reactivity inintact coronary vessels.
Enhanced basal tone or reactivity has been reportedby many investigators in many animal models of hy-percholesterolemia and diabetes (19, 36, 47, 58, 67, 70,72, 73, 78). Factors that lead to increased basal tone
Fig. 11. Kv1.4 channel protein expres-sion in coronary smooth muscle wasnot affected by experimental treat-ments. A: Western blot image probingfor Kv1.4 protein in coronary smoothmuscle from all animals in each group.The strength of this technique is theability to probe for the Kv1.4 protein inall animals on 1 gel. Number of ani-mals from which coronary tissue wasobtained is under the correspondingseries of lanes. B: summary data for allgroups indicate that there were no sta-tistical differences in the Kv1.4 chan-nel protein density.
1189K� CHANNELS IN DIABETIC DYSLIPIDEMIA
J Appl Physiol • VOL 95 • SEPTEMBER 2003 • www.jap.org
by 10.220.33.2 on August 18, 2016
http://jap.physiology.org/D
ownloaded from

and elevated vessel reactivity include an increasedbasal cytosolic Ca2�, increased circulating concentra-tion of agonist, increased number of agonist receptors,a reduction in Ca2� removal mechanisms, or a combi-nation of these factors. C. A. Witczak and M. Sturek(unpublished observations) have studied fura 2-loadedCSM cells dispersed from the same experimentalmodel to determine basal Ca2�. They reported a signif-icantly greater basal Ca2� in diabetic animals fed anatherogenic diet (diabetic dyslipidemia) compared withcontrol animals. Furthermore, basal cytosolic Ca2� lev-els in diabetic dyslipidemic exercise-trained animalswere less than control values, which is consistent withan exercise-induced augmentation of Ca2� efflux (75).
There is little information on the effect of diabetesand diabetic dyslipidemia on Ca2� efflux mechanismsin CSM from conduit arteries. We have reported thatthe activity of the Na�/Ca2� exchanger was blunted inCSM from diabetic dyslipidemic swine and that thisreduction in Na�/Ca2� exchange activity was also pre-vented by endurance exercise training (45). The netresult of reduced Na�/Ca2� exchange activity couldalso lead to the elevated basal Ca2� levels seen in theCSM cells in this animal model. Taken together, theelevation in IK density in the CSM cells from thediseased animals appears to be a compensatory mech-anism in response to elevated basal Ca2� levels.
Immunohistochemistry and Western blotting for KCa
and Kv channel. Two different techniques were used todetermine the amount of KCa channel �-subunit pro-tein that was expressed in the vessels. Analysis of theWestern blots showed that there was no difference inthe density of the bands located at the 125-kDa posi-tion. Furthermore, by using immunocytochemistrytechniques, tissue was stained with a fluorescentlylabeled antibody directed against the KCa channel. Themean pixel intensity for all groups indicated that therewas no difference between the experimental treat-ments.
To date, the distribution of Kv channels in porcinevascular smooth muscle is unknown, largely owing tothe Kv channel dependence on vessel location but alsobecause of the difficulty in identifying all the differentK� channel subtypes. Immunoblotting for the K�
channel subtype Kv1.4 was performed as an initialprobe to begin to determine which K� channel subtypewas responsible for the change in whole cell K� currentin this present animal model. Several investigatorshave confirmed that the mRNA for the �-subunit forKv1.4 are indeed expressed in vascular smooth musclecells (77, 79). The data show that the Kv1.4 member ofthe K� channel family was not affected by the experi-mental treatments. Taken together, these data sug-gests that diabetes, diabetic dyslipidemia, and endur-ance exercise training did not affect the expression ofKCa or the Kv1.4 channel proteins in the coronaryarteries. It is possible, however, that functional gatingor second messenger, i.e., Ca2�, modulation of thechannels was altered. These alternative explanationsrepresent an area for further research.
Caffeine-induced outward current. The application ofa caffeine-containing superfusate (5 mM) was per-formed to activate the KCa channel and also to indi-rectly assess the caffeine-sensitive Ca2� stores. Previ-ous investigators have shown that such an applicationto CSM cells releases the majority of Ca2� from the SR(62, 63). This release of Ca2� from the caffeine-sensi-tive stores causes rapid activation of the KCa channelsand a significant transient increase in current. A largerincrease in the current density on activation by caf-feine in one group compared with another could occurfor several reasons. A greater caffeine-sensitive cur-rent could be the result of greater caffeine-sensitivestores, a greater number of KCa channels, or an alter-ation in the coupling of the SR Ca2� release channels tothe KCa channels such that the same amount of Ca2�
released from the SR activated more KCa channels. It isunlikely that the size of the caffeine-sensitive Ca2�
stores was greater in the diabetic dyslipidemic groupbecause there were no differences in the peak fura 2ratios on caffeine application between groups. Becausethe immunohistochemistry and immunoblotting showedno statistical differences between groups, it is unlikelythat there were a greater number of KCa channel �subunits in the group that exhibited the greatest caf-feine-induced outward current. A more likely explana-tion is that the function of the channels was altered,despite a lack of change in protein expression.
Another plausible explanation for the increase in thecaffeine-activated outward current in CSM cells fromanimals with diabetic dyslipidemia is that the couplingof the ryanodine receptor to the KCa channel was al-tered in a way to cause greater activation of the KCachannel. Such an alteration could stem from a changein the distribution of ryanodine receptor or a differencein the regulation of the activation of the KCa channel.Indeed, Lohn et al. (41) have provided evidence for aregulatory function of the ryanodine receptor isoformthat is expressed predominantly in vascular smoothmuscle, ryanodine receptor 3. They have shown thatmice deficient of this isoform of the ryanodine receptorhave a remarkable change in the voltage dependence ofthe activation of KCa channel, which was measured inSTOCs.
STOC events. It has been clearly demonstrated thatquantal releases of Ca2� from the SR in the form ofCa2� sparks plays a important role in activating theKCa channel, initiating STOCs (18, 24, 33), and alter-ing Vm (12, 38) or vessel lumen diameter (50, 40).Hyperpolarization by activation of the KCa channeldecreases Ca2� influx via VGCC and leads to vesselrelaxation (40, 50). Several groups have shown that, inpathological conditions, a compensatory increase inKCa channel activity or protein expression occurs, in anattempt to augment membrane hyperpolarization (38,55). STOCs were not quantified in these previous stud-ies, however. Importantly, Stehno-Bittel et al. (63)have proposed that STOC frequency is reduced in CSMobtained from exercise-trained miniature swine be-cause of an enhanced ability to extrude Ca2� withoutactivating the KCa channel. Taken together, these
1190 K� CHANNELS IN DIABETIC DYSLIPIDEMIA
J Appl Physiol • VOL 95 • SEPTEMBER 2003 • www.jap.org
by 10.220.33.2 on August 18, 2016
http://jap.physiology.org/D
ownloaded from

studies suggest that STOC events can be altered undercertain conditions, and there may be a functional rolerelated to alterations in STOC frequency.
The functional role of STOCs in conduit-sized arter-ies of the pig is still not entirely known; however, it isclear that there is low basal activity of STOCs. The factthat there is basal activity of STOCs suggests thatthere may be a functional role of STOCs in CSM. Ourwhole cell patch-clamp experiments consisted of twodistinct voltage-step protocols to better elucidate theeffect of hyperlipidemia and hyperglycemia on STOCevents. The first protocol was an SR Ca2� loadingprotocol in which holding potential was set to �40 mVand depolarizing step pulses were applied at 0.2 Hz.The purpose of this protocol was to facilitate Ca2�
influx to gradually load the SR with Ca2� by the slightactivation of VGCC and the reverse mode action of theNa�/Ca2� exchanger. Our goal for the first portion ofthe protocol was to have similar SR Ca2� stores in allgroups, to determine how STOC frequency was affectedby our experimental conditions.
The STOC frequency in the D group was signifi-cantly lower than in C, H, and DD, suggesting thathyperglycemia is partially responsible for the diminu-tion of STOC frequency in these cells. The STOCevents in the DD group were not different from in C orH, but there were significantly fewer STOC events inthe DDX group compared with the DD group. Finally,when animals are diabetic, dyslipidemic, and exercise-trained, the number of STOC events was normalized tolevels seen in the C group. Changes in STOC frequencycan be the result of the amount of KCa channel proteinexpressed in the cells, the activation characteristics ofthe KCa channel, the amount of Ca2� in the SR, thejuxtaposition of the ryanodine receptor to the KCachannel (microdomain sublocalization), and the gatingcharacteristics of the ryanodine receptor. There was areduction in the STOC frequency in the D group de-spite an increase in KCa current, an increase in thecaffeine-sensitive IK, and similar caffeine-induced[Ca2�]i. One explanation for these results is that thecoupling of the ryanodine receptor to the KCa channelwas impaired as a result of chronic diabetes such that,under steady-state activation of the KCa channel (SRCa loading and unloading protocols), fewer KCa chan-nels were activated. This reduction in steady-stateactivation of the KCa channel could be due to fewerquantal Ca2� release events (sparks) or the same num-ber of sparks but a larger physical distance betweenthe two proteins.
The result with the DD group was somewhat puz-zling because we had expected even fewer STOCs inthe DD group than in the D group. Instead, we ob-served an increase in STOC frequency in DD comparedwith D animals. It is possible that the combination ofdiabetes and dyslipidemia resulted in more of an in-crease in the KCa current density that senses sponta-neous quantal release of Ca2� and therefore a relativeincrease in STOC activation. An additional explana-tion involves an improved coupling of the ryanodinereceptor with the KCa channel in the DD group. In this
group, caffeine-sensitive IK was also elevated above Cand DDX, with no between-group differences in thecaffeine-induced myoplasmic Ca2� concentration. A re-localization of the ryanodine receptor with the KCachannel or a relative increase in the amount of vectoralCa2� release toward the subsarcolemmal space mighthave occurred in response to diabetic dyslipidemia.Regardless of the mechanism (changes in KCa protein,ryanodine receptor and KCa channel coupling, or sparkfrequency), the diabetic dyslipidemia-induced increasein STOC events with respect to those induced by dia-betes alone was returned to control conditions afterendurance exercise training. The ryanodine receptorchannel number, spark frequency, or colocalizationstudies were not determined in the present study.
In conclusion, our data indicate that 20 wk of chronichyperlipidemia, diabetes, and diabetic dyslipidemiasignificantly increased whole cell IK in CSM cells inswine. The immunoblotting results suggest that KCaand Kv protein was not changed; instead, functionalactivation of KCa channels was increased. Moderateendurance exercise training appeared to prevent thechanges in whole cell IK and either normalized thealtered coupling of the KCa channel and ryanodinereceptor or rescued the pattern of bolus Ca2� release.
We thank Drs. Joseph Dixon and Ela Wysocka for assistance inperforming the Kv1.4 channel immunoblots and Dr. Nancy Dietz fortechnical assistance.
DISCLOSURES
This work was supported by National Institutes of Health GrantsRR-13223 and HL-62552 (to M. Sturek), HL-47382 (to L. Toro), andIndividual National Research Service Award HL-107094 (to E. A.Mokelke).
REFERENCES
1. Abebe W and MacLeod KM. Protein kinase C-mediated con-tractile responses of arteries from diabetic rats. Br J Pharmacol225: 29–36, 1990.
2. Agrawal DK, Bhimji S, and McNeill HH. Effect of chronicexperimental diabetes on vascular smooth muscle function inrabbit carotid artery. J Cardiovasc Pharmacol 9: 584–593, 1987.
3. Agrawal DK and McNeill JH. Vascular responses to agonistsin rat mesenteric artery from diabetic rats. Can J Physiol Phar-macol 65: 1484–1490, 1987.
4. Barrett-Connor EL, Cohn BA, Wingard DL, and EdelsteinSL. Why is diabetes mellitus a stronger risk factor for fatalischemia disease in women than in men? The Rancho BernardoStudy. JAMA 265: 627–631, 1991.
5. Bell DSH. Diabetic cardiomyopathy: a unique entity or a com-plication of coronary artery disease. Diabetes Care 18: 708–714,1995.
6. Bell DSH. Diabetes mellitus and coronary artery disease. CoronArtery Dis 7: 715–722, 1996.
7. Benham CD and Bolton TB. Spontaneous transient outwardcurrents in single visceral and vascular smooth muscle cells ofthe rabbit. J Physiol 381: 385–406, 1986.
8. Boullion RD, Mokelke EA, Wamhoff BR, Otis CR, WenzelJ, Dixon JL, and Sturek M. Porcine model of diabetic dyslip-idemia: insulin and feed algorithms for mimicking diabetes inhumans. Comp Med 53: 42–52, 2003.
9. Bove AA and Dewey JD. Proximal coronary vasomotor reac-tivity after exercise training in dogs. Circulation 71: 620–625,1985.
10. Bowles DK, Hu Q, Laughlin MH, and Sturek M. Exercisetraining increases L-type calcium current density in coronary
1191K� CHANNELS IN DIABETIC DYSLIPIDEMIA
J Appl Physiol • VOL 95 • SEPTEMBER 2003 • www.jap.org
by 10.220.33.2 on August 18, 2016
http://jap.physiology.org/D
ownloaded from

smooth muscle. Am J Physiol Heart Circ Physiol 275: H2159–H2169, 1998.
11. Bowles DK, Laughlin MH, and Sturek M. Exercise trainingincreases K� channel contribution to regulation of coronaryarterial tone. J Appl Physiol 84: 1225–1233, 1998.
12. Brayden JE and Nelson MT. Regulation of arterial tone byactivation of calcium-dependent potassium channels. Science256: 532–535, 1992.
13. Chaturvedi N, Fuller JH, and The WHO MultinationalStudy Group. Mortality risk by body weight and weight changein people with NIDDM: The WHO Multinational Study of Vas-cular Disease in Diabetes. Diabetes Care 18: 766–774, 1995.
14. Chaturvedi N, Stevens LK, Fuller JH, and The WHO Mul-tinational Study Group. Mortality and morbidity associatedwith body weight in people with IDDM: The WHO MultinationalStudy of Vascular Disease in Diabetes. Diabetes Care 18: 761–765, 1995.
15. Cox RH and Tulenko TN. Altered contractile and ion channelfunction in rabbit portal vein with dietary atherosclerosis. Am JPhysiol Heart Circ Physiol 268: H2522–H2530, 1995.
16. DiCarlo SE, Blair RW, Bishop VS, and Stone HL. Dailyexercise enhances coronary resistance vessel sensitivity to phar-macological activation. J Appl Physiol 66: 421–428, 1989.
17. Dixon JL, Stoops JD, Parker JL, Laughlin MH, WeismanGA, and Sturek M. Dyslipidemia and vascular dysfunction indiabetic pigs fed an atherogenic diet. Arterioscler Thromb VascBiol 19: 2981–2992, 1999.
18. Fay FS. Calcium sparks in vascular smooth muscle: relaxationregulators. Science 270: 588–589, 1995.
19. Fleischhacker E, Esenabhalu VE, Spitaler M, Holzmann S,Skrabal F, Koidl B, Kostner GM, and Graier WF. Humandiabetes is associated with hyperreactivity of vascular smoothmuscle cells due to altered subcellular Ca2� distribution. Diabe-tes 48: 1323–1330, 1999.
20. Gelband CH and Hume JR. Ionic currents in single smoothmuscle cells of the canine renal artery. Circ Res 71: 745–758,1992.
21. Gerrity RG, Natarajan R, Nadler JL, and Kimsey T. Dia-betes-induced accelerated atherosclerosis in swine. Diabetes 50:1654–1665, 2001.
22. Hardingham GE, Chawla S, Johnson CM, and Bading H.Distinct functions of nuclear and cytoplasmic calcium in thecontrol of gene expression. Nature 385: 260–265, 1997.
23. Hattori Y, Kawasaki H, Kanno M, Gando S, and Fukao M.Attenuated contractile response of diabetic rat aorta to caffeinebut not to noradrenaline in Ca2�-free medium. Eur J Pharmacol256: 215–219, 1994.
24. Herrera GM, Heppner TJ, and Nelson MT. Voltage depen-dence of the coupling of Ca2� sparks to BKCa channels in urinarybladder smooth muscle. Am J Physiol Cell Physiol 280: C481–C490, 2001.
25. Hill BJF, Dixon JL, and Sturek M. Effect of atorvastatin onintracellular calcium uptake in coronary smooth muscle cellsfrom diabetic pigs fed an atherogenic diet. Atherosclerosis 159:117–124, 2001.
26. Hodgson JM, Reddy KG, Suneja R, Nair RN, Lesnefsky EJ,and Sheehan HW. Intracoronary ultrasound imaging: correla-tion of plaque morphology with angiography, clinical syndromeand procedural results in patients undergoing coronary angio-plasty. J Am Coll Cardiol 21: 35–44, 1993.
27. Hogikyan RV, Galecki AT, Halter JB, and Supiano MA.Heightened norepinephrine-mediated vasoconstriction in type 2diabetes. Metabolism 48: 1536–1541, 1999.
28. Ikenaga H, Bast JP, Fallet RW, and Carmines PK. Exag-gerated impact of ATP-sensitive K� channels on afferent arte-riolar diameter in diabetes mellitus. J Am Soc Nephrol 11:1199–1207, 2000.
29. Johnson GJ, Griggs TR, and Badimon L. The utility ofanimal models in the preclinical study of interventions to pre-vent human coronary artery restenosis: analysis and recommen-dations. Thromb Haemost 81: 835–843, 1999.
30. Kahn AM and Song T. Insulin inhibits dog vascular smoothmuscle contraction and lowers Ca2�
i by inhibiting Ca2� influx. JNutr Suppl 125: 1732S–1737S, 1995.
31. Kannel WB and McGee DL. Diabetes and cardiovascular dis-ease: the Framingham study. JAMA 241: 2035–2038, 1979.
32. King H, Aubert RE, and Herman WH. Global burden ofdiabetes, 1995–2025. Prevalence, numerical estimates, and pro-jections. Diabetes Care 21: 1414–1431, 1998.
33. Kirber MT, Etter EF, Bellve KA, Lifshitz LM, Tuft RA, FayFS, Walsh JV Jr, and Fogarty KE. Relationship of Ca2�
sparks to STOCs studied with 2D and 3D imaging in felineoesophageal smooth muscle cells. J Physiol 531: 315–327, 2001.
34. Lee DL, Wamhoff BR, Katwa LC, Reddy HK, Voelker DJ,Dixon JL, and Sturek M. Increased endothelin-induced Ca2�
signaling, tyrosine phosphorylation, and coronary artery diseasein diabetic dyslipidemic swine are prevented by atorvastatin.J Pharmacol Exp Ther 306: 132–140, 2003.
35. Lerner DJ and Kannel WB. Patterns of coronary heart diseasemorbidity and mortality in the sexes: a 26-year follow-up of theFramingham population. Am Heart J 111: 383–390, 1986.
36. Levy J, Gavin JR, and Sowers JR. Diabetes mellitus: adisease of abnormal cellular calcium metabolism? Am J Med 96:260–273, 1994.
37. Liu Y, Jones AW, and Sturek M. Ca release and sarcolemmalmechanisms for increased outward K current in aortic smoothmuscle cells of aldosterone-salt hypertensive rats and spontane-ously hypertensive stroke-prone rats (Abstract). Hypertension22: 428, 1993.
38. Liu Y, Jones AW, and Sturek M. Ca2�-dependent K� currentin arterial smooth muscle cells from aldosterone-salt hyperten-sive rats. Am J Physiol Heart Circ Physiol 269: H1246–H1257,1995.
39. Liu Y, Pleyte KA, Knaus HG, and Rusch NJ. Increasedexpression of Ca2�-sensitive K� channels in aorta of hyperten-sive rats. Hypertension 30: 1403–1409, 1997.
40. Liu YP, Hudetz AG, Knaus HG, and Rusch NJ. Increasedexpression of Ca2�-sensitive K� channels in the cerebral micro-circulation of genetically hypertensive rats. Evidence for theirprotection against cerebral vasospasm. Circ Res 82: 729–737,1998.
41. Lohn M, Wolfgang J, Furstenau M, Wellner M, SorrentinoV, Haller H, Luft FC, and Gollasch M. Regulation of calciumsparks and spontaneous transient outward currents by RyR3 inarterial vascular smooth muscle cells. Circ Res 89: 1051–1057,2001.
42. Mathew V and Lerman A. Altered effects of potassium chan-nel modulation in the coronary circulation in experimental hy-percholesterolemia. Atherosclerosis 154: 329–335, 2001.
43. Matveev S, Van der Westhuyzen DR, and Smart EJ. Co-expression of scavenger receptor-BI and caveolin-1 is associatedwith enhanced selective cholesteryl ester uptake in THP-1 mac-rophages. J Lipid Res 40: 1647–1654, 1999.
44. Mistry DK and Garland CJ. Characteristics of single, large-conductance calcium-dependent potassium channels (BKCa)from smooth muscle cells isolated from the rabbit mesentericartery. J Membr Biol 164: 125–138, 1998.
45. Mokelke EA, Wang MF, and Sturek M. Exercise trainingenhances coronary smooth muscle cell sodium-calcium exchangeactivity in diabetic dyslipidemic Yucatan swine. Ann NY AcadSci 976: 1–3, 2002.
46. Nelson MT, Patlak JB, Worley JF, and Standen NB. Cal-cium channels, potassium channels, and voltage dependence ofarterial smooth muscle tone. Am J Physiol Cell Physiol 259:C3–C18, 1990.
47. Ohara T, Sussman KE, and Draznin B. Effect of diabetes oncytosolic free Ca2� and Na�-K�- ATPase in rat aorta. Diabetes40: 1560–1563, 1991.
48. Otis CR, Wamhoff BR, and Sturek M. Hyperglycemia-in-duced insulin resistance in diabetic dyslipidemic Yucatan swine.Comp Med 53: 53–64, 2003.
49. Panepinto LM, Phillips RW, Westmoreland NW, and CleekJL. Influence of genetics and diet on the development of diabetesin Yucatan miniature swine. J Nutr 112: 2307–2313, 1982.
50. Paterno R, Heistad DD, and Faraci FM. Functional activityof Ca2�-dependent K� channels is increased in basilar artery
1192 K� CHANNELS IN DIABETIC DYSLIPIDEMIA
J Appl Physiol • VOL 95 • SEPTEMBER 2003 • www.jap.org
by 10.220.33.2 on August 18, 2016
http://jap.physiology.org/D
ownloaded from

during chronic hypertension. Am J Physiol Heart Circ Physiol272: H1287–H1291, 1997.
51. Perez GJ, Bonev AD, Patlak JB, and Nelson MT. Functionalcoupling of ryanodine receptors to KCa channels in smooth mus-cle cells from rat cerebral arteries. J Gen Physiol 113: 229–237,1999.
52. Phillips RW, Panepinto LM, Spangler R, and Westmore-land N. Yucatan miniature swine as a model for the study ofhuman diabetes mellitus. Diabetes 31: 30–36, 1982.
53. Ramanadham S, Lyness WH, and Tenner TE. Alterations inaortic and tail artery reactivity to agonists after streptozotocintreatment. Can J Physiol Pharmacol 62: 418–423, 1984.
54. Rogers PJ, Miller TD, Bauer BA, Brum JM, Bove AA, andVanhoutte PM. Exercise training and responsiveness of iso-lated coronary arteries. J Appl Physiol 71: 2346–2351, 1991.
55. Rusch NJ, Liu YP, and Pleyte KA. Mechanisms for regulationof arterial tone by Ca2�-dependent K� channels in hypertension.Clin Exp Pharmacol Physiol 23: 1077–1081, 1996.
56. Sakai K, Zhang S, Ureshino H, Tomiyasu S, and SumikawaK. Interaction of isoflurane and cromakalim, a KATP channelopener, on coronary and systemic haemodynamics in chronicallyinstrumented dogs. Acta Anaesthesiol Scand 44: 1122–1127,2000.
57. Scherer PE, Lisanti MP, Baldini G, Sargiacomo M, Ma-stick CC, and Lodish HF. Induction of caveolin during adipo-genesis and association of GLUT4 with caveolin-rich vessels.J Cell Biol 127: 1233–1243, 1994.
58. Sen L, Bialecki RA, Smith E, Smith TW, and Colucci WS.Cholesterol increases the L-type voltage-sensitive calcium chan-nel current in arterial smooth muscle cells. Circ Res 71: 1008–1014, 1992.
59. Sobey CG. Potassium channel function in vascular disease.Arterioscler Thromb Vasc Biol 21: 28–38, 2001.
60. Sowers JR and Lester MA. Diabetes and cardiovascular dis-ease. Diabetes Care 22: C14–C20, 1999.
61. Srere PA. Citrate synthase. Methods Enzymol 13: 3–5, 1982.62. Stehno-Bittel L, Laughlin MH, and Sturek M. Exercise
training alters Ca release from coronary smooth muscle sarco-plasmic reticulum. Am J Physiol Heart Circ Physiol 259: H643–H647, 1990.
63. Stehno-Bittel L, Laughlin MH, and Sturek M. Exercisetraining depletes sarcoplasmic reticulum calcium in coronarysmooth muscle. J Appl Physiol 71: 1764–1773, 1991.
64. Stehno-Bittel L and Sturek M. Spontaneous sarcoplasmicreticulum calcium release and extrusion from bovine, not por-cine, coronary artery smooth muscle. J Physiol 451: 49–78, 1992.
65. Sturek M, Stehno-Bittel L, and Obye P. Modulation of ionchannels by calcium release in coronary artery smooth muscle.In: Electrophysiology and Ion Channels of Vascular SmoothMuscle Cells and Endothelial Cells, edited by Sperelakis N andKuriyama H. New York: Elsevier, 1991, p. 65–79.
66. Sullivan S and Sparks HV. Diminished contractile response ofaortas from diabetic rabbits. Am J Physiol Heart Circ Physiol236: H301–H306, 1979.
67. Tam ESL, Ferguson DG, Bielefeld DR, Lorenz JN, CohenRM, and Pun RYK. Norepinephrine-mediated calcium signal-ing is altered in vascular smooth muscle of diabetic rat. CellCalcium 21: 143–150, 1997.
68. Wamhoff BR, Dixon JL, and Sturek M. Exercise trainingprevents altered coronary smooth muscle L-type calcium chan-nel function in diabetic dyslipidemia (Abstract). Circulation 104:II-157, 2001.
69. Wamhoff BR, Dixon JL, and Sturek M. Atorvastatin treat-ment prevents alterations in coronary smooth muscle nuclearCa2� signaling associated with diabetic dyslipidemia. J Vasc Res39: 208–220, 2002.
70. Wang R, Wu YJ, Tang GH, Wu LY, and Hanna ST. AlteredL-type Ca2� channel currents in vascular smooth muscle cellsfrom experimental diabetic rats. Am J Physiol Heart CircPhysiol 278: H718–H722, 2000.
71. Waring JV and Wendt IR. Effects of streptozotocin-induceddiabetes mellitus on intracellular calcium and contraction oflongitudinal smooth muscle from rat urinary bladder. J Urol163: 323–330, 2000.
72. Weisbrod RM, Griswold MC, Du Y, Bolotina VM, and Co-hen RA. Reduced responsiveness of hypercholesterolemic rabbitaortic smooth muscle cells to nitric oxide. Arterioscler ThrombVasc Biol 17: 394–402, 1997.
73. White RE and Carrier GO. Vascular contraction induced byactivation of membrane calcium ion channels is enhanced instreptozotocin-diabetes. J Pharmacol Exp Ther 253: 1057–1062,1990.
74. Williams JK, Kaplan JR, Suparto I, Fox JL, and ManuckSB. Effects of exercise on cardiovascular outcomes in monkeyswith risk factors for coronary heart disease. Arterioscler ThrombVasc Biol 23: 864–871, 2003.
75. Witczak CA and Sturek M. Exercise training reverses thedecreased coronary smooth muscle calcium buffering seen indiabetic dyslipidemia. Med Sci Sports Exerc 306: 132–140, 2003.
77. Xu C, Lu Y, Tang G, and Wang R. Expression of voltage-dependent K� channel genes in mesenteric artery smooth mus-cle cells. Am J Physiol Gastrointest Liver Physiol 277: G1055–G1063, 1999.
78. Yoo HJ, Kozaki K, Akishita M, Watanabe M, Eto M, Na-gano K, Sudo N, Hashimoto M, Kim S, Yoshizumi M, TobaK, and Ouchi Y. Augmented Ca2� influx is involved in themechanism of enhanced proliferation of cultured vascularsmooth muscle cells from spontaneously diabetic Goto-Kakizakirats. Atherosclerosis 131: 167–175, 1997.
79. Yuan XJ, Wang J, Juhaszova M, Golovina VA, and RubinLJ. Molecular basis and function of voltage-gated K� channelsin pulmonary arterial smooth muscle cells. Am J Physiol LungCell Mol Physiol 274: L621–L635, 1998.
1193K� CHANNELS IN DIABETIC DYSLIPIDEMIA
J Appl Physiol • VOL 95 • SEPTEMBER 2003 • www.jap.org
by 10.220.33.2 on August 18, 2016
http://jap.physiology.org/D
ownloaded from




















