
Application of Proteomics to the Study of Cardiovascular Biology
10
66 TCM Vol. 11, No. 2, 2001 Lander E: 1996. The new genomics: global views of biology. Science 274:536–539. Lilliefors I: 1970. Coronary heart disease in male twins. Hereditary and environmental factors in concordant and discordant pairs. Acta Med Scand Suppl 511. Luc G, Bard JM, Arveiler D, et al.: 1994. Impact of apolipoprotein E polymorphism on lipoproteins and risk of myocardial in- farction. The ECTIM study. Arterioscler Thromb 14:1412–1419. Mansfield TA, Simon DB, Farfel Z, et al.: 1997. Multilocus linkage of familial hyper- kalaemia and hypertension, pseudohypo- aldosteronism type II, to chromosomes 1q31-42 and 17p11-q21. Nat Genet 16:202– 205. Masood E: 1999. As consortium plans free SNP map of human genome. Nature 545–546. Motulsky AG, Brunzell JD: 1992. The genetics of coronary atherosclerosis. In The Genetic Basis of Complex Diseases. Oxford Univer- sity Press, pp 150–169. Nickerson D, Taylor S, Weiss K, et al.: 1998. DNA sequence diversity in a 9.7-kb region of the human lipoprotein lipase gene. Nat Genet 19:233–240. Osler W: 1897. Lectures on Angina Pectoris and Allied States. New York, D. Appleton & Co. Picoult-Newberg L, Ideker TE, Pohl MG, et al.: 1999. Mining SNPs from EST data- bases. Genome Res 9:167–174. Ridker PM, Hennekens CH, Lindpaintner K, et al.: 1995. Mutation in the gene coding for coagulation factor V and the risk of myocar- dial infarction, stroke, and venous throm- bosis in apparently healthy men. N Engl J Med 14:912–917. Rieder M, Taylor S, Clark A, Nickerson D: 1999. Sequence variation in the human angiotensin converting enzyme. Nat Genet 22:59–62. Risch N, Merikangas K: 1996. The future of genetic studies of complex human dis- eases. Science 273:1516–1517. Saunders AM, Strittmatter WJ, Schmechel D, et al.: 1993. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurol- ogy 43:1467–1472. Scaglione L, Bergerone S, Gambino R, et al.: 1999. Role of lipid, apolipoprotein levels and apolipoprotein E genotype in young Italian patients with myocardial infarction. Nutr Metab Cardiovasc Dis 9:118–124. Taillon-Miller P, Gu Z, Li Q, et al.: 1998. Over- lapping genomic sequences: a treasure trove of single-nucleotide polymorphisms. Genome Res 8:748–754. Vauhkonen I, Niskanen L, Ryynanen M, et al.: 1997. Divergent association of apolipopro- tein E polymorphism with vascular disease in patients with NIDDM and control sub- jects. Diabet Med 14:748–756. Winkelmann BR, Nauck M, Klein B, et al.: 1997. Deletion polymorphism of the angio- tensin I-converting enzyme gene is associ- ated with increased plasma angiotensin- converting enzyme activity but not with increased risk for myocardial infarction and coronary artery disease. Ann Intern Med 125:19–25. PII S1050-1738(01)00097-1 TCM Joseph Macri and Stephen T. Rapundalo are at Pfizer Global Research and Development, Ann Arbor, Michigan, USA. *Address correspondence to: Stephen T. Rapundalo, Ph.D., Department of Cardiovas- cular Molecular Sciences, Pfizer Global Research and Development, 2800 Plymouth Road, Ann Arbor, MI 48105. Tel.: 734-622- 5170; fax: 734-622-1480; e-mail: stephen. rapundalo@pfizer.com. © 2001, Elsevier Science Inc. All rights reserved. 1050-1738/01/$-see front matter Application of Proteomics to the Study of Cardiovascular Biology Joseph Macri and Stephen T. Rapundalo* Proteomics involves the integration of a number of technologies with the aim of analyzing the complete complement of proteins expressed by a biological system in response to various stimuli and/or under differ- ent physiological or pathophysiological conditions. Recent technical improvements to the methods employed for protein separation and protein identification have resulted in a dramatic increase in the num- ber of proteomics-based research projects. More importantly, it has become readily apparent that examining changes in the proteome offers insight into understanding cellular and molecular mechanisms that cannot be obtained through genomic analysis. There are numerous examples of cardiovascular functions whose molecular pathways are mediated through post-translational processes such as phosphoryla- tion. The use of proteomics offers the ability to simultaneously moni- tor the changes in protein expression and/or cell signaling pathways in response to such conditions as cardiac hypertrophy and heart failure. Together with complementary genomic data, proteomics-based research can greatly increase our understanding of cardiovascular biology. (Trends Cardiovasc Med 2001;11:66–75). © 2001 Elsevier Science Inc. The list of organisms whose genome has been completely sequenced currently stands at greater than 20 (Tekaia et al. 1999) with the human genome sequenced earlier this year (Venter et al., 2001; Sa- chidanandam et al., 2001). The enor- mous amount of DNA sequence data generated through these sequencing ef- forts and the rapidity of their accumula- tion have already impacted virtually every discipline of biological sciences (Jasny and Hines 1999). At one time unlocking genomic information was thought to be the key to understanding cellular and molecular mechanisms. With the multi- tude of known post-transcriptionally reg- ulatory mechanisms, it is clear that knowl- edge of the DNA sequence is essential but not sufficient (Humphery-Smith et al. 1997). A more dynamic illustration of gene expression can be achieved through the characterization of the products of gene expression, namely proteins. The term proteome was recently
-
Upload
independent -
Category
Documents
-
view
9 -
download
0
Transcript of Application of Proteomics to the Study of Cardiovascular Biology

66
TCM Vol. 11, No. 2, 2001
Lander E: 1996. The new genomics: globalviews of biology. Science 274:536–539.
Lilliefors I: 1970. Coronary heart disease inmale twins. Hereditary and environmentalfactors in concordant and discordantpairs. Acta Med Scand Suppl 511.
Luc G, Bard JM, Arveiler D, et al.: 1994.Impact of apolipoprotein E polymorphismon lipoproteins and risk of myocardial in-farction. The ECTIM study. ArteriosclerThromb 14:1412–1419.
Mansfield TA, Simon DB, Farfel Z, et al.: 1997.Multilocus linkage of familial hyper-kalaemia and hypertension, pseudohypo-aldosteronism type II, to chromosomes1q31-42 and 17p11-q21. Nat Genet 16:202–205.
Masood E: 1999. As consortium plans free SNPmap of human genome. Nature 545–546.
Motulsky AG, Brunzell JD: 1992. The geneticsof coronary atherosclerosis.
In
The GeneticBasis of Complex Diseases. Oxford Univer-sity Press, pp 150–169.
Nickerson D, Taylor S, Weiss K, et al.: 1998.DNA sequence diversity in a 9.7-kb regionof the human lipoprotein lipase gene. NatGenet 19:233–240.
Osler W: 1897. Lectures on Angina Pectoris andAllied States. New York, D. Appleton & Co.
Picoult-Newberg L, Ideker TE, Pohl MG, etal.: 1999. Mining SNPs from EST data-bases. Genome Res 9:167–174.
Ridker PM, Hennekens CH, Lindpaintner K,et al.: 1995. Mutation in the gene coding forcoagulation factor V and the risk of myocar-dial infarction, stroke, and venous throm-bosis in apparently healthy men. N Engl JMed 14:912–917.
Rieder M, Taylor S, Clark A, Nickerson D:1999. Sequence variation in the humanangiotensin converting enzyme. Nat Genet22:59–62.
Risch N, Merikangas K: 1996. The future ofgenetic studies of complex human dis-eases. Science 273:1516–1517.
Saunders AM, Strittmatter WJ, Schmechel D,et al.: 1993. Association of apolipoproteinE allele epsilon 4 with late-onset familialand sporadic Alzheimer’s disease. Neurol-ogy 43:1467–1472.
Scaglione L, Bergerone S, Gambino R, etal.: 1999. Role of lipid, apolipoproteinlevels and apolipoprotein E genotype inyoung Italian patients with myocardialinfarction. Nutr Metab Cardiovasc Dis9:118–124.
Taillon-Miller P, Gu Z, Li Q, et al.: 1998. Over-lapping genomic sequences: a treasuretrove of single-nucleotide polymorphisms.Genome Res 8:748–754.
Vauhkonen I, Niskanen L, Ryynanen M, et al.:1997. Divergent association of apolipopro-tein E polymorphism with vascular disease
in patients with NIDDM and control sub-jects. Diabet Med 14:748–756.
Winkelmann BR, Nauck M, Klein B, et al.:1997. Deletion polymorphism of the angio-tensin I-converting enzyme gene is associ-ated with increased plasma angiotensin-
converting enzyme activity but not withincreased risk for myocardial infarctionand coronary artery disease. Ann InternMed 125:19–25.
PII S1050-1738(01)00097-1 TCM
Joseph Macri and Stephen T. Rapundalo areat Pfizer Global Research and Development,Ann Arbor, Michigan, USA.
* Address correspondence to: Stephen T.Rapundalo, Ph.D., Department of Cardiovas-cular Molecular Sciences, Pfizer GlobalResearch and Development, 2800 PlymouthRoad, Ann Arbor, MI 48105. Tel.: 734-622-5170; fax: 734-622-1480; e-mail: [email protected].
© 2001, Elsevier Science Inc. All rightsreserved. 1050-1738/01/$-see front matter
Application of Proteomics to the Study of Cardiovascular Biology
Joseph Macri and Stephen T. Rapundalo*
Proteomics involves the integration of a number of technologies withthe aim of analyzing the complete complement of proteins expressed bya biological system in response to various stimuli and/or under differ-ent physiological or pathophysiological conditions. Recent technicalimprovements to the methods employed for protein separation andprotein identification have resulted in a dramatic increase in the num-ber of proteomics-based research projects. More importantly, it hasbecome readily apparent that examining changes in the proteome offersinsight into understanding cellular and molecular mechanisms thatcannot be obtained through genomic analysis. There are numerousexamples of cardiovascular functions whose molecular pathways aremediated through post-translational processes such as phosphoryla-tion. The use of proteomics offers the ability to simultaneously moni-tor the changes in protein expression and/or cell signaling pathways inresponse to such conditions as cardiac hypertrophy and heart failure.Together with complementary genomic data, proteomics-based researchcan greatly increase our understanding of cardiovascular biology.
(TrendsCardiovasc Med 2001;11:66–75).
© 2001 Elsevier Science Inc.
The list of organisms whose genome hasbeen completely sequenced currentlystands at greater than 20 (Tekaia et al.1999) with the human genome sequencedearlier this year (Venter et al., 2001; Sa-chidanandam et al., 2001). The enor-
mous amount of DNA sequence datagenerated through these sequencing ef-forts and the rapidity of their accumula-tion have already impacted virtually everydiscipline of biological sciences (Jasnyand Hines 1999). At one time unlockinggenomic information was thought to bethe key to understanding cellular andmolecular mechanisms. With the multi-tude of known post-transcriptionally reg-ulatory mechanisms, it is clear that knowl-edge of the DNA sequence is essentialbut not sufficient (Humphery-Smith etal. 1997). A more dynamic illustration ofgene expression can be achieved throughthe characterization of the products ofgene expression, namely proteins.
The term proteome was recently

TCM Vol. 11, No. 2, 2001
67
coined (Wasinger et al. 1995, Wilkins etal. 1996) to describe all the proteins en-coded from a specific genome. Proteomicanalysis involves the qualitative alter-ations in proteins along with the quanti-tative changes in protein expression lev-els that occur in response to a given setof conditions (Anderson and Anderson1998). The rapid increase in effort di-rected toward proteomic-based studiesstems from the potential benefits associ-ated with the elucidation of cellular mech-anisms. These benefits are based on anumber of key observations, whichstrongly support the contention that un-derstanding the function of gene productscannot be readily obtained solely fromDNA and/or RNA sequence analysis(Klose 1999). A recent study analyzinghuman liver samples determined thecorrelation coefficient between the amountof mRNA present to the correspondingprotein abundance to be 0.48 (Andersonand Seilhamer 1997). This low correlationhighlights the need for proteomic analysis.
The apparent information gap occur-ring between a genome and the cellularprocesses resulting from its gene prod-ucts can be largely attributed to post-translational modifications such as phos-phorylation and glycosylation. Thesemodifications, which cannot be moni-tored by using only genomic data, havebeen shown to modulate pivotal regula-tory processes such as protein turnover,protein activity, and protein localizationwithin a cell (Faux and Scott 1996, Hart1992, Rhoads 1999). In addition, virtu-ally all known cellular signaling pathwaysare largely mediated through a complexcascade of reversible protein phosphoryla-tion (Meek 1998). An appreciation of theincreased complexity associated withthe events occurring within the proteinrealm comes from the realization thatthe information within a single genomegives rise to a constantly changing pro-teome. The proteome is a dynamic feature,subject to changes due to developmentalstage, disease state or environmental (i.e.,physiological, pharmacological, etc.)conditions (Haynes et al. 1998, Williamsand Hochstrasser 1997).
• Components of a Proteomics-Based Approach
The execution of a proteomics-basedstudy involves the integration of a num-ber of technologies (Figure 1) and exper-
tise in biochemistry/molecular biology,physiology, bioinformatics/statistics andanalytical protein chemistry (Patton1999). Although a number of optionsexist within each stage of the proteomicanalysis, the overall approach involvessample preparation, protein separation,imaging and identification (Williams andHochstrasser 1997). Currently, the mostapplied and accepted protocol for pro-teomic sample analysis incorporatestwo-dimensional electrophoresis (2-DE)for protein separation, with protein de-tection accomplished through the use ofautoradiography and/or various stainsor dyes. Analysis of the resulting proteinprofile either can be qualitative (visualgel inspection) or quantitative throughthe use of dedicated 2-DE gel imagingsoftware (Dunn 1987, Patton 1999). Pro-teins of interest are identified by a varietyof analytical techniques, typically withmass spectrometry being widely used(Haynes et al. 1998). One of the goals ofproteomics-based research is to incor-porate the information obtained fromthis type of analysis into a comprehen-sive proteome database containing bothprotein identifications and their corre-sponding functional characteristics.
The success of any proteomics effortis inextricably linked to performing eachaspect of the analysis in a consistent andreproducible fashion, and to minimize the
incorporation of artifacts. Recent techno-logical advances have facilitated the pro-teomics process to the extent that 2-DEand analysis are becoming core tools ofmany research laboratories, rather thana specialized application employed by aselect few.
Sample Preparation
The most important component of aproteomics-based study is sample prep-aration. Artifacts presented at this pointin the analysis can often be magnifiedand potentially jeopardize the validity ofthe results. The excellent resolving powerof 2-DE is a “dual-edged sword” with theability to detect subtle post-translationalchanges such as a single phosphoryla-tion or artifactitious modifications suchas protein carbamylation induced by ex-cessive sample heating or sonication(Rabilloud 1996).
An important issue is the ability tomaximally and consistently solubilizesample proteins without introducingnon-experimental modification. This en-tails disrupting the numerous protein/protein and protein/non-protein interac-tions that occur within tissue or biologi-cal fluid using various combinations ofreducing agents, chaotropic agents anddetergents (Rabilloud and Chevallet 1999).The use of a protease inhibitor cocktailis often included to inhibit the inherentproteolytic activity associated with thesample. However, the presence of rela-tively high concentrations of a chaotropeand detergent can usually minimize pro-teolytic activity for the duration of thesample-processing period (Rabilloud1996).
There have been a number of com-prehensive reviews regarding the prepa-ration and utilization of protein solubili-zation buffer for 2-DE (Herbert 1999,Rabilloud 1996). One of the most com-monly used agents to reduce protein di-sulfide bonds remains dithiothreitol (DTT).Certain phosphine derivatives such astributylphosphine and tris (carboxyethyl)phosphine have recently been utilized asalternatives to DTT (Herbert et al. 1998).
The chaotropic component of solubi-lization buffer is most often urea or acombination of urea and the more highlychaotropic agent thiourea (Rabilloud1998). Owing to the relative incompati-bility of ionic detergents such as SDSwith isoelectric focusing (IEF), only non-
Figure 1. Elements of a proteomics-basedstudy.

68
TCM Vol. 11, No. 2, 2001
ionic detergents (Triton X-100, Nonin-det P-40) and zwitterionic detergentssuch as CHAPS are routinely employed.Membrane-associated proteins pose aparticular challenge with respect to sol-ubilization and are significantly under-represented on 2D gels (Santoni et al.1999). The use of alternative detergentshas been one attempt to improve the poorsolubility associated with membrane-bound proteins (Chevallet et al. 1998). Inthe first attempt to characterize andmap rat cardiac sarcoplasmic reticulumand sarcolemmal proteins, we recentlyhave utilized a new alkylaryl aminosul-fobetaine zwitterionic detergent, C8
∅
,to increase the solubility of these mem-brane-associated proteins such as phos-pholamban, Ca
2
1
-ATPase, and calse-questrin (Macri et al. 2000b). Despitethe increase in the total number of pro-teins detected, the presence of C8
∅
didnot result in the detection of a numberof key membrane-associated proteins.These results highlight the importanceof optimizing the composition of anysolubilization buffer for the specificsample under investigation.
Sample Prefractionation
There are a number of enrichment tech-niques frequently applied to samplesprior to the 2-DE. The focus of sampleprefractionation can range from enrich-ment of a single protein or group of pro-teins, with selectivity based on physicalcharacteristics (pI, molecular weight,hydrophobicity) or biological localiza-tion within a cell or extracellular matrix(Lopez 2000). The isolation of subcellu-lar organelles and/or their constituentcomponents can be utilized as a meansof concentrated proteins which may beunder-represented within the total pro-tein profile (Huber 1995). The sequen-tial use of different detergents has alsobeen used to extract specific proteinfractions (Molloy et al. 1998). Other pro-cedures for sample prefractionation in-clude affinity-based methods such as theuse of heparin (Karlsson et al. 1999), im-munoprecipitation (Fouilitt et al. 2000)and concanavalin A (Iannello and Jef-frey 1990). An alternative method to in-crease the presence of low level proteinswithin a protein profile is to removehighly abundant proteins prior to 2-DE.This approach has been used to removealbumin (Walsh et al. 1984) and the im-
munoglobulin fraction (Lollo et al. 1999)from serum samples. Although the bene-fits of prefractionation are readily ap-parent, none of the above-mentionedtechniques are absolutely ligand specificand therefore confirmatory studies arewarranted in some situations.
• Protein Separation
Two-Dimensional Electrophoresis
The use of 2-DE remains the best tech-nique to separate complex protein mix-tures. One can routinely resolve 2000–3000 proteins from a single sample prep-aration, or up to 10,000 proteins fromlarge-scale custom gels (Jungblut et al.1994). Although other 2-DE systemsare used (Manabe 2000, Monribot andBoucherie 2000), today’s standard 2-DEtechnique is essentially the same as themethod originally described 25 yearsago (Klose 1975, O’Farrell 1975). Thissystem involves protein separation byIEF in the first dimension followedby sodium dodecylsulfate polyacryla-mide gel electrophoresis (SDS-PAGE) inthe second dimension. The IEF/SDS-PAGE combination separates proteinsbased on the orthogonal parameters ofisoelectric point (pI) and molecularweight (Figure 2). A large number of
studies have been performed since thento refine all aspects of this separationprocedure including resolution, repro-ducibility and throughput (Dunn 1987).
First Dimension.
Proteins are amphot-eric molecules and hence will migrate inthe presence of an electric field based onthe sum total of the charges present onthe molecule. When a protein is appliedto a support matrix containing a pH gra-dient, the proteins will migrate in re-sponse to the applied current until thepoint is reached in the gradient whichcorresponds to pI of the protein (Dun-bar 1987). Polyacrylamide tube gelshave traditionally been used as the sup-port matrix for IEF. These gels contain amixture of aliphatic polyaminopolycar-boxylic acids as the carrier ampholytes inorder to establish the pH gradient, whichis generally within a pH range of 3–11(Dunbar 1987). The most recent progresswith respect to IEF has been the intro-duction of immobilized pH gradient(IPG) strips (Bjellqvist et al. 1982). IPGstrips and are composed of acidic andbasic buffering groups covalently linkedto the polyacrylamide matrix. AlthoughIPG strips can be prepared “in-house,”they are also available from a number ofvendors in various lengths and pH ranges(Görg et al. 2000). There are also severaltypes of commercially available IEF units
Figure 2. 2-DE of rat cardiac left ventricle. Tissue was solubilized in buffer containing 9.5 Murea, 2% CHAPS and 1% DTT. A 100-mg sample of protein was subjected to IEF in the first di-mension on an immobilized pH gradient (IPG) strip having a pH range of 4–7. The second-dimension separation was accomplished with a 12% SDS-PAGE gel. A total of 2142 proteinspots were detected following silver staining of the gel. Several key contractile proteins such asmyosin light chain and actin as well as the chaperone protein Hsp72 are indicated.

TCM Vol. 11, No. 2, 2001
69
used for two-dimensional protein elec-trophoresis (Choe and Lee 2000).
The issue of whether to use carrier am-pholytes IEF of IPG strips for 1D proteinseparation continues to be the subject ofconsiderable discussion. The developmentof IPG strips was in response partly tothe problems associated with carrierampholyte IEF (Hanash 2000). Theseproblems include the labor-intensive na-ture associated with casting IEF tubegels, lot-to-lot variability in the pH gra-dients due to difficulties with carrierampholytes production and the instabil-ity of the gradient in the higher pH range(Hanash 2000). An advantage of IPGstrips relative to tube gels is the increasein the protein loading capacity whichmay allow for a higher rate of success ofprotein identification from preparativegels (Sanchez et al. 1997). Whether ornot IPG strips offer a solution to theproblems associated with IEF is open todebate (Fichmann 1999, Lopez 1999a).
Second Dimension.
There are a numberof options available with regard to theconditions surrounding protein separa-tion in the second dimension usingSDS-PAGE. Specific gel characteristicssuch as size, thickness and percentageacrylamide, the inclusion of a stackinggel as well as the accompanying buffer-ing chemistries are all important consid-erations that will vary according to thespecific application. Homogeneous gels(10–12.5% acrylamide) can be used toseparate the protein constituents of mostcellular preparations (Lopez 1999b). Gra-dient gels can also be used to further im-prove separation but are somewhatmore difficult to reproducibly cast. Sec-ond-dimension SDS-PAGE systems areavailable in both a horizontal or verticalconfiguration. Compared to vertical sys-tems, horizontal systems are more con-venient and produce protein profileswith a greater sharpness (Görg et al.1995). Vertical systems are generally de-signed for high throughput and can runup to 10 gels in a single electrophoresistank. In this way large gel batches (10–50) can be processed in a single electro-phoresis run, which can serve to mini-mize the run-to-run variation sometimesassociated with 2-DE.
Alternative Separation Methods
Even with the recent advances in the 2-DE,it remains problematic with respect to
key issues such as quantification andthroughput relative to some of the analo-gous array technologies (Mann 1999). Forthese reasons, a number of researchersare currently developing methods of per-forming proteomics without utilizing 2-DE. Much of this research has focusedon techniques such as capillary electro-phoresis (Foret et al. 1995) or liquid chro-matography (McCormack et al. 1997) inconjunction with mass spectrometry toprovide a means of direct analysis ofprotein mixtures. Recently, global pro-tein expression was determined in yeastwith the use of isotope-coded affinitytags (Gygi et al. 1999). Although verypromising, these methods have yet toequal 2-DE with respect to either resolu-tion or capacity.
• Protein Imaging
Coomassie Stains
The Coomassie Blue Dyes, designated asG-250 and R-250, are routinely used tostain for proteins separated by 2D gel elec-trophoresis. Their popularity continuesdespite the fact that Coomassie stainscannot accurately quantify proteins andhave sensitivity well below silver and fluo-rescent stains (Hames 1990). In manyinstances a high degree of sensitivity isnot required. More importantly, unlikemany silver stain methods, CoomassieBlue staining does not greatly interferewith subsequent protein identification bymass spectrometry and is inexpensiverelative to other detection methods.
Silver Stain
With a sensitivity approximately 10–50times that of Coomassie Blue, silver stainoffers the ability to detect as little as 1ng of protein. The majority of publishedmethods to date have utilized eithersilver diammine (alkaline method) or sil-ver nitrate (acidic) as the silvering agent(Rabilloud 1992). There are currently anumber of commercially available silverstain kits, which are often utilized forreasons of convenience and consistency.Despite the superior sensitivity, thereare a number of drawbacks and limita-tions associated with the use of silverstain. Unlike Coomassie staining, theuse of silver stain is relatively expensiveowing to the high price of reagents andassociated cost of waste disposal. In addi-tion, a high background resulting from a
number of variables may cause poor res-olution of protein spots (Hames 1990).There is also a significant protein-to-protein variability relative to the extentof silver deposited on the protein that isoften a function of the degree of glyco-sylation (Hochstrasser et al. 1988). Quan-tifying protein abundance is difficultwith silver stained gels due to the poorlinearity to protein concentration (Gio-metti et al. 1991). Another major issueis the relative incompatibility of silverstain with subsequent analytical tech-niques (Hames 1990).
Fluorescent Stains and Dyes
Reliable fluorescent stains for proteindetection have become available recentlyand offer an attractive alternative to otherdetection methods. Fluorescence detec-tion technologies have a number distinctof advantages over both Coomassie andsilver stains (Patton 2000). The fact thatseveral of the SYPRO stains actuallybind non-covalently to the SDS mole-cules coating the protein results in reli-able quantitative comparison betweenproteins and also permits subsequentanalysis of the gel by Western blotting(Steinberg et al. 1996). Fluorescent stainscan accurately determine changes in pro-tein expression levels and yield a fivefoldhigher dynamic range and greater sensi-tivity than silver stain. The actual stain-ing procedure is quite simple and the re-sulting fluorescently labeled proteinscan be imaged with a UV transillumina-tor, a blue-light transilluminator or laser-scanning instrument. Several of the newerstains such as SYPRO Ruby are com-pletely compatible with mass spectrom-etry, and SYPRO Ruby has sensitivityequivalent to silver stain (Patton 2000).
Fluorescent dyes such as the cyanine(Cy) dyes have also been used to detectproteins separated by 2D gel electro-phoresis (Unlu et al. 1997). Samples ofinterest are labeled with different Cydyes, combined and subjected to 2-DE.The technique is referred to as differ-ence gel electrophoresis (DIGE) and al-lows the detection of protein differenceswith the use of a single gel, eliminatinggel-to-gel variation. The only drawbackassociated with fluorescent stains is thecost, both for reagents and imaging equip-ment, which can be somewhat prohibi-tive to laboratories running large num-bers of 2D gels.

70
TCM Vol. 11, No. 2, 2001
• Image Analysis
Regardless of the method used to detectand image the proteins, it is essential tohave a system to acquire, analyze andstore the 2-DE gel images. Image acqui-sition is most often accomplished witheither an imaging densitometer, documentscanner, charged-coupled device (CCD)camera, or storage phosphor imager.There is considerable variation in thecharacteristics of an imaging device(Sutherland 1993). Depending on the in-strumentation, the resulting image iseither acquired digitally or can be readilyconverted to a digital format. A numberof Unix, NT and Mac-based computersoftware packages are currently com-mercially available for the analysis of 2Dgel images. The current generation ofsystem packages include the Investiga-tor 2-D, ImageMaster 2D Elite, MelanieIII, PDQUEST, Phoretix 2D and the Z32D-PAGE Analysis System.
In general, image analysis entails de-tection of protein spots on the gel image,quantification of abundance and match-ing protein spots on different gels. Al-though spot detection is an automatedprocedure, inherent variations in the 2-DE process invariably necessitate a con-siderable degree of manual editing. Thedegree of confidence placed on the quan-titative analysis is highly dependent onthe normalization functions associatedwith the imaging software. Gel distor-tions, variations in sample loading anduptake, staining intensities and absorp-tion are all important issues that mustbe compensated by software (Quadroniand James 1999). Such quantitative anal-ysis of matched protein spots will resultin the generation of a substantial amountof data that is generally exported to aspreadsheet program for statistical anal-ysis. Such export files will often containquantitative measurements as well asprotein spots identification data suchas x,y coordinates, area and volume ofspecific protein spots.
• Protein Identification
Analytical techniques such as immuno-blotting, comigration analysis, microse-quencing and amino acid compositionalanalysis have all been used to identifyproteins from 2-DE gels. These techniqueswere crucial to earlier studies and are stillwidely used, but are labor-intensive and
time-consuming procedures. The appli-cation of mass spectrometry (MS) tech-niques for the analysis of proteins andpeptides is considered a major advancein the characterization and identifica-tion of proteins separated by 2-DE(Yates 2000).
Mass determination using MS re-quires conversion of proteins or peptidesinto gas-phase ions with an ionizationsource. The ions are separated based ontheir mass (m) and charge (z), by usinga mass analyzer with detection occurringvia an electronic multiplier (Corthals etal. 2000). The resulting MS spectra arerepresented as ion intensity vs. the m/zvalue. There are a number of differenttypes of mass spectrometers which areclassified according to the ionizationsource and mass analyzer employed(Patterson and Aebersold 1995). Themost widely used ionization sources arematrix-assisted laser desorption ioniza-tion (MALDI) and electrospray ion-ization (ESI) (Gevaert and Vandekerck-hove 2000). MALDI sources are mostfrequently coupled to time-of-flight(TOF) mass analyzer while ESI sourcesare generally linked to a quadrupolemass analyzer (Corthals et al. 2000).
The first technique generally em-ployed to identify proteins purified by 2-DE is peptide mass fingerprinting (PMF).This method involves chemical or enzy-matic cleavage of the protein spot of in-terest to a peptide mass fingerprint. Theindividual peptide constituents are typi-cally identified using MALDI-MS, andcompared to the theoretical mass finger-prints of proteins within various sequencedatabases (Henzel et al. 1993). Such acomparison generates a list of possiblecandidate proteins rated as a percentageof matched peptides. The success of PMFis highly dependent on the existence ofcomprehensive, searchable databases forthe species under investigation. The PMFdata are often insufficient to render aprotein identification with a high de-gree of confidence. For this reason PMFdata are often coupled with additionalMS techniques to obtain further infor-mation regarding the protein under in-vestigation (Gevaert and Vandekerck-hove 2000).
The next level of protein identifica-tion typically often involves the use oftandem mass spectrometry (MS/MS).The design of these instruments is suchthat a peptide isolated in the gas phase
of the first mass scanning stage under-goes collision-induced dissociation (CID).The masses of the peptide fragments aredetermined in the second mass stagewith the resulting data used for sequenceanalysis or database searching (Yates etal. 1995). An example of data obtainedfrom LC/MS/MS analysis of protein iso-lated from our laboratory is shown inFigure 3. Other examples of MS/MS in-strumentation include combination ofMALDI-MS with post-source decay (PSD)or ESI-MS coupled to triple-quadrupoleMS or ion-trap MS.
• Cardiovascular Biology
The ability to investigate the cellular ormolecular “big picture” with the use ofproteomics may be the one of the bestapproaches to elucidate the complex andmulti-factorial basis for many aspects ofcardiovascular biology, especially diseaseprocesses. Determining global changes inprotein expression levels of cardiomyo-cytes in response to states of ischemia,hypertension, hypertrophy, failure, in-farction, or alterations in the hepaticprotein profile associated with dyslipi-demias, obesity and/or diabetes mayprovide unique insight and understand-ing of the respective underlying mecha-nisms. In addition, monitoring the changesin entire cell-signaling pathways throughpost-translational modifications (i.e.,phosphorylation) can be instrumental indelineating the neural and/or hormonalreceptor-mediated processes involved withcardiovascular diseases. Such an ap-proach has recently been utilized to in-vestigate the changes in the cell-signal-ing pathway of mouse fibroblasts inresponse to platelet-derived growth fac-tor (PDGF). Of the 560 phosphorylatedproteins detected, approximately 100were observed to have altered phosphor-ylation state after treatment with PDGF(Soskic et al. 1999). Equally importantis the discovery of novel proteins thatmay play an integral function in normaland disease processes. Such identifica-tion may serve to not only facilitate theelucidation of molecular mechanismsbut may act as targets for novel drug de-signs and/or biomarkers.
Protein Databases
Many of the early proteomic-based studieshave involved the construction of pro-
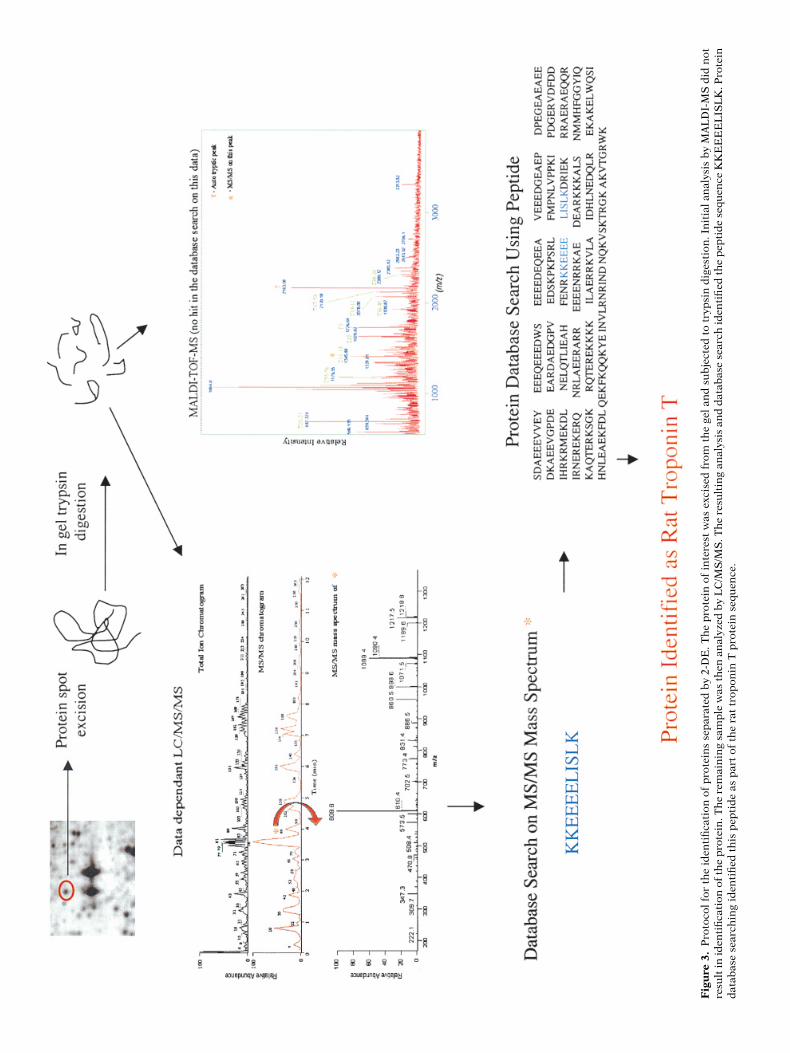
TCM Vol. 11, No. 2, 2001
71Fig
ure
3.
Pro
toco
l fo
r th
e id
enti
fica
tion
of
pro
tein
s se
par
ated
by
2-D
E. T
he
pro
tein
of
inte
rest
was
exc
ised
fro
m t
he
gel
and
su
bje
cted
to
tryp
sin
dig
esti
on. I
nit
ial
anal
ysis
by
MA
LD
I-M
S d
id n
otre
sult
in id
enti
fica
tion
of
the
pro
tein
. Th
e re
mai
nin
g sa
mp
le w
as t
hen
an
alyz
ed b
y L
C/M
S/M
S. T
he
resu
ltin
g an
alys
is a
nd
dat
abas
e se
arch
iden
tifi
ed t
he
pep
tid
e se
quen
ce K
KE
EE
EL
ISL
K. P
rote
ind
atab
ase
sear
chin
g id
enti
fied
th
is p
epti
de
as p
art
of t
he
rat
trop
onin
T p
rote
in s
equ
ence
.

72
TCM Vol. 11, No. 2, 2001
tein databases containing the molecularweight, pI, and, in some instances, func-tional data of proteins identified frommyocardial tissue. The importance ofthese databases cannot be overempha-sized as they form the fundamental ba-sis of proteomic studies. The earliest ref-erences to the construction of myocardial2-DE protein databases focused on iden-tifying proteins from human myocar-dium (Baker et al. 1992, Jungblut et al.1992). Subsequent studies have suc-ceeded in identifying additional proteins,thereby enhancing the list of alreadycharacterized proteins found in respec-tive databases (Corbett et al. 1994, Jung-blut et al. 1994). A recently establishedhuman myocardial 2-DE protein data-base identified 40 proteins, using coelec-trophoresis, immunoblotting, microse-quencing as well as comparative analysisof proteins characterized from previ-ously established cardiac protein data-bases (Kovalyov et al. 1995).
An important step forward in the ap-plication of proteomics to investigatecardiovascular disease was the compar-ative analysis of myocardial protein pro-files from different species. Utilizing in-formation contained within the human2-DE protein database, proteins origi-nating from canine, murine and rat myo-cardial tissue were tentatively identified(Corbett et al. 1995). A recent report de-scribed the identification of 64 proteinsfrom the more than 3300 proteins spe-cies resolved by 2-DE of myocardial tis-sue from the Wistar Kyoto rat (Li et al.1999). The establishment of 2-DE proteindatabases from various animal speciesand comparison to homologous proteinsin human databases, or from direct iden-tification, is of the utmost importance toproteomic-based cardiovascular researchinvolving animal models.
Since the initial reports, a number ofdevelopments have led to an increase inthe content and general accessibility ofmyocardial 2-DE protein databases. Im-
provements in the analytical arena havebeen utilized to improve the throughputand detection limits of myocardial pro-teins separated using 2-DE (Müller et al.1996, Otto et al. 1996, Sutton et al. 1995and 1997). More important has been thedevelopment of myocardial 2-DE proteindatabases that are available via the WorldWide Web: the HSC-2DPAGE (Evans et al.1997) and the HEART-2DPAGE (Pleißneret al. 1996) (see Table 1). The informa-tion from these databases is invaluableto both researchers, novice and expertalike, wishing to obtain indirect proteinidentification on a comparable 2D im-age or validation of a protein previouslyidentified through analytical analysis.There are currently a number of com-puter-imaging tools that allow directcomparison of 2D gel images from indi-vidual laboratories to master gel imageson the internet databases (Lemkin 1999,Pleißner et al. 1997).
The importance of 2-DE protein data-bases has led to a major initiative in ourlaboratory to establish several databasesto support several ongoing functionalstudies. Data compilation is currentlyunderway with respect to the identifica-tion of proteins originating from themyocardial tissue of rabbit, mouse, rat,isolated cardiac rat myocytes, neonatalcardiomyocytes and body fluids. Somedatabases are subdivided to representchamber-specific protein profiles as wellprotein profiles representing subcellu-lar oragnelle fractions. To date, a totalof 284 proteins from myocardial tissue ofthe various species have been identifiedwith the use of either MALDI-MS or MS/MS (unpublished data). Data collectionis ongoing, as is validation of completedprotein identification through databasecomparison and/or orthogonal analysis.
Cardiovascular Diseases
One area of cardiovascular disease re-search that has received considerable at-
tention through proteomics is the charac-terization of cardiomyopathies in human,bovine and canine cardiac tissues. 2-DEanalysis of human dilated cardiomyopa-thy (DCM) biopsy tissue revealed signifi-cant decreases in the abundance of 88proteins when compared to the proteinprofile of tissue from patients withischemic heart disease (IHD) (Corbett etal. 1998). Proteins identified with de-creased expression included myosinlight chain 2, desmin, ATP synthase, cre-atine kinase, HSP60 and HSP70.
The global changes in protein expres-sion accompanying pacing-induced heartfailure in canines was recently investi-gated by 2-DE in conjunction with anumber of analytical techniques and da-tabase comparison (Heinke et al. 1998,1999). The results of these studies indi-cated significant changes in the abundanceof a number of proteins associated withmitochondrial energy production (HSP70,pyruvate dehydrogenase, triosephosphateisomerase) and structural proteins suchas desmin and actin. Similarly, 2-DE anal-ysis of bovine hereditary dilated cardio-myopathy (Weekes et al. 1999) revealedchanges in the abundance of severalmitochondrial-associated proteins. Mostnotable was a disease-associated seven-fold increase in ubiquitin C-terminal hy-drolase, lending support to the proposedinvolvement of ubiquitin-mediated pro-tein degradation in the development ofheart disease (Field and Clark 1997).
The use of an animal model of heartfailure is also at the center of a major in-vestigation currently underway in ourlaboratory. Protein profiles from ventric-ular tissue of Sprague-Dawley rats withmyocardial infarction resulting from cor-onary ligation are currently being charac-terized. Tissue samples originated fromnon-infarcted free wall of the left ventri-cle as well as infarcted tissue, right ven-tricle and septum have been separatelyanalyzed in order to try to maintain ahomogenous cell type and hence ho-
Table 1. Myocardial 2-DE protein databases on the World Wide Web
Tissue source Database Web location Organization
Human, rat, dog HSC-2DPAGE http://www.harefield.nthames.nhs.uk/ Heart Science Centre, Harefield Hospital
Human HEART-2DPAGE http://www.chemie.fu-berlin.de/user/pleiss/ German Heart Institute, Berlin
Human HP-2DPAGE http://www.mdc-berlin.de/
z
emu/heart/ MDC, Berlin
Rat
RAT HEART-2DPAGE
http://gelmatching.inf.fu-berlin.de/
z
pleiss/2d/
German Heart Institute, Berlin

TCM Vol. 11, No. 2, 2001
73
mogenous expression profile. Analysis isfocusing on the changes in protein ex-pression levels as well as identifyingthe specific post-translational modifi-cations associated with the progres-sion of the disease.
Proteomics-Based Cardiovascular Research
A number of other studies have utilizedprotein profiling of myocardial tissue inan effort to elucidate the cellular andmolecular mechanisms associated withvarious pathological states. Arnott etal. (1998) observed that phenylephrine-induced hypertrophy in neonatal car-diac myocytes was associated with pro-tein expression changes. These includedsignificant increases in protein abundancewere observed for several proteins such asMLC 1 and MLC 2 (atrial and ventricu-lar). Recent work in our laboratory hasalso focused proteomic analysis of car-diac hypertrophy. Endothelin (ET)-induced hypertrophy in rat neonatalcardiomyocites resulted in twofold de-crease in 21 proteins compared to thelevels observed in untreated cells. Com-parable to that observed in previousresearch, ET-induced hypertrophy wasalso accompanied by a 30% increase inMLC 1 and MLC 2 (Macri et al. 2000a).Other studies have used a proteomics-based research approach to investigatethe changes in the myocardial proteinprofile accompanying lead exposure(Toraason et al. 1997) and the globalchanges in myocardial proteins accom-panying chronic ethanol consumption(Patel et al. 1997).
• The Future of Proteomics
Despite the fact that proteomics-basedresearch has only recently begun to bewidely used, it is fast approaching a cross-road with respect to future utilization.While recent technological advancementshave dramatically increased the potentialof proteomics, the developments withinthe next several years will dictate the ex-tent of its utility. The success of proteom-ics will greatly depend on the ability toperform the associated technologies in areproducible and high-throughput man-ner, as has occurred with genomics. Al-though automated systems have beendeveloped for protein spot excision andanalysis, areas such as sample prepara-
tion, 2-DE and image analysis continue torequire a considerable amount of manualinput. The expected improvement in the“front-end” of proteomics will requiresystems with the capacity to organize,store, retrieve and analyze the largeamount of data generated from suchstudies. Bioinformatics support to pro-teomics is currently underdeveloped, andneeds to be improved to match the levelsapplied to genomics. Part of the prob-lem stems from the difficulty of inte-grating the diversity of information(gel images, post-translational modifi-cations, functional data) generatedfrom proteomics-based research (Harryet al. 2000).
The enormous investment by indus-try in proteomics as a tool for drug dis-covery is one indication that the field ofproteomics will continue to develop andeventually become a core technology fora wide diversity of scientific laboratories(Page et al. 1999). Many pharmaceuticalcompanies have established collabora-tions with academic institutions to con-duct specific proteomic analyses in areasof interest. In this way, information fromrelated studies can be used to help cre-ate large public proteomic databases. Itis hoped that such databases will eventu-ally undergo the same exponential growthcurves as those witnessed in the field ofgenomics. This will allow the ability tothoroughly mine the data for the com-monality and differences that may existbetween biological systems. It is undersuch circumstances where the potentialof proteomics will truly be realized.
References
Anderson L, Seilhamer J: 1997. A comparisonof selected mRNA and protein abundancesin human liver. Electrophoresis 18:533–537.
Anderson NL, Anderson NG: 1998. Proteomeand proteomics: new technologies, newconcepts, and new words. Electrophoresis19:1853–1861.
Arnott D, O’Connell KL, King KL, Stults JT:1998. An integrated approach to proteomeanalysis: identification of proteins associ-ated with cardiac hypertrophy. Anal Bio-chem 258:1–18.
Baker CS, Corbett JM, May AJ, et al.: 1992. Ahuman myocardial two-dimensional elec-trophoresis database: protein characteri-sation by microsequencing and immuno-blotting. Electrophoresis 13:723–726.
Bjellqvist B, Ek K, Righetti PG, et al.: 1982.
Isoelectric focusing in immobilized pH gra-dients: principle, methodology and someapplications. J Biochem Biophys Methods6:317–339.
Chevallet M, Santoni V, Poinas A, et al.: 1998.New zwitterionic detergents improve theanalysis of membrane proteins by two-dimensional electrophoresis. Electrophore-sis 19:1901–1909.
Choe LH, Lee KH: 2000. A comparison ofthree commercially available isoelectricfocusing units for proteome analysis: themultiphor, the IPGphor and the proteanIEF cell. Electrophoresis 21:993–1000.
Corbett JM, Wheeler CH, Baker CS, et al.:1994. The human myocardial two-dimen-sional gel protein database: update 1994.Electrophoresis 15:1459–1465.
Corbett JM, Wheeler CH, Dunn MJ: 1995.Coelectrophoresis of cardiac tissue fromhuman, dog, rat and mouse. Towards theestablishment of an integrated two-dimen-sional protein database. Electrophoresis16:1524–1529.
Corbett JM, Why HJ, Wheeler CH, et al.:1998. Cardiac protein abnormalities indilated cardiomyopathy detected by two-dimensional polyacryamide gel electro-phoresis. Electrophoresis 19:2031–2042.
Corthals GL, Gygi SP, Aebesold R, PattersonSD: 2000. Identification of proteins bymass spectrometry.
In
Rabilloud T, ed. Pro-teome Research: Two-Dimensional GelElectrophoresis and Identification Meth-ods. Berlin, Springer, pp 197–227.
Dunbar BS: 1987. Basic theories and prin-ciples of electrophoresis.
In
Two-dimen-sional Electrophoresis and ImmunologicalTechniques. New York, Plenum Press, pp1–23.
Dunn MJ: 1987. Two-dimensional gel electro-phoresis of proteins. J Chromatogr 418:145–185.
Evans G, Wheeler CH, Corbett JM, Dunn MJ:1997. Construction of HSC-2DPAGE: atwo-dimensional gel electrophoresis data-base of heart proteins. Electrophoresis18:471–479.
Faux MC, Scott JD: 1996. More on targetwith protein phosphorylation: conferringspecificity by location. Trends Biochem Sci21:312–315.
Fichmann J: 1999. Advantages of immobi-lized pH gradients. Methods Mol Biol112:173–174.
Field ML, Clark JF: 1997. Inappropriate ubiq-uitin conjugation: a proposed mechanismcontributing to heart failure. CardiovascRes 33:8–12.
Foret F, Muller O, Thorne J, et al.: 1995. Anal-ysis of protein fractions by microprepara-tive capillary isoelectric focusing andmatrix-assisted laser desorption time-of-

74 TCM Vol. 11, No. 2, 2001
flight mass spectrometry. J Chromatogr A716:157–166.
Fouillit M, Poirier F, Monostori E, et al.:2000. Analysis of galectin 1-mediated cellsignaling by combined precipitation andelectrophoresis techniques. Electrophore-sis 21:275–280.
Gevaert K, Vandekerckhove J: 2000. Proteinidentification methods in proteomics.Electrophoresis 21:1145–1154.
Giometti CS, Gemmell M, Tollaksen SL, Tay-lor J: 1991. Quantitation of human leuko-cyte proteins after silver staining: a studywith two-dimensional electrophoresis.Electrophoresis 12:536–543.
Görg A, Boguth G, Obermaier C, et al.: 1995.Two-dimensional polyacrylamide gel elec-trophoresis with immobilized pH gradi-ents in the first dimension (IPG-Dalt): thestate of the art and the controversy of verti-cal versus horizontal systems. Electro-phoresis 16:1079–1086.
Görg A, Obermaier C, Boguth G, et al.: 2000.The current state of two-dimensional elec-trophoresis with immobilized pH gradi-ents. Electrophoresis 21:1037–1053.
Gygi SP, Rist B, Gerber SA, et al.: 1999.Quantitative analysis of complex proteinmixtures using isotope-coded affinity tags.Nat Biotechnol 17:994–999.
Hames BD: 1990. One dimensional polyacryl-amide gel electrophoresis. In Hames BD,Rickwood D, eds. Gel Electrophoresis ofProteins: A Practical Approach. New York,Oxford University Press, pp 51–85.
Hanash SM: 2000. Biomedical applicationsof two-dimensional electrophoresis usingimmobilized pH gradients: current status.Electrophoresis 21:1202–1209.
Harry JL, Wilkins MR, Herbert BR, et al.:2000. Proteomics: capacity versus utility.Electrophoresis 21:1071–1081.
Hart GW: 1992. Glycosylation. Curr Opin CellBiol 4:1017–1023.
Haynes PA, Gygi SP, Figeys D, Aebersold R:1998. Proteome analysis: biological assayor data archive? Electrophoresis 19:1862–1871.
Heinke MY, Wheeler CH, Chang D, et al.:1998. Protein changes observed in pacing-induced heart failure using two-dimen-sional electrophoresis. Electrophoresis 19:2021–2030.
Heinke MY, Wheeler CH, Yan JX, et al.: 1999.Changes in myocardial protein expressionin pacing-induced canine heart failure.Electrophoresis 20:2086–2093.
Henzel WJ, Billeci TM, Stults JT, et al.: 1999.Identifying proteins from two-dimensionalgels by molecular mass searching of pep-tide fragments in protein sequence data-bases. Proc Natl Acad Sci USA 90:5011–5015.
Herbert B: 1999. Advances in protein solu-bilisation for two-dimensional electro-phoresis. Electrophoresis 20:660–663.
Herbert BR, Molloy MP, Gooley AA, et al.:1998. Improved protein solubility in two-dimensional electrophoresis using tributylphosphine as reducing agent. Electro-phoresis 19:845–851.
Hochstrasser DF, Patchornik A, Merril CR:1988. Development of polyacrylamide gelsthat improve the separation of proteinsand their detection by silver staining. AnalBiochem 173:412–423.
Huber LA: 1995. Mapping cells and sub-cellu-lar organelles on 2-D gels: “new tricks foran old horse.” FEBS Lett 369:122–125.
Humphery-Smith I, Cordwell SJ, BlackstockWP: 1997. Proteome research: complemen-tarity and limitations with respect to theRNA and DNA worlds. Electrophoresis18:1217–1242.
Iannello RC, Jeffrey PL: 1990. Glycoproteinsof rat skeletal muscle sarcolemma: charac-terization by two-dimensional gel electro-phoresis and effect of denervation. ExpNeurol 108:156–161.
Jasny BR, Hines PJ: 1999. Genome prospect-ing. Science 286:443.
Jungblut P, Otto A, Regitz V, et al.: 1992.Identification of human myocard proteinsseparated by two-dimensional electro-phoresis. Electrophoresis 13:739–741.
Jungblut P, Otto A, Zeindl-Eberhart E, et al.:1994. Protein composition of the humanheart: the construction of a myocardialtwo-dimensional electrophoresis data-base. Electrophoresis 15:685–707.
Karlsson K, Cairns N, Lubec G, FountoulakisM: 1999. Enrichment of human brain pro-teins by heparin chromatography. Electro-phoresis 20:2970–2976.
Klose J: 1975. Protein mapping by combinedisoelectric focusing and electrophoresis ofmouse tissues. A novel approach to testingfor induced point mutations in mammals.Humangenetik 26:231–243.
Klose J: 1999. Genotypes and phenotypes.Electrophoresis 20:643–652.
Kovalyov LI, Shihkin SS, Efimochkin AS, etal.: 1995. The major protein expressionprofile and two-dimensional protein data-base of human heart. Electrophoresis16:1160–1169.
Lemkin PF: 1999. Comparing 2-D electro-phoretic gels across internet databases.Methods Mol Biol 112:393–410.
Li XP, Pleißner K-P, Scheler C, et al.: 1999. A two-dimensional electrophoresis database of ratheart proteins. Electrophoresis 20:891–897.
Lollo BA, Harvey S, Liao J, et al.: 1999.Improved two-dimensional gel electrophore-sis representation of serum proteins by usingProtoClear. Electrophoresis 20:854–859.
Lopez MF: 1999a. Advantages of carrierampholyte IEF. Methods Mol Biol 112:109–110.
Lopez MF: 1999b. Proteome analysis. I. Geneproducts are where the biologocal actionis. J Chromatogr B Biomed Sci Appl 722:191–202.
Lopez MF: 2000. Better approaches to find-ing the needle in a haystack: optimizingproteome analysis through automation.Electrophoresis 21:1082–1093.
Macri J, Dubay T, Matteson D, et al.: 2000a.Characterization of the protein profileassociated with endothelin-induced hyper-trophy in neonatal rat myocytes. J Mol CellCardiol 32:A60.
Macri J, McGee B, Thomas JN, et al.: 2000b.Cardiac sarcoplasmic reticulum and sar-colemmal proteins separated by two-dimensional electrophoresis: surfactanteffects on membrane solubilization. Elec-trophoresis 21:1685–1693.
Manabe T: 2000. Combination of electro-phoretic techniques for comprehensiveanalysis of complex protein systems. Elec-trophoresis 21:1116–1122.
Mann M: 1999. Quantitative proteomics? NatBiotechnol 17:954–955.
Marshall E: 1999. Human genome project:sequencers endorse plan for a draft in 1year. Science 284:1439–1441.
McCormack AL, Schieltz DM, Goode B, et al.:1997. Direct analysis and identification ofproteins in mixtures by LC/MS/MS anddatabase searching at the low-femtomolelevel. Anal Chem 69:767–776.
Meek DW: 1998. Multisite phosphorylationand integration of stress signals at p53.Cell Signal 10:159–166.
Molloy MP, Herbert BR, Walsh BJ, et al.:1998. Extraction of membrane proteins bydifferential solubilization for separationusing two-dimensional gel electrophoresis.Electrophoresis 19:837–844.
Monribot C, Boucherie H: 2000. Two-dimen-sional electrophoresis with carrier ampho-lytes. In Rabilloud T, ed. Proteome Research:Two-dimensional Gel Electrophoresis andIdentification Methods. Berlin, Springer,pp 31–52.
Müller E-C, Thiede B, Zimmy-Arndt U, et al.:1996. High-performance human myocardialtwo-dimensional electrophoresis database:edition 1996. Electrophoresis 17: 1700–1712.
O’Farrell PH: 1975. High resolution two-dimensional electrophoresis of proteins. JBiol Chem 250:4007–4021.
Otto A, Thiede B, Müller E-C, et al.: 1996.Identification of human myocardial pro-teins separated by two-dimensional elec-trophoresis using an effective samplepreparation for mass spectrometry. Elec-trophoresis 17:1643–1650.

TCM Vol. 11, No. 2, 2001 75
Page MJ, Amess B, Rohlff C, et al.: 1999. Pro-teomics: a major new technology for thedrug discovery process. Drug Discov Today4:55–62.
Patel VB, Corbett JM, Dunn MJ et al.: 1997.Protein profiling in cardiac tissue inresponse to the chronic effects of alcohol.Electrophoresis 18:2788–2794.
Patterson SD, Aebersold R: 1995. Mass spec-trometric approaches for the identificationof gel-separated proteins. Electrophoresis16:1791–1814.
Patton WF: 1999. Proteome analysis. II. Pro-tein subcellular redistribution: linkingphysiology to genomics via the proteomeand separation technologies involved. JChromatogr A 722:203–223.
Patton WF: 2000. A thousand points of light:the application of fluorescence detectiontechnologies to two-dimensional gel elec-trophoresis and proteomics. Electrophore-sis 21:1123–1144.
Pleißner K-P, Sander S, Oswald H, et al.:1996. The construction of the World-WideWeb-accessible myocardial two-dimen-sional gel electrophoresis protein database“HEART-2DPAGE”: a pratical approach.Electrophoresis 17:1386–1392.
Pleißner K-P, Sander S, Oswald H, et al.:1997. Towards design and comparison ofWorld Wide Web-accessible myocardialtwo-dimensional gel electrophoresis pro-tein databases. Electrophoresis 18:480–483.
Quandroni M, James P: 1999. Proteomics andautomation. Electrophoresis 20:664–677.
Rabilloud T: 1992. A comparison betweenlow background silver diammine and silvernitrate protein stains. Electrophoresis13:429–439.
Rabilloud T: 1996. Solubilization of proteinsfor electrophoretic analyses. Electrophore-sis 17:813–829.
Rabilloud T: 1998. Use of thiourea to increasethe solubility of membrane proteins intwo-dimensional electrophoresis. Electro-phoresis 19:758–760.
Rabilloud T, Chevallet M: 1999. Solubiliza-tion of proteins in two-dimensional elec-trophoresis. Methods Mol Biol 112:9–29.
Rhoads RE: 1999. Signal transduction path-ways that regulate eukaryotic protein syn-thesis. J Biol Chem 274:30,337–30,340.
Sachidanandam R, Weissman D, SchmidtSC, et al.: 2001. A map of human genomesequence variation containing 1.42 millionsingle nucleotide polymorphisms. Nature409:928–933.
Sanchez JC, Rouge V, Pisteur M, et al.: 1997.Improved and simplified in-gel sampleapplication using reswelling of dry immo-bilized pH gradients. Electrophoresis18:324–327.
Santoni V, Rabilloud T, Doumas P, et al.:1999. Towards the recovery of hydropho-bic proteins on two-dimensional electro-phoresis gels. Electrophoresis 20:705–711.
Soskic V, Gorlach M, Poznanovic S, et al.:1999. Functional proteomics analysis ofsignal transduction pathways of the plate-let-derived growth factor beta receptor.Biochemistry 38:1757–1764.
Steinberg TH, Haughland RP, Singer VL:1996. Applications of SYPRO orange andSYPRO red protein gel stains. Anal Bio-chem 35:238–245.
Sutherland J: 1993. Electronic imaging of elec-trophorectic gels and blots. In ChrambachA, Dunn M, Radola B, eds. Advances in Elec-trophoresis. New York, VCH, pp 1–42.
Sutton CW, Pemberton KS, Cottrell JS, et al.:1995. Identification of myocardial proteinsfrom two-dimensional gels by peptide massfingerprinting. Electrophoresis 16:308–316.
Sutton CW, Wheeler CH, Sally U, Corbett JM,et al.: 1997. The analysis of myocardialproteins by infrared and ultraviolet laserdesorption mass spectrometry. Electro-phoresis 18:424–431.
Tekaia F, Lazcano A, Dujon B: 1999. Thegenomic tree as revealed from whole pro-teome comparisons. Genome Res 9:550–557.
Toraason M, Moorman W, Mathias PI, et al.:1997. Two-dimensional electrophoretic analy-sis of myocardial proteins from lead-exposedrabbits. Electrophoresis 18:2978–2982.
Unlu M, Morgan ME, Minden JS: 1997. Dif-ference gel electrophoresis: a single gelmethod for detecting changes in proteinextracts. Electrophoresis 18:2071–2077.
Velculescu VE: 1999. Tantalizing transcrip-tomes—SAGE and its use in global gene
expression analysis. Science 286:1491–1492.
Venter JC, Adams MD, Myers EW, et al.:2001. The sequence of the human genome.Science 291:1304–1351.
Walsh MJ, Limos L, Tourtellotte WW: 1984.Two-dimensional electrophoresis of cere-brospinal fluid and ventricular fluid pro-teins, identification of enriched and uniqueproteins, and comparison with serum. JNeurochem 43:1277–1285.
Wasinger VC, Cordwell SJ, Cerpa-Pojak A, etal.: 1995. Progress with gene-product map-ping of the Mollicutes: Mycoplasma geni-talium. Electrophoresis 16:1090–1094.
Weekes J, Wheeler CH, Yan JX, et al.: 1999.Bovine dilated cardiomyopathy: proteomicanalysis of an animal model of humandilated cardiomyopathy. Electrophoresis20:898–906.
Wilkins MR, Sanchez JC, Gooley AA, et al.:1996. Progress with proteome projects:why all proteins expressed by a genomeshould be identified and how to do it. Bio-technol Genet Eng Rev 13:19–50.
Williams KL, Hochstrasser DF: 1997. Intro-duction to the proteome. In Wilkins MR,Williams KL, Appel RD, Hochstrasser DF,eds. Proteome Research: New Frontiers inFunctional Genomics. Berlin, Springer, pp1–11.
Yates JR: 2000. Mass spectrometry. Fromgenomics to proteomics. Trends Genet 16:5–8.
Yates JR, Eng JK, McCormack AL, SchieltzD: 1995. Method to correlate tandem massspectra of modified peptides to amino acidsequences in the protein database. AnalChem 67:1426–1436.
PII S1050-1738(01)00088-3 TCM
MEETING REPORTSAre you attending a conference that would be
of interest to other cardiologists?If you are interested in preparing a report of conference
highlights for TCM please contact:
Kenneth R. Chien, MD, PhD
Editor-in-ChiefTrends in Cardiovascular Medicine
Editorial Office, 655 Avenue of the AmericasNew York, NY 10010
Phone: 212-633-3918 Fax: 212-633-3977




















