
Investigation of amino-tail translocation by the conserved YidC ...
124
Investigation of amino-tail translocation by the conserved YidC, Sec and independent pathways DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Sri Karthika Shanmugam Ohio State University Biochemistry Program The Ohio State University 2019 Dissertation Committee Dr. Ross E. Dalbey, Advisor Dr. James Cowan Dr. Natividad Ruiz Dr. Thomas Magliery
-
Upload
khangminh22 -
Category
Documents
-
view
3 -
download
0
Transcript of Investigation of amino-tail translocation by the conserved YidC ...

1
Investigation of amino-tail translocation by the
conserved YidC, Sec and independent pathways
DISSERTATION
Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy
in the Graduate School of The Ohio State University
By
Sri Karthika Shanmugam
Ohio State University Biochemistry Program
The Ohio State University
2019
Dissertation Committee
Dr. Ross E. Dalbey, Advisor
Dr. James Cowan
Dr. Natividad Ruiz
Dr. Thomas Magliery

2
Copyrighted by
Sri Karthika Shanmugam
2019

ii
Abstract
Chapter 1 of this dissertation reviews existing knowledge of the areas of protein
translocation and membrane insertion. The core machineries involved in membrane protein
biogenesis are remarkably conserved. Proteins that fold within the cell prior to export are
translocated by the Tat system. Canonical substrates of this pathway possess signal
sequences with a twin-arginine motif which interacts with the TatABC membrane
translocon complex to facilitate substrate translocation. The majority of the secreted and
membrane proteins are translocated by the Sec machinery in an unfolded state. It consists
of the SecY, SecE and SecG proteins which form an hour-glass shaped channel with a
lateral gate opening into the membrane. Substrate targeting to the Sec translocon occurs
either post-translationally or co-translationally. Most exported proteins in E. coli are post-
translationally targeted by the SecA/B pathway. The SecB is a molecular chaperone that
delivers a subset of substrate proteins in an unfolded state to SecA. The SecA motor
ATPase powers the movement of substrates through the SecYEG channel. Co-translational

iii
targeting typically involves the association of the translating ribosome with the translocase
directly, or with the translocase after delivery by the signal-recognition particle (SRP) and
its receptor (SR). Certain substrates of the SRP pathway are targeted to another translocon,
YidC. YidC plays a pivotal role in the membrane integration, folding and assembly of a
subset of proteins including energy-transducing and respiratory complexes. It functions
both autonomously and in concert with the SecYEG channel in bacteria. The YidC family
of proteins are widely conserved in all domains of life with new members recently
identified in the eukaryotic ER membrane. Bacterial and organellar members share the
conserved 5 TM core which forms a unique hydrophilic cavity in the inner leaflet of the
bilayer accessible from the cytoplasm and the lipid phase. The work presented here
investigates the pathway-determining factors for amino-terminal translocation in E. coli.
In addition, the conserved function of the YidC family of proteins to insert a single-
spanning protein into the membrane was explored using biophysical methods.
Different attributes of membrane protein substrates have been proposed and characterised
as translocation-pathway determinants. However, several gaps in our understanding of the
mechanism of targeting, insertion and assembly of inner membrane proteins exist.
Specifically, the role played by hydrophilic N-terminal tails in pathway selection is unclear.
In Chapter 2, we have evaluated length and charge density as translocase determinants
using model proteins. Strikingly, the 36 residue N-tail of 2Pf3-Lep translocates
independent of YidC-Sec. This is the longest N-tail region that is translocated by this
pathway. We confirmed this using a newly constructed YidC-Sec double-depletion strain.
Increasing its N-tail length with uncharged spacer peptides led to YidC dependence and

iv
eventually YidC-Sec dependence, hence establishing that length has a linear effect on
translocase dependence. Tails longer than 60 residues were not inserted, however an MBP-
2Pf3-Lep fusion protein could be translocated. This suggests that longer N-tails can be
translocated if it can engage SecA. In addition, we have examined how the positioning of
charges within the translocated N-tail affects the insertion pathway. Additional charges can
be translocated by the Lep TM when the charges are distributed across a longer N-tail. We
tested charge density as a translocase determinant and confirmed that the addition of
positive or negatives charges led to a greater dependence on YidC-Sec when they were
placed close to each other than away. Findings from this work make an important advance
in our existing knowledge about the different insertion mechanisms of membrane proteins
in E. coli.
The YidC family of proteins share structural homology and engage in the process of
membrane protein biogenesis of various cellular and organellar membranes. Chapter 4
explores the functional conservation amongst the eukaryotic YidC homologs by testing the
insertion of the YidC-only model substrate Pf3 coat protein. Fluorescence correlation
spectroscopy technique was utilized to follow the insertion process at the single-molecule
level in vitro. The thylakoid membrane of chloroplasts contains 2 YidC-paralogs: Alb3 and
Alb4. Although both have distinct set of substrates in the chloroplasts, we found that they
can insert Pf3 coat substrate with comparable efficiencies to YidC. This is in agreement
with the fact that these insertases can complement YidC in E. coli. We also tested the
mitochondrial homolog Oxa1 and the newly proposed ER-resident member Get1. Oxa1 is
a bonafide member of the YidC family; both YidC and Oxa1 can complement one another.

v
However, experimental evidence is lacking to confirm the placement of Get1 in the YidC
family. Interestingly, both Oxa1 and Get1 can insert Pf3 coat protein into reconstituted
proteoliposomes. This suggests that Get1 and the other homologs tested are functionally
conserved. This study provides fundamental information about the evolutionarily
conserved role of YidC in membrane protein biogenesis.
vi
Dedication
This document is dedicated to my family.

vii
Acknowledgments
Firstly, I would like to thank my advisor, Dr. Ross E. Dalbey, for his mentorship, support
and encouragement throughout the course of my graduate studies.
Additionally, I am grateful for the valuable advice and feedback that were provided by our
collaborators Dr. Andreas Kuhn and Dr. Gregory Phillips. I would like to thank my
committee members Dr. James Cowan, Dr. Natividad Ruiz and Dr. Thomas Magliery for
their support and guidance.
I would also like to thank former and current lab members Dr. Bala Subramani Hariharan,
Dr. Yuanyuan Chen, Haoze He and Margaret Steward for their advice and friendship. I am
also grateful to the Kuhn lab for their support.
Most importantly, I would like to thank my father Dr. K. R. Shanmugam for his counsel
and encouragement. I am grateful to my sister Sakthi Indra Shanmugam and my
grandmother Rukmani Rangasamy for their constant love and support.
Finally, I would like to thank my friends Sharadhi Sukumaran, Karthic Subramanian,
Anusha Kumar, Suriya Subramanian, Dr. Nidhi Seethapathi and Gowtham Venkatraman
for their kindness and motivation during this journey.

viii
Vita
May 26, 1992 .................................................Born in Erode, India
2013................................................................B. Tech. Industrial Biotechnology,
Anna University, India
2013 - Present ...............................................Graduate Teaching and Research Associate,
Department of Chemistry and Biochemistry,
The Ohio State University
Publications
Chen, Y., Soman, R., Shanmugam, S. K., Kuhn, A., and Dalbey, R.E. (2014) The role of
the strictly conserved positively charges residue differs amongst Gram-positive, Gram-
negative and chloroplast YidC homologs. J. Biol Chem. 289, 35656 - 35667.
Fields of Study
Major Field: Ohio State University Biochemistry Program

ix
Table of Contents
Abstract ............................................................................................................................... ii
Dedication .......................................................................................................................... vi
Acknowledgments............................................................................................................. vii
Vita ................................................................................................................................... viii
Publications ...................................................................................................................... viii
Fields of Study ................................................................................................................. viii
Table of Contents ............................................................................................................... ix
List of Tables ..................................................................................................................... xi
List of Figures ................................................................................................................... xii
Chapter 1 ............................................................................................................................. 1
Introduction ..................................................................................................................... 1
1.1 Overview of bacterial membrane protein translocation ................................... 1
1.2 Tat pathway ...................................................................................................... 4
1.3 Sec pathway ...................................................................................................... 8
1.4 YidC family of proteins .................................................................................. 17
1.5 Figures ............................................................................................................ 25
Chapter 2 ........................................................................................................................... 29
New insights into amino-terminal translocation as revealed by the use of YidC and Sec
depletion strains ............................................................................................................ 29

x
2.1 Introduction .................................................................................................... 29
2.2 Results ............................................................................................................ 32
2.3 Discussion ....................................................................................................... 39
2.4 Materials and methods .................................................................................... 43
2.5 Tables.............................................................................................................. 49
2.6 Figures ............................................................................................................ 50
Chapter 3 ........................................................................................................................... 69
FCS analysis of Pf3 coat insertion by reconstituted YidC homologs ........................... 69
3.1 Introduction .................................................................................................... 69
3.2 Results ............................................................................................................ 73
3.3 Discussion ....................................................................................................... 75
3.4 Materials and methods .................................................................................... 78
3.5 Figures ............................................................................................................ 82
Chapter 4 ........................................................................................................................... 90
Conclusion .................................................................................................................... 90
4.1 Summary of work performed ......................................................................... 90
4.2 Figures ............................................................................................................ 93
References ......................................................................................................................... 95

xi
List of Tables
Table 2.1 - Oligonucleotide primers used in NB167 strain construction. ........................ 49

xii
List of Figures
Figure 1.1 - YidC family of proteins. ............................................................................... 25 Figure 1.2 - Model of YidC-mediated membrane insertion of Pf3 coat protein. .............. 27 Figure 1.3 - Model of YidC-Sec insertion pathway. ......................................................... 28 Figure 2.1 - Construction of model substrates. ................................................................. 50 Figure 2.2 - N-tail length requirement for translocase dependence. ................................. 52
Figure 2.3 - Increasing N-tail length directs YidC-only N-tails to YidC-Sec pathway. ... 54
Figure 2.4 - Translocation of MBP in the N-terminal direction. ...................................... 56
Figure 2.5 - The translocation of MBP-2Pf3-Lep is blocked when Arg residues are added
to its TM and analysis of its SecA-dependence. ............................................................... 58
Figure 2.6 - Testing negative charge distribution on N-terminal tail as a pathway-
determinant. ...................................................................................................................... 60
Figure 2.7 - Testing positive charge distribution on N-terminal tail as pathway-
determinant. ...................................................................................................................... 62 Figure 2.8 - Confirming non-promiscuous insertion of 2Pf3-Lep using YidC-Sec double-
depletion. ........................................................................................................................... 64 Figure 2.9 - Detection of YidC and Sec depletion using western blot. ............................ 66
Figure 2.10 - Amino acid sequence of the model protein constructs................................ 68 Figure 3.1 - Single molecule studies using FCS. .............................................................. 82
Figure 3.2 - Comparison of Pf3 insertion efficiencies by the chloroplast Alb3 and Alb4
paralogs. ............................................................................................................................ 84
Figure 3.3 - Comparison of Pf3 insertion efficiencies by the eukaryotic homologs Oxa1
and Get1. ........................................................................................................................... 86 Figure 3.4 - Representational purification of Alb4. .......................................................... 88
Figure 4.1 - Model for N-tail translocation pathway based on length and charge density.
........................................................................................................................................... 93

1
Chapter 1
Introduction
1.1 Overview of bacterial membrane protein translocation
Biological membranes are protective barriers that separate the cytosol of a cell or enclose
organelles within a cell. The cellular membrane is primarily constituted by phospholipids
arranged as a bilayer and is highly dynamic in nature. It is decorated by an assortment of
integral or peripherally-associated proteins that perform essential functions for the cell like
signal transport, enzymatic activity and cell-cell communication. It is selectively
permeable; however, the membrane is fluidic, allowing the lateral diffusion of its
phospholipid and protein components to perform their different functions. In addition to
the cell membrane, eukaryotes have evolved to contain multiple endomembranes with
unique compositions that border the sub-cellular organelles like mitochondria, lysosome,

2
chloroplast, endoplasmic reticulum, golgi body, peroxisome and nucleus. Some organelles
have multiple surrounding membranes as found in the mitochondria and chloroplasts.
In bacteria, two distinct subgroups exist that differ in their membrane organization and
composition. Gram-positive bacteria contain only the cytoplasmic membrane surrounded
by peptidoglycan cell wall with a narrow periplasmic space in between. A
lipopolysaccharide-rich outer membrane is present in Gram-negative bacteria that is
separated from the cytoplasmic one by a large periplasmic space. Within the periplasmic
space, there is a thin layer of peptidoglycan making up the cell wall. The phospholipid
composition of membranes varies between different species and its environment (1).
Typically, the phospholipids are amphipathic with a hydrophilic head group facing the
aqueous cytosol or periplasm, followed by a tail region made of two fatty acid chains that
form the hydrophobic core of the membrane.
Around 30% of all genes encode membrane proteins and about 20% of the E. coli genome
codes for its cytoplasmic membrane proteins (2). Membrane proteins vary greatly in size,
topology and composition enabling them to perform their different functions either in or at
the membrane. Membrane proteins are responsible for mediating several important
processes like respiration, electron chain transfer, ATP synthesis, cell division, signal
transduction and controlled transport of ions, nutrients and proteins. Not surprisingly, about
60% of all known drug targets are membrane proteins owing to their ease of access and
prominent roles in the cell. Membrane proteins typically possess hydrophobic
transmembrane domains and hydrophilic cytoplasmic and periplasmic domains. In E. coli,

3
the inner membrane proteins have alpha-helical secondary structure domains whereas beta-
barrel proteins reside in the outer membrane.
The hydrophobic nature of the membrane poses a challenge for translocation of the polar
domains of membrane proteins. Proteins need to be shuttled to their destined location by
moving into different organellar or the plasma membrane. In bacteria, proteins synthesized
in the cytosol are required to translocate across one or both membranes in order to reach
its final destination outside the cytosol. The movement of molecules across the membrane
bilayer is regulated by specialized machineries. In particular, the molecular devices that
facilitate the insertion and translocation of proteins across the membrane are conserved in
all domains of life. Remarkably, the proteins are inserted/exported via two different
targeting pathways, co-translational and post-translational. Further upon reaching their
desired location, the substrate proteins are assisted in the folding of the polypeptide chain
to enable it to achieve its final functional form.
Recent advancements in technology has fueled research on membrane protein biogenesis
and the different secretory systems. The fundamental steps involved in the essential
pathway of protein trafficking across the membrane is an active area of study. In this
chapter, we will focus on what is known about the different molecular machineries that
facilitate the membrane translocation of proteins in E. coli so far.

4
1.2 Tat pathway
Many essential exported proteins require the cellular environment for their proper folding
and co-factor association. This includes proteins that require binding to divalent metal ions
or those that are secreted into conditions that are not conducive for folding such as in case
of extremophilic organisms (3, 4). Prominent examples are certain redox proteins involved
in anaerobic respiration, virulence factors and members of the iron and phosphate nutrition
pathway. Cell solves the challenge of translocating bulky globular proteins across the
membrane without disrupting the membrane integrity using the twin-arginine translocation
(Tat) system (5, 6). This pathway has the special ability to transport prefolded proteins
along with their cofactors across the membrane (7). It is broadly conserved across all
domains of life (8, 9).
Substrates of this pathway possess a signal peptide which directs it to the Tat system (10).
The Tat signal peptide has a tripartite structure: a positively charged N-terminus followed
by a moderately hydrophobic segment and a C-terminal region. Signal Peptidase enzyme
(SP1) cleaves off the signal once the substrate is translocated. Sequencing analysis revealed
that the consensus sequence S/T-R-R-X-F-L-K (X is any polar amino acid) is present in
the N-terminal region of the signal sequence of most Tat pathway substrates. The
conserved nature of the double-Arg in the signal led to the term twin-arginine translocation
for this system.

5
In some cases, either one of the Arginine residues could be replaced by a Lysine residue
and still be targeted to the Tat system with varying efficiencies (11-13). However, the other
residues in the consensus sequence are more compliant with substitutions (14). While a
subset of the proteins that contain this twin arginine signal sequence are strictly exported
by the Tat system, there are some that are said to be able to promiscuously translocate by
either the Tat or the Sec translocon, which is the major protein translocase that is discussed
in detail later in this chapter (15). Interestingly, certain proteins that do not have this signal
sequence are exported by Tat as a part of a multimeric complex where at least one protein
has the Tat-specific signal and the other proteins “hitchhike” along with it (16). There are
no known targeting factors to the Tat system, however, the signal sequence has been shown
to bind to Trigger factor (TF), DnaK, SlyD and REMP proteins which may assist in folding
of the substrate before targeting to Tat for translocation (17-19).
Most organisms have a TatA and TatC pair forming the translocase complex. TatC is the
major component of the Tat translocon. The E. coli version has 6 TM segments with a Nin-
Cin orientation (20). X-ray studies revealed that it forms a curved wall structure with a
periplasmic cap that covers the central groove of the concave domain (21). This
periplasmic cap region is formed by the first two periplasmic loops of the protein. TatC is
said to directly engage with the twin-arginine motif of the signal peptide, acting as an initial
substrate docking site (22). It is also shown to interact with other TatC proteins (23).
Suppressor mutation studies revealed that the N-tail and cytoplasmic loop 1 regions are
involved in signal peptide recognition (24). In addition, the C-terminal region is shown to
be critical for protein translocation in B. subtilis (25).

6
The other more abundant member of the complex is TatA, which has an Nout-Cin
conformation with an N-terminal TM segment ending in a hinge region, followed by a
predicted amphipathic helix that lies along the cytoplasmic interface and ending with an
unstructured C-tail (26). In certain Gram-negative and chloroplast systems, an additional
TatA-like protein called TatB is present and essential for translocation (27). Despite the
structural similarity, TatB has distinct functions in the cell, indicating that this could be the
result of an early duplication event (28). Interestingly, TatA from Gram-positive B. subtilis
could functionally replace TatB and TatA of the TatABC complex in E. coli (29). It is also
shown that TatB contacts TatA in its TM region and the substrate signal peptide in the
presence of TatC (30, 31). Thus, it could be serving as an intermediary, handing over the
substrate from TatC to TatA. It is also shown to stabilize the TatBC complex formation
(32).
The mechanism of Tat-mediated translocation is an active field of study. Different models
have been proposed and tested but the process is incompletely understood at the present
time. According to the current model, there are three steps involved: substrate recruitment,
TatA oligomerization and cargo translocation. In the first step, the signal peptide of the
substrate is docked onto the signal peptide recognition site on TatC (30, 33). TatB (TatA-
like protein) interacts with TatC and the substrate to form the TatBC complex (32). A recent
study suggests that TatB proteins form a dome-like structure surrounded by TatC proteins
which form the substrate-binding cavity in the membrane (34). This initiates the formation
of a translocation complex in the next step by recruiting several TatA subunits to the TatBC
complex in a proton-motive-force dependent manner (35, 36). In the second step, TatBC

7
complex binds to TatA proteins making contacts with TatB where the substrate is handed
over for translocation.
For the final step, there are two different models that attempt to explain the translocation
process: the pore model and the membrane-destabilization model. Evidence for the pore
model emerged through a single-particle EM study that showed that TatA proteins can
assemble to form pore-like structures through which the substrates could be translocated
across (26). In keeping with this, TatA oligomers run at different sizes on Native-gels,
suggesting that TatA composition in the complex can be altered to accommodate the folded
cargo of varying diameters (37). It is suggested that the amphipathic helix region of TatA
protein folds or twists inside in such a way to translocate the substrate through the TatA
complex pore (38, 39). However, the presence of an aqueous translocation pore lined by
the amphipathic helix is yet to be confirmed and how this pore prevents the leakage of
cellular contents needs further investigation.
In contrast to the pore model, the membrane destabilization model interprets the EM results
as TatA protein aggregates that form destabilized regions on the membrane that enables
the passage of substrate cargo (40). In addition to this, the TM segment of TatA is not long
enough to span the membrane bilayer (41). The amphipathic helical region is not flexible,
so it was suggested that the movement of the helix into the membrane would be disruptive
(42). Lastly, phage shock protein PspA is linked with the Tat pathway, which suggests that
Tat-mediated translocation might induce stress in the cell (43). PspA is also implicated in
reducing proton leakage in the cell (44) and has been shown to interact with TatA in E. coli

8
(45). This evidence combined with the observation that PspA improves Tat-cargo export
support the membrane destabilization model (46). Nevertheless, a clear picture of what the
transportation device looks like and how the transport occurs needs to be elucidated to fully
understand the system.
1.3 Sec pathway
Majority of the proteins that need to insert or translocate across the membrane in E. coli
utilize the Sec machinery to accomplish this task (47). The core complex is composed of
the heterotrimeric SecYEG proteins which together forms the protein-conducting channel.
Owing to the unique structure of the channel, substrates can be both vertically exported
across the membrane as well as laterally inserted into the membrane (48). Substrates of this
pathway are translocated in an unfolded state either co-translationally or post-
translationally. The Sec machinery is conserved in all domains of life, serving as the major
translocase in the plasma membrane of prokaryotes and in the thylakoid and endoplasmic
reticulum (ER) membranes of eukaryotes (49).
E. coli Sec-dependent substrates typically contain an N-terminal hydrophobic signal
sequence or signal anchor which is utilized for targeting through the interaction of a
number of binding partners. Membrane protein substrates are targeted co-translationally as
ribosome nascent chain complexes (RNCs) where the signal recognition particle (SRP)
binds to the N-terminal signal anchor as it emerges from the ribosome exit tunnel and

9
directs it to its membrane-associated receptor FtsY for translocation (50). In contrast, most
secretory proteins are targeted post-translationally as pre-proteins that are stabilized in the
unfolded state by chaperones like SecB and by engaging with the motor ATPase SecA
protein (51). However, SecA is also involved in the translocation of large periplasmic
domains of membrane proteins (52). These targeting pathways will be discussed in detail
later in this chapter.
The Sec channel is composed of the three integral proteins SecY, SecE and SecG (53). The
first crystal structure of the archaeal SecY complex was solved in a ground-breaking study
published in 2004 (48). It revealed, along with existing experimental data, that the channel
is functional in its monomeric form. SecY protein is the largest, with 10 transmembrane
(TM) segments that forms a pseudo-symmetrical crab-claw like structure where the two
halves (5 TM each) are connected by a hinge formed by the loop between TM 5 & 6. On
the opposite side of the hinge, a lateral gate that can allow membrane proteins to exit into
the lipid bilayer exists (54). The two halves of SecY can further separate to widen the
lateral gateway. SecE consists of 1 to 3 TMs and a horizontal amphipathic helix on the
cytoplasmic membrane surface. It stabilizes SecY by wrapping around the two halves.
SecG contains 1 or 2 TM segments and is found at the periphery of the complex. Although
SecG is non-essential, it improves the translocation efficiency (55).
In addition to revealing the architecture of the channel, the structure provided a deeper
understanding of how the channel functions to move proteins inside or across the
membrane without compromising the membrane integrity. It was shown that protein

10
translocation was feasible because it contained an hour-glass shaped channel which is open
to both the cytoplasm and the periplasm. The translocating polypeptides moved through a
tight pore of 3 - 5 A in the center that is surrounded by ring of hydrophobic residues (56).
TM 2a of SecY acts as a plug helix on the periplasmic side that seals the pore when the
channel inactive (48). The plug helix can be in the center of the channel or at the periphery,
suggesting that it moves away during the translocation process to allow the nascent chain
to pass through (57). However, the plug is non-essential for the E. coli SecY channel as the
neighboring loops may take its place (58, 59).
The lateral gate opening was further characterized by later studies that captured the SecY
complex in a “semi-open” state (60, 61). It was shown that TM 7 and TM 2b of SecY move
away from each other upon SecA binding, revealing a gap opening to the membrane
bilayer. In addition to this, the plug helix was shown to be slightly displaced at this stage,
but not completely open as it is during protein translocation. The translocation process is
believed to occur via three steps: i) Activation of the channel by cytoplasmic SecA with a
substrate bound in an unfolded confirmation. ii) Insertion of the substrate into the channel
where the signal sequence interacts at the lateral gate and the C-terminal is translocated
across via the SecY pore. iii) ATP hydrolysis catalyzed by SecA powers the translocation
of the protein into the periplasmic space, while the substrate TM segments exit the channel
via the lateral gate. The observation that a signal sequence added in trans is sufficient for
protein translocation and the identification of protein localization (prl) mutations on the
lateral gate that allows the export of preproteins with defective signals further substantiate
this model (62, 63).

11
The ancillary elements SecDFYajC and YidC are involved in the Sec-mediated membrane
insertion process in E. coli (64). However, in eukaryotes a different set of proteins assist in
the process like the translocating chain-associated membrane protein (TRAM) and the
translocon-associated protein (TRAP) (65). SecD and SecF that function in bacteria and
archaea are integral proteins with 6 TM segments each. SecDF structure revealed a mobile
periplasmic domain and its proton-conducting function (66). It is proposed that the proton
movement through the complex prompts conformational changes in its periplasmic
domain. It is speculated that these changes result in a pulling action on the translocating
substrate from the periplasmic side and prevent its back-sliding. YajC has a single TM with
a cytoplasmic domain and is a shown to form a complex with SecDF, but its function is
unknown. It is proposed that SecDFYajC complex recruits the membrane insertase YidC
to form the Sec holocomplex, which will be discussed in detail in later in this chapter.
SecA-mediated targeting
SecA is a highly dynamic nanomotor that drives the export of majority of the secretory
proteins post-translationally in E. coli in association with the Sec translocon (67). It
catalyzes the protein translocation process via two distinct modes of action: i) As a
targeting factor that guides preproteins synthesized in the cytoplasm to reach their destined
location at the SecYEG channel. ii) Energizes the channel to translocate the substrate
protein using ATP hydrolysis to power the process (68). Perhaps to enable these dual roles,
SecA exists as both membrane-associated as well as cytoplasmically diffusing. In order to
maintain the preprotein substrates in an unfolded state until translocation, SecA is also

12
believed to recruit the preprotein that is bound to chaperone proteins like SecB or TF (69,
70), but the complex network of interactions that facilitate this process is not fully
understood.
The structural organization of SecA has been elucidated using crystallographic and
biochemical techniques (61, 71). The studies revealed that it has a helicase-like DEAD
motor composed of two RecA-like nucleotide binding domains (NBD1 & NBD2) and an
Intramolecular Regulator of ATP hydrolysis 2 (IRA2) domain, which together binds and
hydrolyses ATP to energize SecA function for translocation (72). In addition to the DEAD
motor, it possesses a preprotein binding domain (PBD) (73) and a C-terminal domain
(CTD), which has the motile IRA1 subdomain (also called as the two-helix finger (74))
and a zinc finger region that is known to interact with SecB (75). PBD binds to the signal
peptide of the substrate protein (76), but it may also bind to certain regions of the mature
domain (73, 77). SecA is capable of dimerizing under certain conditions but it is unclear if
it is the native state (78, 79).
The mechanism of SecA mediated targeting has the following proposed steps. First, the
preprotein that is being synthesized in the cytoplasm is recognized by SecA and/or
chaperone proteins. Recognition is facilitated by the interaction of SecA with the substrate
N-terminal signal as well as certain regions of the mature domain. Recent studies suggest
that the ribosome exit tunnel protein L23 binds SecA (80). In addition, cryo-EM studies by
Singh et al proposed another binding site near L22 and L24, which could allow the binding
of two SecA molecules at the same time (81). TF is also proposed to bind L23 protein,

13
however, superimposing the structures showed no steric clash for binding (82). This
suggests that SecA likely scans the emerging polypeptide for a signal sequence with
moderate hydrophobicity and binds to it along with chaperone proteins to carry out post
translational targeting of the substrate to SecYEG (83). In addition, SecA also has the
capacity to bind preproteins that are completely released from the ribosome and located in
the cytoplasm.
Once the SecA binds the preprotein, it undergoes conformational changes including dimer
to monomer conversion to activate the channel (84). The structure of SecYEG complexed
with monomeric SecA (72, 80) revealed a groove at the interface through which the
preprotein can enter the channel. The signal peptide unlocks the channel as observed with
the prl mutations (85) and the translocation process begins. SecA binds and catalyzes
multiple rounds of ATP hydrolysis while simultaneously moving the preprotein through
the SecYEG pore using its “two-helix finger” region. Consistent with this, Rapoport’s
group showed that the model secretory substrate proOmpA contacts both the SecA two-
helix finger and the SecY pore ring (74). SecA could be crosslinked to the pore ring as
well. About 20-30 residues of the substrate protein are translocated through the channel
per ATP hydrolysis cycle (86). PMF is hypothesized to play a role in orienting the signal
sequence at the start of the translocation step. Once the substrate is translocated, the signal
peptide is cleaved off by SP1 enzyme, whose catalytic domain is on the periplasmic side
of the membrane. Finally, the mature protein either folds in the periplasmic (Gram-negative
bacteria) or extracellular space (Gram-positive bacteria) or is inserted into the outer
membrane of Gram-negative bacteria via other specialized machineries.

14
SecB chaperone
Sec-dependent exported proteins synthesized in the cytoplasm need to be maintained in an
unfolded state. Cytosolic chaperone proteins like SecB and TF assist in this process and
also prevent the aggregation and degradation of substrate proteins. TF is ubiquitous in
bacteria, however SecB is prevalent in proteobacteria only (87). But in E. coli a group of
proteins require SecB for proper secretion. This suggests that there must be other proteins
with functional overlap that take up this role in other organisms. General chaperones like
DnaK and DnaJ are proposed to carry out this function in bacteria that lacks SecB (88).
The functional unit of SecB is homo-tetrameric (89). The crystal structure of SecB revealed
a 70 A channel on either side of the tetramer which is proposed to be the preprotein binding
site. These binding sites had deep cleft regions lined with bulky hydrophobic side chains
that could bind to unfolded hydrophobic regions of the substrate protein. In addition, it has
a shallow groove that could potentially bind to β-sheet regions of the polypeptide. The Zn-
binding domain of SecA functions to interact with SecB at its C-terminus electrostatically
(90, 91). It has a higher binding affinity to SecA when it is associated with SecYEG (92).
This later study suggests that SecB binding induces conformational changes in SecA and
leads to the transfer of the substrate protein from SecB to the channel-associated SecA for
translocation. Further, Crane et al showed that SecB has a higher binding affinity to SecA
in its dimeric form than in monomeric form, suggesting that SecB dissociates when SecA
monomerizes during translocation (91).

15
SRP-mediated targeting
Membrane protein targeting is a crucial step in the biogenesis of membrane proteins and
the machineries that regulate this process are universally conserved. The dynamic
interactions between the Signal Recognition Particle (SRP) and its receptor FtsY (also
known as SRP receptor or SR) facilitate the co-translational delivery of substrate
membrane proteins as ribosome-nascent chain (RNC) complexes to their destined location
in the membrane (93). Chloroplast SRP is an exception to this since it operates post-
translationally to deliver substrate proteins to the thylakoidal membrane (94). SRP
typically targets proteins to the Sec translocon for membrane insertion (95), however new
evidences have emerged suggesting that certain substrates are targeted to another
translocon YidC (96).
The composition of SRP varies across different species, having some combination of
proteinaceous and/or RNA domains. Bacterial SRP is the simplest homologue which
remarkably contains one domain each of the protein and RNA component (97). The protein
component is a GTPase containing domain called the Fifty fourth homolog of the
eukaryotic SRP54 (Ffh) (98). It also has a 4.5S RNA component that has been shown to be
essential for SRP stability and its function (97). The SRP receptor is usually membrane-
associated and has a similar GTPase domain as the SRP (99). The bacterial SRP receptor
homologue lacks the TM domain found in the eukaryotic SR. This could explain the
appearance of SR as evenly distributed between the membrane and the cytoplasm in
bacteria, however the role of cytosolic SR is unclear (100). Amazingly, the bacterial SRP

16
and SR can replace their sophisticated mammalian counterparts to mediate successful
targeting of substrate proteins to the ER indicating the evolutionarily conserved function
of this pathway (101).
Bacterial Ffh has two functionally distinct domains: a methionine-rich region called the M
domain and the NG domain that contains the amino-terminal and the GTPase domain (102,
103). Crystallographic evidence showed that the M-domain has a flexible signal peptide
groove that is lined with methionine residues that can interact with various hydrophobic
sidechains (104, 105). The studies also revealed the presence of a flexible fingerloop
structure which may stabilize the substrate signal sequence in the groove, but this remains
to be tested (106). Residues on this fingerloop have been demonstrated to be critical for
GTPase activity and SR binding (107). SR consists of an A domain that is believed to
involve in membrane binding and a GTPase containing NG domain (108, 109). The A
domain of SR can be crosslinked to the SecY cytoplasmic loop regions, which are also said
to contact the ribosome (110, 111).
SRP mediated substrate targeting and delivery consists of the following steps. Recognition
and binding of the N-terminal signal sequence of substrate proteins emerging from the
ribosome exit tunnel by SRP, which initiates conformational changes in SRP and facilitates
binding to SR. Lastly, GTP hydrolysis powered dissociation of SRP and SR and the
successful transfer of RNC complex to the translocon (112). E. coli SRP differentiates
between cytoplasmic and membrane-destined proteins based on the increased
hydrophobicity of the signal sequence, which is typically 20-30 residues long with a

17
tripartite structure described before and may be cleaved off by SP1 on reaching the desired
membrane location (113). Interestingly, SRP can bind to non-translating ribosomes
however the interaction is much more stable in the presence of the signal sequence (114,
115).
Once SRP binds to the ribosome in such a way that the M domain is positioned to receive
the nascent signal peptide emerging from its exit tunnel, a series of conformational changes
are initiated in the G domain and the SRP-RNC complex is directed to the SRP receptor
(116, 117). A “closed” state conformation is achieved where the NG domains of both SRP
and SR are surrounded by GTP molecules (118). The SRP RNA Tetraloop region of the
4.5S RNA has been shown to interact with SR in the presence of RNC initially (119).
However, data presented in (118) showed that the GTPase domains of SRP and SR are
displaced towards the opposite end of the 4.5S RNA which may facilitate the increased
GTPase activity. The RNC complex is transferred to the Sec translocon and the SRP-SR
complex disassembles upon GTP hydrolysis enabling the recycling of components.
However, the mechanistic details of RNC complex hand-over to the different translocons
and how specificity is achieved is not clear.
1.4 YidC family of proteins
Membrane proteins constitute about 30% of the cellular proteome (120) and perform
critical functions like signal transduction, molecular transport and cell adhesion. The

18
molecular machineries that catalyze their targeting, insertion and assembly in the different
cellular and subcellular membranes are remarkably conserved. Sec translocon is
responsible for moving the majority of the proteins across/into the bacterial, archaeal,
thylakoidal and ER membranes in an unfolded state (49). In bacteria, it is proposed to form
a holo-complex composed of the heterotrimeric protein channel SecYEG, and the
accessory elements SecDFYajC, SecA ATPase and YidC (121).
As part of the holo-complex, YidC operates in various capacities ranging from assisting in
the membrane insertion process and the lateral clearance of the substrate TM segments
from the channel to serving as a foldase for Sec-dependent proteins (122). In addition to
this, YidC facilitates the membrane insertion of small membrane protein substrates
independently (123). While larger proteins are typically targeted by the SRP-FtsY
partnership to the Sec holotranslocon, smaller substrates that cannot engage SRP are post-
translationally delivered to YidC (124). However, certain YidC-only substrates like MscL
(96) and the tail-anchored proteins TssL (125), DjlC and Flk (126) employ SRP for
targeting.
YidC/Alb3/Oxa1 family proteins are highly conserved insertases that operate in the
bacterial, thylakoidal and mitochondrial inner membrane respectively (127). Structurally,
they are helical bundles formed by 5 core TM segments (Fig 1.1). YidC is required for the
insertion and assembly of several respiratory and energy-transducing proteins (128) like
the subunits of the F1FOATPase (129), Cytochrome o Oxidase (130) and NADH
dehydrogenase (131). In Gram-negative bacteria, YidC has an additional N-terminal TM

19
segment that acts as a membrane anchor followed by a large beta-sandwich fold within the
first periplasmic domain (132). Although these regions are largely non-essential for
function (133), they have contact sites to SecY (134) and SecDF (135), suggesting a kinetic
role in the protein insertion and substrate folding process. Most Gram-positive bacteria
possess two paralogs: YidC1 and YidC2. While YidC1 is constitutively expressed, YidC2
gene expression is controlled by a MifM sensor protein in B. subtilis (136). Though the
paralogs are functionally exchangeable, YidC1 is specifically required for the sporulation
process (137).
In archaea, DUF106 protein has a three-TM core with a low structural homology to the
bacterial YidC, but its protein insertion function remains to be tested (138). Eukaryotes
contain multiple YidC paralogs and some of them can replace E. coli YidC at least partially,
indicating shared functionality in the cell (139-141). In plants, the paralogs Alb3 and Alb4
exist in the thylakoid membrane of chloroplasts (142, 143). The primary substrates of Alb3
are a subset of the light-harvesting chlorophyll binding protein subunits (144), whereas
Alb4 is involved in the biogenesis of chloroplast F1FOATPase assembly (145). A
prominent feature of Alb3 is the presence of a long cytoplasmic C-terminal domain which
acts as an anchor for SRP43 (146). Both post-translational and co-translational targeting
occurs and Alb3 is known to interact with the chloroplast SecYE translocon like its
bacterial counterparts (147). Oxa1 and Oxa2 paralogs are found in the mitochondrial inner
membrane of eukaryotic cells (148, 149). Sec is absent in this membrane, so Oxa1 is
believed to facilitate the insertion of all mitochondrial DNA encoded membrane proteins
independently (150). Oxa1 has a C-terminal extension which is the ribosome-docking site

20
for translating substrates that are co-translationally inserted (151). Oxa2 performs similar
insertion function for certain respiratory proteins post-translationally (152).
Until recently the presence of YidC homologs in the ER was unknown (153). Anghel et al
(154) employed phylogenic homology studies and identified three Oxa1-like highly
conserved proteins: TMCO1, EMC3 and Get1 which are all involved in the ER membrane
protein translocation process in eukaryotes. The study found that Get1 and EMC3 proteins
were evolutionarily related to the DUF106 group of proteins. Get1 is a part of the tail-
anchored protein insertion complex and substrates of this pathway have a C-terminally
located TM segment that is post-translationally targeted to the ER membrane (155). The
ER Membrane Complex 3 (EMC3) promotes the co-translational membrane insertion of
multi-pass ER proteins with charged TM segments (156, 157). TMCO1 is predicted to
insert newly synthesized ER membrane proteins co-translationally, but it also engages with
the Sec translocon like YidC (154).
YidC-only pathway
YidC’s function was first annotated in 2000 (123); it was shown to be essential in E.coli
and required for the insertion of phage proteins Pf3 coat and M13 Procoat which were
previously thought to insert by an unassisted mechanism. The minimal functional unit is
monomeric (158) even though YidC can dimerize under certain conditions (159). It was
shown using reconstituted proteoliposomes that YidC is sufficient for the membrane
integration of Pf3 (160). In addition to this, YidC is responsible for the membrane insertion
of subunit c of ATP synthase (129), the mechanosensitive channel protein MscL (96) and

21
the C-terminal tail-anchored proteins TssL (161), DjlC and Flk (126). A common feature
of the YidC-only pathway substrates is that they contain short translocated regions
followed by one or two TM segments (162).
Crystal structures of YidC from Gram-positive (163) and Gram-negative bacteria (164)
uncovered important mechanistic details about its function. The conserved 5 TM core of
YidC forms a unique hydrophilic cavity in the inner leaflet facing the cytoplasm but is
closed from the periplasmic side. The groove contains a conserved positive charge which
was shown to be critical for function in gram-positive bacteria but not in the gram-negative
homolog (165). Kumazaki et al showed that MifM substrate could be crosslinked to the
groove (163). Hence it is proposed that the positive charge interacts electrostatically with
the charges on the substrate hydrophilic regions to recruit it into the groove and reduce its
membrane-crossing distance (Fig 1.2). Consistent with this, negative charges on the
substrate N-tail or TM segment have been proposed to act as YidC-only pathway
determinants (166, 167). The proton motive force (PMF) is implied to play a role in
releasing the hydrophilic domain from the groove but it is unclear whether this occurs and,
if so, how it occurs. Further reduction in membrane crossing distance for the substrate was
suggested by MD simulation studies (168) which found thinning of the membrane region
around YidC.
The major substrate contact sites of YidC are the hydrophobic residues found in TM3 and
TM5 that were shown by crosslinking studies to bind the substrate TM segment of Pf3 coat
(169) and MscL (170). This suggests that YidC facilitates substrate insertion through

22
hydrophobic interactions via a greasy-sliding mechanism (Fig 1.2). In line with this, Cryo-
EM studies showed that the TM segment of the FOc substrate is in proximity to the greasy
slide (171). Substrate insertion kinetics was studied in real-time using time resolved single-
molecule FRET analysis (172) which showed that the entire process of substrate contact,
insertion and separation from YidC occurred within 20 ms and Pf3 inserted into
reconstituted YidC proteoliposomes at the rate of 500 molecules per second.
Another feature of YidC is the cytosolic loops C1, C2 and the C-terminal tail region, of
which the latter two constitute the protein docking sites for receiving its translating
substrates. C1 loop forms a helical hairpin that is essential for function (165) and is believed
to be highly dynamic based on their relative positions in the crystal structures. Crosslinking
studies performed by Koch’s group show that the C1 loop interacts with SRP and FtsY,
highlighting its role in recruiting substrates (134). Similarly, Driessen et al found that the
C2 loop and C-terminal region of YidC provide stable docking sites for ribosome nascent
chain complexes (173). These studies define the role played by the different regions of
YidC leading to a better understanding of the mechanism of its insertion function.
YidC-Sec pathway
Substrate specificity studies indicate that YidC has limited potential to function
independently and the translocation of energetically unfavourable regions of substrates
require both YidC and Sec (174). Several essential inner membrane proteins like ATP
synthase subunit a, b (124, 175) and subunit II of Cytochrome b0 oxidase (130, 176), TatC
(177, 178) and anaerobic respiratory protein NuoK (167) are inserted by the combined

23
efforts of YidC and Sec. This phenomenon may also occur in higher eukaryotes in the ER
and thylakoidal membrane where YidC and Sec homologs are known to interact. The
bacterial holo-translocon (HTL), made up of SecYEG, SecDFYajC and YidC, is proposed
to be an efficient insertion machine for the membrane protein substrates of the YidC-Sec
pathway (179).
SecYEG forms a channel through which substrate polar domains are exported across the
membrane whereas the TM segments exit the channel with the help of YidC via a lateral
gate formed by TM 2b and TM7 of SecY (48, 180) (Fig 1.3). Consistent with this, lateral
gate of SecY can be photo-crosslinked to YidC (181). It is predicted that the greasy slide
of YidC might contact the SecY lateral gate and move the substrate TM segment via
hydrophobic interactions from within the channel and into the lipid bilayer. Recent insight
into how this partnership works has revealed that the first TM of E.coli YidC contacts SecY
and SecG (134). It is proposed that this TM may enter the channel and draw the TM
segments out through the lateral gate, but this remains to be tested. The study also reported
C1 loop as a contact site for SecY.
In addition to this, YidC is also known to act as a folding and packaging site for Sec-
dependent proteins (182). Nagamori et al (183) found that LacY protein required YidC to
achieve its functional folded form using monoclonal antibodies recognizing specific
conformational domains. Strikingly, the translocation of the six periplasmic domains of
LacY required only SecYEG while the folding of the protein was dependent on YidC (184).
YidC’s role in folding LacY was further explored by Serduik et al by using single molecule

24
force spectroscopy (185). A mechanical pulling force was applied on a single LacY
molecule to unfold it and extract it from a membrane using the stylus of a cantilever. This
protein was then slowly allowed to refold into another membrane in the presence of YidC.
The study showed that only in the presence of YidC, LacY could fold back to its stable
form in the membrane.
The accessory elements SecDFYajC is believed to promote YidC’s interaction with the
Sec channel (64). SecDF was shown to contact the periplasmic domain of E. coli YidC
using affinity pull-down experiments (135). This interaction may indicate the shared
functional role of SecDF and periplasmic domain of YidC in the substrate folding process.
The crystal structure of SecDF and electrophysiological experiments revealed a proton-
transport mechanism which could provide the energetic driving force for pulling the
substrate out of the Sec channel during translocation and prevent its back-sliding (66).
Substrates of this pathway are targeted to the holotranslocon by SRP and its membrane-
associated receptor FtsY for co-translational insertion (124).
25
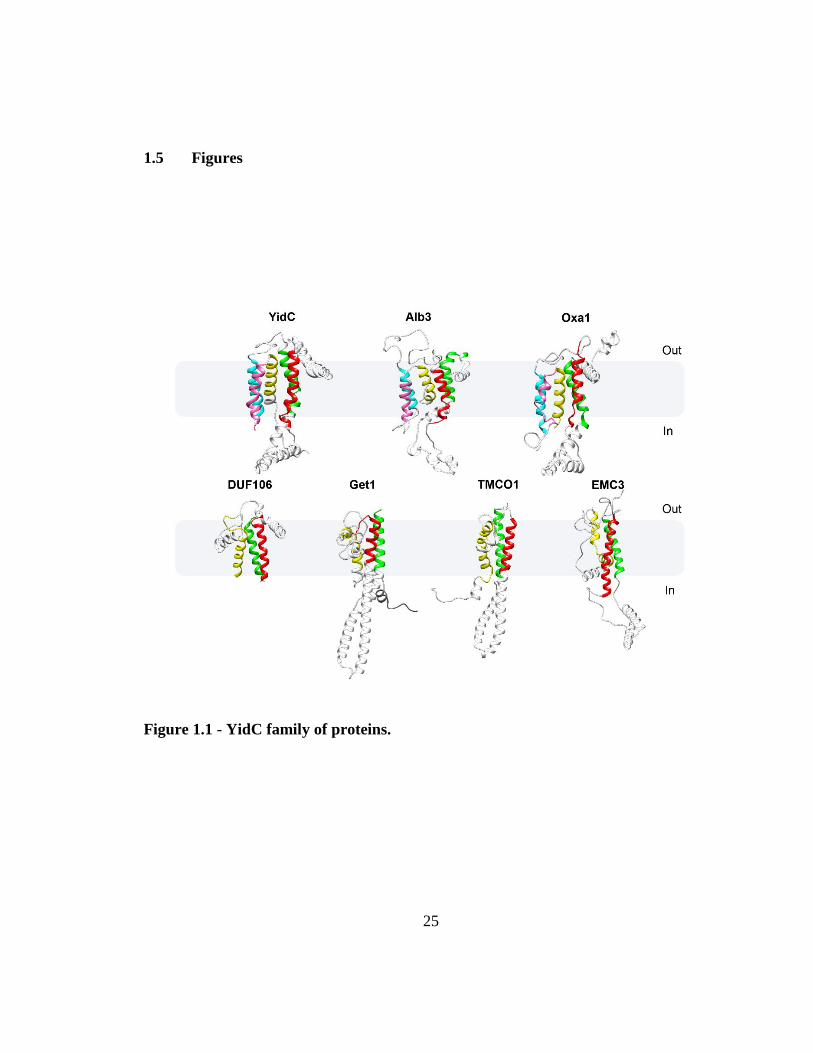
1.5 Figures
Figure 1.1 - YidC family of proteins.

26
Top panel: Structural homology in the YidC/Alb3/Oxa1 family shown by highlighting the
conserved TMs in Green (TM1), Red (TM2), Cyan (TM3), Purple (TM4) and Yellow
(TM5) respectively. YidC structure is adapted from the crystal structure solved in B.
halodurans (PDB: 3WO7); Alb3 and Oxa1 structures are 3D computational models made
using SWISS-MODEL workspace as described in [186]. Bottom panel: Newly identified
members of Oxa1 superfamily highlighting the conserved three TM segments in Green
(TM1), Red (TM2) and Yellow (TM3) respectively. Archaeal DUF106 is adapted from the
crystal structure solved in M. jannaschii (PDB: 5C8J); Yeast Get1, Human TMCO1 and
Human EMC3 structures are evolutionary covariance-based 3D models adapted from [153,
154]. The cytoplasmic regions of these models were modified as described in [153].

27
Figure 1.2 - Model of YidC-mediated membrane insertion of Pf3 coat protein.
This figure is adapted from a review by Kiefer et al [187] (A) Binding of Pf3 coat protein
to YidC. (B) Pf3 TM segment interacts with the cytoplasmic part of the greasy slide and
the N-terminal tail of Pf3 (blue) enters the hydrophilic cavity of YidC possessing the
conserved Arg residue (red). (C) Pf3 coat TM segment inserts across the YidC “greasy
slide” formed by TM3 and TM5 (purple) and release of the N-tail into the periplasmic
space. (D) Release of Pf3 into the bilayer.
28
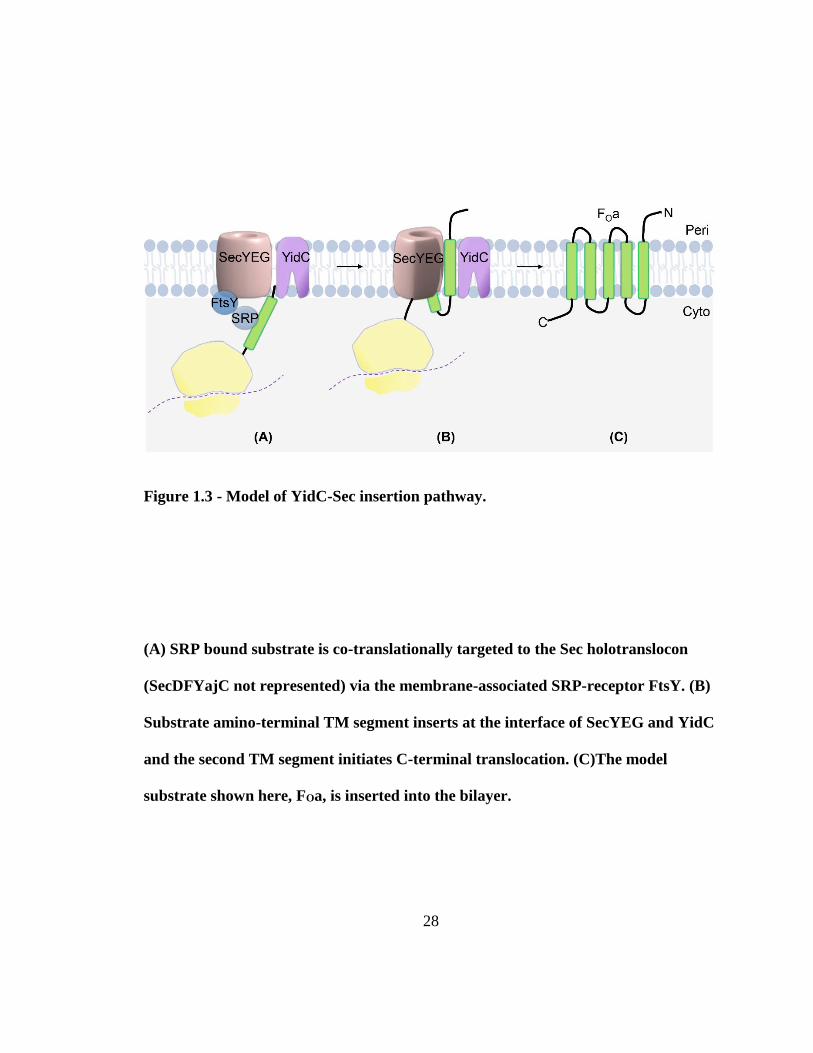
Figure 1.3 - Model of YidC-Sec insertion pathway.
(A) SRP bound substrate is co-translationally targeted to the Sec holotranslocon
(SecDFYajC not represented) via the membrane-associated SRP-receptor FtsY. (B)
Substrate amino-terminal TM segment inserts at the interface of SecYEG and YidC
and the second TM segment initiates C-terminal translocation. (C)The model
substrate shown here, FOa, is inserted into the bilayer.

29
Chapter 2
New insights into amino-terminal translocation as revealed by the use of
YidC and Sec depletion strains
2.1 Introduction
The bio-machineries responsible for membrane protein biogenesis are universally
conserved. Facilitated insertion and assembly of inner membrane proteins are catalyzed by
two known protein translocases in E. coli, Sec and YidC (186). However, some proteins
like KdpD (187), KscA (188), Pf3-Lep (166) have been proposed to insert by an unassisted
pathway. The question of what features of a membrane protein substrate determines its
translocation-pathway is incompletely answered. In this study, we have addressed
important gaps in this area to improve our understanding of the fundamental mechanism
of membrane protein insertion and assembly. Specifically, we have studied the features of

30
the substrate N-terminal tails that dictate its translocase requirements, which heretofore
have been difficult to address experimentally.
Sec is the membrane integration site for majority of the inner membrane proteins. The
holotranslocon consists of the SecYEG channel, and the accessory elements SecDF, YajC
and YidC (179). Majority of its substrates are targeted to SecYEG as ribosome nascent-
chain complexes (RNCs) by the SRP-FtsY pathway (189) and the substrate hydrophilic
domain is translocated across the membrane in an unfolded state through a pore, while the
TM segment exits the channel through a lateral gate to integrate into the membrane (48).
SecA is an associated ATPase that powers the movement of periplasmic proteins, and large
hydrophilic domains of membrane proteins across the channel (190). YidC is responsible
for the insertion of a smaller subset of proteins like Pf3 coat and M13 procoat (123), subunit
c of FoF1ATPase (129), MscL (96) and SciP (161). In addition, it also assists in the folding,
assembly and membrane partitioning of certain Sec-dependent proteins like MalF (191)
and LacY (183, 184). Recent studies showed that YidC possesses an aqueous cavity in the
inner leaflet that likely can host a substrate N-tail and reduce its membrane crossing
distance (163). YidC and Sec can also work together to insert substrates, presumably at the
YidC TM greasy slide and Sec lateral gate interface via the YidC-Sec pathway (181).
Substrates of this pathway include NuoK (167), subunit a of FOa FOF1ATPase (124, 192)
and CyoA (130, 176).
It is unclear how substrates are destined for translocation by these pathways. Previous
studies suggest that YidC has limited potential to function independently and recruits Sec
for the translocation of more sophisticated proteins in terms of the size, charges and

31
hydrophilicity (166, 174, 193). Typical substrates of the YidC-only pathway have short
translocated N-tails like Pf3 whereas Sec is required for membrane insertion and
translocation of large periplasmic domains of substrate proteins like leader peptidase (194),
-lactamase (195) and alkaline phosphatase (196). Exception to this is ProW, which has
been proposed to insert by a Sec-independent mechanism (197). Cao et al. showed that the
Sec-independent protein Pf3-Lep requires Sec on increasing its N-tail length (198).
However, it is unclear what is the size-limit for N-translocation by YidC-Sec independent,
YidC-only and YidC-Sec pathways. Pathway-selection based on charges have been
controversial and needs further investigation. It has been shown that negative charges on
the N-tail and TM segment can act as YidC determinants and positive charges as Sec
determinants (166, 167). But the unfavorable distribution of positive charges have also
been proposed to act as YidC-determinants (199).
To further our understanding of N-terminal tail translocation, we have examined length
and charge features as pathway determinants by employing single-spanning model
substrates. We have evaluated the critical length required for YidC-only and YidC-Sec
mediated substrate insertion using a new strategy to regulate expression of two essential
genes (yidC and secE) in the same cell. On increasing the N-tail length, we find that
substrates switch from independent to YidC-only to YidC-Sec. Beyond 60-residue N-tail
length, the substrates were not translocated. However, the large periplasmic protein
maltose-binding protein (MBP) could be translocated in the N-terminal direction likely
because it can engage SecA. We also find that longer N-tails could translocate additional
charges, both positive and negative, if they are distributed away from each other. This led

32
to the hypothesis that N-tail charge density plays a role in necessitating translocase
dependency. Our results show that crowding of charges causes a switch in translocation
pathway from YidC-Sec independent to dependent.
2.2 Results
N-tail length requirement for translocase dependence
To study if N-tail length is a determinant for the translocase requirement for insertion, we
used Pf3-Lep, based on the YidC-Sec independent model protein used in (166). Pf3-Lep
has the 18-residue long Pf3 coat N-tail with two negative charges, followed by leader
peptidase positions 4-323. A positive charge added at position 79 renders its TM2 defective
for insertion (198). This prevents the translocation of the C-terminal P2 domain and allows
us to monitor the translocation of the amino-terminus alone. To study the length
requirement, the Pf3 tail segment was first doubled by adding another Pf3 N-tail ahead of
the TM segment to make 2Pf3-Lep (Fig. 2.1A). To further increase the N-tail length,
uncharged spacer residues used in (198) were inserted between residue 36 and 37 of 2Pf3-
Lep (Fig. 2.1B).
The translocase requirements for these substrates were studied using the YidC-depletion
strain JS7131 (123) and the SecE-depletion strain CM124 (200) that has either the yidC or
the secE gene under the araBAD promoter, respectively. SecE depletion has been shown
to affect SecYEG dependent substrates since SecE is required for the stabilization of SecY
(201). The depletion of the respective translocases was confirmed using western blot
analysis (Fig. 2.9A), which showed a steep decline in the translocase levels under

33
conditions where transcription of secE and yidC was blocked. To test YidC-dependence
for membrane insertion, the substrates were expressed in JS7131 and labelled using [35S]
methionine for 1 min under YidC expression (0.2% arabinose) and YidC depletion (0.2%
glucose) growth conditions. N-tail translocation was studied using a protease-accessibility
assay as described in (202). Briefly, spheroplasts were generated using lysozyme to allow
access to the inner membrane and then treated with Proteinase K for 30 min. When the N-
tail of the substrate protein (full length indicated by P in Fig. 2.2) was translocated across
the membrane, the N-tail was digested by the externally added protease (PK) and a smaller
band corresponding to the protease-resistant fragment was observed (indicated by F in Fig
2.2); whereas when the amino-terminal region of the substrate was not translocated, it is
not protease-accessible, so a band whose size corresponds to the full-length protein was
seen. Similarly, to determine Sec dependence of the various constructs, CM124 cells
expressing the model proteins were labelled with [35S] methionine for 1 min under SecE
expression (0.2% arabinose) and SecE depletion conditions (0.2% glucose), and the
protease mapping assay was carried out as described above.
To assess the efficiency of the formation of spheroplasts, degradation of outer membrane
protein A (OmpA) was used as a positive control as it can be digested by Proteinase K from
the periplasmic side of the membrane in properly prepared spheroplasts but cannot be
accessed in intact cells. OmpA controls were performed for all the experiments reported in
this study but are only shown once for representative purposes.
When the substrate N-tail was doubled to 2Pf3-Lep, the 36-residue long N-tail was found
to be translocated efficiently under YidC or Sec depletion conditions and hence it was still

34
YidC-Sec independent (Fig. 2.2A). Upon extending the N-tail with spacer pentapeptides
to 41 (2Pf3+5-Lep) and 46 (2Pf3+10-Lep) residues, we observed a gradual increase in
YidC dependence; but these substrates were still largely Sec-independent (Fig. 2.2B, 2.2C).
Upon further increasing the N-tail length to 51 residues (2Pf3+15-Lep), the efficiency of
insertion was reduced but it continued to be inserted by the YidC-only pathway and did not
require Sec (Fig. 2.2D). At 56 residue N-tail length (2Pf3+20-Lep), the substrate required
both YidC and Sec, albeit the insertion was not efficient (Fig. 2.2E). Further elongation of
the N-tail to 61 residues (2Pf3+25-Lep) prevented its translocation completely (Fig. 2.2F).
The substrate percentage translocated in each condition was measured by quantifying the
bands using ImageJ (see “Materials and Methods”). As a control, we confirmed that OmpA
(indicated by M in Fig. 2.2) was completely digested by the protease indicating good
spheroplast formation (Fig. 2.2G). Additionally, OmpA export required Sec-only, so its
protease-protected precursor form Pro-OmpA (indicated by P in Fig. 2.2) accumulated in
Sec depletion condition but not in YidC.
Sec-dependence on increasing N-tail length of the YidC-only substrate Pf3-23Lep
To test if increasing the N-tail length of a YidC-dependent substrate will cause a pathway-
switch to require Sec, the model substrate Pf3-23Lep was employed. Pf3-23Lep contains
the N-tail and TM segment of Pf3 coat protein fused to the 23rd residue of Lep and is a
well-characterized YidC-only substrate (165). Doubling the N-tail of Pf3-23Lep to 2Pf3-
23Lep (Fig. 2.1B) did not cause a change in its translocation pathway; the substrate
continued to insert by YidC-only mechanism (Fig. 2.3A). However, a gradual Sec
dependence was observed in addition to the YidC dependence, when its N-tail length was

35
increased to 41 (2Pf3+5-23Lep) and 46 (2Pf3+10-Lep) amino acids in length using the
uncharged spacer residues used previously (Fig. 2.3B, 2.3C) (198). Further increases in the
N-tail length to 51 (2Pf3+15-23Lep) and 56 residues (2Pf3+20-23Lep) resulted in a strict
dependence on both YidC and Sec, but the substrate was not translocated efficiently (Fig.
2.3D, 2.3E), as seen in the previous study. The longer N-tail of 61 residues length
(2Pf3+25-23Lep) was not translocated (Fig. 2.3F).
Translocation of the mature domain of MBP in the N-terminal direction
The model substrates tested above, which contained the duplicated Pf3 tails and spacers
peptides, did not insert beyond a size of 60 residues length. This may have to do with the
fact that there is something inherent about these sequences that prevent export, such as the
lack of recognition sites for SecA/B machineries. Therefore, we examined whether a
protein domain that is normally exported, i.e., in the C-terminal direction, could be
exported in the amino-terminal direction. We also wanted to examine whether
translocation of a long protein segment in the amino-terminal would require YidC. To test
this, we fused the secretory protein MBP to the N-terminus of 2Pf3-Lep (MBP-2Pf3-Lep,
Fig. 2.4A) and its translocation was studied as described above. The mature domain
(indicated by M in Fig. 2.4) was translocated efficiently by Sec pathway without its
cleavable signal sequence (Fig. 2.4B). However, YidC was dispensable as the translocated
domain of the substrate was translocated and cleaved by the external protease to produce
the protease-resistant fragment (indicated by F in Fig. 2.4), even when YidC was depleted.
Based on the size of the protease-protected fragment of MBP-2Pf3-Lep, we predict that it
consists of the Lep portion of MBP-2Pf3-Lep, that has 7 Met residues, as compared to the

36
full length MBP-2Pf3-Lep which that has 16 Met residues. The data suggests that the
substrate C-terminal TM behaves as a reverse signal to open the SecYEG channel. We
confirmed this by adding a pair of positive charges in the TM region (MBP-2Pf3-Lep
L41R, L48R) that disabled MBP translocation (Fig. 2.5A). We also analyzed the SecA
requirement for MBP-2Pf3-Lep using a sodium azide study and found that the translocation
was SecA dependent (Fig. 2.5B). Next, we truncated the periplasmic region of this
substrate by deleting all but the first 60 residues of the MBP mature domain (MBP-2Pf3-
Lep ∆61-325, Fig. 2.4A). At this intermediate N-tail length of 96 residues, we observed
that the substrate was inserted partially by Sec and YidC (Fig. 2.4C). PreMBP-2Pf3Lep
that still has the N-terminal signal sequence of MBP attached, was also tested as a positive
control. The precursor protein (indicated by P in Fig. 2.4D) accumulates under Sec
depletion conditions whereas it doesn’t require YidC.
Testing charge density hypothesis
Previously it was shown by Zhu et al. that charges within the N-tail can act as translocase
determinants (166). When a negative or a positive charge was inserted in the Pf3-Lep N-
tail, it caused a switch in translocase requirement from YidC-Sec independent to YidC-
only or YidC-Sec dependent respectively. However, we observed that additional charges
can be translocated when they are distributed across a longer N-tail in the 2Pf3-Lep
construct, which has twice as many charges (Fig. 2.2A). This led us to propose and test the
hypothesis that it is the charge density of the N-tail rather than just the presence of charges
per se that determines its translocation pathway.

37
First, we introduced additional negative charges to the 2Pf3-Lep N-tail (Fig. 2.6A) and
examined its translocase preference. Addition of one negative charge at position 15 on the
first N-tail (2Pf3-Lep V15D), or 15’ that is on the N-tail closer to the TM (2Pf3-Lep
V15’D), did not influence the translocation pathway (Fig. 2.6B, 2.6C); the substrates
continued to insert independent of YidC and Sec. When two negative charges were added
at positions 15 and 15’ (2Pf3-Lep V15D, V15’D), the substrate showed partial dependence
on both YidC and Sec (Fig. 2.6D). To increase the local density of charges in one of the
two N-tails, the two negative charges were either added at positions 12 and 15 in the first
N-tail (2Pf3-Lep L12E, V15D) or at the same positions in the second N-tail (2Pf3-Lep
L12’E, V15’D). The charges introduced in the first N-tail resulted in a greater dependence
on both YidC and Sec (Fig. 2.6E). However, when the charges were positioned closer to
the TM, the N-tail was not efficiently translocated and strictly required YidC and Sec (Fig.
2.6F).
Similarly, to evaluate the effect of adding positive charges, an Arg residue was substituted
in the first and second N-tail of 2Pf3-Lep at the same positions (Fig. 2.7A) as with the
previous study. 2Pf3-Lep V15R was largely independent of YidC and Sec (Fig. 2.7B), but
2Pf3-Lep V15’R that bears the substitution closer to the TM segment was partially
dependent on both YidC and Sec (Fig. 2.7C). On adding two Arg residues at positions 15
and 15’ (2Pf3-Lep V15R, V15’R), the substrate became strictly dependent on YidC and
Sec (Fig. 2.7D). When the two positive charges were placed close to each other in the first
N-tail (2Pf3-Lep L12R, V15R) the substrate was poorly inserted and required both YidC

38
and Sec (Fig. 2.7E). When the charges were substituted in the second N-tail (2Pf3-Lep
L12’R, V15’R), it was not inserted (Fig. 2.7F).
2Pf3-Lep is YidC-Sec independent in double-depletion conditions
In vitro membrane insertion studies have shown that some Sec-dependent substrates
display promiscuity and can be also translocated by the YidC-only pathway (178). This
promiscuity may explain why 2Pf3-Lep can insert under YidC depletion conditions, where
it may go by the Sec pathway, and vice-versa. Therefore, it is critical to evaluate its
insertion under YidC-Sec double depletion conditions to rule out the possibility of a
promiscuous insertion pathway. For this, we utilized CRISPR interference (CRISPRi)
(203) to repress expression of secE in the YidC depletion strain JS7131 and the
translocation of 2Pf3-Lep was assayed using the protease-mapping assay as described
above. The results showed that 2Pf3-Lep was fully inserted when both YidC and Sec were
depleted at the same time (Fig. 2.8A).
To further characterize YidC/SecE depletion strain, we tested the insertion of YidC-Sec
dependent protein subunit a of the FOF1 ATPase with a C-terminal P2 epitope of Lep that
served as a cytoplasmic tag for immunoprecipitation (FOa-P2). The substrate was largely
blocked under YidC or Sec depletion conditions (Fig. 2.8B). However, it remained
uninserted when both YidC and Sec were depleted simultaneously. We also evaluated the
insertion of the YidC-only model protein wild-type Procoat-Lep (PC-Lep) and a YidC-Sec
dependent mutant protein ARGRR-Procoat-Lep (ARGRR-PC-Lep) (174) as controls. The
YidC-only substrate PC-Lep (indicated by P in Fig. 2.8) was converted to the mature coat
protein (indicated by C in Fig. 2.8) by the action of signal peptidase only upon insertion

39
under YidC expression conditions (Fig. 2.8C). In contrast, PC-Lep was largely uninserted
when YidC was depleted and only slightly affected by Sec depletion (Fig. 2.8C). However,
the YidC-Sec substrate ARGRR PC-Lep mutant was largely blocked in any translocase
depletion condition, as expected (Fig. 2.8D). Next, we evaluated the export of the Sec-
only substrate OmpA and found it was affected as seen by ProOmpA accumulation under
Sec- depletion and YidC-Sec depletion conditions (Fig. 2.8E). However, the export was
not completely inhibited, which could be because OmpA has a higher affinity for the Sec
apparatus. To further characterize the double-depletion strain, immunoblotting analysis
was performed (Fig. 2.9B). We observed a dramatic decrease in the translocase levels to
under 5% of the levels measured in their wild type conditions (i.e., over 20 fold decrease),
thus demonstrating the depletion of both YidC and SecE under the conditions tested. We
also verified that 2Pf3-Lep was fully inserted (data not shown) under a more stringent
depletion condition (over 40 fold depletion) of both YidC and SecE. Based on this study,
we favor the idea that 2Pf3-Lep can translocate by a Sec/YidC independent mechanism.
2.3 Discussion
Typically, substrates that are inserted by the YidC-only pathway have short translocated
segments, like Pf3 coat (18 residues), Procoat (20 residues), SciP (11 residues), DjlC (1
residue), Flk (1 residue), FoF1 ATPase subunit c (8 residues), MscL (29 residues), and the
N-region of CyoA (26 residues) (96, 123, 126, 129, 161, 204). Longer translocated regions
typically require the Sec system (195, 196). YidC-Sec independent insertion is rare and
observed in proteins with short translocated regions like KdpD (10 residues), Pf3-Lep (18

40
residues) model protein, and KscA (30 residues) (166, 187, 188). In this study, we report
that a 36-residue tail can be translocated independent of Sec and YidC. This result was
confirmed for the first time using a new strategy to deplete both YidC and SecE in the same
E. coli strain to confirm that the substrate is not promiscuously being inserted by Sec when
we deplete YidC, and vice-versa. Under the YidC/Sec depletion conditions, where the
insertion of 2Pf3-Lep was unaffected, we confirmed that YidC was depleted over 30 fold
and SecY over 20 fold. The combined data suggests the possibility that unassisted
translocation of polar domains across the membrane is feasible in nature and it is
conceivable that the hydrophobicity of the TM segment of the protein fuels this process.
The hydrophobicity of TM1 of 2Pf3-Lep is very high, much higher than the 2Pf3-23Lep
construct that inserts by the YidC-only pathway. However, the presence of residual levels
of translocases under depletion conditions and the involvement of any other unknown
translocation mechanism needs to be considered as well and tested in future studies.
On evaluating size of the translocated region as a pathway-determinant, we found that
increasing the length of the YidC-Sec independent substrate N-tail with uncharged residues
led to YidC dependence and eventually Sec dependence as well. Similarly, increasing the
length of a YidC-dependent substrate N-tail caused a switch in its translocase requirement
to YidC-Sec mode of insertion. This suggests that these substrates insert into the membrane
at the YidC-Sec interface. In both cases, the N-tails were poorly inserted beyond a certain
length and substrate N-tails of about 60-residue length was found to be the upper limit of
translocation capacity for these model proteins. Thus, we observe a correlation between
length of the translocating region and the number of translocating devices employed by the

41
substrate. This agrees with previous studies suggesting a limited role for YidC to function
independently and often require the assistance of Sec for more sophisticated proteins in
terms of the energy barrier of translocation (174).
We should point out that this length study here is a reinvestigation of amino-terminal
translocation. Previously, we reported that short tails up to 38 residues are efficiently
translocated in a SecA and SecY-independent manner whereas longer tails are poorly
inserted (198). This agrees with the results reported in this paper. In Cao and Dalbey
(1994), we could obtain translocation of a long N-tail only when we added a leader
sequence to the N-terminus of the protein. However, the N tail translocation of -lactamase
and alkaline phosphatase were reported in later studies to occur even in the absence of an
amino-terminal signal peptide (205, 206). Therefore, we wanted to reinvestigate the
properties of the mature domain that can be translocated in the N-terminal direction.
Specifically, we wanted to look at whether this requires YidC, which was discovered in
2000 (123), and also examine Sec-dependency of insertion using the SecE depletion strain
that was constructed in 1996 (200).
Interestingly here, we found that longer N-tails can be exported by the Sec system if the
substrate has a downstream reverse signal. The mature domain of MBP fused to the N-
terminus of the model substrate 2Pf3-Lep was translocated even when not preceded by its
N-terminal signal sequence. One hypothesis is, Sec A/B engages with the MBP domain
and directs it to the SecYEG channel, which is then opened by the C-terminal TM segment
of the model protein, enabling the translocation of the large periplasmic domain. In keeping
with this, we found that MBP-2Pf3-Lep is SecA dependent (Fig. 2.5B). Previously, the

42
mature alkaline phosphatase has been shown to be exported without an attached signal
peptide if a signal sequence is added in trans (62). Later studies showed that regions of the
translocated segment need also to contain SecA targeting signals that allow this region to
bind to SecA, enabling SecA assisted translocation (77).
In addition to length, we have also tested charge density of the N-tail region as a translocase
determinant. It was previously shown that the addition of charges to N-tail region of the
model substrate Pf3-Lep resulted in a switch in its translocation pathway (166). However,
we observed that doubling this protein’s N-tail length, hence also the number of charged
residues it carries, did not change its translocase requirements. This was surprising and
suggested that additional charges on the N-tail could be translocated without altering the
mechanism if they are distributed across a longer N-tail. This was confirmed by
strategically placing charged residues either close to each other or away and studying the
changes in its translocation pathway. While we found that positive charges had a greater
effect on the translocation mechanism than negative charges, all mutants showed equal
dependence on both YidC and Sec. This is in contrary to the previous study that proposed
negative charges as YidC determinant and positive charges as YidC-Sec determinants
(166). We hypothesize that this can explained as an effect of charge crowding over the
short N-tail of Pf3-Lep substrate that was tested. Although the negatively charged
substrates tested in the previous study are YidC-dependent, our data suggests that
increasing the number of negative charges appear to make it more Sec-dependent as well.
Based on this, we propose that YidC has limited potential to insert charged substrates
independently and requires Sec for more complex substrates.

43
In conclusion, we observed that both N-tail length and charge density can specify the
insertion pathway of substrates. Shorter tails are translocated independently, whereas
longer ones first recruit YidC and beyond a certain threshold, need both YidC and Sec. For
these substrates, we hypothesize that insertion occurs at the YidC-Sec interface, between
the greasy-slide and the lateral gate. Longer periplasmic domains are exported when
followed by a reverse signal by the Sec apparatus. Additionally, crowding of charges in the
translocated region, increases the energy barrier of translocation and thus requires the
assistance of translocases for its membrane clearance.
2.4 Materials and methods
Materials
Isopropyl 1-thio-β-D-galactopyranoside was purchased from Research Products
International Corp. Tran35S-label (mixture of 85% [35S] methionine and 15% [35S]
cysteine at 1000 Ci/mmol concentration) was purchased from PerkinElmer Life Sciences.
Anhydrotetracycline hydrochloride was purchased from ACROS Organics. Lysozyme was
purchased from Sigma, Proteinase K (PK) from Qiagen and PMSF from United States
Biochemical (Affymetrix). Antisera to leader peptidase (anti-Lep) and outer membrane
protein A (anti-OmpA) were from our lab collection. Restriction endonucleases, T4 DNA
ligase, Q5 polymerase, Monarch PCR and DNA Cleanup kits and NEBuilder HiFi DNA

44
Assembly Master Mix were purchased from New England Biolabs. PCR primers were
synthesized by Integrated DNA Technologies (IDT).
Plasmids and Site-directed mutagenesis
To express the 2Pf3-Lep and 2Pf3-23Lep derivatives and mutants in JS7131, CM124 and
YidC-Sec double depletion strain NB167, the genes were cloned into the pLZ1 vector (177)
under the control of T7/lacUV5 promoter. The amino acid sequences of the model proteins
are shown in (Fig 2.10). The techniques described previously (207) were used for DNA
manipulations. Site-directed mutations were made by QuikChange or Fusion PCR method.
N-tail length of the substrates was increased by stepwise insertion of neutral spacer residues
used in (198) (TQVLNAPTSGGQSLNPGTSAQGNLS). DNA sequencing of the entire
gene verified all mutations. The N-terminal addition of MBP and Pre-MBP to 2Pf3Lep was
done by sub-cloning the respective genes amplified from the plasmid pMAL-c2X
(Addgene #75286), a gift from Paul Riggs.
Bacterial Strains
The E.coli YidC depletion strain JS7131 is from our collection (123). The SecE depletion
strain CM124 was obtained from Beth Traxler (200). These strains have either the yidC or
the secE genes expressed under the control of the araBAD promoter while their endogenous
yidC or secE genes are inactivated. The YidC-Sec double depletion strain NB167 is a
derivative of JS7131 with the secE gene repressed using CRISPRi (203) by inducing the
expression of a catalytically defective Cas9 (dCas9). This strain was constructed by

45
synthesizing dCas9 (IDT), originally from Streptococcus pyogenes, with codons optimized
for expression in E. coli. This synthetic construct was introduced into a modified pAH63
vector (208) containing a tetracycline inducible promoter to yield pTR-dCas9. The tetR –
dcas9 region from this plasmid was PCR-amplified using P1 and P2 primers (Supp. Table
S1.) and cloned into the Tn7-based vector pMS26 by USER cloning as described (209),
yielding plasmid pTn7-TR-dCas9. This plasmid was then transformed into JS7131 and
ampicillin resistant transformants were streaked on LB agar at 42C for plasmid curing, as
described (209). Colony PCR (primers P3 and P4) was performed on individual, ampicillin
sensitive colonies to confirm successful chromosomal integration of tetR-dCas9 into
attTn7. To target dCas9 to secE a guide RNA array was constructed by first identifying
three distinct 20-bp pair proto-spacer target sequences adjacent to a NGG PAM site within
the secE promoter region. These 20-bp sequences were each introduced into pgRNA (203)
by inverse PCR. Each individual guide RNA was then assembled as an array into pDLC29,
a low copy ColE1-like plasmid compatible with other vectors used in this study (210)
(primers P11-P16 and P17-P18) using the NEBuilder HiFi DNA Assembly Master Mix.
The resulting plasmid, psgRNA-secE123 was transformed into JS7131 strain harboring
tetR-dCas9 at the attTn7 site to yield NB167.
Growth conditions
The YidC depletion strain JS7131 was cultured at 37 °C for 3.5 h in LB media with 0.2%
arabinose (YidC expression conditions) or 0.2% glucose (YidC depletion conditions). The
SecE depletion strain CM124 was cultured in M9 media with 0.2% arabinose plus 0.4%

46
glucose (SecE expression conditions) or 0.4% glucose (SecE depletion conditions) for 8 h
at 37 °C. YidC-Sec double depletion strain NB167 was cultured at 37 °C for 4 h in LB
media with 0.2% arabinose (YidC-Sec expression conditions) or 0.2% arabinose plus 0.02
μg/ml ATc (SecE depletion conditions) or 0.2% glucose (YidC depletion conditions) or
0.02% glucose plus 0.02 μg/ml ATc (YidC-Sec depletion conditions). For all conditions
tested, the cells were exchanged into fresh M9 media and shaken for 30 min at 37 °C before
inducing the plasmid-encoded substrate protein. SecA dependence was evaluated by
treating the samples with 3 mM sodium azide 5 min prior to induction.
Protease Accessibility Studies
Protein substrates were expressed by induction using 1 mM IPTG (final concentration) for
5 min at 37 °C. Cells were labelled with [35S] methionine for 1 min and converted to
spheroplasts. Briefly, after labelling, the cells were collected by centrifugation and
resuspended in spheroplast buffer (33mM Tris-HCl, pH 8.0, 40% (m/v) sucrose). The
resuspended cells were then treated with 1 mM EDTA (pH 8.0) and 10 μg/ml Lysozyme
on ice for 30 min. To an aliquot of this, Proteinase K (0.75 mg/ml) was added. After 30
min digestion on ice, the reaction was quenched by the addition of 5mM PMSF for 5 min.
Another aliquot was not treated with PK. An equal volume of ice-cold 20% (m/v) TCA
was added to the solution and incubated on ice for 1 h. The total protein was then spun
down at 13000 rpm for 10 min and washed with the sample volume of ice-cold acetone.
The protein pellet was solubilized in Tris-SDS buffer (10mM Tris-HCl, pH 8.0, 2% (m/v)
SDS) overnight at room temperature. The samples were then immunoprecipitated with

47
antiserum to leader peptidase, which precipitates the Lep derivative substrates, or
precipitated with antiserum to OmpA. The samples were analyzed by SDS-PAGE and
phosphorimaging.
Signal Peptidase Processing Assay
Substrates were expressed using 1mM IPTG (final concentration) for 5 minutes at 37°C
and labelled with [35S] methionine for 1 min. The total protein was precipitated on ice for
1 h with an equal volume of ice-cold 20% (m/v) TCA. The samples were spun down at
13000 rpm for 10 min and washed with an equal volume of ice-cold acetone. The pellet
was solubilized in Tris-SDS buffer (10mM Tris-HCl, pH 8.0, 2% (m/v) SDS) overnight at
room temperature. The samples were then immunoprecipitated with antiserum to leader
peptidase, which precipitates the Lep derivative substrates, or precipitated with antiserum
to OmpA. The samples were analyzed by SDS-PAGE and phosphorimaging.
Western blot. E. coli strains JS7131, CM124 and NB167 were grown for various times
(JS7131 and NB167 for 3.5 h, and CM124 for 7 h) in the presence of arabinose or glucose
(with or without ATc for NB167 in each condition) at 37 °C. The cells were then collected
by centrifugation and washed with ice-cold PBS buffer (137 mM NaCl, 2.7 mM KCl, 10
mM Na2HPO4, 2 mM KH2PO4 (pH 7.4)). After normalizing the cells to A600 of 1.2, the
cell samples were pelleted, washed, and dissolved in 40 μl of SDS gel loading buffer, and
5 μl of the protein samples were loaded on a 15% (SecY visualization) and a 10% (YidC
visualization) SDS-polyacrylamide gel. YidC was analyzed by a Western blot using the
total protein. To analyse SecY expression, membrane vesicles were prepared from cells

48
grown in arabinose and glucose conditions, adjusted to A600 of 1.2, respectively and the
proteins resolved on the 15% SDS-polyacrylamide gel. Antisera to YidC and SecY (each
diluted 1:5000) and secondary antibody (goat-to-rabbit IgG horseradish peroxidase,
1:10,000) were used for the Western blot.
Quantification of Membrane Insertion Data
The percent of substrate translocation efficiency was determined by quantifying the
intensities of the protease-protected bands in each condition using ImageJ, a tool developed
by National Institutes of Health as described in (165). The intensities of the cleaved and
cleaved substrate bands were divided by the predicted number of methionine residues they
contain and substituted in the Equation below. For example, 2Pf3-Lep uncleaved substrate
contains 9 methionine residues, where the cleaved product has 7. Hence, the equation to be
used becomes:
% 𝑡𝑟𝑎𝑛𝑠𝑙𝑜𝑐𝑎𝑡𝑖𝑜𝑛 = [9
7𝑐𝑙𝑒𝑎𝑣𝑒𝑑 2𝑃𝑓3𝐿𝑒𝑝
{9
7𝑐𝑙𝑒𝑎𝑣𝑒𝑑 2𝑃𝑓3𝐿𝑒𝑝+𝑢𝑛𝑐𝑙𝑒𝑎𝑣𝑒𝑑 2𝑃𝑓3𝐿𝑒𝑝}
] ∗ 100%

49
2.5 Tables
Oligonucleotide primers used for construction of strain NB167.
Primer and description Sequence (5’ to 3’)
P1 USER ends of dCas9 For. GGGAAAGTUCGCTTTGATACGGAGTAGAG
P2 USER ends of dCas9 Rev. GGAGACATUGTCTCATGAGCGGATACATATT
P3 Tn7-9TR integration For. GATATGCCGGTTATTGTTGTTG
P4 Tn7-9TR integration Rev. AGAGCATCAAGTCGCTAAAG
P5 secE Target-1 For. (5Phos) AACTTCTGACGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGG
P6 secE Target-1 Rev. TTCTACAAACAGACCGCTAAACTGAAAGTTACTAGTATTATAC
P7 secE Target-2 For. (5Phos) GCGCACTAAAGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGG
P8 secE Target-2 Rev. ATTGCGTCAAAGACCGCTAAACTGAAAGTTACTAGTATTATAC
P9 secE Target-3 For. (5Phos) GCTCTGTTGAGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGG
P10 secE Target-3 Rev. CTGTCTCAGCAGACCGCTAAACTGAAAGTTACTAGTATTATAC
P11 secE Target-1 Assembly For. CATTACTCGCATCCATTCTCGCTTTCGCTAAGGATGATTTCTG
P12 secE Target-1 Assembly Rev. TCCGTCTACGAACTCCCAGCAGTCAGTGAGCGAGGAAG
P13 secE Target-2 Assembly For. GCTGGGAGTTCGTAGACGGAGCTTTCGCTAAGGATGATTTCTG
P14 secE Target-2 Assembly Rev. CGATTGAGGACCTTCAGTGCAGTCAGTGAGCGAGGAAG
P15 secE Target-3 Assembly For. GCACTGAAGGTCCTCAATCGGCTTTCGCTAAGGATGATTTCTG
P16 secE Target-3 Assembly Rev. TATTGCTGGCAGGAGGTCAGAGTCAGTGAGCGAGGAAG
P17 DLC29 vector Assembly For. CTGACCTCCTGCCAGCAATATTCCAGTCGGGAAACCT
P18 DLC29 vector Assembly Rev GAGAATGGATGCGAGTAATGCGCTCCTTTCGCTTTCTT
Table 2.1 - Oligonucleotide primers used in NB167 strain construction.
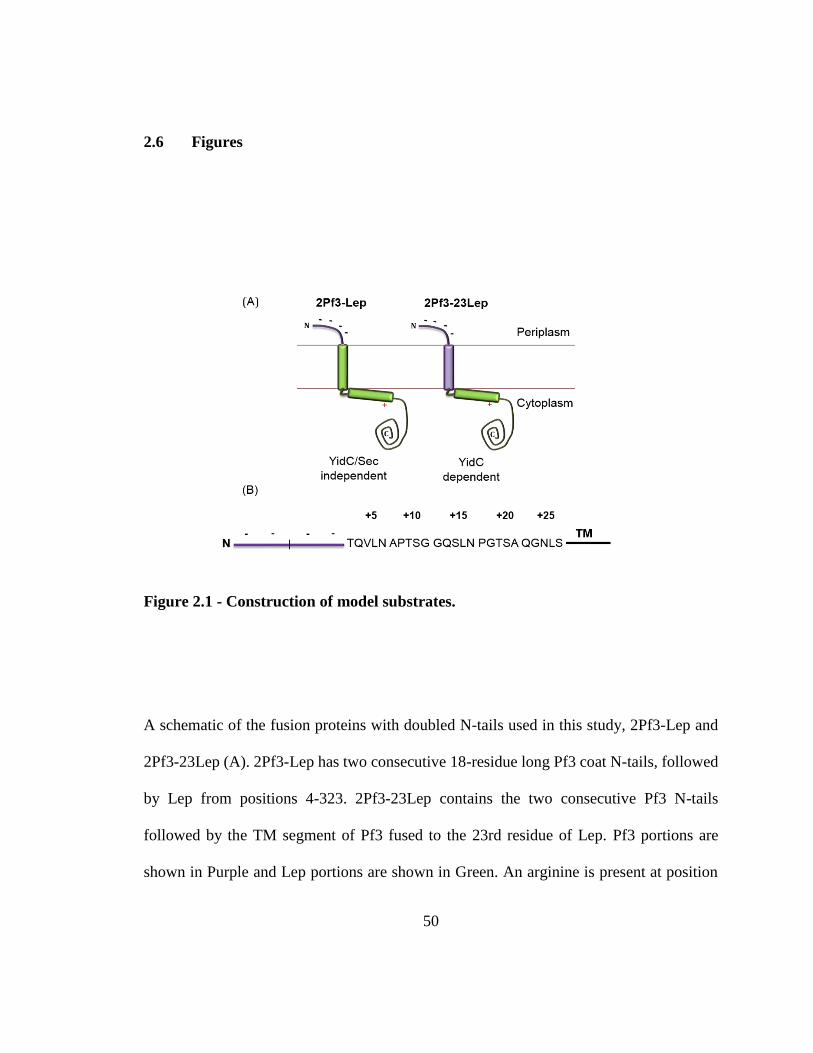
50
2.6 Figures
Figure 2.1 - Construction of model substrates.
A schematic of the fusion proteins with doubled N-tails used in this study, 2Pf3-Lep and
2Pf3-23Lep (A). 2Pf3-Lep has two consecutive 18-residue long Pf3 coat N-tails, followed
by Lep from positions 4-323. 2Pf3-23Lep contains the two consecutive Pf3 N-tails
followed by the TM segment of Pf3 fused to the 23rd residue of Lep. Pf3 portions are
shown in Purple and Lep portions are shown in Green. An arginine is present at position

51
79 of Lep following TM2 in both proteins. The amino acid sequence of the uncharged
pentapeptides (B) added between the N-tail and the TM segments of the model proteins
used to construct the substrates 2Pf3+5-Lep, 2Pf3+10-Lep, 2Pf3+15-Lep, 2Pf3+20-Lep
and 2Pf3+25-Lep with 2Pf3-Lep; 2Pf3+5-23Lep, 2Pf3+10-23Lep, 2Pf3+15-23Lep,
2Pf3+20-23Lep and 2Pf3+25-23Lep with 2Pf3-23Lep.

52
Figure 2.2 - N-tail length requirement for translocase dependence.
E. coli JS7131 cells bearing different plasmids were grown for 3.5 h under YidC expression
(0.2% arabinose) or YidC depletion conditions (0.2% glucose). The substrates 2Pf3-Lep
(A), 2Pf3+5-Lep (B), 2Pf3+10-Lep (C), 2Pf3+15-Lep (D), 2Pf3+20-Lep (E) and 2Pf3+25-
Lep (F) were expressed from pLZ1 plasmid using 1mM IPTG for 5 min and labelled with
[35S] methionine for 1 min. Translocation of the substrate N-tail was analyzed using the
protease-accessibility assay, where the cells were converted into spheroplasts, and a portion
was treated with Proteinase K (PK) (see “Material and Methods”). E. coli CM124 bearing
the same plasmids (A, B, C, D, E, F) were grown for 8 h under SecE expression (0.2%

53
arabinose + 0.2% glucose) or SecE depletion conditions (0.4% glucose) and analyzed using
the protease-accessibility assay. A representational OmpA immunoprecipitation data is
included for both studies (G). The percent translocation of the substrate was quantified as
described in “Methods and Materials”. P denotes the full-length protein, whereas F denotes
the protease-resistant fragment. In panel (G) M denotes the mature OmpA whereas P
denotes the precursor protein.

54
Figure 2.3 - Increasing N-tail length directs YidC-only N-tails to YidC-Sec pathway.
E. coli JS7131 cells bearing different plasmids were grown under YidC expression or
depletion conditions. The substrates 2Pf3-23Lep (A), 2Pf3+5-23Lep (B), 2Pf3+10-23Lep
(C), 2Pf3+15-23Lep (D), 2Pf3+20-23Lep (E) and 2Pf3+25-23Lep (F) were expressed from
pLZ1 plasmid using 1mM IPTG for 5 min and labelled with [35S] methionine for 1 min.
Translocation of the substrate N-tail was analyzed using the protease-accessibility assay,
where the cells were converted into spheroplasts, and a portion was treated with Proteinase
K (PK) (see “Material and Methods”. E. coli CM124 expressing the same substrates (A, B,
C, D, E, F) were grown under SecE expression or depletion conditions and analyzed using
the protease-accessibility assay. Representational OmpA data is included at the bottom for

55
both studies (G). The percent translocation of the substrate was quantified as described in
“Methods and Materials”. P denotes the full-length protein, whereas F denotes the
protease-resistant fragment. In panel (G) M denotes the mature OmpA whereas P denotes
the precursor protein.

56
Figure 2.4 - Translocation of MBP in the N-terminal direction.
A schematic of MBP-2Pf3-Lep, MBP-2Pf3-Lep ∆61-375 and PreMBP-2Pf3-Lep
substrates (A). Maltose-binding protein (shown in Orange) was fused to the N-terminus of
2Pf3-Lep either with its SP (PreMBP-2Pf3-Lep) or without (MBP-2Pf3-Lep). All but the

57
first 60 residues of MBP were deleted to make the truncated MBP-2Pf3-Lep ∆61-375
substrate. E. coli JS7131 cells bearing pLZ1 plasmids expressing MBP-2Pf3-Lep (B),
MBP-2Pf3-Lep ∆61-375 (C) or PreMBP-2Pf3-Lep (D) were grown under YidC expression
or depletion conditions. The substrates were expressed using 1mM IPTG for 5 min and
labelled with [35S] methionine for 1 min. Translocation of the periplasmic domain was
analyzed using the protease-accessibility assay, where the cells were converted into
spheroplasts, and a portion was treated with Proteinase K (PK) (see “Material and
Methods”). E. coli CM124 bearing the same substrate plasmids (B, C, D) were grown under
SecE expression or depletion conditions and analyzed using the protease-accessibility
assay. The percent translocation of the substrate was quantified as described in “Methods
and Materials”. M denotes the mature domain region of MBP fused to 2Pf3-Lep, whereas
F denotes the protease-resistant fragment. P denotes the full-length precursor protein in
panel (D).
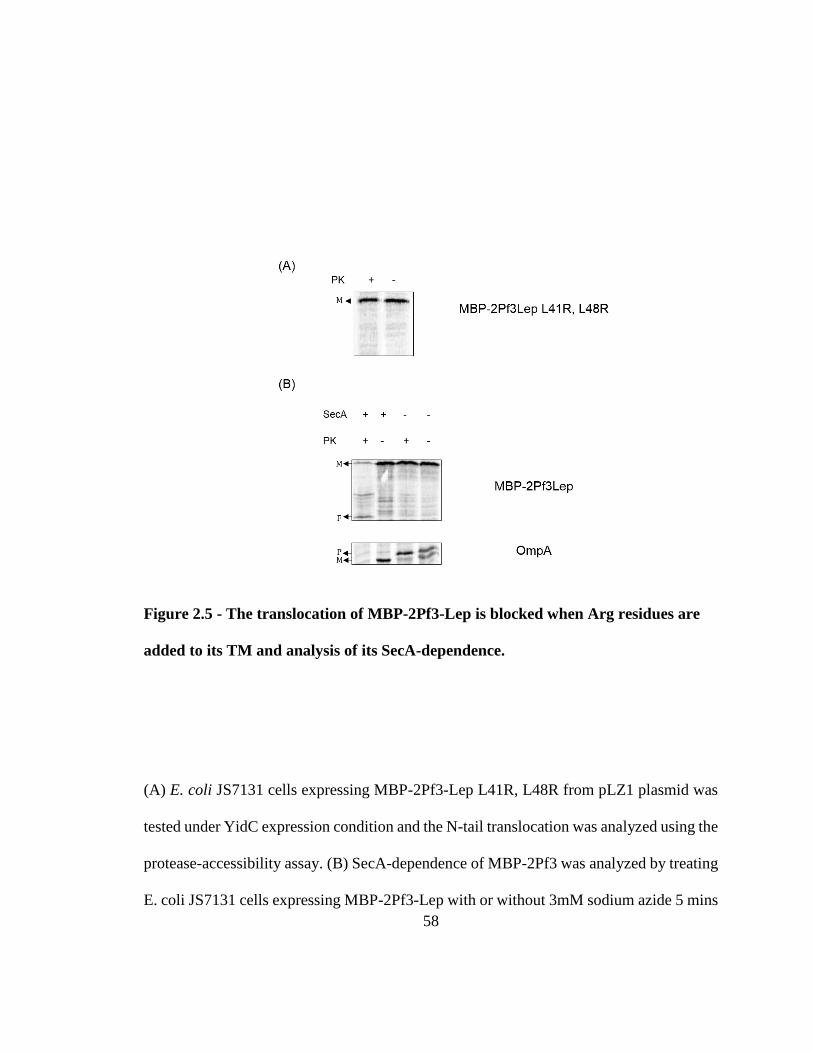
58
Figure 2.5 - The translocation of MBP-2Pf3-Lep is blocked when Arg residues are
added to its TM and analysis of its SecA-dependence.
(A) E. coli JS7131 cells expressing MBP-2Pf3-Lep L41R, L48R from pLZ1 plasmid was
tested under YidC expression condition and the N-tail translocation was analyzed using the
protease-accessibility assay. (B) SecA-dependence of MBP-2Pf3 was analyzed by treating
E. coli JS7131 cells expressing MBP-2Pf3-Lep with or without 3mM sodium azide 5 mins

59
prior to substrate induction under YidC expression conditions and the N-tail translocation
was evaluated using the protease-accessibility assay. OmpA export was analyzed under the
same conditions as a control.

60
Figure 2.6 - Testing negative charge distribution on N-terminal tail as a pathway-
determinant.
The positions of the negative charges introduced on the N-tail of 2Pf3-Lep (A). E. coli
JS7131 cells bearing different plasmids were grown under YidC expression or depletion

61
conditions. The substrates were expressed using 1mM IPTG for 5 min and labelled with
[35S] methionine for 1 min. Substrate N-tail translocation was analyzed using the protease-
accessibility assay, where the cells were converted into spheroplasts, and a portion was
treated with Proteinase K (PK) (see “Material and Methods”). The plasmids encoded the
proteins 2Pf3-Lep V15D (B), V15’D (C), V15D/V15’D (D), L12E/V15D (E) and
L12’E/V15’D (F) (mutations positioned in the second Pf3 N-tail ahead of the TM segment
are indicated by (’)). E. coli CM124 expressing the same substrate proteins (B, C, D, E, F)
were grown under SecE expression or depletion conditions and analyzed using the
protease-accessibility assay. Representational OmpA data is included at the bottom for
both studies (G). The percent translocation of the substrate was quantified as described in
“Methods and Materials”. P denotes the full-length protein, whereas F denotes the
protease-resistant fragment. In panel (G) M denotes the mature OmpA whereas P denotes
the precursor protein.

62
Figure 2.7 - Testing positive charge distribution on N-terminal tail as pathway-
determinant.
The positions of the positive charges introduced on the N-tail of 2Pf3-Lep (A). E. coli
JS7131 cells bearing different plasmids were grown under YidC expression or depletion

63
conditions. The plasmids encoded the proteins 2Pf3-Lep V15R (B), V15’R (C),
V15R/V15’R (D), L12’R/V15’R (E) and L12R/V15R (F). The substrates were expressed
using 1mM IPTG for 5 min and labelled with [35S] methionine for 1 min. Translocation of
the substrate N-tail was analyzed using the protease-accessibility assay, where the cells
were converted into spheroplasts, and a portion was treated with Proteinase K (PK) (see
“Material and Methods”). E. coli CM124 expressing the same substrate proteins (B, C, D,
E, F) were grown under SecE expression or depletion conditions and analyzed using the
protease-accessibility assay. Representational OmpA data is included at the bottom for
both studies (G). The percent translocation of the substrate was quantified as described in
“Methods and Materials”. P denotes the full-length protein, whereas F denotes the
protease-resistant fragment. In panel (G) M denotes the mature OmpA whereas P denotes
the precursor protein.

64
Figure 2.8 - Confirming non-promiscuous insertion of 2Pf3-Lep using YidC-Sec
double-depletion.
E. coli NB167 cells expressing 2Pf3-Lep (A) from pLZ1 plasmid and FOa-P2 (B) from
pMS119 plasmid were grown under YidC-Sec expression or YidC/Sec/YidC-Sec depletion
conditions. The substrate was expressed using 1mM IPTG for 5 min and labelled with [35S]
methionine for 1 min. Translocation of the substrate regions was analyzed using the
protease-accessibility assay, where the cells were converted into spheroplasts, and a portion

65
was treated with Proteinase K (PK) (see “Material and Methods”). NB167 cells bearing the
pLZ1 plasmid expressing either the YidC-only substrate PC-Lep (C) or the YidC-Sec
substrate ARGRR PC-Lep mutant (D) were grown under YidC-Sec expression or
YidC/Sec/YidC-Sec depletion conditions and the substrate membrane insertion was
analyzed using the signal peptidase processing assay, where the substrate labelled with
[35S] methionine for 1 min and precipitated with TCA were analyzed for signal peptidase
processing (see “Material and methods”). OmpA signal processing assay data for the same
conditions are also included (E). P denotes the full-length protein, whereas F denotes the
protease-resistant fragment. C denotes the signal peptide processed mature coat protein in
panel (C, D). In panel (E), M denotes the mature OmpA whereas P denotes the precursor
protein.

66
Figure 2.9 - Detection of YidC and Sec depletion using western blot.
(A) Western blot analysis of YidC and SecY protein expression under YidC/Sec expression
and depletion conditions respectively, in strains JS7131 and CM124. (B) The YidC and

67
SecY levels were evaluated in the double-depletion strain NB164 using western blot. The
percent YidC/Sec remaining under depletion conditions were quantified for each case and
reported as fold decrease relative to the corresponding wild-type levels. For details, see
“Material and Methods” section.

68
Figure 2.10 - Amino acid sequence of the model protein constructs.
(A) The 2Pf3-Lep model protein contains two consecutive Pf3 N-tails followed by the
LepTM segment. (B) In 2Pf3-23Lep model protein the Lep TM segment is replaced by a
Pf3 TM segment instead. Pf3 portions are shown in Purple whereas Lep portions are shown
in Green. The TM segment of each construct is followed by residues 23–323 of Lep, where
an arginine is present at position 79 of Lep following TM2.

69
Chapter 3
FCS analysis of Pf3 coat insertion by reconstituted YidC homologs
3.0 Contributions
The work reported in this chapter was carried out by Maximilian Haase and Sri Karthika
Shanmugam in the laboratory of Dr. Andreas Kuhn in University of Hohenheim, Germany.
Both authors contributed equally to the data collection and interpretation process. The
proteins used in the study were purified by Gisela Nagler, Renate Hess, Eleni Silioni,
Maximilian Haase and Sri Karthika Shanmugam.
3.1 Introduction
Membrane proteins have vital functions for the cell and are attractive drug targets.
Therefore, it is critical to understand how proteins are inserted and assembled into the
membrane. The fundamental process of membrane protein biogenesis is a well-

70
orchestrated interplay between several key bio-machineries that are highly conserved in
nature. These molecular devices ensure that substrates reach their destined location and
achieve their folded functional form with proper orientation in the membrane. The YidC
family of proteins represent a novel class of insertases that operate in the bacterial cell
membrane and eukaryotic organellar membranes of chloroplasts and mitochondria (127).
Additional members have been proposed to reside in the ER membrane (154). Although
these proteins display structural conservation, less is known about the functional overlap
between them. Of these, the best understood is the bacterial homolog YidC that is
responsible for the membrane insertion of a number of essential proteins including those
involved in respiration and energy-transduction (128).
The YidC paralogs Alb3 and Alb4 are found to be present in the chloroplast thylakoid
membranes of higher plants and algae (142, 143). They play essential roles in the
biogenesis of chloroplast enzyme complex. Alb3 (Albino3) was first identified to be
essential for thylakoid biogenesis in a pigmentless-mutant A. thaliana (142). Specifically,
Alb3 is required for membrane assembly of post-translationally imported light-harvesting
chlorophyll-binding proteins (211, 212). Interestingly, Alb3 has been shown to interact
with the chloroplast SecY (cpSecY) protein (147). However, it is currently unknown if they
constitute a holotranslocon comprised of SecYE and Alb3 for membrane protein substrate
integration and stabilization similar to its bacterial counterparts. Another common feature
between YidC and Alb3 is their chaperone activity; Alb3 has been suggested to assist in
the folding of D1 protein, a component of the chloroplast photosystem II complex (213).

71
Alb4 protein was more recently discovered to be localized in the thylakoid membrane
(143). Though Alb4 depletion had a minor effect on growth, it was required for the efficient
assembly of chloroplast ATP synthase complex (CF1FO-ATPase) (145). Further, the
subunits CF1β and CFOII were shown to interact with Alb4 but not Alb3 or cpSecY. This
suggests that though both Alb3 and Alb4 are involved in thylakoid membrane biogenesis,
it is likely that they have evolved to promote the membrane assembly and insertion of
distinctive substrate groups. Interestingly, the conserved regions of both Alb3 and Alb4
can functionally replace the bacterial YidC in E. coli (139, 145).
In contrast to the thylakoid membrane of chloroplasts, the mitochondrial inner membrane
does not contain a SecY-like protein translocon. Instead, Oxa1 (oxidase assembly protein
1) insertase facilitates the membrane assembly of mitochondrion-encoded subunits of the
respiratory complexes like cytochrome c oxidase (Cox 1 and 2) and NADH reductase
(214). Certain nuclear-encoded proteins that are imported from the cytoplasm by other
transport machineries, also utilize Oxa1 to insert into the mitochondrial membrane (215).
It is closely related to YidC; it can complement E. coli YidC and shares the conserved 5
TM core region (216). Additionally, it possesses a C-terminal helical domain that can bind
to the translating ribosomes and recruit it for co-translational insertion (151, 217, 218).
Controversially, Oxa1 has been shown to form a dynamic pore in the membrane bilayer
that responds to the membrane potential and substrate signals (219).
Get1 (guided-entry tail-anchored protein 1) is a newly identified member of the YidC
superfamily (220). Until recently, the ER resident YidC homologs were unknown. A
systematic homology study by the Keenan group (154) revealed that the Get1 along with

72
two others proteins EMC3 and TMCO1 share structural homology with the mitochondrial
homolog Oxa1 protein. Like the other members of this family, Get1 also plays a role in the
ER membrane biogenesis. It assists in the membrane insertion of tail-anchored (TA)
proteins by recognizing the substrate C-terminal TM anchor. However, it remains to be
seen if these ER homologs can functionally substitute for the bacterial YidC.
To demonstrate a conserved role for YidC in the substrate membrane insertion process, we
have taken advantage of the highly sensitive biophysical technique, fluorescence
correlation spectroscopy (FCS) (221). This method can be used to trace individual
fluorescently-labelled proteins within small confocal volumes in millisecond time-scales.
A strong excitation light is employed to excite the sample placed as a droplet on a coverslip
of an inverted confocal microscope with a water-immersive objective (Fig 3.1A). The
labelled molecules diffuse through the detection volume and variations in the fluorescence
intensities are measured as single photons to determine the diffusion time of the molecule.
It is critical to sufficiently dilute the number of particles in the sample in order to detect
single events and separate it from the background signals.
In this study, we have evaluated the insertion efficiency of the model protein Pf3 coat by
the eukaryotic YidC homologs Alb3, Alb4, Oxa1 and Get1 in vitro. The different homologs
were purified and reconstituted into proteoliposomes in sub-nanomolar concentrations. We
uncoupled the substrate translation step from membrane insertion by adding purified and
fluorescently labelled Pf3 coat protein to the proteoliposomes and the insertion event was
followed using FCS (Fig. 3.1A). Upon excitation of the sample, the quencher-resistant
fluorescent signals were counted using an avalanche photo diode (APD) after filtering out

73
the excitation photons using a beam splitter. The process was followed for 10 minutes and
the software “Burst Analyzer 2.0” by Becker and Hickl was used to identify inserted Pf3
molecules within the proteoliposomes using their diffusion properties determine in the
work by Ernst et al (222). Potassium iodide (KI) was used to quench the fluorescence of
uninserted Pf3 molecules (Fig. 3.1B). For each homolog tested, so far 30 bursts were
analyzed and normalized to determine the rate of insertion. Our preliminary results suggest
that all the tested homologs could successfully facilitate the insertion of Pf3 with
comparable efficiencies. This study sheds light on the conserved nature of YidC function
across these homologs.
3.2 Results
Insertion of Pf3 coat protein by the chloroplast YidC homologs Alb3 and Alb4
To study the insertion of the YidC substrate Pf3 using FCS, a mutant version (Pf3 16C)
was expressed and purified as described in (172). The substrate was kept in a partially
unfolded state using an organic solvent. Pf3 N-tail was labelled using the fluorescent dye
Atto520 and the free dye was separated from the labelled protein using gel-filtration
chromatography. The mature region of Alb3 was expressed and purified using affinity
chromatography with Nickel-resin, followed by a gel-filtration step. YidC translocase
encompassing a C-terminal 10 His-tag was also purified using affinity chromatography to
compare the substrate insertion efficiencies in amongst the homologs. Alb3 and YidC were
reconstituted into DOPC liposomes at the final concentration of 0.4 nM.

74
The substrate protein was diluted and added to the proteoliposomes along with the
fluorescence quencher KI. If the substrate N-tail was translocated within 10 mins into the
lumen of the liposome by the action of the insertase, then the molecule will continue to
fluoresce as KI is membrane impermeable (Fig. 3.1B). This mixture was analyzed for the
diffusion of fluorescent molecules using the FCS setup (Fig 3.1A). The excited signals
from the sample were scanned for “burst” events which correspond to the diffusion time
previously calculated for fluorescent proteins within a liposome (Fig. 3.1C). We observed
that Pf3 coat was inserted into both YidC and Alb3 proteoliposomes. The number burst
events in each condition was normalized and plotted along with its standard deviations (Fig
3.2A). The insertion efficiency into Alb3 proteoliposomes was about 75% in comparison
to the YidC proteoliposomes. As a negative control, we also monitored Pf3 insertion using
DOPC liposomes under the same conditions. The substrate was confirmed to be largely
uninserted in the absence of the insertase, since it had none, or significantly lesser number
of bursts recorded during each trial.
Alb4 protein was also tested for its ability to insert the YidC substrate Pf3 protein. The
mature region of Alb4 preceded by the first TM segment of YidC was expressed and
purified similar to Alb3. The pure protein was reconstituted into DOPC liposomes with a
final concentration of 0.4 nM. The substrate was diluted to follow the individual molecules
within the focal volume and mixed with the proteoliposomes and the quencher KI. Pf3
insertion into DOPC liposomes and YidC proteoliposomes was also followed as controls.
Our results show that Alb4 can also insert Pf3 and the efficiency was either comparable or
slightly more than that of YidC (110%) (Fig 3.2B). In the insertase-free DOPC conditions,

75
some burst-like events were observed (8%), which could be due to fluorescent noises in
the background.
Functional conservation in Oxa1 and Get1 eukaryotic homologs
The mature domains of both Oxa1 and Get1 proteins were expressed with an N-terminal
TM1 of YidC. Oxa1 and Get1 proteins were purified using affinity chromatography
(Nickel resin), followed by size exclusion chromatography. The proteins were
reconstituted into DOPC liposomes and assayed as described before to monitor Pf3 coat
insertion. YidC proteoliposomes and DOPC liposomes were used as controls. The
experiments were performed in similar conditions and the number of bursts recorded in
each condition was normalized and plotted with standard deviations (Fig 3.3A & 3.3B).
The insertion was efficient in both Oxa1 and Get1 reconstituted liposomes. They displayed
comparable efficiencies to that of YidC proteoliposomes. However, in the conditions we
tested, Get1 displayed better insertion rate (115%) compared to YidC. The insertion
efficiency by Oxa1 was about 93% compared to that of YidC. We confirmed that there
were only a small number of burst-like events in DOPC liposomes conditions in all the
trials.
3.3 Discussion
YidC is a unique insertase that assists in the membrane insertion and folding of substrate
proteins both autonomously and in association with the Sec machinery by forming the holo-
translocase complex (162). It is remarkably conserved in all domains of life. Previously, it

76
was thought to exist in endosymbiotic organellar membranes of chloroplasts and
mitochondria only. But recent evidences indicate that there are homologs present in the ER
membrane as well (154). However, it is currently unknown if these new members can
functionally complement YidC. Pf3, the 44-residue long bacteriophage coat protein is a
well-studied model substrate of the E. coli YidC insertase. Here, we utilized FCS analysis
to monitor Pf3 insertion efficiencies by the different YidC homologs, Alb3, Alb4, Oxa1
and Get1.
Alb3 and Alb4 in the thylakoid membranes of chloroplasts are proposed to be involved in
the membrane biogenesis pathway, like YidC. Previously it has been shown that both Alb3
and Alb4 can functionally replace E. coli YidC. However, Alb3 is responsible for the
membrane insertion of light-harvesting chloroplast-binding substrate proteins, whereas
Alb4 is responsible for non-chlorophyll binding photosynthetic proteins (145, 154, 172).
Structural predictions show that Alb3 has a unique C-terminal extension that Alb4 lacks.
This region has been proposed to be involved in the recruitment of SRP for the co-
translational insertion of its substrate proteins (223). But Alb3 can facilitate post-
translational insertion as well. In this study, we followed the insertion efficiency of Pf3
substrate by Alb3 and Alb4. Functional conservation in terms of substrate insertion has not
been visualized for these homologs before. Using FCS, we monitored Pf3 insertion into
reconstituted proteoliposomes by the action of Alb3 and Alb4 and compared its efficiency
to that of YidC at similar concentrations. Our results show that both homologs can
effectively insert Pf3 with slightly different efficiencies when compared to YidC-mediated
insertion rate.

77
Next, we demonstrated that the other eukaryotic homologs Oxa1 and Get1 can also assist
in the translocation of the Pf3 N-tail. The mitochondrial Oxa1 and E. coli YidC can
functionally complement each other (216). Since SecY channel is absent in the
mitochondria, it is suggested that Oxa1 can only carry out the independent insertion
function of YidC. In line with this, it was shown that Oxa1 can insert the YidC-only
substrates M13 Procoat and FOc in E. coli, but it could not be crosslinked to the Sec-
dependent substrate FtsQ like YidC can. We have confirmed that Oxa1 can insert in vitro
a YidC-only substrate Pf3, showing indeed it has a similar insertion function. The ER
resident Get1 was recently proposed to share structural homology with the YidC family of
proteins (154). Tail-anchored protein are more common in eukaryotes than bacteria.
However, the tail-anchored TssL protein is a substrate of the E. coli YidC (161). In this
work, we have demonstrated for the first time that Get1 can efficiently insert the YidC
pathway substrate Pf3 coat, which has the opposite topology of its normal substrates which
are C-tail anchored. Since the SecY channel is present in the ER membrane, it would be
interesting to explore in future if Get1 also cooperates with Sec for Sec-dependent
substrates.
Substrate translocation is commonly assessed in vivo using protease-accessibility assays.
However, these involve the construction of depletion strains and it cannot be used to
visualize insertion in real-time. In vitro methods serve as useful tools to monitor protein
translocation in the absence of other components that may influence the process. YidC has
been shown to be the minimal translocase unit, making it an ideal system for biophysical
studies. In this study, we have established the experimental setup to study the various YidC

78
homologs in purified reconstituted systems. Fluorescence based assays offer the advantage
to follow the insertion in real-time with single-molecule resolution. We have used DOPC
lipids for our reconstitution assays, however, the lipid composition of the different
membranes varies greatly in size and charge status and could have an effect on the insertion
efficiencies. In (188), the authors showed that the lipid composition could affect the rate of
insertion as it could play a role in the recruitment of substrate proteins. It could be
hypothesized that the slight differences we observed in the insertion rates could be due to
the change in the lipid composition between the native membranes of these homologs.
Taken together, this work provides biophysical evidence for the functionally conserved
nature of YidC across its different homologs.
3.4 Materials and methods
Materials
Ni2+-NTA beads were obtained from Qiagen. DOPC lipids and extruder were purchased
from Avanti Polar Lipids Inc. Activated charcoal was from Merck. The fluorescence label
Atto520 maleimide was purchased from Atto-tec. All other chemicals were supplied from
Sigma.
Expression and purification of the YidC homologs
The mature region of Alb3 was expressed with a C-terminal 6 His-tag from a pET29b
vector (224). The mature regions of the other YidC homologs, Alb4, Oxa1 and Get1 were
expressed with an upstream E. coli YidC TM segment 1 (1-45 residues) and a C-terminal
10 His-tag from a pET22b vector. The plasmids were transformed into E. coli LEMO

79
(DE3) cells and induced with 0.5mM IPTG at mid-log phase. The cells were harvested 1-
hour post-induction and resuspended in 20 mM Tris-HCl buffer (pH was maintained at 7.5
for YidC and Alb4, whereas Get1, Alb3 and Oxa1 purifications were carried out at pH 8)
with 300 mM NaCl and lysed using Oneshot. The membranes were isolated and separated
overnight using a sucrose gradient. The inner membrane fraction obtained from the
gradient was diluted 5 times in 20 mM Tris-HCl buffer with 300 mM NaCl at appropriate
pH values and solubilized with 1% DDM in the presence of 0.5 mM PMSF for 2 h. The
proteins were purified using a Ni2+ affinity column and eluted in 20 mM Tris-HCl buffer
containing 300 mM Imidazole, 300 mM NaCl and 0.01% DDM. The samples were
analyzed using SDS-PAGE and the eluates containing the protein was further purified by
gel filtration chromatography using a Superdex 200 column with 20 mM Tris-HCl buffer
(pH 7.5 or 8 respectively) with 300 mM NaCl and 0.01% DDM. The fractions containing
the protein was run on SDS-PAGE to confirm its purity and the concentration of the
proteins were estimated using Nanodrop2000. A representational purification of Alb4
protein is shown in (Fig. 3.4A & 3.4B).
Pf3 coat mutant purification and labelling
Pf3 16C mutant protein was purified as described in (222). Briefly, E. coli BL21 (DE3)
cells bearing pMS119-Pf3 16C gene was grown in LB media at 37°C until 0.6 OD was
reached, and then protein expression was induced using 1mM IPTG for 3 h. The cells were
harvested and resuspended in 20 mM Tris-HCl buffer (pH 8) containing 10% sucrose per
gram of cell pellet before flash freezing in liquid N2 and storing at -20°C. The cells were
disrupted using Oneshot and the membranes were isolated and homogenized in 100 mM

80
Tris-HCl buffer (pH 8) containing 8 M Urea. For the purification of Pf3 coat mutant, the
extract was first diluted with an equal volume of 100 mM Tris-HCl buffer (pH 8) with 10%
isopropanol. Next, it was fractionated using reversed phase chromatography that was run
with a buffer gradient of 5% to 80% isopropanol in 100 mM Trsi-HCl (pH 8) with 0.1%
trifluoroethanol. The substrate was further purified by two steps of gel filtration using a
Superdex 200 and a Superdex 75 column run with 100 mM Tris-HCl (pH 7.5) containing
10% isopropanol. The purified protein was mixed with Atto520 maleimide dye in dimethyl
sulfoxide solution in a ratio of 1:1.3 protein to dye concentration, in a dark room for 1 h.
Finally, the free dye was removed by gel filtration using a Superdex 200 column with 100
mM Tris-HCl (pH 7.5) with 10% isopropanol.
Liposome preparation and reconstitution of the YidC homologs
The liposomes and proteoliposomes used in this study were prepared using 1,2-Dioleoyl-
sn-glycero-3-phosphocholine (DOPC) lipids as described in (222). Briefly, the lipids were
dissolved in dichloromethane and dried by rotation under vacuum for 8 h to get a thin lipid
film. This was dissolved in 100 μl water and diluted with an equal volume of 20 mM Tris-
HCl buffer containing 300 mM NaCl (pH 7.5). To form the unilamellar liposomes, the
lipids solution was extruded 31 times through a nitrocellulose membrane of 0.4 μm pore
size. Reconstituted proteoliposomes were prepared by adding purified YidC homologs to
the lipid solution in the ratio of 1:25,000 with a final protein concentration of 0.4 nM and
extruded as described above. The mean particle diameter of the liposomes and the
proteoliposomes were evaluated to be 250 nm using dynamic light scattering technique
(DLS).

81
Confocal fluorescence correlation spectroscopy (FCS) measurement of Pf3 insertion
To evaluate the insertion efficiency of Pf3 substrate protein into the reconstituted
proteoliposomes, FCS measurements were performed as described in (188). At room
temperature, the liposomes or the reconstituted proteoliposomes with the different YidC
homologs were diluted in 20 mM Tris-HCl buffer (pH 7.5) to ensure that the protein
concentrations were as similar as possible in all measurements. 0.1 ng of labelled Pf3
protein was mixed into it along with 200 mM potassium iodide (fluorescence quencher)
and placed as a droplet (45 μl) on the coverslip of the lab-built confocal microscope,
Olympus IX71 (Fig. 3.1A). It has a water immersion objective (UPlanSApo 60X, N.A.1.2;
Olympus) which was used to focus a 105 μW attenuated laser beam to excite the
fluorophore on Pf3 protein in the droplet. A dichroic beam splitter separated the excited
signal from the source signal. An avalanche photodiode detected the single photons from
the excited signal that passed through an interference filter and the photons were counted
using a TCSPC card. In each trial, the fluorescent signals were recorded for 600 s. Photon
bursts with a minimum threshold of 35 counts per millisecond that last for at least 40 ms
were separated from background signals and analyzed using an automated algorithm called
“Burst Analyzer 2” by Becker and Hickl. A sample burst is shown in (Fig. 3.1B).
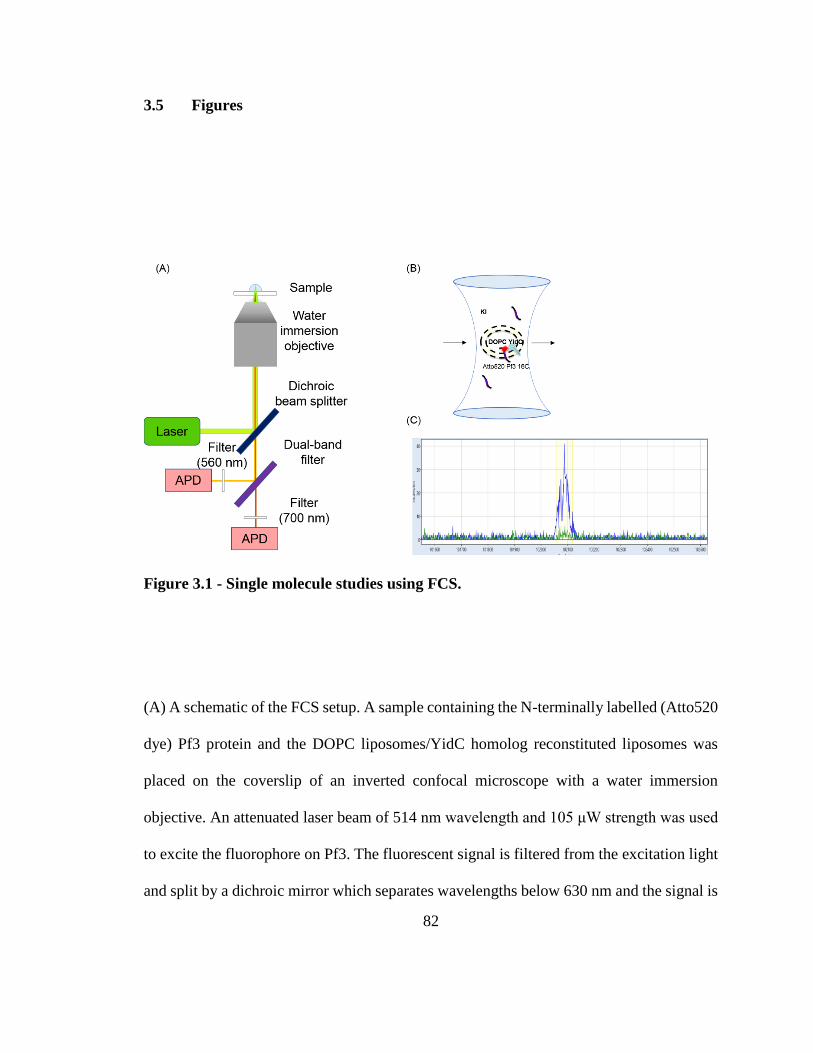
82
3.5 Figures
Figure 3.1 - Single molecule studies using FCS.
(A) A schematic of the FCS setup. A sample containing the N-terminally labelled (Atto520
dye) Pf3 protein and the DOPC liposomes/YidC homolog reconstituted liposomes was
placed on the coverslip of an inverted confocal microscope with a water immersion
objective. An attenuated laser beam of 514 nm wavelength and 105 μW strength was used
to excite the fluorophore on Pf3. The fluorescent signal is filtered from the excitation light
and split by a dichroic mirror which separates wavelengths below 630 nm and the signal is

83
further passed through another filter of 560 nm and detected by the APD (avalanche
photodiode). The wavelengths higher than 630 nm are filtered and detected by another
APD. The signals from the APDs are counted as single photons by synchronized TCSPC
(time-correlated single photon counting) cards. (B) A schematic representation of the freely
diffusing molecule within the excitation focal volume. The Atto520 dye on uninserted Pf3
is quenched by KI, but N-tails that are translocated are fluorescent. The fluorescent signals
from the diffusing liposome is recorded as single photons and analyzed. (C) A
representative burst that is recorded by the Becker & Hickl “Burst Analyzer 2” program.
The single photons detected by the APDS were recorded. The excited signals specific to
the Pf3 Atto520 dye was only observed in the APD which received the signals from the
560 nm filter (blue) and not in the other one (green). The burst events with a minimum
threshold of 35 counts/ms with a diffusion time of at least 40 ms which corresponds to the
fluorescent liposomes were estimated in each trial.
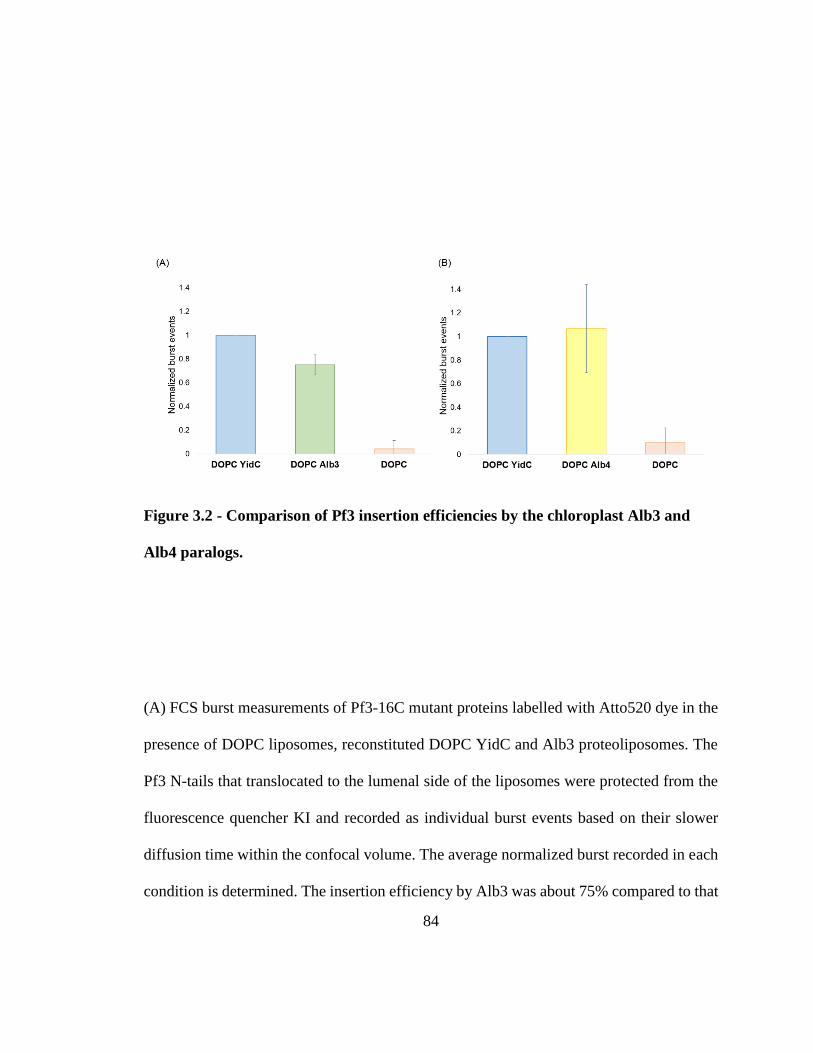
84
Figure 3.2 - Comparison of Pf3 insertion efficiencies by the chloroplast Alb3 and
Alb4 paralogs.
(A) FCS burst measurements of Pf3-16C mutant proteins labelled with Atto520 dye in the
presence of DOPC liposomes, reconstituted DOPC YidC and Alb3 proteoliposomes. The
Pf3 N-tails that translocated to the lumenal side of the liposomes were protected from the
fluorescence quencher KI and recorded as individual burst events based on their slower
diffusion time within the confocal volume. The average normalized burst recorded in each
condition is determined. The insertion efficiency by Alb3 was about 75% compared to that

85
of YidC. In the negative control, marginal to no insertion was observed in the absence of
the insertase. (B) Insertion efficiency of labelled Pf3 proteins examined in the presence of
DOPC liposomes, reconstituted DOPC YidC and Alb4 proteoliposomes. The burst events
corresponding to translocated Pf3-N tails within the liposomes were averaged for each
condition and plotted in comparison with DOPC YidC condition. Alb4 showed a slightly
higher efficiency rate (110%) for Pf3 coat insertion. In comparison, only a small number
of burst-like events were observed in DOPC liposomes conditions.
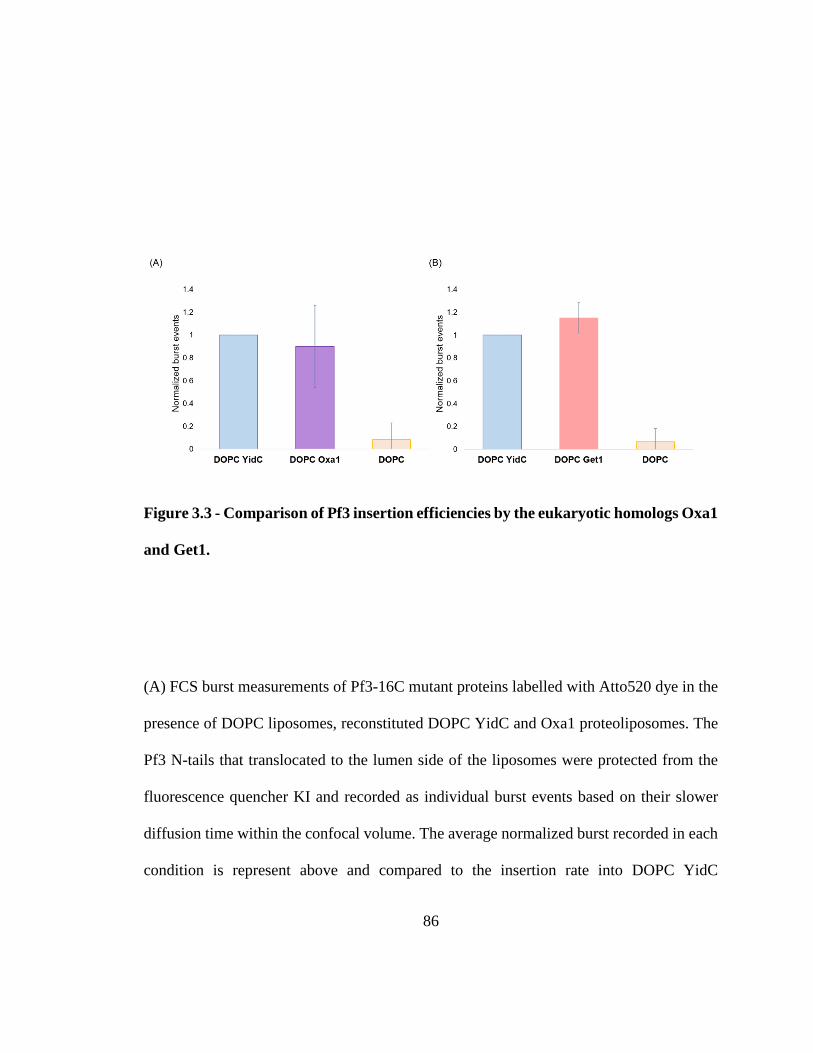
86
Figure 3.3 - Comparison of Pf3 insertion efficiencies by the eukaryotic homologs Oxa1
and Get1.
(A) FCS burst measurements of Pf3-16C mutant proteins labelled with Atto520 dye in the
presence of DOPC liposomes, reconstituted DOPC YidC and Oxa1 proteoliposomes. The
Pf3 N-tails that translocated to the lumen side of the liposomes were protected from the
fluorescence quencher KI and recorded as individual burst events based on their slower
diffusion time within the confocal volume. The average normalized burst recorded in each
condition is represent above and compared to the insertion rate into DOPC YidC

87
liposomes. The insertion efficiency by Oxa1 was about 93% compared to that of YidC. In
the negative control with DOPC liposomes, a small number of burst-like events were
observed. (B) Insertion efficiency of labelled Pf3 proteins examined in the presence of
DOPC liposomes, reconstituted DOPC YidC and Get1 proteoliposomes. The burst events
corresponding to translocated Pf3-N tails within the liposomes were averaged for each
condition and plotted. Get1 showed 115% Pf3 insertion efficiency rate compared to that of
YidC. In comparison, only a small number of burst-like events were observed in DOPC
liposomes conditions.
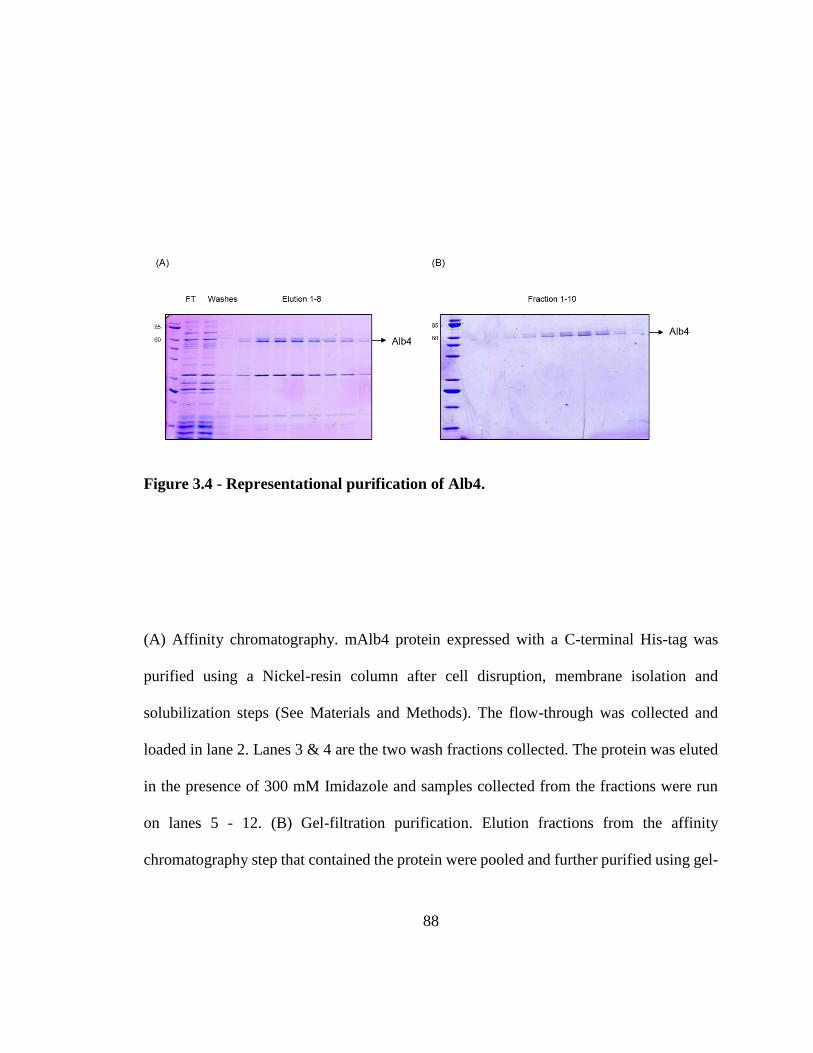
88
Figure 3.4 - Representational purification of Alb4.
(A) Affinity chromatography. mAlb4 protein expressed with a C-terminal His-tag was
purified using a Nickel-resin column after cell disruption, membrane isolation and
solubilization steps (See Materials and Methods). The flow-through was collected and
loaded in lane 2. Lanes 3 & 4 are the two wash fractions collected. The protein was eluted
in the presence of 300 mM Imidazole and samples collected from the fractions were run
on lanes 5 - 12. (B) Gel-filtration purification. Elution fractions from the affinity
chromatography step that contained the protein were pooled and further purified using gel-

89
filtration chromatography. The samples taken from fractions 1 - 10 were analyzed on SDS-
PAGE for purity.

90
Chapter 4
Conclusion
4.1 Summary of work performed
The primary findings of the work are described below:
(i) The length of the translocated N-terminal tails of membrane protein substrates
acts as a translocase-determinant. YidC and Sec independent translocation of a
short amino-terminal tail was confirmed in vivo using a novel double depletion
strain. Upon increasing its length, the substrate required the YidC translocase.
Further increase led to YidC-Sec dependence and eventually could not be
inserted.

91
(ii) The large mature region of Maltose-binding protein (MBP) was translocated by
the Sec system when it was N-terminally fused to a YidC-Sec independent
model membrane protein. In the absence of its signal sequence, we hypothesize
that MBP translocation was feasible since the C-terminal TM segment of the
protein could open the channel. In line with this, we confirmed that
destabilizing the TM segment with positive charges blocks its translocation.
(iii) The location of charges within the N-tail has an effect on the translocation
mechanism. When the charged residues, positive or negative, were positioned
in proximity to each other, the need for translocase involvement increased.
YidC-Sec independent substrates switched to the dependent pathway when
additional charges were introduced close to each other. Positively charged
residues had a greater effect than negatively charged residues, which could be
explained a greater need for a translocase to move the positively charged
regions to the positive side of the membrane due to the membrane electrical
potential.
(iv) The conserved function of YidC homologs in membrane protein insertion was
investigated using single-molecule methods. The eukaryotic YidC homologs
Oxa1, Get1 and the chloroplast paralogs Alb3 and Alb4 were reconstituted into
proteoliposomes and their ability to insert the fluorescently-labelled YidC-
substrate Pf3 was analyzed using fluorescence correlation spectroscopy. Our

92
results show that all the tested YidC homologs can insert the substrate with near
similar efficiencies as the E. coli YidC protein.
The findings from this work provide a framework to understand the mechanistic
features of the substrate amino-tail that determine its insertion pathway. An increase in
length or charge density necessitates translocase assistance. YidC has limited potential
to act independently and serves as a secondary site for insertion of proteins with shorter
translocated regions while Sec tackles the more energy-intensive substrates (Fig. 4.1A).
Our study suggests that YidC insertase is functionally conserved in all walks of life.
This dissertation builds upon existing knowledge on how protein translocation occurs
in E. coli and in similar pathways in higher organisms.
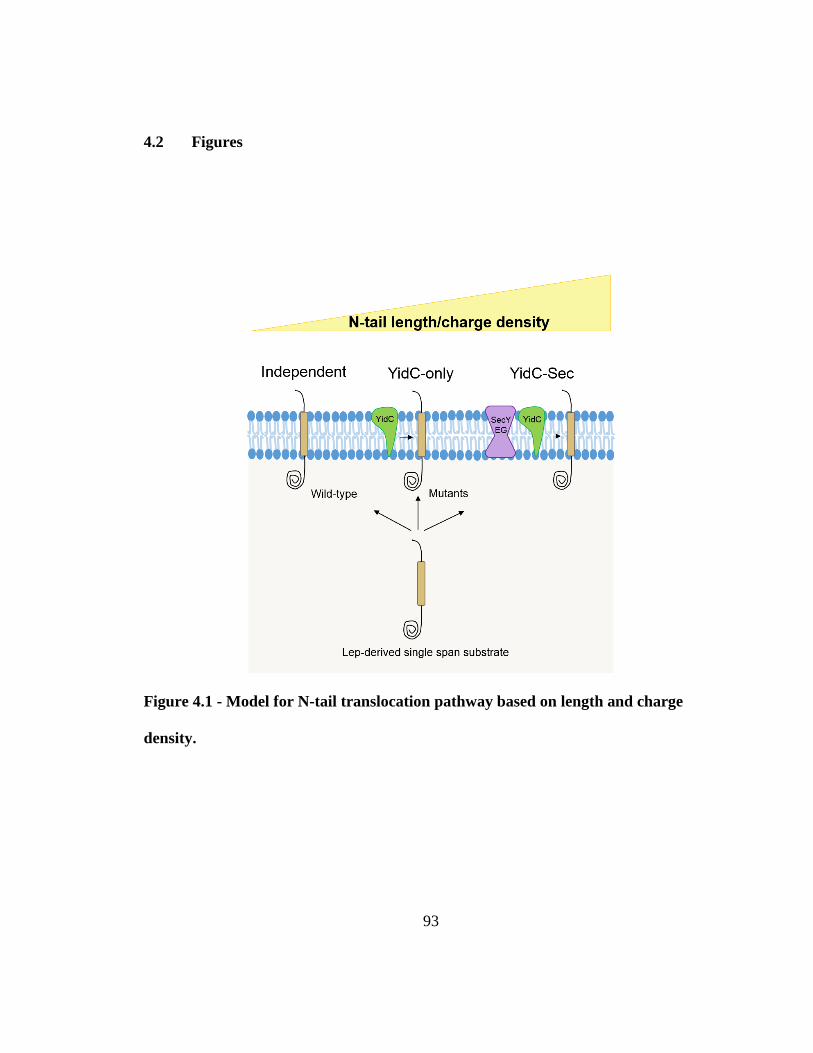
93
4.2 Figures
Figure 4.1 - Model for N-tail translocation pathway based on length and charge
density.

94
The wild-type model substrate inserts independent of YidC and Sec. An increase in the
length and/or charge density of the translocating amino-terminal switches its translocation
pathway from independent to YidC-only, to YidC-Sec dependent.

95
References 1. Sohlenkamp C, Geiger O. Bacterial membrane lipids: diversity in structures and
pathways. FEMS microbiology reviews. 2016;40(1):133-59. Epub 2015/04/12.
2. Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane
protein topology with a hidden Markov model: application to complete genomes. Journal
of molecular biology. 2001;305(3):567-80. Epub 2001/01/12.
3. Tottey S, Waldron KJ, Firbank SJ, Reale B, Bessant C, Sato K, et al. Protein-
folding location can regulate manganese-binding versus copper- or zinc-binding. Nature.
2008;455(7216):1138-42. Epub 2008/10/25.
4. Bolhuis A. Protein transport in the halophilic archaeon Halobacterium sp. NRC-1:
a major role for the twin-arginine translocation pathway? Microbiology. 2002;148(Pt
11):3335-46. Epub 2002/11/13.
5. Sargent F, Bogsch EG, Stanley NR, Wexler M, Robinson C, Berks BC, et al.
Overlapping functions of components of a bacterial Sec-independent protein export
pathway. The EMBO journal. 1998;17(13):3640-50. Epub 1998/07/03.
6. Berks BC, Palmer T, Sargent F. Protein targeting by the bacterial twin-arginine
translocation (Tat) pathway. Current opinion in microbiology. 2005;8(2):174-81. Epub
2005/04/02.
7. Wickner W, Schekman R. Protein translocation across biological membranes.
Science. 2005;310(5753):1452-6. Epub 2005/12/03.
8. Yen MR, Tseng YH, Nguyen EH, Wu LF, Saier MH, Jr. Sequence and
phylogenetic analyses of the twin-arginine targeting (Tat) protein export system.
Archives of microbiology. 2002;177(6):441-50. Epub 2002/05/25.
9. Bogsch EG, Sargent F, Stanley NR, Berks BC, Robinson C, Palmer T. An
essential component of a novel bacterial protein export system with homologues in
plastids and mitochondria. The Journal of biological chemistry. 1998;273(29):18003-6.
Epub 1998/07/11.
10. Sambasivarao D, Turner RJ, Simala-Grant JL, Shaw G, Hu J, Weiner JH.
Multiple roles for the twin arginine leader sequence of dimethyl sulfoxide reductase of
Escherichia coli. The Journal of biological chemistry. 2000;275(29):22526-31. Epub
2000/05/10.
11. Ize B, Gerard F, Zhang M, Chanal A, Voulhoux R, Palmer T, et al. In vivo
dissection of the Tat translocation pathway in Escherichia coli. Journal of molecular
biology. 2002;317(3):327-35. Epub 2002/04/02.
12. Buchanan G, Sargent F, Berks BC, Palmer T. A genetic screen for suppressors of
Escherichia coli Tat signal peptide mutations establishes a critical role for the second
arginine within the twin-arginine motif. Archives of microbiology. 2001;177(1):107-12.
Epub 2002/01/18.
13. DeLisa MP, Samuelson P, Palmer T, Georgiou G. Genetic analysis of the twin
arginine translocator secretion pathway in bacteria. The Journal of biological chemistry.
2002;277(33):29825-31. Epub 2002/05/22.

96
14. Stanley NR, Palmer T, Berks BC. The twin arginine consensus motif of Tat signal
peptides is involved in Sec-independent protein targeting in Escherichia coli. The Journal
of biological chemistry. 2000;275(16):11591-6. Epub 2000/04/15.
15. Cristobal S, de Gier JW, Nielsen H, von Heijne G. Competition between Sec- and
TAT-dependent protein translocation in Escherichia coli. The EMBO journal.
1999;18(11):2982-90. Epub 1999/06/05.
16. Rodrigue A, Chanal A, Beck K, Muller M, Wu LF. Co-translocation of a
periplasmic enzyme complex by a hitchhiker mechanism through the bacterial tat
pathway. The Journal of biological chemistry. 1999;274(19):13223-8. Epub 1999/05/01.
17. Jong WS, ten Hagen-Jongman CM, Genevaux P, Brunner J, Oudega B, Luirink J.
Trigger factor interacts with the signal peptide of nascent Tat substrates but does not play
a critical role in Tat-mediated export. European journal of biochemistry / FEBS.
2004;271(23-24):4779-87. Epub 2004/12/21.
18. Graubner W, Schierhorn A, Bruser T. DnaK plays a pivotal role in Tat targeting
of CueO and functions beside SlyD as a general Tat signal binding chaperone. The
Journal of biological chemistry. 2007;282(10):7116-24. Epub 2007/01/12.
19. Jack RL, Buchanan G, Dubini A, Hatzixanthis K, Palmer T, Sargent F.
Coordinating assembly and export of complex bacterial proteins. The EMBO journal.
2004;23(20):3962-72. Epub 2004/09/24.
20. Behrendt J, Standar K, Lindenstrauss U, Bruser T. Topological studies on the
twin-arginine translocase component TatC. FEMS microbiology letters.
2004;234(2):303-8. Epub 2004/05/12.
21. Rollauer SE, Tarry MJ, Graham JE, Jaaskelainen M, Jager F, Johnson S, et al.
Structure of the TatC core of the twin-arginine protein transport system. Nature.
2012;492(7428):210-4. Epub 2012/12/04.
22. de Leeuw E, Granjon T, Porcelli I, Alami M, Carr SB, Muller M, et al.
Oligomeric properties and signal peptide binding by Escherichia coli Tat protein
transport complexes. Journal of molecular biology. 2002;322(5):1135-46. Epub
2002/10/09.
23. Punginelli C, Maldonado B, Grahl S, Jack R, Alami M, Schroder J, et al. Cysteine
scanning mutagenesis and topological mapping of the Escherichia coli twin-arginine
translocase TatC Component. Journal of bacteriology. 2007;189(15):5482-94. Epub
2007/06/05.
24. Holzapfel E, Eisner G, Alami M, Barrett CM, Buchanan G, Luke I, et al. The
entire N-terminal half of TatC is involved in twin-arginine precursor binding.
Biochemistry. 2007;46(10):2892-8. Epub 2007/02/16.
25. Eijlander RT, Kolbusz MA, Berendsen EM, Kuipers OP. Effects of altered TatC
proteins on protein secretion efficiency via the twin-arginine translocation pathway of
Bacillus subtilis. Microbiology. 2009;155(Pt 6):1776-85. Epub 2009/04/23.
26. Gohlke U, Pullan L, McDevitt CA, Porcelli I, de Leeuw E, Palmer T, et al. The
TatA component of the twin-arginine protein transport system forms channel complexes
of variable diameter. Proceedings of the National Academy of Sciences of the United
States of America. 2005;102(30):10482-6. Epub 2005/07/20.

97
27. Sargent F, Stanley NR, Berks BC, Palmer T. Sec-independent protein
translocation in Escherichia coli. A distinct and pivotal role for the TatB protein. The
Journal of biological chemistry. 1999;274(51):36073-82. Epub 1999/12/14.
28. Goosens VJ, van Dijl JM. Twin-Arginine Protein Translocation. Current topics in
microbiology and immunology. 2017;404:69-94. Epub 2016/04/29.
29. Barrett CM, Freudl R, Robinson C. Twin arginine translocation (Tat)-dependent
export in the apparent absence of TatABC or TatA complexes using modified Escherichia
coli TatA subunits that substitute for TatB. The Journal of biological chemistry.
2007;282(50):36206-13. Epub 2007/09/21.
30. Alami M, Luke I, Deitermann S, Eisner G, Koch HG, Brunner J, et al. Differential
interactions between a twin-arginine signal peptide and its translocase in Escherichia coli.
Molecular cell. 2003;12(4):937-46. Epub 2003/10/29.
31. Barrett CM, Mangels D, Robinson C. Mutations in subunits of the Escherichia
coli twin-arginine translocase block function via differing effects on translocation activity
or tat complex structure. Journal of molecular biology. 2005;347(2):453-63. Epub
2005/03/03.
32. Lausberg F, Fleckenstein S, Kreutzenbeck P, Frobel J, Rose P, Muller M, et al.
Genetic evidence for a tight cooperation of TatB and TatC during productive recognition
of twin-arginine (Tat) signal peptides in Escherichia coli. PloS one. 2012;7(6):e39867.
Epub 2012/07/05.
33. Robinson C, Matos CF, Beck D, Ren C, Lawrence J, Vasisht N, et al. Transport
and proofreading of proteins by the twin-arginine translocation (Tat) system in bacteria.
Biochimica et biophysica acta. 2011;1808(3):876-84. Epub 2010/12/04.
34. Blummel AS, Haag LA, Eimer E, Muller M, Frobel J. Initial assembly steps of a
translocase for folded proteins. Nature communications. 2015;6:7234. Epub 2015/06/13.
35. Mori H, Cline K. A twin arginine signal peptide and the pH gradient trigger
reversible assembly of the thylakoid [Delta]pH/Tat translocase. The Journal of cell
biology. 2002;157(2):205-10. Epub 2002/04/17.
36. Dabney-Smith C, Mori H, Cline K. Oligomers of Tha4 organize at the thylakoid
Tat translocase during protein transport. The Journal of biological chemistry.
2006;281(9):5476-83. Epub 2006/01/13.
37. Beck D, Vasisht N, Baglieri J, Monteferrante CG, van Dijl JM, Robinson C, et al.
Ultrastructural characterisation of Bacillus subtilis TatA complexes suggests they are too
small to form homooligomeric translocation pores. Biochimica et biophysica acta.
2013;1833(8):1811-9. Epub 2013/04/10.
38. Walther TH, Gottselig C, Grage SL, Wolf M, Vargiu AV, Klein MJ, et al. Folding
and self-assembly of the TatA translocation pore based on a charge zipper mechanism.
Cell. 2013;152(1-2):316-26. Epub 2013/01/22.
39. Gouffi K, Gerard F, Santini CL, Wu LF. Dual topology of the Escherichia coli
TatA protein. The Journal of biological chemistry. 2004;279(12):11608-15. Epub
2004/01/01.
40. Bruser T, Sanders C. An alternative model of the twin arginine translocation
system. Microbiological research. 2003;158(1):7-17. Epub 2003/03/01.

98
41. Rodriguez F, Rouse SL, Tait CE, Harmer J, De Riso A, Timmel CR, et al.
Structural model for the protein-translocating element of the twin-arginine transport
system. Proceedings of the National Academy of Sciences of the United States of
America. 2013;110(12):E1092-101. Epub 2013/03/09.
42. Walther TH, Grage SL, Roth N, Ulrich AS. Membrane alignment of the pore-
forming component TatA(d) of the twin-arginine translocase from Bacillus subtilis
resolved by solid-state NMR spectroscopy. Journal of the American Chemical Society.
2010;132(45):15945-56. Epub 2010/10/28.
43. Vrancken K, Van Mellaert L, Anne J. Characterization of the Streptomyces
lividans PspA response. Journal of bacteriology. 2008;190(10):3475-81. Epub
2008/03/11.
44. Kobayashi R, Suzuki T, Yoshida M. Escherichia coli phage-shock protein A
(PspA) binds to membrane phospholipids and repairs proton leakage of the damaged
membranes. Molecular microbiology. 2007;66(1):100-9. Epub 2007/08/30.
45. Mehner D, Osadnik H, Lunsdorf H, Bruser T. The Tat system for membrane
translocation of folded proteins recruits the membrane-stabilizing Psp machinery in
Escherichia coli. The Journal of biological chemistry. 2012;287(33):27834-42. Epub
2012/06/13.
46. DeLisa MP, Lee P, Palmer T, Georgiou G. Phage shock protein PspA of
Escherichia coli relieves saturation of protein export via the Tat pathway. Journal of
bacteriology. 2004;186(2):366-73. Epub 2004/01/02.
47. Driessen AJ, Nouwen N. Protein translocation across the bacterial cytoplasmic
membrane. Annual review of biochemistry. 2008;77:643-67. Epub 2007/12/15.
48. Van den Berg B, Clemons WM, Jr., Collinson I, Modis Y, Hartmann E, Harrison
SC, et al. X-ray structure of a protein-conducting channel. Nature. 2004;427(6969):36-
44. Epub 2003/12/09.
49. Pohlschroder M, Prinz WA, Hartmann E, Beckwith J. Protein translocation in the
three domains of life: variations on a theme. Cell. 1997;91(5):563-6. Epub 1997/12/11.
50. Steinberg R, Knupffer L, Origi A, Asti R, Koch HG. Co-translational protein
targeting in bacteria. FEMS microbiology letters. 2018;365(11). Epub 2018/05/24.
51. Chatzi KE, Sardis MF, Economou A, Karamanou S. SecA-mediated targeting and
translocation of secretory proteins. Biochimica et biophysica acta. 2014;1843(8):1466-74.
Epub 2014/03/04.
52. Facey SJ, Kuhn A. Membrane integration of E. coli model membrane proteins.
Biochimica et biophysica acta. 2004;1694(1-3):55-66. Epub 2004/11/18.
53. du Plessis DJ, Nouwen N, Driessen AJ. The Sec translocase. Biochimica et
biophysica acta. 2011;1808(3):851-65. Epub 2010/08/31.
54. du Plessis DJ, Berrelkamp G, Nouwen N, Driessen AJ. The lateral gate of
SecYEG opens during protein translocation. The Journal of biological chemistry.
2009;284(23):15805-14. Epub 2009/04/16.
55. Hanada M, Nishiyama KI, Mizushima S, Tokuda H. Reconstitution of an efficient
protein translocation machinery comprising SecA and the three membrane proteins,
SecY, SecE, and SecG (p12). The Journal of biological chemistry. 1994;269(38):23625-
31. Epub 1994/09/23.

99
56. Park E, Rapoport TA. Preserving the membrane barrier for small molecules
during bacterial protein translocation. Nature. 2011;473(7346):239-42. Epub 2011/05/13.
57. Harris CR, Silhavy TJ. Mapping an interface of SecY (PrlA) and SecE (PrlG) by
using synthetic phenotypes and in vivo cross-linking. Journal of bacteriology.
1999;181(11):3438-44. Epub 1999/05/29.
58. Maillard AP, Lalani S, Silva F, Belin D, Duong F. Deregulation of the SecYEG
translocation channel upon removal of the plug domain. The Journal of biological
chemistry. 2007;282(2):1281-7. Epub 2006/11/10.
59. Li W, Schulman S, Boyd D, Erlandson K, Beckwith J, Rapoport TA. The plug
domain of the SecY protein stabilizes the closed state of the translocation channel and
maintains a membrane seal. Molecular cell. 2007;26(4):511-21. Epub 2007/05/29.
60. Tsukazaki T, Mori H, Fukai S, Ishitani R, Mori T, Dohmae N, et al.
Conformational transition of Sec machinery inferred from bacterial SecYE structures.
Nature. 2008;455(7215):988-91. Epub 2008/10/17.
61. Zimmer J, Nam Y, Rapoport TA. Structure of a complex of the ATPase SecA and
the protein-translocation channel. Nature. 2008;455(7215):936-43. Epub 2008/10/17.
62. Gouridis G, Karamanou S, Gelis I, Kalodimos CG, Economou A. Signal peptides
are allosteric activators of the protein translocase. Nature. 2009;462(7271):363-7. Epub
2009/11/20.
63. Emr SD, Hanley-Way S, Silhavy TJ. Suppressor mutations that restore export of a
protein with a defective signal sequence. Cell. 1981;23(1):79-88. Epub 1981/01/01.
64. Nouwen N, Driessen AJ. SecDFyajC forms a heterotetrameric complex with
YidC. Molecular microbiology. 2002;44(5):1397-405. Epub 2002/06/19.
65. Hegde RS, Kang SW. The concept of translocational regulation. The Journal of
cell biology. 2008;182(2):225-32. Epub 2008/07/23.
66. Tsukazaki T, Mori H, Echizen Y, Ishitani R, Fukai S, Tanaka T, et al. Structure
and function of a membrane component SecDF that enhances protein export. Nature.
2011;474(7350):235-8. Epub 2011/05/13.
67. Lee C, Beckwith J. Cotranslational and posttranslational protein translocation in
prokaryotic systems. Annual review of cell biology. 1986;2:315-36. Epub 1986/01/01.
68. Chatzi KE, Sardis MF, Karamanou S, Economou A. Breaking on through to the
other side: protein export through the bacterial Sec system. The Biochemical journal.
2013;449(1):25-37. Epub 2012/12/12.
69. Oh E, Becker AH, Sandikci A, Huber D, Chaba R, Gloge F, et al. Selective
ribosome profiling reveals the cotranslational chaperone action of trigger factor in vivo.
Cell. 2011;147(6):1295-308. Epub 2011/12/14.
70. Weiss JB, Ray PH, Bassford PJ, Jr. Purified secB protein of Escherichia coli
retards folding and promotes membrane translocation of the maltose-binding protein in
vitro. Proceedings of the National Academy of Sciences of the United States of America.
1988;85(23):8978-82. Epub 1988/12/01.
71. Sharma V, Arockiasamy A, Ronning DR, Savva CG, Holzenburg A, Braunstein
M, et al. Crystal structure of Mycobacterium tuberculosis SecA, a preprotein
translocating ATPase. Proceedings of the National Academy of Sciences of the United
States of America. 2003;100(5):2243-8. Epub 2003/02/28.

100
72. Yang Q, Jankowsky E. ATP- and ADP-dependent modulation of RNA unwinding
and strand annealing activities by the DEAD-box protein DED1. Biochemistry.
2005;44(41):13591-601. Epub 2005/10/12.
73. Papanikou E, Karamanou S, Baud C, Frank M, Sianidis G, Keramisanou D, et al.
Identification of the preprotein binding domain of SecA. The Journal of biological
chemistry. 2005;280(52):43209-17. Epub 2005/10/26.
74. Erlandson KJ, Miller SB, Nam Y, Osborne AR, Zimmer J, Rapoport TA. A role
for the two-helix finger of the SecA ATPase in protein translocation. Nature.
2008;455(7215):984-7. Epub 2008/10/17.
75. Breukink E, Nouwen N, van Raalte A, Mizushima S, Tommassen J, de Kruijff B.
The C terminus of SecA is involved in both lipid binding and SecB binding. The Journal
of biological chemistry. 1995;270(14):7902-7. Epub 1995/04/07.
76. Gelis I, Bonvin AM, Keramisanou D, Koukaki M, Gouridis G, Karamanou S, et
al. Structural basis for signal-sequence recognition by the translocase motor SecA as
determined by NMR. Cell. 2007;131(4):756-69. Epub 2007/11/21.
77. Chatzi KE, Sardis MF, Tsirigotaki A, Koukaki M, Sostaric N, Konijnenberg A, et
al. Preprotein mature domains contain translocase targeting signals that are essential for
secretion. The Journal of cell biology. 2017;216(5):1357-69. Epub 2017/04/14.
78. Driessen AJ. SecA, the peripheral subunit of the Escherichia coli precursor
protein translocase, is functional as a dimer. Biochemistry. 1993;32(48):13190-7. Epub
1993/12/07.
79. Jilaveanu LB, Zito CR, Oliver D. Dimeric SecA is essential for protein
translocation. Proceedings of the National Academy of Sciences of the United States of
America. 2005;102(21):7511-6. Epub 2005/05/18.
80. Huber D, Rajagopalan N, Preissler S, Rocco MA, Merz F, Kramer G, et al. SecA
interacts with ribosomes in order to facilitate posttranslational translocation in bacteria.
Molecular cell. 2011;41(3):343-53. Epub 2011/02/05.
81. Singh R, Kraft C, Jaiswal R, Sejwal K, Kasaragod VB, Kuper J, et al. Cryo-
electron microscopic structure of SecA protein bound to the 70S ribosome. The Journal
of biological chemistry. 2014;289(10):7190-9. Epub 2014/01/21.
82. Merz F, Boehringer D, Schaffitzel C, Preissler S, Hoffmann A, Maier T, et al.
Molecular mechanism and structure of Trigger Factor bound to the translating ribosome.
The EMBO journal. 2008;27(11):1622-32. Epub 2008/05/24.
83. Wu ZC, de Keyzer J, Kedrov A, Driessen AJ. Competitive binding of the SecA
ATPase and ribosomes to the SecYEG translocon. The Journal of biological chemistry.
2012;287(11):7885-95. Epub 2012/01/24.
84. Or E, Boyd D, Gon S, Beckwith J, Rapoport T. The bacterial ATPase SecA
functions as a monomer in protein translocation. The Journal of biological chemistry.
2005;280(10):9097-105. Epub 2004/12/25.
85. Gouridis G, Karamanou S, Sardis MF, Scharer MA, Capitani G, Economou A.
Quaternary dynamics of the SecA motor drive translocase catalysis. Molecular cell.
2013;52(5):655-66. Epub 2013/12/18.
86. Vassylyev DG, Mori H, Vassylyeva MN, Tsukazaki T, Kimura Y, Tahirov TH, et
al. Crystal structure of the translocation ATPase SecA from Thermus thermophilus

101
reveals a parallel, head-to-head dimer. Journal of molecular biology. 2006;364(3):248-58.
Epub 2006/10/25.
87. van der Sluis EO, Driessen AJ. Stepwise evolution of the Sec machinery in
Proteobacteria. Trends in microbiology. 2006;14(3):105-8. Epub 2006/02/24.
88. Wild J, Altman E, Yura T, Gross CA. DnaK and DnaJ heat shock proteins
participate in protein export in Escherichia coli. Genes & development. 1992;6(7):1165-
72. Epub 1992/07/01.
89. Xu Z, Knafels JD, Yoshino K. Crystal structure of the bacterial protein export
chaperone secB. Nature structural biology. 2000;7(12):1172-7. Epub 2000/12/02.
90. Fekkes P, de Wit JG, Boorsma A, Friesen RH, Driessen AJ. Zinc stabilizes the
SecB binding site of SecA. Biochemistry. 1999;38(16):5111-6. Epub 1999/04/23.
91. Crane JM, Mao C, Lilly AA, Smith VF, Suo Y, Hubbell WL, et al. Mapping of
the docking of SecA onto the chaperone SecB by site-directed spin labeling: insight into
the mechanism of ligand transfer during protein export. Journal of molecular biology.
2005;353(2):295-307. Epub 2005/09/20.
92. Crane JM, Suo Y, Lilly AA, Mao C, Hubbell WL, Randall LL. Sites of interaction
of a precursor polypeptide on the export chaperone SecB mapped by site-directed spin
labeling. Journal of molecular biology. 2006;363(1):63-74. Epub 2006/09/12.
93. Akopian D, Shen K, Zhang X, Shan SO. Signal recognition particle: an essential
protein-targeting machine. Annual review of biochemistry. 2013;82:693-721. Epub
2013/02/19.
94. Li X, Henry R, Yuan J, Cline K, Hoffman NE. A chloroplast homologue of the
signal recognition particle subunit SRP54 is involved in the posttranslational integration
of a protein into thylakoid membranes. Proceedings of the National Academy of Sciences
of the United States of America. 1995;92(9):3789-93. Epub 1995/04/25.
95. Ulbrandt ND, Newitt JA, Bernstein HD. The E. coli signal recognition particle is
required for the insertion of a subset of inner membrane proteins. Cell. 1997;88(2):187-
96. Epub 1997/01/24.
96. Facey SJ, Neugebauer SA, Krauss S, Kuhn A. The mechanosensitive channel
protein MscL is targeted by the SRP to the novel YidC membrane insertion pathway of
Escherichia coli. Journal of molecular biology. 2007;365(4):995-1004. Epub 2006/11/23.
97. Bernstein HD, Poritz MA, Strub K, Hoben PJ, Brenner S, Walter P. Model for
signal sequence recognition from amino-acid sequence of 54K subunit of signal
recognition particle. Nature. 1989;340(6233):482-6. Epub 1989/08/10.
98. Romisch K, Webb J, Herz J, Prehn S, Frank R, Vingron M, et al. Homology of
54K protein of signal-recognition particle, docking protein and two E. coli proteins with
putative GTP-binding domains. Nature. 1989;340(6233):478-82. Epub 1989/08/10.
99. Voigts-Hoffmann F, Schmitz N, Shen K, Shan SO, Ataide SF, Ban N. The
structural basis of FtsY recruitment and GTPase activation by SRP RNA. Molecular cell.
2013;52(5):643-54. Epub 2013/11/12.
100. Luirink J, ten Hagen-Jongman CM, van der Weijden CC, Oudega B, High S,
Dobberstein B, et al. An alternative protein targeting pathway in Escherichia coli: studies
on the role of FtsY. The EMBO journal. 1994;13(10):2289-96. Epub 1994/05/15.

102
101. Bernstein HD, Zopf D, Freymann DM, Walter P. Functional substitution of the
signal recognition particle 54-kDa subunit by its Escherichia coli homolog. Proceedings
of the National Academy of Sciences of the United States of America. 1993;90(11):5229-
33. Epub 1993/06/01.
102. Keenan RJ, Freymann DM, Walter P, Stroud RM. Crystal structure of the signal
sequence binding subunit of the signal recognition particle. Cell. 1998;94(2):181-91.
Epub 1998/08/08.
103. Focia PJ, Shepotinovskaya IV, Seidler JA, Freymann DM. Heterodimeric GTPase
core of the SRP targeting complex. Science. 2004;303(5656):373-7. Epub 2004/01/17.
104. Janda CY, Li J, Oubridge C, Hernandez H, Robinson CV, Nagai K. Recognition
of a signal peptide by the signal recognition particle. Nature. 2010;465(7297):507-10.
Epub 2010/04/07.
105. Hainzl T, Huang S, Merilainen G, Brannstrom K, Sauer-Eriksson AE. Structural
basis of signal-sequence recognition by the signal recognition particle. Nature structural
& molecular biology. 2011;18(3):389-91. Epub 2011/02/22.
106. Hainzl T, Huang S, Sauer-Eriksson AE. Interaction of signal-recognition particle
54 GTPase domain and signal-recognition particle RNA in the free signal-recognition
particle. Proceedings of the National Academy of Sciences of the United States of
America. 2007;104(38):14911-6. Epub 2007/09/12.
107. Bradshaw N, Walter P. The signal recognition particle (SRP) RNA links
conformational changes in the SRP to protein targeting. Molecular biology of the cell.
2007;18(7):2728-34. Epub 2007/05/18.
108. Parlitz R, Eitan A, Stjepanovic G, Bahari L, Bange G, Bibi E, et al. Escherichia
coli signal recognition particle receptor FtsY contains an essential and autonomous
membrane-binding amphipathic helix. The Journal of biological chemistry.
2007;282(44):32176-84. Epub 2007/08/30.
109. Weiche B, Burk J, Angelini S, Schiltz E, Thumfart JO, Koch HG. A cleavable N-
terminal membrane anchor is involved in membrane binding of the Escherichia coli SRP
receptor. Journal of molecular biology. 2008;377(3):761-73. Epub 2008/02/19.
110. Angelini S, Boy D, Schiltz E, Koch HG. Membrane binding of the bacterial signal
recognition particle receptor involves two distinct binding sites. The Journal of cell
biology. 2006;174(5):715-24. Epub 2006/08/23.
111. Kuhn P, Weiche B, Sturm L, Sommer E, Drepper F, Warscheid B, et al. The
bacterial SRP receptor, SecA and the ribosome use overlapping binding sites on the SecY
translocon. Traffic. 2011;12(5):563-78. Epub 2011/01/25.
112. von Loeffelholz O, Knoops K, Ariosa A, Zhang X, Karuppasamy M, Huard K, et
al. Structural basis of signal sequence surveillance and selection by the SRP-FtsY
complex. Nature structural & molecular biology. 2013;20(5):604-10. Epub 2013/04/09.
113. Peterson JH, Woolhead CA, Bernstein HD. Basic amino acids in a distinct subset
of signal peptides promote interaction with the signal recognition particle. The Journal of
biological chemistry. 2003;278(46):46155-62. Epub 2003/09/02.
114. Bornemann T, Jockel J, Rodnina MV, Wintermeyer W. Signal sequence-
independent membrane targeting of ribosomes containing short nascent peptides within

103
the exit tunnel. Nature structural & molecular biology. 2008;15(5):494-9. Epub
2008/04/09.
115. Zhang X, Rashid R, Wang K, Shan SO. Sequential checkpoints govern substrate
selection during cotranslational protein targeting. Science. 2010;328(5979):757-60. Epub
2010/05/08.
116. Halic M, Blau M, Becker T, Mielke T, Pool MR, Wild K, et al. Following the
signal sequence from ribosomal tunnel exit to signal recognition particle. Nature.
2006;444(7118):507-11. Epub 2006/11/07.
117. Buskiewicz IA, Jockel J, Rodnina MV, Wintermeyer W. Conformation of the
signal recognition particle in ribosomal targeting complexes. RNA. 2009;15(1):44-54.
Epub 2008/11/26.
118. Ataide SF, Schmitz N, Shen K, Ke A, Shan SO, Doudna JA, et al. The crystal
structure of the signal recognition particle in complex with its receptor. Science.
2011;331(6019):881-6. Epub 2011/02/19.
119. Estrozi LF, Boehringer D, Shan SO, Ban N, Schaffitzel C. Cryo-EM structure of
the E. coli translating ribosome in complex with SRP and its receptor. Nature structural &
molecular biology. 2011;18(1):88-90. Epub 2010/12/15.
120. Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane
protein topology with a hidden Markov model: application to complete genomes. Journal
of molecular biology. 2001/01/12 ed2001. p. 567-80.
121. Duong F, Wickner W. Distinct catalytic roles of the SecYE, SecG and SecDFyajC
subunits of preprotein translocase holoenzyme. The EMBO journal. 1997;16(10):2756-
68. Epub 1997/05/15.
122. Dalbey RE, Kuhn A. YidC family members are involved in the membrane
insertion, lateral integration, folding, and assembly of membrane proteins. The Journal of
cell biology. 2004;166(6):769-74. Epub 2004/09/15.
123. Samuelson JC, Chen M, Jiang F, Moller I, Wiedmann M, Kuhn A, et al. YidC
mediates membrane protein insertion in bacteria. Nature. 2000;406(6796):637-41. Epub
2000/08/19.
124. Yi L, Celebi N, Chen M, Dalbey RE. Sec/SRP requirements and energetics of
membrane insertion of subunits a, b, and c of the Escherichia coli F1F0 ATP synthase.
The Journal of biological chemistry. 2004;279(38):39260-7. Epub 2004/07/21.
125. Pross E, Soussoula L, Seitl I, Lupo D, Kuhn A. Membrane Targeting and
Insertion of the C-Tail Protein SciP. Journal of molecular biology. 2016;428(20):4218-
27. Epub 2016/09/08.
126. Peschke M, Le Goff M, Koningstein GM, Karyolaimos A, de Gier JW, van Ulsen
P, et al. SRP, FtsY, DnaK and YidC Are Required for the Biogenesis of the E. coli Tail-
Anchored Membrane Proteins DjlC and Flk. Journal of molecular biology.
2018;430(3):389-403. Epub 2017/12/17.
127. Hennon SW, Soman R, Zhu L, Dalbey RE. YidC/Alb3/Oxa1 Family of Insertases.
The Journal of biological chemistry. 2015;290(24):14866-74. Epub 2015/05/08.
128. van der Laan M, Urbanus ML, Ten Hagen-Jongman CM, Nouwen N, Oudega B,
Harms N, et al. A conserved function of YidC in the biogenesis of respiratory chain

104
complexes. Proceedings of the National Academy of Sciences of the United States of
America. 2003;100(10):5801-6. Epub 2003/05/02.
129. van der Laan M, Bechtluft P, Kol S, Nouwen N, Driessen AJ. F1F0 ATP synthase
subunit c is a substrate of the novel YidC pathway for membrane protein biogenesis. The
Journal of cell biology. 2004;165(2):213-22. Epub 2004/04/21.
130. du Plessis DJ, Nouwen N, Driessen AJ. Subunit a of cytochrome o oxidase
requires both YidC and SecYEG for membrane insertion. The Journal of biological
chemistry. 2006;281(18):12248-52. Epub 2006/03/04.
131. Price CE, Driessen AJ. YidC is involved in the biogenesis of anaerobic
respiratory complexes in the inner membrane of Escherichia coli. The Journal of
biological chemistry. 2008;283(40):26921-7. Epub 2008/07/19.
132. Ravaud S, Stjepanovic G, Wild K, Sinning I. The crystal structure of the
periplasmic domain of the Escherichia coli membrane protein insertase YidC contains a
substrate binding cleft. The Journal of biological chemistry. 2008;283(14):9350-8. Epub
2008/02/01.
133. Jiang F, Chen M, Yi L, de Gier JW, Kuhn A, Dalbey RE. Defining the regions of
Escherichia coli YidC that contribute to activity. The Journal of biological chemistry.
2003;278(49):48965-72. Epub 2003/09/25.
134. Petriman NA, Jauss B, Hufnagel A, Franz L, Sachelaru I, Drepper F, et al. The
interaction network of the YidC insertase with the SecYEG translocon, SRP and the SRP
receptor FtsY. Scientific reports. 2018;8(1):578. Epub 2018/01/14.
135. Xie K, Kiefer D, Nagler G, Dalbey RE, Kuhn A. Different regions of the
nonconserved large periplasmic domain of Escherichia coli YidC are involved in the
SecF interaction and membrane insertase activity. Biochemistry. 2006;45(44):13401-8.
Epub 2006/11/01.
136. Chiba S, Ito K. MifM monitors total YidC activities of Bacillus subtilis, including
that of YidC2, the target of regulation. Journal of bacteriology. 2015;197(1):99-107.
Epub 2014/10/15.
137. Errington J, Appleby L, Daniel RA, Goodfellow H, Partridge SR, Yudkin MD.
Structure and function of the spoIIIJ gene of Bacillus subtilis: a vegetatively expressed
gene that is essential for sigma G activity at an intermediate stage of sporulation. Journal
of general microbiology. 1992;138(12):2609-18. Epub 1992/12/01.
138. Borowska MT, Dominik PK, Anghel SA, Kossiakoff AA, Keenan RJ. A YidC-
like Protein in the Archaeal Plasma Membrane. Structure. 2015;23(9):1715-24. Epub
2015/08/11.
139. Jiang F, Yi L, Moore M, Chen M, Rohl T, Van Wijk KJ, et al. Chloroplast YidC
homolog Albino3 can functionally complement the bacterial YidC depletion strain and
promote membrane insertion of both bacterial and chloroplast thylakoid proteins. The
Journal of biological chemistry. 2002;277(22):19281-8. Epub 2002/03/14.
140. Preuss M, Ott M, Funes S, Luirink J, Herrmann JM. Evolution of mitochondrial
oxa proteins from bacterial YidC. Inherited and acquired functions of a conserved protein
insertion machinery. The Journal of biological chemistry. 2005;280(13):13004-11. Epub
2005/01/18.

105
141. van Bloois E, Koningstein G, Bauerschmitt H, Herrmann JM, Luirink J.
Saccharomyces cerevisiae Cox18 complements the essential Sec-independent function of
Escherichia coli YidC. The FEBS journal. 2007;274(21):5704-13. Epub 2007/10/10.
142. Sundberg E, Slagter JG, Fridborg I, Cleary SP, Robinson C, Coupland G.
ALBINO3, an Arabidopsis nuclear gene essential for chloroplast differentiation, encodes
a chloroplast protein that shows homology to proteins present in bacterial membranes and
yeast mitochondria. The Plant cell. 1997;9(5):717-30. Epub 1997/05/01.
143. Gerdes L, Bals T, Klostermann E, Karl M, Philippar K, Hunken M, et al. A
second thylakoid membrane-localized Alb3/OxaI/YidC homologue is involved in proper
chloroplast biogenesis in Arabidopsis thaliana. The Journal of biological chemistry.
2006;281(24):16632-42. Epub 2006/04/06.
144. Woolhead CA, Thompson SJ, Moore M, Tissier C, Mant A, Rodger A, et al.
Distinct Albino3-dependent and -independent pathways for thylakoid membrane protein
insertion. The Journal of biological chemistry. 2001;276(44):40841-6. Epub 2001/08/29.
145. Benz M, Bals T, Gugel IL, Piotrowski M, Kuhn A, Schunemann D, et al. Alb4 of
Arabidopsis promotes assembly and stabilization of a non chlorophyll-binding
photosynthetic complex, the CF1CF0-ATP synthase. Molecular plant. 2009;2(6):1410-
24. Epub 2009/12/10.
146. Falk S, Ravaud S, Koch J, Sinning I. The C terminus of the Alb3 membrane
insertase recruits cpSRP43 to the thylakoid membrane. The Journal of biological
chemistry. 2010;285(8):5954-62. Epub 2009/12/19.
147. Klostermann E, Droste Gen Helling I, Carde JP, Schunemann D. The thylakoid
membrane protein ALB3 associates with the cpSecY-translocase in Arabidopsis thaliana.
The Biochemical journal. 2002;368(Pt 3):777-81. Epub 2002/09/10.
148. Bonnefoy N, Chalvet F, Hamel P, Slonimski PP, Dujardin G. OXA1, a
Saccharomyces cerevisiae nuclear gene whose sequence is conserved from prokaryotes to
eukaryotes controls cytochrome oxidase biogenesis. Journal of molecular biology.
1994;239(2):201-12. Epub 1994/06/03.
149. Funes S, Nargang FE, Neupert W, Herrmann JM. The Oxa2 protein of
Neurospora crassa plays a critical role in the biogenesis of cytochrome oxidase and
defines a ubiquitous subbranch of the Oxa1/YidC/Alb3 protein family. Molecular biology
of the cell. 2004;15(4):1853-61. Epub 2004/02/10.
150. Ott M, Herrmann JM. Co-translational membrane insertion of mitochondrially
encoded proteins. Biochimica et biophysica acta. 2010;1803(6):767-75. Epub
2009/12/08.
151. Jia L, Dienhart M, Schramp M, McCauley M, Hell K, Stuart RA. Yeast Oxa1
interacts with mitochondrial ribosomes: the importance of the C-terminal region of Oxa1.
The EMBO journal. 2003;22(24):6438-47. Epub 2003/12/06.
152. Fiumera HL, Broadley SA, Fox TD. Translocation of mitochondrially synthesized
Cox2 domains from the matrix to the intermembrane space. Molecular and cellular
biology. 2007;27(13):4664-73. Epub 2007/04/25.
153. Chen Y, Dalbey RE. Oxa1 Superfamily: New Members Found in the ER. Trends
in biochemical sciences. 2018;43(3):151-3. Epub 2018/01/10.

106
154. Anghel SA, McGilvray PT, Hegde RS, Keenan RJ. Identification of Oxa1
Homologs Operating in the Eukaryotic Endoplasmic Reticulum. Cell reports.
2017;21(13):3708-16. Epub 2017/12/28.
155. Srivastava R, Zalisko BE, Keenan RJ, Howell SH. The GET System Inserts the
Tail-Anchored Protein, SYP72, into Endoplasmic Reticulum Membranes. Plant
physiology. 2017;173(2):1137-45. Epub 2016/12/08.
156. Guna A, Volkmar N, Christianson JC, Hegde RS. The ER membrane protein
complex is a transmembrane domain insertase. Science. 2018;359(6374):470-3. Epub
2017/12/16.
157. Shurtleff MJ, Itzhak DN, Hussmann JA, Schirle Oakdale NT, Costa EA, Jonikas
M, et al. The ER membrane protein complex interacts cotranslationally to enable
biogenesis of multipass membrane proteins. eLife. 2018;7. Epub 2018/05/29.
158. Spann D, Pross E, Chen Y, Dalbey RE, Kuhn A. Each protomer of a dimeric
YidC functions as a single membrane insertase. Scientific reports. 2018;8(1):589. Epub
2018/01/14.
159. Boy D, Koch HG. Visualization of distinct entities of the SecYEG translocon
during translocation and integration of bacterial proteins. Molecular biology of the cell.
2009;20(6):1804-15. Epub 2009/01/23.
160. Serek J, Bauer-Manz G, Struhalla G, van den Berg L, Kiefer D, Dalbey R, et al.
Escherichia coli YidC is a membrane insertase for Sec-independent proteins. The EMBO
journal. 2004;23(2):294-301. Epub 2004/01/24.
161. Aschtgen MS, Zoued A, Lloubes R, Journet L, Cascales E. The C-tail anchored
TssL subunit, an essential protein of the enteroaggregative Escherichia coli Sci-1 Type
VI secretion system, is inserted by YidC. MicrobiologyOpen. 2012;1(1):71-82. Epub
2012/09/06.
162. Dalbey RE, Kuhn A, Zhu L, Kiefer D. The membrane insertase YidC. Biochimica
et biophysica acta. 2014;1843(8):1489-96. Epub 2014/01/15.
163. Kumazaki K, Chiba S, Takemoto M, Furukawa A, Nishiyama K, Sugano Y, et al.
Structural basis of Sec-independent membrane protein insertion by YidC. Nature.
2014;509(7501):516-20. Epub 2014/04/18.
164. Kumazaki K, Kishimoto T, Furukawa A, Mori H, Tanaka Y, Dohmae N, et al.
Crystal structure of Escherichia coli YidC, a membrane protein chaperone and insertase.
Scientific reports. 2014;4:7299. Epub 2014/12/04.
165. Chen Y, Soman R, Shanmugam SK, Kuhn A, Dalbey RE. The role of the strictly
conserved positively charged residue differs among the Gram-positive, Gram-negative,
and chloroplast YidC homologs. The Journal of biological chemistry.
2014;289(51):35656-67. Epub 2014/11/02.
166. Zhu L, Wasey A, White SH, Dalbey RE. Charge composition features of model
single-span membrane proteins that determine selection of YidC and SecYEG translocase
pathways in Escherichia coli. The Journal of biological chemistry. 2013;288(11):7704-
16. Epub 2013/01/29.
167. Price CE, Driessen AJ. Conserved negative charges in the transmembrane
segments of subunit K of the NADH:ubiquinone oxidoreductase determine its

107
dependence on YidC for membrane insertion. The Journal of biological chemistry.
2010;285(6):3575-81. Epub 2009/12/05.
168. Chen Y, Capponi S, Zhu L, Gellenbeck P, Freites JA, White SH, et al. YidC
Insertase of Escherichia coli: Water Accessibility and Membrane Shaping. Structure.
2017;25(9):1403-14 e3. Epub 2017/08/29.
169. Klenner C, Yuan J, Dalbey RE, Kuhn A. The Pf3 coat protein contacts TM1 and
TM3 of YidC during membrane biogenesis. FEBS letters. 2008;582(29):3967-72. Epub
2008/11/11.
170. Neugebauer SA, Baulig A, Kuhn A, Facey SJ. Membrane protein insertion of
variant MscL proteins occurs at YidC and SecYEG of Escherichia coli. Journal of
molecular biology. 2012;417(4):375-86. Epub 2012/02/09.
171. Kedrov A, Wickles S, Crevenna AH, van der Sluis EO, Buschauer R,
Berninghausen O, et al. Structural Dynamics of the YidC:Ribosome Complex during
Membrane Protein Biogenesis. Cell reports. 2016;17(11):2943-54. Epub 2016/12/16.
172. Winterfeld S, Ernst S, Borsch M, Gerken U, Kuhn A. Real time observation of
single membrane protein insertion events by the Escherichia coli insertase YidC. PloS
one. 2013;8(3):e59023. Epub 2013/03/26.
173. Geng Y, Kedrov A, Caumanns JJ, Crevenna AH, Lamb DC, Beckmann R, et al.
Role of the Cytosolic Loop C2 and the C Terminus of YidC in Ribosome Binding and
Insertion Activity. The Journal of biological chemistry. 2015;290(28):17250-61. Epub
2015/05/30.
174. Soman R, Yuan J, Kuhn A, Dalbey RE. Polarity and charge of the periplasmic
loop determine the YidC and sec translocase requirement for the M13 procoat lep protein.
The Journal of biological chemistry. 2014;289(2):1023-32. Epub 2013/11/28.
175. Kol S, Majczak W, Heerlien R, van der Berg JP, Nouwen N, Driessen AJ. Subunit
a of the F(1)F(0) ATP synthase requires YidC and SecYEG for membrane insertion.
Journal of molecular biology. 2009;390(5):893-901. Epub 2009/06/06.
176. Celebi N, Yi L, Facey SJ, Kuhn A, Dalbey RE. Membrane biogenesis of subunit
II of cytochrome bo oxidase: contrasting requirements for insertion of N-terminal and C-
terminal domains. Journal of molecular biology. 2006;357(5):1428-36. Epub 2006/02/21.
177. Zhu L, Klenner C, Kuhn A, Dalbey RE. Both YidC and SecYEG are required for
translocation of the periplasmic loops 1 and 2 of the multispanning membrane protein
TatC. Journal of molecular biology. 2012;424(5):354-67. Epub 2012/10/13.
178. Welte T, Kudva R, Kuhn P, Sturm L, Braig D, Muller M, et al. Promiscuous
targeting of polytopic membrane proteins to SecYEG or YidC by the Escherichia coli
signal recognition particle. Molecular biology of the cell. 2012;23(3):464-79. Epub
2011/12/14.
179. Schulze RJ, Komar J, Botte M, Allen WJ, Whitehouse S, Gold VA, et al.
Membrane protein insertion and proton-motive-force-dependent secretion through the
bacterial holo-translocon SecYEG-SecDF-YajC-YidC. Proceedings of the National
Academy of Sciences of the United States of America. 2014;111(13):4844-9. Epub
2014/02/20.
180. Egea PF, Stroud RM. Lateral opening of a translocon upon entry of protein
suggests the mechanism of insertion into membranes. Proceedings of the National

108
Academy of Sciences of the United States of America. 2010;107(40):17182-7. Epub
2010/09/22.
181. Sachelaru I, Petriman NA, Kudva R, Kuhn P, Welte T, Knapp B, et al. YidC
occupies the lateral gate of the SecYEG translocon and is sequentially displaced by a
nascent membrane protein. The Journal of biological chemistry. 2013;288(23):16295-
307. Epub 2013/04/24.
182. Beck K, Eisner G, Trescher D, Dalbey RE, Brunner J, Muller M. YidC, an
assembly site for polytopic Escherichia coli membrane proteins located in immediate
proximity to the SecYE translocon and lipids. EMBO reports. 2001;2(8):709-14. Epub
2001/07/21.
183. Nagamori S, Smirnova IN, Kaback HR. Role of YidC in folding of polytopic
membrane proteins. The Journal of cell biology. 2004;165(1):53-62. Epub 2004/04/07.
184. Zhu L, Kaback HR, Dalbey RE. YidC protein, a molecular chaperone for LacY
protein folding via the SecYEG protein machinery. The Journal of biological chemistry.
2013;288(39):28180-94. Epub 2013/08/10.
185. Serdiuk T, Mari SA, Muller DJ. Pull-and-Paste of Single Transmembrane
Proteins. Nano letters. 2017;17(7):4478-88. Epub 2017/06/20.
186. Xie K, Dalbey RE. Inserting proteins into the bacterial cytoplasmic membrane
using the Sec and YidC translocases. Nature reviews Microbiology. 2008;6(3):234-44.
Epub 2008/02/05.
187. Facey SJ, Kuhn A. The sensor protein KdpD inserts into the Escherichia coli
membrane independent of the Sec translocase and YidC. European journal of
biochemistry / FEBS. 2003;270(8):1724-34. Epub 2003/04/16.
188. Altrichter S, Haase M, Loh B, Kuhn A, Leptihn S. Mechanism of the Spontaneous
and Directional Membrane Insertion of a 2-Transmembrane Ion Channel. ACS chemical
biology. 2017;12(2):380-8. Epub 2016/12/15.
189. Jomaa A, Boehringer D, Leibundgut M, Ban N. Structures of the E. coli
translating ribosome with SRP and its receptor and with the translocon. Nature
communications. 2016;7:10471. Epub 2016/01/26.
190. Economou A, Wickner W. SecA promotes preprotein translocation by undergoing
ATP-driven cycles of membrane insertion and deinsertion. Cell. 1994;78(5):835-43.
Epub 1994/09/09.
191. Wagner S, Pop OI, Haan GJ, Baars L, Koningstein G, Klepsch MM, et al.
Biogenesis of MalF and the MalFGK(2) maltose transport complex in Escherichia coli
requires YidC. The Journal of biological chemistry. 2008;283(26):17881-90. Epub
2008/05/06.
192. Yi L, Jiang F, Chen M, Cain B, Bolhuis A, Dalbey RE. YidC is strictly required
for membrane insertion of subunits a and c of the F(1)F(0)ATP synthase and SecE of the
SecYEG translocase. Biochemistry. 2003;42(35):10537-44. Epub 2003/09/03.
193. Kuhn A. Alterations in the extracellular domain of M13 procoat protein make its
membrane insertion dependent on secA and secY. European journal of biochemistry /
FEBS. 1988;177(2):267-71. Epub 1988/11/01.

109
194. Lee JI, Kuhn A, Dalbey RE. Distinct domains of an oligotopic membrane protein
are Sec-dependent and Sec-independent for membrane insertion. The Journal of
biological chemistry. 1992;267(2):938-43. Epub 1992/01/15.
195. Pradel N, Delmas J, Wu LF, Santini CL, Bonnet R. Sec- and Tat-dependent
translocation of beta-lactamases across the Escherichia coli inner membrane.
Antimicrobial agents and chemotherapy. 2009;53(1):242-8. Epub 2008/11/05.
196. Yang YB, Lian J, Tai PC. Differential translocation of protein precursors across
SecY-deficient membranes of Escherichia coli: SecY is not obligatorily required for
translocation of certain secretory proteins in vitro. Journal of bacteriology.
1997;179(23):7386-93. Epub 1997/12/11.
197. Whitley P, Zander T, Ehrmann M, Haardt M, Bremer E, von Heijne G. Sec-
independent translocation of a 100-residue periplasmic N-terminal tail in the E. coli inner
membrane protein proW. The EMBO journal. 1994;13(19):4653-61. Epub 1994/10/03.
198. Cao G, Dalbey RE. Translocation of N-terminal tails across the plasma
membrane. The EMBO journal. 1994;13(19):4662-9. Epub 1994/10/03.
199. Gray AN, Henderson-Frost JM, Boyd D, Sharafi S, Niki H, Goldberg MB.
Unbalanced charge distribution as a determinant for dependence of a subset of
Escherichia coli membrane proteins on the membrane insertase YidC. mBio. 2011;2(6).
Epub 2011/11/24.
200. Traxler B, Murphy C. Insertion of the polytopic membrane protein MalF is
dependent on the bacterial secretion machinery. The Journal of biological chemistry.
1996;271(21):12394-400. Epub 1996/05/24.
201. Matsuyama S, Akimaru J, Mizushima S. SecE-dependent overproduction of SecY
in Escherichia coli. Evidence for interaction between two components of the secretory
machinery. FEBS letters. 1990;269(1):96-100. Epub 1990/08/20.
202. Dalbey RE, Wickner W. The role of the polar, carboxyl-terminal domain of
Escherichia coli leader peptidase in its translocation across the plasma membrane. The
Journal of biological chemistry. 1986;261(29):13844-9. Epub 1986/10/15.
203. Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, et al.
Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene
expression. Cell. 2013;152(5):1173-83. Epub 2013/03/05.
204. van Bloois E, Haan GJ, de Gier JW, Oudega B, Luirink J. Distinct requirements
for translocation of the N-tail and C-tail of the Escherichia coli inner membrane protein
CyoA. The Journal of biological chemistry. 2006;281(15):10002-9. Epub 2006/02/17.
205. Mitsopoulos C, Hashemzadeh-Bonehi L, Broome-Smith JK. N-tail translocation
of mature beta-lactamase across the Escherichia coli cytoplasmic membrane. FEBS
letters. 1997;419(1):18-22. Epub 1998/01/13.
206. McMurry JL, Kendall DA. An artificial transmembrane segment directs SecA,
SecB, and electrochemical potential-dependent translocation of a long amino-terminal
tail. The Journal of biological chemistry. 1999;274(10):6776-82. Epub 1999/02/26.
207. Maniatis T, Fritsch EF, Sambrook J. Molecular cloning: a laboratory manual:
Cold spring harbor laboratory Cold Spring Harbor, NY; 1982.

110
208. Haldimann A, Wanner BL. Conditional-replication, integration, excision, and
retrieval plasmid-host systems for gene structure-function studies of bacteria. Journal of
bacteriology. 2001;183(21):6384-93. Epub 2001/10/10.
209. Sibley MH, Raleigh EA. A versatile element for gene addition in bacterial
chromosomes. Nucleic acids research. 2012;40(3):e19. Epub 2011/11/30.
210. Phillips GJ, Park SK, Huber D. High copy number plasmids compatible with
commonly used cloning vectors. BioTechniques. 2000;28(3):400-2, 4, 6 passim. Epub
2000/03/21.
211. Bellafiore S, Ferris P, Naver H, Gohre V, Rochaix JD. Loss of Albino3 leads to
the specific depletion of the light-harvesting system. The Plant cell. 2002;14(9):2303-14.
Epub 2002/09/07.
212. Moore M, Harrison MS, Peterson EC, Henry R. Chloroplast Oxa1p homolog
albino3 is required for post-translational integration of the light harvesting chlorophyll-
binding protein into thylakoid membranes. The Journal of biological chemistry.
2000;275(3):1529-32. Epub 2000/01/15.
213. Ossenbuhl F, Gohre V, Meurer J, Krieger-Liszkay A, Rochaix JD, Eichacker LA.
Efficient assembly of photosystem II in Chlamydomonas reinhardtii requires Alb3.1p, a
homolog of Arabidopsis ALBINO3. The Plant cell. 2004;16(7):1790-800. Epub
2004/06/23.
214. Bonnefoy N, Fiumera HL, Dujardin G, Fox TD. Roles of Oxa1-related inner-
membrane translocases in assembly of respiratory chain complexes. Biochimica et
biophysica acta. 2009;1793(1):60-70. Epub 2008/06/05.
215. Hell K, Herrmann JM, Pratje E, Neupert W, Stuart RA. Oxa1p, an essential
component of the N-tail protein export machinery in mitochondria. Proceedings of the
National Academy of Sciences of the United States of America. 1998;95(5):2250-5. Epub
1998/04/16.
216. van Bloois E, Nagamori S, Koningstein G, Ullers RS, Preuss M, Oudega B, et al.
The Sec-independent function of Escherichia coli YidC is evolutionary-conserved and
essential. The Journal of biological chemistry. 2005;280(13):12996-3003. Epub
2005/01/27.
217. Szyrach G, Ott M, Bonnefoy N, Neupert W, Herrmann JM. Ribosome binding to
the Oxa1 complex facilitates co-translational protein insertion in mitochondria. The
EMBO journal. 2003;22(24):6448-57. Epub 2003/12/06.
218. Keil M, Bareth B, Woellhaf MW, Peleh V, Prestele M, Rehling P, et al. Oxa1-
ribosome complexes coordinate the assembly of cytochrome C oxidase in mitochondria.
The Journal of biological chemistry. 2012;287(41):34484-93. Epub 2012/08/21.
219. Kruger V, Deckers M, Hildenbeutel M, van der Laan M, Hellmers M, Dreker C,
et al. The mitochondrial oxidase assembly protein1 (Oxa1) insertase forms a membrane
pore in lipid bilayers. The Journal of biological chemistry. 2012;287(40):33314-26. Epub
2012/07/26.
220. Wang F, Chan C, Weir NR, Denic V. The Get1/2 transmembrane complex is an
endoplasmic-reticulum membrane protein insertase. Nature. 2014;512(7515):441-4. Epub
2014/07/22.

111
221. Elson EL. Fluorescence correlation spectroscopy: past, present, future.
Biophysical journal. 2011;101(12):2855-70. Epub 2012/01/03.
222. Ernst S, Schonbauer AK, Bar G, Borsch M, Kuhn A. YidC-driven membrane
insertion of single fluorescent Pf3 coat proteins. Journal of molecular biology.
2011;412(2):165-75. Epub 2011/07/30.
223. Moore M, Goforth RL, Mori H, Henry R. Functional interaction of chloroplast
SRP/FtsY with the ALB3 translocase in thylakoids: substrate not required. The Journal of
cell biology. 2003;162(7):1245-54. Epub 2003/10/01.
224. Dunschede B, Bals T, Funke S, Schunemann D. Interaction studies between the
chloroplast signal recognition particle subunit cpSRP43 and the full-length translocase
Alb3 reveal a membrane-embedded binding region in Alb3 protein. The Journal of
biological chemistry. 2011;286(40):35187-95. Epub 2011/08/13.




















