
Analysis of the DNA adducts of phenyl glycidyl ether in a calf thymus DNA hydrolysate by capillary...
10
Carcinogenesis vol.19 no.6 pp.1077–1086, 1998 Analysis of the DNA adducts of phenyl glycidyl ether in a calf thymus DNA hydrolysate by capillary zone electrophoresis– electrospray mass spectrometry: evidence for phosphate alkylation Dieter L.D.Deforce 1 , Filip Lemie `re 2 , Ilse Hoes 2 , Rebecca E.M.Millecamps 1 , Eddy L.Esmans 2 , Andreas De Leenheer 1 and Elfriede G.Van den Eeckhout 1,3 1 Laboratory for Pharmaceutical Biotechnology, University of Ghent, Harelbekestraat 72, B-9000 Ghent, and 2 Department of Chemistry, Nucleoside Research and Mass Spectrometry Unit, University of Antwerp, Groenenborgerlaan 171, B-2020 Antwerp, Belgium 3 To whom correspondence should be addressed Email: [email protected] Calf thymus DNA was reacted in vitro with phenyl glycidyl ether (PGE) and was hydrolysed enzymatically, to the 59-monophosphate nucleotides using deoxyribonuclease I (DNA-ase I) and nuclease P1. The adducts were concen- trated using solid phase extraction (SPE), on a polystyrene divinylbenzene copolymer in order to remove the unmodi- fied nucleotides. The adducts could be identified using capillary zone electrophoresis–electrospray tandem mass spectrometry (CZE ES–MS/MS), using sample stacking. In addition to the base alkylated 29-deoxynucleotides present in the DNA-hydrolysate, also phosphate alkylated 29-deoxy- nucleotide adducts were identified for TMP and dAMP. An additional adduct, dUMP alkylated on the uridine moiety was found originating from the hydrolytic deamin- ation of dCMP alkylated on N 3 of the cytosine moiety. Enzymatic hydrolysis using nuclease P1 was incomplete as shown by the presence of dinucleotides alkylated on the base moiety. They were successfully hydrolysed to the corresponding 29-deoxynucleotides by snake venom phos- phodiesterase (SVP). Data are shown indicating that alkylations on the pyrimidine bases were more resistant to enzymatic hydrolysis with nuclease P1 than the purine alkylated products. Introduction The interaction of xenobiotics with DNA and the resulting adducts have been studied extensively, in order to understand how they exhibit their cytotoxic and carcinogenic properties (1,2). It is within this philosophy that our group has focused on simple unsaturated epoxides on which until now only little research has been performed (3–6). As lead compound phenyl glycidyl ether (PGE*) was chosen, which is an important industrial epoxide mainly used in the paint and the resin industry. We have investigated the application of CZE ES– MS (Capillary Zone Electrophoresis-Electrospray Mass Spectrometry) (7) for the detection of nucleotides modified with PGE as an alternative to the popular 32 P post-labelling technique. Although the 32 P post-labelling (8,9) technique is very sensitive, one of its major drawbacks is that it gives no structural information. The combination of CZE as a high *Abbreviations: PGE, phenyl glycidyl ether; CZE ES–MS, capillary zone electrophoresis–electrospray mass spectrometry; SPE, solid phase extraction. © Oxford University Press 1077 efficiency separation technique to electrospray tandem mass spectrometry (ES–MS/MS) proved to be very promising in the field of DNA-adduct research, enabling identification and structure elucidation of unknown adducts (7,10). In order to be able to load enough sample on the capillary to obtain ES– MS/MS spectra, sample stacking was performed as described by Wolf, Vouros and ourselves (7,10,11). The identification of unknown adducts and their structure can be important to explain or better understand the working mechanism of cyto- toxic agents. In the field of DNA adduct research, most research is focused on the adducts formed by the interaction of the alkylating agent with the purine or pyrimidine moiety (12). In a previous report (13) we were able to identify alkylation on the phosphate backbone of the DNA structure. Subsequent spontaneous hydrolysis of this alkylated backbone was shown to be responsible for the breakdown of the DNA structure into shorter oligonucleotides. Phosphate alkylation was also found in the reaction of mustards with DNA (14–16) and nitrosourea with RNA. In the latter case it was held responsible for the degradation of the RNA string (17). In our former experiments no phosphate alkylated adducts were detected in the enzymatically hydrolysed DNA pellet (7). In this report, a sample pretreatment step, using solid phase extraction (SPE), was elaborated in order to concentrate the modified nucleotides and to remove the unmodified 29- deoxynucleotides, resulting in better stacking efficiency and loadability. The enzymatic hydrolysate of the DNA pellet was investigated especially for phosphate alkylated adducts because the presence of phosphate alkylated products in the DNA hydrolysate would be indicative for splicing of the DNA molecule at this site (13). This would result in an alkylated oligonucleotide which was then hydrolysed by the enzymatic procedure. Materials and methods DNA adduct formation and enzymatic hydrolysis Calf thymus DNA (highly polymerized DNA Type I from calf thymus, Sigma, St. Louis, MO) was reacted, during 48 h, with PGE (Aldrich, Steinheim, Germany) and hydrolysed enzymatically to the nucleotide level as described in our previous work (7). This DNA-hydrolysate was subjected to a sample clean up procedure in order to remove the salts and the excessive amount of unreacted nucleotides. This was accomplished by using Chromabond HR-P SPE columns (Macherey-Nagel, Du ¨ren, Germany) of which the amount of solid phase was reduced to 125 mg. 200 μl of the DNA-hydrolysate was applied to the column and rinsed with 6 ml of HPLC-grade water. The nucleotide adducts were then eluted using 2 ml of methanol/water 50/50, followed by 5 ml of methanol. The eluate was evaporated to dryness using a stream of nitrogen gas, and redissolved in 500 μl HPLC-grade water. Since the presence of adducted dinucleotides revealed incomplete (7) hydrolysis using nuclease P1, 200 μl of the DNA-hydrolysate was incubated at 37°C for 4 h with 5 units SVP (Snake Venom Phosphodiesterase, from Crotalus amandateus, Pharmacia Biotech, Uppsala, Sweden), in order to investigate hydrolysis of the remaining dinucleotides. Capillary zone electrophoresis CZE separations were done on a Lauerlab Prince system equipped for on- column detection with a Kontron UV detector (type HPLC 332). Data
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Analysis of the DNA adducts of phenyl glycidyl ether in a calf thymus DNA hydrolysate by capillary...

Carcinogenesisvol.19 no.6 pp.1077–1086, 1998
Analysis of the DNA adducts of phenyl glycidyl ether in a calfthymus DNA hydrolysate by capillary zone electrophoresis–electrospray mass spectrometry: evidence for phosphate alkylation
Dieter L.D.Deforce1, Filip Lemiere2, Ilse Hoes2,Rebecca E.M.Millecamps1, Eddy L.Esmans2,Andreas De Leenheer1 andElfriede G.Van den Eeckhout1,3
1Laboratory for Pharmaceutical Biotechnology, University of Ghent,Harelbekestraat 72, B-9000 Ghent, and2Department of Chemistry,Nucleoside Research and Mass Spectrometry Unit, University of Antwerp,Groenenborgerlaan 171, B-2020 Antwerp, Belgium3To whom correspondence should be addressedEmail: [email protected]
Calf thymus DNA was reactedin vitro with phenyl glycidylether (PGE) and was hydrolysed enzymatically, to the59-monophosphate nucleotides using deoxyribonuclease I(DNA-ase I) and nuclease P1. The adducts were concen-trated using solid phase extraction (SPE), on a polystyrenedivinylbenzene copolymer in order to remove the unmodi-fied nucleotides. The adducts could be identified usingcapillary zone electrophoresis–electrospray tandem massspectrometry (CZE ES–MS/MS), using sample stacking. Inaddition to the base alkylated 29-deoxynucleotides presentin the DNA-hydrolysate, also phosphate alkylated 29-deoxy-nucleotide adducts were identified for TMP and dAMP.An additional adduct, dUMP alkylated on the uridinemoiety was found originating from the hydrolytic deamin-ation of dCMP alkylated on N3 of the cytosine moiety.Enzymatic hydrolysis using nuclease P1 was incomplete asshown by the presence of dinucleotides alkylated on thebase moiety. They were successfully hydrolysed to thecorresponding 29-deoxynucleotides by snake venom phos-phodiesterase (SVP). Data are shown indicating thatalkylations on the pyrimidine bases were more resistant toenzymatic hydrolysis with nuclease P1 than the purinealkylated products.
Introduction
The interaction of xenobiotics with DNA and the resultingadducts have been studied extensively, in order to understandhow they exhibit their cytotoxic and carcinogenic properties(1,2). It is within this philosophy that our group has focusedon simple unsaturated epoxides on which until now only littleresearch has been performed (3–6). As lead compound phenylglycidyl ether (PGE*) was chosen, which is an importantindustrial epoxide mainly used in the paint and the resinindustry. We have investigated the application of CZE ES–MS (Capillary Zone Electrophoresis-Electrospray MassSpectrometry) (7) for the detection of nucleotides modifiedwith PGE as an alternative to the popular32P post-labellingtechnique. Although the32P post-labelling (8,9) technique isvery sensitive, one of its major drawbacks is that it gives nostructural information. The combination of CZE as a high
*Abbreviations: PGE, phenyl glycidyl ether; CZE ES–MS, capillary zoneelectrophoresis–electrospray mass spectrometry; SPE, solid phase extraction.
© Oxford University Press 1077
efficiency separation technique to electrospray tandem massspectrometry (ES–MS/MS) proved to be very promising in thefield of DNA-adduct research, enabling identification andstructure elucidation of unknown adducts (7,10). In order tobe able to load enough sample on the capillary to obtain ES–MS/MS spectra, sample stacking was performed as describedby Wolf, Vouros and ourselves (7,10,11). The identification ofunknown adducts and their structure can be important toexplain or better understand the working mechanism of cyto-toxic agents.
In the field of DNA adduct research, most research isfocused on the adducts formed by the interaction of thealkylating agent with the purine or pyrimidine moiety (12). Ina previous report (13) we were able to identify alkylation onthe phosphate backbone of the DNA structure. Subsequentspontaneous hydrolysis of this alkylated backbone was shownto be responsible for the breakdown of the DNA structure intoshorter oligonucleotides. Phosphate alkylation was also foundin the reaction of mustards with DNA (14–16) and nitrosoureawith RNA. In the latter case it was held responsible for thedegradation of the RNA string (17).
In our former experiments no phosphate alkylated adductswere detected in the enzymatically hydrolysed DNA pellet (7).
In this report, a sample pretreatment step, using solid phaseextraction (SPE), was elaborated in order to concentratethe modified nucleotides and to remove the unmodified 29-deoxynucleotides, resulting in better stacking efficiency andloadability. The enzymatic hydrolysate of the DNA pellet wasinvestigated especially for phosphate alkylated adducts becausethe presence of phosphate alkylated products in the DNAhydrolysate would be indicative for splicing of the DNAmolecule at this site (13). This would result in an alkylatedoligonucleotide which was then hydrolysed by the enzymaticprocedure.
Materials and methods
DNA adduct formation and enzymatic hydrolysis
Calf thymus DNA (highly polymerized DNA Type I from calf thymus, Sigma,St. Louis, MO) was reacted, during 48 h, with PGE (Aldrich, Steinheim,Germany) and hydrolysed enzymatically to the nucleotide level as describedin our previous work (7). This DNA-hydrolysate was subjected to a sampleclean up procedure in order to remove the salts and the excessive amount ofunreacted nucleotides. This was accomplished by using Chromabond HR-PSPE columns (Macherey-Nagel, Du¨ren, Germany) of which the amount ofsolid phase was reduced to 125 mg. 200µl of the DNA-hydrolysate wasapplied to the column and rinsed with 6 ml of HPLC-grade water. Thenucleotide adducts were then eluted using 2 ml of methanol/water 50/50,followed by 5 ml of methanol. The eluate was evaporated to dryness using astream of nitrogen gas, and redissolved in 500µl HPLC-grade water.
Since the presence of adducted dinucleotides revealed incomplete (7)hydrolysis using nuclease P1, 200µl of the DNA-hydrolysate was incubatedat 37°C for 4 h with 5 units SVP (Snake Venom Phosphodiesterase, fromCrotalus amandateus, Pharmacia Biotech, Uppsala, Sweden), in order toinvestigate hydrolysis of the remaining dinucleotides.
Capillary zone electrophoresis
CZE separations were done on a Lauerlab Prince system equipped for on-column detection with a Kontron UV detector (type HPLC 332). Data

D.L.D.Deforce et al.
Fig. 1. (A) Electropherogram of the DNA hydrolysate using nuclease P1before the sample clean up procedure (injection was 30 mbar during0.22 min, followed by sample stacking, conditions see Materials andmethods). (B) Electropherogram of the DNA hydrolysate using nuclease P1after the sample clean up procedure (injection was 120 mbar during0.22 min, followed by sample stacking, conditions see Materials andmethods). In both panels zone 1 represents the alkylated 29-deoxynucleotides, zone 2 represents the alkylated dinucleotides and zone 3represents the unmodified 29-deoxynucleotides. (The identity of all theproducts was confirmed by CZE ES–MS/MS.)
collection was performed with the PC Integration Pack version 3.90 (KontronInstruments). The CZE capillary was a 75µm i.d. fused silica capillary. Thedistance of the inlet of the CZE capillary to the UV detector was 61.5 cm.Detection was performed at a wavelength of 270 nm. The total length was91.5 cm. The buffer system used was a 100 mM ammonium carbonate buffersystem (pH 9.68). Electrophoresis was performed using a constant voltage of17 kV. In order to load sufficient sample on the capillary, sample stackingwas performed as described in our earlier work (7). The samples were injectedapplying a pressure of 120 mbar during 0.22 min prior to sample stacking.
Coupling CZE to electrospray mass spectrometry.In order to obtain more structural information, the CZE system was coupledon-line to ES–MS (electrospray mass spectrometry) and ES–MS/MS (VGQuattro II triple-quadrupole system, Micromass) as previously published (7).The sample obtained as a result of the sample clean up procedure describedabove was concentrated ten times by evaporating the volume to 50µl usinga stream of nitrogen gas. The buffer system used to perform electrophoresiswas a 100 mM ammonium carbonate buffer system (pH 9.68). Electrophoresiswas performed using a constant voltage of 13 kV. The ES-capillary(Electrospray) was set at –4.2 kV. The cone voltage was set at 30 V and thesource temperature was kept constant at 75°C. A make-up flow of 80% 2-propanol, 15% water and 5% 0.01 M ammonium carbonate buffer (pH 9.68)was introduced using the sheath capillary at a flow rate of 10µl/min. Theflow rate of the N2 bath gas was adjusted between 50 and 100 l/h, and theflow rate of the N2 nebulizing gas was set at 30 l/h.
CZE ES–MS data were obtained by pressure injection, applying 60 mbarduring 0.2 min, prior to sample stacking. A scan range of 75 to 800 Da wascovered at a scan speed of 725 Da/s. CZE ES–MS/MS data were obtained bypressure injection, applying 120 mbar during 1.2 min, prior to sample stacking.Product ion spectra were obtained of [M-H]– ions. The collision gas (Ar)pressure was 2310–3 mbar and the collision energy was optimized for eachcomponent. The optimal collision energy varied between 23 and 33 eV.
1078
Fig. 2. Electropherogram of the DNA hydrolysate using both nuclease P1and Snake Venom Phosphodiesterase (conditions see Materials andmethods). Zone 1 represents the group of alkylated 29-deoxynucleotides andzone 2 represents the unmodified 29-deoxynucleotides. (The identity of allthe products was determined by CZE ES–MS/MS.)
Results
CZE results using UV detectionUsing SPE-sample clean up the salts present in the DNA-hydro-lysate were efficiently removed. As a result not only the adductswere efficiently concentrated by selective removal of theunmodified nucleotides but also a low conductive sample plugwas obtained which led to a better stacking efficiency of theanalytes (Figure 1). Furthermore, by removing the former com-pounds prior to CZE analysis a larger amount of adducts couldbe injected by the sample stacking technique (see Materials andmethods) without overloading the CZE column. The recoveryof the modified 29-deoxynucleotides was ~99% (as determinedwith a reference reaction mixture of TMP with PGE).
The adducted dinucleotides were efficiently hydrolysedusing an additional digestion with SVP (Figure 2). However,since no new adducts were found after this additional hydrolyticprocedure, we decided to perform the analysis of the DNA-hydrolysate after nuclease P1 treatment.
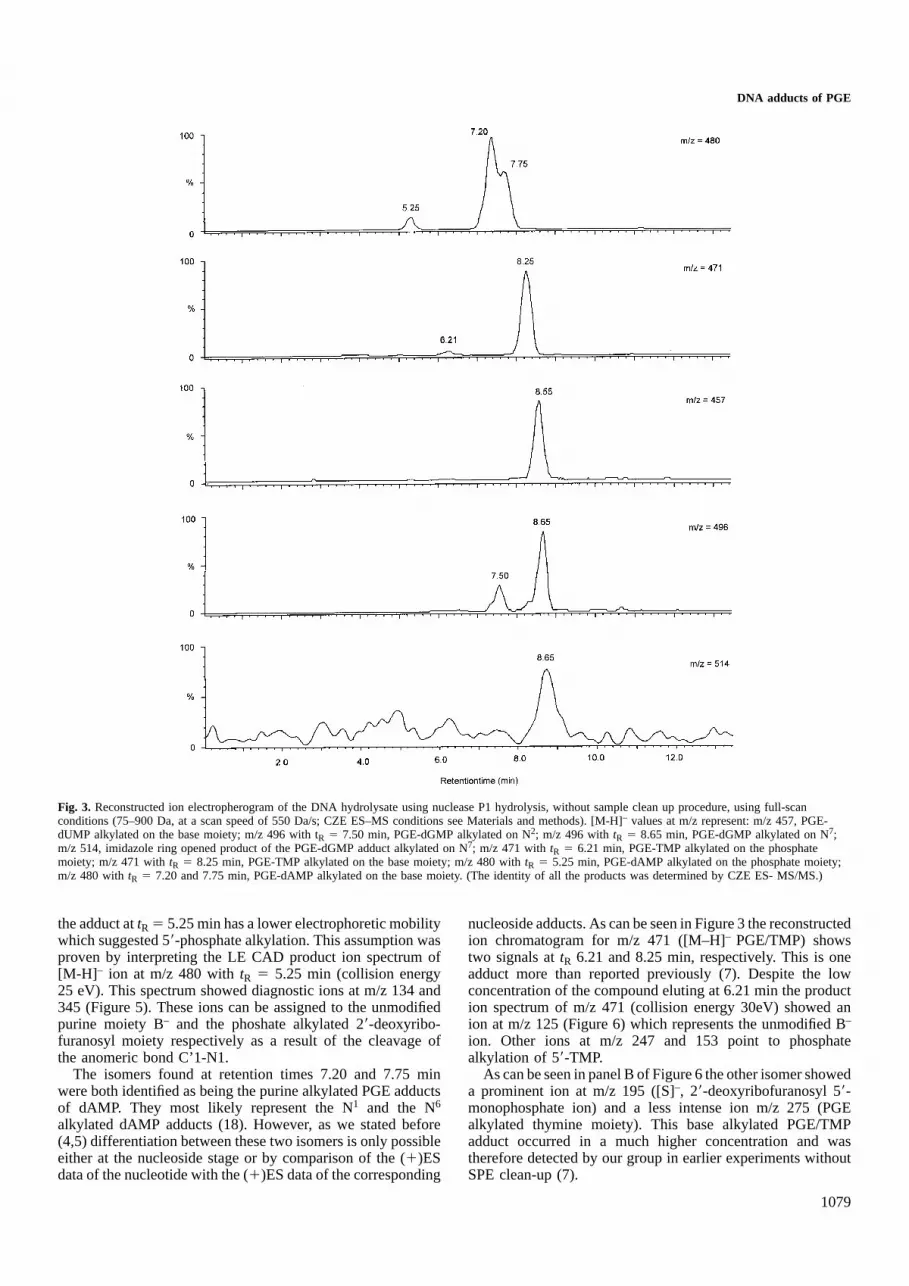
CZE ES–MS and CZE ES–MS/MSresultsCompared to previously reported data (7) this approachrevealed the presence of additional adducts hitherto not detected(Figure 3).
A compound was found attR 5 8.55 min characterized bythe presence of a [M-H]– at m/z 457. This corresponds to amolecular mass of 458 which could correspond to the mono-alkylated PGE adduct of dUMP. Although dUMP does notoccur in Calf thymus DNA a study published by Lemie`reet al. (4) proved that the N3 mono-alkylated adduct of PGEwith dCyd underwent hydrolytic deamination and producedthe corresponding PGE adduct of dUrd. A product with [M-H]– at m/z 456, corresponding to the mono-alkylated PGE-dCMP was not found in the DNA-hydrolysate. This wouldsignify that all initially formed N3 PGE/dCMP was deaminated.
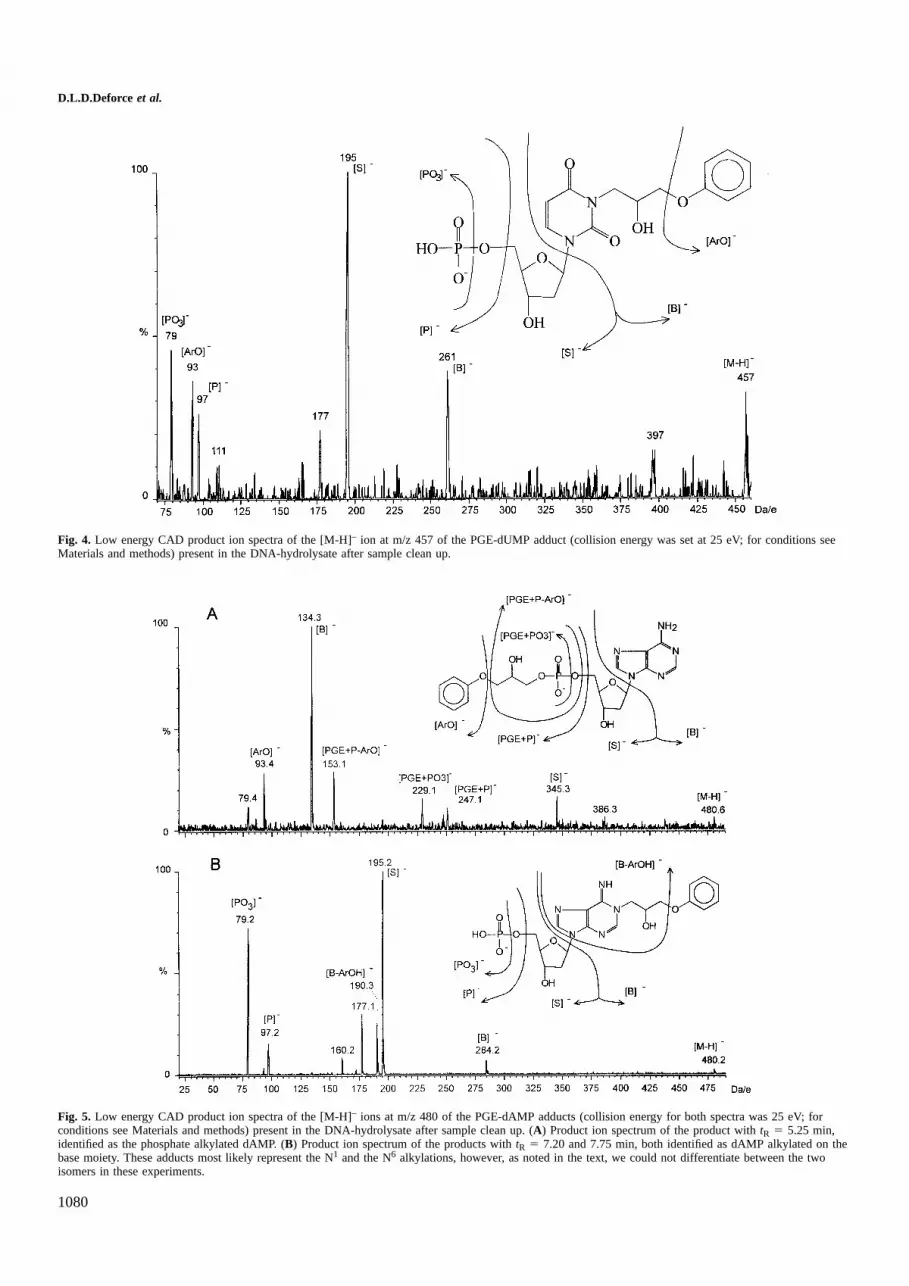
In order to prove the structure of this PGE/dUMP adduct,the low energy collisional activated decomposition (LE CAD)product ion spectrum of m/z 457 was recorded (Figure 4). Theion observed at m/z 261 represents the N3 alkylated fragmentof PGE with uracil. For the mono-alkylated PGE-adduct ofdAMP ([M-H]– at m/z 480) three signals were observed atrespectivelytR 5 5.25, 7.20 and 7.75 min. One of them, i.e.
DNA adducts of PGE
Fig. 3. Reconstructed ion electropherogram of the DNA hydrolysate using nuclease P1 hydrolysis, without sample clean up procedure, using full-scanconditions (75–900 Da, at a scan speed of 550 Da/s; CZE ES–MS conditions see Materials and methods). [M-H]– values at m/z represent: m/z 457, PGE-dUMP alkylated on the base moiety; m/z 496 with tR 5 7.50 min, PGE-dGMP alkylated on N2; m/z 496 withtR 5 8.65 min, PGE-dGMP alkylated on N7;m/z 514, imidazole ring opened product of the PGE-dGMP adduct alkylated on N7; m/z 471 withtR 5 6.21 min, PGE-TMP alkylated on the phosphatemoiety; m/z 471 withtR 5 8.25 min, PGE-TMP alkylated on the base moiety; m/z 480 withtR 5 5.25 min, PGE-dAMP alkylated on the phosphate moiety;m/z 480 withtR 5 7.20 and 7.75 min, PGE-dAMP alkylated on the base moiety. (The identity of all the products was determined by CZE ES- MS/MS.)
the adduct attR 5 5.25 min has a lower electrophoretic mobilitywhich suggested 59-phosphate alkylation. This assumption wasproven by interpreting the LE CAD product ion spectrum of[M-H] – ion at m/z 480 withtR 5 5.25 min (collision energy25 eV). This spectrum showed diagnostic ions at m/z 134 and345 (Figure 5). These ions can be assigned to the unmodifiedpurine moiety B– and the phoshate alkylated 29-deoxyribo-furanosyl moiety respectively as a result of the cleavage ofthe anomeric bond C’1-N1.
The isomers found at retention times 7.20 and 7.75 minwere both identified as being the purine alkylated PGE adductsof dAMP. They most likely represent the N1 and the N6
alkylated dAMP adducts (18). However, as we stated before(4,5) differentiation between these two isomers is only possibleeither at the nucleoside stage or by comparison of the (1)ESdata of the nucleotide with the (1)ES data of the corresponding
1079
nucleoside adducts. As can be seen in Figure 3 the reconstructedion chromatogram for m/z 471 ([M–H]– PGE/TMP) showstwo signals attR 6.21 and 8.25 min, respectively. This is oneadduct more than reported previously (7). Despite the lowconcentration of the compound eluting at 6.21 min the production spectrum of m/z 471 (collision energy 30eV) showed anion at m/z 125 (Figure 6) which represents the unmodified B–
ion. Other ions at m/z 247 and 153 point to phosphatealkylation of 59-TMP.
As can be seen in panel B of Figure 6 the other isomer showeda prominent ion at m/z 195 ([S]–, 29-deoxyribofuranosyl 59-monophosphate ion) and a less intense ion m/z 275 (PGEalkylated thymine moiety). This base alkylated PGE/TMPadduct occurred in a much higher concentration and wastherefore detected by our group in earlier experiments withoutSPE clean-up (7).
D.L.D.Deforce et al.
Fig. 4. Low energy CAD product ion spectra of the [M-H]– ion at m/z 457 of the PGE-dUMP adduct (collision energy was set at 25 eV; for conditions seeMaterials and methods) present in the DNA-hydrolysate after sample clean up.
Fig. 5. Low energy CAD product ion spectra of the [M-H]– ions at m/z 480 of the PGE-dAMP adducts (collision energy for both spectra was 25 eV; forconditions see Materials and methods) present in the DNA-hydrolysate after sample clean up. (A) Product ion spectrum of the product withtR 5 5.25 min,identified as the phosphate alkylated dAMP. (B) Product ion spectrum of the products withtR 5 7.20 and 7.75 min, both identified as dAMP alkylated on thebase moiety. These adducts most likely represent the N1 and the N6 alkylations, however, as noted in the text, we could not differentiate between the twoisomers in these experiments.
1080

DNA adducts of PGE
Fig. 6. Low energy CAD product ion spectra of the [M-H]– ions at m/z 471 of the PGE-TMP adducts (collision energy for both spectra was 30 eV; forconditions see Materials and methods) present in the DNA-hydrolysate after sample clean up. (A) Product ion spectrum of the product withtR 5 6.21 min,identified as the phosphate alkylated TMP. (B) Product ion spectrum of the product withtR 5 8.25 min, identified as TMP alkylated on the base moiety.
Table I. AUC (Area Under the Curve) ratios of the different adducts, to unmodified dAMP present in the DNA-hydrolysate, before and after addition ofSVPa (conditions see text)
Before SVP hydrolysis After SVP hydrolysisAUC ratio AUC ratio
dAMP phosphate alkylation tR 5 5.25 min 0.0019 0.0444b 0.0020 (3 1.0)a 0.0092b
dAMP base alkylation tR 5 7.20;7.75 min 0.0433 0.2134 (3 4.9)a
TMP base alkylation tR 5 8.25 min 0.0467 0.3852 (3 8.3)a
dUMP base alkylation tR 5 8.55 min 0.0131 0.1999 (3 15.2)a
dGMP-N2 alkylation tR 5 7.50 min 0.0083 0.1949c 0.04452 (3 5.3)a 1.6434c
dGMP-N7 alkylation tR 5 8.65 min 0.0428 0.0271 (3 0.6)a
Imidazole ring opened dGMP adduct 0.0048 0.0119 (3 2.5)a
aRelative increase of the adduct detected after SVP hydrolysis.bRatio of phosphate alkylation to base alkylation.cRatio of dGMP-N2 alkylation to dGMP-N7 alkylation.
Finally, as can be seen from Figure 3, two products with[M-H] – at m/z of 496 were found (tR 5 7.50 min andtR 58.65 min). In Table I we can see that the ratio of theirabundance increases by a factor of 8.43 after additionalhydrolysis of the dinucleotide adducts with SVP. This is dueto the increase of the abundance of the product withtR 5 7.50min and the simultaneous decrease of the product withtR 58.65 min. This decrease can be explained by the observationof Vanhoutteet al. (6) that the N7 alkylated adduct of dGuowith bisphenol A, undergoes hydrolytic imidazole ring opening.
1081
This hypothesis is confirmed by our experimental data wherewe can observe an increase in the imidazole ring openedproduct of the N7 alkylation of dGMP with PGE ([M-H]– atm/z 514, [tR 5 8.65 min]). Both products have identicalelectrophoretic behavior. These data indicate that the productwith tR 5 8.65 min is the N7 alkylated adduct of dGMP withPGE. The product withtR 5 7.50 min, which is according tothese data, not susceptible to imidazole ring opening, couldbe either the N2 or the O6-alkylated adduct of dGMP withPGE, in reference to the data obtained on the nucleoside

D.L.D.Deforce et al.
Fig. 7. Low energy CAD product ion spectra of the [M-H]– ions at m/z 496 of the PGE-dGMP adducts (collision energy for both spectra was 23 eV; forconditions see Materials and methods) present in the DNA-hydrolysate after sample clean up. (A) Product ion spectrum of the product withtR 5 7.50 min,identified as the N2 alkylated adduct of dGMP. (B) Product ion spectrum of the product withtR 5 8.65 min, identified as the N7 alkylated adduct of dGMP.
adducts, published by Van den Eeckhout (18). The CZE ES–MS/MS data obtained of the compounds with [M-H]– at m/z496 (collision energy 23 eV) indeed revealed two distinctadducts (Figure 7). The product ion spectrum of the adductwith tR 5 8.65 min is characterized by the presence of theions at m/z 195 and 300, originating from the cleavage of theglycosidic bond. In the N7 alkylated dGMP this glycosidicbond is prone to cleavage because of the vicinity of thequaternary N7 (Figure 8). Bearing in mind the data on theimidazole ring opening we could identify this product as beingthe N7 alkylated dGMP. In the product ion spectrum of theadduct with tR 5 7.50 min, only the ions at m/z 79 and 97were observed, both originating from the phosphate group.This indicates that alkylation could be present on N2. In thiscase the glycosidic bond is stronger due to the absence of aquaternary N7, which explains the absence in this spectrum ofthe ion at m/z 300, and the very low abundancy of the ion atm/z 195.
In order to investigate whether phosphate- or base alkylationwas responsible for the incomplete digestion of dinucleotideadducts, the abundance of the phosphate- and base alkylatedadducts was investigated before and after additional hydrolysiswith SVP. Both samples were analysed by CZE ES–MSwithout sample clean up so that the unmodified nucleotideswere present in the sample and could serve as an internalstandard. The ratio of the alkylated adducts versus dAMP wascalculated, before and after SVP hydrolysis (Table I). Because
1082
Fig. 8. Proposed fragmentation scheme for the N7 alkylated adduct ofdGMP with PGE.
only the ratio of the base alkylated adducts increased it wasobvious that the base alkylation and not phosphate alkylationwas responsible for the incomplete digestion by the nucleaseP1 enzyme. Hence, these data suggest that the dinucleotideadducts are all modified on their base moiety which is consistentwith the reactivity of SVP (19).

DNA adducts of PGE
Fig. 9. Reconstructed ion electropherogram of the DNA hydrolysate using nuclease P1 hydrolysis, without sample clean up procedure, using full-scanconditions (75–900 Da, at a scan speed of 550 Da/s; CZE ES–MS conditions see Materials and methods). [M-H]– values at m/z represent: m/z 784, dAMP-TMP dinucleotide alkylated on the base moiety; m/z 775, TMP-TMP dinucleotide alkylated on the base moiety; m/z 760, dCMP-TMP dinucleotide alkylatedon the base moiety. (The identity of all the products was determined by CZE ES–MS/MS.)
Fig. 10. Low energy CAD product ion spectrum of the [M-H]– ion at m/z 784. Identified as a mixture of two co-eluting adducts of pApT(PGE), structure (A)the specific fragments for molecule A are denoted in the spectrum with A and pTpA(PGE), structure (B), with specific fragments (collision energy was 33 eV;for conditions see Materials and methods) present in the DNA-hydrolysate after sample clean up.
1083

D.L.D.Deforce et al.
Fig. 11. General fragmentation scheme for the [M-H]– ions of the adducted dpNdpN dinucleotides under LE-CAD.
In order to get additional evidence the CZE ES–MS/MSdata were obtained for the most abundant dinucleotide adducts.LE CAD spectra (collision energy 33 eV), were obtained forthe products with deprotonated molecules [M-H]– at m/z 760,775 and 784. The reconstructed ion chromatogram (RIC)obtained for these ions in a CZE ES–MS run is shown inFigure 9. The dinucleotide with molecular mass 785 ([M-H]–
at m/z 784) corresponds to a mono-alkylated dinucleotidepApT or pTpA. Analysis of the LE CAD product ion spectrumof [M-H] – 784 (Figures 10 and 11) revealed the presence oftwo co-eluting isomers. The product ions at m/z 284 and 275were assigned to the alkylated [B]– ions of the Ade- and Thy-moiety respectively. Other ions were found at m/z 321 (b-type
1084
ion) and m/z 401 (d-type ion) proving the presence of apTpA(PGE) dinucleotide while m/z 330 (b-type ion) and 410(d-type ion) could be explained out of the pApT(PGE) isomer,which is the most abundant.
The product with [M-H]– at m/z 775, was identified as themono-alkylated adduct of pTpT. In this case two possibleisomers were possible, i.e. pT(PGE)pT and pTpT(PGE).Because of the presence of product ions at m/z 321 (b-typeion) and 401 (d-type ion) and because of the absence of ionsat 471 (b-type ion) and m/z 551 (d-type ion) the production spectrum was assigned to the pTpT(PGE) isomer. Basealkylation was proven by the occurrence of the B– ion atm/z 275.

DNA adducts of PGE
The LE CAD product ion spectrum from the adduct with[M-H] – at m/z 760 was far more complex compared to theothers. A molecular mass of 761 corresponds to either apCpT(PGE), a pC(PGE)pT, a pT(PGE)pC or a pTpC(PGE)-dinucleotide. If we look at the product ion spectrum, ions werefound at m/z 306 (b-type ion) and m/z 386 (d-type ion) whichexplain the sequence pCpT(PGE). At the same time a B– ionwas found at m/z 275 corresponding to an alkylated Thy-moiety. Because of the absence of product ions at m/z 456(b-type ion) and 536 (d-type ion) the occurrence of thepC(PGE)pT isomer was ruled out. Also present in the spectrumwere ions at m/z 321 (b-type ion) and 401 (d-type ion) whichpoint at the same time to the presence of a pTpC(PGE)-dinucleotide. If the pT(PGE)pC-dinucleotide had been formedions at m/z 471 (b-type ion) and 551 (d-type ion) should havebeen detected. However if such a pTpC(PGE)-dinucleotide ispresent we should at least expect a B– ion at m/z 260 (alkylatedCyt-moiety). Unfortunately, this ion is not present, but anintense one is found at m/z 261. This ion, together with theobservation of an ion at m/z 761, suggests the presence of apTpU(PGE)-dinucleotide resulting from the pTpC(PGE)adduct by hydrolytic deamination (20).
Discussion
By introducing a SPE sample clean up we were able to identifyphosphate alkylated adducts of TMP and dAMP in DNAhydrolysates of calf thymus DNA reactedin vitro with PGEby using CZE ES–MS/MS. Since inin vivo samples generally1 modification occurs per 107 nucleotides (21) a sampleclean up procedure to remove unmodified nucleotides is aprerequisite. The reaction of PGE on the phosphate backbone,producing unstable phosphotriesters (17) is probably of majorbiological significance, since we were able to prove that thesealkylations are responsible for the hydrolysis of DNA resultingin strand breaks (13).
Additional hydrolysis with SVP removed the dinucleotideswhich could not be completely hydrolysed by the nuclease P1enzyme. This indicates that the SVP enzyme is less sensitiveto inhibition by modified nucleotides. In the field of DNAadduct research it may therefore be interesting to include thisadditional enzyme in order to achieve complete digestion ofmodified DNA to the 59-mononucleotides. The ratios of changeof the signal intensity before and after hydrolysis with theSVP enzyme, as depicted in Table I, indicate that alkylationson the pyrimidine bases (TMP and dCMP converted to dUMPby hydrolytic deamination) are more resistant to hydrolysiswith the nuclease P1 enzyme than the purine alkylated adducts(dGMP and dAMP). This was confirmed by the CZE ES–MS/MS data obtained for the most abundant mono-alkylateddinucleotides, where most alkylations were located on thepyrimidine bases. These data also confirmed that the basealkylations and not the phosphate alkylated adducts wereresponsible for the incomplete hydrolysis by nuclease P1.
Alkylation on the base moiety of dCMP resulted in hydrolyticdeamination to PGE modified dUMP, in the DNA hydrolysateno dCMP adducts were observed, this indicates that all dCMPadducts were hydrolysed to the dUMP adduct. Since thisreaction occurs only to a small extent in the reaction mixtureof the dCMP nucleotide with PGE, it is possible that thishydrolytic deamination is favoured by the conditions used toenzymatically hydrolyse the modified DNA.
For dGMP two alkylation products were found, a N7
1085
alkylated adduct and a N2 or O6 alkylated adduct. The N7
alkylated adduct is susceptible to imidazole ring opening, andto subsequent deglycosylation (6). By this mechanism the N7
modified guanine would be removed from the DNA chain andcould thus be repaired by repair enzymes (22). The N2 or O6
alkylated adduct is not susceptible to imidazole ring openingand deglycosylation and may result in more cytotoxic lesionsthan the N7 alkylated product. This N2 or O6 alkylated productwas not formed in the reaction between dGMP and PGE andis thus specific for interaction of PGE with DNA, in which itis the most abundant species (see Table I).
In summary, CZE in combination with ES–MS and ES–MS/MS proves to be very promising in the field of DNA-adduct research, enabling structure elucidation of unknownadducts and contributing to the elucidation of cytotoxic reactionmechanisms.
Acknowledgements
We wish to thank the FWO-Vlaanderen for grant number 32013394. One ofus (E.E.) is indebted to the Flemish Government for financial support(GOA-action).
References
1.Bartsch,H. (1996) DNA adducts in human carcinogenesis: etiologicrelevance and structure-activity relationship.Mutat. Res., 340, 67–79.
2.Van Zeeland,A.A. (1996) Molecular dosimetry of chemical mutagens.Relationship between DNA adduct formation and genetic changes analysedat the molecular level.Mutat. Res., 353, 123–150.
3.Van den Eeckhout,E., Coene,J., Claereboudt,J., Borremans,F., Claeys,M.,Esmans,E. and Sinsheimer,J.E. (1991) Comparison of the isolation ofadducts of 29-deoxycytidine and 29-deoxyguanosine with phenylglycidylether by high-performance liquid chromatography on a reversed-phasecolumn and a polystyrene-divinylbenzene column.J. Chromatogr., 541,317–331.
4.Lemiere,F., Esmans,E.L., Van Dongen,W., Van den Eeckhout,E. and VanOnckelen,H. (1993) Evaluation of liquid chromatography-thermospraymass spectrometry in the determination of some phenylglycidyl ether-29-deoxynucleoside adducts.J. Chromatogr., 647, 211–218.
5.Vanhoutte,K., Van Dongen,W., Esmans,E.L., Van den Eeckhout,E.L. andVan Onckelen,H.O. (1995) A strategy for the identification of 29-deoxynucleoside and 29-deoxynucleotide adducts using electrospraytandem mass spectrometry.Eur. Mass Spectrom., 2, 181–192.
6.Vanhoutte,K., Joos,P., Lemie`re,F., Van Dongen,W., Claeys,M., Van denEeckhout,E. and Esmans,E.L. (1995) Thermospray liquid chromatography-mass spectrometry of the DNA adducts formed between 29-deoxynucleosides and bisphenol A diglcidyl ether.J. Mass Spectrom., 30,1453–1461.
7.Deforce,D.L.D., Ryniers,F.P.K., Lemie`re,F., Esmans,E.L. and Van denEeckhout,E.G. (1996) Analysis of DNA adducts in DNA hydrolysatesby capillary zone electrophoresis and capillary zone electrophoresis-electrospray mass spectrometry.Anal. Chem., 68, 3575–3584.
8.Beach,A.C. and Gupta,R.C. (1992) Human biomonitoring and the32P-postlabelling assay.Carcinogenesis, 13, 1053–1074.
9.Farmer,P.B. (1994) Carcinogen adducts: use in diagnosis and riskassessment.Clin. Chem., 40/7, 1438–1443.
10.Barry,J.P., Norwood,C. and Vouros,P. (1996) Detection and identificationof benzo[a]pyrene diol epoxide adducts to DNA utilizing capillaryelectrophoresis-electrospray mass spectrometry.Anal. Chem., 68, 1432–1438.
11.Wolf,S.M. and Vouros,P. (1995) Incorporation of sample stackingtechniques into the capillary electrophoresis CF-FAB mass spectrometricanalysis of DNA adducts.Anal. Chem., 67, 891–900.
12.Dizdaroglu,M. (1994) Chemical determination of oxidative DNA damageby gas chromatography-mass spectrometry.Methods Enzym., 234, 3–16.
13.Deforce,D.L.D., Lemie`re,F., Esmans,E.L., De Leenheer,A. and Van denEeckhout,E. (1998) Analysis of the DNA-damage induced by phenylglycidyl ether using capillary zone electrophoresis-electrospray massspectrometry.Anal. Biochem.(in press).
14.Lindemann,H. (1984) Interaction of cyclophosphamide with DNA inisolated rat liver cell nuclei.Anticancer Res., 4, 53–58.

D.L.D.Deforce et al.
15.Lindemann,H. and Harbers,E. (1980) In vitro reaktion der drei alkylierendenpharmaka Cyclophosphamid, Ifosfamid und Trofosfamid mit DNS undDNS-Bausteinen.Arzneim.-Forsch./Drug. Res., 30, 2075–2080.
16.Maccubbin,A.E., Caballes,L., Riordan,J.M., Huang,D.H. and Gurtoo,H.L.(1991) A cyclophosphamide/DNA phosphoester adduct formedin vitroand in vivo. Cancer Res., 51, 886–892.
17.Singer,B., Sun,L. and Fraenkel-Conrat,A. (1975) Effects of alkylation ofphosphodiesters and of bases on ineffectivity and stability of Tobaccomosaic virus RNA.Proc. Natl Acad. Sci. USA, 72, 2232–2236.
18.Van den Eeckhout,E.G. (1991) Reaction between phenylglycidyl ether and29-deoxynucleosides: identification of adducts. In Van den Eeckout,E.G.(ed.) Aggregaatsthesis: Development of Analytical Methodology in theStructure–genotoxicity Relationship of Some Aliphatic Epoxides.Universityof Gent, Belgium, pp. 165–262.
19.Miller,P.S., Fang,K.N., Kondo,N.S. and Ts’o,P.O.P. (1971) Syntheses andproperties of adenine and thymine nucleoside alkyl phosphotriesters, theneutral analogs of dinucleoside monophosphates.J. Am. Chem. Soc., 93,6657–6665.
20.Lemiere,F., Joos,P., Vanhoutte,K., Esmans,E.L., De Groot,A., Claeys,M.and Van den Eeckhout,E.L. (1996) Phenylglycidyl ether adducts of 29-deoxyadenosine and 29-deoxycytidine: stability in solution and structureanalysis by electrospray tandem mass spectrometry.J. Am. Soc. MassSpectrom., 7, 682–691.
21.Wellemans,J., Cerny,R.L. and Gross,M.L. (1994) Tandem mass spectro-metry for the determination of deoxyribonucleic acid damage by polycyclicaromatic hydrocarbons.Analyst, 119, 497–503.
22.Boulikas,T. (1996) DNA lesion-recognizing proteins and the p53connection.Anticancer Res., 16, 225–242.
Received on December 8, 1997; revised on February 5, 1998; accepted onFebruary 5, 1998
1086




















