
Structure-miscibility relationships in weakly interacting ...
Upload
khangminh22Category
view
1download
0
NUCLEAR TRANSLOCATION OF WT1-INTERACTING PROTEIN IN
RESPOSE TO PODOCYTE INJURY
by
MARIBEL RICO
Submitted in partial fulfillment of the requirements
for the degree of Doctor of Philosophy
Dissertation Adviser: John R. Sedor, M.D.
Department of Physiology and Biophysics
CASE WESTERN RESERVE UNIVERSITY
May 2005

CASE WESTERN RESERVE UNIVERSITY
SCHOOL OF GRADUATE STUDIES
We hereby approve the thesis/dissertation of
Maria Isabel Rico-Salas
Candidate for the Doctor of Phylosophy degree *.
(signed) Richard L. Eckert, Ph.D. (chair of the committee) John R. Sedor, M.D. Catheleen Carlin, Ph.D. Richard T. Miller, M.D. Robert Harvey. Ph.D. Frank Sonnichsen, Ph.D. (date) April 5th 2005 *We also certify that written approval has been obtained for any proprietary material contained therein.

i
TABLE OF CONTENTS
List of Tables iv
List of Figures v
Acknowledgements vi
Abstract vii
Chapter 1 Introduction 9
1.1 Kidney disease 9
1.1.1 The Glomerulus 10
1.2 Physiology of the Podocyte 13
1.2.1 The Podocyte Cyoskeleton 14
1.2.2 The Slit Diaphragm 15
1.2.3 Signalling at the Slit Diaphragm 17
1.2.4 Differentiation of the Podocyte 18
1.3 The Conditionally Immortalized Podocyte 20
1.4 Purpose of the Study 21
1.5 Hypothesis 22
1.6 Table and Figures 23
Chapter 2 Experimental Procedures 36
2.1 Cell lines 35
2.2 Immunoluorescence Microscopy 36
2.3 WTIP antibody generation 38
2.4 Podocyte BSA Filter Assay 38

ii
2.5 Induction of Podocyte Injury by PAN 39
2.6 Quantification of Nuclear Fluorescence 39
2.7 Isolation of Nuclear Extracts 40
2.8 Western Blot 41
2.9 Immunoprecipitation 41
2.10 Generation of Adenoviral Expression Vector 42
2.11 Statistics 42
Chapter 3 WT1-interacting protein Translocates to the Podocyte Nucleus
after PAN Injury: Translating Junction Injury into Gene Expression. 43
3.1 Introduction 43
3.2 Results 45
3.2.1 WTIP Antibody Specificity 45
3.2.2 Localization of endogenous WTIP 46
3.2.3 Co-localization of ZO-1 and WTIP 47
3.2.4 Assesment of Podocyte Function by Albumin Diffussion
Assay 48
3.2.5 Podocyte injury withPpuromycin Aminonucleoside (PAN)
Causes Cytoskeletal Reorganization and Loss of
Synaptopodin Expression 49
3.2.6 PAN Induces Translocation of WTIP and ZO-1 from Cell
Junctions to the Nucleus 50.

iii
3.2.7 Figures 53
3.3 Discussion 62
Chapter 4 Future directions 68
4.1 Introduction 68
4.2 Experimental Design 69
4.2.1 Molecular Mechanisms that Regulate WTIP Nuclear
Translocation 67
4.2.2 In Vivo Studies of WTIP localization 78
Appendix I Myosin Heavy Chain Kinase B Participates in the
Regulation of Myosin Assembly into the Cytoskeleton 92
Appendix II Dictyostelium discoideum has a single Diacylglycerol
Kinase Gene with Similarity to Mammalian Theta Isoforms 101
Bibliography 109

iv
List of Tables
Table 1. WTIP Specifically Interacts with WT1 in Yeast

v
List of Figures
Figure 1 The Bowmann Capsule
Figure 2. The Podocyte
Figure 3. The Podocyte
Figure 4. The Slit Diaphragm
Figure 5 Molecular Organization of the Slit Diaphragm
Figure 6 WTIP phylogenetic tree. Anti-WTIP antibody characterization
Figure 7 WTIP and ZO-1 co-localization. Albumin diffusion assay
Figure 8 Injury model, cytoskeletal changes and de-differentiation.
Figure 9 Nuclear Translocation, and Rbbp7 downregulation
Figure 10 WTIP sequence and structure
Figure 11 Phosphorylation map of theWTIP moiety

vi
Acknowledgements This work would not have been possible without the continuous support of
Dr. Antonio Scarpa, Dr. Richard Eckert and Dr. John Sedor. Also significant has
been the support of Dr. Thomas. T. Egelhoff, Dr. Antonio Gualberto, and Dr.
Meredith Bond. The guidance provided by the members of my thesis committee
has been invaluable. I acknowledge Dr. Cathy Carlin, Dr.Frank Soennichsen,
Dr.Gary Landreth, Dr. Tyler Miller, Dr. Robert Harvey, Dr. JP Jin and Dr. George
Dubyak for their contributions to my development as a scientist.
The staff and students of the Department of Physiology and Biophysics as
well as the community of Case Western Research University provided a unique
environment that undoubtedly shaped my professional future. I would like to
make special mention to the labor of Dean Leonore Kola, now retired, and to the
labor of Dr. Glenn Nichols, Vicepresident for Students Affairs. Also, President
Hundrert and his magnificent vision of Case Western Reserve University deserve
special mention here for the impact that his management had made and will
certainly make in my career.

vii
Nuclear Translocation of WT1-Interacting Protein in Respose to Podocyte Injury
Abstract
By
Maribel Rico
In kidney diseases the initial insult is to the glomerulus. Glomerular Injury causes
structural abnormalities that conclude in the progressive loss of glomerular
function, scarring and segmental sclerosis, which progress towards global
sclerosis, degeneration of the tubule and interstitial fibrosis. Sclerotic glomeruli
are not functional and effacement of the podocyte foot process is commonly
observed. Recent studies have shown that foot process effacement is a
consequence of a step down in the differentiated phenotype of the podocyte.
Moreover, mutations in the Wilm’s Tumor suppressor gene (wt-1), which controls
podocyte differentiation, have been identified in patients with nephritic
syndromes. Also, animal models with deletions or lower genetic doses of the wt-1
gene developed proteinuria and glomerulosclerosis. Indicating that podocyte
differentiation is critical for the function of the blood-to-urine barrier. In support
of this, expression and localization of proteins that are elements of the slit
diaphragm, a modified adherens junction, are altered in animal models of
proteinuria. However, the molecular mechanisms underlying the initial steps of
podocyte detachment and foot process effacement are obscure. We have

viii
previously identified a WT1 Interacting Protein (WTIP) that is a co-modulator of
WT1 transcriptional activity. Here, we demonstrate that WTIP localizes to the
adherens junction of podocytes and translocates to the nucleus upon injury with
PAN. The junctional protein ZO-1, a member of the slit diapragm, also
translocates to the nucleus of the podocyte because of this treatment. We observe
downregulation of the WT1-induced Rbbp7 protein. To compliment our studies
functionally, we developed an albumin diffusion assay and observed that
diffusion of albumin in this system is dependent of podocyte differentiation.
Treatment with PAN increased albumin diffusion to levels not distinguishable
from undifferentiated podocytes, while this treatment did not affect albumin
diffusion when other epithelial cell lines were used as control. We propose that
WTIP is an environmental sensor at the slit diaphragm junction which, upon
injury, translocates to the nucleus of the podocyte to repress WT1-dependent gene
expression thus driving dedifferentiation. This would promote changes in the
actin cytoskeleton that promote disassembly of the slit diaphragm, proteinuria,
glomerular hypertrophy and ultimately glomerulosclerosis and kidney failure.

9
Chapter 1
Introduction
1.1 Kidney Disease
As an osmoregulatory organ, the kidney coordinates filtration of water and
solutes from the blood stream, secretion of hormones that regulate water and
electrolyte balance, and excretion of salts and waste products. The filtration unit
of the kidney is the glomerulus, and its structure and function will be discussed
later in this chapter. Each nephron contains a single glomerulus surrounded by
Bowman’s capsule, which gives origin to the proximal tubule (Figure 1). In the
majority of kidney diseases, the initial insult is to the glomerulus. Generally, the
insult causes structural abnormalities that conclude in the progressive loss of
glomerular function, scarring and segmental sclerosis of the kidney which
progress towards global sclerosis, degeneration of the corresponding tubule and
local interstitial fibrosis1,2. Diabetes mellitus and hypertension are the two major
conditions that cause renal failure and progression in renal disease3,4. Conversely,
patients with Chronic Kidney Disease (CKD) are more likely to develop
cardiovascular disease (CVD) than non-CDK patients5. Thus understanding the
initial steps of kidney disease may also help reducing the incidence rate of other

10
diseases of epidemic magnitude.
Progression of kidney disease is the sequential loss of nephrons. This,
however, is not the result of a transfer of the disease between adjacent nephrons
but an independent degeneration of each nephron probably due to increased single
nephron glomerular filtration rate (SNGFR) upon failure of adjacent nephrons.
Alterations in glomerular hemodynamics, increased pressure and flow in the
glomerulus leading to a higher SNGFR, are thought to be a major factor in the
induction of glomerular injury. Glomerular hypertrophy, which may cause
podocyte injury, is associated with intraglomerular hypertension, setting the early
stage in the progression of renal disease.
1.1.1 The Glomerulus.
The glomerulus is a vascular epithelial organ. It consists of a tuft of
capillaries arranged in lobules, a supporting framework of extracellular matrix
and cells in centrolobular position called the mesangium to which the capillaries
are attached. A layer of podocytes, also called visceral epithelial cells, lines the
external surfaces of the capillaries by extending numerous cellular processes that
establish specialized adherens junctions between each other. These junctions are
called the Slit diaphragm. The urinary space, continuous with the proximal tubule
lumen, is the gap area between the podocyte layer and the parietal epithelium
which coats the Bowman’s capsule, a spherical bag of basement membrane that
contains the capillary tuft and its supporting and coating tissues (Figure 2)

11
The function of this intricate organ is to generate the blood-to-urine
barrier, which is considered to be composed of three major components: the
fenestrated endothelium, the glomerular basal membrane (GBM), and the slit
diaphragm (SD) (Figure 3). Classical renal physiology describes the filtration
barrier as being highly permeable to water but selective for size and charge of the
solutes. Thus, filtration occurs through extracellular space opened by the 100 nm-
diameter fenestrations in the capillary endothelium which lead into the
multilayered GBM, composed of negatively charged glycoproteins and
proteoglycans, and finally through the slit diaphragm, a specialized adherens
junction established between the cellular processes of neighboring podocytes6.
The fenestrae of the endothelium are too large to effectively sieve
macromolecules. However, the endothelial glycocalix is thought to play an
important role in size sieving and charge selectivity7. The physical-chemical
properties of the GBM fit the predictive hydraulic models of swelling porous
media, but the pressure imposed on the protein-fiber matrix by the slit diaphragm
controls the degree of GBM hydration. Moreover, though having little effect on
macromolecular sieving in vivo8, the GBM is thought to be responsible for 50-
70% of the resistance to filtrate flow9. Hence, the podocytes accelerate the drain
at the same time that imposes a size restriction due to the limiting 14-nm gap of
the modified adherens junction10
Developmentally, the glomerulus originates from the metanephric
mesenchyme. It is thought that factors controlling podocyte differentiation are
also critical for glomerular morphogenesis11. Differentiated phenotypes are seen

12
in mouse podocytes at the capillary-loop stage12 on developmental day E16. Only
differentiated podocytes express specific components of the slit diaphragm such
as nephrin, which are critical for its function. After differentiation, podocytes
dramatically reduce their rate of proliferation as shown by using the Proliferating
Cell Nuclear Antigen (PCNA)13. Therefore, the number of podocytes per
glomerulus at E16 is believed to remain fairly constant or decrease throughout
life. Hence, the pathological consequences of podocyte loss have root in the
limited number of cells found at the blood-to-urine barrier.
Podocyte loss is considered to be the major cause of glomerular
hypertrophy. Violent changes in hemodynamics or toxic agents may cause
podocyte detachment from the GBM or apoptosis. As explained above, the SD of
the podocyte layer counteracts the resistance to filtrate flow. Loss of adhesion to
other podocytes or to the GBM favors further synthesis of GBM fibers, such as
lamin, by the remaining healthy podocytes. Thus generating glomerular sclerosis
and increased resistance to filtrate flow, which may lead to glomerular
hypertrophy. Glomerulosclerosis is the result of a process whereby glomeruli
become progressively fibrosed. Fibrotic glomeruli are not functional and
remaining nephrons are forced to maintain volume and electrolyte homeostasis,
thus developing hypertrophy in structure and function. Many factors are important
in the progression of glomerular injury and scarring but increased glomerular
pressure is capable of causing injury in all three types of glomerular cells. Other
factors such as hyperlipidemia and activation of the coagulation may also
contribute to glomerular injury. The initial injury may vary in etiology.

13
Detachment of the podocytes might be potentiated by multiple metabolic and
genetic factors; microthrombosis triggers fibrosis at the GBM and loss of the
capillary loops resulting in a small number of glomerular cells. Mesangial cell
proliferation might be caused by infiltrating macrophages, which release
cytokines and growth factors and can directly induce hypertrophy and cellular
injury in the glomerulus. Glomerular injury and its manifestation, proteinuria,
may not have a single origin. All three layers (fenestrated endothelium, GBM and
Slit diaphragm) must function in concert to yield the ultrafiltrate that will be
subjected to reabsorptive and secretory processes in the tubular epithelium.
1.2 Physiology Of The Podocyte
Podocytes are highly specialized cells with a unique morphology. The cell
body, contained into the urinary space, projects long primary processes that divide
into secondary processes which further branch into foot processes (Figure 3). The
primary processes extend to the capillaries where secondary processes and foot
processes attach to the GBM and to adjacent processes. The podocyte is a
polarized cell containing an apical membrane domain, a brief but distinctive
lateral membrane domain and a basal membrane domain. The apical or luminal
domain of the podocyte displays a thick glycocalix that is rich in
sialoglycoproteins including podocalyxin and podoendin14,15, thought to be
important for maintaining cell polarity. The lateral domain is densely occupied by
lipid rafts and serves as the display platform for the molecular components of the
slit diaphragm. Podocytes proteins such as podocyn, nephrin and neph1 localize

14
to this domain. Other proteins that are components of adherens junction as well as
scaffolding proteins are also localized to this area. There is no classic
physiological function that can be irrefutably assigned to the podocyte. However,
the slit diaphragm is a unique type of adherens junction that has a critical role in
the physiology of the blood to urine barrier. The basal membrane attaches to
laminin 11 and collagen IV in the GBM via α-3β1 integrins16 and dystroglycans.
This domain is rich in focal adhesions, which are thought to be closely
coordinated with the slit diaphragm to prevent podocyte loss.
The luminal cell body contains a prominent nucleus, a well developed
golgi and endoplasmic reticulum, numerous lysosomes and many mitochondria,
indicating a high level of metabolic activity as corresponds to a cell type that is
responsible for the synthesis of most of the GBM components (Figure 4).
1.2.1 Podocyte Cytoskeleton
The cell processes, which contain almost no organelles, are supported by a
robust cytoskeleton (Figure 4). The cytoskeleton maintains communication
between the metabolic apparatus and the cell periphery through trafficking of
vesicles and macromolecular structures but also works to counteract distensive
forces in the glomerular capillary wall originated by normal hemodynamics.
Microtubules and intermediate filaments such as vimentin and desmin accumulate
in the cell body whereas the secondary and foot processes are filled with a
complex contractile apparatus. The actin cytoskeleton integrates integrin function

15
with the slit diaphragm. Parallel actin bundles arranged in a branching fashion, are
attached to the basal membrane through focal adhesions, matrix receptors and
anchoring proteins. The F-actin network could be modified to allow process
growth, branching and disassembly. The microfilament is coated with bundles of
myosin II filaments and microtubules. Other actin binding proteins such as α-
actinin-4 and synaptopodin are associated with filamentous actin in the
differentiated podocyte.
Podocyte loss starts with process detachment. In podocyte injury models
the foot processes efface from the GBM causing proteinuria. The actin bundels
rearrange in a less organized manner, distribution of actin binding proteins is
altered and expression of podocyte differentiation markers is lost.
1.2.2 The Slit Diaphragm
The slit diaphragm consists of rod-like units, connected in the center to a
linear bar, together forming a zipper-like pattern17. The foot process cytoskeleton
is in close coordination with the molecules of the slit diaphragm such that
components of the slit diaphragm are capable of directing foot process growth and
retraction. Interdigitating foot processes form a filtration slit and are connected by
a specialized adherens junction that results from the transformation of an earlier
tight junction during development. Proteins that comprise this junction are ZO-1,
Nephrin, NEPH1, Podocin, CD2AP, FAT, and P-cadherin (Figure 5).
The most influential study on the structure of the slit diaphragm has been
that of Rodewald and Karnovsky17. Their electronic micrographs showed a

16
zipper-like structure in which a central fiber, parallel to the plasma membrane,
connected by alternating perpendicular fibers. The open space between these
filaments has approximately the same dimensions of an albumin molecule. P-
cadherin and FAT1, a giant protocadherin, nephrin and Neph1 are the molecular
component of the filaments seen on electron micrographs.
Nephrin, a member of the Ig superfamily, is probably the most critical
component of the slit diaphragm. The congenital nephrotic syndrome of the Finish
type, a disease that affects 1:10,000 of the Finish population, is caused by a
mutation in the Nphs1 gene, which codifies for nephrin. In animal models,
deletions in the nphs1gene cause proteinuria and foot process effacement.
Similarly, the injection of the Mab 5-1-6, which recognizes nephrin, causes
proteinuria. Nephrin oligomers associate within lipid rafts and disruption of these
membrane microdomains with specific antibodies also causes proteinuria and
nephrin mislocalization to the apical domain18,19. Nephrin cannot be detected
during development of immature podocytes and appears only as the podocytes
become fully differentiated. However mRNA of the nphs1gene can be detected at
the s-shape body stage.
Another member of the Ig superfamily, NEPH1, is strongly expressed by
podocytes and localizes to the slit diaphragm. Disruption of neph-1 gene also
results in heavy proteinuria and foot process effacement. Like nephrin NEPH1 is
a transmembrane protein containing a single transmembrane domain and a short
cytosolic tail. Nephrin and NEPH1 form hetero-oligomers and interact with
podocin and ZO-120,21.

17
ZO-1 is a membrane associated, 225-kDa protein, localized at the
cytoplasmic face of the junction, precisely at the points of insertion of the slit
diaphragms into the lateral membrane domain22. ZO-1 is a member of the
MAGUK protein family and contains several protein-protein interaction motifs,
including SH3 domains and PDZ domains. Given its domain structure, ZO-1 is
thought to serve as a scaffolding protein, orchestrating the molecular organization
of the slit diaphragm and connecting the plasma membrane and transmembrane
proteins with actin filaments and signaling molecules that regulate the physiology
of the junction. Munich-Wistar-Froemter rats develop proteinuria spontaneously.
They have apparently normal foot processes and slit diaphragms except for
mislocalization of ZO-1. Treatment with ACE inhibitors ameliorates the
proteinuria and restores the normal localization of ZO-1.
ZO-1 appears apically during early development, at the time of the first
epithelial-to-mesenchimal transition. Later, ZO-1 migrates laterally and associates
through its first PDZ domain with nephrin, which had migrated in opposite
direction from the basal membrane domain.
Podocin is the product of the Nphs2 gene. Mutations in this gene are the
cause of autosomal recessive steroid-resistant nephrotic syndrome and may also
cause sporadic focal segmental glomerulosclerosis. Podocin is required for
nephrin signaling and serves as a scaffolding protein organizing the molecular
structure of the cytosolic multiprotein complex subjacent to the slit junction.
CD2AP was originally discovered in T-lymphocytes where it associates with the
CD2 receptor. In the kidney, CD2AP is localized exclusively in the podocyte and

18
CD2AP -/- mice die of massive peroteinuria shortly after birth23. CD2AP
associates directly with actin24, nephrin23, and podocin25.
1.2.3 Signaling At The Slit Diaphragm
Our understanding of the architecture of the slit diaphragm has advanced
greatly in the past few years. Structural proteins of the slit diaphragm that are
necessary for maintaining a suitable glomerular filter have been shown to act as
signaling molecules also. Thus, the slit diaphragm regulates complex biologic
programs in the podocyte such as cytoskeletal rearrangements, vesicle trafficking,
polarized sorting and endocytosis, cell differentiation, suppression of
proliferation, survival and mechanotransducction.
Tyrosine phosphorylation at the cytoplasmic face of the slit diaphragm had
been shown previously. These phosphorylation events seem to be tightly
regulated by a member of the src family of non-receptor tyrosine kinase family,
Fyn26. Interaction of the tyrosine-phosphorylated cytosolic tail of nephrin with
podocin facilitated nephrin signalling by stimulating the activation of a MAP
kinase module that ultimately activated the nuclear factor AP-127. AP-1 stimulates
both survival and differentiation but depending on its molecular components and
on the particular pathways that control its activity, may also trigger apoptosis or
cell division. In this regard, elements of the slit diaphragm such as nephrin,
expressed only in the differentiated podocyte, may perpetuate the differentiated
state of the podocyte by creating a positive feedback through the control of AP-1

19
activity. Podocyte injury, slit diaphragm disassembly or specific mutations may
allow regulation of AP-1 by other cellular signaling pathways.
Mutations that prevent nephrin expression do not affect ZO-1 expression
or its localization at the cell periphery but injection of the anti-nephrin Mab 5-1-6
antibody decreased ZO-1 expression dramatically suggesting that nephrin may
influence gene expression in the differentiated podocyte28. ZO-1 also interacts
with NEPH1 in a phosphorylation dependent manner, increasing tyrosine
phosphorylation of the NEPH1 cytoplasmic tail and facilitating NEPH1
signalling21. ZO-1 also binds F-actin thus contributing to the organization of the
foot processes. Likewise, CD2AP binds actin at the slit diaphragm and could
modify actin dynamics by association with the ARP2/3 complex, WASP and
CAPZ or cortactin29-31.
The slit diaphragm is not the only podocyte membrane domain where
signaling that is relevant for the physiology of the blood-to-urine barrier occurs.
Growth factors, chemokines, and integrin signaling are other major regulators of
podocyte physiology.
1.2.4 Differentiation Of The Podocyte
The importance of podocyte specification and differentiation becomes
clear upon analysis of data from patients with familial chronic kidney disease and
genetically modified mice used as laboratory models of glomerulosclerosis.
Factors that control podocyte differentiation are mutated or non-functional in
these individuals 32,33. Several transcription factor genes have been identified that

20
are necessary for podocyte specification and differentiation. Pax-2, a mammalian
homeobox gene is essential for the induction of the renal vesicle from the
metanephric mesenchyme. Podocytes maturation requires a primary mesenchymal
to epithelial transition (MTE) that allows expression of epithelial-specific genes
and a secondary epithelial to mesenchymal transition (ETM), in which PAX-2
expression diminishes and WT1 expression increases34. Podocyte differentiation
is controlled by the transcription factor WT1 but downregulation of PAX-2
appears to be a pre-requisite for this differentiation35. Other homeobox genes and
transcription factors are also involved in podocyte differentiation but WT1 is
strongly expressed in the podocyte throughout life and constitutes a podocyte-
specific marker36. Dominant mutations in WT1 are associated with the Denys-
Drash (DDS) and Frasier (FS) syndromes37,38 where glomerulosclerosis is a key
feature of the diagnosis. The transcription factor WT1 was first identified as a
tumor suppressor gene. Its genetic locus at chromosome 11p13 is a common area
of deletions. Also, duplications giving origin to the Beckwith-Wiedemann
Syndrome (BWS) have been identified in its imprinted locus situated
telomerically at 11 p1539,40.
The WT1 protein is expressed in at least 24 distinct isoforms, the main 4
isoforms result from two alternate splicing regions at exon 5 and exon 9 and all of
them are expressed and exist in a temporarily, spatially and evolutionarily stable
ratio with respect to each other6. The first alternative splicing site either includes
or excludes 17 amino acids of unknown function encoded by exon 5 and is only
expressed in mammals41. The second alternative splicing site at exon 9 includes or

21
excludes 3 amino acids, KTS in the nucleic acid binding domain. WT1 contains
four tandem C-terminal zinc finger motifs (ZF) closely related to those of the
early response gene 1 (erg1). WT1- ZF can bind either DNA or RNA depending
on the presence or absence of exon 9 (- KTS or + KTS isoforms respectively)42.
Moreover, WT1 protein has shown to be associated with either transcriptional
multiprotein complex or spliceosomes indicating that WT1 controls gene
expression both at the transcriptional and post-transcriptional levels and can elicit
either gene repression or activation43,44. However, factors that control WT1
activity remain obscure. Our group has dedicated partial effort to identify co-
regulators of WT1. Screening an adult mice kidney cDNA library with a full-
length wt-1 bait containing exon 5 and lacking the KTS insertion, we encountered
WTIP (WT1 Interacting Protein)26. The interaction with WT1 is mediated by the
LIM domain only region of WTIP and did not require the KTS insertion or the
fourth WT1-ZF as showed by two-hybrid assays using the LIM sequences of
WTIP and the DDS WT1 mutant (WT1396) (Table1)
1.3 The Conditionally Immortalized Podocyte Cell Line.
Glomerular podocytes from the immortomouse were isolated by Peter
Mundel and co-workers in 199734. These cells express a temperature sensitive T7
antigen that allows them to be propagated at 33 degrees centigrade (permissive
temperature) in their cobblestone morphology when γ-interferon is added to the
culture media. Switching of the cultured cells to 37 degrees (non-permissive

22
temperature) suppresses expression of the T7 antigen and triggers the
differentiation program. Thus, podocytes growing at non-permissive temperature
express markers of differentiated podocytes in vivo, including the actin binding
protein synaptopodin, which is strongly expressed by the conditionally
immortalized cell line. Other proteins expressed by this cell line are WT1, ZO-1,
P-cadherin, β- and γ-catenin, CD2AP35. Nephrin and podocin are only expressed
inconsistently in this cell line.
The immortalized podocyte cell line has contributed greatly to advancing
the understanding of the physiology of the podocyte. However, as occurs with
other cell types, many specific cellular properties may change during cell culture
and results obtained from cultured immortalized podocyte should be confirmed in
animal models.
1.4 Purpose of the Study
Although we anticipated finding a nuclear co-factor of WT1, WTIP is
localized in the cytosol. At the C-terminus WTIP contains a PDZ-binding domain,
three LIM domains, two SH3-binding domains followed by consensus
phosphorylation sites for important signaling molecules such as GSK3β, Casein
Kinase II, CDK2 and CDK 5 and a Nuclear Export Signal (NES) at the N-
terminal. Exogenous expression of WTIP in Cos-7 cells demonstrated that full
length WTIP localized to the cytosol whereas a truncated version lacking the NES
was exclusively nuclear. Therefore, the purpose of this study is to elucidate the

23
natural localization of endogenous WTIP in immortalized podocytes. Given its
protein domain structure, does endogenous WTIP localize at the slit diaphragm or
does it localize to the podocyte nucleus or both? Under which circumstances does
localization change? Additionally, we aim to confirm that, as in the data obtained
from Cos-7 cells exogenously expressing WTIP26, endogenous WTIP also
downregulates WT1 transcriptional activity. Additionally, we expect to make
novel observations in the structure-function of the podocyte specialized cell
junction and its control over podocyte gene expression patterns.
1.5 Hypothesis
We hypothesize that WTIP is a member of the multiprotein complex
subjacent to the specialized podocyte junction that upon podocyte injury
translocate into the nucleus to modify WT1-dependent gene expression.
1.6 Table And Figures

24
Table 1. Two-hybrid assays demonstrated that WTIP and WT1
specifically interact. This partial WTIP sequence did not self-activate or interact
with a negative control yeast GAL4 binding domain fusion protein or irrelevant
transcription factor LMX1B. From Srichiai et al. J. Biol. Chem.,279, (14), 2004.

25

26
Figure 1. The Nephron. Schematic drawing of the structure of a nephron,
the functional unit of the kidney. The afferent capillaries enter the Bowman´s
capsule (in red) to form the capillary tuft in the glomerulus. The Bowman´s
capsule contains the glomerulus and collects the blood ultrafiltrate that will be
processed into urinein the tubular structures of the nephron.

27
http://www.kdj.org.sg/healthtopics/kidneyfunction1

28
Figure 2. The Bowman’s Capsule. Histological section of a rat kidney
showing the glomerulus in the Bowman’s capsule. M: mesangium, C:capillaries,
P: podocytes, L: lumen, Pa: parietal cells of the Bowman´s capsule wall. From
http://www.meddean.luc.edu/meded/histo/Histoimages

29

30
Figure 3. The Slit Diphragm. A. Transversal electron micrograph of the
blood-to-urine barrier illustrating its organization. CL: capillary lumen, GBM:
glomerular basement membrane, FP: foot process, SD: slit diaphragm.
http://trc.ucdavis.edu/mjguinan/apc100/modules/Urinary/mammal/vasc1/vasc.html.
B. Longitudinal electron micrograph of the blood to urine barrier illustrating the
interdigitation of podocyte foot processes. The arrow indicates the area where the
specialized adherens junction is formed. FP: foot process.
http://www.pathology.vcu.edu/education/renal/lab1.b.html
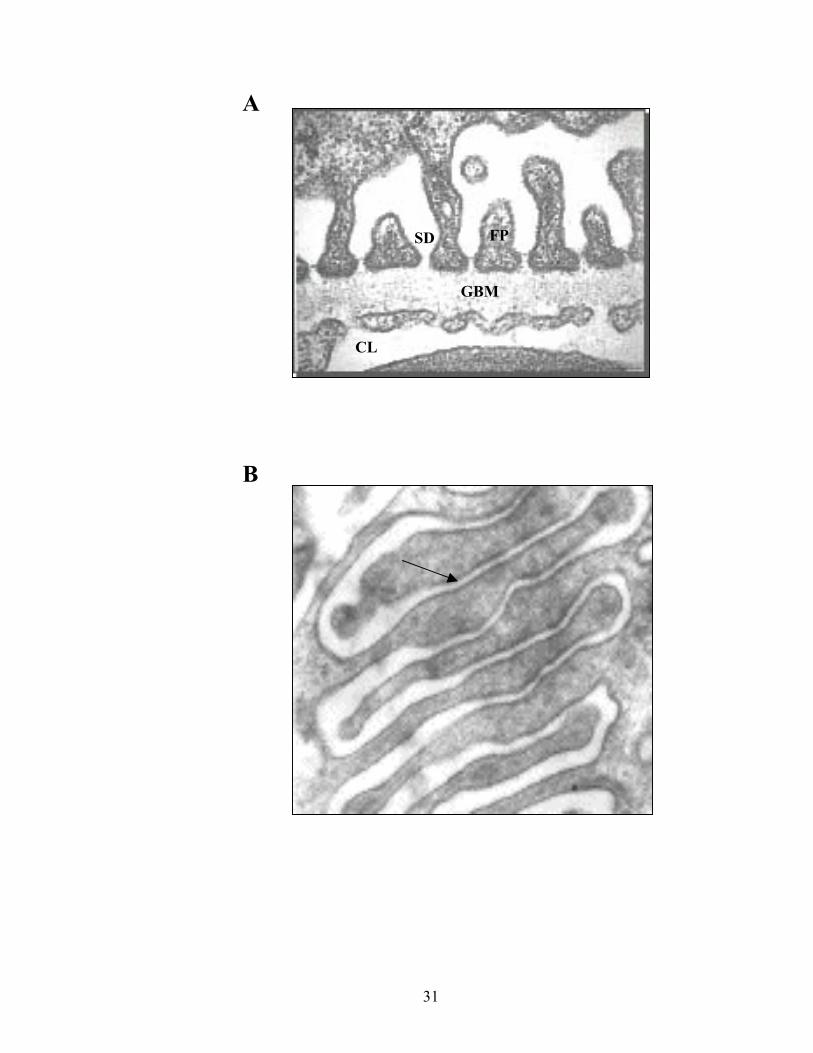
31
SD
GBM
CL
A
FP
B

32
Figure 4. The Podocyte. A. Electron micrograph of a podocyte illustraing
its primary and secondary processes or foot processes around the capillary. From
http://www.nephcure.org. B. Drawing of a histological section showing the
subcellular organization of a podocyte coating a fenestrated capillary.
FI:intermediate filaments, FiS: Filtration slits, CL: capillary lumen, Fen:
fenestrae. From http://www.bioeng.auckland.ac.nz/physiome/ontologies/urinary/cells.php

33
A
B

34
Figure 5. Molecular organization of the slit diphragm. Schematic
illustrating the molecular organization of the slit diaphragm. Two neigboring
podocytes are shown with their corresponding apical, lateral and basal membrane
domains and the particular proteins that are expressed in them. The specialized
adherens junction and its components are organized bades on the current
understanding of the slit diaphragm structure. From http://www.nature.com

35

36
Chapter 2
Materials and Methods 2.1 Cell Lines.
The conditionally immortalized podocyte cell line MPC was a generous
gift of Dr. Peter Mundel (Albert Einstein Medical College, The Bronx, NY). Cells
were maintained in RPMI-1640 medium (Cambrex. Walkersville, MD)
supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, and
100 mg/ml streptomycin. To propagate podocytes, cells were cultivated at 5%
CO2 and 33°C (permissive conditions), and culture medium was supplemented
with 10 U/ml mouse recombinant γ-interferon (Sigma Chemical. St. Louis, MO)
to enhance expression of the SV40 Large T-antigen. To induce differentiation,
podocytes were maintained on type I collagen at 5% CO2 and 37°C without γ-
interferon (non-permissive conditions) for at least 10 days. A detailed
characterization of these cells has been published previously45. All podocytes used
for differentiation expressed the podocyte-selective transcription factor, WT1.
Differentiated podocytes, as indicated by the expression of the differentiation
marker synaptopodin, between passages 10 and 25 were used in these
experiments. MDCK cells (clone 8) were a generous gift of Dr. Bingcheng Wang
(Case School of Medicine) and were maintained in 10% DMEM supplemented

37
with 10% FBS, 100 U/ml penicillin, and 100 mg/ml streptomycin at 5% CO2 and
370C. Cos-7 cells (ATCC CRL-1651; American Type Culture Collection,
Manassas, VA) were maintained at 5% CO2 and 37 °C in DMEM containing 10%
FBS; 100 U/ml penicillin, and 100 mg/ml streptomycin.
2.2 Immunofluorescense Microscopy.
Differentiated podocytes grown on collagen-coated slides were analyzed
by immunofluorescence as we have published26. Briefly, cells were fixed with 4%
paraformaldehyde for 30 minutes at room temperature and then permeabilized
with PBS containing 0.2% Triton X-100 and 2% BSA for additional 30 minutes at
room temperature. After blocking, cells were incubated with primary antibodies at
1/50 dilution in permeabilizing buffer for 1h at 37oC or overnight at 4oC. Primary
antibodies included mouse monoclonal anti-myc (Santa Cruz Biotechnology.
Santa Cruz, CA), rabbit polyclonal anti-P-cadherin (Zymed Laboratories, San
Francisco, CA), rat monoclonal anti-ZO-1 (Chemicon International. Temecula,
CA), mouse monoclonal anti-synaptopodin (Maine Biotechnology. Portland,
MA), mouse monoclonal anti-TFII-β, and both rabbit polyclonal and mouse
monoclonal anti-WT1 (Santa Cruz Biotechnology. Santa Cruz, CA). Antibody
binding was detected with anti-rabbit, anti-mouse, (Molecular Probes. Eugene,
OR), and anti-rat (Jackson Laboratories. West Groove, PA) antibodies conjugated
with either FITC or rhodamine. Antibody staining was visualized using a Nikon
epifluorescence E600 microscope, and photographs were taken with a SPOT

38
Digital System camera model 2.3.0. Confocal images were obtained with a Leica
TCS SP2 Confocal system (Leica Mycrosystems. Wetzlar, Germany). Digital
images were processed and grouped using Adobe Photoshop v 6.0 (Adobe
Systems Inc., San Jose, CA).
2.3 WTIP Antibody Generation.
A WTIP antibody was raised in rabbit using purified glutathione S-
transferase (GST)-WTIP fusion protein as described26, which contained the LIM
domain region of WTIP (N∆WTIP, aa 186-430). The crude anti-serum was
purified against GST-N∆WTIP after preclearing on a GST-only affinity column.
To eliminate residual anti-GST antibodies, the antibody was further affinity-
purified using a 6 x His-WTIP (His-WTIP) fusion protein conjugated to cyanogen
bromide (CNBr)-activated beads (Amersham Biosciences, Piscataway, NJ).
Antibody was eluted from the CNBr column by using 0.1M glycine (pH 2.4)
followed immediately by neutralization with 1M Tris base (pH 9) and
subsequently dialyzed against PBS. In some experiments, anti-WTIP specificity
was assessed by incubating the affinity purified anti-WTIP antibody with the His-
WTIP fusion protein.
2.4 Podocyte BSA Filter Assay.
Corning Transwell-Col 3 µM pore PTFE filters (Corning Inc. New York,

39
NY) were seeded with 3x105 podocytes/filter and cultured under differentiating or
permissive conditions. Cell density was monitored by cell counting previous to
filter seeding and by immunomicroscopy or fluorescent microscopy of parallel
filters stained with cell tracker. After 10-12 days, synaptopodin expression was
assayed by immunofluorescence of parallel filters. Upon confirmation of
differentiation, the cells were washed twice with PBS supplemented with 1 mM
MgCl2 and 1mM CaCl2 in order to preserve the cadherin-based junctions. The
upper compartment was then refilled with 0.5 mL 1640 RPMI and the lower
compartment with 1 mL BSA-media (1640 RPMI supplemented with 40 mg/ml
BSA) and incubated for two hours or as indicated at 37oC. Protein concentration
in the upper compartment was determined using a Bio-Rad protein assay (Bio-
Rad Laboratories, Inc., Hercules, CA). In some experiments, podocytes were
treated with PAN, as described below.
2.5 Induction of Podocyte Injury by PAN Treatment.
Podocytes differentiatied for 10 to 18 days on coverslips or Transwell
filters were incubated with 100 µg/mL PAN (Sigma Chemical. St. Louis, MO).
After 24h at 37oC, cells were analyzed by immunofluorescence microscopy or by
the BSA filter assay described above.
2.6 Quantification of Nuclear Fluorescence.

40
Differentiated podocytes, fixed on coverslips and stained with the nuclear
dye Tropo (Molecular Probes) and either of the specified antibodies, were
scanned horizontally in non-overlapping 3 µM-thick photographic slides with the
Leica TCS SP2 confocal system. Slides covering the nuclear area were projected
on a single photograph. Twenty to 50 projections of control and PAN-treated
podocytes were quantified for nuclear area fluorescence as determined by a
Region Of Interest (ROI) that corresponds to the Tropo nuclear dye signal with
Leica TCS SP2 confocal system software (Leica Mycrosystems. Wetzlar,
Germany).
2.7 Isolation of Nuclear Extracts.
Podocytes (5x104) were treated with 100 µg/mL PAN or vehicle for 24 h
at 37oC, washed with PBS and scrapped into 1ml PBS containing Protease
Inhibitor Cocktail (Sigma-Aldrich, St. Louis, MO). Nuclear protein extracts were
obtained as described46. Briefly, cells were pelleted at 250 x g at 4oC for 2 min
and immediately resuspended in 400 µL chilled Buffer A (10 mM HEPES pH 7.9;
10 mM KCl; 0.1 mM EDTA; 0.1 mM EGTA; 1 mM DTT; 0.5 mM PMSF).
Resuspended cells were incubated on ice for 15 minutes without vortexing, then
25 µL 10% NP-40 was added. This mixture was vortexed vigorously for 10 sec
and centrifuged at 1500 x g for 1 minute at 4oC. The remaining nuclear pellet was
resuspended in 50 µL ice-cold Buffer B (20 mM HEPES pH 7.9; 400 mM NaCl;
1 mM EDTA; 1 mM EGTA; 1 mM DTT; 0.5 mM PMSF), rocked vigorously at

41
4oC for 15 min and then centrifuged at 14,000 x g for 10 min at 4oC. Nuclear
proteins were separated by 4–20% SDS-PAGE, transferred to Immobilon
(Millipore Corp., Billerica, MA) membranes, and analyzed by Western blotting.
2.8 Western Blot:
Primary antibodies used in western blot only were rabbit polyclonal anti-
RbAp46 (Affinity Bioreagents. Golden, CO), rabbit polyclonal anti-myc (Santa
Cruz Biotechnology. Santa Cruz, CA) and mouse monoclonal anti-FLAG (Sigma
Chemical. St. Louis, MO). Immobilon membranes were blocked with 5% milk for
30 minutes at room temperature, then with primary antibody (1:2000 dilution in
5% milk) overnight at 4oC. Next day, membranes were washed in TBST buffer
(10 mM Tris 7.5; 150 NaCl and 0.05% Tween). Washed membranes were
incubated with protein A-horseradish peroxidase conjugate secondary antibody
(Sigma) diluted 1:5000 in 5% milk for 1 h. at room temperature and then washed
in TBST. Bound antibody was detected by chemiluminescence (Western
Lightning; PerkinElmer Life Sciences).
2.9 Immunoprecipitation Experiments.
Cells were scrapped in chilled PBS containing Protease Inhibitor
Cocktail (Sigma), pelleted down by centrifugation and resuspended in RIPA
buffer for lysis. Lysates were cleared by centrifugation and then incubated

42
overnight at 4 °C with equal amounts of antibody and 6x His-WTIP fusion
protein. Proteins antibody were precipitated from the lysates using protein A-
conjugated sepharose beads. After washing, ZO-1 was detected by
immunoblotting using polyclonal anti-ZO-1 antibody (1:1000 dilution).
2.10 Generation of Adenoviral Expression Vector
The WTIP was cloned in from with the GFP gene in pEGFP-C2 (Clontech
Laboratories, Palo Alto, CA) and then the GFP-WTIP fusion protein gene was
amplified by PCR using specific primers. The PCR product was subcloned into
pShuttle-CMV, an AdEasyTM transfer plasmids for recombinant adenovirus
construction: recombinant transfer vector was linearized and co-transformation
with pAdEasy-1 DNA into BJ5183 according to the manufacturer’s instructions.
Bacteria were selected on LB plates containing kanamycin. Plasmids were
amplified, purified (Qiagen, Valencia, CA), linearized and transfected into 293
cells (American Type Culture Collection, Manassas, VA) for viral particles
generation. The recombinant viral particles were then amplified and purified
using BD Adeno-X virus purification kit and titered. Infecting podocytes with
200-300 pfu/cell was sufficient to get uniform WTIP expression.
Statistics. Data are presented as the mean + standard error (S.E) of at least
three experiments unless otherwise specified. Statistical analysis was performed
using the Student’s T test; P<0.05 was considered to be statistically significant.

43
Chapter 3
WT1-Interacting Protein and ZO-1 Translocate into Podocyte Nuclei
after Puromycin Aminonucleoside Treatment: Translating Cell Junction
Disassembly into Altered Gene Expression
3.1 Introduction
The glomerular filtration barrier is composed of a highly fenestrated
endothelium, the glomerular basement membrane (GBM) and the podocyte. After
a tightly orchestrated differentiation program, podocytes develop foot process and
assemble a specialized adherens junction, the slit diaphragm, that mediates
contact between adjacent cells 6. In proteinuric diseases, regardless of the
etiology, podocytes undergo marked morphologic change. The actin cytoskeleton
rearranges into a cytoskeletal mat below the plasma membrane opposed to the
GBM, slit diaphragm structures are lost and the podocyte assumes a cuboidal
shape. At a molecular level in both human biopsies and experimental models, this
stereotypical morphological response is associated with changes in cytoplasmic,
plasma membrane and nuclear podocyte differentiation marker expression47. In
addition, podocytes in some glomerular diseases also revert to a less differentiated
phenotype, more characteristic of the developing rather than the fully
differentiated glomerulus. Appropriate treatment can restore normal podocyte
structure and filtration barrier function, suggesting that regulation of foot process

44
architecture is plastic and dynamic. Given its unique microenvironment with
exposure to hemodynamic forces and high ultrafiltrate flow, the podocyte must
rapidly respond to changes in physical forces or soluble signals that occur with
injury. We have hypothesized that slit diaphragm-associated proteins monitor the
podocyte microenvironment and may trigger signaling cascades that alter in
podocyte differentiation state 26.
Cell-cell junction molecules can transmit extracellular cues by shuttling
into the nucleus to regulate gene expression 48-50. Here, we describe two candidate
podocyte molecules for this important adaptive role. The Wilm’s tumor gene 1
(WT1) is a zinc finger transcription factor, whose function is required for normal
nephrogenesis 51 and podocyte differentiation 40. We previously reported that a
WT1 co-regulator, the LIM domain protein WTIP, localized to nascent cell-cell
contacts and interacted with the actin binding proteins Mena and CD2AP 26. A
truncated WTIP, containing only its LIM domains, co-localized with WT1 in
nuclei, co-precipitated with WT1, and inhibited WT1-dependent transcriptional
activation of the amphiregulin promoter. Although full-length WTIP was
excluded from cell nuclei, it accumulated in the nucleus and co-precipitated with
WT1 after the addition of an inhibitor of Crm1-mediated nuclear export,
leptomycin B. These data suggest that WTIP may function to transfer information
from the slit diaphragm microenvironment into the nucleus. After wounding, the
MAGUK family member zonula occludens-1 (ZO-1) translocates into epithelial
cell nuclei from tight junctions52 with its cognate Y-box transcription factor
ZONAB to regulate proliferation and to disrupt cell-cell contacts31,53. An

45
analogous ZO-1 function has not been described in the podocyte, where ZO-1
normally is localized to adherens junctions. In this study, we test the hypothesis
that WTIP and ZO-1, components of podocyte cell-cell junctions, translocate into
the nucleus after injury. As an in vitro model of damage, podocyte morphological
characteristics and functional response, measured in an albumin diffusion assay,
were determined after puromycin aminonucleoside (PAN) treatment. Both WTIP
and ZO-1 translocate to the nucleus after PAN treatment, a finding associated
with downregulated expression of a WT1 target gene, Rbbp7
3.2 Results
3.2.1 WTIP Antibody Specificity.
WTIP is a member of the zyxin family of LIM domain-containing proteins
that appear to have evolved from Drosphila zyxin (figure 6A). WTIP is most
closely related to a zyxin subfamily, defined by the Drosophila zyxin paralogue
CG1106341 and among these, most closely related to ajuba, which is contained in
cadherin-based cell-cell contacts and translocates into nucleus to regulate mitotic
commitment. WTIP LIM domains share significant sequence homology with
other zyxin family orthologues26, although ajuba mRNA is not detected by RT-
PCR in isolate glomeruli (A. Padiyar and J.R. Sedor, unpublished data). To test
the specificity of the affinity purified WTIP antibody, we immunoblotted lysates
from Cos-7 cells transiently expressing either the immunogen (myc-epitope

46
tagged N∆WTIP), a myc-epitope tagged full length WTIP, FLAG-epitope tagged
ajuba or myc-epitope tagged zyxin (figure 6B). The WTIP antibody detected a
single band of the appropriate size in for N∆WTIP and full length WTIP. No
cross-reactivity with ajuba or zyxin, other LIM domain proteins that also
translocate from sites of cell adhesion into the cell nucleus27,37,54 was observed.
Both differentiated and undifferentiated podocytes expressed WTIP (figure 6C).
In this case, antibody specificity was verified by competition of anti-WTIP
binding with the 6 x His-WTIP fusion protein (figure 6C). The same single band
was detected in rat glomerular lysates(not shown). To further characterize the
antibody for immunocytochemistry, we transiently transfected Cos-7 cells with
myc-WTIP and showed that the signal from our affinity purified antibody (green
channel, figure 6D) overlapped completely with that of a commercial anti-myc
antibody (red channel, figure 6D), indicating that the affinity purified anti-WTIP
antibody specifically recognizes WTIP and does not cross-react with other
cellular proteins.
3.2.2 Localization of Endogenous WTIP
Since the anti-WTIP antibody detected endogenous podocyte WTIP by
Western blotting (figure 6C), we next localized WTIP in cultured podocytes. In
undifferentiated podocytes, WTIP was detected in the nucleus as well as at the
plasma membranes (figure 6E). In differentiated podocytes, nuclear localization
of WTIP is reduced and WTIP localization is relatively more prominent at cell-

47
cell junctions (Figure 6E). Significant perinuclear WTIP expression was observed
in both differentiated and undifferentiated podocytes.
3.2.3 Co-Localization of ZO-1 And WTIPaAt Podocyte Adherens
Junctions.
Since endogenous WTIP is localized in plasma membrane and we have
hypothesized that it would be expressed at the slit diaphragm as part of a
cadherin-based, cell-cell contact, we next characterize cell-cell contacts in
differentiated podocytes. As previously reported by Mundel and co-workers, P-
cadherin is the major cadherin isoform expressed in cultured, immortalized
podocytes. E-cadherin does not localize at cell-cell junctions but is distributed
diffusely throughout cytoplasm6. ZO-1 localizes to the cytoplasmic side of
filtration slits55, where it has been shown to co-localize with P-cadherin and to
interact with Neph1, a slit diaphragm protein of the immunoglobulin superfamily
with a PDZ binding site21. Differentiating podocytes form interdigitating cell-cell
contacts, which can be visualized with Cell Tracker (figure 7A) and contain both
ZO-1 and a cadherin identified by an anti-pan-cadherin antibody. A specific anti-
P cadherin antibody confirmed that the cadherin expressed at cell-cell contacts
was P cadherin (not shown). High-resolution immunofluorescence micrographs
revealed that P-cadherin and ZO-1 are juxtaposed at most cell-cell junctions in
differentiated podocytes (figure 7B).
As reported, WTIP contains a PDZ binding domain, suggesting it may

48
associate with ZO-1 at sites of cell-cell contact. To test this hypothesis, cells were
seeded on collagen-coated permeable Transwell filters to maximize epithelial
polarity, differentiated for 8-12 days and assessed for co-localization of WTIP
with ZO-1 by confocal microscopy. WTIP and ZO-1 co-localize precisely (figure
7C), suggesting that ZO-1 may organize WTIP in a multi-protein complex of slit
diaphragm proteins. MDCK cells abundantly express ZO-1, and
immunoprecipitation assays, using a 6 x His-WTIP fusion protein, confirmed
WTIP physically associated with ZO-1 (figure 7D).
3.2.4 Assessment of Podocyte Function by Albumin Diffusion Assay.
In vivo, podocyte differentiation state is critical for establishment and
maintenance of the slit diaphragm, which functions to exclude proteins from the
ultrafiltrate. WTIP localizes to podocyte cell-cell junctions, suggesting that it may
regulate slit diaphragm function. We developed a method to measure vectorial
bovine serum albumin diffusion across podocytes, which were maintained on
permeable supports, as an assay of slit diaphragm function. Using this system,
undifferentiated podocytes permitted much greater albumin flux over time
compared to differentiated podocytes (figure 7E, left panel). At 2 hr, albumin
diffusion was significant across filters on which undifferentiated podocytes were
cultured compared to Transwell filters on which no cells were cultured (figure 7E,
right panel). In contrast, albumin transit across Transwell filters which contained
differentiated podocytes was significantly less. Podocyte slit diaphragms allow

49
significant and selective passage of plasma constituents. In contrast, MDCK cells
express tight junctions, which act as primary barrier to diffusion of solutes.
Consistent with assembly of tight junctions, virtually no albumin flux was
observed in MDCK cells after 12 hours (not shown).
3.2.5 Podocyte injury withPpuromycin Aminonucleoside (PAN) Causes
Cytoskeletal Reorganization and Loss of Synaptopodin Expression.
To test the hypothesis that WTIP is a reporter of environmental cues and
contributes to the regulation of the podocyte phenotype, we disrupted podocyte
cell junctions with PAN, which causes proteinuria in animal models56,57. We
detected podocyte morphology changes with 100 µg/ml PAN, which correlates
with local concentrations achieved with 1 mg/g body weight dose used in rat
models43,58 . Figure 8A shows that differentiated podocytes robustly express
filamentous actin, whereas podocytes treated with PAN displayed remarkable
rearrangement of actin filaments and a rounded, smaller size. These data are
consistent with a prior report that PAN promotes cytoskeletal changes in
podocytes43 . Upon closer examination of adherens junctions, actin filaments were
highly organized in control, differentiated podocytes (figure 8B, upper panel).
Conversely, after treatment with PAN, actin filaments were distributed in a less
organized, actin mat (Figure 8B, lower panel), consistent with observations of
podocytes in situ from PAN-treated animal models. Tubulin arrangement
paralleled actin patterns in control cells, but was not dramatically affected by

50
treatment with PAN (figure 8B, lower panel). Withdrawal of PAN permitted
podocytes to revert to a normal morphology (not shown), suggesting that PAN did
not stimulate irreversible cell deathfor the 24 hour treatment. PAN also led to
enhanced intercellular albumin diffusion (figure 8C, right panel). In contrast to
untreated differentiated podocytes, albumin easily diffused across Transwell
filters containing PAN-treated podocytes and filters without cells. Phalloiding
staining of parallel filters demonstrated similar cell number and distribution in
both conditions, suggesting that increased diffusion of albumin in these conditions
is not due to cell loss and supporting the role of the podocyte junction preventing
proteinuria 59. The actin-binding protein, synaptopodin, was highly expressed in
cultured, differentiated podocytes (figures 8D and 8F), in agreement with
previous reports 33. After PAN treatment, synaptopodin distribution became
punctuate (figure 8D), consistent with actin cytoskeletal rearrangement, and
expression was reduced (figure 8F). As expected, undifferentiated podocytes did
not express synaptopodin (not shown).
3.2.6 PAN Induces Translocation of WTIP and ZO-1 from Cell Junctions
to the Nucleus.
Slit diaphragm injury causes disassemblyof ZO-1 from cell junctions 39,60,
and injury to non-podocyte epithelial cells causes ZO-1 to traffic from cell
junctions to the nucleus 42,52. Similarly, PAN treatment caused ZO-1 to
disassemble from the podocyte cell-cell junctions and relocate to the nucleus

51
(figure 8A, upper panels). P-cadherin also disappeared from cell-cell contact
areas, but did not relocate to the podocyte nucleus (not shown). We previously
demonstrated that WTIP contains a nuclear export signal and shuttled between the
nucleus and cytosol, a process that was disrupted by treatment with the nuclear
export inhibitor leptomycin B 26. Consistent with data from Figures 6 and 7,
WTIP localized in perinuclear regions and at cell-cell contacts in differentiated
podocytes. However, upon PAN treatment, WTIP moved to the nucleus, similar
to the re-distribution pattern for ZO-1 (figure 9A, lower panels). Quantification
of the nuclear fluorescence signal showed that nuclear WTIP content increased
significantly after PAN treatment as did ZO-1 however, the increase in the
nuclear facto TFII-β was not statistically significant (Figure 9 B). Similar results
were observed by immunoblotting nuclear lysates derived from untreated and
PAN-treated podocytes for endogenous ZO-1 and WTIP. Podocytes transfected
with an adenoviral construct that encoded a GFP-tagged full length WTIP
confirmed this result (Figure 9C).
Our prior work in WTIP-expressing 3T3 and HeLa cells showed that
WTIP inhibited WT1-dependent transcription using an amphiregulin promoter-
luciferase reporter assay26. In the current studies, we examined the effect of PAN
on expression of the retinoblastoma binding protein Rbbp7 (RbAP46), an
endogenous podocyte protein and known WT1 target gene 61. PAN-driven
translocation of WTIP was associated with reduced expression of RbAp46 protein
(figure 9D). The data suggest that WTIP may play a pathophysiological role in the
podocyte, by translocating from cell junctions to the nucleus, where it regulates

52
WT1-dependent podocyte differentiation

53
3.2.7 Figures
Figure 6. A. Predicted phylogenetic relationship between the indicated
zyxin family, LIM domain-containing proteins. Protein sequences were analyzed
using the ClustalW algorithm with the PAM 250 residue weight table within the
Lasergene MegAlign module (DNASTAR). Human and mouse WTIP sequences
have been reported 26. GenBankTM sequences include NP 116265.1 (human
ajuba), AAF48328.2 (CG11063), NP 055055.1 (human LIM domains containing
protein 1 [LIMD1]), NP 005569.1 (human lipoma partner protein (LPP)), NP
003293.1 (human thyroid receptor-interacting protein 6 [TRIP6]), and Q15942
(human zyxin). The units at the bottom of the tree indicate the number of
substitution events. B. Cell lysates from Cos-7 cells transiently expressing a myc-
tagged full length WTIP (myc-WTIP), myc-tagged N∆WTIP (myc-N∆WTIP),
FLAG-tagged ajuba (FLAG-Ajuba) or myc-tagged zyxin (myc-zyxin) were
separated by 4–20% SDS-PAGE and incubated with either anti-myc, anti-FLAG
or affinity purified, anti-WTIP antibodies as indicated. C. Western blots
demonstrate that WTIP is an endogenous protein in lysates from both
undifferentiated (1) and differentiated (2) podocytes. D. Confocal image of a Cos-
7 cell line transiently expressing myc-WTIP. Cells on slides were fixed and
incubated with both monoclonal anti-myc antibody (red) and affinity purified,
anti-WTIP antibody (green). Signal intensity was quantified using Leica TCS SP2
confocal system software and both antibodies recognized exclusively the same
protein. Bar, 10 µm. E. Immunolocalzation of endogenous WTIP in of both

54
undifferentiated and differentiated podocytes using affinity purified anti-WTIP
antibody. In the undifferentiated podocytes, WTIP localizes in the nucleus (arrow
head) as well as in the incipient cell-cell junctions (arrows) whereas in the
differentiated podocyte nuclear localization is markedly reduced and WTIP
localizes strongly at the cell-cell junctions. Both micrographs reveal significant
perinuclear localization. Podocyte cell size increase significantly with
differentiation34. Bar, 50 µm


56
Figure 7. A. Confocal microscopy images showing two podocytes
differentiated for 15 days and stained Cell Tracker (upper panels) to show cell
morphology, anti-ZO-1 antibody (middle panels) and anti-cadherin antibody
(lower panels) that recognizes E- and P-cadherin. Whole cells are shown on the
left (Bar, 50 µm) and magnified images of cell-cell contacts on the right of each
panel (Bar, 10 µm). B. Confocal zoom image of cell-cell contact between two
podocytes differentiated for 15 days demonstrating that ZO-1 and P-cadherin are
in close spatial association but do not necessarily overlap. Bar, 10 µm. C.
Podocytes were seeded on collagen-coated Transwell filters and then analyzed by
confocal microscopy for localization of WTIP (green, panel on left) and ZO-1
(red, middle panel). Panel on left shows merged inmages and demonstrates close
association between both molecules. Bar, 50 µm. D. WTIP and ZO-1 physically
associate in a pull down assay described in Methods. E, Left panel. A
representative graph of time course BSA-diffusion across collagen-coated
Transwell filters alone or seeded with podocytes. F ( ), collagen-coated filter
only; UND ( ), collagen-coated filter seeded with 5x103 undifferentiated
podocytes; ( ) collagen-coated filter seeded with 5x103 podocytes and
differentiated for 8-12 days. Right panel. After 2 hr, BSA diffusion was
quantified in Transwell assays using collagen coated filters (F), collagen-coated
filters seeded with 5x103 undifferentiated podocytes (UND), collagen filter seeded
with 5x103 (8-12)-days-differentiated podocytes. N=5 independent experiements.
Data presented as mean + S.E.


58
Figure 8 A. Differentiated podocytes treated with vehicle (upper panels)
or with 100 µg/mL PAN for 24h (lower panels), which promoted dramatic
changes in actin rearrangements and in overall cell morphology. Bar, 50 µm. B.
Confocal, zoom image of a cell-cell contact between two podocytes differentiated
for 15 d showing actin filaments (left panel) and tubulin filaments (right panels)
arrangement in untreated (top panels) and PAN (100 µg/mL, 24h)-treated cells
(bottom panels). Bar, 10 µm.. C. Left Panel fluorescence photograph of
Phalloidin stained collagen-coated Transwell filters with control podocytes
(upper) or podocytes treated with 100 µg/mL PAN for 24 h showing similar cell
density and distribution for both conditions. Right panel. Albumin-diffusion
assay collagen-coated filters alone (F), collagen-coated filters coated seeded with
5 x 103 podocytes and differentiated for 10-days in the absence (D) or presence of
PAN (P) for 24 h. Albumin diffusion was allowed to proceed for 2 hr and protein
in the upper quantified as described in the Methods. Data presented as mean +
S.E. p<0.05 using the Student’s t test. D. Confocal image of an untreated or PAN-
treated, differentiated podocyte stained for the differentiation marker
synaptopodin. Bar, 50 µm E. Western blot for synaptopodin demonstrating that
reduced expression of the protein levels after treatment with PAN (100 µg/mL,
24h).


60
Figure 9 A. Confocal images (representative of n = 4) of podocytes
(seeded at 5x103/filter) and differentiated for 10-days. Top panels, ZO-1
distribution in control (left) and PAN (100 µg/mL, 24h, right)-treated cells.
Bottom panels, WTIP distribution in control (left) and PAN (100 µg/mL, 24h,
right)-treated cells. After PAN treatment, both proteins show translocation into
nuclei and diminished localization at cell contacts. Bar, 50 µm. B. Quantification
of nuclear fluorescence intensity of 20-50 projections of nuclei of each condition
as defined by Tropo nuclear dye (see methods for details). Both, WTIP and ZO-1
reveal significant increase in fluorescence intensity but not the nuclear
transcription factor TFIIβ. Data presented as mean + S.E. p<0.05 using the
Student’s t test C. Western blot of nuclear extracts from control and PAN-treated
podocytes. Both ZO-1 and WTIP protein are increased. In contrast, WT1
expression is constant. Nuclear extraxts from podocytes expressing GFP-tagged
WTIP confirmed the same result. C. Western blot analysis demonstrating reduced
expression of the WT1 target gene Rbbp7 (RbAP46) in total lysates of control and
PAN-treated podocytes. In contrast, WT1 levels remain constant, suggesting that
reduced Rbbp7 expression is specific and not a reflection of cytotoxicity.


62
3.3 Discussion
Differentiation of the podocyte is critical for the filtration function of the
glomerulus since only the differentiated podocyte phenotype expresses molecules
that are specific of the slit diaphragm6. The tumor suppressor gene WT1 regulates
podocyte differentiation34 and is mutated in syndromes of familial
glomerulosclerosis, suggesting WT1 dysfunction may contribute to more common
causes of nephropathy51,62,63. We previously identified WT1 Interacting Protein
(WTIP) and reported that it functions as a co-repressor of WT1 transcriptional
activity. Another WT1 interacting protein (WTAP) was identified through yeast
two-hybrid screening and localizes in nuclear spliceosomes but its function in
kidney is as yet unknown28. Although persistent expression of WT1 protein in
podocyte nuclei suggests that podocyte differentiation requires ongoing
transcription of WT1-dependent genes, none of the WT1 protein partners known
before WTIP explained how WT1 activity might be regulated in the podocyte.
WTIP contains three LIM domains that are similar to the LIM domains in zyxin,
the prototype for the LIM-domain protein family, which localizes to focal
adhesions37,38. The LIM domain is a conserved zinc finger protein-interaction
motif, and proteins containing LIM domains mediate cytoskeletal organization,
cell lineage specification, organogenesis and oncogenesis64-67. A number of LIM
domain-containing zyxin paralogues shuttle from sites of cell-cell contacts to the
nucleus and can regulate cell differentiation state 27,29,36. Our present data supports
the model predicting that WTIP functions both as a scaffold for slit diaphragm

63
proteins and as a co-repressor of WT1-transcriptional activity by shuttling from
cell-cell adhesions to nucleus after slit diaphragm injury 26. Other junctional
proteins that do not contain LIM domains also translocate to the nuclei of
epithelial or endothelial cells and bind to transcription factors including ZO-1,
PECAM-1, and β-catenin, 42,50,52,68-70. Analogously, WTIP as well as ZO-1
translocated from podocyte adherens junctions into the nuclei of PAN-treated
cells. Re-localization of these proteins associated with loss of the podocyte
differentiation marker synaptopodin, increased albumin diffusion across a
podocyte layer, and reduced protein expression of the WT1-induced gene Rbbp7,
a member of the histone modifying complexes Sin3 and NuRD71. Taken together,
this study and our published data suggest that WTIP monitors slit diaphragm
protein assembly and shuttles into the nucleus after podocyte injury, translating
environmental information into changes in the slit diaphragm structure and
altering gene expression to promote a less differentiated phenotype. Similarly,
ZO-1 has been reported previously to translocate from adherens junctions into
MDCK cells nuclei and to modify gene expression 72. Although its role in
podocyte gene expression is unknown, in the nuclei of MDCK cells ZO-1
promotes cell proliferation and reduces cell density by binding to the Y-box
transcription factor ZONAB 73. Other proteins of the slit diaphragm or members
of focal adhesion complexes may have similar behavior. Thus, the integration of
their activities would determine the severity of the phenotypic change.
Differentiated cultured podocytes develop cell-cell junctions that are
modified adherens junctions. In particular, these junctions present a podocyte

64
specific cadherin, P-cadherin, and the MAGUK-family member, ZO-1 6. WTIP
co-localizes with ZO-1 at these cell junctions and they physically interact. In vivo,
differentiation of the podocytes prevent proteinuria and mutations or
environmental agents that affect podocyte differentiation cause proteinuria 51,62.
Consistently, diffusion of albumin through a podocyte layer was inversely
proportional to podocyte differentiation in our functional assay. We used this
assay to confirm injury of cultured podocytes by PAN, an agent that causes
proteinuria in animal models30. PAN has been proposed to cause changes in
podocyte morphology by the same mechanisms as Fibroblast Growth Factor-2
(FGF-2) 44. Cells treated with PAN undertook major rearrangements of the
cytoskeleton consisting on an unorganized mesh of actin filaments at the places of
cell-cell contact and the development of a subcortical ring of F-actin. Generaly,
microtubules paralleled F-acting rearrangements. However, the differentiation
marker synaptopodin, which is a F-actin-bindin protein 33,34 adquired a punctuated
pattern typical of early stages of podocyte differentiation. Moreover, expression,
as analyzed by western blot, was markedly reduced suggesting that the podocyte
gene expression pattern had distanced from the differentiation program.
Consistently, albumin diffusion levels were comparable to undifferentiated
podocytes, matching the published data on PAN-treated animal models 1.
When WTIP is retained in the nucleus, our previous data in
overexpressing cell lines suggested that it functions as a transcriptional repressor
of WT1 activity26. We determined here that translocation of endogenous WTIP
into the nuclei of PAN-treated podocytes was associated with decreased

65
expression of the WT1-induced gene Rbbp7 (also known as RbAp46). Rbbp7 is a
retinoblastoma (Rb)-associated protein, which was reported to be upregulated in
WT1 overexpressing cell lines and is co-expressed with WT1 in developing
kidney 61. Reduction in Rbbp7 protein expression might have important
consequences for the physiology of the podocyte. Rbbp7 is a transcriptional
repressor and can inhibit the AP-1 component c-Fos 74. Typically, Ras-dependent
Erk2/p38/MAPK pathways stimulate AP-175. However, in the podocyte, Nephrin
and NEPH-1, both components of the slit diaphragm, trigger AP-1 activity via
Tec-kinase family members 20. Moreover, podocytes from normal human
glomeruli did not express any isoform of Ras in a study that revealed upregulation
of all Ras isoforms in podocytes from sclerotic glomeruli 76. Suggesting that Ras-
dependent and NEPH-1/Neprin-dependent signaling pathways may be competing
for the control of the nuclear factor AP-1. Since Rbbp7 is a member of the NuRD
and Sin3 histone-modifying complexes, and its C. elegans orthologs have been
shown to inhibit Ras-dependent signaling during worm development 77,78, we
speculate that Rbbp7 expression, enhanced by WT1, would favor slit diaphragm
control over AP-1 by down-regulating Ras-signalling pathways. Conversely,
nuclear WTIP would do the opposite by suppressing WT1-dependent expression
of Rbbp7. It is not clear to date whether Rbbp7 down-regulation of Ras-signalling
pathways involves suppression of Ras/p21 gene expression or other mechanisms.
However, down-regulation of Rbbp7 may change the composition of histone
modifying complexes in the podocyte, which may lead to a modification of the
gene expression pattern and to increased Ras protein expression. In support of

66
this, Ras-dependent signaling pathways triggered by the FGF-receptor have been
implicated in cytoskeletal changes leading to disassembly of adherens junctions,
loss of cell polarization and cell proliferation 79
In Conclusion, WTIP translocates into the nucleus after treatment with
PAN, where it represses WT1-dependent gene expression and deregulates
podocyte phenotype. This Change in localization of WTIP from its cytosolic
location may promote reorganization of the cytoskeleton, disassembly of slit
diaphragm proteins and proteinuria. We suggest that WTIP regulates podocyte
phenotype by monitoring slit diaphragm protein integrity, ultimately translating
changes in slit diaphragm structure or function into altered expression of podocyte
differentiation genes.

67
Chapter 4
Future Directions
4.1 Introduction
The study described in the previous section supports our hypothesis that
WTIP may act as an environmental reporter carrying signals from the cell
periphery into the nucleus in order to modify WT1-dependent gene expression
and provide the necessary plasticity to the cell for survival and adaptation to its
environment. From the perspective of cell physiology, this study generates a point
of disjunction; one direction leading to the analysis of the molecular mechanisms
that control WTIP translocation into the nucleus and the other to the exploration
of the WTIP/WT1/Rbbp7 interactions and their physiological consequences.
An aspect of our future studies ought to be concerned with animal models
since the podocyte exists in a unique microenvironment. Due to its exposure to
hemodynamic forces and high flow of ultrafiltrate, the podocyte must be able to
rapidly respond to changes in physical forces or soluble signals, a process that
probably requires WTIP nuclear translocation. This microenvironment is hardly
simulated in experimental conditions using cell lines. Thus, as we understand the
mechanisms by which WTIP modulates WT1 activity in cultured cells, it would
be necessary to confirm such mechanisms in animal models and their in vivo
relevance to proteinuria. It would be important to analyze three models that

68
faithfully recapitulate common causes of human kidney disease due to podocyte
injury: primary focal glomerulosclerosis, HIVAN and diabetic nephropathy.
In this chapter I discuss some of the studies that might derive directly from
my previous work, described in Chapter 3; leaving the broader project outside of
the body of my dissertation. I will not propose any studies of WTIP-WT1
interactions to define mechanism of transcriptional repression or the interacting
domains. We decided that these experiments are secondary studies to defining
aspects of the structure-function of WTIP and to confirm that full-length WTIP
shuttles in response to a physiological stimulus in vivo using animal models.
When this is established, further studies of the WTIP-WT1 interaction would then
seem warranted. We would pursue two lines of investigation. First we should
establish the domains of WTIP and WT1 that interact. Preliminary data suggest
that WTIP LIM domains are necessary and that the third and fourth zinc fingers of
WT1 are not necessary for WT1-WTIP interaction. The design of these studies
would be similar in concept to experiments presented in this chapter, Second, we
would determine the mechanism of transcriptional repression. WTIP may
compete for the WT1 coactivator CBP/p300, which binds to the first WT1 zinc
finger 80.
The following is a detail experimental plan to explore some of the
possibilities mentioned above.
4.2 Experimental Design

69
4.2.1. Molecular mechanisms that regulate WTIP nuclear translocation.
An ever increasing number of components of cell adhesion structures are
shown to be able of translocating into the nucleus27,65. Of these, only a few are
known to have a nuclear binding partner and, rarely, a physiological gene which
expression may be altered by their nuclear localization has been identified. WTIP
is a member of an adhesive structure that could be associated directly with
changes in expression of a physiologically relevant gene, Rbbp7, by association
with the Wilm’s tumor suppressor protein, WT1.
The molecular mechanisms by which WTIP modulates WT1 activity have
been the subject of previous studies from this laboratory: WTIP binds the DNA-
binding isoform of WT1 (- KTS) by its C-terminal LIM domains thereby
suppressing WT1 transcriptional activity26. However, other functional and
mechanistic aspects remain to be studied.
Our previous studies26 revealed that myc-tagged full-length WTIP
localized in the cytoplasm of Cos-7 cells, whereas a N-terminal deletion mutant
that contained only the three LIM domains and the C-terminal PDZ-binding
domain, colocalized and interacted with recombinant nuclear Flag-WT1
repressing its transcriptional activity. WTIP junctional localization and its
physical interaction with the junctional protein ZO-1 suggest a role for the PDZ-
binding domain in the cytoplasm. Nonetheless, ARVCF another cytoplasmic
protein that localizes in adhesive structures, has been reported to utilize its PDZ-

70
binding motif to be carried into the nucleus81-83. Only a weak putative nuclear
localization signal (NLS) was found in an earlier analysis of WTIP domain
organization. This NLS hasn’t been confirmed in secondary screening of WTIP
domain structure. Therefore, WTIP may conceivably be using its C-terminal
PDZ-binding domain for its nuclear translocation also. Alternatively, localizes to
similar structures as actinin-4, which also shuttles to the nucleus, suggesting it
could carry WTIP as adherens junctions reorganize. In cases, the role of putative
phosphorylation and/or association with cytosolic or junctional complexes may
become relevant for its cytosolic retention.
Initial analysis of the WTIP full protein sequence by PhosphoBase
(http://www.cbs.dtu.dk/databases/PhosphoBase) and by NetPhos 2.0
(http://www.cbs.dtu.dk/services/Netphos) revealed that WTIP contains several
consensus phosphorylation sites for various kinases involved in important
signaling pathways including PKA, PKC and GSK3β. The highest probabilities,
as well as the highest number of events, fall on a stretch of the WTIP moiety
between amino acid 100 and amino acid 150. This area corresponds with the
segment between the NES and the first SH3-binding domain, which is located N-
terminally to the LIM domains, as shown in the WTIP domain map (figure 11).
This segment is deleted in the NDWTIP mutant used in our previous studies,
which is retained into the nucleus of transfected Cos-7 cells. Nuclear retention is
likely due to deletion of the NES. However, the presence of this mutant in the
nucleus, suggests that phosphorylation may not be necessary for nuclear
translocation. Nonetheless, phosphorylation events may be important for cytosolic

71
retention, junctional localization, and/or degradation. Localization of WTIP to
specific cellular structures may be necessary for its incorporation into a molecular
machinery that may define nuclear translocation conditions for WTIP in the
podocyte and that may be absent in Cos-7 cells. On a different perspective, a
weak putative NLS for WTIP overlaps with a consensus CDK2 kinase
phosphorylation site. Both serine/threonine phosphorylation and tyrosine
phosphorylation near the NLS have been associated with activation of a weak
NLS 84-87). WTIP nuclear translocation could also occur by release of WTIP LIM
domains after enzymatic cleavage88or an intranuclear acetylation/deacetylation
event, which promotes intranuclear retention as WTIP shuttles89. Other second
order studies will confirm that NLS and NES sequences are functionally
important by site-directed mutagenesis.
We hypothesize that deleting specific domains or phosphorylation sites in
the WTIP moiety will prevent WTIP translocation into the nucleus and/or
association with WT1. In order to test this hypothesis, we will dissect the
molecular elements that contribute to the nuclear localization of WTIP. We will
organize our strategy in two categories: 1. Structure-function studies of the WTIP
moiety. 2. Studies of phosphorylation and its effects on WTIP localization. In
both categories, differentiated podocytes will be infected with recombinant WTIP
mutants and their nuclear translocation upon treatment with PAN will be
monitored.

72
Experiment 1. Deletion of the PDZ-binding domain (PBD) in the WTIP
moiety. Both mouse and human WTIP contain a PDZ-binding domain (VTEL) at
the carboxyl terminus (Figure 10), a motif that distinguishes these gene products
from other zyxin family members and potentially mediates functions specific for
WTIP. Using PCR we will generate two PBD deletion mutants: N∆WTIP∆PBD,
containing only the three LIM domains and WTIP∆PBD, containing all the N-
terminal domains (NES, SH3 and putative phosphorylation sites) and the three
LIM domains but missing the PDZ-binding domain. Other combinations of
deletion mutants may be considered at the time of the experiments.
We will digest our previously used pCMV-based vectors conatining
recombinant cDNA for full-length WTIP and N∆WTIP3 with appropriate
restriction enzimes and purify the corresponding cDNA insert. Then, 25 ng of
each cDNA will be added to a PCR mixture (dNTPs, 15 pmol of each primer,
PCR buffer, Mg2+, loading buffer and 0.5 U of Taq polymerase) and run in a
termocycler. Conditions will be determined empirically. The upstream primer (5’
primer) and downstream primer (3’ primer) will be about 20 bases-long
oligonucleotides. The 3’ primer will be designed to eliminate the VTEL amino
acids of the PDZ-binding domain. Both primers will be chosen to include or
engineer restriction digestion sites for subsequent cloning into expression vector.
PCR reactions will be run in a 1% agarose gel by electrophoresis, the product
purified using Qiagen PCR purification spin columns and sequenced for
verification.

73
Because differentiated podocytes are very sensitive to conventional
transfection protocols, the corresponding mutant cDNAs will be cloned first into a
pCMV-myc vector to engineer myc-tagged mutants and then into an adenoviral
vector (see Chapter 2 for technical details). In our previous studies (Chapter 3),
infecting podocytes with 200-300 pfu/cell was sufficient to get uniform WTIP
expression after 3 days of infection.
Podocytes, conditionally immortalized with a thermosensitive SV40
antigen transgene, cultured less than 10 days under non-permissive conditions are
considered to be differentiating podocytes and cells cultures longer than 10 to 14
days after the thermoshift are considered differentiated podocytes when they
express synaptopodin34 (see Chapter 3). The adenoviral constructs will be added
to 15-days differentiated podocyte media and analyzed at day 18th of
differentiation. On day 17th 100 mg/ml PAN or vehicle will be added to each of
the infected podocyte wells to promote nuclear translocation of the mutants,
which will be monitored by three different approaches: western blot analysis of
nuclear extracts, immunoprecipitation with WT1, and co-localization with WT1
followed by quantification of nuclear anti-myc antibody signal using confocal
microscopy. Podocytes infected with empty adenovirus will be used as control
and synaptopodin expression will be assayed for all different conditions.
Expected Results. We expect that deletion of the PBD will cause WTIP to
mislocalize to diverse cytosolic structures. We will probably detect dramatic
reduction of WTIP in cell-cell junctions and increased perinuclear signal. Perhaps

74
the absence of the PBD may intensify the role of the LIM domains allowing
WTIP association with new cytosolic proteins or with the actin cytoskeleton and
structures like focal adhesions where other LIM-domain proteins, like zyxin,
localize. Upon treatment with PAN we expect to detect a significant reduction in
the amount of WTIP translocated to the nucleus and no quantitative changes in
the expression of Rbbp7 protein.
Experiment 2. Engineering point mutations in putative GSK3β
phosphorylation sites. First, we will verify phosphorylation in the WTIP moiety
by western blot. Total cell lysates of untreated and PAN-treated podocytes will be
analyzed using specific anti-phospho amino acid (serine, threonine and tyrosine)
antibodies. Then, we will use the information obtained from the probabilistic
analysis of phosphorylation sites by Phosphobase and Netphos 2.0 to create serine
to alanine mutations in serine 104 and serine 146. These amino acids showed a
probability of phosphorylation close to 1 in both programs and sit on a GSK3β
consensus sequence. GSK3β role in β-catenin stabilization and degradation is
well understood and may serve as a guide for our initial steps in trying to
elucidate the role of phosphorylation in WTIP function. For the engineering of the
point mutations we will use the technique of mutagenesis by megaprimer, which
is a simple and versatile method requiring three primers and two rounds of PCR.
One primer introduces the specific mutation, while the other two primers flank the
region of interest, and can be used in the generation of a set of mutants. The first
round of PCR is performed using the mutant primer, which in our experiments

75
will introduce either a serine to aspartic acid mutation, or a serine to alanine
mutation and one of the flanking primers. The double-stranded product will be
purified as explained in experiment 1 and used as a "megaprimer" in the second
round along with the primer flanking the region on the other end. The wild type
DNA is used as template in both PCRs. Once synthesized, the mutant cDNAs
will be purified and subcloned into pCMV-myc and processed for incorporation
into the adenoviral vector as explained earlier (chapter 2 and experiment 1).
Subsequently, we will infect immortalized podocytes with WTIP mutant and wild
type species using the experimental approaches described in experiment 1. We
will analyze the cellular localization of these mutants using immunocytochemical
techniques and immunomicroscopy (see chapter 2) Then we will injure infected
cells with PAN and monitor the nuclear translocation of the mutants following the
strategy decribed in experiment 1 of this section.
Expected results: We expect that PAN injury will cause a detectable
quantitative variation of the overall phosphorylation of endogenous WTIP.
However, changes in WTIP phosphorylation may be qualitative only and
therefore not detectable in our first approach. Notably, GSK3β is implicated in the
regulation of β−catenin nuclear translocation by targeting it for degradation90.
When GSK3β phosphorylation of β-catenin is blocked, β-catenin is stabilized and
translocates into the cell nucleus to modify gene expression. We expect that this
same mechanism might be used by the podocyte for regulating WTIP junctional-
nuclear translocation. Therefore, the unphosphorylatable mutants may show
increased nuclear translocation, while mutants containing serine to aspartic acid

76
mutations, which mimics the negative charge of the phosphate group, would be
expected to join proteolytic pathways and not be found in the nucleus. Secondary
experiments using proteosome inhibitors may be considered in order to verify this
hypothesis.
Preliminary experiments using infection techniques in the laboratory,
have demonstrated that, in undifferentiated podocytes, WTIP localizes first to
focal adhesions, as suggested by observations from immunomicroscopy
micrographs where WTIP co-localizes with vimentin, and later to cell-cell
junctions. Indicating that WTIP association with ZO-1 is not uniquely dependent
on the presence of the PDZ-binding domain. It is possible that a phosphorylation
event may be required previously to binding to ZO-1, and that protein kinases
associated with focal adhesion structures may fulfill this function. Therefore we
may observe accumulation of unphosphorylatable WTIP mutants in focal
adhesions.
Despite a growing knowledge of many phosphorylation consensus
sequences, this post-translational modification cannot usually be predicted
accurately from the protein sequence alone. Thus, the experimental determination
of sites of phosphorylation is an important task. Various methods for protein
phosphorylation site determination have been developed through the years.
Conventional methods involving the analysis of 32P labeled phosphoproteins by
Edman degradation and 2D phosphopeptide mapping have been useful but due to
technical limitations, including the inconveniences of working with radioactivity,
mass spectrometry eventually became the consensually favored method for

77
determination of phosphorylation sites91. Phosphorylation analysis by mass
spectrometry is based on two main steps: first, the phosphoprotein of interest is
digested with trypsin, and the tryptic peptides are analyzed to determine which are
phosphorylated. Then those phosphopeptides are further analyzed, usually by
tandem mass spectrometry (MS/MS) to determine the precise location of the
phosphorylation site. Comparison of the expected sizes with the actual values
would reveal mass increases of 80 Da (the added mass of the phosphate group) for
the phosphorylated residues. We will proceed with the mass spectrometric
analysis after our molecular biology approach in order to confirm the
GSK3β phosphorylation sites in the WTIP moiety.
4.2.2. In vivo studies of WTIP localization.
By northern analysis, WTIP is a 2.2 kb and a 1.8 to 2.0 kb transcript in
human and mouse kidney, respectively3. Species differences in transcript size
reflect differences in the 3’-UTR sequence. Northern blots and RT-PCR showed
cultured murine podocytes contain WTIP mRNA, a finding confirmed by in situ
hybridization. WTIP is most robustly expressed during mouse nephrogenesis in a
temporal pattern similar to WT1. Peak WTIP mRNA expression is at E16-17 and
persists into adulthood. In E15 kidneys, WTIP transcripts can be localized using
in situ hybridization to S-shaped bodies and the podocytes of developing
glomeruli, which stain for WT1. In summary, our previous studies have shown
that the tissue expression pattern of WTIP and WT1 during development is

78
similar, and that WTIP is expressed in podocytes. Since in vitro WTIP inhibits
WT1 function3, we did not expect WTIP to parallel WT1 expression during
nephrogenesis. We believe WTIP is excluded from podocyte nuclei in intact cells,
and these data suggest that WTIP is necessary for normal podocyte
differentiation, a process requiring adherens junction assembly.
Loss of WTIP from its cytosolic location promotes actin rearrangement
characteristic of foot process effacement and its nuclear translocation promotes
podocyte phenotype dysregulation by inhibiting WT1-dependent transcription
(Chapter 3). In these studies, we will determine whether WTIP protein
localization changes during glomerulosclerosis in vivo and if it translocates to the
podocyte nucleus upon injury. Both genetic and animal model evidence
demonstrate that, in some diseases, podocyte dysfunction initiates and/or
propagates glomerular scarring. We have chosen to analyze three models that
faithfully recapitulate common causes of human kidney disease due to podocyte
injury- primary focal glomerulosclerosis (FSGS), HIVAN and diabetic
nephropathy. First, infusion of the podocyte toxin adriamycin in BALB/c mice
causes proteinuria, glomerulosclerosis and renal insufficiency similar to
progressive human FSGS92,93 . Second, we will characterize changes in WTIP
protein localization in a mouse model of diabetic nephropathy, the C57BL/KsJ-
db/db. Both human and animal studies suggest that podocyte injury occurs early
in the course of diabetic nephropathy94,95. The C57BL/KsJ-db/db strain develops
many pathologic features of human diabetic nephropathy96,97. Finally HIV-
associated nephropathy (HIVAN) is characterized by a unique histologic lesion,

79
collapsing sclerosis98, and podocyte phenotype99. Given the evidence that the
podocyte is a renal HIV reservoir100 and the diminished expression of WT1 in
human and murine HIVAN98,99, we feel characterization of WTIP is warranted in
this model.
Animal models: Adriamycin nephrotoxicity. To induce FSGS, BALB/c
mice (Jackson Labs; 6-8 weeks old, 20-25 g body weight) will receive a single tail
vein injection of adriamycin (Sigma, 10 mg/kg) or vehicle (1% DMSO) as
published93 and then will be sacrificed on day 1 and after 2 and 6 weeks for the
functional, WTIP mRNA expression and histological analyses indicated below.
Adriamycin-induced FSGS requires an appropriate genetic background, and
several groups have demonstrated that BALB/c animals develop a stable and
reproducible murine model of FSGS, which is characterized by proteinuria,
reduced GFR, glomerulosclerosis and tubulointerstitial infiltrates93. Animals are
sacrificed on day 1 to evaluate acute effects of adriamycin, at the end of week 2
when proteinuria is peaking and at week 6 when glomerular scarring has
developed in approximately 20% of glomeruli. In the initial studies, 8 to 10
adriamycin- and 8 to 10 DMSO-injected mice will be studied at each time point.
Diabetes. The renal phenotype of the db/db mouse closely resembles
human diabetic nephropathy from type-2 diabetes, the most common cause of
ESRD. Db/db mice have a G/T point mutation in the leptin receptor, which
generates a premature stop codon in the cytosolic domain of the leptin receptor97.
C57BL/KsJ-db/db mice develop early onset (6-8 weeks of age) obesity,
hyperinsulinemia and hyperglycemia, followed by pancreatic islet cell atrophy

80
and hypoinsulinemia, weight loss and death from renal failure at 6-10 months.
The C57BL/KsJ-db/db strain develops many pathologic features of diabetic
nephropathy, including early (8-10 weeks of age) mesangial matrix expansion and
hyperfiltration, and subsequent (16 weeks and beyond) diffuse and nodular
glomerulosclerosis, glomerular basement membrane thickening, proteinuria and
progressive renal insufficiency96. Eight db/db and db/m heterozygotes
(heterozygote control, identical genetic background, without diabetes or diabetic
nephropathy phenotype) will be purchased from Jackson Labs and evaluated at 9
and 18 weeks using the phenotyping and molecular assays described below. These
mice do not need insulin therapy.
HIVAN. The transgenic mouse line TgN(pNL43d14)26Lom to be used in
these studies contains ten copies of the HIV-1 proviral DNA pNL4-3d1443,
generated by deleting the gag and pol genes from the infectious molecular clone
NL4-3101. Bruggeman and Barisoni have previously demonstrated the
homozygous and heterozygous mice develop multi-organ pathology, including a
progressive renal disease clinically and pathologically similar to HIVAN99,102. Dr.
Bruggeman maintains this colony, which was obtained from the original NIH
colony and has been maintained by inbreeding (which has increased penetrance of
the renal disease to 100% of adult mice102 in the animal facility in the
Rammelkamp Center. These mice are housed in microisolator cages within a
pathogen-defined, mouse hepatitis virus-free facility one floor above our
laboratories. Animals are fed standard chow (Purina), allowed free access to
water, and cycled for 14 hr daylight and 10 hr darkness. Mice will be observed

81
daily for signs of illness. Since mice homozygous for the transgene rarely survive
to weaning, heterozygous mice will be used in these studies. Although
heterozygotes can be distinguished from normal littermates by the appearance of
cataracts (Bruggeman, personal communication), transgene genotypes are
determined by Southern analysis of tail DNA as previously described103. Initially,
eight heterozygotes and eight wild type mice will be analyzed at age 6 to 8 weeks
using the phenotyping and molecular assays described below, when the
heterozygotes are proteinuric and azotemic99,103.
Experimental design: When albuminuria is detected, animals will be
sacrificed immediately and at 2 weekly intervals. At the time of sacrifice, blood
will be collected by retro-orbital puncture for determination of serum creatinine
and the kidneys, heart, and lungs (tissues in which both WTIP and WT1 are
coexpressed) are harvested and processed for light, immunohistochemical and
ultrastructural evaluations. We will initially focus on kidney pathology. PAS-
stained 4 µm sections will be assessed qualitatively for segmental scars and
increases in GBM thickness. If qualitative changes are not detected, quantitative
assessment will be undertaken with the assistance of the Imaging Core.
Specifically, mean foot-process width and mean slit-diaphragm width104will be
measured on transmission electron micrographs by blinded observers from three
animals at each time point. Expression and localization of WTIP, nephrin,
podocin, CD2AP and synaptopodin will be determined by immunohistochemistry
and quantification of fluorescence (Chapter 2). Future analyses would assay slit
diaphragm protein-protein interactions using isolated glomeruli25 and determine if

82
the fraction of nephrin, which is F-actin-associated, is decreased105.
Expected results. We expect to be able to confirm our cellular data
(Chapter 3) in all or some of these in vivo models. In control animals, we expect
to find glomerular expression of WTIP, but not tubular. We expect that this
expression be specific for podocytes as indicated by co-localization with
synaptopodin. We don’t expect to see tubular or mesangial expression of WTIP.
In injured animals, or amimals that have developed proteinuria and
glomerulosclerosis we expect to find WTIP signal apparently reduced, due to
relocalization. Electron microscopy studies will be needed to confirm
delocalization of WTIP from the slit diaphragm and its increased localization in
the podocyte nucleus. O We also expect to see reduction in the expression of
WT1 dependent genes like podocalyxin. A WTIP -/- mice will confirm that this
reduction in podocalyxin expression is a consequence of WTIP/WT1 interaction

83
Figure 10. WTIP sequence and structure
A, alignment of mouse and human WTIP LIM domains with related LIM domain-
containing proteins was generated using the ClustalW algorithm with the PAM
250 residue weight table within the Lasergene MegAlign module (DNASTAR).
Zinc-coordinating residues are boxed in pink (LIM domain 1), yellow (LIM
domain 2), and blue (LIM domain 3). Human and mouse WTIP contain a
conserved five-amino acid insertion (underlined, (F/Y)SGFQ). The PDZ domain
binding sequence in WTIP is indicated in red. Fully and strongly conserved
residues are indicated in boldface type; weakly conserved groups are in boldface
and italic type. GenBankTM sequences include NP 116265.1 (human ajuba),
AAF48328 [GenBank] 2 (CG11063), NP 055055.1 (human LIMD1), NP
005569.1 (human lipoma partner protein (LPP)), NP 003293.1 (human thyroid
receptor-interacting protein 6), and Q15942 [GenBank] (human zyxin). B,
predicted domain structure for WTIP. The position of a putative nuclear export
sequence (NES) is shown in the blue box. Alignment of the WTIP nuclear export
sequence with validated nuclear export sequence in the fragile X mental
retardation protein (FMRP) and the hdm2 oncogene is shown in the box along
with the nuclear export sequence consensus. Putative SH3 binding domains (P183
(GPAPFPLPALPLPPG) and P189 (LPALPLPPGREGGPS) in human WTIP) are
indicated as green ovals, and the PDZ domain binding group is shown as a red
triangle. The yellow boxes indicate the three C-terminal LIM domains.

84

85
Figure 11. Phosphorylation map of theWTIP moiety.
Analysis of the WTIP full protein sequence by PhosphoBase
(http://www.cbs.dtu.dk/databases/PhosphoBase) and by NetPhos 2.0
(http://www.cbs.dtu.dk/services/Netphos) revealed that WTIP contains several
consensus phosphorylation sites for various kinases involved in important
signaling pathways including PKA, PKC and GSK3β. The highest probabilities,
as well as the highest number of events, fall on a stretch of the WTIP moiety
between amino acid 100 and amino acid 150. This area corresponds with the
segment between the NES and the first SH3-binding domain, which is located N-
terminally to the LIM domains,

86

Appendix I
The following project in Myosin Heavy Chain Kinase B (MHCK B) from the
amoeba Dictyostelium discoideum was part of my graduate work in the
laboratory of Dr. Thomas T. Egelhoff. The study was not part of the research in
WTIP, therefore is not placed in the text of the dissertation but was added as an
appendix. However, my work in this project trained me in diverse techniques
such as chromatography, biochemistry, cell signaling and molecular biology
that greatly contributed to my understanding of podocyte biology and to my
later work with WTIP. I would like to Thank Dr. Egelhoff for his effort on this
project.
88

89

90

91

92

93
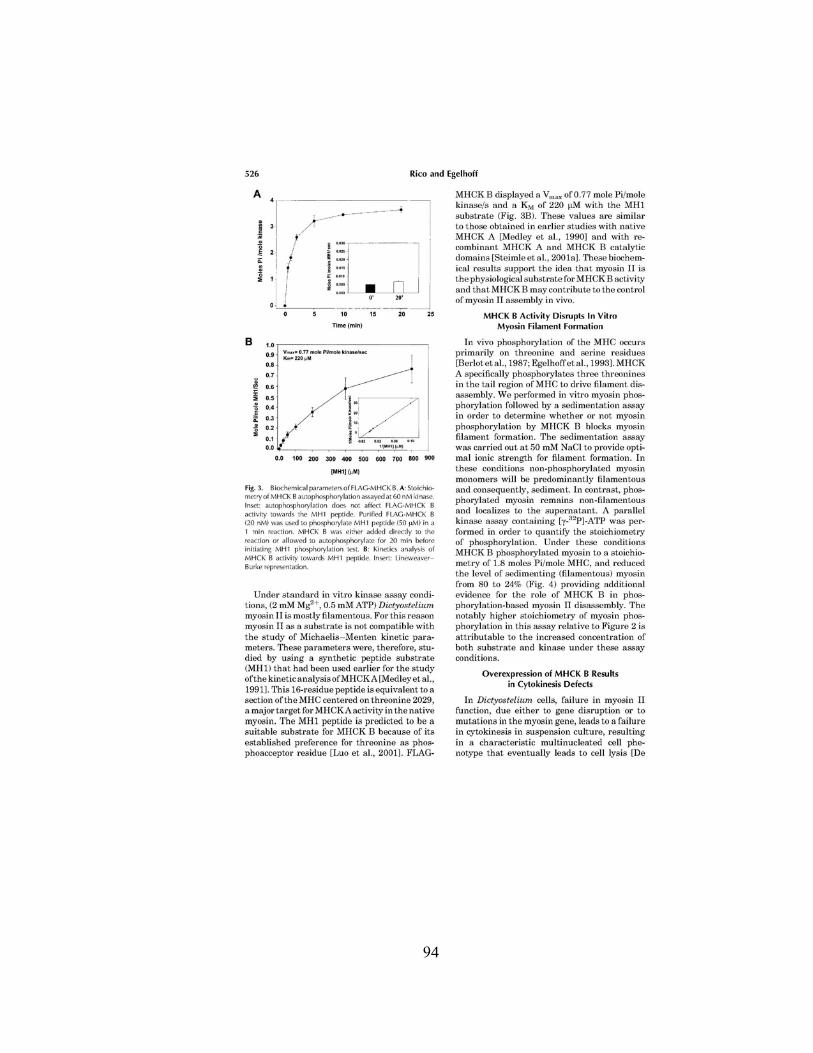
94
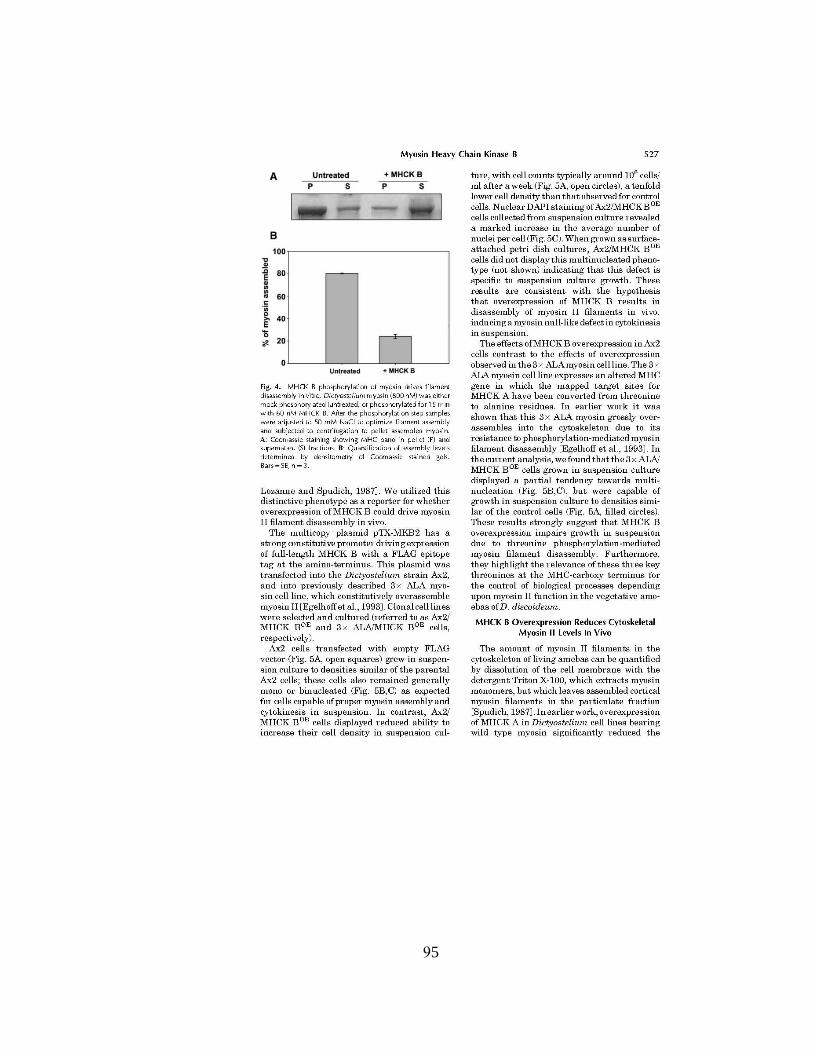
95

96

97

98

99

100

Appendix
The following project on the Dictyostelium discoideum Diacylglycerol Kinase A
(DGKA) was part of my graduate work in the laboratory of Dr. Thomas T. Egelhoff.
The study was not part of the research in WTIP, therefore is not placed in the text of
the dissertation but was added as an appendix. The study on the signaling pathways
that control MHCK B activity led me into the study of this lipid kinase and to a
collaboration with Dr. Isabel Mérida from the Spanish National Center for
Biotechnology, CSIC. I would like to thank Dr. Egelhoff and Dr. Mérida for their
effort in this project.
101

102

103

104

105

106

107

108

Bibliography
1. Kriz,W. Podocyte is the major culprit accounting for the progression of chronic renal disease. Microsc. Res. Tech. 57, 189-195 (2002).
2. Vleming,L.J., Bruijn,J.A. & van Es,L.A. The pathogenesis of progressive renal failure. Neth. J. Med. 54, 114-128 (1999).
3. McGill,J.B., Brown,W.W., Chen,S.C., Collins,A.J. & Gannon,M.R. Kidney Early Evaluation Program (KEEP). Findings from a community screening program. Diabetes Educ. 30, 196-2, 206 (2004).
4. Levin,A. Identification of patients and risk factors in chronic kidney disease--evaluating risk factors and therapeutic strategies. Nephrol. Dial. Transplant. 16 Suppl 7, 57-60 (2001).
5. Weiner,D.E. et al. Chronic kidney disease as a risk factor for cardiovascular disease and all-cause mortality: a pooled analysis of community-based studies. J. Am. Soc. Nephrol. 15, 1307-1315 (2004).
6. Reiser,J., Kriz,W., Kretzler,M. & Mundel,P. The glomerular slit diaphragm is a modified adherens junction. J. Am. Soc. Nephrol. 11, 1-8 (2000).
7. Haraldsson,B. & Sorensson,J. Why do we not all have proteinuria? An update of our current understanding of the glomerular barrier. News Physiol Sci. 19, 7-10 (2004).
8. Rossi,M. et al. Heparan sulfate chains of perlecan are indispensable in the lens capsule but not in the kidney. EMBO J. 22, 236-245 (2003).
9. Deen,W.M., Lazzara,M.J. & Myers,B.D. Structural determinants of glomerular permeability. Am. J. Physiol Renal Physiol 281, F579-F596 (2001).
10. Tryggvason,K. & Pettersson,E. Causes and consequences of proteinuria: the kidney filtration barrier and progressive renal failure. J. Intern. Med. 254, 216-224 (2003).
11. Tomari,S. et al. Glomerular differentiation in p27 and p57 double-mutant metanephroi. Anat. Embryol. (Berl) 206, 31-36 (2002).
12. Mundel,P. & Kriz,W. Structure and function of podocytes: an update. Anat. Embryol. (Berl) 192, 385-397 (1995).
13. Kihara,I., Yatoita,E., Kawasaki,K. & Yamamoto,T. Limitations of podocyte adaptation for glomerular injury in puromycin aminonucleoside nephrosis. Pathol. Int. 45, 625-634 (1995).
109

14. Huang,T.W. & Langlois,J.C. Podoendin. A new cell surface protein of the podocyte and endothelium. J. Exp. Med. 162, 245-267 (1985).
15. Sawada,H., Stukenbrok,H., Kerjaschki,D. & Farquhar,M.G. Epithelial polyanion (podocalyxin) is found on the sides but not the soles of the foot processes of the glomerular epithelium. Am. J. Pathol. 125, 309-318 (1986).
16. Kagami,S. & Kondo,S. Beta1-integrins and glomerular injury. J. Med. Invest 51, 1-13 (2004).
17. Rodewald,R. & Karnovsky,M.J. Porous substructure of the glomerular slit diaphragm in the rat and mouse. J. Cell Biol. 60, 423-433 (1974).
18. Barletta,G.M., Kovari,I.A., Verma,R.K., Kerjaschki,D. & Holzman,L.B. Nephrin and Neph1 co-localize at the podocyte foot process intercellular junction and form cis hetero-oligomers. J. Biol. Chem. 278, 19266-19271 (2003).
19. Simons,M. et al. Involvement of lipid rafts in nephrin phosphorylation and organization of the glomerular slit diaphragm. Am. J. Pathol. 159, 1069-1077 (2001).
20. Sellin,L. et al. NEPH1 defines a novel family of podocin interacting proteins. FASEB J. 17, 115-117 (2003).
21. Huber,T.B. et al. The carboxyl terminus of Neph family members binds to the PDZ domain protein zonula occludens-1. J. Biol. Chem. 278, 13417-13421 (2003).
22. Schnabel,E., Anderson,J.M. & Farquhar,M.G. The tight junction protein ZO-1 is concentrated along slit diaphragms of the glomerular epithelium. J. Cell Biol. 111, 1255-1263 (1990).
23. Shih,N.Y. et al. CD2AP localizes to the slit diaphragm and binds to nephrin via a novel C-terminal domain. Am. J. Pathol. 159, 2303-2308 (2001).
24. Lehtonen,S., Zhao,F. & Lehtonen,E. CD2-associated protein directly interacts with the actin cytoskeleton. Am. J. Physiol Renal Physiol 283, F734-F743 (2002).
25. Schwarz,K. et al. Podocin, a raft-associated component of the glomerular slit diaphragm, interacts with CD2AP and nephrin. J. Clin. Invest 108, 1621-1629 (2001).
26. Srichai,M.B. et al. A WT1 co-regulator controls podocyte phenotype by shuttling between adhesion structures and nucleus. J. Biol. Chem. (2004).
27. Wang,Y. & Gilmore,T.D. Zyxin and paxillin proteins: focal adhesion plaque LIM domain proteins go nuclear. Biochim. Biophys. Acta 1593, 115-120 (2003).
110

28. Lee,S.B. & Haber,D.A. Wilms tumor and the WT1 gene. Exp. Cell Res. 264, 74-99 (2001).
29. Li,H.Y. et al. Translocation of a human focal adhesion LIM-only protein, FHL2, during myofibrillogenesis and identification of LIM2 as the principal determinants of FHL2 focal adhesion localization. Cell Motil. Cytoskeleton 48, 11-23 (2001).
30. Lowenborg,E.K., Jaremko,G. & Berg,U.B. Glomerular function and morphology in puromycin aminonucleoside nephropathy in rats. Nephrol. Dial. Transplant. 15, 1547-1555 (2000).
31. Li,L. et al. Bcl-2 expression decreases cadherin-mediated cell-cell adhesion. J. Cell Sci. 116, 3687-3700 (2003).
32. Lu,X. & Horvitz,H.R. lin-35 and lin-53, two genes that antagonize a C. elegans Ras pathway, encode proteins similar to Rb and its binding protein RbAp48. Cell 95, 981-991 (1998).
33. Mundel,P., Gilbert,P. & Kriz,W. Podocytes in glomerulus of rat kidney express a characteristic 44 KD protein. J. Histochem. Cytochem. 39, 1047-1056 (1991).
34. Mundel,P., Reiser,J. & Kriz,W. Induction of differentiation in cultured rat and human podocytes. J. Am. Soc. Nephrol. 8, 697-705 (1997).
35. Mundel,P. et al. Rearrangements of the cytoskeleton and cell contacts induce process formation during differentiation of conditionally immortalized mouse podocyte cell lines. Exp. Cell Res. 236, 248-258 (1997).
36. Ng,E.K. et al. Interaction of the heart-specific LIM domain protein, FHL2, with DNA-binding nuclear protein, hNP220. J. Cell Biochem. 84, 556-566 (2002).
37. Nix,D.A. & Beckerle,M.C. Nuclear-cytoplasmic shuttling of the focal contact protein, zyxin: a potential mechanism for communication between sites of cell adhesion and the nucleus. J. Cell Biol. 138, 1139-1147 (1997).
38. Nix,D.A. et al. Targeting of zyxin to sites of actin membrane interaction and to the nucleus. J. Biol. Chem. 276, 34759-34767 (2001).
39. Patek,C.E. et al. Murine Denys-Drash syndrome: evidence of podocyte de-differentiation and systemic mediation of glomerulosclerosis. Hum. Mol. Genet. 12, 2379-2394 (2003).
40. Reeves,W., Caulfield,J.P. & Farquhar,M.G. Differentiation of epithelial foot processes and filtration slits: sequential appearance of occluding junctions, epithelial polyanion, and slit membranes in developing glomeruli. Lab Invest 39, 90-100 (1978).
111

41. Renfranz,P.J., Siegrist,S.E., Stronach,B.E., Macalma,T. & Beckerle,M.C. Molecular and phylogenetic characterization of Zyx102, a Drosophila orthologue of the zyxin family that interacts with Drosophila Enabled. Gene 305, 13-26 (2003).
42. Riesen,F.K., Rothen-Rutishauser,B. & Wunderli-Allenspach,H. A ZO1-GFP fusion protein to study the dynamics of tight junctions in living cells. Histochem. Cell Biol. 117, 307-315 (2002).
43. Saleem,M.A. et al. Co-localization of nephrin, podocin, and the actin cytoskeleton: evidence for a role in podocyte foot process formation. Am. J. Pathol. 161, 1459-1466 (2002).
44. Sasaki,T., Hatta,H. & Osawa,G. Cytokines and podocyte injury: the mechanism of fibroblast growth factor 2-induced podocyte injury. Nephrol. Dial. Transplant. 14 Suppl 1, 33-34 (1999).
45. Mundel,P. et al. Rearrangements of the cytoskeleton and cell contacts induce process formation during differentiation of conditionally immortalized mouse podocyte cell lines. Exp. Cell Res. 236, 248-258 (1997).
46. Schreiber,E., Matthias,P., Muller,M.M. & Schaffner,W. Rapid detection of octamer binding proteins with 'mini-extracts', prepared from a small number of cells. Nucleic Acids Res. 17, 6419 (1989).
47. Schnabel,E., Anderson,J.M. & Farquhar,M.G. The tight junction protein ZO-1 is concentrated along slit diaphragms of the glomerular epithelium. J. Cell Biol. 111, 1255-1263 (1990).
48. Gama-Carvalho,M. & Carmo-Fonseca,M. The rules and roles of nucleocytoplasmic shuttling proteins. FEBS Lett. 498, 157-163 (2001).
49. Conti,E. & Izaurralde,E. Nucleocytoplasmic transport enters the atomic age. Curr. Opin. Cell Biol. 13, 310-319 (2001).
50. Gorlich,D. & Kutay,U. Transport between the cell nucleus and the cytoplasm. Annu. Rev. Cell Dev. Biol. 15, 607-660 (1999).
51. Hastie,N.D. Dominant negative mutations in the Wilms tumour (WT1) gene cause Denys-Drash syndrome--proof that a tumour-suppressor gene plays a crucial role in normal genitourinary development. Hum. Mol. Genet. 1, 293-295 (1992).
52. Gottardi,C.J., Arpin,M., Fanning,A.S. & Louvard,D. The junction-associated protein, zonula occludens-1, localizes to the nucleus before the maturation and during the remodeling of cell-cell contacts. Proc. Natl. Acad. Sci. U. S. A 93, 10779-10784 (1996).
112

53. Balda,M.S., Garrett,M.D. & Matter,K. The ZO-1-associated Y-box factor ZONAB regulates epithelial cell proliferation and cell density. J. Cell Biol. 160, 423-432 (2003).
54. Kanungo,J., Pratt,S.J., Marie,H. & Longmore,G.D. Ajuba, a cytosolic LIM protein, shuttles into the nucleus and affects embryonal cell proliferation and fate decisions. Mol. Biol. Cell 11, 3299-3313 (2000).
55. Schnabel,E., Anderson,J.M. & Farquhar,M.G. The tight junction protein ZO-1 is concentrated along slit diaphragms of the glomerular epithelium. J. Cell Biol. 111, 1255-1263 (1990).
56. Kim,Y.H. et al. Podocyte depletion and glomerulosclerosis have a direct relationship in the PAN-treated rat. Kidney Int. 60, 957-968 (2001).
57. Yamazaki,T. Podocytic degeneration and regeneration in puromycin aminonucleoside nephropathy in the rat. Pathol. Int. 45, 465-472 (1995).
58. Gloy,J. et al. Amino acid transport in podocytes. Am. J. Physiol Renal Physiol 278, F999-F1005 (2000).
59. Asanuma,K. & Mundel,P. The role of podocytes in glomerular pathobiology. Clin. Exp. Nephrol. 7, 255-259 (2003).
60. Macconi,D. et al. Effect of angiotensin-converting enzyme inhibition on glomerular basement membrane permeability and distribution of zonula occludens-1 in MWF rats. J. Am. Soc. Nephrol. 11, 477-489 (2000).
61. Guan,L.S., Rauchman,M. & Wang,Z.Y. Induction of Rb-associated protein (RbAp46) by Wilms' tumor suppressor WT1 mediates growth inhibition. J. Biol. Chem. 273, 27047-27050 (1998).
62. Barisoni,L., Kriz,W., Mundel,P. & D'Agati,V. The dysregulated podocyte phenotype: a novel concept in the pathogenesis of collapsing idiopathic focal segmental glomerulosclerosis and HIV-associated nephropathy. J. Am. Soc. Nephrol. 10, 51-61 (1999).
63. Barisoni,L. et al. Podocyte cell cycle regulation and proliferation in collapsing glomerulopathies. Kidney Int. 58, 137-143 (2000).
64. Akazawa,H. et al. A novel LIM protein Cal promotes cardiac differentiation by association with CSX/NKX2-5. J. Cell Biol. 164, 395-405 (2004).
65. Dawid,I.B., Breen,J.J. & Toyama,R. LIM domains: multiple roles as adapters and functional modifiers in protein interactions. Trends Genet. 14, 156-162 (1998).
66. Khurana,T., Khurana,B. & Noegel,A.A. LIM proteins: association with the actin cytoskeleton. Protoplasma 219, 1-12 (2002).
113

67. Krause,A. et al. Tbx5 and Tbx4 transcription factors interact with a new chicken PDZ-LIM protein in limb and heart development. Dev. Biol. 273, 106-120 (2004).
68. Benmerah,A., Scott,M., Poupon,V. & Marullo,S. Nuclear functions for plasma membrane-associated proteins? Traffic. 4, 503-511 (2003).
69. Conacci-Sorrell,M. et al. Autoregulation of E-cadherin expression by cadherin-cadherin interactions: the roles of beta-catenin signaling, Slug, and MAPK. J. Cell Biol. 163, 847-857 (2003).
70. Ilan,N. & Madri,J.A. PECAM-1: old friend, new partners. Curr. Opin. Cell Biol. 15, 515-524 (2003).
71. Zhang,Y. et al. Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. 13, 1924-1935 (1999).
72. Balda,M.S. & Matter,K. The tight junction protein ZO-1 and an interacting transcription factor regulate ErbB-2 expression. EMBO J. 19, 2024-2033 (2000).
73. Balda,M.S., Garrett,M.D. & Matter,K. The ZO-1-associated Y-box factor ZONAB regulates epithelial cell proliferation and cell density. J. Cell Biol. 160, 423-432 (2003).
74. Yang,J., Kiefer,S. & Rauchman,M. Characterization of the gene encoding mouse retinoblastoma binding protein-7, a component of chromatin-remodeling complexes. Genomics 80, 407-415 (2002).
75. Eferl,R. & Wagner,E.F. AP-1: a double-edged sword in tumorigenesis. Nat. Rev. Cancer 3, 859-868 (2003).
76. Kocher,H.M. et al. Expression of Ras GTPases in normal kidney and in glomerulonephritis. Nephrol. Dial. Transplant. 18, 2284-2292 (2003).
77. Lu,X. & Horvitz,H.R. lin-35 and lin-53, two genes that antagonize a C. elegans Ras pathway, encode proteins similar to Rb and its binding protein RbAp48. Cell 95, 981-991 (1998).
78. Solari,F. & Ahringer,J. NURD-complex genes antagonise Ras-induced vulval development in Caenorhabditis elegans. Curr. Biol. 10, 223-226 (2000).
79. Furthauer,M., Lin,W., Ang,S.L., Thisse,B. & Thisse,C. Sef is a feedback-induced antagonist of Ras/MAPK-mediated FGF signalling. Nat. Cell Biol. 4, 170-174 (2002).
80. Richard,D.J., Schumacher,V., Royer-Pokora,B. & Roberts,S.G. Par4 is a coactivator for a splice isoform-specific transcriptional activation domain in WT1. Genes Dev. 15, 328-339 (2001).
114

81. Islas,S., Vega,J., Ponce,L. & Gonzalez-Mariscal,L. Nuclear localization of the tight junction protein ZO-2 in epithelial cells. Exp. Cell Res. 274, 138-148 (2002).
82. Hsueh,Y.P., Wang,T.F., Yang,F.C. & Sheng,M. Nuclear translocation and transcription regulation by the membrane-associated guanylate kinase CASK/LIN-2. Nature 404, 298-302 (2000).
83. Kausalya,P.J., Phua,D.C. & Hunziker,W. Association of ARVCF with zonula occludens (ZO)-1 and ZO-2: binding to PDZ-domain proteins and cell-cell adhesion regulate plasma membrane and nuclear localization of ARVCF. Mol. Biol. Cell 15, 5503-5515 (2004).
84. Briggs,L.J. et al. The cAMP-dependent protein kinase site (Ser312) enhances dorsal nuclear import through facilitating nuclear localization sequence/importin interaction. J. Biol. Chem. 273, 22745-22752 (1998).
85. Zhang,F., White,R.L. & Neufeld,K.L. Phosphorylation near nuclear localization signal regulates nuclear import of adenomatous polyposis coli protein. Proc. Natl. Acad. Sci. U. S. A 97, 12577-12582 (2000).
86. Xiao,Z., Watson,N., Rodriguez,C. & Lodish,H.F. Nucleocytoplasmic shuttling of Smad1 conferred by its nuclear localization and nuclear export signals. J. Biol. Chem. 276, 39404-39410 (2001).
87. Xiao,Z., Liu,X., Henis,Y.I. & Lodish,H.F. A distinct nuclear localization signal in the N terminus of Smad 3 determines its ligand-induced nuclear translocation. Proc. Natl. Acad. Sci. U. S. A 97, 7853-7858 (2000).
88. Logeat,F. et al. The Notch1 receptor is cleaved constitutively by a furin-like convertase. Proc. Natl. Acad. Sci. U. S. A 95, 8108-8112 (1998).
89. Smith,D.S. & Tsai,L.H. Cdk5 behind the wheel: a role in trafficking and transport? Trends Cell Biol. 12, 28-36 (2002).
90. Rubinfeld,B. et al. Binding of GSK3beta to the APC-beta-catenin complex and regulation of complex assembly. Science 272, 1023-1026 (1996).
91. Loyet,K.M., Stults,J.T. & Arnott,D. Mass spectrometric contributions to the practice of phosphorylation site mapping through 2003: A literature review. Mol. Cell Proteomics. (2005).
92. Chen,A. et al. Experimental focal segmental glomerulosclerosis in mice. Nephron 78, 440-452 (1998).
93. Wang,Y., Wang,Y.P., Tay,Y.C. & Harris,D.C. Progressive adriamycin nephropathy in mice: sequence of histologic and immunohistochemical events. Kidney Int. 58, 1797-1804 (2000).
115

94. Aaltonen,P. et al. Changes in the expression of nephrin gene and protein in experimental diabetic nephropathy. Lab Invest 81, 1185-1190 (2001).
95. Pagtalunan,M.E. et al. Podocyte loss and progressive glomerular injury in type II diabetes. J. Clin. Invest 99, 342-348 (1997).
96. Velasquez,M.T., Kimmel,P.L. & Michaelis,O.E. Animal models of spontaneous diabetic kidney disease. FASEB J. 4, 2850-2859 (1990).
97. Ziyadeh,F.N. et al. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-beta antibody in db/db diabetic mice. Proc. Natl. Acad. Sci. U. S. A 97, 8015-8020 (2000).
98. Barisoni,L. & Kopp,J.B. Modulation of podocyte phenotype in collapsing glomerulopathies. Microsc. Res. Tech. 57, 254-262 (2002).
99. Barisoni,L., Bruggeman,L.A., Mundel,P., D'Agati,V.D. & Klotman,P.E. HIV-1 induces renal epithelial dedifferentiation in a transgenic model of HIV-associated nephropathy. Kidney Int. 58, 173-181 (2000).
100. Winston,J.A. et al. Nephropathy and establishment of a renal reservoir of HIV type 1 during primary infection. N. Engl. J. Med. 344, 1979-1984 (2001).
101. Kopp,J.B. & Klotman,P.E. Transgenic animal models of renal development and pathogenesis. Am. J. Physiol 269, F601-F620 (1995).
102. Bruggeman,L.A. et al. Nephropathy in human immunodeficiency virus-1 transgenic mice is due to renal transgene expression. J. Clin. Invest 100, 84-92 (1997).
103. Kopp,J.B. et al. Nephropathy in HIV-transgenic mice. Contrib. Nephrol. 107, 194-204 (1994).
104. Wharram,B.L. et al. Altered podocyte structure in GLEPP1 (Ptpro)-deficient mice associated with hypertension and low glomerular filtration rate. J. Clin. Invest 106, 1281-1290 (2000).
105. Yuan,H. et al. Nephrin dissociates from actin, and its expression is reduced in early experimental membranous nephropathy. J. Am. Soc. Nephrol. 13, 946-956 (2002).
116
Copyright © 2022 FDOKUMEN




















