
Application of Computational Kinetic Mechanism Generation to Model the Autocatalytic Pyrolysis of...
11
Application of Computational Kinetic Mechanism Generation to Model the Autocatalytic Pyrolysis of Methane Jeffrey M. Grenda,* Ioannis P. Androulakis, and Anthony M. Dean ² Corporate Strategic Research, ExxonMobil Research & Engineering Company, Annandale, New Jersey 08801 William H. Green, Jr. Department of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139 An automated computational mechanism-generation technique is applied to construct elementary- step chemical kinetic reaction models for the pyrolysis of methane at 1038 K and 0.58 atm. Under these conditions, the pyrolysis process is extremely complex and exhibits autocatalysis. The mechanism-generation approach constructs a detailed set of elementary reactions, retrieves or estimates required reaction rates and thermochemistry, and constructs a kinetic model that gives excellent agreement with experimental data for several species. Key to the success is a newly developed capability of the algorithm to identify pressure-dependent chemically activated reactions. A rate-based species-selection methodology is used to determine kinetically significant species, and the algorithm is demonstrated to identify critical low-concentration byproducts. Multistep chemically activated reactions involving the formation of cyclopentadiene and subsequent hydrogen atom production are found to be important reactions, agreeing with previous literature findings. The present work demonstrates that computer-generated kinetic models can quantitatively predict experimental behavior for conditions where reaction rate constants and thermochemistry are reasonably well established. Several topics are also presented that outline areas of ongoing research. Introduction For about two decades, it has been recognized that the process of constructing chemical kinetic models could be computationally automated and that this would be extremely helpful in predicting or simulating the complex free-radical chemistry of pyrolysis and combus- tion. This ongoing research has been primarily moti- vated by the fact that manually constructed mecha- nisms are extremely time- and labor-intensive to develop even for simple model fuels. This is not surprising given the large number of species and reactions that can occur, the requirement of accurate estimations of thermody- namic properties and reaction rates, and the need to determine which species and reaction pathways are significant. Several groups have developed kinetic-model-genera- tion approaches 1-10 and used them for a variety of homogeneous gas-phase chemistry applications. Similar work has also been used extensively and successfully to model catalytic processes 11 for complex feeds typical of petroleum applications. Given the tremendous num- ber of potentially important species and kinetic reac- tions for representative fuel feeds, the increasing use of computer-generated kinetic models will likely be required if we are ever to model the elementary chemi- cal reaction details of processes involving important complex materials such as gasoline and jet fuel. A major conceptual and algorithmic challenge in applying computational kinetic modeling techniques is determining which chemical species and reactions should be included in the kinetic model. Clearly, this challenge is analogous to the difficulties faced by the kineticist pursuing manual mechanism model development. It is highly desirable to keep the model as small as possible to speed computations and improve understanding. However, it is far from clear how the computer should determine whether particular species or reactions be- long in the mechanism. Our rate-based selection algo- rithm 12 appears to be a viable proposal for handling this difficult problem. In the current work, we present an application to methane pyrolysis, a difficult system with extremely nonlinear multistep autocatalysis 13 that pro- vides a severe test of the robustness of the rate-based algorithm. Typical existing kinetic-model-construction algo- rithms are based on a series of rules that describe known elementary reaction steps, and structural algo- rithms that identify and manage reactant and product species. Given a generated reaction, the parameters required to compute the reaction rate k(T) as a function of temperature are needed. These are often expressed in a modified Arrhenius form on the basis of experi- mental data, quantum chemical calculations, or Evans- Polanyi correlations. For combustion chemistry applications, chemically activated reactions often play a significant role in the system reaction kinetics. Such reactions involve highly energized nonthermalized species that undergo rapid internal isomerization or dissociation before collisional stabilization. The energized nonthermal intermediates are sufficiently short-lived that these reactions appear to be direct elementary reactions between the thermal- ized species, but the chemical changes between reac- * Corresponding author. Tel.: (908)730-2545. Fax: (908)- 730-3344. E-mail: [email protected]. ² Present address: Department of Chemical Engineering, Colorado School of Mines, Golden, CO 80401. 1000 Ind. Eng. Chem. Res. 2003, 42, 1000-1010 10.1021/ie020581w CCC: $25.00 © 2003 American Chemical Society Published on Web 01/28/2003
Transcript of Application of Computational Kinetic Mechanism Generation to Model the Autocatalytic Pyrolysis of...

Application of Computational Kinetic Mechanism Generation toModel the Autocatalytic Pyrolysis of Methane
Jeffrey M. Grenda,* Ioannis P. Androulakis, and Anthony M. Dean†
Corporate Strategic Research, ExxonMobil Research & Engineering Company, Annandale, New Jersey 08801
William H. Green, Jr.
Department of Chemical Engineering, Massachusetts Institute of Technology,Cambridge, Massachusetts 02139
An automated computational mechanism-generation technique is applied to construct elementary-step chemical kinetic reaction models for the pyrolysis of methane at 1038 K and 0.58 atm.Under these conditions, the pyrolysis process is extremely complex and exhibits autocatalysis.The mechanism-generation approach constructs a detailed set of elementary reactions, retrievesor estimates required reaction rates and thermochemistry, and constructs a kinetic model thatgives excellent agreement with experimental data for several species. Key to the success is anewly developed capability of the algorithm to identify pressure-dependent chemically activatedreactions. A rate-based species-selection methodology is used to determine kinetically significantspecies, and the algorithm is demonstrated to identify critical low-concentration byproducts.Multistep chemically activated reactions involving the formation of cyclopentadiene andsubsequent hydrogen atom production are found to be important reactions, agreeing with previousliterature findings. The present work demonstrates that computer-generated kinetic models canquantitatively predict experimental behavior for conditions where reaction rate constants andthermochemistry are reasonably well established. Several topics are also presented that outlineareas of ongoing research.
Introduction
For about two decades, it has been recognized thatthe process of constructing chemical kinetic modelscould be computationally automated and that this wouldbe extremely helpful in predicting or simulating thecomplex free-radical chemistry of pyrolysis and combus-tion. This ongoing research has been primarily moti-vated by the fact that manually constructed mecha-nisms are extremely time- and labor-intensive to developeven for simple model fuels. This is not surprising giventhe large number of species and reactions that can occur,the requirement of accurate estimations of thermody-namic properties and reaction rates, and the need todetermine which species and reaction pathways aresignificant.
Several groups have developed kinetic-model-genera-tion approaches1-10 and used them for a variety ofhomogeneous gas-phase chemistry applications. Similarwork has also been used extensively and successfullyto model catalytic processes11 for complex feeds typicalof petroleum applications. Given the tremendous num-ber of potentially important species and kinetic reac-tions for representative fuel feeds, the increasing useof computer-generated kinetic models will likely berequired if we are ever to model the elementary chemi-cal reaction details of processes involving importantcomplex materials such as gasoline and jet fuel.
A major conceptual and algorithmic challenge inapplying computational kinetic modeling techniques is
determining which chemical species and reactions shouldbe included in the kinetic model. Clearly, this challengeis analogous to the difficulties faced by the kineticistpursuing manual mechanism model development. It ishighly desirable to keep the model as small as possibleto speed computations and improve understanding.However, it is far from clear how the computer shoulddetermine whether particular species or reactions be-long in the mechanism. Our rate-based selection algo-rithm12 appears to be a viable proposal for handling thisdifficult problem. In the current work, we present anapplication to methane pyrolysis, a difficult system withextremely nonlinear multistep autocatalysis13 that pro-vides a severe test of the robustness of the rate-basedalgorithm.
Typical existing kinetic-model-construction algo-rithms are based on a series of rules that describeknown elementary reaction steps, and structural algo-rithms that identify and manage reactant and productspecies. Given a generated reaction, the parametersrequired to compute the reaction rate k(T) as a functionof temperature are needed. These are often expressedin a modified Arrhenius form on the basis of experi-mental data, quantum chemical calculations, or Evans-Polanyi correlations.
For combustion chemistry applications, chemicallyactivated reactions often play a significant role in thesystem reaction kinetics. Such reactions involve highlyenergized nonthermalized species that undergo rapidinternal isomerization or dissociation before collisionalstabilization. The energized nonthermal intermediatesare sufficiently short-lived that these reactions appearto be direct elementary reactions between the thermal-ized species, but the chemical changes between reac-
* Corresponding author. Tel.: (908)730-2545. Fax: (908)-730-3344. E-mail: [email protected].
† Present address: Department of Chemical Engineering,Colorado School of Mines, Golden, CO 80401.
1000 Ind. Eng. Chem. Res. 2003, 42, 1000-1010
10.1021/ie020581w CCC: $25.00 © 2003 American Chemical SocietyPublished on Web 01/28/2003

tants and products reflect the multistep nature of theprocess. The typical approach of building the mechanismcomputationally considers only classical single-stephigh-pressure-limit elementary reactions and fails toidentify these important pathways, potentially leadingto significant errors. Because each of the energizedintermediate adducts involves a coupled system ofcompeting multistep isomerization and deactivationpathways, the overall rate constants for the apparentreactions exhibit complex pressure and temperaturedependencies. If the pressure dependence due to falloffor chemical activation is known for a particular reaction,k(T,P) can be given in one of several complicated forms.For a computer algorithm to construct accurate kineticmodels, these chemically activated processes should beincluded in the construction process. The availabilityof accurate rate constant libraries and methods forestimating unknown rate constants is critical andremains the primary challenge in employing automatedmechanism-generation techniques. The current focus ison demonstrating that one can construct pressure-dependent reaction pathways using mechanism genera-tion to capture the kinetics of autocatalytic systems.
To facilitate comparison with previous literature andmanually developed mechanisms, we have deliberatelyused equivalent thermodynamic parameters of existinganalyses.13 The methane pyrolysis problem serves as agood validation example case that, although apparentlysimple, exhibits highly nonlinear and surprisingly com-plex kinetics. Recent modeling14 has modified the re-ported thermochemical properties of cyclopentadienyl,which impacts the accepted understanding of pyrolysiskinetics and reaction pathways. Using improved valuesfor the thermodynamic parameters and some of the ratecoefficients, important new reaction channels in meth-ane pyrolysis have been identified.15 In the currentwork, we demonstrate an example for methane pyrolysiswhere the omission of chemically activated pathwaysleads to substantial disagreement with experimentaldata. The results described should be viewed as avalidation of the approach used to construct mecha-nisms rather than an attempt to validate a particularmechanism. The current work is the first demonstrationof an automated mechanism approach constructingchemically activated reaction pathways in the courseof the mechanism-generation process.
Computational Mechanism-GenerationAlgorithm
The computational approach is a structure-basedmethod that represents molecular structures and chemi-cal reactions using planar graphs and matrix manipula-tion algorithms.9,16,17 The industrial version of the codeknown as XMG (ExxonMobil mechanism-generationcode)18,19 is used in the model development research.This approach is based on previous work of Klein andBroadbelt,9,10 and many of the core species identificationand manipulation routines are taken from this previouswork. The current version includes newly developedalgorithms for the construction of pressure-dependentchemically activated reaction pathways, detailed ther-modynamic reversibility and reaction rate consistency,automatic character-based nomenclature, structuralstring generation, and coupling with group additivitythermochemistry estimation,20 as well as coupling withadvanced mechanism reduction techniques.21 Althoughnot the focus here, the code can also be used in stand-
alone mode to explore and compute k(T,P) for individu-ally specified pressure-dependent reaction channels.22,23
The purpose of the current work is to incorporate thepressure-dependent channel algorithm into the overallmechanism-generation algorithm and to demonstratethe importance of including chemical activation chan-nels in the predictive capability of the algorithm.
Mathematical Representation of Species andChemically Activated Reactions. Existing kinetic-model-construction algorithms fail to accurately modelpressure-dependent chemically activated reactions. Theprimary reason for this deficiency is that most chemi-cally activated reactions consist of a series of elementarysteps that form a coupled system. The methyl radicalrecombination reaction plays an important role in themethane pyrolysis system and is an example of achemically activated approach where the initial reac-tants form a unimolecular product with a single exitchannel. Matrix species representations and the methodof computing chemical reactions for this case are shownin Figure 1. Further details can be found in theliterature9,10 for high-pressure reactions. The followingdiscussion emphasizes the unique ability of the currentapproach to consider chemically activated reactions. Thebond and electron (BE) matrix16 representation formethyl radical is shown in the upper left-hand sidematrix in Figure 1. The atoms are arbitrarily numbered,and each atom corresponds to a row and column of aconnectivity matrix. Atom connectivity is given bynonzero entries equal to the bond order in the off-diagonal locations of the matrix. Radicals include anentry on the diagonal of the matrix element containingthe unpaired electron. The numbering for methyl radicalis shown above the matrix in the top left of Figure 1.Different initial atom numberings result in differentmatrices but identical bonding. The algorithms used todetermine unique string nomenclatures for each speciesare built on canonical structures, and details can befound in the literature.9,16,17
Chemical reactions are performed by permuting en-tries in the reactant matrix bonding. A list of possiblereaction families is maintained, and proposed reactantsare tested systematically to determine whether therequired atomic reactive sites exist for the given reactionfamily. The key to the approach is the fact that chemicalreactions typically involve only a small number ofreactive atomic sites relative to the total number ofatoms in the reactants. A reaction matrix can berepresented compactly for large arbitrarily bondedspecies and is a key advantage in performing compu-tational reactions.
Returning to the chemically activated methyl radicalrecombination reaction shown in Figure 1, in the upperleft corner, the two methyl radical matrices are com-bined to form a single reactant matrix. Inspection of thismatrix indicates that the entries are such that the twooriginal distinct species are retained, with differentatom numbering. A query of the possible reactionfamilies indicates that radical recombination is a plau-sible reaction for the initial reactant matrix. To performa chemical reaction, the participating atom sites arecollected in the upper left-hand corner of the matrix.Matrix manipulation routines taken from Klein andBroadbelt9,10 are used to perform the required row andcolumn permutations. Each reaction family can beassigned a concise matrix that will perform the requiredconnectivity changes to model the reaction. The reaction
Ind. Eng. Chem. Res., Vol. 42, No. 5, 2003 1001

matrix for radical recombination is noted above thereactant matrix in Figure 1.
To perform the reaction, the reaction matrix is addedto the upper left corner of the reactant matrix (ef-fectively a padded matrix addition), and the resultingmatrix shown to the right indicates that the energizedethane adduct C2H6* has been formed from the recom-bination. The species structure is noted by the algorithmas energetically excited, and the algorithm proceeds toexplore for possible further isomerization or decomposi-tion reactions. The possible reaction pathways are firstdetermined with no consideration of reaction ratevalues. A search of the possible reaction families indi-cates that this species can undergo scission of either thecarbon-carbon bond, or one of the six identical carbon-hydrogen bonds. The reaction matrix for C-C bondscission is shown above the adduct matrix in Figure 1,and application of the matrix will result in a reversereaction to the original reactant matrices. The chemicalactivation matrix is reorganized to collect a representa-tive C-H site in the upper left corner of the matrix toperform the C-H bond scission. This is shown in thetop right matrix of Figure 1. Application of the reactionmatrix forms the product matrix shown in the bottomright of Figure 1, and the algorithm deconvolves theproduct matrix into the product matrices ethyl radical+ H. The final reaction pathway for the energizedadduct is that of collisional stabilization, which is anidentity matrix transformation to a stabilized product,or C2H6
* f C2H6. The overall set of chemically activatedreaction pathways is summarized in the bottom left ofFigure 1. The set of overall apparent reactions involvingthe thermalized species are then predicted to be CH3 +CH3 ) C2H6, CH3 + CH3 ) H + C2H5, and C2H6 ) H +C2H5. In the current approach, reaction rate constantsfor the overall reactions in the system are determinedfrom a database of QRRK,25 RRKM, or experimental
reaction rate constants as functions of pressure andtemperature.
The previous example illustrates the systematic searchmethod used to identify all chemically activated reactionpathways. The algorithm maintains a connected treestructure of chemically activated adducts, and the treestructure is developed on-line as the reaction mecha-nism is constructed. When a unimolecular product isformed from an entrance reaction, a series of proposedsubsequent unimolecular reactions is queried and con-sidered. If determined to be possible, the reactions areexecuted, analyzed, and stored in the developing treestructure. If further unimolecular isomerization is real-ized, then a new excited adduct is formed and isrecursively handled as an entry in the chemicallyactivated reaction system. A set of bimolecular products,such as from a â-scission reaction, form an exit channeland are not considered further. The assumption is thatmuch of the chemical energy released in exothermicreactions forming multiple products goes into the ex-ternal rotational modes and the relative translation ofthe products, so that the product species are notthemselves chemically activated. Once the entire systemhas been explored and all possible energized adductshave been formed, a complete set of overall apparentreaction channels can be written.
An example considering a more complex multistepchemical activation reaction is the radical additionreaction of allyl to acetylene, which is illustrated inFigure 2. As before, the initial reactant species arecombined to form a reactant matrix. The reaction matrixcontains three reactive atom sites: the radical site ofthe attacking radical and the unsaturated carbon-carbon bond. The addition reaction matrix is shownabove the reactant matrix and is applied to form acarbon-carbon bond between the radical site and oneof the unsaturated acetylene carbons, reducing the order
Figure 1. Matrix operations for the chemically activated recombination of methyl radical. A temporary reactant matrix is formed, andseveral reaction matrices are applied to the unimolecular energized adduct. The product species are deconvolved from the final productmatrix to form the system of reactions. For this simple system, the energized adduct can either return to reactants, further react toproduce ethyl radical + H, or be collisionally stabilized. The overall reaction pathway structure is shown in the inset at left.
1002 Ind. Eng. Chem. Res., Vol. 42, No. 5, 2003

of the bond and appropriately moving the radical site.The product matrix to the right of the reactant matrixin Figure 2 now contains the representation for theenergized product 1,4-pentadien-1-yl radical (the nota-tion CdCCCdC*‚ is used where d represents a doublebond and ‚ denotes the radical site). Under lower-temperature and higher-pressure conditions, the colli-sional stabilization of the initially formed adduct islikely dominant, and the overall reaction would be C2H2+ CdCC‚ ) CdCCCdC‚. This is the method used inconventional mechanism-generation approaches. At lowerpressures and higher temperatures, however, isomer-ization of the adduct can lead to a series of additionalreaction pathways before the radical is thermalized bycollisions with the bath gas. As before, the currentalgorithm treats the formed adduct CdCCCdC*‚ asenergetically excited and then explores for furtherisomerization or decomposition reactions.
A search of the reaction templates indicates that theactivated adduct can be collisionally quenched; decom-pose to the initial reactants; isomerize through hydrogenshifting; or form cyclic intermediates through intraradi-cal addition, where the radical site can add to eitherunsaturated carbon atom. Addition to the primarycarbon involves five atoms, the radical site and attachedcarbon backbone terminating with an unsaturated bond,as seen in the 1,5 intraradical addition matrix shownat the top of Figure 2. Application of this reaction matrixto the chemically activated matrix produces the ener-gized cyclopenten-4-yl radical CY(CdCCC‚C)*, shownin the top right of Figure 2. This species is similarlynoted as an energized adduct, and the algorithm againrecursively explores for possible further isomerization
or decomposition reactions. This search reveals a viableâ-scission pathway, and the matrix is reorganized in thelower right of Figure 2 to apply the given â-scissionreaction operator to the C-H site. The resulting productmatrix is shown to produce the bimolecular channelcyclopentadiene CY(CdCCCdC) + H.
Numerous other coupled channels are computed andincluded but not shown in Figure 2 for simplicity.Hydrogen shift reactions from the CdCCCdC*‚ adductproduce CdCC‚CdC* and CdCCC‚dC* as possibleintermediates, with vinyl radical CdC‚ and propadiene(allene) CdCdC as examples of possible products.Similarly, intraradical addition to the secondary unsat-urated carbon forms cyclobutenylcarbinyl radical C‚CY-(CCdCC)*, a much less thermodynamically favorablepathway relative to that shown in Figure 2. All of theexcited intermediate adducts can also be collisionallystabilized and are included and stored in the coupledsystem. It is important to note that the algorithmcaptures all of the possible pathways and maintainsthem in a connected tree structure for the estimationof the reaction rate constants of the overall apparentreactions.
Having identified the chemically activated reactionchannels for this system, we query available reactiondatabases for reaction rate constant estimates of k(T,P),as many of the kinetically important reactions formethane pyrolysis have previously been compiled. Inthe present work, we discard chemically activatedreactions whose reaction rate constants are unavailablein the database. This implies that the pathways werejudged insignificant in the off-line manual QRRK analy-ses used to compile the database. This approach can
Figure 2. Matrix operations for the chemically activated radical addition of allyl to acetylene. The multistep chemically activated pathwaysequence of allyl + acetylene producing cyclopentadiene + H is shown. Numerous additional channels resulting from isomerization of theintermediate energized adducts are not shown for brevity. The developing chemically activated pathway structures are used to generateoverall apparent reactions that are included in the mechanism-generation procedure.
Ind. Eng. Chem. Res., Vol. 42, No. 5, 2003 1003

work effectively when the database contains accurateestimates of the important reactions, but it relies on theaccuracy of the database systems to ensure that missingchannels are unimportant.
It would certainly be better if the k(T,P) values couldbe estimated during the mechanism-construction pro-cedure. Our ongoing research is focused on directlycoupling a k(T,P) estimation procedure into the model-construction algorithm15 and will allow us to test thereliability of the current procedure. For larger chemi-cally activated adducts, an enormous number of isomer-izations are possible, making it impractical to directlyand accurately compute all of the k(T,P) reaction rates.To enable this, we have constructed a numerical screen-ing procedure to avoid including isomers and reactionsthat are negligible under the conditions of interest22 anda criterion for determining whether k(T,P) will differsignificantly from k∞(T) for a reaction through a largeadduct without requiring the full k(T,P) calculation.These techniques will serve as effective screeningmethods to enable the computation of large chemicallyactivated systems.
Estimation of Thermochemistry. Accurate esti-mations of the enthalpy and entropy are critical inobtaining accurate reaction rates estimates, obtainproper energy balances, and ensuring reaction micro-scopic reversibility. From a computational perspective,this accuracy is especially important because it elimi-nates the need for reaction rate estimate rules in bothreaction directions, thus simplifying the approach. Forreactions written identically in both directions (such asabstractions), a preferred reaction direction is specifiedon the basis of exothermicity.
The computational mechanism-generation procedureprovides challenges in that the number of speciesconsidered can be large and the structure of the productspecies cannot be determined ahead of time and mightbe complex. A fast and reasonably accurate estimationprocedure is required for those species that cannot belocated in appropriate literature database libraries. Thisis accomplished through coupling with an automatedgroup additivity approach to estimate thermochemicaland physical properties of molecules and radicals.20,24
The approach utilizes a large existing database of groupcontributions24 required for the accurate estimation ofradicals critical to combustion chemistry applications.Representative examples of the thermodynamic datautilized by the mechanism-generation procedure are
shown in Table 1. The additivity approach makesestimates of the heat and entropy of formation and theheat capacity at several discrete temperatures for usewith reaction rate constant estimation rules describedbelow. The calculation of reduced frequencies for usewith QRRK25 analyses is also automatically performed.
Estimation of Reaction Rates k∞(T). The kinetic-model-construction process depends on the ability toidentify kinetically significant reactions for the condi-tions of interest and to estimate their high-pressure-limit rate constants k∞(T). Reaction-identification andrate-estimation rules of different levels of complexityhave been included for a wide variety of reactionfamilies important in hydrocarbon pyrolysis and oxida-tion systems. Important reaction families include inter-and intramolecular abstraction, intramolecular addi-tion, recombination/dissociation, and addition/â-scission.Rate estimates for several dozen of these reactionfamilies have been detailed in recent publications.26-32
Newly developed rate-estimation rules that are usedin the current approach incorporate substantial materialfrom the literature and include a significant amount ofstructural detail in assembling the hierarchy of rulesfor reactant rate constant estimation. This informationincludes examples such as the bond type and locationwithin the species, the effect of resonant stabilizationand bond strength, the number and effect of rotor losseson reaction intermediates, and ring-strain in the transi-tion state. The rate constant rules include specialestimation rules for reactions involving unique radicalspecies such as H, O, OH, CH3, etc. Reactions that havebeen studied with great detail and have experimentalor ab initio modeling estimates available can be re-trieved from a reaction rate constant archive.
Automatic Generation of Species Nomenclature.As shown earlier in Figures 1 and 2, the molecularstructure is represented as a connected graph anddecomposed iteratively into unique substituents thatcan be reconstructed systematically to generate a uniquelexigraphical and parenthetical string nomenclatureused to identify species uniquely.9 Simple examples ofunique molecular strings include C(H3)C(H3) for ethane,C[C(H2)C(H3)H] for propene, and C(H)C(H)C(H)C(H)C-(H2) for 1,3-cyclopentadiene. Although such stringsserve the critical purpose of identifying species uniquely,this nomenclature is extremely burdensome for thekinetic modeler to interpret manually. To address thisproblem, we have developed an automated species
Table 1. Example Thermodynamic Predictions Obtained from the Group Additivity Estimation Procedure CoupledDirectly to the Mechanism-Generation Algorithma
species Hfb Sfb Cp300 400 500 600 800 1000 1500
CCC -25.33 64.50 17.88 22.63 27.05 30.93 37.11 41.88 49.36 C3 H8CCC‚ 23.67 69.29 17.11 21.27 25.14 28.53 33.95 38.14 44.70 C3 H7CC‚C 21.02 70.31 16.38 20.30 23.95 27.54 33.36 37.43 44.16 C3 H7CCCC -30.26 73.92 23.38 29.58 35.30 40.28 48.18 54.22 63.56 C4 H10CC(C)C -32.50 70.43 23.11 29.52 35.37 40.42 48.37 54.36 63.92 C4 H10C*CCC -0.11 73.61 20.57 26.09 31.04 35.28 41.96 46.97 54.75 C4 H8CC*CC -3.22 72.39 20.70 25.74 30.42 34.58 41.34 46.44 54.40 C4 H8C*CC‚C 33.39 69.80 19.03 24.27 28.96 32.96 39.21 43.83 50.90 C4 H7C*CCC‚ 48.89 78.40 19.80 24.73 29.13 32.88 38.80 43.23 50.09 C4 H7C*CC(C)C -6.53 80.34 25.80 32.98 39.46 44.77 53.22 59.45 69.25 C5 H10C*CC‚(C)C 24.77 76.65 24.01 30.60 36.72 41.80 49.94 55.90 65.18 C5 H9CY(CCCCC) -18.74 70.00 19.93 28.24 35.86 42.32 52.29 59.48 70.25 C5 H10CY(C*CCCC) 7.82 70.62 19.56 26.79 33.22 38.57 46.73 52.57 61.31 C5 H8CY(C*CC*CC) 31.26 66.87 18.14 24.69 30.22 34.66 41.21 45.78 52.56 C5 H6a Data used to compute NASA coefficients for determining required temperature-dependent thermodynamic properties. Group
contribution values are taken from the NJIT Database24 of Bozzelli et al. b Units: enthalpy, kcal/mol; entropy, cal/mol/K; specific heat,cal/mol/K.
1004 Ind. Eng. Chem. Res., Vol. 42, No. 5, 2003

nomenclature generator based on the convention ofBozzelli24 that parses arbitrary species into readablestrings. Our computer-generated nomenclature givesCC, CCdC, and CY(CdCCdCC) for ethane, propene,and cyclopentadiene, respectively. We maintain a checkthat prevents two isomers from being assigned the sameuser-readable name. Common species names are usedif defined in a database, such as the nomenclature C2H6and C2H5 for ethane and ethyl radical, respectively. Ashort list of representative reactions is given in Figure3, illustrating the format of the elementary reactionsgenerated and output of the approach, and highlightsthe relative ease by which the reactions can be inter-preted for use with manual mechanism-constructionefforts. It is our experience that the capability toconstruct and output a detailed, human-readable mech-anism transparent to differences between manual andautomated construction is a key modeling capability andcan be used as a powerful modeling tool.
Species Selection and Model Construction. Achallenging aspect of kinetic model construction isdevising species-selection and model-generation algo-rithms that capture important species and reactions butthat avoid including unnecessary species or reactions.In the current work, the model is constructed iteratively,using a modified version of the rate-based algorithm.12
Kinetically significant species (called “reacted”) appearas reactants and products in the model, and “unreacted”species appear only as products. To construct themechanism, the algorithm selects the species from theunreacted species pool that has the largest kineticproduction rate. The algorithm then generates all pos-sible reactions of the species with itself and withprevious reacted species, increasing the reaction modelsize and adding any new unknown species to theunreacted species pool. For each single-product reaction,the algorithm is extended as described previously totreat that reaction as chemically activated, and so itundergoes an arbitrary series of unimolecular isomer-ization reactions, terminated either by collisional quench-ing or by the production of exit products. Once allreactions are found, the original reactant species is thenredesignated a reacted species. The new larger reactionscheme is solved, and the resulting temporal species
concentrations are used to repeat the entire processuntil all of the remaining unreacted species are formedat rates below user-specified tolerances. One definedmodel-construction procedure convergence criterion canbe written as12
where Rj is the formation rate of the jth unreactedspecies computed using the current kinetic model, τ isthe user-specified time scale of the simulation, ε is theuser-specified tolerance, and the symbols indicate thatspecies j is included from the reacted species set. In thepresent work, we defined the characteristic rate Rcharas the formation rate of the species with the maximumcurrent rate of production.
Note that, as the algorithm progresses and reactsavailable species together to generate chemical reac-tions, species whose production rates are initiallydetermined to be unimportant are continually beingreevaluated to assess whether they have evolved to besignificant. The rate-based algorithm has the importantadvantage of converging to a model that includeskinetically significant species but relatively few unim-portant species. In contrast, most model-constructionalgorithms presented in the literature do not includeany rate-based significance test and might miss impor-tant species and reactions and include unimportantpathways. However, tight tolerances might be requiredto ensure that all of the minor species important in astiff nonlinear autocatalytic system are included. Thisis particularly true for cases where the autocatalysisloop involves several elementary steps, because thekinetic importance of this loop will not become evidentuntil several of the species involved have been identifiedas significant.
The methane pyrolysis case has such character andprovides an excellent test of the robustness of thespecies-selection criterion used by the algorithm, as wellas of the importance of including the chemically acti-vated channels. The kinetics of this system are alsoextremely nonlinear during the induction phase, provid-
Figure 3. Representative output of generated elementary reactions and overall apparent chemically activated reactions from the automatedchemical reaction generation algorithm. Internal matrix representations of species are translated into readable string nomenclature.Simple common species names such as methane or hydrogen are taken from a database. Other more complex species are named using anewly developed species-nomenclature-generation algorithm.
Rj(t) < εRchar(t) ∀j ∈{reacted species}to < t < to + τ (1)
Ind. Eng. Chem. Res., Vol. 42, No. 5, 2003 1005

ing a particularly stringent test of the rate-basedspecies-selection criterion. To illustrate the effects ofchanging the tolerance ε and the importance of identify-ing the chemically activated channels, a variety ofsimulations are presented in the following section.
Model Predictions and Comparison with Ex-perimental Data. The process for mechanism con-struction of methane pyrolysis is illustrated in Figure4. In the first step in the top right of Figure 4, theoriginal neat methane has undergone C-H initiationreactions to produce methyl radicals and H atoms. Atthis point, the species lists include CH4 as a reactedspecies and H and CH3 as unreacted species. After twoquick iterations, H and CH3 have been included in themodel, generating the new product species ethane, H2,and ethyl radical (C2H5). The lines leading to C2H5 andH are drawn differently to indicate that this productset is only formed by a multistep chemically activatedreaction; conventional high-pressure-limit-based model-generation programs would omit this step. After twomore iterations, ethane and H have been included asreactive species, significantly improving the model.However, in the following iteration, the ethyl radical isincorporated into the model, and this opens up manychannels leading to new species, including severaladditional chemically activated channels. The growthto higher-molecular-weight species begins through re-combination reactions to form propane and butane andchemically activated routes to propyl radical and butylradical. At this point, a major product, ethene, is firstidentified. It is clear that the numbers of generatedspecies and reactions grow rapidly. The mechanism-generation process is iterated several dozen more timesbefore the completeness criterion of eq 1 is satisfied toour specified precision.
The predictions of these computer-generated modelsare compared with the experimental data in Figures5-7. The operating conditions of T ) 1058 K and P )0.58 atm were selected to be consistent with availableexperimental data.33,34 The solid lines show the numer-ical predictions for different product species as a func-
tion of time, and the symbols represent availableexperimental data.
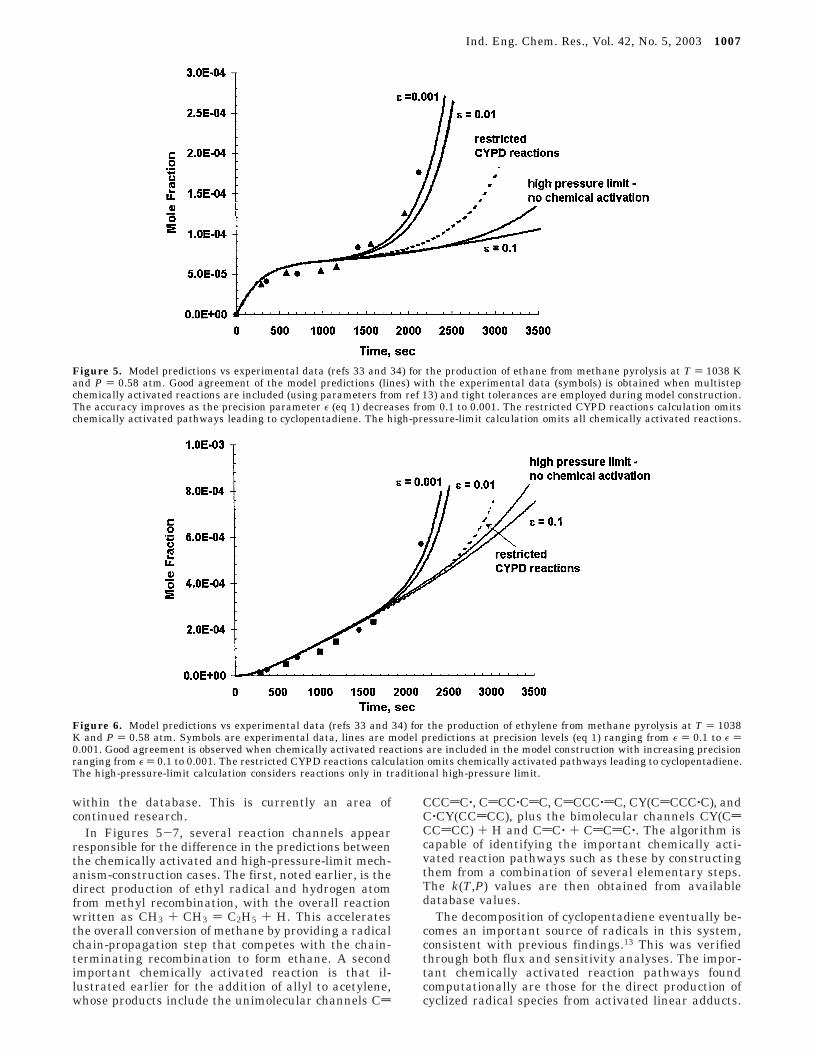
Figure 5 shows a comparison between numericalpredictions and experimental measurements for theproduction of ethane versus time. The significant induc-tion period, after which the production of ethane beginsto accelerate nonlinearly, can be readily seen in theexperimental data. Several numerical predictions areshown in Figure 5. The second curve from the bottomlabeled “no chemical activation” considers only reactionswritten in the high-pressure limit and ignores channelsthat are chemically activated. In this case, which ischaracteristic of existing computational mechanism-generation approaches, the induction time is not cap-tured. The ethane production growth occurs very slowlyand does not reach the levels of the experiment evenover a long time period. Several additional curves areshown in Figure 5 that include the new capability ofincorporating chemically activated reactions. A discus-sion of the restricted reactions (dashed) curve is post-poned temporarily. The remaining numerical predic-tions (solid lines) in Figure 5 vary the level of precisionselected as defined by eq 1. For the precision level of ε
) 0.1 (larger precision level magnitudes are equivalentto less strict tolerances in eq 1), the temporal predictionsof the ethane levels still remain in disagreement withthe experimental values, as, within this tolerance level,the growth of the mechanism to include higher-molec-ular-weight species has not occurred. Only a smallnumber of species (17) have been included in themechanism at this termination criterion. Numericaltests have shown that, for a variety of systems, precisionlevels usually less than 1% of the maximum speciesproduction rate are required to adequately capture theunderlying detail of the radical chemistry. When thetolerance used is ε ) 0.01, the agreement between thepredicted and experimental values improves signifi-cantly. The acceleration of ethane is captured, as higher-molecular-weight species have begun to be included inthe mechanism. As the tolerance is tightened furtherby an order of magnitude, some additional improvementis seen in the agreement. Further restriction of precisionresults in effectively nonobservable macroscopic changesin the results. Clearly, the agreement of the modelpredictions with experiment requires the incorporationof chemically activated channels in the mechanism-construction procedure.
The ethylene yields obtained from each of the modelpredictions are compared with experiment in Figure 6.In the shorter time frame, the agreement between allmodels is close, but neither the low-precision nor high-pressure-limit approaches are able to capture the gradualincrease in production at later times, consistent withthe underprediction of the acceleration of ethane shownearlier in Figure 5. Some additional improvements areagain seen in the agreement between the model andexperiment with tightening levels of precision rangingfrom ε ) 0.1 to ε ) 0.001. The predicted propylene yieldsare presented in Figure 7 and show gradually improvingagreement to the experimental data over ranges ofprecision varying over 3 orders of magnitude from ε )0.1 to ε ) 0.001. No significant adjustment of rateconstants from our existing database was performed inan effort to improve the fit of the data. It is also possiblethat disagreements with experiment might also beattributed to missing chemical activation pathways
Figure 4. Iterative construction of the kinetic mechanism for thepyrolysis of methane. Species that appear as both reactants andproducts (reacted) are enclosed by solid box. Edge (unreacted)product species are continually evaluated for inclusion as reactantsin the developing model. Dashed lines indicate multistep chemi-cally activated reactions that would be missed by conventionalmodel-construction algorithms. Some arrows have been omittedfor clarity.
1006 Ind. Eng. Chem. Res., Vol. 42, No. 5, 2003
within the database. This is currently an area ofcontinued research.
In Figures 5-7, several reaction channels appearresponsible for the difference in the predictions betweenthe chemically activated and high-pressure-limit mech-anism-construction cases. The first, noted earlier, is thedirect production of ethyl radical and hydrogen atomfrom methyl recombination, with the overall reactionwritten as CH3 + CH3 ) C2H5 + H. This acceleratesthe overall conversion of methane by providing a radicalchain-propagation step that competes with the chain-terminating recombination to form ethane. A secondimportant chemically activated reaction is that il-lustrated earlier for the addition of allyl to acetylene,whose products include the unimolecular channels Cd
CCCdC‚, CdCC‚CdC, CdCCC‚dC, CY(CdCCC‚C), andC‚CY(CCdCC), plus the bimolecular channels CY(CdCCdCC) + H and CdC‚ + CdCdC‚. The algorithm iscapable of identifying the important chemically acti-vated reaction pathways such as these by constructingthem from a combination of several elementary steps.The k(T,P) values are then obtained from availabledatabase values.
The decomposition of cyclopentadiene eventually be-comes an important source of radicals in this system,consistent with previous findings.13 This was verifiedthrough both flux and sensitivity analyses. The impor-tant chemically activated reaction pathways foundcomputationally are those for the direct production ofcyclized radical species from activated linear adducts.
Figure 5. Model predictions vs experimental data (refs 33 and 34) for the production of ethane from methane pyrolysis at T ) 1038 Kand P ) 0.58 atm. Good agreement of the model predictions (lines) with the experimental data (symbols) is obtained when multistepchemically activated reactions are included (using parameters from ref 13) and tight tolerances are employed during model construction.The accuracy improves as the precision parameter ε (eq 1) decreases from 0.1 to 0.001. The restricted CYPD reactions calculation omitschemically activated pathways leading to cyclopentadiene. The high-pressure-limit calculation omits all chemically activated reactions.
Figure 6. Model predictions vs experimental data (refs 33 and 34) for the production of ethylene from methane pyrolysis at T ) 1038K and P ) 0.58 atm. Symbols are experimental data, lines are model predictions at precision levels (eq 1) ranging from ε ) 0.1 to ε )0.001. Good agreement is observed when chemically activated reactions are included in the model construction with increasing precisionranging from ε ) 0.1 to 0.001. The restricted CYPD reactions calculation omits chemically activated pathways leading to cyclopentadiene.The high-pressure-limit calculation considers reactions only in traditional high-pressure limit.
Ind. Eng. Chem. Res., Vol. 42, No. 5, 2003 1007
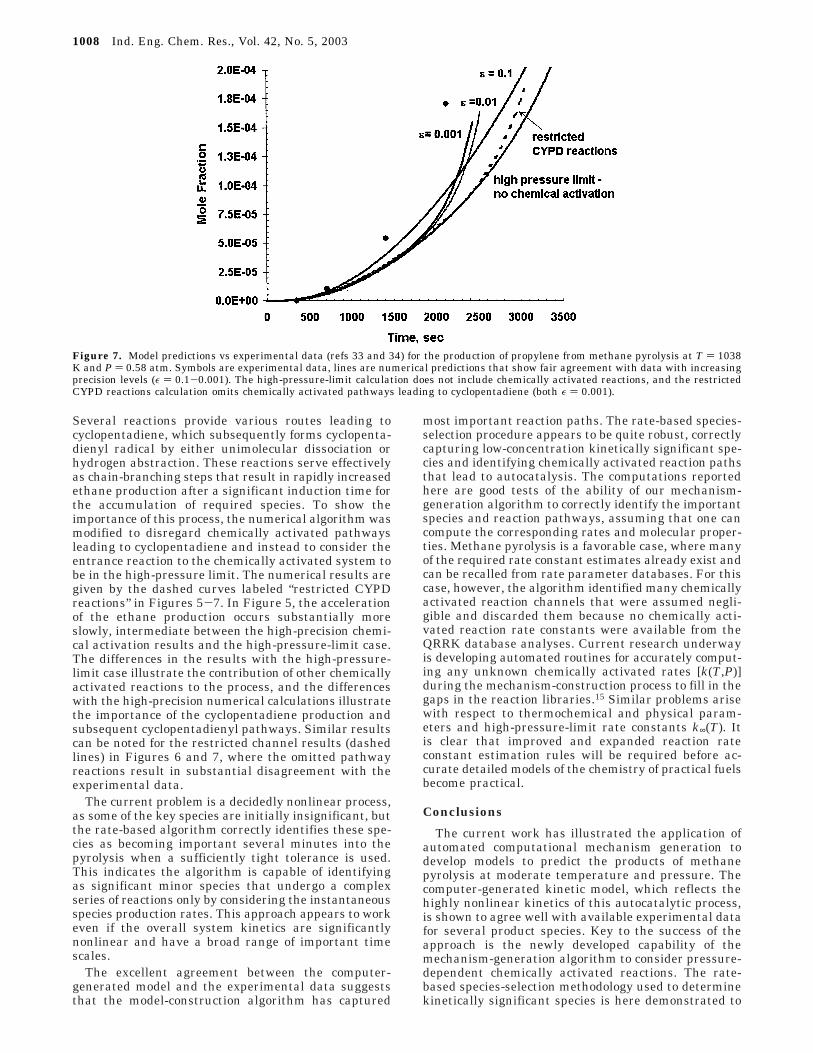
Several reactions provide various routes leading tocyclopentadiene, which subsequently forms cyclopenta-dienyl radical by either unimolecular dissociation orhydrogen abstraction. These reactions serve effectivelyas chain-branching steps that result in rapidly increasedethane production after a significant induction time forthe accumulation of required species. To show theimportance of this process, the numerical algorithm wasmodified to disregard chemically activated pathwaysleading to cyclopentadiene and instead to consider theentrance reaction to the chemically activated system tobe in the high-pressure limit. The numerical results aregiven by the dashed curves labeled “restricted CYPDreactions” in Figures 5-7. In Figure 5, the accelerationof the ethane production occurs substantially moreslowly, intermediate between the high-precision chemi-cal activation results and the high-pressure-limit case.The differences in the results with the high-pressure-limit case illustrate the contribution of other chemicallyactivated reactions to the process, and the differenceswith the high-precision numerical calculations illustratethe importance of the cyclopentadiene production andsubsequent cyclopentadienyl pathways. Similar resultscan be noted for the restricted channel results (dashedlines) in Figures 6 and 7, where the omitted pathwayreactions result in substantial disagreement with theexperimental data.
The current problem is a decidedly nonlinear process,as some of the key species are initially insignificant, butthe rate-based algorithm correctly identifies these spe-cies as becoming important several minutes into thepyrolysis when a sufficiently tight tolerance is used.This indicates the algorithm is capable of identifyingas significant minor species that undergo a complexseries of reactions only by considering the instantaneousspecies production rates. This approach appears to workeven if the overall system kinetics are significantlynonlinear and have a broad range of important timescales.
The excellent agreement between the computer-generated model and the experimental data suggeststhat the model-construction algorithm has captured
most important reaction paths. The rate-based species-selection procedure appears to be quite robust, correctlycapturing low-concentration kinetically significant spe-cies and identifying chemically activated reaction pathsthat lead to autocatalysis. The computations reportedhere are good tests of the ability of our mechanism-generation algorithm to correctly identify the importantspecies and reaction pathways, assuming that one cancompute the corresponding rates and molecular proper-ties. Methane pyrolysis is a favorable case, where manyof the required rate constant estimates already exist andcan be recalled from rate parameter databases. For thiscase, however, the algorithm identified many chemicallyactivated reaction channels that were assumed negli-gible and discarded them because no chemically acti-vated reaction rate constants were available from theQRRK database analyses. Current research underwayis developing automated routines for accurately comput-ing any unknown chemically activated rates [k(T,P)]during the mechanism-construction process to fill in thegaps in the reaction libraries.15 Similar problems arisewith respect to thermochemical and physical param-eters and high-pressure-limit rate constants k∞(T). Itis clear that improved and expanded reaction rateconstant estimation rules will be required before ac-curate detailed models of the chemistry of practical fuelsbecome practical.
Conclusions
The current work has illustrated the application ofautomated computational mechanism generation todevelop models to predict the products of methanepyrolysis at moderate temperature and pressure. Thecomputer-generated kinetic model, which reflects thehighly nonlinear kinetics of this autocatalytic process,is shown to agree well with available experimental datafor several product species. Key to the success of theapproach is the newly developed capability of themechanism-generation algorithm to consider pressure-dependent chemically activated reactions. The rate-based species-selection methodology used to determinekinetically significant species is here demonstrated to
Figure 7. Model predictions vs experimental data (refs 33 and 34) for the production of propylene from methane pyrolysis at T ) 1038K and P ) 0.58 atm. Symbols are experimental data, lines are numerical predictions that show fair agreement with data with increasingprecision levels (ε ) 0.1-0.001). The high-pressure-limit calculation does not include chemically activated reactions, and the restrictedCYPD reactions calculation omits chemically activated pathways leading to cyclopentadiene (both ε ) 0.001).
1008 Ind. Eng. Chem. Res., Vol. 42, No. 5, 2003

recognize low concentration but kinetically significantspecies under extremely nonlinear temporal productionconditions.
The new algorithm provides increased confidence thatthe predictions are not in error because particularspecies or reactions were arbitrarily left out. Thealgorithm is capable of identifying significant speciesby considering the instantaneous species production rateas a function of time even if its concentration is smalland the overall kinetics are significantly nonlinear. Thealgorithm monitors species that are numerically sig-nificant and solves the system of differential equationsto a user-specified level of precision. Essentially, theuser need only input the initial concentrations, thedesired degree of conversion and precision, and a set ofestimation rules, and the computer will generate theset of significant reactions, intermediates, and productsand their concentrations as a function of time.
A critical aspect of the calculations is the accuracy towhich species thermochemistry and reaction rate con-stant values can be assigned. Many of these values areunknown or have significant uncertainty, and these areundoubtedly the largest sources of error. Our ability toquantitatively predict overall kinetics is limited nowprimarily by the accuracy with which the individual rateparameters are known or can be estimated, rather thanother computational modeling issues. The advent ofgeneral-purpose reaction-scheme-generation programsplaces a renewed emphasis on the importance of devel-oping accurate rate-estimation techniques and providesan efficient way to use those rate-estimation rules thatare already available. It is hoped that improved empiri-cal and theoretical estimates for k(T,P) will soon bedeveloped for additional reaction systems, allowingconstruction of more accurate chemical kinetic modelsfor a broader range of application.
Acknowledgment
J.M.G. acknowledges the generous time and discus-sions spent with Prof. Joseph Bozzelli regarding thedetails of chemically activated reaction systems. All ofthe detailed group additivity values used in the auto-mated thermochemical estimation procedures have beendeveloped through his research group at the New JerseyInstitute of Technology. All authors acknowledge thecomputer programming assistance of Dr. Pawel Peczakof ExxonMobil Research and Engineering and Mr. DavidM. Matheu of MIT. W.H.G. acknowledges financialsupport from the Division of Chemical Sciences, Officeof Basic Energy Sciences, Office of Energy Research,U.S. Department of Energy, under Grant DE-FG02-98ER14914.
Literature Cited
(1) Chevalier, C.; Pitz, W. J.; Warnatz, J.; Westbrook, C. K.;Melenk, H. Hydrocarbon Ignition: Automatic Generation of Reac-tion Mechanisms and Applications to Modeling of Engine Shock.Proc. Combust. Inst. 1992, 24, 1362-1367.
(2) Chinnick, S. J.; Baulch, D. L.; Ayscough, P. B. An ExpertSystem for Hydrocarbon Pyrolysis Reactions. Chemom. Intell. Lab.Syst. 1988, 5, 39-52.
(3) DiMaio, F. P.; Lignola, P. G. KING, a Kinetic NetworkGenerator. Chem. Eng. Sci. 1992, 51, 2713-2718.
(4) Dente, M.; Pierucci, S.; Ranzi, S. E.; Bussani, G. NewImprovements in Modeling Kinetic Schemes for HydrocarbonPyrolysis Reactors. Chem. Eng. Sci. 1992, 47, 2629-2634.
(5) Blurock, E. S. Reaction: System for Modeling ChemicalReaction. J. Chem. Inf. Comput. Sci. 1995, 35, 607-616.
(6) Prickett, S. E.; Mavrovouniotis, M. L. Construction ofComplex Reaction Systems III. An Example: Alkylation of Olefins.Comput. Chem. Eng. 1997, 21, 1325-1337.
(7) Tomlin, A. S.; Turanyi, T.; Pilling, M. J. Mathematical Toolsfor the Construction, Investigation and Reduction of CombustionMechanisms. In Low-Temperature Combustion and Autoignition;Pilling, M. J., Ed.; Comprehensive Chemical Kinetics Series;Elsevier: Amsterdam, 1997; Vol. 35, Chapter 4.
(8) Glaude P. A.; Warth, V.; Fournet, R.; Battin-Leclerc, F.;Scacchi, G.; Come, G. M. Modelling of the Oxidation of n-Octaneand n-Decane Using Automatic Generation of Mechanisms. Int.J. Chem. Kinet. 1998, 30, 949-959.
(9) Broadbelt, L. J.; Stark, S. M.; Klein, M. T. ComputerGenerated Pyrolysis Modeling: On-the-Fly Generation of Species,Reactions, and Rates. Ind. Eng. Chem. Res. 1994, 33, 790-799.
(10) Broadbelt, L. J.; Stark, S. M.; Klein, M. T. Termination ofComputer-Generated Reaction Mechanisms: Species Rank-BasedConvergence Criterion. Ind. Eng. Chem. Res. 1995, 34, 2566-2573.
(11) Quann, R. J.; Jaffe, S. B. Building Useful Models ofComplex Reaction Systems in Petroleum Refining. Chem. Eng. Sci.1996, 51 (10), 1615.
(12) Susnow, R. G.; Dean, A. M.; Green, W. H., Jr.; Peczak, P.;Broadbelt, L. J. Rate-Based Construction of Kinetic Models forComplex Systems. J. Phys. Chem. A 1997, 101 (20), 3731-3740.
(13) Dean, A. M. Detailed Kinetic Modeling of Autocatalysisin Methane Pyrolysis. J. Phys. Chem. 1990, 94, 1432-1439.
(14) Kiefer, J. H.; Tranter, R. S.; Wang, H.; Wagner, A. F.Thermodynamic Functions for the Cyclopentadienyl Radical: TheEffect of Jahn-Teller Distortion. Int. J. Chem. Kinet. 2001, 33,834-845.
(15) Matheu, D. M.; Aghalayam, P.; Green, W. H., Jr.; Grenda,J. M. Capturing Pressure-Dependence in Automated MechanismGeneration for Pyrolysis and Combustion. In Book of Abstracts:17th International Symposium on Gas Kinetics; IPTC, UniversitatEssen: Essen, Germany, 2002.
(16) Ugi, I.; Bauer, J.; Brandt, J.; Freidrich, J.; Gasteiger, J.;Jochum, C.; Schubert, W. New Applications of Computers inChemistry. Angew. Chem., Int. Ed. Engl. 1979, 18, 111-123.
(17) Tarjan, R. E. Graph Algorithms in Chemical Computation.In Algorithms for Chemical Computations; Christofferson, R. E.,Ed.; American Chemical Society: Washington, DC, 1977.
(18) Grenda, J. M.; Dean, A. M.; Green, W. H., Jr.; Peczak, P.K. Recent Advances in Automated Kinetic Mechanism Generation.Presented at the Work-in-Progress Poster (WIPP) Session, at the27th International Symposium on Combustion, Boulder, CO, Aug2-7, 1998.
(19) Grenda, J. M.; Bozzelli, J. W.; Dean, A. M. AutomatedMethods of Treating Chemically Activated Reactions in KineticMechanism Generation. In Book of Abstracts: Eighth InternationalConference on Numerical Combustion; Presented at the EightSIAM International Conference on Numerical Combustion. AmeliaIsland, FL.
(20) Grenda, J. M.; Bozzelli, J. W.; Dean, A. M. AutomatedGroup Additivity Estimation of Thermodynamics for Use inComputational Mechanism Generation. Presented at the Work-in-Progress Poster (WIPP) Session, at the 27th InternationalSymposium on Combustion, Boulder, CO, Aug 2-7, 1998.
(21) Grenda, J. M.; Androulakis, I. P.; Bozzelli, J. W. CombinedUse of Automated Kinetic Mechanism Generation and Reductionin the Development of Chemical Reaction Models. In Proceedingsof 2nd Joint Meeting of the U.S. Sections of the CombustionInstitute; The Combustion Institute: Pittsburgh, PA, 2001.
(22) Matheu, D. M.; Lada, T. A.; Green, W. H., Jr.; Dean, A.M.; Grenda, J. M. Rate-Based Screening of Pressure-DependentReaction Networks. Comput. Phys. Commun. 2001, 138, 237-249.
(23) Matheu, D. M.; Green, W. H., Jr.; Grenda, J. M. Compu-tational Application of Automated Pressure-Dependent Mecha-nism Generation: Reaction through Cycloalkyl Intermediates. Int.J. Chem. Kinet., 2003, 35 (3), 95-119.
(24) Lay, T. H.; Bozzelli, J. W.; Dean, A. M.; Ritter, E. R. J.Phys. Chem. 1995, 99, 145-85.
(25) Bozzelli, J. W.; Chang, A. Y.; Dean, A. M. Combust. Sci.Technol. 1991, 80, 63-85.
Ind. Eng. Chem. Res., Vol. 42, No. 5, 2003 1009

(26) Green, W. H.; Barton, P. I.; Bhattacharjee, B.; Matheu,D. M.; Schwer, D. A.; Song, J.; Sumathi, R.; Carstensen, H. H.;Dean, A. M.; Grenda, J. M. Computer Construction of DetailedChemical Kinetic Models for Gas-Phase Reactors. Ind. Eng. Chem.Res. 2001, 40, 5362-5370.
(27) Sumathi, R.; Green, W. H. A priori Rate Constants forKinetic Modeling. Theor. Chem. Acc. 2002, 108, 187-213.
(28) Sumathi, R.; Carstensen, H. H.; Green, W. H. ReactionRate Prediction via Group Additivity, Part 2: H Abstraction fromAlkenes, Alkynes, Alcohols, and Acids by H Atoms. J. Phys. Chem.A 2001, 105, 8969-8984.
(29) Curran, H. J.; Gaffuri, P.; Pitz, W. J.; Westbrook, C. K. Acomprehensive modeling study of n-heptane oxidation. Combust.Flame 1998, 114 (1-2), 149-177.
(30) Truong, T. N. Reaction Class Transition State Theory:Hydrogen Abstraction Reactions by Hydrogen Atoms as TestCases. J. Chem. Phys. 2000, 113 (12), 4957-4964.
(31) Ranzi, E.; Dente, M.; Faravelli, T.; Pennati, G. Predictionof Kinetic-Parameters for Hydrogen Abstraction Reactions. Com-bust. Sci. Technol. 1994, 95 (1-6), 1-50.
(32) Forst, W. Theory of Unimolecular Reactions; AcademicPress: New York, 1973.
(33) Back, M. H.; Back, R. A. In Pyrolysis: Theory andIndustrial Practice; Albright, L. F., Crynes B. L., Corcoran, W.H., Eds.; Academic Press: New York, 1983; pp 1-24.
(34) Chen, C. J.; Back, M. H.; Back, R. A. Can. J. Chem. 1976,54, 3175-3184.
Received for review July 30, 2002Revised manuscript received December 16, 2002
Accepted December 16, 2002
IE020581W
1010 Ind. Eng. Chem. Res., Vol. 42, No. 5, 2003




















