
A fluorescence assay for peptide translocation into mitochondria

CORRESPONDENCE
Hepatocellular Carcinoma and Gene Profiling: Back from Perspective to Reality
To the Editor:
I can hardly share the passionate enthusiasm of Breuhahn et al. forthe ‘‘dramatic’’ improvements in understanding of molecular pathoge-nesis of hepatocellular carcinoma (HCC) and the claim for ‘‘furtherrationally designed clinical trials based on molecular evidence’’.1
Among the causes of HCC, they cite aflatoxins and hemochro-matosis but failed, as too many do, to cite tobacco, which repre-sents the cause of one-third of the cases.2 Despite the success ofthe hepatitis B vaccine and the cure for hepatitis C, HCC remainsa growing epidemic due to alcohol, tobacco, and processed foods(obesity and diabetes).3 Here are the three agents of the modernepidemics.
Considering ‘‘molecular evidence’’, a PubMed search for ‘‘gene-expression profiling’’ and ‘‘cancer’’ provides more than 17,000references since 1999. However, out of hundreds of biomarkers,KRAS (v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog) isthe only one integrated into clinical practice. It is limited to meta-static colorectal cancer, which constitutes half of all patients withcolorectal cancer. Evidence was obtained in 2008 from a post hocanalysis of the CRYSTAL trial (study EMR 62202-013) withcetuximab, and the first trial designed with an intention-to-treatanalysis, PRIME (study 20050203), has just been published.4 Theeffect, although statistically significant, has very limited relevance:in wild-type KRAS, panitumumab–FOLFOX4 (infusional fluo-rouracil, leucovorin, and oxaliplatin) increases progression-free sur-vival by 1.6 months compared with FOLFOX4 alone.
Medicine must avoid ‘‘sciensationalism’’ (sensationalism in sci-ence).5 If more research is needed, it must be concerned with howto improve the implementation of evidence-based care and publicpolicies against the leading avoidable causes of cancer worldwide:tobacco, alcohol, and obesity. A focus on molecular biology thatignores medical practice, interventional epidemiology, and socialand political sciences will improve neither patient care norprevention.
ALAIN BRAILLON, M.D., PH.D.Gres. 27 rue Voiture
Amiens, France
References1. Breuhahn K, Gores G, Schirmacher P. Strategies for HCC therapy and
diagnostics — lessons learned from high throughput and profilingapproaches. HEPATOLOGY 2011;53:2112-2121.
2. Braillon A. Screening for hepatocellular carcinoma: from lack of evi-dence to common sense. HEPATOLOGY 2010;52:1863-1864.
3. Starley BQ, Calcagno CJ, Harrison SA. Nonalcoholic fatty liver diseaseand hepatocellular carcinoma: a weighty connection. HEPATOLOGY
2010;51:1820-1832.4. Douillard JY, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel M,
et al. Randomized, phase III trial of panitumumab with infusional fluo-rouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4alone as first-line treatment in patients with previously untreated meta-static colorectal cancer: the PRIME study. J Clin Oncol 2010;28:4697-4705.
5. Braillon A. Sciensationalism. Am J Med 2011;124:e13.
CopyrightVC 2011 by the American Association for the Study of Liver Diseases.View this article online at wileyonlinelibrary.com.DOI 10.1002/hep.24365Potential conflict of interest: Nothing to report.
Reply:
We thank Dr. Braillon for his comments on our review ‘‘Strat-egies for HCC therapy and diagnostics - lessons learned from highthroughput and profiling approaches’’,1 although we have to saythat they are neither correct nor related to the focus of our article.If he was more familiar with hepatocellular carcinoma (HCC)research, he would have recognized the dramatic increase in knowl-edge over the last decade. We do not want to comment on theaccusation of ‘‘sensationalism’’, but instead will get some pointsstraight:• The intention of the review was to provide some integrativeoverview about the many molecular studies, especially profilinganalyses, relating to the various molecular alterations that occurduring HCC development and how they may, or should,impact on novel approaches to diagnosis and therapy. We nei-ther addressed epidemiology nor prevention. Nevertheless, thereis no question that hepatitis B and C viruses are the main eti-ologies, and they are by no means eradicated by vaccination orantiviral treatment.2 In addition, HCC is epidemic where nei-ther alcohol, tobacco, nor obesity and diabetes represent a sig-nificant factor.• Knowledge about predictive molecular markers needs more thanan ignorant Medline search. Clinical oncology has integrated manypredictive markers—breast cancer (ER [estrogen receptor], PR[progesterone receptor], HER2/ERBB2/NEU), non–small-cell lungcancer (epidermal growth factor receptor mutations), gastric cancer(HER2/ERBB2/NEU), gastrointestinal stromal tumor (KIT) toname a few and many more (BRAF, ELM4-ALK, and others) tocome soon. We are sure that testing for these markers is also avail-able to French cancer patients.• Of course, it could always be better, but there is significantfunding and research in epidemiology and prevention of cancer.Those who understand the topic know that molecular cancerresearch is not an opponent but a friend and significant driver ofcancer epidemiology and prevention (and of course vice versa),which holds true also specifically in HCC. Otherwise, without mo-lecular research, how would we know about hepatitis B virus, howit causes HCC, and that this can be prevented by vaccination3; orread the seminal articles about specific p53 mutations caused byaflatoxins, a paradigm of molecular cancer epidemiology4,5;and…much more to come.
KAI BREUHAHN, PH.D.1
GREGORY GORES, M.D.2
PETER SCHIRMACHER, M.D.11Institute of Pathology, Heidelberg University Hospital
Heidelberg, Germany2Mayo Medical School, Mayo Clinic College of Medicine
Rochester, MN
References1. Breuhahn K, Gores G, Schirmacher P. Strategies for HCC therapy and
diagnostics - lessons learned from high throughput and profilingapproaches. HEPATOLOGY 2011;53:2112-2121.
2. El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiologyand molecular carcinogenesis. Gastroenterology 2007;132:2557-2576.
3. Yang JD, Roberts LR. Hepatocellular carcinoma: A global view. NatRev Gastroenterol Hepatol 2010;7:448-458.
738

4. Hsu IC, Metcalf RA, Sun T, Welsh JA, Wang NJ, Harris CC. Muta-tional hotspot in the p53 gene in human hepatocellular carcinomas.Nature 1991;350:427-428.
5. Ozturk M. p53 mutation in hepatocellular carcinoma after aflatoxin ex-posure. Lancet 1991;338:1356-1359.
CopyrightVC 2011 by the American Association for the Study of Liver Diseases.View this article online at wileyonlinelibrary.com.DOI 10.1002/hep.24425Potential conflict of interest: Nothing to report.
Apply Sound Scientific Methodology to the Evaluation of New Medical Treatments
To the Editor:
A recent HEPATOLOGY article by Lindor and Lindor1 suggeststhat we need to reconsider the status of randomized controlledclinical trials as the gold standard of evidence for evaluating newmedical treatments. They suggest that observational studies for theevaluation of medical therapy are faster and cheaper and are moreeasily performed, and they ultimately lead to faster approval, areduction of medication costs, and better patient care. However,Lindor and Lindor ignore the well-known fallibility of observatio-nal studies that have supported the therapeutic value of a treatmentwithout evidence from well-developed randomized trials. There aremany notable contemporary examples of widely used treatmentsthat were accepted on the basis on observational studies and weresubsequently proved to be ineffective or harmful when controlledclinical trials were performed. These treatments include the following:
1. High-dose chemotherapy with bone marrow transplantationfor metastatic breast cancer. This treatment produced high rates ofresponse in phase 2 trials but was proved to be no more effectivethan standard chemotherapy and was more toxic.2–4
2. Hormone replacement in menopausal females, which waseventually shown to be not beneficial to cardiovascular health andto be associated with multiple serious adverse outcomes.5,6
3. Erythropoietin for the treatment of anemia due to chronickidney disease. Observational studies had shown increased survivaland higher hemoglobin levels; however, randomized trials wereunable to show such benefits and found an association withincreased rates of death, nonfatal heart attacks, strokes, and heartfailure.7
4. Early initiation of hemodialysis versus late initiation. A num-ber of observational studies suggested that starting dialysis earlycould improve the survival and quality of life of patients. Althoughthis practice was widely employed by nephrologists throughout theUnited States with substantial financial advantages, a recentrandomized controlled trial has found that the early initiation ofdialysis is not associated with improvements in survival or clinicaloutcomes.8
In each of these contemporary examples, observational studiessuggested a beneficial effect that was widely promoted in medicinebut was proven false by a randomized controlled trial.
Even more bothersome is the recent report by Zuckermanet al.,9 who reported that the US Food and Drug Administration(FDA) recalled 113 medical devices between 2005 and 2009because they were found to pose a high risk to patients and werenot rigorously studied before they were cleared for sale by theFDA. Most of these devices were approved only on the basis ofobservational studies and were not subjected to sound scientificmethodology.
Although observational studies often are cheaper, quicker, andless difficult to perform, we should not lose sight of the simple factnoted by Pocock and Elbourne10: ‘‘ignorance calls for careful
experimentation.’’ This means high-quality randomized controlledtrials and not observations that reflect personal choices or beliefsthat often rely on bogus assumptions and mathematical argumentsto prove what is not true to be true. In this era of money-basedmedicine and FDA laxity, when the ethics of pharmaceutical com-panies are continually being questioned and the costs of treatmentcontinue to skyrocket, let us continue to evaluate health care inter-ventions by the most scientifically sound and rigorous methodsavailable.
RUSSELL H. WIESNER, M.D.Mayo Clinic
Rochester, MN
References1. Lindor RA, Lindor KD. The value of observational research in liver
disease. HEPATOLOGY 2011;53:1-3.2. Echt DS, Liebson PR, Mitchell LB, Peters RW, Obias-Manno D,
Barker AH, et al. Mortality and morbidity in patients receiving encai-nide, flecainide, or placebo. The Cardiac Arrhythmia Suppression Trial.N Engl J Med 1991;324:781-788.
3. Stadtmauer EA, O’Neill A, Goldstein LJ, Crilley PA, Mangan KF, IngleJN, et al. Conventional-dose chemotherapy compared with high-dosechemotherapy plus autologous hematopoietic stem-cell transplantationfor metastatic breast cancer. Philadelphia Bone Marrow TransplantGroup. N Engl J Med 2000;342:1069-1076.
4. Rettig RA, Jacobson PD, Farquhar CM, Aubry WM. False Hope:Bone Marrow Transplantation for Breast Cancer. Oxford, United King-dom: Oxford University Press; 2007.
5. Moseley JB, O’Malley K, Petersen NJ, Menke TJ, Brody BA, Kuyken-dall DH, et al. A controlled trial of arthroscopic surgery for osteoarthri-tis of the knee. N Engl J Med 2002;347:81-88.
6. Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Ste-fanick ML, et al. Risks and benefits of estrogen plus progestin in healthypostmenopausal women: principal results from the Women’s Health Initia-tive randomized controlled trial. JAMA 2002;288:321-333.
7. Food and Drug Administration. Public advisory: erythropoiesis-stimu-lating agents including Procrit, Epogen, and Aranesp.
8. Cooper BA, Branley P, Bulfone L, Collins JF, Craig JC, Fraenkel MB,et al. A randomized controlled trial of early versus late initiation of di-alysis. N Engl J Med 2010;363:609-619.
9. Zuckerman DM, Brown P, Nissen SE. Medical device recalls and theFDA approval process. Arch Intern Med; doi:10.1001/archinternmed.2011.30.
10. Pocock SJ, Elbourne DR. Randomized trials or observational tribula-tions? N Engl J Med 2000;342:1907-1909.
CopyrightVC 2011 by the American Association for the Study of Liver Diseases.View this article online at wileyonlinelibrary.com.DOI 10.1002/hep.24375Potential conflict of interest: Nothing to report.
HEPATOLOGY, Vol. 54, No. 2, 2011 CORRESPONDENCE 739

Reply:
We appreciate the response to our January 2011 editorial inHEPATOLOGY
1 and welcome the opportunity to continue the discus-sion about the relative roles of observational studies and random-ized clinical trials (RCTs). Importantly, we would like to reiterateour support for broader use of observational research as a comple-ment to traditional clinical trials, not as a replacement. We agreewith Wiesner that there are some issues, such as questions of basicefficacy and safety, that are best evaluated in the setting of anRCT.2 However, we also recognize that the controlled setting in-herent to RCTs limits their utility in evaluating measures of real-world effectiveness, which can be significantly affected by patients’social circumstances and biological variation. In addition, steadfastadherence to traditional RCTs hinders the evaluation of a numberof rare diseases, for which logistical barriers make it difficult toaccrue sufficient numbers of patients within the confines of a tradi-tional clinical trial.
Wiesner’s response includes a number of examples in whichtreatments based on observational studies were later demonstratedthrough clinical trials to be ineffective or harmful. However, ineach of these cases—including high dose chemotherapy with bonemarrow transplant for breast cancer, estrogen replacement for men-opause, erythropoietin for anemia of chronic kidney disease, andearly stage hemodialysis—initial questions about the effectivenessof the interventions were clouded by conflicting RCT data as well.Therefore, these examples demonstrate the potential fallibility ofboth types of trials and speak more to the need for careful trialdesign rather than the superiority of a given trial type.
Similarly, the criticism of recalls by the Food and Drug Admin-istration warrants further scrutiny. Wiesner is correct in noting thatmore than a hundred devices were recalled by the Food and DrugAdministration after having been cleared for marketing withoutundergoing clinical trials. However, the same study referenced bythe article he cites actually shows that devices evaluated by RCTsprior to marketing were far more likely to be recalled than devices
that went through a less stringent evaluation pathway, again illus-trating the limitations of traditional clinical trials.3
Together, the two sets of examples provided by Wiesner reflectmany of the common criticisms of observational studies. At the sametime, however, these examples also illustrate the limitations of tradi-tional clinical trials, which often arrive at conflicting conclusions andfail to capture sufficient data to ensure patient safety. We do not tryto identify here where RCTs or observational studies are more appro-priate, but rather hope to prompt further discussion about the needto reconsider the evidence base on which we make clinical decisions.In the future, both observational studies and RCTs are sure to influ-ence clinical practice, and the sooner we understand their respectivestrengths and weaknesses, the more effective our care will be.
RACHEL A. LINDOR, J.D.KEITH D. LINDOR, M.D.Mayo Medical School and
Division of Gastroenterology and HepatologyMayo ClinicRochester, MN
References1. Lindor RA, Lindor KD. The value of observational research.
HEPATOLOGY 2011;53:1-3.2. Wiesner RH. Apply sound scientific methodology to evaluating new
medical treatment. HEPATOLOGY 2011; doi:10.1002/hep.24375.3. United States Government Accountability Office. Medical devices:
FDA should take steps to ensure that high-risk device types areapproved through the most stringent premarket review process. GAO-09-109. Washington, DC: GAO; 2009.
CopyrightVC 2011 by the American Association for the Study of Liver Diseases.View this article online at wileyonlinelibrary.com.DOI 10.1002/hep.24426Potential conflict of interest: Nothing to report.
The Ethics of Placebo Treatment for Patients With Acute Exacerbation of Chronic Hepatitis B
To the Editor:
I read with interest the article by Garg et al.,1 who showed thattenofovir improves the outcome in patients with spontaneous reac-tivation of hepatitis B virus (HBV) presenting as acute-on-chronicliver failure (ACLF). As indicated by the authors, the short-termprognosis of patients with spontaneous severe acute exacerbation ofchronic hepatitis B leading to ACLF-like presentation is extremelypoor, with a mortality rate ranging from 30%-70%. The currentstudy showed that mortality rate was 43% in the tenofovir groupand up to 85% in the placebo group. Prior to the start of the trialby Garg et al., Chien et al.2 demonstrated that the use of lamivu-dine is definitely beneficial for these patients, with an improvedsurvival compared to historic controls. Moreover, this study showedthat patients with serum bilirubin lower than 20 mg/dL could usu-ally be rescued with the use of lamivudine. As a consequence, theHBV management guidelines proposed by organizations such asthe Asian Pacific Association for the Study of the Liver (APASL)3
as well as the American Association for the Study of Liver Diseases(AASLD)4 consistently recommend that when patients with HBVwho have ACLF are treated, antiviral drugs should be promptlyinstituted. The trial by Garg et al., which used placebo drug as acontrol, leading to an appreciably high mortality in this group, inorder to demonstrate the efficacy of tenofovir in treating patientswith HBV who have ACLF, was apparently medically unethical.Similar studies should be strongly discouraged.
GIN-HO LO, M.D.Department of Medical Education
Digestive CenterE-Da Hospital, I-Shou UniversityKaohsiung City, Taiwan
References1. Garg H, Sarin SK, Kumar M, Garg V, Sharma BC, Kumar A. Tenofovir
improves the outcome in patients with spontaneous reactivation of hepatitisB presenting as acute-on-chronic liver failure. HEPATOLOGY 2011;53:774-780.
2. Chien RN, Lin CH, Liaw YF. The effect of lamivudine therapy inhepatic decompensation during acute exacerbation of chronic hepatitis B.J Hepatol 2003;38:322-327.
3. Liaw YF, Leung N, Guan R, Lau GK, Merican I, McCaughan G,et al.; Asian-Pacific consensus update working party on chronic hepati-tis B. Asian-Pacific consensus statement on the management of chronichepatitis B: a 2005 update. Liver Int 2005;25:472-489.
4. Lok AS, McMahon BJ. Chronic hepatitis B. HEPATOLOGY 2007;45:507-539.
CopyrightVC 2011 by the American Association for the Study of Liver Diseases.View this article online at wileyonlinelibrary.com.DOI 10.1002/hep.24392Potential conflict of interest: Nothing to report.
740 CORRESPONDENCE HEPATOLOGY, August 2011

Table 1. Published Studies Prior to 2007 on Antiviral Treatment for Decompensated Severe Acute Reactivation of Hepatitis B
Year Author No. of Patients Treatment Severity Scores Mortality (%) Remarks/Conclusions
2001 Tsang SW et al.6 24 Lamivudine NA 67 Lamivudine treatment should be instituted early
to prevent decompensation and fulminant
liver failure. This is likely to reduce morbidity
and mortality.
2002 Chan HL et al.7 28 Lamivudine versus
historic controls
NA 22 versus 28 Lamivudine confers no survival benefit to con-
ventional treatment in severe exacerbations
of chronic hepatitis B
2003 Chien RN et al.9 60 Lamivudine versus
historic controls
CTP-11 (6-13) 38 versus 29 Lamivudine may prevent fatality in chronic hep-
atitis B patients with hepatic decompensa-
tion if therapy starts early enough or before
a serum bilirubin level rise over 20 mg/dL,
but helps little if serum bilirubin has already
risen over that level
2005 Tsubota A et al.8 25 Lamivudine versus
historic controls
NA 12 versus 20 Lamivudine monotherapy conferred no signifi-
cant protection against rapid progression of
the disease to hepatic failure and death
NA, not applicable; NS, not significant.
Reply:
We read with interest the letter by Dr. Lo where he raisedconcerns about placebo treatment for patients with acute exac-erbation of chronic hepatitis B. We appreciate his concerns.However, we would like to clarify that Dr. Lo has probablynot considered the widely accepted definition of acute-on-chronic liver failure (ACLF)1 and has misunderstood it asmerely acute exacerbation of chronic hepatitis B. ACLF is verydifferent from the commonly encountered acute reactivation ofchronic hepatitis B. The patients are very sick and the 3-month mortality is >76%, especially when the Model forEnd-Stage Liver Disease score is >20.2 In contrast of ACLF,the acute reactivation of hepatitis B has a low mortality rate of�6%.3 Lamivudine may be helpful in acute reactivation ofhepatitis B as it may prevent decompensation. Therefore, theguidelines from Asian Pacific Association for the Study of theLiver and the American Association for the Study of Liver Dis-eases had recommended antiviral for acute reactivation of hep-atitis B.4,5 When this reactivation has led to decompensationpresenting as ACLF, the patients become very sick and requireliver transplantation. Use of antiviral treatment for these sickpatients was not clear, until conceptualization of our study.There were four published studies prior to 2007 on decom-pensated severe acute reactivation of hepatitis B, which did notshow any definite advantage of antiviral treatment in thesepatients6-9 (Table 1). Only Chien et al. could show survivalbenefit in a subgroup of patients on lamivudine who had bili-rubin levels <342 lmol/L (20 mg/dL). Keeping in mind thelimited benefits attained with lamivudine in previous trials andin our own earlier study,10 in the present study, we assessedthe efficacy of a potent antiviral such as tenofovir against pla-cebo. We wish to dispel any misconception in the minds ofreaders such as Dr. Lo about the ethical approach on whichthis study was based. We do believe that this well-designedand ethically conducted randomized clinical trial has madeseminal observations which would serve as benchmark for clin-ical practice and help design future antiviral strategies forpatients presenting with ACLF due to hepatitis B virusreactivation.
HITENDRA GARG, M.D., D.M.1,2
SHIV KUMAR SARIN, M.D., D.M.1,2,3
MANOJ KUMAR, M.D., D.M.2
VISHAL GARG, M.D.1
BARJESH CHANDER SHARMA, M.D., D.M.1
ASHISH KUMAR, M.D., D.M.2,31Department of Gastroenterology, G.B. Pant Hospital, New Delhi, India2Department of Hepatology, Institute of Liver and Biliary Sciences
(ILBS), New Delhi, India3Special Centre for Molecular Medicine, Jawaharlal Nehru University,
New Delhi, India
References1. Sarin SK, Kumar A, Almeida JA, Chawla YK, Fan ST, Garg H, et al.
Acute-on-chronic liver failure: consensus recommendations of the AsianPacific Association for the Study of the Liver (APASL). Hepatol Int2009;3:269-282.
2. Sen S, Williams R, Jalan R. The pathophysiological basis of acute-on-chronic liver failure. Liver 2002;22:5-13.
3. Levy P, Marcellin P, Martinot-Peignoux M, Degott C, Nataf J, Benha-mou JP. Clinical course of spontaneous reactivation of hepatitis B virusinfection in patients with chronic hepatitis B. HEPATOLOGY 1990;12:570-574.
4. Liaw YF, Leung N, Guan R, Lau GK, Merican I, McCaughan G, et al.Asian-Pacific consensus statement on the management of chronic hepa-titis B: a 2005 update. Liver Int 2005;25:472-489.
5. Lok ASF, McMahon BJ. Chronic hepatitis B. HEPATOLOGY 2007;45:507-539.
6. Tsang SW, Chan HL, Leung NW, Chau TN, Lai ST, Chan FK, et al.Lamivudine treatment for fulminant hepatic failure due to acute exacer-bation of chronic hepatitis B infection. Aliment Pharmacol Ther 2001;15:1737-1744.
7. Chan HL, Tsang SW, Hui Y, Leung NW, Chan FK, Sung JJ. The roleof lamivudine and predictors of mortality in severe flare-up of chronichepatitis B with jaundice. J Viral Hepat 2002;9:424-428.
8. Tsubota A, Arase Y, Suzuki Y, Suzuki F, Sezaki H, Hosaka T,et al. Lamivudine monotherapy for spontaneous severe acute exac-erbation of chronic hepatitis B. J. Gastroenterol Hepatol 2005;20:426-432.
HEPATOLOGY, Vol. 54, No. 2, 2011 CORRESPONDENCE 741

9. Chien RN, Lin CH, Liaw YF. The effect of lamivudine therapy inhepatic decompensation during acute exacerbation of chronic hepatitis B.J Hepatol 2003;38:322-327.
10. Kumar M, Satapathy S, Monga R, Das K, Hissar S, Pande C, et al. Arandomized controlled trial of lamivudine to treat acute hepatitis B.HEPATOLOGY 2007;45:97-101.
CopyrightVC 2011 by the American Association for the Study of Liver Diseases.View this article online at wileyonlinelibrary.com.DOI 10.1002/hep.24438Potential conflict of interest: Nothing to report.
Hepatitis C Virus Therapeutics: Editing Enzymes Promising Therapeutic Targets?
To the Editor:
We read with great interest the recent article by Peng et al.,1 inwhich the authors investigated the role of APOBEC3G (apolipo-protein B mRNA editing enzyme, catalytic polypeptide-like 3G),also named hA3G, in the regulation of hepatitis C virus (HCV)replication. In particular, the authors demonstrated that silencingof hA3G increased the rate of HCV replication in infected Huh7.5cells. Furthermore, the study highlighted that hA3G stabilizationby RN-5 [N,N0-(dimethylbipheny1-4, 40-diyl) dibenzenesulfona-mide] or IMB-26 [([3-(a-bromopropyl) amino-4-methoxy benzene]formyl (30,40,50-trimethoxybenzene) amide] increased the expres-sion of the intracellular hA3G, resulting in the inhibition of HCVreplication.
The hA3G belongs to the APOBEC3 family of proteins, whichare the best-characterized type of editing enzymes able to restrictthe infectivity of human immunodeficiency virus-1.2 However, themost frequent type of editing in humans is mediated by threeadenosine deaminases acting on RNA (ADARs), ADAR1-3, whichconvert adenosine (A) to inosine (I) in double-stranded RNA.3
Interestingly, the ADAR enzymes also play important roles duringviral infection by acting as proviral or antiviral factors.3
It has been reported that ADAR1 silencing in hepatoma cellsexpressing HCV replicons may stimulate the expression of HCVRNA.4 In addition, it has been suggested that ADAR1 may act asan antiviral factor in the context of HCV infection through a com-bination of direct and indirect mechanisms.3
We recently investigated the expression of ADAR1 in Huh7.5cells infected using the in vitro HCV infection and replicationJFH1 (Japanese fulminant hepatitis-1) model. We demonstrated(Fig. 1) that HCV infection specifically modulates the ratiobetween the two major ADAR1 isoforms: p150-kDa and p110-kDa. In particular, HCV up-regulated the 150-kDa isoform, whichis the classical interferon-inducible isoform, whereas the 110-kDaconstitutive isoform seems not significantly modified.
Taken together, these studies demonstrate that during HCVinfection at least two host factors, hA3G and ADAR1, are acti-vated. Despite the antiviral action of hA3G that has been recentlyshown by Peng et al., we suggest that hA3G and ADARs requirefurther investigation, because they can disclose additive or oppositeeffects during HCV infection.
The involvement of other host editing enzymes, such asADARs, can introduce an additional step of complexity in theHCV infection and its related fibrogenic and oncogenic properties.Moreover, the comprehension of the specific role played by theseenzymes in the HCV life-cycle, and in the regulation of virus–hostintracellular interactions, can provide relevant information indesigning novel potential safe and efficient molecular therapies.
ANNA ALISI, PH.D.1
SARA TOMASELLI, PH.D.2
CLARA BALSANO, M.D.3
ANGELA GALLO, PH.D.21Liver Unit and2RNA Editing Laboratory–Oncohaematology Department
Bambino Gesu Children’s Hospital and Research InstituteRome, Italy
3Department of Internal MedicineUniversity of L’Aquila, L’Aquila, Italy
References1. Peng ZG, Zhao ZY, Li YP, Wang YP, Hao LH, Fan B, et al. Host apo-
lipoprotein b messenger RNA-editing enzyme catalytic polypeptide-like3G is an innate defensive factor and drug target against hepatitis Cvirus. HEPATOLOGY 2011;53:1080-1089.
2. Albin JS, Harris RS. Interactions of host APOBEC3 restriction factors withHIV-1 in vivo: implications for therapeutics. Expert Rev Mol Med 2010;12:e4.
3. Samuel CE. Adenosine deaminases acting on RNA (ADARs) are bothantiviral and proviral. Virology 2011;411:180-193.
4. Taylor DR, Puig M, Darnell ME, Mihalik K, Feinstone SM. New antivi-ral pathway that mediates hepatitis C virus replicon interferon sensitivitythrough ADAR1. J Virol 2005;79:6291-6298.
CopyrightVC 2011 by the American Association for the Study of Liver Diseases.View this article online at wileyonlinelibrary.com.DOI 10.1002/hep.24409Potential conflict of interest: Nothing to report.
Reply:
We thank Alisi et al.1 for their interested in our research on theanti–hepatitis C virus (HCV) activity of human apolipoprotein Bmessenger RNA–editing enzyme catalytic polypeptide (APO-BEC3G, also known as hA3G),2 and believe that their considera-tion is valuable for our investigation. Basically, adenosine deami-nases that act on RNA (ADARs) and cytidine deaminases are twogeneric classes of deaminases. Homology between hA3G andADAR at neither messenger RNA nor protein level is recognized.
Fig. 1. ADAR1 expression levels in control and HCV-infected Huh7.5cells. Western blotting of p150 and p110 isoforms of ADAR1 in con-trol (C) and JFH1 HCV–infected (HCV) cells. Glutaraldehyde-3-phos-phate dehydrogenase (GAPDH) was used as loading control.
742 CORRESPONDENCE HEPATOLOGY, August 2011

ADARs act at double-stranded (ds) RNA. They catalyze the A-to-Iediting reaction on dsRNA, causing nucleotide substitution and dsRNAdestabilization. The effect of ADARs on viruses is complicated withvariability, such as ADAR concentration, viral strains, among others.3
Transgenic mice overexpressing wild-type or deaminase-deficient ADAR2protein exhibited metabolic alterations characterized with hyperphagia andadult onset obesity;4 knockout of the ADAR gene in mice caused highlethality.5-7 Thus, ADARs might not be an ideal drug target for virus.
hA3G belongs to the family of cytidine deaminases. It acts ateither RNA or DNA, and mediates hydrolytic deamination at theC4 position of the cytidine (C) or deoxycytidine (dC) base, con-verting C to uracil (U) (or dC to dU). hA3G inhibits replicationof RNA as well as DNA viruses.8 The hA3G-mediated viral restric-tion is not simply via editing, and other mechanisms such as inhi-bition on tRNA3Lys annealing to viral RNA, or on reverse tran-scription, often play key roles.8 The anti-HCV activity of hA3Gappeared not to be mediated through its RNA editing function.2 Aproviral effect of hA3G has not been documented. Furthermore,APOBEC3 is not essential for development, survival, or fertility inmouse experiments;9 overexpression of hA3G did not alter cellgrowth,2 suggesting its potential as a drug target.
ZONG-GEN PENG, PH.D.XUE-FU YOU, M.D.JIAN-DONG JIANG, M.D.Institute of Medicinal Biotechnology
Chinese Academy of Medical SciencesBeijing, China
References1. Alisi A, Tomaselli S, Balsano C, Gallo A. Hepatitis C virus (HCV)
therapeutics: Editing enzymes promising therapeutic targets? HEPATO-
LOGY 2011; doi:10.1002/hep.24409.
2. Peng ZG, Zhao ZY, Li YP, Wang YP, Hao LH, Fan B, et al. Host apo-lipoprotein b messenger RNA-editing enzyme catalytic polypeptide-like3G is an innate defensive factor and drug target against hepatitis C vi-rus. HEPATOLOGY 2011;53:1080-1089.
3. Samuel CE. Adenosine deaminases acting on RNA (ADARs) are bothantiviral and proviral. Virology 2011;411:180-193.
4. Singh M, Kesterson RA, Jacobs MM, Joers JM, Gore JC, EmesonRB. Hyperphagia-mediated obesity in transgenic mice misexpressingthe RNA-editing enzyme ADAR2. J Biol Chem 2007;282:22448-22459.
5. XuFeng R, Boyer MJ, Shen H, Li Y, Yu H, Gao Y, et al. ADAR1 isrequired for hematopoietic progenitor cell survival via RNA editing.Proc Natl Acad Sci U S A 2009;106:17763-17768.
6. Wang Q, Miyakoda M, Yang W, Khillan J, Stachura DL, Weiss MJ,et al. Stress-induced apoptosis associated with null mutation ofADAR1 RNA editing deaminase gene. J Biol Chem 2004;279:4952-4961.
7. Higuchi M, Maas S, Single FN, Hartner J, Rozov A, Burnashev N,et al. Point mutation in an AMPA receptor gene rescues lethality inmice deficient in the RNA-editing enzyme ADAR2. Nature 2000;406:78-81.
8. Bransteitter R, Prochnow C, Chen XS. The current structural andfunctional understanding of APOBEC deaminases. Cell Mol Life Sci2009;66:3137-3147.
9. Mikl MC, Watt IN, Lu M, Reik W, Davies SL, Neuberger MS, et al.Mice deficient in APOBEC2 and APOBEC3. Mol Cell Biol 2005;25:7270-7277.
CopyrightVC 2011 by the American Association for the Study of Liver Diseases.View this article online at wileyonlinelibrary.com.DOI 10.1002/hep.24462Potential conflict of interest: Nothing to report.
Bacterial Translocation Attenuates the Systemic Hemodynamic Improvement Producedby Terlipressin in Patients with Cirrhosis
To the Editor:
We read with interest the article by Bellot et al., who foundan association between bacterial translocation (BT) and systemichemodynamic derangement in patients with cirrhosis.1 BT wor-sens splanchnic arterial vasodilation in cirrhosis by stimulatingnitric oxide (NO) production in the splanchnic vasculature, ei-ther directly or through the cytokine cascade.2 Indeed, intestinaldecontamination reduces BT2 and serum NO levels,3 andimproves systemic hemodynamics3 in patients with cirrhosis.However, increased NO synthesis could also adversely affect sys-temic hemodynamics by decreasing mesenterial arterial reactivityto endogenous4 or exogenous vasoconstrictors, such asterlipressin.5
We present preliminary data on the impact of BT on systemichemodynamic effects of terlipressin in 17 patients with decompen-sated cirrhosis (male, n ¼ 13; mean age ¼ 52 6 3 years; Child-Pugh score ¼ 10.1 6 0.5). Plasma endotoxin levels were detectedby the Limulus amebocyte lysate chromogenic endpoint assay(Hycult biotech, Uden, the Netherlands) at baseline. Mean arterialpressure, cardiac output by Doppler ultrasound, and systemic vas-cular resistance (SVR) as the ratio mean arterial pressure/cardiacoutput were evaluated at baseline and 30 minutes after bolus intra-venous administration of terlipressin (1 mg). SVR increased signifi-cantly after terlipressin (1768 6 101 versus 1404 6 91 dyn/sec-ond/cm�5; P < 0.001). Endotoxin levels were correlated inversely
and significantly with baseline SVR values (r ¼ �0.587; P ¼0.01) and SVR changes after terlipressin (Fig. 1).
Fig. 1. Correlation between plasma endotoxin levels (in endotoxinunits [EU]) and systemic vascular resistance changes (DSVR) after ter-lipressin infusion.
HEPATOLOGY, Vol. 54, No. 2, 2011 CORRESPONDENCE 743

Endotoxin is detectable in all patients6 whereas bacterial DNA isdetectable in only 58% of patients with decompensated cirrhosis1
and was preferred as a marker of BT in our study. Furthermore,bacterial DNA was not correlated with systemic hemodynamics inthe study by Bellot et al.1 The present results support the linkbetween BT and systemic circulatory dysfunction in cirrhosis,suggesting that intestinal decontamination could enhance thehemodynamic effects of terlipressin and contribute to a decrease inrebleeding events in patients with variceal bleeding taking antibioticprophylaxis.7
GEORGIOS N. KALAMBOKIS, M.D.1
ATHANASIA MOUZAKI, M.D., PH.D.3
MARIA RODI, M.D.3
KONSTANTINOS PAPPAS, M.D.2
EPAMEINONDAS V. TSIANOS, M.D., PH.D.11First Division of Internal Medicine and Hepato-Gastroenterology Unit,
Ioannina, Greece2Department of Cardiology, Medical School of Ioannina, Ioannina,
Greece3Division of Hematology, Department of Internal Medicine, Medical
School, University of Patras, Patras, Greece
References1. Bellot P, Garcıa-Pagan JC, Frances R, Abraldes JG, Navasa M, Perez-
Mateo M, et al. Bacterial DNA translocation is associated with systemiccirculatory abnormalities and intrahepatic endothelial dysfunction inpatients with cirrhosis. HEPATOLOGY 2010;52:2044-2052.
2. Garcia-Tsao G. Gut microflora in the pathogenesis of the complicationsof cirrhosis. Best Pract Res Clin Gastroenterol 2004;18:353-372.
3. Albillos A, de la Hera A, Gonzalez M, Moya JL, Calleja JL, MonserratJ, et al. Increased lipopolysaccharide binding protein in cirrhoticpatients with marked immune and hemodynamic derangement. HEPA-
TOLOGY 2003;37:208-217.4. Lefilliatre P, Sogni P, Bertrand V, Del Soldato P, Pateron D, Moreau R,
et al. Aortic hyporeactivity to norepinephrine induced by lipopolysac-charide in cirrhotic rats: beneficial effects of a non-steroidal anti-inflam-matory drug coupled with a nitric oxide donor. J GastroenterolHepatol 2001;16:70-78.
5. Heinemann A, Stauber RE. Effect of terlipressin on in vitro vascularhyporeactivity of portal hypertensive rats. J Hepatol 1996;24:739-746.
6. Vlachogiannakos J, Daikos G, Thalheimer U, Burroughs AK, Ladas SD. Isbacterial DNA a better marker of bacterial translocation than endotoxin indecompensated cirrhosis? HEPATOLOGY 2011; doi:10.1002/hep.24303
7. Chavez-Tapia NC, Barrientos-Gutierrez T, Tellez-Avila FI, Soares-Weiser K, Uribe M. Antibiotic prophylaxis for cirrhotic patients withupper gastrointestinal bleeding. Cochrane Database Syst Rev 2010;(9):CD002907.
CopyrightVC 2011 by the American Association for the Study of Liver Diseases.View this article online at wileyonlinelibrary.com.DOI 10.1002/hep.24424Potential conflict of interest: Nothing to report.
Reply:
We appreciate the comments by Kalambokis et al. Bacterialtranslocation (BT) is associated with systemic hemodynamicderangement in patients with cirrhosis, because it worsens theinflammatory response and increases nitric oxide bioavailability.1
The preliminary data reported by Kalambokis et al. are interesting,and we consider that their findings are in line with our previousresults, although they used different methods to assess BT.
It has been shown by different authors that BT appears inapproximately 40% of patients with advanced cirrhosis. Albilloset al.2 showed that lipopolysaccharide (LPS)-binding protein(LBP), a reliable surrogate marker of LPS release, is increased in42% of patients, and Cirera et al.3 showed that BT is present in30.8% of patients with Child-Pugh C cirrhosis. Our own datahas repeatedly supported this proportion. There are several rea-sons for not considering LPS measurement as a robust tool indetecting BT: the Limulus amebocyte lysate was originallydesigned to measure LPS in water, and protocols are not specifi-cally directed to quantify LPS in biological samples. Furthermore,LPS levels, which have a short half-life, may be influenced byseveral factors, such as the concentration of LPS transporters,antibodies, high-density lipoprotein, and numerous other physio-logical variables, and are hampered by the need for using endo-toxin-free systems for sample collection. These drawbacks arereflected in the rate of endotoxin detection in cirrhosis, whichranges widely, from 0%-93% in several studies.2 This is whyLBP was proposed as a marker instead. Besides, it is obvious thatbacterial DNA detection, but not LPS or LBP measurements, canidentify BT from gram-positive cocci, which elicit a similarinflammatory response as BT by gram-negative organisms. Wecan therefore consider bacterial DNA as the ‘‘broadest range’’ sys-tem to detect BT.
Thus, although research on LPS in patients with cirrhosis isinteresting, relevant, and worth doing, measurements of LPS arefar from being a good method for its assessment. In our opinion,measurements of LPS, due to its limitations, are not even a reliablemarker of BT. When studying LPS in biological samples, one mustbe aware that the available method is not designed for this pur-pose, that different variables may confound the validity of the dataobtained, and finally, that LPS measurement cannot detect gram-positive translocation, a fact increasingly observed in the clinicalsetting. Results based solely on LPS measurement are hampered bythese reasons. Paraphrasing Dr. Groszmann, ‘‘anything worth doingshould be done right’’.
PABLO BELLOT, M.D.
RUBEN FRANCES, M.D.
JOSE SUCH, M.D.
JAIME BOSCH, M.D., PH.D.Hepatic Hemodynamics LaboratoryHospital Clınic, Barcelona, Spain
References1. Bellot P, Garcıa-Pagan JC, Frances R, Abraldes JG, Navasa M,
Perez-Mateo M, et al. Bacterial DNA translocation is associatedwith systemic circulatory abnormalities and intrahepatic endothe-lial dysfunction in patients with cirrhosis. HEPATOLOGY 2010;52:2044-2052.
2. Albillos A, de la Hera A, Gonzalez M, Moya JL, Calleja JL, MonserratJ, et al. Increased lipopolysaccharide binding protein in cirrhoticpatients with marked immune and hemodynamic derangement. HEPA-
TOLOGY 2003;37:208-217.3. Cirera I, Bauer TM, Navasa M, Vila J, Grande L, Taura P, et al. Bacterial
translocation of enteric organisms in patients with cirrhosis. J Hepatol2001;34:32-37.
CopyrightVC 2011 by the American Association for the Study of Liver Diseases.View this article online at wileyonlinelibrary.com.DOI 10.1002/hep.24444Potential conflict of interest: Nothing to report.
744 CORRESPONDENCE HEPATOLOGY, August 2011

Mechanisms by Which TLR4 and Endotoxin Promote Hepatocellular Carcinoma Require FurtherInvestigation
To the Editor:
Despite being impressed by the interesting hypothesis and theabundant amount of data, we would like to raise serious doubtsabout the validity and conclusions of the study ‘‘Endotoxin accu-mulation prevents carcinogen-induced apoptosis and promotes livertumorigenesis in rodents’’ by Yu and colleagues.1
Although the title of this study states that endotoxin accumula-tion prevents carcinogen-induced apoptosis, the authors showexactly the opposite in Fig. 7B and Supporting Fig. 8A in whichreduction of endotoxin by antibiotic treatment prevents carcino-gen-induced liver injury and apoptosis. The findings in Fig. 7Band Supporting Fig. 8A not only directly contradict the title ofthe article, but are also opposite to findings in Figs. 3A and 6A,and Supporting Figs. 3 and 7A in which the authors show thatdeletion of the lipopolysaccharide receptor Toll-like receptor 4(TLR4) increases carcinogen-induced liver injury. It is virtuallyimpossible that inhibition at the level of the ligand (i.e., reductionof endotoxin by antibiotics as shown in Fig. 7A) and at the levelof the receptor (deletion of TLR4) have opposite effects on theliver. If this were the case, then antibiotics should promote hepa-tocellular carcinoma (HCC), and TLR4 deletion should preventHCC. However, Yu et al. show similar effects on HCC develop-ment with both TLR4 deletion and treatment with antibiotics.Because of this conflicting data, one not only has to doubt thevalidity of the title but of the presented mechanisms and the pro-posed role of diethylnitrosamine (DEN)-induced liver injury andapoptosis. These doubts are further substantiated when consider-ing that Karin and coworkers have shown that inhibition of nu-clear factor-jB increases liver injury after DEN, and that thisincrease in injury translates to enhanced carcinogenesis in theDEN model.2 In contrast to the well-established concept, Yuet al. argue that decreased injury after DEN leads to an increasein HCC.
Because of the conflicts between the title of the study andparts of the presented data, the authors need to make a definitestatement whether (1) endotoxin accumulation prevents carcin-ogen-induced apoptosis or (2) whether the reduction of endo-toxin accumulation by treatment with antibiotics prevents car-cinogen-induced apoptosis. A study with a wrongly stated titleand conflicting data will not only confuse the readership of HE-
PATOLOGY, but also presents an obstacle to scientific progress inthis relevant research area. If the authors cannot demonstratethe validity of their title and the mechanism suggested by boththe title and their article, they should take additional time toinvestigate the role of endotoxin in carcinogen-inducedapoptosis.
ALI MENCIN, M.D.GEUM-YOUN GWAK, M.D., PH.D.ROBERT F. SCHWABE, M.D.Department of Medicine, Columbia University, New York, NY
References1. Yu LX, Yan HX, Liu Q, Yang W, Wu HP, Dong W, et al. Endo-
toxin accumulation prevents carcinogen-induced apoptosis and pro-motes liver tumorigenesis in rodents. HEPATOLOGY 2010;52:1322-1333.
2. Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples he-patocyte death to cytokine-driven compensatory proliferation that pro-motes chemical hepatocarcinogenesis. Cell 2005;121:977-990.
CopyrightVC 2011 by the American Association for the Study of Liver Diseases.View this article online at wileyonlinelibrary.com.DOI 10.1002/hep.24408Potential conflict of interest: Nothing to report.
Reply:
We thank Dr. Schwabe and colleagues for interest in our arti-cle.1,2 We have carefully reviewed our published results and alsothe original data. Our experiments showed that carcinogen-inducedapoptosis in mice can be protected by lipopolysaccharide (LPS), anendotoxin (Fig. 1), or other factors induced by diethylnitrosamine(DEN). This figure was originally included in the manuscript, butdeleted in revision due to page limitations. The main questionconcerns why reduction of LPS by antibiotics (figure 7B)1 led tolower liver injury but LPS receptor Toll-like receptor 4 (TLR4)deficiency potentiated liver injury (figure 3A).1 Possible explana-tions are as follows: (1) The alanine aminotransferase level usuallyindicates liver damage and does not exclusively represent the degreeof cell apoptosis. LPS protection as previously described takes effectby a certain time period (e.g., at 48 hours after injection; figure6C-F),1 although it even enhances liver injury at 24 hours afterDEN injection. We did not claim that no or low LPS levels andantibiotic suppression of LPS will increase liver injury. (2) Becausetreatment with antibiotics prevented the increase of plasma LPS(figure 7A),1 inhibition of LPS-induced inflammation upon DENexposure might be responsible for the reduction of liver injury. Wefound that antibiotic-induced reduction of LPS and lack of TLR4reduced the number and volume of tumors with the decrease oftumor necrosis factor a and interleukin-6.
We had the same concerns about this discrepancy, because itappears to contradict the hypothesis that TLR4�/� mice should haveless liver injury. However, in terms of the acute liver damage inducedby DEN, the TLR4 knockout mice are different from wild-typemice in response to the short-term antibiotics treatment. In additionto LPS, TLR4 interacts with other endogenous ligands, such as highmobility group box 1 (HMGB1) and heat shock proteins (HSPs).TLR4 deficiency therefore ablated the protective effects of both LPSand endogenous ligands on hepatocytes, thus leading to more severeliver damage. Treatment with antibiotics mainly reduced the acuteinflammatory responses to LPS in nonparenchymal cells, which isthought to be the major contributing factor to acute hepatocyteinjury. It is conceivable that reduced production of proinflammatorycytokines such as tumor necrosis factor a was responsible for allevia-tion of DEN-induced acute liver damage. On the other hand, duringthe multistage process of hepatocarcinogenesis, accumulation of en-dotoxin promotes chronic tumorigenic inflammation and alleviatesapoptosis in precancerous and cancerous cells. So, loss of TLR4 isnot equivalent to LPS ablation in terms of the responsive cell typesand inflammation status.
As cited by Dr. Schwabe and colleagues, Karin’s group at Uni-versity of California San Diego reported an increase in DEN-induced hepatocellular carcinoma development when inhibitor of
HEPATOLOGY, Vol. 54, No. 2, 2011 CORRESPONDENCE 745
nuclear factor kappa B kinase subunit beta (IKKb) was deleted inhepatocytes. However, the same group also found that hepatocellu-lar carcinoma was suppressed in mice with IKKb ablated in bothhepatocytes and Kupffer cells.3 Furthermore, Pikarsky et al. foundthat inhibiting nuclear factor jB signaling suppressed hepatocarci-nogenesis.4 Resolving these differences will lead to better under-standing of hepatocarcinogenesis.
LE-XING YU, PH.D.1
HE-XIN YAN, PH.D.1
WEN YANG, PH.D.1
HONG-YANG WANG, M.D.1,21International Cooperation Laboratory
Liver Centre of SMMUEastern Hepatobiliary Surgery HospitalSecond Military Medical UniversityShanghai, China
2National Laboratory for Oncogene and Related GenesCancer Institute of Shanghai Jiao Tong UniversityShanghai, China
References
1. Yu LX, Yan HX, Liu Q, Yang W, Wu HP, Dong W, et al. Endo-toxin accumulation prevents carcinogen-induced apoptosis andpromotes liver tumorigenesis in rodents. HEPATOLOGY 2010;52:1322-1333.
2. Mencin A, Gwak GY, Schwabe RF. None. HEPATOLOGY 2011.
3. Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta cou-ples hepatocyte death to cytokine-driven compensatory prolifera-tion that promotes chemical hepatocarcinogenesis. Cell 2005;121:977-990.
4. Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, et al.NF-kappaB functions as a tumour promoter in inflammation-associatedcancer. Nature 2004;431:461-466.
CopyrightVC 2011 by the American Association for the Study of Liver Diseases.View this article online at wileyonlinelibrary.com.DOI 10.1002/hep.24463Potential conflict of interest: Nothing to report.
Detection of Acetaminophen–Cysteine Adducts in Cases of Indeterminate Liver Failure Is NotDiagnostic of Intentional Overdose
To the Editor:
Khandelwal and colleagues have elegantly demonstrated throughthe detection of acetaminophen–cysteine adducts that a significantproportion of indeterminate acute liver failure (ALF) is due toacetaminophen (N-acetyl-p-aminophenol; APAP) toxicity.1 How-ever, I have concerns regarding the authors’ suggestion that the useof adducts to recognize APAP overdosing might be used to influ-ence decisions concerning transplant candidacy.
We have recently highlighted cases of ALF that have occurredas a result of administration of APAP at the maximum recom-mended daily dose in adults with malnutrition and/or low bodyweight. We also demonstrated through an internal audit at ourcenter that most practitioners are unaware that these patients haveincreased susceptibility to APAP toxicity and that biochemical evi-dence of subclinical liver injury is not infrequent.2 This has led usto suspect that APAP toxicity following ‘‘therapeutic’’ doses inhigh-risk patients contributes to the number of cases labeled as
COLOR
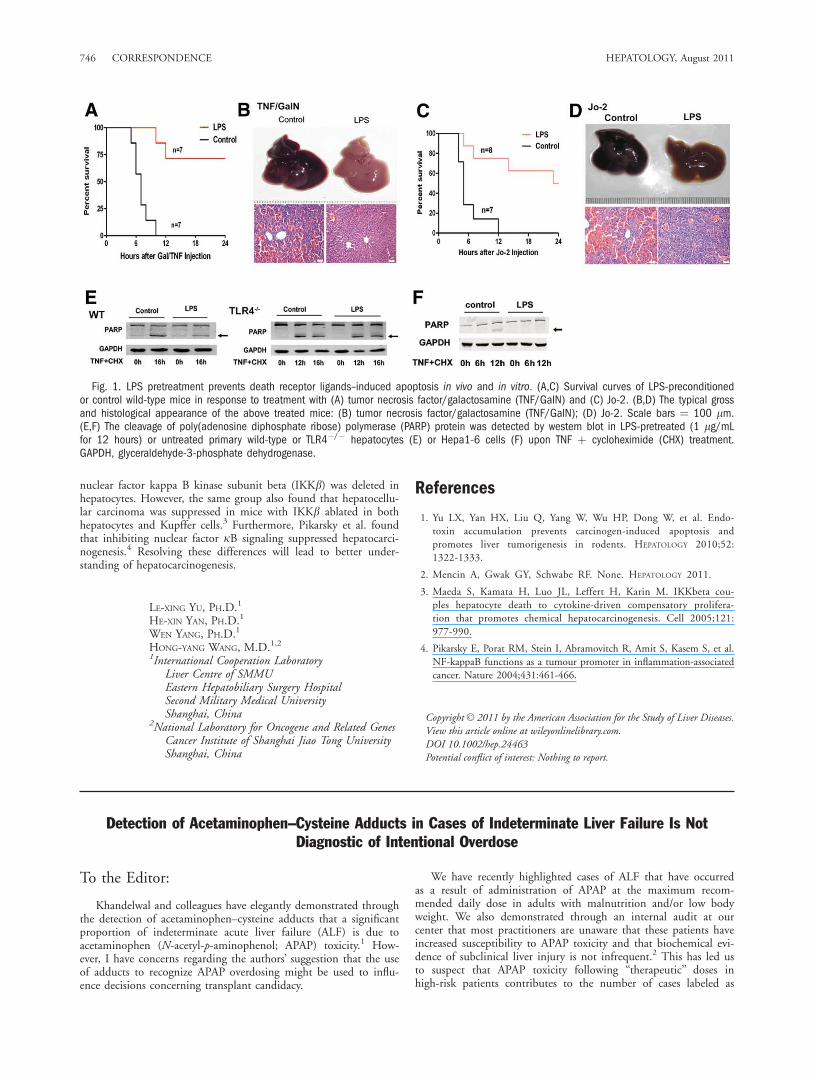
Fig. 1. LPS pretreatment prevents death receptor ligands–induced apoptosis in vivo and in vitro. (A,C) Survival curves of LPS-preconditionedor control wild-type mice in response to treatment with (A) tumor necrosis factor/galactosamine (TNF/GaIN) and (C) Jo-2. (B,D) The typical grossand histological appearance of the above treated mice: (B) tumor necrosis factor/galactosamine (TNF/GalN); (D) Jo-2. Scale bars ¼ 100 lm.(E,F) The cleavage of poly(adenosine diphosphate ribose) polymerase (PARP) protein was detected by western blot in LPS-pretreated (1 lg/mLfor 12 hours) or untreated primary wild-type or TLR4�/� hepatocytes (E) or Hepa1-6 cells (F) upon TNF þ cycloheximide (CHX) treatment.GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
746 CORRESPONDENCE HEPATOLOGY, August 2011

indeterminate ALF. Although previous studies have shown thatadduct levels are low in subjects receiving therapeutic doses,3 thesewere carried out in healthy subjects. Adducts are likely to be signif-icantly elevated in those with low body mass whose peak plasmaconcentration of APAP reaches toxic levels, and in malnourishedindividuals with glutathione deficiency and diminished capacity toneutralize N-acetyl-p-benzoquinone imine. Furthermore, in theUnited States, up to 50% of APAP-induced ALF is thought tooccur as a result of unintentional overdose,4 leading to the recentdecision by the U.S. Food and Drug Administration to limit thedosage unit of APAP to 325 mg in combination prescription prod-ucts. Thus, unless there is a clear psychiatric history, transplanta-tion must not be precluded on the basis of positive acetamino-phen–cysteine adducts.
The use of these adducts may help confirm APAP toxicity as thecause of ALF, providing more accurate epidemiological data. Yet,unless the levels can be correlated with prognosis, it is difficult tosee how they will change clinical practice. There is already evidencefor the efficacy of N-acetylcysteine (NAC) in non-APAP-inducedALF,5 and it is therefore surprising that only 40% of adduct-posi-tive and 17.8% of adduct-negative patients with indeterminate ALFreceived NAC. The most important message we should take fromthis study is that all patients with indeterminate ALF should betreated with NAC.
LEE C. CLARIDGE, BM,BS (HONS), MRCP (UK)Centre for Liver Research, University of Birmingham
Birmingham, United Kingdom
References
1. Khandelwal N, James LP, Sanders C, Larson AM, Lee WM; the AcuteLiver Failure Study Group. Unrecognized acetaminophen toxicity as acause of indeterminate acute liver failure. HEPATOLOGY 2011;53:567-576.
2. Claridge LC, Eksteen B, Smith A, Shah T, Holt AP. Acute liver failureafter administration of paracetamol at the maximum recommendeddaily dose in adults. BMJ 2010;341:c6764.
3. James LP, Simpson P, Russo M, Watkins PB. Detection of acetamino-phen protein adducts in serum during therapeutic exposure to acet-aminophen in healthy volunteers [Abstract]. HEPATOLOGY 2007;46(Suppl 1):812A.
4. Larson AM, Polson J, Fontana RJ, Davern TJ, Lalani E, Hynan LS,et al. Acetaminophen-induced acute liver failure: results of a UnitedStates multicenter, prospective study. HEPATOLOGY 2005;42:1364-1372.
5. Lee WM, Hynan LS, Rossaro L, Fontana RJ, Stravitz RT, Larson AM,et al. Intravenous N-acetylcysteine improves transplant-free survival inearly stage non-acetaminophen acute liver failure. Gastroenterology2009;137:856-864.
CopyrightVC 2011 by the American Association for the Study of Liver Diseases.View this article online at wileyonlinelibrary.com.DOI 10.1002/hep.24251Potential conflict of interest: Nothing to report.
Proper Antiviral Therapy for Hepatitis B Virus–Associated Acute-on-Chronic Liver Failure
To the Editor:
Convincing evidence that antiviral therapy contributes toreduce short-term mortality of patients with hepatitis B virus(HBV)-associated acute-on-chronic liver failure (ACLF) is stillunavailable.1,2 I read with great interest the article by Garget al.,3 who demonstrated that tenofovir can reduce the mortal-ity of patients with severe spontaneous reactivation of chronichepatitis B presenting as ACLF. Their optimistic results give ussome evidence about antiviral therapy in HBV-associatedACLF. However, a recent prospective cohort study by Wonget al.4 demonstrated that entecavir treatment, as compared tolamivudine treatment, is associated with increased short-termmortality of patients with severe acute exacerbation of chronichepatitis B.
Both tenofovir and entecavir are now the most effective antivi-ral agents for chronic hepatitis B. But why did they have converseeffects upon the short-term mortality of HBV-associated ACLF?Here, we would like to offer some possible reasons for this interest-ing question. First, Garg et al. identified patients with HBV-associ-ated ACLF by a high HBV DNA level (>105 copies/mL).3 Thecriterion on which their previous study is based may be useful, butit is still immature with only 14 patients enrolled in the tenofovirtreatment group. There are obviously distinct prognoses betweenacute hepatitis B–related liver failure and HBV-associated ACLF.Therefore, enrollment of any patients with acute HBV-related liverfailure may interfere with the results. Besides, genotype D was thepredominant HBV genotype in the Indian study subjects (85.2%),but genotype B or C is the main HBV genotype in China. Also, itis still unknown whether HBV genotype may affect the results ofantiviral treatment in ACLF. In addition, Wong et al. speculated
that the problem of lactic acidosis or an exaggerated immuneresponse due to rapid virological suppression may lead to and exac-erbate liver injury among patients with severe acute exacerbation ofchronic hepatitis B.4 However, the latter explanation is contradic-tory to the opinion of Garg et al., who proposed that rapid reduc-tion in HBV DNA levels independently predicted a good short-term survival rate. Thus, larger prospective and multicentric studiesare encouraged to further evaluate the affect of tenofovir and ente-cavir on short-term mortality of patients with HBV-associatedACLF.
TAO LIU, M.D.1,2
CHUNMEI ZHANG, M.D.1
YINGJIE WANG, M.D.21Department of Internal Medicine III
The Northern Region of No. 401 HospitalQingdao, Shandong, China
2Department of Infectious Diseases, Southwest HospitalThird Military Medical University, Chongqing, China
References1. Roche B, Samuel D. The difficulties of managing severe hepatitis B vi-
rus reactivation. Liver Int 2011;31(Suppl 1):104-110.2. Cui YL, Yan F, Wang YB, Song XQ, Liu L, Lei XZ, et al. Nucleoside
analogue can improve the long-term prognosis of patients with hepatitisB virus infection-associated acute on chronic liver failure. Dig Dis Sci2010;55:2373-2380.
3. Garg H, Sarin SK, Kumar M, Garg V, Sharma BC, Kumar A. Tenofo-vir improves the outcome in patients with spontaneous reactivation ofhepatitis B presenting as acute-on-chronic liver failure. HEPATOLOGY;doi:10.1002/hep.24109.
HEPATOLOGY, Vol. 54, No. 2, 2011 CORRESPONDENCE 747

4. Wong VW, Wong GL, Yiu KK, Chim AM, Chu SH, Chan HY, et al.Entecavir treatment in patients with severe acute exacerbation ofchronic hepatitis B. J Hepatol 2011;54:236-242.
CopyrightVC 2011 by the American Association for the Study of Liver Diseases.View this article online at wileyonlinelibrary.com.DOI 10.1002/hep.24266Potential conflict of interest: Nothing to report.
Genetic Background of Hepatocyte Cell Lines: Are InVitro Hepatitis C Virus Research Data Reliable?
To the Editor:
The results of independent genome-wide association studieshave recently been reported, describing the identification of sin-gle-nucleotide polymorphisms (SNPs) strongly associated withnatural hepatitis C virus (HCV) clearance during acute infectionand cure of chronic HCV infection on antiviral therapy withpegylated interferon (IFN)-a and ribavirin.1-4 These SNPs, inchromosomal region 19q13 upstream of the interleukin-28B(IL28B; IFN-k3) gene, appear to be markers of cell responsive-ness to type 1 IFNs. Nevertheless, the underlying mechanismsremain unknown. Several articles have been subsequently pub-lished that confirmed the association and provided valuable infor-mation on the predictive value of the so-called ‘‘IL28B genotype’’on the outcomes of acute HCV infection, antiviral treatment ofchronic infection, and recurrence of HCV infection after livertransplantation. These findings have been recently reviewed byAfdhal et al.5
To date, most of the current knowledge on HCV biologyand new HCV drug development is based on in vitro studiesusing hepatoma cell lines, principally Huh7 cells and their deriv-atives. These cells are permissive for HCV entry, and harbor rep-lication of subgenomic and genomic replicons of various geno-types, as well as the full life cycle of the genotype 2a JFH1(Japanese fulminant hepatitis 1) infectious clone and derivatives.Nevertheless, their IL28B genotype remains uncharacterized, de-spite its likely effects on numerous intracellular biological proc-esses relevant to HCV infection. We therefore genotyped theIL28B rs12979860 SNP of hepatoma cell lines used in HCVresearch, including Huh7, Huh7.5, Huh7.5.1, and HepG2 cells,as well as the commonly used non-hepatoma HEK293 (humanembryonic kidney 293) and Hela cell lines. We used ultra-deeppyrosequencing to sequence 292 nucleotides flanking thers12979860 locus, using a GS FLX Titanium Sequencing Kit inconjunction with a Genome Sequencer FLX (Roche MolecularSystems, Pleasanton, CA). Data was analyzed using two originalin-house softwares: Pyroclass and Pyromute, with the resultsshown in Table 1.
Huh7 cells and derivatives, and even two Huh7 cell linesoriginating from different laboratories, demonstrated differentallelic frequencies. The ‘‘non-Mendelian’’ distribution of thesepolymorphisms is likely a consequence of the polyploidal natureof hepatoma cells, but may indicate the presence of multipleclonal populations arising under different environmentalpressures.
Data obtained from in vitro cell culture systems may oftenbe related to the genetic background of the cell line(s) used.Our results emphasize the need for the genetic characterizationof hepatoma cell lines used for HCV research, especially whenthese studies involve innate immunity, IFN responsiveness, theassessment of antiviral compound efficacy, or experimentsaimed at cell cure. This conclusion may be extended to anumber of other in vitro experiments in which the geneticbackground of the cell line has an influence on the propertiesstudied.
PAUL BENSADOUN, M.D.1
CHRISTOPHE RODRIGUEZ, M.D.1,2
ALEXANDRE SOULIER, M.D.1,2
MARTIN HIGGS, M.D.1
STEPHANE CHEVALIEZ, PHARM.D., PH.D.1,2
JEAN-MICHEL PAWLOTSKY, M.D., PH.D.1,21Institut National de la Sante et de la Recherche
Medicale, Unite U955, Creteil, France2National Reference Center for Viral Hepatitis B, C and
Delta, Department of Virology, Henri Mondor HospitalUniversity of Paris-Est, Creteil, France
References1. Ge D, Fellay J, Thompson AJ, Simon JS, Shianna KV, Urban TJ, et al.
Genetic variation in IL28B predicts hepatitis C treatment-induced viralclearance. Nature 2009;461:399-401.
2. Suppiah V, Moldovan M, Ahlenstiel G, Berg T, Weltman M, AbateML, et al. IL28B is associated with response to chronic hepatitis Cinterferon-alpha and ribavirin therapy. Nat Genet 2009;41:1100-1104.
3. Tanaka Y, Nishida N, Sugiyama M, Kurosaki M, Matsuura K, Saka-moto N, et al. Genome-wide association of IL28B with response topegylated interferon-alpha and ribavirin therapy for chronic hepatitisC. Nat Genet 2009;41:1105-1109.
4. Thomas DL, Thio CL, Martin MP, Qi Y, Ge D, O’Huigin C, et al.Genetic variation in IL28B and spontaneous clearance of hepatitis Cvirus. Nature 2009;461:798-801.
5. Afdhal NH, McHutchison JG, Zeuzem S, Mangia A, Pawlotsky JM,Murray JS, et al. Hepatitis C pharmacogenetics: state of the art in2010. HEPATOLOGY 2011;53:336-345.
CopyrightVC 2011 by the American Association for the Study of Liver Diseases.View this article online at wileyonlinelibrary.com.DOI 10.1002/hep.24278Potential conflict of interest: Nothing to report.
Table 1. Percentage Presence of rs12979860 Alleles in Selected Hepatoma and Nonhepatoma Cell Lines
rs12979160 Allele Distribution Huh7* Huh7** Huh7.5 Huh7.5.1 HepG2 HEK293 Hela
%C 100% 20% 56% 0% 61% 17% 0%
%T 0% 80% 44% 100% 39% 83% 100%
IL28B genotype CC CT CT TT CT CT TT
The percentage presence of each allele was calculated from approximately 8000 sequences of each cell type generated by ultra-deep pyrosequencing. Huh7*
and Huh7** were two Huh7 cell lines originating from two separate laboratories.
748 CORRESPONDENCE HEPATOLOGY, August 2011

Silibin’s Mode of Action Against Hepatitis C Virus: A Controversy Yet to Be Resolved
To the Editor:
Wagoner et al.1,2 suggested that Legalon-SIL (SIL), a commer-cially available intravenous preparation of silibinin, has an effect onhepatitis C virus (HCV) entry and cell-to-cell spread in vitro withonly marginal suppression of HCV nonstructural protein 5B RNA–dependent RNA polymerase (RdRp) activity, an observation that isin contrast with the findings of Ahmed-Belkacem et al.3 Three clini-cal studies have reported the viral response during SIL therapy.4-6
The protocols were similar and consisted of daily injection of SIL for7 days followed by pegylated interferon plus ribavirin; however, inBiermer and Berg,5 ribavirin was administered before and duringsilibinin treatment. Viral decline after the initiation of SIL wasmonophasic until day 7 in the two case reports and in the majorityof subjects in the study by Ferenci et al.4 (Fig. 1, red squares). Inter-estingly, a monophasic pattern of viral decline (Fig. 1, blue circles)was also observed in some patients given 14 days of monotherapywith RG7128, a nucleoside HCV-RdRp inhibitor (unpublisheddata), and in 3 subjects (N ¼ 5) in Le Pogam et al. (figure 1A in thatarticle).7 This monophasic decline is strikingly different from thebiphasic viral decline typically observed in patients treated with pro-tease inhibitors or (pegylated)interferon-a–based therapies8 (Fig. 1,triangles). The fact that both SIL and RG7128 led to a monophasicHCV decline in some patients is interesting and tends to support thefindings of Ahmed-Belkacem et al.3
According to the standard HCV infection model,9 a monophasicviral decline pattern results when viral infection is blocked, whichtends to support the results of Wagoner et al.1,2 On the other hand,one can also predict a monophasic decline of virus if one assumes inthe standard viral kinetic model a gradual reduction in viral produc-tion (unpublished observation), rather than an immediate high anti-viral effectiveness in reducing viral production, as is the case withinterferon-a or protease inhibitors. This gradual reduction in viralproduction could be related to drug pharmacokinetic and pharmaco-dynamic properties, which could explain the similarity in the patternof viral decline observed under treatment with SIL and RG7128 andpossibly will shed light on why some patients treated with either ofthese two agents had a monophasic viral decline pattern.
In summary, to further investigate this controversy we suggestthat pharmacokinetic and pharmacodynamic studies of SIL areneeded to better understand the nature of the observed monophasicviral decline in treated patients. If SIL-resistant strains can be identi-fied, the nature of the resistance mutations would provide informa-
tion about the mechanism of action. If resistance mutations arefound in the HCV polymerase, it would favor an HCV-RdRp inhib-itor mechanism, whereas if resistance mutations exist in HCV E1/E2, it would support an entry inhibitor mechanism. Further in vitroexperiments10 that include detailed kinetics of both intracellular andextracellular HCV RNA during treatment with SIL are likely to pro-vide more insights into its mechanism(s) of action against HCV.
HAREL DAHARI, PH.D.1,2
JEREMIE GUEDJ, PH.D.1
ALAN S. PERELSON, PH.D.11Theoretical Biology and Biophysics
Los Alamos National LaboratoryLos Alamos, NM
2Department of MedicineUniversity of Illinois at ChicagoChicago, IL
References1. Wagoner J, Negash A, Kane OJ, Martinez LE, Nahmias Y, Bourne N,
et al. Multiple effects of silymarin on the hepatitis C virus lifecycle.Hepatology 2010;51:1912-1921.
2. Wagoner J, Morishima C, Graf TN, Oberlies NH, Teissier E, Pecheur EI,et al. Differential in vitro effects of intravenous versus oral formulations of sil-ibinin on the HCV life cycle and inflammation. PLoS One 2011;6:e16464.
3. Ahmed-Belkacem A, Ahnou N, Barbotte L, Wychowski C, Pallier C,Brillet R, et al. Silibinin and related compounds are direct inhibitors ofhepatitis C virus RNA-dependent RNA polymerase. Gastroenterology2010;138:1112-1122.
4. Ferenci P, Scherzer TM, Kerschner H, Rutter K, Beinhardt S, Hofer H,et al. Silibinin is a potent antiviral agent in patients with chronic hepa-titis C not responding to pegylated interferon/ribavirin therapy. Gastro-enterology 2008;135:1561-1567.
5. Biermer M, Berg T. Rapid suppression of hepatitis C viremia induced byintravenous silibinin plus ribavirin. Gastroenterology 2009;137:390-391.
6. Payer BA, Reiberger T, Rutter K, Beinhardt S, Staettermayer AF, Peck-Radosavljevic M, et al. Successful HCV eradication and inhibition ofHIV replication by intravenous silibinin in an HIV-HCV coinfectedpatient. J Clin Virol 2010;49:131-133.
7. Le Pogam S, Seshaadri A, Ewing A, Kang H, Kosaka A, Yan JM, et al.RG7128 alone or in combination with pegylated interferon-a2a and riba-virin prevents hepatitis C virus (HCV) replication and selection of resistantvariants in HCV-infected patients. J Infect Dis 2010;202:1510-1519.
8. Guedj J, Rong L, Dahari H, Perelson AS. A perspective on modellinghepatitis C virus infection. J Viral Hepat 2010;17:825-833.
9. Neumann AU, Lam NP, Dahari H, Gretch DR, Wiley TE, Layden TJ,et al. Hepatitis C viral dynamics in vivo and the antiviral efficacy ofinterferon-alpha therapy. Science 1998;282:103-107.
10. Dahari H, Barretto N, Sainz B Jr, Guedj J, Perelson AS, Uprichard SL.Modeling interferon-alpha mediated inhibition kinetics of intracellularand extracellular HCV RNA during HCV infection in vitro. J Hepatol2011;54(Suppl 1):S312.
CopyrightVC 2011 by the American Association for the Study of Liver Diseases.View this article online at wileyonlinelibrary.com.DOI 10.1002/hep.24310This work was performed under the auspices of the U.S. Department of Energyunder contract DE-AC52-06NA25396, and supported by National Institutesof Health grants RR06555-19, P20-RR018754-6, AI065256-5,R56-AI078881-01, and AI28433-20, the National Science Foundationunder grant NSF PHY05- 51164, and by the University of Illinois WalterPayton Liver Center GUILD. Potential conflict of interest: Nothing to report.
COLOR
Fig. 1. Representative serum HCV RNA decline from baseline duringthe first week of treatment with silibinin monotherapy (digitized fromPayer et al.6, red squares), RG7128 1500-mg BID (blue circles; manu-script in preparation), daily 10 MIU IFN9 (black triangles) and telapre-vir+PegIFN8 (gray triangles). Solid lines were used to emphasizeplausible phases of viral decline.
HEPATOLOGY, Vol. 54, No. 2, 2011 CORRESPONDENCE 749

Liver Biopsy Interpretation in Liver Cancer
To the Editor:
Having the privilege of working closely with a liver pathologist,I fully agree with the comments of Brunt and Gores.1 Today, thewidespread ignorance of both the medical and scientific commun-ities about the role of liver pathologists has led to a misapprehen-sion of the complexity of the pathology underlying hepatocellularcarcinoma (HCC).
There is a paradox in most specialized (tertiary) liver centers.Radiologists often perform liver biopsy to prove that imagingapproaches can effectively replace this invasive procedure fordiagnosing HCC; however, clinicians seldom use biopsies forwhat they can bring to the understanding of the disease. As aresult of this paradigm shift, the time is long gone when path-ologists were the most influential in the understanding of dis-eases. I do not, however, share the pessimism of Brunt andGores.1 Indeed, having the chance to work with a molecular bi-ologist and a liver pathologist, I know that pathologists havealso made great progress from their interactions with molecularbiologists in the understanding of what they are used to seeing(but not necessarily understanding).
This is clearly illustrated by recent studies of hepatocellularadenoma (HCA) from our group. HCA is a benign hepatocellu-lar tumor with the potential to transform into HCC. Until2007, HCA was described as a single benign liver tumor entityin all the textbooks. Although HCA phenotypes present clearcharacteristics that could have led to a phenotypic classificationof these tumors, it was only around 2006 that molecular biol-ogy–based approaches demonstrated HCA to be an heterogene-ous entity with three major classes driven by genetic mutations(i.e., hepatocyte nuclear factor 1a, b-catenin, and inflamma-tion).2,3 The reason that liver pathologists ignored these pheno-types for more than 30 years is probably that they did notthink that HCA could be due to genetic disorders. A similarexample has been observed with the diagnostic correctionbrought by molecular biology approaches in cases of inflamma-tory HCA wrongly diagnosed as telangiectatic focal nodularhyperplasia.4 Pathologists definitely have learned from molecularbiologists and will learn more from them.
I doubt, however, that molecular biologists alone, without apathological background in HCC, will be able to identify rele-vant subgroups of HCC in terms of diagnosis, prognosis, and/or
clinical management. However, I have no doubt that liver path-ologists, once they have examined the liver tissue provided tomolecular biologists and have efficiently collaborated with them,will be able in the near future to identify relevant HCCsubtypes.
HCC is too complex an entity to be left in the hands of asingle group of liver specialists, whoever they might be. Themultidisciplinary approach to tackling HCC pathology in termsof diagnosis, prognosis, and clinical management should becomereal. In that context, liver pathologists not only should be add-ing tags to samples but also should be actively participating inthe characterization of the samples. In the mean time, liverpathologists should keep faith in what they do best: preservingtissue for molecular studies and establishing up-to-date andmeaningful pathology reports.
CHARLES BALABAUD, M.D.Universite Bordeaux 2, Bordeaux, France
References1. Brunt EM, Gores G. ‘‘Pardon the interruption’’: comments on the
International Liver Cancer Association Fourth Annual Conference,2010. Is there really no role for liver biopsy interpretation in liver can-cer? HEPATOLOGY 2011;53:721-722.
2. Bioulac-Sage P, Rebouissou S, Thomas C, Blanc JF, Saric J, Sa CunhaA, et al. Hepatocellular adenoma subtype classification using molecularmarkers and immunohistochemistry. HEPATOLOGY 2007;46:740-748.
3. Zucman-Rossi J, Jeannot E, Nhieu JT, Scoazec JY, Guettier C,Rebouissou S, et al. Genotype-phenotype correlation in hepatocellularadenoma: new classification and relationship with HCC. HEPATOLOGY
2006;43:515-524.4. Bioulac-Sage P, Rebouissou S, Sa Cunha A, Jeannot E, Lepreux S,
Blanc JF, et al. Clinical, morphologic, and molecular features definingso-called telangiectatic focal nodular hyperplasias of the liver. Gastroen-terology 2005;128:1211-1218.
CopyrightVC 2011 by the American Association for the Study of Liver Diseases.View this article online at wileyonlinelibrary.com.DOI 10.1002/hep.24320Potential conflict of interest: Nothing to report.
No Transfats
To the Editor:
The recent report of nonalcoholic steatohepatitis–like featuresand liver fibrosis in mice fed a diet high in saturated fats and high-fructose corn syrup by Kohli et al.1 is another important additionto our understanding of the pathogenesis of nonalcoholic steatohe-patitis. However, readers should be aware that there is an error inthis article’s title, which indicates that the mice were fed a diet con-taining transfats. The fat fed to the mice in these experimentscame from fully hydrogenated coconut oil. According to materials
available from the manufacturer, Research Diets, the naturally lowmonounsaturated and polyunsaturated fat content of coconut oil(8.2%) is reduced to 0.8% through hydrogenation, and the rest(99.2%) is saturated fat. The unsaturated fat (0.8%) is monounsat-urated fatty acid oleic acid, so the diet does not contain transfats.This error in describing the composition of the diet highlights theimportance of including as much detail as possible in the Materialsand Methods section with respect to the sources of fat, carbohy-drates, and protein in animal diets used to induce features of non-alcoholic steatohepatitis.
750 CORRESPONDENCE HEPATOLOGY, August 2011

BRENT A. NEUSCHWANDER-TETRI, M.D.Division of Gastroenterology and Hepatology, St. Louis University
St. Louis, MO
Reference1. Kohli R, Kirby M, Xanthakos SA, Softic S, Feldstein AE, Saxena V,
et al. High-fructose, medium chain trans fat diet induces liver fibro-
sis and elevates plasma coenzyme Q9 in a novel murine model ofobesity and nonalcoholic steatohepatitis. HEPATOLOGY 2010;52:934-944.
CopyrightVC 2011 by the American Association for the Study of Liver Diseases.View this article online at wileyonlinelibrary.com.DOI 10.1002/hep.24321Potential conflict of interest: Nothing to report.
HEPATOLOGY, Vol. 54, No. 2, 2011 CORRESPONDENCE 751
Copyright © 2022 FDOKUMEN




















