
analisis spektroskopik kompleks bis-4- heksiloksibenzilamin ...
79
1 ANALISIS SPEKTROSKOPIK KOMPLEKS BIS-4- HEKSILOKSIBENZILAMIN TEMBAGA(I) DENGAN METODE AB INITIO SKRIPSI Diajukan untuk memenuhi salah satu syarat mencapai gelar Sarjana Sains (S.Si) Program Studi Ilmu Kimia pada Fakultas Matematika dan Ilmu Pengetahuan Alam Universitas Islam Indonesia Jogjakarta Diajukan oleh : DEWI KUMUDANINGSIH SISWOYO No Mhs : 03612003 JURUSAN ILMU KIMIA FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM UNIVERSITAS ISLAM INDONESIA JOGJAKARTA 2011
-
Upload
khangminh22 -
Category
Documents
-
view
3 -
download
0
Transcript of analisis spektroskopik kompleks bis-4- heksiloksibenzilamin ...

1
ANALISIS SPEKTROSKOPIK KOMPLEKS BIS-4-HEKSILOKSIBENZILAMIN TEMBAGA(I) DENGAN
METODE AB INITIO
SKRIPSI
Diajukan untuk memenuhi salah satu syarat mencapai gelar Sarjana Sains (S.Si) Program Studi Ilmu Kimia pada Fakultas Matematika dan Ilmu Pengetahuan Alam
Universitas Islam Indonesia Jogjakarta
Diajukan oleh :
DEWI KUMUDANINGSIH SISWOYO No Mhs : 03612003
JURUSAN ILMU KIMIA FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM
UNIVERSITAS ISLAM INDONESIA JOGJAKARTA
2011

2

3
PERSEMBAHAN
“Dan Dia menundukkan untuk kamu apa yang ada di langit
dan apa yang ada di bumi semuanya, sebagai suatu rahmat daripada-Nya.
Sesungguhnya pada yang demikian itu, benar-benar terdapat ayat (bukti-bukti
kekuasaan Allah) bagi orang-orang yang berpikir”
(QS. Al-Jatsiyah: 13)
“…. Allah akan meninggikan orang-orang yang beriman dan orang-orang yang
diberi ilmu pengetahuan beberapa derajat. . . . . “
(QS. Al-Mujaadilah: 11 )
Kupersembahkan untuk Ayah dan Ibu yang
tersayang yang selalu mendukung aktivitas
ananda, suami dan adik-adik serta sahabatku
tersayang.

4
KATA PENGANTAR
Alhamdulillahirobbil’alamin, segala puji hanya bagi Allah SWT yang
telah melimpatkan rahmat dan karunia serta ampunan-Nya sehingga penulis dapat
menyelesaikan penelitian dan penulisan skripsi yang berjudul “Analisis
Spektroskopik Kompleks bis-4-heksiloksibenzilamin tembaga(I) Dengan Metode
Ab Initio” yang menjadi salah satu persyaratan untuk mencapai program studi
strata-1 di Fakultas Matematika dan Ilmu Pengetahuan Alam Universitas Islam
Indonesia. Penulis menyadari bahwa banyak terdapat kekurangan dalam penulisan
ini karena keterbatasan ilmu yang dimiliki penulis. Namun penulis bersyukur
karena akhirnya skripsi ini dapat terselesaikan.
Penulis menyadari bahwa terselesaikannya skripsi ini tidak lepas dari
bantuan banyak pihak yang telah menyediakan waktu, tenaga dan fikiran untuk
memberikan motivasi, nasihat atau saran kepada penulis. Oleh karena itu, dalam
kesempatan ini ingin mengucapkan terima kasih sebesar-besarnya kepada :
1. Bapak Yandi Syukri, S.Si, M.Si, Apt, selaku Dekan F-MIPA Universitas
Islam Indonesia Jogjakarta.
2. Bapak Dr. Ria Armunanto, selaku Dosen Pembimbing I yang dengan
sangat sabar membimbing, memberikan saran, serta arahan selama
penulisan skripsi sehingga skripsi ini dapat terselesaikan dengan baik.
3. Bapak Dwiarso Rubiyanto, M.Si, selaku Dosen Pembimbing II yang selalu
memberikan motivasi, kritik, saran dan pengarahan dalam membantu
menyelesaikan skripsi.

5
4. Laboratorium Pusat Kimia Indonesia-Austria yang berkenan menyediakan
fasilitas penelitian.
5. Semua pihak, baik dari Fakultas MIPA UII maupun lainnya yang telah
berkenan membantu penulis dalam proses penyelesaian skripsi ini yang
tidak dapat disebut satu persatu.
Penulis menyadari bahwa skripsi ini jauh dari sempurna, oleh karena itu
penulis sangat mengharapkan kritik dan saran yang bersifat membangun. Penulis
harap karya tulis ini berguna bagi masyarakat dan perkembangan ilmu
pengetahuan.
Wassalamualaikum Wr.Wb.
Jogjakarta, Januari 2011
Penulis

6
DAFTAR ISI
HALAMAN JUDUL i
HALAMAN PENGESAHAN iii
KATA PENGANTAR iv
DAFTAR ISI vi
DAFTAR GAMBAR ix
DAFTAR TABEL xi
DAFTAR LAMPIRAN xii
INTISARI xiii
ABSTRACT xiv
BAB I PENDAHULUAN
1.1 Latar Belakang 1
1.2 Rumusan Masalah 4
1.3 Tujuan Penelitian 4
1.4 Manfaat Penelitian 4
1.5 Batasan Masalah 5
BAB II TINJAUAN PUSTAKA 6
BAB III DASAR TEORI
3.1 Mekanika Kuantum 12
3.1.1 Persamaan Schrödinger 13
3.1.2 Metode Ab-Initio 14
3.1.3 Basis Set 20

7
3.2 Pemodelan Molekul 22
3.3 Optimasi Geometri 24
3.4 Celah Energi 25
3.5 Spektroskopi Molekul 27
3.5.1 Spektroskopi Ultraviolet dan Tampak 27
3.5.2 Spektroskopi Inframerah 30
3.6 Hipotesis Penelitian 32
BAB IV METODOLOGI PENELITIAN
4.1 Alat 33
4.1.1 Perangkat Keras 33
4.1.2 Perangkat Lunak 33
4.2 Bahan 34
4.3 Cara Kerja 35
4.3.1 Optimasi Geometri dengan Metode MM+ 35
4.3.2 Optimasi Geometri dengan Metode Ab-Initio 36
4.3.3 Perhitungan Transisi Elektronik dengan Spektra UV 37
4.3.4 Perhitungan Spektra Vibrasi menggunakan Perhitungan Ab
initio 37
BAB V HASIL DAN PEMBAHASAN
5.1 Optimasi Geometri 39
5.2 Celah Energi 42
5.3 Serapan Ultraviolet 46
5.4 Spektra Inframerah 49

8
BAB VI KESIMPULAN DAN SARAN
6.1 Kesimpulan 53
6.2 Saran 54
DAFTAR PUSTAKA 55
LAMPIRAN 56

9
DAFTAR GAMBAR
Gambar 1. [Ag(NH2-CH2-C6H4-OC6H13)2]+ 6
Gambar 2. Spektra inframerah CuPc dengan menggunakan basis set STO-3G 9
Gambar 3. Spektra inframerah CuPc dengan menggunakan basis set STO-6G 9
Gambar 4. Porifirin yang dikonjugasikan dengan Cu 11
Gambar 5. Diagram alir yang disederhanakan untuk perhitungan SCF 17
Gambar 6. Pita konduksi dan pita valensi pada berbagai material 25
Gambar 7. Struktur kompleks bis-4-heksiloksibenzilamin tembaga(I) 34
Gambar 8. Diagram alir prosedur optimasi geometri struktur kompleks bis-4-
heksiloksibenzilamin tembaga(I) dengan metode MM+ 35
Gambar 9. Diagram alir prosedur optimasi geometri struktur kompleks bis-4-
heksiloksibenzilamin tembaga(I) dengan metode ab initio 36
Gambar 10. Struktur 2 dimensi bis-4-heksiloksibenzilamin 39
Gambar 11. Struktur 2 dimensi kompleks bis-4-heksiloksibenzilamin tembaga(I)
39
Gambar 12. Senyawa bis-4- heksiloksibenzilamin teroptimasi 42
Gambar 13. Senyawa kompleks bis-4-heksiloksibenzilamin tembaga(I)
teroptimasi 42
Gambar 14. Diagram energi gap bis-4-heksiloksibenzilamin menggunakan basis
set STO-3G 44
Gambar 15. Diagram energi gap kompleks bis-4-heksiloksibenzilamin tembaga(I)
menggunakan basis set STO-3G 45

10
Gambar 16. Spektra inframerah bis-4-heksiloksibenzilamin 51
Gambar 17. Spektra inframerah kompleks bis-4-heksiloksibenzilamin tembaga(I)
51

11
DAFTAR TABEL
Tabel 1. Energi Hasil Optimasi Geometri 41
Tabel 2. Energi HOMO-LUMO 44
Tabel 3. Spektra Transisi Elektronik 48

12
DAFTAR LAMPIRAN
Lampiran 1. Koordinat kartesian atom-atom penyusun bis-4-
heksiloksibenzilamin dari hasil seleksi program HyperChem 7.0 for
Windows 57
Lampiran 2. Koordinat kartesian atom-atom penyusun kompleks bis-4-
heksiloksibenzilamin tembaga(I) dari hasil seleksi program
HyperChem 7.0 for Windows 58
Lampiran 3. Log File energi HOMO-LUMO senyawa bis-4-
heksilloksibenzilamin 59
Lampiran 4. Log File energi HOMO-LUMO senyawa kompleks bis-4-
heksiloksibenzilamin tembaga(I) 60
Lampiran 5. Spektrum transisi elektronik berupa panjang gelombang dan
intensitas serapan senyawa bis-4-heksiloksibenzilamin 62
Lampiran 6. Spektrum transisi elektronik berupa panjang gelombang dan
intensitas serapan senyawa kompleks bis-4-heksiloksibenzilamin
tembaga(I) 62
Lampiran 7. Input Frekuensi Senyawa bis-4-heksiloksibenzilamin dalam
Gaussian 98 for Windows 63
Lampiran 8. Input frekuensi senyawa kompleks bis-4-heksiloksibenzilamin
tembaga(I) dalam Gaussian 98 for Windows 64

13
ANALISIS SPEKTROSKOPIK KOMPLEKS BIS-4-HEKSILOKSIBENZILAMIN TEMBAGA(I) DENGAN
METODE AB INITIO
INTISARI
Oleh
DEWI KUMUDANINGSIH SISWOYO 03612003
Telah dilakukan kajian mengenai analisis spektroskopi pada kompleks bis-
4-heksiloksibenzilamin tembaga(I) dengan perhitungan komputasi. Metode yang digunakan adalah ab initio tingkat Hartree- Fock (HF) dengan basis set minimal Slater Type Orbital-3 Gaussian (STO-3G). Tujuan penelitian ini adalah memperoleh celah energi, transisi elektron, dan serapan gugus C=C, C-O, C-N pada senyawa kompleks bis-4-heksiloksibenzilamin tembaga(I). Kajian dilakukan dengan pembuatan model senyawa dan dilakukan langkah optimasi geometri menggunakan metode ab initio dilanjutkan dengan perhitungan transisi elektronik dan inframerah.
Hasil perhitungan menunjukkan bahwa celah energi pada senyawa kompleks bis-4-heksiloksibenzilamin tembaga(I) adalah 1,23 eV. Hasil perhitungan komputasi tersebut menunjukkan bahwa senyawa kompleks bis-4-heksiloksibenzilamin tembaga(I) termasuk semikonduktor organik. Transisi elektronik senyawa kompleks bis-4-heksiloksibenzilamin tembaga(I) mengalami pergeseran merah dengan selisih panjang gelombang 2,72 nm sedangkan pada spektra inframerah mengalami pergeseran merah dengan perbedaan frekuensi serapan gugus C-C sebesar 6,75 cm-1, gugus C-O sebesar 102,22 cm-1 dan gugus C-N sebesar 32,8 cm-1. Kata kunci : kompleks bis-4-heksiloksibenzilamin tembaga(I), celah energi, transisi elektronik, serapan inframerah, ab initio.

14
SPECTROSCOPIC ANALYSIS OF BIS-4-HEXYLOXYBENZYLAMINE COPPER(I) COMPLEX WITH
AB INITIO METHOD
ABSTRACT
DEWI KUMUDANINGSIH SISWOYO 03612003
Spectroscopic analysis of bis-4-hexyloxybenzylamine copper(I) complex with computational calculations has been done. The method used is the ab initio level Hartree-Fock (HF) with a minimal Slater basis set 3-Gaussian Type Orbital (STO-3G). The purpose of this study is to get the energy gap, electron transitions, and absorption of group C = C, C-O, C-N bis-4-hexyloxybenzylamine copper(I) complex. Modeling compound was done and geometry optimization on the next steps. This study was performed by using ab initio method followed by the calculate of electronic and infrared transition.
The result showed band gap bis-4-hexyloxybenzylamine copper(I) complex is 1,23 eV. Based on this result, the bis-4-hexyloxybenzylamine copper(I) complex is considered as an organic semiconductor. Electronic transition of bis-4-hexyloxybenzylamine copper(I) complex compound have red shifted with difference wavelength at 2,72 nm while in the infrared spectra red shifted with differences in absorption frequency C-C group at 6,75 cm-1, C-O group at 102,22 cm-1 and C-N group at 32,8 cm-1.
Keyword : bis-4-hexyloxybenzylamine copper(I) complex, the energy gap, electronic transition, infrared absorption, ab initio.

15
BAB I
PENDAHULUAN
1.1 Latar Belakang
Spektroskopi didefinisikan sebagai ilmu yang mempelajari interaksi antara
cahaya dan materi. Dalam catatan sejarah, spektroskopi mengacu kepada cabang
ilmu di mana cahaya tampak digunakan dalam teori-teori struktur materi serta
analisa kualitatif dan kuantitatif. Dalam masa modern, definisi spektroskopi
berkembang seiring teknik-teknik baru yang dikembangkan untuk memanfaatkan
tidak hanya cahaya tampak, tetapi juga bentuk lain dari radiasi elektromagnetik
dan non-elektromagnetik.
Dewasa ini, bahan semikonduktor organik yang dikonjugasikan sangat
menarik untuk beberapa aplikasi optik dan elektronik. Senyawa tersebut
menggabungkan kemudahan pembuatannya dengan struktur kimianya yang
fleksibel yang dapat dirancang untuk maksud khusus. Semikonduktor organik
menyediakan keuntungan salah satunya dalam sintesis kimia bahan organik yang
tidak terbatas, variasi dalam modifikasi struktur molekul organik, dan yang paling
potensial adalah biaya fabrikasi yang lebih rendah dalam pembuatan piranti
elektronik. Karakteristik bahan semikonduktor didasarkan pada perhitungan
selisih energi HOMO-LUMO.
Senyawa kompleks bis-4-heksiloksibenzilamin tembaga(I) merupakan
bahan semikonduktor organik yang mampu menyerap sinar gelombang
elektromagnetik. Struktur senyawa kompleks bis-4-heksiloksibenzilamin

16
tembaga(I) mempunyai ikatan rangkap terkonjugasi yang memungkinkan
terjadinya proses serapan gelombang elektromagnetik untuk mengeksitasi
elektron-elektron dari tingkat dasar ke kondisi eksitasi. Beda energi antara tingkat
eksitasi dan tingkat dasar ini disebut dengan celah energi (band gap). Panjang
gelombang yang diserap oleh senyawa kompleks bis-4-heksiloksibenzilamin
tembaga(I) terkait dengan beda energi kedua tingkat tersebut. Pada saat elektron-
elektron dari tingkat eksitasi kembali ke tingkat dasar akan memancarkan sinar
sesuai dengan panjang gelombang yang telah diserap. Panjang gelombang tersebut
mencirikan suatu tipe radiasi elektromagnetik, seperti gelombang radio,
ultraviolet, inframerah, tampak dan lain-lain yang secara langsung menjelaskan
bahwa senyawa tersebut dapat menyerap salah satu dari radiasi elektromagnetik
tersebut.
Eksperimen komputer memainkan peranan yang sangat penting dalam
perkembangan sains. Perkembangan komputasi yang sangat pesat, dimulai pada
tahun 1950 telah mengubah diskripsi suatu sistem kimia-fisika dengan masuknya
unsur baru diantara eksperimen dan teori yaitu eksperimen komputer, yang lebih
dikenal dengan nama komputasi sains (Pranowo, 2001).
Perkembangan komputer mengubah secara substansial hubungan tradisional
antara teori dan eksperimen. Simulasi komputer membutuhkan suatu metode yang
akurat dalam memodelkan suatu sistem yang dikaji. Simulasi sering dapat
dilakukan dengan kondisi yang sangat mirip dengan eksperimen sehingga hasil
dari perhitungan komputasi dapat dibandingkan secara langsung dengan
eksperimen. Selain itu juga, dapat mengkaji bagian yang tidak dapat dijangkau

17
secara eksperimen karena keterbatasan peralatan yang ada, serta waktu dan biaya
penelitian dapat ditekan seminimal mungkin.
Program yang digunakan dalam komputasi sains didasarkan pada berbagai
metode mekanika kuantum, salah satu metodenya yang berkembang adalah
metode ab initio. Metode ab initio ini mendiskripsikan sifat atom dan molekul
berdasarkan penyelesaian persamaan Schrödinger dengan pendekatan Born-
Oppenheimer. Metode ini menyelesaikan semua persamaan secara eksak dan
semua elektron yang ada diperhitungkan, sehingga memerlukan waktu
perhitungan yang lama. Oleh karena itu, sebelum memilih basis set yang akan
digunakan, diperhitungkan terlebih dahulu waktu untuk mengerjakannya.
Dalam bidang pengetahuan dan industri kompleks bis-4-
heksiloksibenzilamin tembaga(I) digunakan sebagai bahan kimia untuk
pembuatan pengolahan bahan celup, pigmen, tekstil, bahan kimia pertanian, asam
amino dan senyawa organik lainnya. Selain itu dapat digunakan sebagai pelarut
dan katalisator.
Pada penelitian ini akan dilakukan analisis spektroskopik kompleks bis-4-
heksiloksibenzilamin tembaga(I) untuk mengetahui sifat bahan terhadap radiasi
ultraviolet dan inframerah. Dalam penelitian ini akan dilakukan perhitungan
parameter berupa energi HOMO, energi LUMO, spektra inframerah dan spektra
transisi elektronik dengan metode ab initio.

18
1.2 Rumusan Masalah
1. Berapa energi gap dari senyawa kompleks bis-4-heksiloksibenzilamin
tembaga(I) menggunakan metode ab-initio?
2 Bagaimana transisi elektron dari senyawa kompleks bis-4-
heksiloksibenzilamin tembaga(I) analisis dengan UV-Visible?
3. Bagaimana pengaruh serapan gugus C=C, C-O, C-N pada struktur
senyawa kompleks bis-4-heksiloksibenzilamin tembaga(I) berdasarkan
spektra inframerah hasil perhitungan dengan metode ab initio?
1.3 Tujuan Penelitian
1. Memprediksi celah energi struktur kompleks bis-4-heksiloksibenzilamin
tembaga(I) menggunakan metode ab initio.
2. Mengkaji serapan sinar ultraviolet struktur kompleks bis-4-
heksiloksibenzilamin tembaga(I) dari celah energi yang diperoleh
berdasarkan hasil perhitungan dengan metode ab initio.
3. Mengkaji serapan gugus C=C, C-O, C-N, pada struktur senyawa
kompleks bis-4-heksiloksibenzilamin tembaga(I) berdasarkan spektra
inframerah hasil perhitungan dengan metode ab initio.
1.4 Manfaat Penelitian
Dari penelitian ini dapat diketahui bahwa sifat spektroskopik pada tingkat
molekular dapat dimanfaatkan untuk penentuan sifat bahan sensitif terhadap
radiasi inframerah dan Ultraviolet.

19
1.5 Batasan Masalah
Penelitian ini mengkaji tentang serapan ultraviolet dan inframerah, metode
simulasi yang digunakan dibatasi pada metode ab initio tingkat Hartree-Fock
dengan basis set minimal STO-3G.

20
BAB II
TINJAUAN PUSTAKA
Penelitian tentang turunan metallomesogens ion dari perak(I) bis-amine
kompleks : struktur dan mesogenik dilakukan oleh Lequerica, Baena dan Espinet
pada tahun 2007. Dalam jurnal menuliskan mesogens ion menggabungkan sifat
yang tidak dapat dipisahkan pada kristal cair dan senyawa ion, dan telah
digunakan sebagai pelarut, katalisator, template untuk sintesis, optik dan sisitem
ferroelektrik, elektronik atau konduktor ion dan sebagai selaput. Penelitian ini
menggunakan perak kompleks amina primer dengan ligan amina (anilin dan
benzilamin) dan rantai alkil atau alkoksi untuk menguji pengaruh anion pada sifat-
sifat dari material. Metode komputasi yang digunakan adalah program gaussian
98.11 dengan basis set LANL2DZ.
Gambar 1. [Ag(NH2-CH2-C6H4-OC6H13)2]+
Dari gambar 1 kemudian digunakan dalam penelitian selanjutnya dengan
menggunakan atom Cu sebagai atom pusat.
Perhitungan komputasi dapat digunakan untuk memprediksikan spektra
inframerah dan UV-Vis, sehingga dari data hasil perhitungan komputasi dapat
dijadikan pemandu dalam menerjemahkan spektra inframerah dan UV-Vis hasil

21
eksperimen. Untuk mendapatkan konformasi yang paling stabil dari struktur
molekul suatu senyawa harus dilakukan langkah optimasi dengan menggunakan
metode yang sesuai. Jika struktur teroptimasi telah didapatkan maka data
spektroskopi seperti inframerah dan UV-Vis tertentu dapat dihitung (Sudanti,
2006). Spektra inframerah yang diperoleh akan membantu mengenali gugus
fungsi dan ikatan antar atom, sedangkan UV-Vis dapat dikenali gugus berikatan
rangkap, juga energi HOMO dan LUMO yang bermanfaat untuk mengenali sifat
semikonduktor berdasarkan gap energi pita valensi (HOMO) dan pita konduksi
(LUMO) (Bintarti, 2008). Semakin kecil celah energi maka konduktifitas
listriknya semakin baik. Celah energi untuk semikonduktor berkisar pada rentang
antara 1-3,5 eV (Brutting, 2005).
Semikonduktor adalah sebuah bahan dengan konduktivitas listrik yang
berada di antara isolator dan konduktor. Pada temperatur yang sangat rendah,
sebuah semikonduktor bersifat sebagai isolator namun pada temperatur ruangan
bersifat konduktor. Bahan semikonduktor dapat digunakan untuk sel surya. Sel
surya (solar cell) terdiri dari dua semikonduktor yaitu semikonduktor tipe p dan
semikonduktor tipe n. Dasar penggunaan semikonduktor adalah terbentuknya
sambungan p-n (p-n juncktion) apabila semikonduktor tipe-p dan tipe-n
digabungkan. Sambungan ini yang merupakan dasar terjadinya revolusi industri
akibat ditemukan transisistor oleh Wiliam Shocklye, John Barden dan Walter
Brattain di laboratorium Bell pada tahun 1948.
Eksitasi elektron dari pita valensi ke pita konduksi disebabkan pula oleh
energi foton dari sinar yang mengenai permukaan semikonduktor. Dapatnya

22
elektron-elektron tereksitasi dari pita valensi ke pita konduksi akibat energi foton
yang mengenai permukaan semikonduktor memungkinkan semikonduktor
digunakan untuk membuat foto sel (photo cell).
Penelitian terdahulu tentang perhitunan celah energi (energi gap) dengan
perhitungan komputasi telah dilakukan oleh Astin Bintarti (2008). Metode yang
digunakan adalah ab initio tingkat Hartree-Fock (HF) dengan basis set minimal
Slater Type Orbital-3 Gaussian (STO-3G) dan basis set minimal Slater Type
Orbital-6 Gaussian (STO-6G).
Penelitian tersebut menggunakan senyawa Phthalocyanine. Senyawa
Phthalocyanine merupakan bahan semikonduktor organik yang mampu menyerap
sinar gelombang elektromagnetik. Perhitungan frekuensi vibrasi inframerah
struktur Copper Phthalocyanine (CuPc) teroptimasi menghasilkan nilai berupa
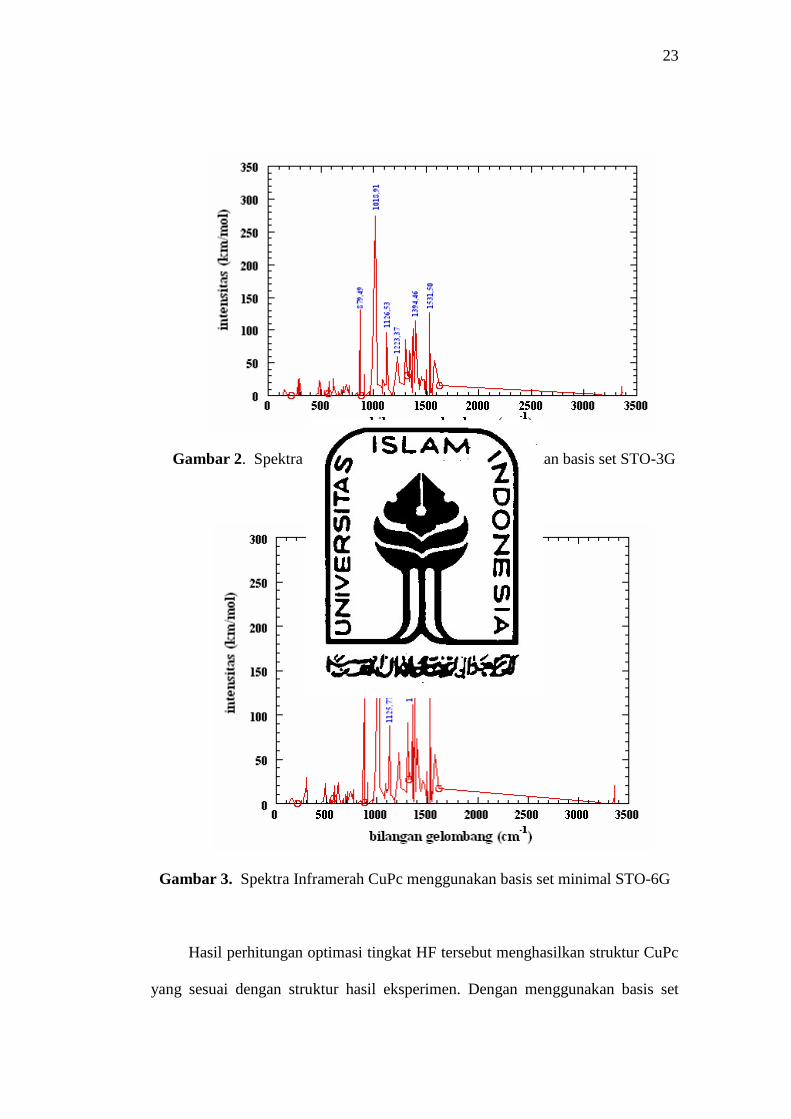
bilangan gelombang dan intensitas inframerah. Hasil tersebut diplot ke dalam
grafik spektrum inframerah yang diperoleh ditampilkan dalam gambar 2 dan
gambar 3. Frekuensi vibrasi inframerah dihitung pada tingkat Hartree-Fock
menggunakan basis set minimal STO-3G dan basis set minimal STO-6G, dan
skala frekuensi dikalikan dengan faktor koreksi 0,89. Faktor koreksi tersebut
digunakan untuk membetulkan kesalahan yang dibuat oleh pendekatan harmonik
dan oleh teori HF determinan tunggal.
23
Gambar 2. Spektra inframerah CuPc dengan menggunakan basis set STO-3G
Gambar 3. Spektra Inframerah CuPc menggunakan basis set minimal STO-6G
Hasil perhitungan optimasi tingkat HF tersebut menghasilkan struktur CuPc
yang sesuai dengan struktur hasil eksperimen. Dengan menggunakan basis set

24
minimal STO-6G diperoleh energi gap CuPc sebesar 1,50 eV, dimana
perhitungan dengan tingkat HF memberikan sedikit perbedaan sekitar 0,1 – 0,2
eV dari hasil eksperimen. Hasil perhitungan komputasi tersebut menunjukan
bahwa senyawa CuPc termasuk semikonduktor organik. Di samping itu, senyawa
CuPc menyerap sinar inframerah dekat dengan panjang gelombang serapan
maksimum sebesar 827 nm. Pada perhitungan frekuensi vibrasi inframerah
senyawa CuPc, pita serapan terkuat molekul CuPc dengan menggunakan dua
basis set yang berbeda tersebut berada pada daerah frekuensi vibrasi rentangan
ikatan C – C, dengan energi serapan sebesar 0,012 eV.
Penelitian yang berjudul Kajian Teoritis untuk Menentukan Celah Energi
Porfirin Terkonjugasi Atom Perak dan Tembaga dengan Menggunakan Metode
Mekanika Kuantum Semiempiris Zindo/1 dilakukan oleh Sundanti (2006).
Penelitian tersebut menggunakan Porfirin dasar (C27H26N4) dan kemudian
dikonjugasikan dengan Ag dan Cu yang ditunjukkan pada gambar 4.
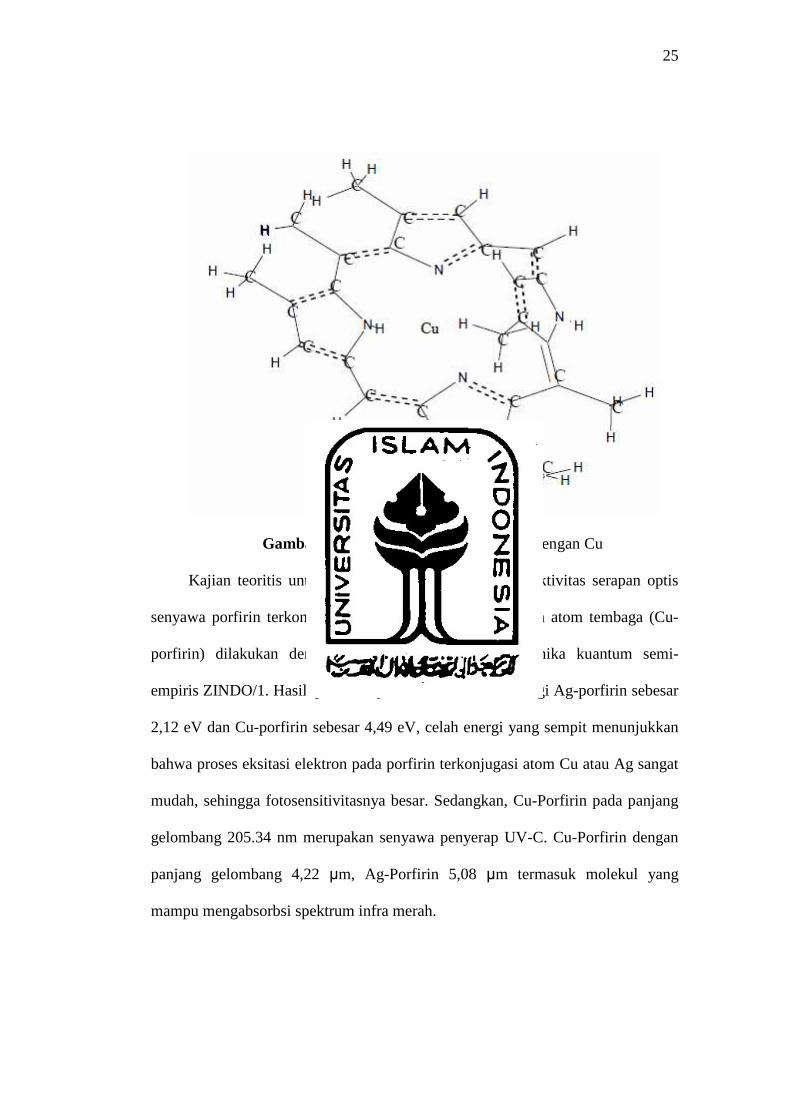
25
Gambar 4. Porfirin yang dikonjugasikan dengan Cu
Kajian teoritis untuk menentukan celah energi dan aktivitas serapan optis
senyawa porfirin terkonjugasi atom perak (Ag-porfirin) dan atom tembaga (Cu-
porfirin) dilakukan dengan menggunakan metode mekanika kuantum semi-
empiris ZINDO/1. Hasilnya menunjukkan bahwa celah energi Ag-porfirin sebesar
2,12 eV dan Cu-porfirin sebesar 4,49 eV, celah energi yang sempit menunjukkan
bahwa proses eksitasi elektron pada porfirin terkonjugasi atom Cu atau Ag sangat
mudah, sehingga fotosensitivitasnya besar. Sedangkan, Cu-Porfirin pada panjang
gelombang 205.34 nm merupakan senyawa penyerap UV-C. Cu-Porfirin dengan
panjang gelombang 4,22 µm, Ag-Porfirin 5,08 µm termasuk molekul yang
mampu mengabsorbsi spektrum infra merah.

26
BAB III
DASAR TEORI
3.1 Mekanika Kuantum
Mekanika kuantum adalah cabang dasar fisika yang menggantikan mekanika
klasik pada tataran atom dan sub atom. Ilmu ini memberikan kerangka
matematika untuk berbagai cabang fisika dan kimia termasuk fisika atom, fisika
molekuler, kimia komputasi, kimia kuantum, fisika partikel dan fisika nuklir.
Mekanika kuantum adalah bagian dari teori medan kuantum dan fisika kuantum
umumnya yang bersama relativitas umum merupakan salah satu pilar fisika
modern. Dasar dari mekanika kuantum adalah bahwa energi itu tidak kontinyu,
tapi diskrit berupa paket atau kuanta. Konsep ini revolusioner bertentangan
dengan fisika klasik yang berasumsi bahwa energi itu berkesinambungan.
Mekanika kuantum sangat berguna untuk menjelaskan apa yang terjadi di
mikroskopik level, misalnya elektron di dalam atom. Atom biasanya digambarkan
sebagai sebuah sistem di mana elektron (yang bermuatan listrik negatif) beredar
seputar nucleus (yang bermuatan listrik positif). Menurut mekanika kuantum
ketika sebuah elektron berpindah dari energi level yang lebih tinggi (misalnya n =
2) ke energi level yang lebih rendah (misalnya n = 1), energi berupa sebuah
cahaya partikel, foton, dilepaskan seperti pada persamaan (1) :
E = hυ (1)
Dimana : E adalah energi (J)
h adalah tetapan planck, (Js), h = 6,63 ×10 -34

27
υ adalah frekuensi dari cahaya (Hz) (Sartika, 2007)
Dalam mekanika kuantum keadaan suatu sistem digambarkan melalui fungsi
koordinat partikel dalam sistem yang disebut dengan fungsi gelombang atau
fungsi keadaan. Fungsi ini dapat diperoleh melalui penyelesaian persamaan
Schrödinger. Mekanika kuantum dalam prakteknya terbagi atas dua metode yaitu
ab-initio dan semiempirik (Pranowo, 2001).
3.1.1 Persamaan Schrödinger
Perhitungan kimia kuantum didasarkan pada teori orbital molekul yang
menetapkan prinsip-prinsip mekanika kuantum dari suatu sistem kimia, berupa
yang menggambarkan inti dan elektron-elektron yang terdistribusi mengelilingi
inti. Energi dan fungsi gelombang dalam keadaan stationer diberikan dengan
penyelesaian persamaan Schrödinger (2) berikut :
Ĥψ = Eψ (2)
Dimana Ĥ adalah operator Hamilton yang menyatakan energi kinetik dan
potensial dari sistem yang mengandung elektron dari inti atom. Energi ini analog
dengan energi kinetik mekanika klasik dari partikel dan interaksi elektrostatik
Coulombic antara inti dan elektron. Simbol ψ adalah fungsi gelombang, yang
merupakan fungsi koordinat (posisi) inti dan elektron dan berisikan semua
informasi mengenai sistem. E adalah energi total dari sistem. Sifat molekul yang
dapat dihitung melalui penyelesaian tersebut adalah geometri molekul, stabilitas
relatif, dipol dan muatan atomik (Dogra, 1990).

28
Empat pendekatan yang biasanya diterapkan adalah
1. Tak gayut waktu, sistem dalam keadaan stationer terhadap waktu.
2. Mengabaikan efek relativitas, hal ini memberikan garansi bahwa elektron
bergerak tidak akan lebih lambat dari kecepatan cahaya. Koreksi perlu
dilakukan untuk atom yang mempunyai muatan inti yang sangat besar.
3. Pendekatan Born-Oppenheimer, pemisahan gerakan inti dan elektron.
4. Pendekatan orbital, elektron berada/menempati daerah dalam ruang tertentu
di sekitar inti (Pranowo, 2001).
3.1.2 Metode Ab-Initio
Teori ab initio adalah sebuah konsep perhitungan yang bersifat umum dari
penyelesaian persamaan Schrödinger yang secara praktis dapat diprediksi tentang
keakuratan dan kesalahannya.
Ab initio digunakan untuk menerangkan bagaimana persamaan Schrödinger
diselesaikan. Dalam teori ab initio, pendefenisian Hamiltonian dan pembentukan
fungsi gelombang dilakukan secara fungsional. Untuk memperoleh fungsi
gelombang terbaik dengan energi yang paling minimum dilakukan dengan
penerapan metode varisional. Langkah pertama untuk menyelesaikan persamaan
Schrödinger adalah pemisahan gerakan inti dan elektron melalui pendekatan
Born-Oppenheimer. Pendekatan Born-Oppenheimer yaitu didasarkan pada
permukaan energi potensial pada tingkat inti atom. Pendekatan ini diterapkan
karena elektron-elektron lebih ringan daripada inti, sehingga menyebabkan

29
gerakan inti lebih lambat atau dengan kata lain dibandingkan dengan gerakan
elektron, inti relatif tidak bergerak (rigid).
Langkah selanjutnya adalah penyelesaian fungsi gelombang melalui
pembahasan atom dan molekul kulit tertutup dan pendekatan umum yang sering
digunakan dalam penyelesaian persamaan Schrödinger. Fungsi gelombang ψ
diasumsikan sebagai suatu fungsi koordinat n-elektron dengan koordinat inti yang
tetap dan inti didekati melalui fungsi n-satu elektron yang mewakili orbital.
Ab initio menyelesaikan semua persamaan secara eksak dan semua elektron
yang ada diperhitungan, sehingga memerlukan waktu perhitungan yang lama.
Fock dan Slater secara serempak dan terpisah mengembangkan suatu persamaan
yang sekarang disebut persamaan Hartree-Fock. Untuk sistem kulit tertutup
biasanya digunakan pendekatan Hartree-Fock terhalang (Restricted Hartree-Fock,
RHF) masing-masing orbital berisi pasangan elektron dinyatakan dalam suatu
bentuk determinan tunggal (Leach, 1996). Sistem kulit tertutup digunakan untuk
mendeskripsikan molekul dengan jumlah elektron genap. Pada sistem kulit
terbuka yaitu sistem yang mempunyai satu atau lebih elektron yang tidak
berpasangan. Untuk sistem ini biasanya digunakan pendekatan Hartree-Fock tak
terhalang (Unrestricted Hartree-Fock, UHF) dengan determinan Slater tidak
terhalang untuk menggambarkan fungsi gelombang (Goodman, 1998).
Determinan ini disusun oleh himpunan orbital spin tidak terhalang. Orbital spin
tidak terhalang mempunyai orbital spasial yang berbeda untuk elektron dengan
spin berbeda. Sistem kulit terbuka digunakan untuk mendiskripsikan molekul
dengan jumlah elektron ganjil.

30
Persamaan Hartree-Fock adalah suatu pasangan persamaan integro
differensial yang dapat diselesaikan hanya melalui metode berulang atau iteratif.
Untuk menyelesaikan persamaan Hartree-Fock, suatu rangkaian perhitungan awal
dilakukan dengan pemilihan orbital, diikuti pembentukan operator Fock dan
selanjutnya adalah penyelesaian persamaan yang digunakan untuk memperoleh
orbital baru. Orbital yang terhitung digunakan untuk menentukan operator Fock
baru. Prosedur ini diulang sampai suatu kriteria konvergensi dicapai. Kriteria
konvergensi biasanya didasarkan pada perubahan energi dari suatu orbital.
Prosedur ini dikenal sebagai metode medan keajegan diri (SCF-Self-Consistent-
Field), karena prosedur berulang terus-menerus dilakukan sampai medan
elektrostatik efektif tidak mengalami perubahan. Diagram perhitungan SCF
ditunjukan dalam gambar 5 (Pranowo, 2001).
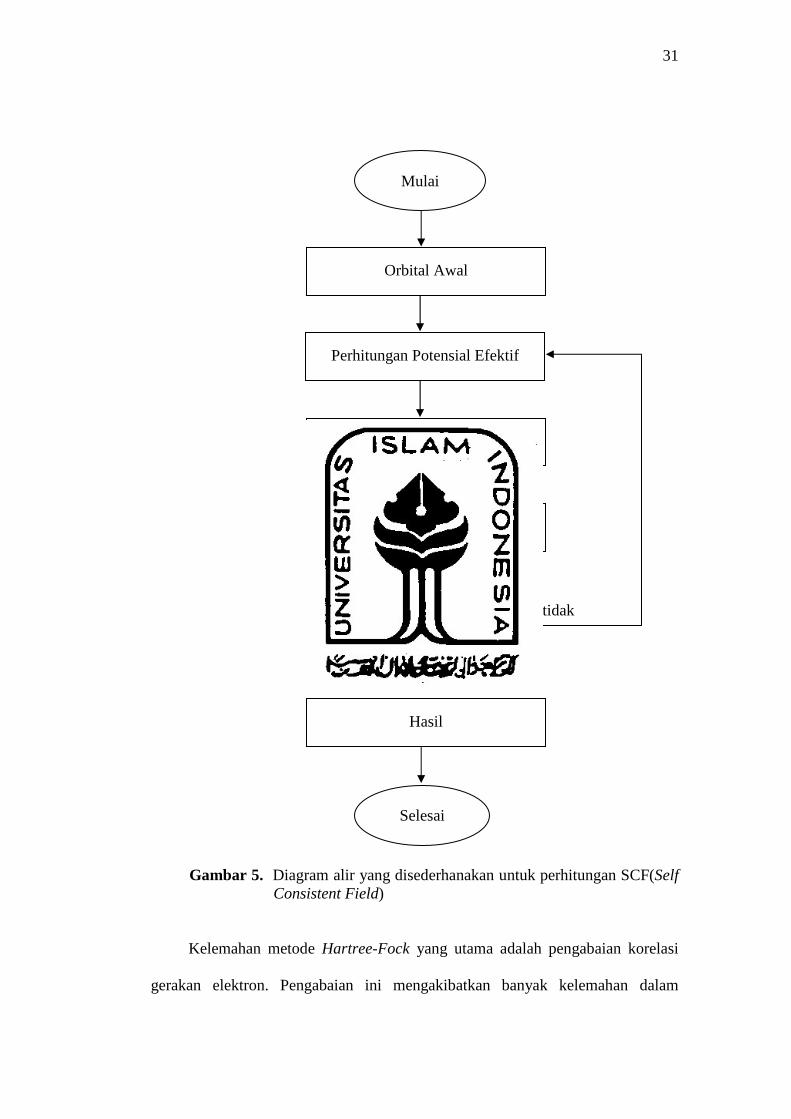
31
Gambar 5. Diagram alir yang disederhanakan untuk perhitungan SCF(Self Consistent Field)
Kelemahan metode Hartree-Fock yang utama adalah pengabaian korelasi
gerakan elektron. Pengabaian ini mengakibatkan banyak kelemahan dalam
Penyelesaian persamaan orbital
Konvergen ?
Hasil
Selesai
Mulai
Orbital Awal
Perhitungan Potensial Efektif
Pembentukan operator Fock
tidak
Ya

32
deskripsi struktur elektronik. Salah satu akibat yang penting pada peristiwa
disosiasi, metode Hartree-Fock sel tertutup sering tidak dapat mendiskripsikan
secara tepat ketika inti bergerak kepemisahan tak terhingga.
Beberapa pendekatan yang digunakan menghitung energi korelasi setelah
perhitungan Hartree-Fock (Post-HF) adalah
1. Interaksi konfigurasi (Configuration Interaction, CI)
Konsep dari CI adalah penyusunan kembali determinan Slater yang
melibatkan orbital virtual tidak terisi dari perhitungan Hartree-Fock.
Keunggulan metode CI adalah sifatnya yang variasional sehingga energi
terhitung selalu lebih besar daripada energi eksak.Kelemahan metode ini
adalah tidak memungkinkan melakukan perhitungan CI penuh untuk sistem
yang besar, mungkin hanya dapat dilakukan pada sistem kecil yang berisi
beberapa atom.
2. Teori Perturbasi Møller-Plesset (Møller-Plesset Pertur-bation Theory)
Untuk mengaplikasikan teori perturbasi, maka Hamilton yang tidak
mengalami perturbasi harus dipilih. Pemilihan yang paling umum adalah
menjadikannya sebagai penjumlahan dari operator-operator Fock. Metode ini
disebut metode Møller-Plesset. Metode yang paling popular adalah korelasi
pada tingkat yang paling rendah yaitu MP2. Dalam prakteknya keunggulan dari
metode MP2 adalah cepat (secepat perhitungan SCF) dan bersifat taat-ukuran
sedangkan kelemahannya tidak bersifat varisioanal sehingga estimasi energi
korelasi bias terlalu besar.

33
3. Multikonfigurasi medan keajekan mandiri (Multi-configuration Self
Consistent Field, MCSCF atau Complete Active Space Self Consistent Field,
CASSCF)
Multi configuration SCF (MCSCF) dan Complete Active SCF (CASSCF)
adalah metode yang perhitungan orbital HF teroptimasi dilakukan secara
simultan dengan prosedur CI. Hal ini berguna dalam mempelajari
permasalahan yang tidak dapat diselesaikan hanya dengan SCF, misalnya pada
sistem dalam keadaan tereksitasi, atau untuk menghasilkan eigenvalue awal
yang akan digunakan dalam perhitungan CI selanjutnya. Metode MCSCF
memerlukan kehati-hatian dalam menentukan himpunan basis dan secara
khusus dalam menentukan ruang aktifnya, dan pada umumnya metode ini tidak
digunakan dalam perhitungan energi secara rutin. Metode ini sangat
bermanfaat untuk mempelajari proses yang melibatkan peralihan permukaan
energi potensial seperti yang terjadi pada reaksi fotokimia.
Perkembangan metode khusus yang dikenal dengan teori fungsional kerapatan
(Density Fungtional Theory, DFT) sangat pesat. Dalam DFT integral elektron
ganda dihitung tidak menggunakan metode konvensional. Metode ini juga
menjanjikan dari segi kecepatan proses, sehingga nampaknya metode ini akan
memberikan sumbangan yang besar dikemudian hari terutama jika kita
berhadapan dengan sistem yang besar.

34
3.1.3 Basis Set
Basis set merupakan deskripsi matematik orbital dalam suatu sistem yang
digunakan dalam perhitungan teoritis. Pemilihan basis set yang paling tepat dalam
metode ab initio menjadi bagian yang sangat penting untuk akurasi dan tingkat
kepercayaan dalam hasil perhitungan. Semakin besar basis set menunjukkan
semakin kecil ketidakleluasaan elektron dan lebih akuratnya perkiraan eksak
orbital molekuler.
Basis set tersusun atas fungsi atom. Terdapat dua tipe fungsi basis yang
umum digunakan dalam perhitungan struktur elektronik, yaitu orbital tipe Slater
(STO) dan orbital tipe Gaussian (GTO). Orbital tipe Slater berbentuk :
χζ,n,l,m(r,θ,φ) = NYl,m(θ,φ)rn-1exp(-ζr) (3)
N adalah konstanta normalisasi dan ζ adalah eksponen orbital, Yl,m adalah fungsi
harmonik sferis, l dan m adalah bilangan kuantum momentum anguler.
Keunggulan utama pada fungsi-fungsi basis Slater adalah kemampuannya
menerangkan kelakuan orbital pada jarak pendek dan panjang. Selanjutnya, secara
variasional penentuan orbital akan mempunyai suatu “ekor” eksponensial dengan
laju penurunan yang terlalu cepat, juga “perpotongan” pada inti hanya akan benar
untuk kombinasi linier khusus orbital Slater.
Selain dalam bentuk koordinat polar, orbital tipe Slater dapat juga ditulis
dalam koordinat kartesian, yaitu :
χkmn = Nxkymzn exp(-ζr) (4)
STO biasanya digunakan untuk sistem atom dan diatom yang membutuhkan
akurasi yang tinggi dan metode semiempirik yang integral tiga dan empatnya

35
diabaikan. Orbital tipe gaussian dapat ditulis dalam koordinat polar ataupun dalam
koordinat kartesian, sebagai berikut :
χζ,n,l,m(r,θ,φ) = NYl,m(θ,φ)r(2n-2-1)exp(-αr2) (5)
χkmn = Nxkymzn exp(-αr2) (6)
k, m, dan n mementukan tipe orbital dan α adalah eksponen, yang menentukan
energi elektronik orbital.
Untuk mendapatkan akurasi yang tinggi dengan waktu perhitungan yang
cepat dilakukan kombinasi linear tertentu dari suatu himpunan yang lengkap dari
fungsi basis primitif (Primitive Gaussian Function, PGF), yang dikenal sebagai
fungsi Gaussian terluaskan (CGF –Contructed Gaussian Function)
χ CGF = ΣαiχiPGF (7)
αi menyatakan koefisien perluasan. Basis set yang terluaskan akan selalu
menghasilkan energi terhitung lebih besar dari harga energi sebelumnya, akibat
adanya keterbatasan jumlah parameter variasional dan berkurangnya fleksibilitas
basis set. Keuntungan dari penggunaan basis set terluaskan adalah bertambahnya
efisiensi komputasi secara signifikan. Basis set yang dipilih untuk perhitungan
kimia komputasi mempunyai batasan bahwa basis set harus mempunyai
kemampuan untuk meningkatkan efisiensi komputasionalnya dan besarnya
akurasi yang hilang masih dalam batas yang bisa diterima.
Jenis basis set yang digunakan untuk perhitungan molekul terdapat beberapa
macam. Pada umumnya basis set diturunkan oleh Pople dan Huzinaga. Himpunan
basis yang dikembangkan oleh Pople adalah minimal basis set STO-LG, dengan L

36
adalah Gaussian primitif yang diperluaskan menjadi satu fungsi. Lebih besar L
yang digunakan, hasilnya lebih akurat. Semua persamaan basis set dalam bentuk
STO-LG merupakan basis set minimal, yang mana basis set tersebut hanya
menggambarkan aspek yang paling mendasar dari orbital-orbital. Basis set
minimal hanya mengandung sejumlah fungsi yang diperlukan untuk
mengakomodasi semua orbital terisi dalam setiap atom. Paling sedikit dibutuhkan
tiga fungsi Gaussian untuk menyatakan secara baik setiap orbital tipe Slater. Basis
set hanya mengandung satu perluasan untuk setiap orbital atomik sehingga
bersifat kurang fleksibel (Pranowo, 2003).
3.2 Pemodelan Molekul
Pemodelan molekul merupakan suatu cara untuk menggambarkan atau
menampilkan perilaku molekul atau sistem molekul sebagaimana keadaan
sesungguhnya. Melalui pemodelan ini diharapkan memudahkan dalam
mempelajari dan memahami bangun molekul, sifat-sifat serta perilaku molekul
maupun sistem molekul tersebut, terutama sifat-sifat yang sulit teramati dalam
eksperimen. Selain itu juga dapat mendukung penelitian maupun studi terhadap
molekul tersebut.
Pemodelan molekul merupakan kumpulan atau teknik-teknik untuk
memperoleh, menggambarkan dan memanipulasi struktur-struktur dan reaksi-
reaksi dari suatu molekul dan sifat-sifatnya yang bergantung pada struktur tiga
dimensinya. Pemodelan molekul dapat dilakukan dengan menggunakan berbagai
metode, seperti Mekanika Kuantum, Mekanika Molekul, minimasi energi,

37
simulasi analisis konformasi serta berbagai metode lainnya yang dapat digunakan
untuk mempelajari dan memprediksi perilaku dari suatu sistem molekul.
Model molekul yang umum dikenal ada dua macam, yaitu model molekul
dalam bentuk stick and ball yang dibuat oleh Dreiding dan model space filling
yang dibuat oleh Corey, Pauling, dan Koltun. Melalui pemodelan molekul akan
mudah dipahami bangun molekul, sifat-sifat serta perilaku molekul (Leach, 1996).
Model digunakan untuk membangun suatu teori yang merupakan jalan
sederhana untuk menggambarkan dan memprediksi hasil ilmiah. Model molekul
merupakan penggambaran yang tidak persis sama dengan kenyataan dan tidak
lengkap. Model dapat berupa penggambaran matematika sederhana atau non
matematika penuh untuk memprediksi dan mengerti fenomena tanpa harus
bekerja dengan manipulasi matematika yang kompleks.
Pemodelan molekul yang dilakukan dengan tujuan untuk memberikan
gambaran tentang perilaku molekul, yang akhirnya digunakan untuk melakukan
perhitungan-perhitungan terhadap sifat-sifat fisika dan kimia molekul tersebut.
Saat ini pemodelan molekul telah berkembang pesat dan memiliki kaitan erat
dengan penggunaan komputer, pemodelan molekul dengan menggunakan
komputer memberikan manfaat yang lebih besar daripada pemodelan molekul
secara mekanis. Karena dengan komputer ini parameter-parameter yang terdapat
pada suatu molekul akan dapat dengan mudah dibuat, perhitungan numeris dapat
dilakukan dengan cepat meskipun memerlukan iterasi yang banyak serta
penggambaran model yang relatif lebih rumit dan kompleks akan lebih mudah
dilakukan (Bintarti, 2008).

38
3.3 Optimasi Geometri
Optimasi geometri merupakan metode untuk menghitung dan menampilkan
struktur molekul dengan energi potensial minimum dan gaya-gaya atomik
terkecil. Optimasi geometri dilakukan untuk menentukan struktur molekul yang
stabil yang memiliki energi potensial rendah.
Optimasi geometri untuk mempersiapkan molekul-molekul agar dapat
dilakukan perhitungan secara single point. Program hyperchem melalui geometri
optimasi dengan set-up koordinat suatu molekul dan berusaha untuk menemukan
koordinat baru yang mempunyai energi potensial yang paling rendah.
Optimasi dalam istilah matematika dimaksudkan untuk menyatakan bahwa
suatu struktur didapatkan dengan proses perhitungan dengan cara membandingkan
struktur yang terhitung dengan struktur sebelumnya. Struktur dimodifikasi agar
konsisten dengan informasi parameter yang ada dalam program.
Beberapa prosedur matematika telah digunakan untuk menentukan
bagaimana geometri akan berubah dari satu langkah ke langkah berikutnya. Setiap
perubahan geometri akan diikuti dengan perhitungan energi. Program yang
tersedia akan menyimpan perubahan harga geometri sampai harga spesifik cut-off
dicapai, pada saat ini molekul dinyatakan telah teroptimasi. Harga cut-off spesifik
dikenal dengan istilah konvergensi. kriteria konvergensi yang umum adalah
perubahan dalam energi, antara struktur terhitung terakhir dengan struktur terakhir
kedua yang harus lebih kecil dari 0,5 kjoule (Pranowo, 2001).

39
3.4 Celah Energi
Interaksi antara atom pada zat padat (solid) membentuk orbital molekul.
Orbital ini menghasilkan daerah tingkat energi yang terikat kuat dinamakan pita
(band) karena prinsip eksklusi Pauli menjaga agar dua elektron dalam atom tidak
mempunyai bilangan kuantum yang sama. Energi elektron terendah menempati
pita valensi dan terikat pada atom. Energi elektron tertinggi menempati pita
konduksi dan mempunyai energi yang cukup untuk melepaskan dari atom dan
dapat bergerak bebas. Perbedaan energi antara orbitalnya (contohnya, orbital 1s
dan orbital 2s) membentuk jurang pita (band gap) dimana pada daerah terlarang
ini tidak ditempati oleh elektron (Carter, 2003).
(a) (b) (c)
Gambar 6. Pita konduksi dan pita valensi pada berbagai material (a) Isolator (b) Semikonduktor (c) Konduktor
Pada material organik, karena molekul berinteraksi hanya oleh interaksi
lemah dari gaya Van der Waals, bagian atas dari tingkat valensi yang terisi
elektron (pita valensi) dan tingkat energi terendah yang tidak terisi elektron (pita
konduksi) biasanya dilokalisasi pada tiap molekulnya. Tingkat energi teratas yang
terisi dan tingkat energi bawah yang tidak terisi biasanya sering ditulis sebagai
HOMO (Highest Occupied Molecular Orbital) dan LUMO (Lowest Unoccupied
Pita Konduksi
Pita Valensi
Eg
Pita Konduksi
Pita Valensi
Pita Konduksi
Pita Valensi
Eg

40
Molecular Orbital). HOMO dan LUMO diukur berdasarkan aras vakum atau
vacuum level (VL), yaitu energi teratas dimana elektron dapat lepas dari atom,
biasanya digunakan untuk energi referensi (Triyana, 2004).
Pada material semikonduktor mempunyai celah energi yang kecil, sekitar
berorde 1 eV (Sze, 2001) dan kemungkinan elektron dapat tereksitasi dari HOMO
ke LUMO (Leach, 2001). Celah energi yang begitu kecil, akan sangat menentukan
sekali daya serapan yang begitu besar, sehingga senyawa ini akan menjadi
detektor inframerah organik dan cahaya tampak yang menghasilkan spektra UV-
Vis dan inframerah (Sudanti, 2006). Celah energi dapat diukur dari kurva serapan
optikal, sesuai data pada jarak energi tampak.
Berdasarkan eksperimen dalam laboratorium, energi ionisasi material
didefinisikan sebagai selisih energi antara vacuum level dan ujung energi ikat
yang rendah dari HOMO, sedangkan afinitas elektron didefinisikan sebagai selisih
energi antara vacuum level dan ujung energi ikat yang tinggi dari LUMO.
Berdasarkan perhitungan komputasi, HOMO dan LUMO dapat diidentifikasikan
dengan menemukan titik dimana pada bagian simetri terdapat perubahan dari O
(occupied) ke V (virtual). Data komputasi yang dihasilkan berupa energi HOMO
yang bernilai negatif (-). Hal ini menunjukkan bahwa orbital tersebut terisi oleh
elektron, sedangkan energi LUMO bernilai positif (+), yang menunjukkan bahwa
orbital tersebut tidak terisi oleh elektron. Oleh karena itu, untuk perhitungan celah
energi dihitung dari titik 0 eV yang diasumsikan sebagai vacuum level (VL).
Energi ionisasi dan afinitas elektron dari zat padat didefinisikan sebagai
pemisahan energi dari HOMO dan LUMO dari VL (Triyana, 2004).

41
Dengan hasil eksperimen dan perhitungan komputasi, energi HOMO dan
LUMO dapat dihitung, sehingga diperoleh celah energi yang dapat dirumuskan
pada persamaan
Eg = EHOMO – ELUMO (8)
dengan Eg adalah energi gap (celah energi).
3.5 Spektroskopi Molekul
Spektroskopi adalah studi mengenai interaksi cahaya dengan atom atau
molekul. Radiasi cahaya adalah suatu radiasi elektromagnet yang memiliki sifat
ganda, yaitu sifatnya sebagai partikel dan sebagai gelombang. Sifat gelombang
yang terpenting adalah panjang gelombang (λ). Tanda λ menyatakan jarak yang
ditempuh oleh gelombang selama satu siklus. Selain itu gelombang juga memiliki
amplitudo (A), periode (τ) atau waktu untuk satu siklus sempurna dan frekuensi
(υ) yaitu jumlah siklus dalam tiap detik. Hubungan antara panjang gelombang dan
frekuensi ditunjukkan pada persamaan
υ = c
λ (9)
dimana c adalah kecepatan cahaya.
Cahaya juga dapat dipandang sebagai paket energi yang bergerak dengan
kecepatan tinggi, yaitu 3,0 x 108 m/s. Paket energi ini disebut dengan foton. Besar
energi foton menurut persamaan Planck adalah
E = h υ (10)
dimana h adalah tetapan Planck yang nilainya 6,63 x 10-34 Joule detik. Apabila
cahaya kontinyu (cahaya dengan semua panjang gelombang yang mungkin)

42
dilewatkan melalui sebuah prisma, maka cahaya tersebut akan terdispersi. Jika
cahaya yang terdispersi ini dilewatkan melalui sel yang mengandung sampel atau
molekul, maka cahaya yang keluar menjadi tidak kontinyu lagi. Beberapa
gelombang cahaya berinteraksi dengan molekul atau atom-atom sampel dan
terabsorbsi. Panjang gelombang yang hilang dapat dideteksi dengan menjatuhkan
cahaya yang keluar dari sel sampel pada plat fotografi. Energi molekul dinyatakan
dalam energi translasi, rotasi, vibrasi, dan elektronik (Sastrohamidjojo,1991).
3.5.1 Spektroskopi Ultraviolet dan Tampak
Spektrum ultraviolet adalah suatu gambar antara panjang gelombang atau
frekuensi serapan lawan intensitas serapan (transmitasi atau absorbansi). Cahaya
yang dapat dilihat oleh manusia disebut cahaya terlihat/tampak. Spektrum tampak
terentang dari sekitar 400 nm (ungu) sampai 750 nm (merah), sedangkan
spektrum ultraviolet terentang dari 100 sampai 400 nm.
Serapan cahaya oleh molekul dalam daerah spektrum ultraviolet dan terlihat
tergantung pada struktur elektronik dari molekul. Spektra ultraviolet dan terlihat
dari senyawa-senyawa organik berkaitan erat transisi-transisi diantara tingkatan-
tingkatan tenaga elektronik. Disebabkan karena hal itu, maka serapan radiasi
ultraviolet/terlihat sering dikenal sebagai spektroskopi elektronik. Karena elektron
dalam molekul memiliki tenaga yang tak sama, maka tenaga yang diserap dalam
proses eksitasi dapat mengakibatkan terjadinya satu atau lebih transisi tergantung
pada jenis elektron yang terlihat. Transisi-transisi tersebut diklasifikasikan seperti
berikut :

43
1. Transisi π → σ → ionisasi
Transisi ini sedikit dipelajari oleh orang-orang organik karena terjadi
dalam ultraviolet jauh yaitu 180 nm dan untuk mempelajarinya membutuhkan
alat khusus. Daerah ini dikenal daerah Schuman atau ultraviolet vakum. Pada
serapan ini elektron-elektron dipromosi ke tingkat tenaga yang lebih tinggi
hingga akhirnya ionisasi terjadi hingga memberikan jalur-jalur serapan.
2. Transisi π → π∗
Serapan dari jenis ini disebabkan penterapan tenaga oleh elektron-elektron
- π dan bergerak dari orbital ikatan ke orbital anti ikatan. Transisi ini
menunjukkan pergeseran merah dengan adanya substitusi gugus-gugus yang
memberi atau menarik elektron dan dengan kenaikan dalam tetapan dielektrik
dari pelarut. Dalam kedua keadaan ini menstabilkan tingkatan tereksitasi polar.
3. Transisi n→ π∗
Transisi jenis ini meliputi transisi elektron-elektron hetero atom tak
berikatan ke orbital anti ikatan π∗ . serapan ini terjadi pada panjang gelombang
dan intensitas rendah. Transisi ini menunjukkan pergeseran hipsokromik (biru)
dalam pelarut-pelarut yang lebih polar dan dengan substituen-substituen yang
bersifat pemberi elektron.
4. Transisi n → σ∗
Senyawa-senyawa jenuh yang mengandung hetero atom seperti oksigen,
nitrogen, belerang atau halogen, memiliki (elektron-elektron n – atau –p)
disamping elektron-elektron -σ .senyawa-senyawa hetero atom menunjukkan
jalur serapan yang kemungkinan disebabkan oleh transisi elektron-elektron dari

44
orbital tak berikatan atom-atom hetero ke orbital anti ikatan σ∗. Transisi n - σ∗
membutuhkan tenaga yang lebih sedikit daripada transisi σ - σ∗. Namun
demikian senyawa-senyawa dalam klas ini tidak menunjukkan serapan dalam
daerah ultraviolet dekat (Sastrohamidjojo, 2001).
3.5.2 Spektroskopi Inframerah
Spektroskopi inframerah merupakan suatu metode yang mengamati
interaksi molekul dengan radiasi elektromagnetik yang berada pada daerah
panjang gelombang 0.75 – 1.000 µm atau pada bilangan gelombang 13.000 – 10
cm-1. Penyerapan gelombang elektromagnetik dapat menyebabkan terjadinya
eksitasi tingkat-tingkat energi dalam molekul. Dapat berupa eksitasi elektronik,
vibrasi, atau rotasi.
Transisi yang terjadi di dalam serapan inframerah berkaitan dengan
perubahan-perubahan vibrasi di dalam molekul. Banyak para kimiawan yang
menggunakan satuan radiasi dalam daerah vibrasi inframerah yang disebut
bilangan gelombang (ν ). Bilangan gelombang dinyatakan sebagai cm-1
(kebalikan cm), yang merupakan kebalikan dari panjang gelombang (λ ) yang
dinyatakan dalam cm. Penggunaan spektroskopi inframerah pada bidang kimia
organik hampir menggunakan daerah dari 650 – 4000 cm-1 (15,4 – 2,5 µ m).
Daerah dengan frekuensi lebih rendah 650 cm-1 disebut inframerah jauh dan
daerah dengan frekuensi lebih tinggi dari 4000 cm-1 disebut inframerah dekat.
Masing-masing daerah tersebut lebih jauh dan lebih dekat dengan spektrum

45
tampak inframerah dekat terutama menunjukkan serapan-serapan harmonic
overtones dari vibrasi pokok yang terdapat dalam daerah normal.
Metode Spektroskopi inframerah ini dapat digunakan untuk
mengidentifikasi suatu senyawa yang belum diketahui, karena spektrum yang
dihasilkan spesifik untuk senyawa tersebut. Metode ini banyak digunakan karena:
1. Dapat digunakan untuk mengidentifikasi gugus fungsional dalam molekul.
2. Spektrum inframerah yang dihasilkan oleh suatu senyawa adalah khas dan
oleh karena itu dapat menyajikan sebuah fingerprint (sidik jari) untuk
senyawa tersebut.
Ada dua jenis vibrasi yaitu :
1. Vibrasi ulur (Stretching Vibration), yaitu vibrasi yang mengakibatkan
perubahan panjang ikatan suatu ikatan.
2. Vibrasi tekuk (Bending Vibrations), yaitu vibrasi yang mengakibatkan
perubahan sudut ikatan antara dua ikatan. Vibrasi tekuk itu sendiri dibagi
lagi menjadi empat : Scissoring, Rocking, Wagging, Twisting.
Bentuk scissoring, di mana atom-atom yang terikat pada atom pusat
bergerak saling mendekat dan menjauh satu sama lain, sedangkan dalam
rocking atom-atomnya bergerak bolak balik dalam bidang. Untuk wagging,
atom-atomnya bergerak bolak balik keluar bidang atau molekul. Dan
akhirnya twisting, atom-atom yang terikat pada molekul yang diam, berotasi
di sekitar ikatannya (Khopkar, 2003).

46
3.6 Hipotesis Penelitian
Energi HOMO, energi LUMO, spektra inframerah dan spektra transisi
elektonik dari senyawa kompleks bis-4-heksiloksibenzilamin tembaga(I) dapat
dihitung dengan metode ab initio dengan perangkat lunak simulasi dan pemodelan
molekul yang dikeluarkan oleh HyperChem untuk windows versi 7.0.

47
BAB IV
METODOLOGI PENELITIAN
Penelitian ini dilaksanakan di laboratorium Kimia Komputasi AIC FMIPA
Universitas Gadjah Mada. Pada penelitian ini digunakan perangkat komputer serta
dilakukan pengambilan data dengan metode ab initio tingkat HF.
4.1 Alat yang digunakan
4.1.1 Perangkat Keras
Peralatan yang digunakan adalah perangkat komputer dengan spesifikasi
sebagai berikut
1. Prosesor Intel Pentium 4 CPU 3.00 GHZ
2. Random Access Memory (RAM) 512 MB
3. Hardisk 40 GB
4.1.2 Perangkat lunak
Prosedur penelitian meliputi pemodelan struktur kompleks bis-4-
heksiloksibenzilamin tembaga(I), optimasi geometri, spektra ultraviolet serta
spektra inframerah menggunakan program-program
1. Hyperchem 7.0 untuk Windows
Digunakan untuk memodelkan molekul kompleks bis-4-
heksiloksibenzilamin tembaga(I) dan optimasi geometri struktur kompleks
bis-4-heksiloksibenzilamin tembaga(I) sehingga diperoleh koordinat
kartesian atom-atom penyusunnya.

48
2. Gaussian 98 untuk Windows
Digunakan untuk optimasi geometri dan vibrasi struktur kompleks bis-4-
heksiloksibenzilamin tembaga(I) sehingga diperoleh struktur kompleks
bis-4-heksiloksibenzilamin tembaga(I) yang stabil dan intensitas serapan
inframerah.
3. Chemcraft
Digunakan untuk mengeplotkan data frekuensi vibrasi kompleks bis-4-
heksiloksibenzilamin tembaga(I) menjadi spektra inframerah (intensitas
vs bilangan gelombang).
4.2 Bahan yang digunakan
Pada penelitian ini digunakan struktur senyawa kompleks bis-4-
heksiloksibenzilamin tembaga(I) yang diperoleh dari Penelitian yang dilakukan
Lequerica, Baena dan Espinet pada tahun 2007. Struktur senyawa kompleks bis-4-
heksiloksibenzilamin tembaga(I) ditunjukkan pada gambar 7.
C
HC CH
C
CHHC
HC
C
HC CH
C
CH
H2C
H2C
CH2
H2C
CH2
OH3C
CH2
NH2
CH2
H2C
CH2
H2CO
CH2
CH3
H2C
H2N
Cu+
Gambar 7. Struktur kompleks bis-4-heksiloksibenzilamin tembaga(I)

49
4.3 Cara Kerja
4.3.1 Optimasi Geometri dengan Metode MM+
Dilakukan optimasi geometri MM+ dengan pendekatan kimia kuantum
dengan menggunakan program HyperChem untuk Windows versi 7.0 diatur
sebagai berikut :
1. Set up : MM+, option : electrostatic = bond dipoles, cutoffs= none,
components = bond, angle, torsion, non-bonded, electrostatic, hydrogen
bonded.
2. Compute, Geometry optimization : Algorithm = Polak Ribiere, RMS (Root
Mean Square), Gradien of 0.01 kcal(Å/mol)
Gambar 8. Diagram alir prosedur optimasi geometri struktur kompleks bis-4-heksiloksibenzilamin tembaga(I) dengan metode MM+.
SET UP PROGRAM HYPER CHEM MM+
GAMBAR STRUKTUR SENYAWA (2D) YANG AKAN DIPERGUNAKAN
BUILD : ADD & MODEL BUILD (3D)
COMPUTE : GEOMETRY OPTIMIZATION

50
4.3.2 Optimasi Geometri dengan Metode Ab-Initio
Dilakukan optimasi geometri ab Initio dengan pendekatan kimia kuantum
dengan menggunakan program HyperChem untuk Windows versi 7.0 diatur
sebagai berikut :
1. Set up : ab initio, option : Convergence Limit = 1e-5, Iteration Limit =
10000, Total Charge = 1, Spin Multiplicity = 1, Spin Pairing = RHF,
Advenced Option = projected huckel, Extra Basis Function, Apply Basis
Set, acceleration curve = yes (x).
2. Compute, Geometry optimization : Algorithm = Polak Ribiere, RMS (Root
Mean Square), Gradien of 0.1 kcal (Å/mol)
Gambar 9. Diagram alir prosedur optimasi geometri struktur kompleks bis-4-heksiloksibenzilamin tembaga(I) dengan metode ab initio.
SIMPAN DALAM FILE LOG
COMPUTE : GEOMETRY OPTIMIZATION
STRUKTUR SENYAWA HASIL OPTIMASI GEOMETRI METODE MM+
STAR LOG
SET UP PROGRAM HYPERCHEM AB INITIO

51
4.3.3 Perhitungan Transisi Elektronik dengan Spektra UV
Dilakukan perhitungan spektra transisi elektronik dari struktur senyawa
kompleks bis-4-heksiloksibenzilamin tembaga(I) yang telah teroptimasi
menggunakan program HyperChem untuk windows versi 7.0 dengan cara
compute, kemudian perhitungan single point menggunakan metode ab initio.
Perhitungan dilakukan dengan menjalankan secara bersamaan Restricted Hartree-
Fock (RHF) dengan Configuration Interaction (CI)-single excited dengan batasan
energi HOMO LUMO. Setelah perhitungan dijalankan akan dihasilkan spektrum
transisi elektronik berupa panjang gelombang dan intensitas serapan berupa
kekuatan osilasi yang berupa diagram spektra diskontinyu. Data hasil perhitungan
disimpan dalam file log.
4.3.4 Perhitungan Spektra Vibrasi menggunakan Perhitungan Ab initio
Dilakukan perhitungan spektra vibrasi dari struktur senyawa kompleks bis-
4-heksiloksibenzilamin tembaga(I) yang telah teroptimasi menggunakan program
gaussian 98 W diatur sebagai berikut :
1. Job type : Frequency
2. Method : Ground State, Hartree Fock, Restricted, Charge=1,
Spin=singlet
3. Basis set : STO-3G
4. General Option : - Mix HOMO LUMO in initial guess
- Write connectivity
5. Submit

52
BAB V
HASIL DAN PEMBAHASAN
Pada penelitian ini dilakukan penentuan spesifikasi struktur awal kompleks
bis-4-heksiloksibenzilamin tembaga(I). Struktur awal kompleks bis-4-
heksiloksibenzilamin tembaga(I) di optimasi geometri dengan metode ab initio
pada tingkat HF dengan basis set STO-3G, dilanjutkan dengan perhitungan
transisi elektronik dan frekuensi vibrasi inframerah. Dari hasil perhitungan
tersebut kemudian dapat ditentukan celah energi kompleks bis-4-
heksiloksibenzilamin tembaga(I). Penggunaan metode ab initio tingkat HF dengan
basis set minimal didasarkan pada akurasi dan waktu hitung yang relatif pendek.
Senyawa kompleks bis-4-heksiloksibenzilamin tembaga(I) digambar dalam
bentuk 2D, kemudian dilanjutkan menjadi struktur 3D menggunakan program
Hyperchem 7.0. Pemodelan molekul kompleks bis-4-heksiloksibenzilamin
tembaga(I) dilakukan untuk memberikan gambaran tentang perilaku molekul
tersebut, yang pada akhirnya digunakan untuk melakukan perhitungan-
perhitungan terhadap sifat-sifat fisika molekul tersebut. Pemodelan molekul
tersebut mencerminkan bentuk nyata dari molekul itu sendiri dilihat dari bentuk
dan tampilan struktur 3 dimensinya. Adapun contoh struktur 2D senyawa
kompleks bis-4-heksiloksibenzilamin tembaga(I) sesuai gambar sebagai berikut :

53
C
HC CH
C
CHHC
HC
C
HC CH
C
CH
O
CH2
H2C
CH2
H2C
CH2
CH3
CH2
H2N
O
CH2
H2C
CH2
H2C
CH2
CH3
CH2
H2N
Gambar 10. Struktur 2 dimensi bis-4-heksiloksibenzilamin
C
HC CH
C
CHHC
HC
C
HC CH
C
CH
H2C
H2C
CH2
H2C
CH2
OH3C
CH2
NH2
CH2
H2C
CH2
H2CO
CH2
CH3
H2C
H2N
Cu+
Gambar 11. Struktur 2 dimensi kompleks bis-4-heksiloksibenzilamin tembaga(I)
5.1 Optimasi Geometri
Dalam penelitian ini dilakukan perhitungan energi total dari optimasi
geometri struktur kompleks bis-4-heksiloksibenzilamin tembaga(I). Tujuan
optimasi geometri ini adalah untuk menghitung energi terendah dan gaya-gaya
atomik terkecil serta untuk menampilkan struktur molekul, sedemikian rupa
sehingga mendekati struktur yang sebenarnya atau paling stabil di alam dengan
energi yang minimal.
Untuk mendapatkan konformasi yang stabil dari senyawa kompleks bis-4-
heksiloksibenzilamin tembaga(I) maka terlebih dahulu dilakukan optimasi
geometri dengan menggunakan metode MM+. Pada prinsipnya tujuan dari MM+

54
adalah untuk meramalkan energi yang berkaitan dengan konformasi tertentu dari
molekul. Waktu perhitungan MM+ tidak lama, karena hanya dilakukan oleh
medan gaya didasarkan pada pendekatan born-openheimer, yaitu memisahkan
sumbangan gerak elektron terhadap energi potensial molekul. Hanya atom-atom
hidrogen terkoneksi pada heteroatom yang diikutkan dalam perhitungan. Metode
MM+ merupakan metode sederhana yang digunakan untuk memudahkan
perhitungan metode selanjutnya yaitu ab initio.
Senyawa kompleks bis-4-heksiloksibenzilamin tembaga(I) yang telah
dioptimasi geometri dengan MM+ selanjutnya dilakukan optimasi dengan
menggunakan metode ab initio dengan basis set STO-3G.
Perintah optimasi geometri dengan nilai batas gradien yang lebih kecil dari
0,1 kkal/(Å.mol) akan memberikan hasil yang lebih bagus tetapi memakan waktu
yang lebih lama karena senyawa kompleks bis-4-heksiloksibenzilamin tembaga(I)
merupakan senyawa makro molekul yang terdiri dari 73 atom, maka untuk
efisiensi waktu ditentukan batas energi gradien adalah 0,1 kkal/(Å.mol).
Semakin besar muatan ion maka semakin mudah mempolarisasikan elektron
pada ligan sehingga strukturnya semakin stabil. Struktur yang stabil adalah
struktur yang gaya tarik menarik dan gaya tolak menolak pada keadaan seimbang,
sehingga energi interaksinya menjadi minimum atau energi potensialnya semakin
kecil. Semakin besar penurunan energi potensialnya semakin besar pula energi
dissosiasi yang dibutuhkan untuk memutuskan ikatan antar atom pada keadaan
yang stabil.

55
Hasil optimasi geometri adalah suatu kumpulan data di dalam sebuah berkas
yang disebut log files, di dalamnya tercantum data-data perhitungan yang penting
termasuk data energi hasil optimasi geometri dalam penelitian ini dapat disajikan
dalam tabel 1. Prinsip dasar dalam perhitungan energi total untuk molekul cu-bis-
4-heksiloksibenzilamin adalah sebagai berikut
∆Ε total = Ε produk – Ε reaktan (11)
di mana ∆Ε mencerminkan total energi potensial minimum struktur molekul,
Εproduk adalah energi total senyawa produk dan Εreaktan adalah energi total senyawa
reaktan.
Tabel 1. Energi Hasil Optimasi Geometri
Energi minimal serta keadaan yang paling stabil dari kompleks bis-4-
heksiloksibenzilamin tembaga(I) ditunjukkan oleh besar energi total hasil
optimasi geometri sebesar -7219,67 kkal/mol.
Optimasi geometri dengan menggunakan metode ab initio tingkat HF basis
set minimal STO-3G menghasilkan kompleks bis-4-heksiloksibenzilamin
tembaga(I) seperti pada gambar berikut :
ECu bis-4-heksiloksibenzilamin
(kkal/mol) E(bis 4-heksiloksibenzilamin)
(kkal/mol) ECu
(kkal/mol) ∆Ε
(kkal/mol) -1802610,35 -785777,81 -1009612,87 -7219,67
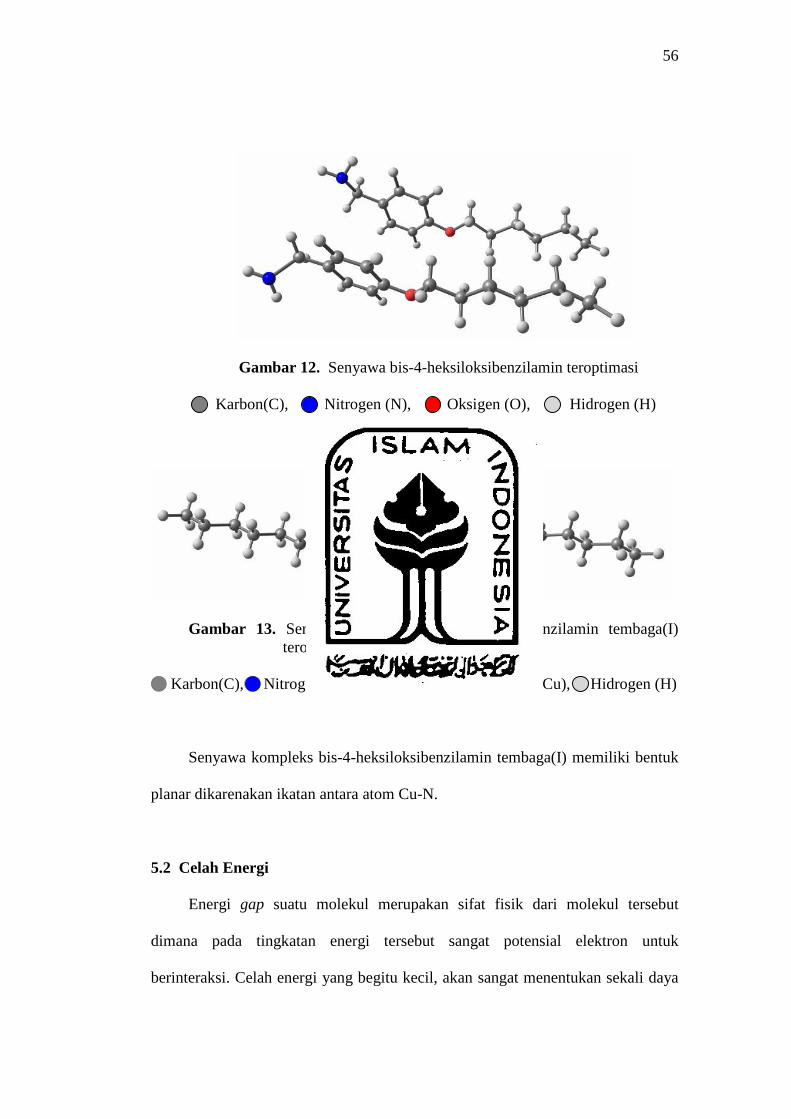
56
Gambar 12. Senyawa bis-4-heksiloksibenzilamin teroptimasi
Karbon(C), Nitrogen (N), Oksigen (O), Hidrogen (H)
Gambar 13. Senyawa kompleks bis-4-heksiloksibenzilamin tembaga(I) teroptimasi
Karbon(C), Nitrogen (N), Oksigen (O), Tembaga(Cu), Hidrogen (H)
Senyawa kompleks bis-4-heksiloksibenzilamin tembaga(I) memiliki bentuk
planar dikarenakan ikatan antara atom Cu-N.
5.2 Celah Energi
Energi gap suatu molekul merupakan sifat fisik dari molekul tersebut
dimana pada tingkatan energi tersebut sangat potensial elektron untuk
berinteraksi. Celah energi yang begitu kecil, akan sangat menentukan sekali daya

57
serapan yang begitu besar, sehingga senyawa ini akan menjadi detektor
inframerah organik dan cahaya tampak yang menghasilkan spektra ultraviolet dan
inframerah.
HOMO adalah orbital tertinggi pada pita valensi yang ditempati elektron.
LUMO adalah orbital terendah pada pita konduksi yang tidak terisi elektron.
Energi HOMO adalah energi ionisasi bahan yang membatasi perbedaan energi
antara tingkat vakum (Vacuum Level) dengan tepi bawah energi ikat dari orbital
HOMO sedangkan energi LUMO adalah afinitas elektron yang membatasi
perbedaan energi antara tingkat vakum (Vacuum Level) dengan tepi atas energi
ikat dari orbital LUMO.
Energi HOMO dan energi LUMO bermanfaat untuk mengenali sifat
semikonduktor berdasarkan gap energi pita valensi (HOMO) dan pita konduksi
(LUMO). Semikonduktor merupakan bahan dengan konduktivitas listrik yang
berada pada isolator dan konduktor. Pada temperatur yang sangat rendah, sebuah
semikonduktor bersifat sebagai isolator namun pada temperatur ruangan bersifat
konduktor. Semakin kecil celah energi maka sifat konduktifitas listrik semakin
baik.
∆H (-) disebabkan proses eksotermis akibat dari energi ionisasi yang terjadi
pada orbital HOMO sedangkan pada orbital LUMO terjadi proses endotermis
akibat dari afinitas elektron sehingga ∆H (+).
Berdasarkan hasil optimasi geometri, diperoleh output data orbital-orbital
molekul dimana dari hasil perhitungan komputasi tersebut dapat diketahui nilai

58
energi HOMO-LUMO struktur kompleks bis-4-heksiloksibenzilamin tembaga(I)
teroptimasi sehingga energi gapnya pun dapat ditentukan.
Nilai energi HOMO-LUMO kompleks bis-4-heksiloksibenzilamin
tembaga(I) berdasarkan perhitungan komputasi dapat dilihat pada tabel di bawah
ini.
Tabel 2. Energi HOMO-LUMO
Struktur E HOMO E LUMO Celah energi (Eg)
Bis-4-heksiloksibenzilamin -6,38 7,39 1,01
Cu-bis-4-heksilloksibenzilamin -0,95 2,18 1,23
Gambar 14. Diagram energi gap bis-4-heksiloksibenzilamin menggunakan basis set STO-3G
HOMO
LUMO-6,38 eV
7,39 eV
1,01 eV
VL

59
Gambar 15. Diagram energi gap kompleks bis-4-heksiloksibenzilamin tembaga(I) menggunakan basis set STO-3G
Dalam hasil perhitungan komputasi, orbital molekul energi HOMO berada
di bawah 0 eV sedangkan untuk energi LUMO berada di atas 0 eV. Menurut Hill
dan Kahn (1998), energi ionisasi suatu material didefinisikan sebagai selisih
energi antara VL dengan ujung pita energi dari HOMO, sedangkan afinitas
elektron didefinisikan sebagai selisih energi antara VL dengan ujung pita energi
dari LUMO.
Berdasarkan hasil perhitungan komputasi, VL berada pada nilai 0 eV. Oleh
karena itu, berdasarkan tabel tersebut dapat dideskripsikan sebagai berikut : pada
senyawa bis-4-heksilloksibenzilamin diperlukan energi sebesar -6,38 eV untuk
melepaskan elektron sampai VL dan untuk menangkap elektron sampai VL
dilepas energi sebesar 7,39 eV, sehingga diperoleh energi gap sebesar 1,01 eV.
Sedangkan pada senyawa kompleks bis-4-heksiloksibenzilamin tembaga(I)
diperlukan energi sebesar -0,95 eV untuk melepaskan elektron sampai VL dan
untuk menangkap elektron sampai VL dilepas energi sebesar 2,18 eV, sehingga
diperoleh energi gap sebesar 1,23 eV. Berdasarkan hasil penelitian dengan
HOMO
LUMO
VL
-0,95 eV
2,18 eV
1,23 eV

60
menggunakan perhitungan komputasi senyawa kompleks bis 4-
heksiloksibenzilamin tembaga(I) termasuk semikonduktor organik.
5.3 Serapan Ultraviolet
Selisih energi HOMO-LUMO meggambarkan kemudahan suatu sistem
molekul untuk mengalami eksitasi ke keadaan elektronik yang lebih tinggi. Selisih
energi HOMO-LUMO yang lebih rendah akan mencerminkan kemudahan dalam
proses terjadinya eksitasi elektron sehingga sifat kepekaannya terhadap cahaya
(fotosensitivitas) akan cenderung lebih kuat.
Sensitivitas suatu senyawa terhadap radiasi sinar ultraviolet vissible
dipengaruhi oleh transisi elektronik yang terjadi. Jika senyawa tersebut dikenai
sinar dengan panjang gelombang yang sesuai maka akan terjadi transisi elektronik
dari orbital molekul yang ditempati elektron menuju ke tingkat orbital yang tidak
ditempati elektron.
Transisi di daerah tampak atau ultraviolet adalah transisi elektronik
(electronic transition). Hal ini dikaitkan dengan lompatan elektron dari orbital
molekul terisi penuh (terisi) ke orbital molekul yang kosong yang lebih tinggi
energinya. Kebolehjadian transisi ∆E yang paling mungkin akan timbul pada
promosi satu elektron dari orbital molekul terisi yang paling tinggi ke orbital tak
terisi yang ada yang terendah.
Senyawa kompleks bis-4-heksiloksibenzilamin tembaga(I) mempunyai
ikatan rangkap terkonjugasi yang memungkinkan terjadinya proses serapan
gelombang elektromagnetik untuk mengeksitasi elektron-elektron dari tingkat

61
dasar ke kondisi eksitasi. Panjang gelombang yang diserap oleh senyawa
kompleks bis-4-heksiloksibenzilamin tembaga(I) terkait dengan beda energi
kedua tingkat tersebut. Pada saat elektron-elektron dari tingkat eksitasi kembali ke
tingkat dasar akan memancarkan sinar sesuai dengan panjang gelombang yang
telah diserap.
Senyawa kompleks bis-4-heksiloksibenzilamin tembaga(I) memiliki
kemampuan menyerap sinar ultraviolet pada panjang gelombang tertentu sebagai
akibat adanya gugus-gugus fungsional yang dapat menghasilkan transisi
elektronik yang besar energinya sesuai dengan rentang energi sinar ultraviolet.
Tiap transisi memiliki intensitas berbeda dalam menyerap sinar ultraviolet dan
menjadi acuan dari kemampuan suatu kompleks bis-4-heksiloksibenzilamin
tembaga(I) dalam menyerap sinar ultraviolet adalah panjang gelombang serapan
sinar ultraviolet dengan intensitas maksimal atau yang biasa disebut panjang
gelombang serapan maksimal.
Data yang dihasilkan adalah spektra diskontinyu berupa pita-pita diskret
pada panjang gelombang tertentu yang menunjukkan intensitas serapan akibat
terjadinya eksitasi elektronik dari suatu senyawa. Panjang gelombang maksimum
ditunjukkan dengan intensitas yang paling tinggi yang berarti semakin besar
intensitasnya maka semakin banyak foton yang diserap akibat radiasi
elektromagnetik.
Radiasi ultraviolet yang dipancarkan adalah merupakan paket-paket energi
yang menyerupai partikel atau yang disebut foton/kuantum. Energi suatu foton

62
berbanding langsung dengan frekuensinya (lebih banyak gelombang persatuan
waktu berarti lebih tinggi energinya).
Data spektra transisi elektronik ditunjukkan pada tabel di bawah ini.
Tabel 3. Spektra Transisi Elektronik
Spektra transisi elektronik Senyawa
λ maks υ (Hz) E (eV)
Bis-4-heksiloksibenzilamin 105,70 2,83x1015 11,72
Cu-4-heksiloksibenzilamin 108,42 2,76x1015 11,42
Energi cahaya terkuantitasikan atau energi foton ini untuk menunjukkan
adanya efek fotolistrik di dalam suatu molekul. Efek fotolistrik adalah proses
terpentalnya elektron dari permukaan logam oleh cahaya, dengan adanya efek
fotolistrik ini menyebabkan terjadinya proses konduksi listrik pada
semikonduktor. Karena pengaruh medan listrik elektron tersebut akan memiliki
energi kinetik dan dapat mengalirkan arus listrik. Semakin rendah energi foton
maka semakin mudah suatu elektron terpental dari pita valensi ke pita konduksi
dari suatu logam.
Energi serapan radiasi (foton) yang didapat untuk senyawa bis-4-
heksiloksibenzilamin adalah sebesar 11,72 eV sedangkan senyawa kompleks bis-
4-heksiloksibenzilamin tembaga(I) sebesar 11,42 eV. Terlihat bahwa energi
sebagai fungsi panjang gelombang, akan semakin besar dengan semakin
pendek/kecilnya panjang gelombang dari absorbsi foton, disamping itu
intensitaspun juga akan semakin besar dengan banyaknya foton yang diserap.

63
Panjang gelombang yang diperoleh untuk senyawa bis-4-
heksilloksibenzilamin adalah sebesar 105,70 nm sedangkan kompleks bis-4-
heksiloksibenzilamin tembaga(I) sebesar 108,42 nm. Terlihat bahwa senyawa
kompleks bis-4-heksiloksibenzilamin tembaga(I) mengalami pergeseran merah
yakni pergeseran serapan ke arah panjang gelombang yang lebih panjang, dengan
selisih panjang gelombang sebesar 2,72 nm. Pada pergeseran tersebut terjadi
proses eksitasi yang mengakibatkan terjadinya transisi elektron yaitu transisi π →
π∗ dan memiliki energi yang lebih rendah sehingga lebih mudah terpental dari
keadaan dasar ke keadaan terekeksitasi dan lebih mudah menghantarkan arus
listrik.
5.4 Spektra Inframerah
Spektroskopi inframerah merupakan suatu teknik pengukuran absorpsi
molekul yang didasarkan pada transisi vibrasi gugus fungsi pada molekul tersebut.
Karena sifat dasar perhitungan komputasi dilibatkan, perhitungan frekuensi
vibrasi inframerah dikatakan akurat jika hanya pada titik minimum global pada
energi potensial permukaan. Jadi, pada perhitungan frekuensi vibrasi infamerah
harus dilakukan pada struktur teroptimasi. Oleh karena itu, sebelum melakukan
perhitungan frekuensi vibrasi inframerah dilakukan optimasi geometri sehingga
diperoleh spektra inframerah dari struktur molekul yang stabil.
Atom N sebagai atom donor elektron yang berinteraksi langsung dengan
logam sebagai atom pusatnya yang merupakan atom akseptor elektron. Perubahan
perbedaan frekuensi vibrasi terjadi karena penambahan logam sebagai atom pusat.

64
Spektra vibrasi juga bermanfaat sebagai petunjuk yang sensitif tentang perubahan
baik pada geometri maupun struktur elektronik akibat adanya interaksi molekul.
Semakin besar perubahan spektra inframerah maka semakin mudah molekul
tersebut untuk bervibrasi dan juga sebaliknya.
Spektra vibrasi inframerah untuk kompleks bis-4-heksiloksibenzilamin
tembaga(I) telah dihitung pada pendekatan harmonik menggunakan metode ab
initio tingkat Hartree-Fock. Perhitungan frekuensi vibrasi inframerah kompleks
bis-4-heksiloksibenzilamin tembaga(I) teroptimasi menghasilkan nilai berupa
bilangan gelombang dan intensitas inframerah.
Kedudukan pita serapan dinyatakan dalam bilangan gelombang υ yang
mempunyai hubungan dengan panjang gelombang λ, yaitu υ = 1/ λ, dimana υ
adalah bilangan gelombang yang dinyatakan dengan satuan cm-1, dan λ adalah
panjang gelombang yang dinyatakan dalam satuan cm.
Gugus C=C, C-O, dan C-N yang diperoleh ditampilkan dalam gambar 16
dan 17. Gugus ini dipilih karena merupakan gugus utama dari senyawa kompleks
bis-4-heksiloksibenzilamin tembaga(I).
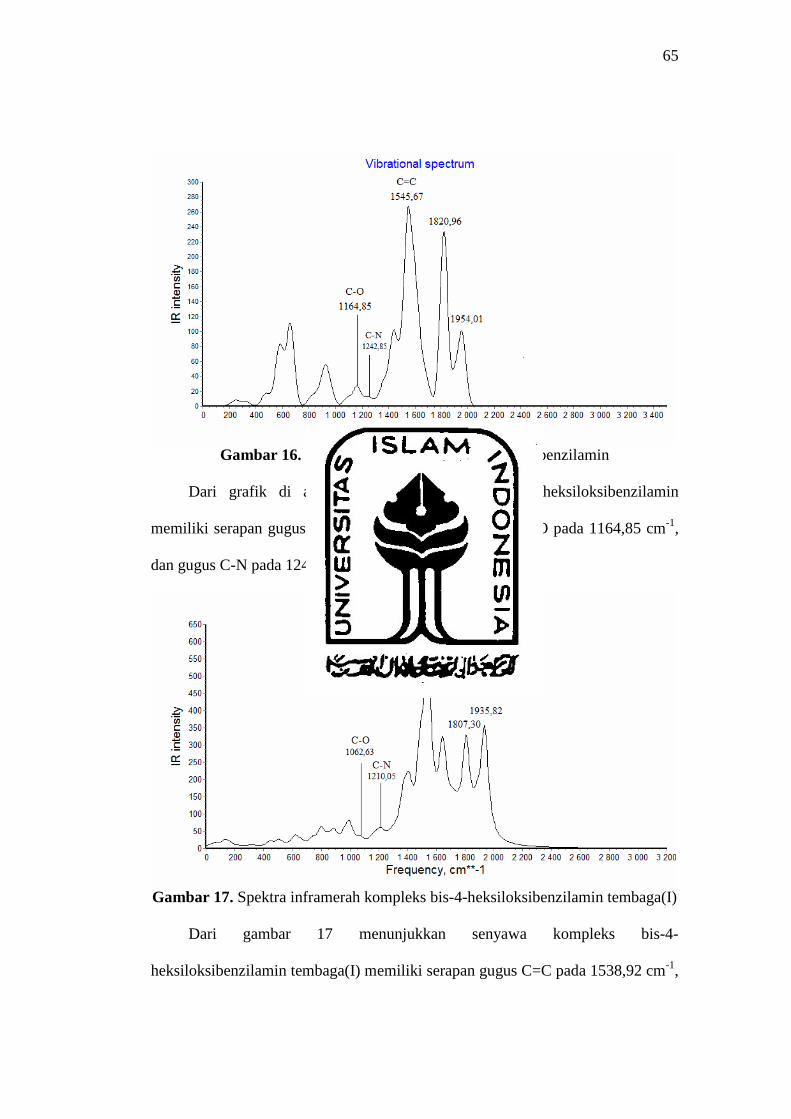
65
Gambar 16. Spektra inframerah bis-4-heksiloksibenzilamin
Dari grafik di atas menunjukkan senyawa bis-4-heksiloksibenzilamin
memiliki serapan gugus C=C pada 1545,67 cm-1, gugus C-O pada 1164,85 cm-1,
dan gugus C-N pada 1242,85 cm-1.
Gambar 17. Spektra inframerah kompleks bis-4-heksiloksibenzilamin tembaga(I)
Dari gambar 17 menunjukkan senyawa kompleks bis-4-
heksiloksibenzilamin tembaga(I) memiliki serapan gugus C=C pada 1538,92 cm-1,

66
gugus C-O pada 1062,63 cm-1 dan gugus C-N pada 1210,05 cm-1. Dari
perbandingan kedua spektra inframerah di atas dapat disimpulkan bahwa senyawa
kompleks bis-4-heksiloksibenzilamin tembaga(I) mengalami pergeseran merah
yakni pergeseran ke frekuensi yang lebih rendah, dengan perbedaan frekuensi
gugus C=C sebesar 6,75 cm-1, gugus C-O sebesar 102,22 cm-1 dan gugus C-N
sebesar 32,8 cm-1. Pergeseran tersebut menghasilkan energi yang rendah dan
memiliki stuktur yang lebih stabil.

67
BAB VI
KESIMPULAN DAN SARAN
6.1 Kesimpulan
Berdasarkan penelitian yang dilakukan dengan menggunakan metode ab
initio tingkat HF diperoleh kesimpulan sebagai berikut :
1. Energi gap dari senyawa kompleks bis-4-heksiloksibenzilamin tembaga(I)
adalah 1,23 eV sedangkan bis-4-heksiloksibenzilamin sebesar 1,01 eV.
Semakin kecil energi gap sifat konduktifitas listriknya semakin baik. Hasil
penelitian tersebut senyawa kompleks bis-4-heksiloksibenzilamin
tembaga(I) termasuk semikonduktor organik.
2. Berdasarkan hasil analisis ultraviolet senyawa kompleks bis-4-
heksiloksibenzilamin tembaga(I) mengalami pergeseran merah
dibandingkan dengan senyawa bis-4-heksiloksibenzilamin, dengan selisih
panjang gelombang sebesar 2,72 nm. Pada pergeseran tersebut terjadi
proses eksitasi yang mengakibatkan terjadinya transisi elektron yaitu
transisi π →π∗ dan memiliki energi yang lebih rendah sehingga lebih
mudah terpental dari keadaan dasar ke keadaan terekeksitasi dan lebih
mudah menghantarkan arus listrik.
3. Berdasarkan hasil analisis inframerah senyawa kompleks bis-4-
heksiloksibenzilamin tembaga(I) mengalami pergeseran merah
dibandingkan dengan senyawa bis-4-heksiloksibenzilamin, dengan
perbedaan frekuensi gugus C=C sebesar 6,75 cm-1, gugus C-O sebesar

68
102,22 cm-1 dan gugus C-N sebesar 32,8 cm-1. Pergeseran tersebut
menghasilkan energi yang rendah dan memiliki stuktur yang lebih stabil.
6.2 Saran
1. Perlu dilakukan perolehan parameter sifat optik dari senyawa kompleks
bis-4-heksiloksibenzilamin tembaga(I) dengan metode eksperimen di
laboratorium.
2. Perlu dipelajari kembali kompleks bis-4-heksiloksibenzilamin tembaga(I)
ini dengan memakai metode yang lain.

69
DAFTAR PUSTAKA
Ardhian, A., 2008, Kajian Teoritis Kompleks Cobalt(II) dan Copper(II) Porphyrin Sebagai Aplikasi Semikonduktor Organik dengan Menggunakan Metode Mekanika Kuantum AB Initio, Skripsi FMIPA UGM, Jogjakarta.
Bintarti, A., 2008, Perhitungan Celah Energi Struktur Copper Phthalocyanine
dengan Menggunakan Metode Ab Initio, Skripsi FMIPA UGM, Jogjakarta. Brutting, W., 2005, Physics of Organic Semiconductor, Wiley-vc Vcrlag Gmbh
and Co kgaA, Weinheim. Carter, A.R., 2003, Optimizing Polymeric Field-Effect Devices, Department of
Physics, The College of Wooster, USA. Dogra, S.K., 1990, Kimia Fisika dan Soal-Soal, UI-Press, Jakarta.
Goodman, J.M., 1998, Chemical Applications of Molecular Modelling, The Royal Society of Chemistry Cambridge.
Harnowo, 2002, Studi Mekanisme Migrasi Proton Ganda pada Ikatan Hidrogen dengan Metode Ab Initio, Skripsi FMIPA UGM, Jogjakarta.
Hill, I.G., and Kahn, A., 1998, Energy Level Alignment at Interfaces of Organic Semiconductor Heterostructures, Journal of Applied Physic, Vol 84 number 10.
Khopkar, S.M., 2003, Konsep Dasar Kimia Analitik, UI-Press, Jakarta.
Leach, A.R., 1996, Molecular Modelling : Principles and Applications, Addison Wesley Longman, London.
Leach, A.R., 2001, Molecular Modeling, Prinsiple and application 2nd Ed,
Longman, Singapore.
Lequerica, M.C., Baena, M.J., and Espinet, P., 2007, Ionic metallomesogens derived from silver(I) bis-amine complexes: Structure and mesogenic behavior, IU CINQUIMA/Química Inorgánica, Facultad de Ciencias, Universidad de Valladolid. E-47071 Valladolid, Spain
Pranowo, H.D., 2001, Kimia Komputasi, Pusat Kimia Komputasi Indonesia
Austria, Kimia FMIPA UGM, Jogjakarta.
Sartika, D., 2007, Analisis HKSA Senyawa Penghambat GAO dari Turunan 4-substitusi 3-hidroksi-1H-pirol-2,5-dion dengan Deskriptor Muatan є HOMO,

70
є LUMO dan Momen Dipol Menggunakan Perhitungan Metode AM1, Skripsi FMIPA UII, Jogjakarta.
Sastrohamidjojo, H., 2001, Spektroskopi, Liberty, Jogjakarta.
Sudanti, S., 2006, Kajian Teoritis Untuk Menentukan Celah Energi Porfirin Terkonjugasi Atom Perak dan Tembaga dengan Menggunakan metode Mekanika Kuantum Semiempiris ZINDO/1, Skripsi FMIPA UGM, Jogjakarta.
Sze, S.M., 2001, Semiconductor Devices, Physics and Technology 2nd Edition, John Wiley & Sons, INC.
Tahir, I., Wijaya, K., Roto, 2007, Analisis Spektra Transisi Elektronik Senyawa
Tabir Surya MAA’S GLY pada Konfigurasi Dimer dan Konfigurasi Solut-Etanol, Pusat Kimia Komputasi Indonesia Austria, Kimia FMIPA UGM, Jogjakarta.
Triyana, K., 2004, Organic Photovoltaic Devices Based on Phthalocyanine and Perylene, Ph. D Thesis, Kyushu University, Jepang.
Yahya, U., Setyopratiwi, A., Tahir, I., 2001, Ikatan Kimia, Jurusan Kimia
Fakultas Matematika dan Ilmu Pengetahuan Alam UGM, Jogjakarta.

71
Lampiran 1. Koordinat Kartesian Atom-Atom Penyusun Bis-4-heksiloksibenzilamin dari Hasil Seleksi Program HyperChem 7.0 for Windows
Atom Z Coordinates(Angstrom) x y z 1 6 -4.86660000 -0.02250000 -1.39400000 2 6 -4.50450000 1.26550000 -1.00150000 3 6 -3.20670000 1.70670000 -1.12910000 4 6 -2.21990000 0.86310000 -1.65480000 5 6 -2.57260000 -0.42100000 -2.05350000 6 6 -3.88740000 -0.84600000 -1.92220000 7 6 -6.31540000 -0.49180000 -1.27280000 8 7 -7.08420000 -0.13900000 -2.50210000 9 8 -0.94470000 1.42620000 -1.73340000 10 6 0.09610000 0.51720000 -2.12000000 11 6 1.43070000 1.29050000 -2.04590000 12 6 2.64020000 0.39380000 -2.37840000 13 6 3.97100000 1.17140000 -2.31530000 14 6 5.19250000 0.27100000 -2.59300000 15 6 6.51810000 1.05160000 -2.53940000 37 6 -3.01770000 -1.79850000 2.32770000 38 6 -2.89520000 -0.44180000 2.62270000 39 6 -4.39060000 -2.47370000 2.30920000 40 7 -4.44700000 -3.55120000 1.28440000 41 6 -1.65960000 0.16590000 2.65890000 42 6 -0.49990000 -0.57260000 2.39570000 43 6 -0.61400000 -1.92600000 2.09940000 44 6 -1.86620000 -2.52259900 2.07140000 45 8 0.69060000 0.15680000 2.45650000 46 6 1.87820000 -0.60780000 2.20800000 47 6 3.08020000 0.35530000 2.31130000 48 6 4.41800000 -0.36680000 2.05390000 49 6 5.62080000 0.59560000 2.12480000 50 6 6.96070000 -0.12330000 1.86510000 51 6 8.16220000 0.83660000 1.92430000 31 1 4.07570000 1.62330000 -1.33100000 32 1 5.21910000 -0.53300000 -1.86040000 33 1 5.08530000 -0.19080000 -3.57240000 34 1 7.35970000 0.39000000 -2.72080000 35 1 6.53350000 1.83550000 -3.29000000 36 1 6.65270000 1.51210000 -1.56550000 16 1 -5.25220000 1.92820000 -0.58300000 17 1 -2.92470000 2.70440000 -0.82090000 18 1 -1.83940000 -1.09740000 -2.46410000 19 1 -4.14740000 -1.84790000 -2.24100000 20 1 -6.34640000 -1.57700000 -1.16790000 21 1 -6.76400000 -0.06060000 -0.37180000 22 1 -8.07080000 -0.39210000 -2.32470000 23 1 -7.09140000 0.89230000 -2.56970000 24 1 0.12800000 -0.34810000 -1.44930000 25 1 -0.06320000 0.14450000 -3.13770000 26 1 1.39060000 2.12930000 -2.73740000 27 1 1.54000000 1.69870000 -1.04340000 28 1 2.67530000 -0.43710000 -1.67660000 29 1 2.51640000 -0.03000000 -3.37300000 30 1 3.94800000 1.98260000 -3.03980000 52 1 -3.78260000 0.14560000 2.82340000 53 1 -4.58830000 -2.93700000 3.28000000 54 1 -5.16160000 -1.71150000 2.15700000 55 1 -5.40660000 -3.93420000 1.29970000 56 1 -4.36390000 -3.09770000 0.35980000 57 1 -1.56410000 1.21930000 2.88450000 58 1 0.25760000 -2.52570000 1.89020000 59 1 -1.94940000 -3.57660000 1.83720000 60 1 1.98470000 -1.41740000 2.93830000 61 1 1.85260000 -1.06150000 1.21140000 62 1 2.94920000 1.15730000 1.58860000 63 1 3.08720000 0.80580000 3.30140000 64 1 4.54690000 -1.16100000 2.78650000 65 1 4.39070000 -0.83610000 1.07240000 66 1 5.48930000 1.38810000 1.39080000 67 1 5.64770000 1.06680000 3.10510000 68 1 7.09330000 -0.91290000 2.60210000 69 1 6.92870000 -0.59900000 0.88690000 70 1 9.08840000 0.30180000 1.73830000

72
71 1 8.06600000 1.61870000 1.17730000 72 1 8.23080000 1.30660000 2.90030000
Lampiran 2. Koordinat Kartesian Atom-Atom Penyusun Kompleks bis-4-heksiloksibenzilamin tembaga(I) dari Hasil Seleksi Program HyperChem 7.0 for Windows
Atom Z Coordinates(Angstrom) x y z 73 29 -7.71150000 -2.57080000 -2.28350000 2 6 -5.93890000 0.27990000 0.76210000 3 6 -4.63860000 0.68950000 0.91570000 4 6 -3.60080000 -0.25550000 1.02960000 5 6 -3.91760000 -1.61160000 0.99580000 6 6 -5.23990000 -2.00760000 0.83930000 7 6 -7.70950000 -1.51140000 0.55870000 8 8 -2.33630000 0.29060000 1.16460000 9 7 -8.23070000 -1.24660000 -0.85460000 10 6 -1.24880000 -0.65150000 1.20780000 11 6 0.05800000 0.16750000 1.27130000 12 6 1.30960000 -0.73280000 1.24870000 13 6 2.60950000 0.09510000 1.30690000 14 6 3.87190000 -0.79000000 1.26590000 15 6 5.16690000 0.03810000 1.33970000 37 6 -3.55830000 -1.48280000 -3.15910000 38 6 -3.34160000 -0.15400000 -2.79720000 39 6 -2.16420000 0.47840000 -3.11980000 40 6 -1.14820000 -0.21400000 -3.79630000 41 6 -1.37060000 -1.53500000 -4.17870000 42 6 -2.57040000 -2.15200000 -3.86170000 43 6 -4.80070000 -2.24690000 -2.71170000 44 7 -6.09420000 -1.46030000 -2.85860000 45 8 0.01180000 0.51130000 -4.02840000 46 6 1.17670000 -0.29220000 -4.29720000 47 6 2.41970000 0.60450000 -4.11160000 48 6 3.71980000 -0.22480000 -4.10310000 49 6 4.96410000 0.64690000 -3.84010000 50 6 6.24930000 -0.19700000 -3.71870000 51 6 7.49120000 0.66720000 -3.44010000 1 6 -6.25860000 -1.08020000 0.70940000 32 1 3.84530000 -1.49270000 2.09620000 33 1 3.86980000 -1.37590000 0.34880000 34 1 6.03650000 -0.61070000 1.30650000 35 1 5.22810000 0.73170000 0.50660000 36 1 5.20450000 0.61050000 2.26140000 16 1 -6.72490000 1.02270000 0.69490000 17 1 -4.38640000 1.74080000 0.96200000 18 1 -3.15110000 -2.36450000 1.09980000 19 1 -5.47060000 -3.06740000 0.83160000 20 1 -7.83040000 -2.57790000 0.74900000 21 1 -8.35769900 -0.96070000 1.24430000 22 1 -9.25990000 -1.31450000 -0.79750000 23 1 -8.05940000 -0.24790000 -1.05830000 24 1 -1.33490000 -1.30290000 2.08400000 25 1 -1.25550000 -1.28350000 0.31310000 26 1 0.07930000 0.85030000 0.42450000 27 1 0.05000000 0.76950000 2.17700000 28 1 1.27840000 -1.41860000 2.09310000 29 1 1.30390000 -1.33550000 0.34260000 30 1 2.62870000 0.79050000 0.47020000 52 1 -4.08790000 0.39660000 -2.23870000 53 1 -1.99020000 1.50720000 -2.83360000 54 1 -0.61610000 -2.09160000 -4.71380000 55 1 -2.72460000 -3.18360000 -4.15540000 56 1 -4.92360000 -3.16310000 -3.28850000 57 1 -4.73050000 -2.51980000 -1.65720000 58 1 -6.19020000 -1.23240000 -3.86200000 59 1 -5.95240000 -0.54600000 -2.40240000 60 1 1.22380000 -1.13560000 -3.60130000 61 1 1.14330000 -0.69730000 -5.31440000 62 1 2.45020000 1.34790000 -4.90480000 63 1 2.31950000 1.13600000 -3.16780000 64 1 3.64780000 -0.98720000 -3.32980000 65 1 3.83000000 -0.74030000 -5.05480000 66 1 5.07660000 1.36850000 -4.64660000 67 1 4.81700000 1.21100000 -2.92130000
73
68 1 6.12640000 -0.91980000 -2.91460000 69 1 6.39900000 -0.76060000 -4.63730000 70 1 8.37590000 0.04460000 -3.35060000 71 1 7.65390000 1.37720000 -4.24490000 72 1 7.37190000 1.22320000 -2.51520000 31 1 2.61430000 0.68890000 2.21850000
Lampiran 3. Log File Energi HOMO-LUMO senyawa Bis-4-heksiloksibenzilamin
EIGENVALUES(eV) Symmetry: 1 A 2 A 3 A 4 A 5 A Eigenvalue: -552.048087 -552.016191 -416.758643 -416.705424 -301.962035 Symmetry: 6 A 7 A 8 A 9 A 10 A Eigenvalue: -301.951998 -301.633895 -301.524258 -301.474305 -301.364720 Symmetry: 11 A 12 A 13 A 14 A 15 A Eigenvalue: -300.598193 -300.575107 -300.543670 -300.541385 -300.481897 Symmetry: 16 A 17 A 18 A 19 A 20 A Eigenvalue: -300.460609 -300.433461 -300.426821 -300.269022 -300.251178 Symmetry: 21 A 22 A 23 A 24 A 25 A Eigenvalue: -300.127371 -300.123425 -300.069543 -300.001433 -299.931577 Symmetry: 26 A 27 A 28 A 29 A 30 A Eigenvalue: -299.918852 -299.882395 -299.879074 -299.768914 -299.763294 Symmetry: 31 A 32 A 33 A 34 A 35 A Eigenvalue: -35.784256 -35.762443 -30.716214 -30.665446 -29.548815 Symmetry: 36 A 37 A 38 A 39 A 40 A Eigenvalue: -29.454155 -28.523709 -28.500528 -27.211115 -27.193025 Symmetry: 41 A 42 A 43 A 44 A 45 A Eigenvalue: -26.131207 -26.112349 -26.061922 -25.972568 -24.929913 Symmetry: 46 A 47 A 48 A 49 A 50 A Eigenvalue: -24.896630 -23.110769 -23.028125 -22.547876 -22.537595 Symmetry: 51 A 52 A 53 A 54 A 55 A Eigenvalue: -21.417406 -21.342624 -20.777319 -20.741105 -20.717314 Symmetry: 56 A 57 A 58 A 59 A 60 A Eigenvalue: -20.605476 -19.949422 -19.827813 -17.671123 -17.611228 Symmetry: 61 A 62 A 63 A 64 A 65 A Eigenvalue: -17.416728 -17.267282 -16.604234 -16.524863 -16.454880 Symmetry: 66 A 67 A 68 A 69 A 70 A Eigenvalue: -16.418271 -16.319098 -16.293874 -15.573130 -15.544889 Symmetry: 71 A 72 A 73 A 74 A 75 A Eigenvalue: -15.289549 -15.257055 -15.141635 -15.085698 -14.976731 Symmetry: 76 A 77 A 78 A 79 A 80 A Eigenvalue: -14.684719 -14.627389 -14.613326 -14.498921 -14.338380 Symmetry: 81 A 82 A 83 A 84 A 85 A Eigenvalue: -14.087770 -13.911143 -13.780203 -13.678569 -13.565590 Symmetry: 86 A 87 A 88 A 89 A 90 A Eigenvalue: -13.552472 -13.234029 -13.218539 -12.743753 -12.694780 Symmetry: 91 A 92 A 93 A 94 A 95 A Eigenvalue: -12.645713 -12.632550 -12.195894 -12.137255 -12.030657 Symmetry: 96 A 97 A 98 A 99 A 100 A Eigenvalue: -11.984293 -11.895697 -11.764191 -11.762654 -11.712557 Symmetry: 101 A 102 A 103 A 104 A 105 A Eigenvalue: -11.660308 -11.597680 -10.864551 -10.834433 -10.561066 Symmetry: 106 A 107 A 108 A 109 A 110 A Eigenvalue: -10.487849 -10.383482 -10.342719 -8.979055 -8.922267 Symmetry: 111 A 112 A 113 A 114 A 115 A Eigenvalue: -7.773780 -7.655064 -6.505116 ----6.389845 7.3950266.389845 7.3950266.389845 7.3950266.389845 7.395026 Symmetry: 116 A 117 A 118 A 119 A 120 A Eigenvalue: 7.439096 7.517770 7.601380 13.556868 13.778663 Symmetry: 121 A 122 A 123 A 124 A 125 A Eigenvalue: 13.897722 13.938704 15.623258 15.758958 15.864535 Symmetry: 126 A 127 A 128 A 129 A 130 A Eigenvalue: 16.029068 16.126697 16.256516 16.476989 16.683115 Symmetry: 131 A 132 A 133 A 134 A 135 A Eigenvalue: 16.925558 17.013641 17.159168 17.248467 17.467866 Symmetry: 136 A 137 A 138 A 139 A 140 A Eigenvalue: 17.482620 17.568245 17.823735 18.066420 18.165657 Symmetry: 141 A 142 A 143 A 144 A 145 A Eigenvalue: 18.234255 18.312710 18.389124 18.469225 18.783289 Symmetry: 146 A 147 A 148 A 149 A 150 A Eigenvalue: 18.816339 18.878284 18.884256 19.118851 19.222975 Symmetry: 151 A 152 A 153 A 154 A 155 A Eigenvalue: 19.522154 19.629009 19.807705 19.837290 19.884007 Symmetry: 156 A 157 A 158 A 159 A 160 A Eigenvalue: 19.960915 20.021056 20.227701 20.311284 20.356875 Symmetry: 161 A 162 A 163 A 164 A 165 A

74
Eigenvalue: 20.413301 20.443977 20.594910 20.637080 20.737572 Symmetry: 166 A 167 A 168 A 169 A 170 A Eigenvalue: 20.802705 20.868751 21.019340 21.158675 21.359635 Symmetry: 171 A 172 A 173 A 174 A 175 A Eigenvalue: 21.481108 21.552916 21.602006 21.653885 21.678922 Symmetry: 176 A 177 A 178 A 179 A 180 A Eigenvalue: 21.826139 21.855976 21.960438 22.652719 23.132224 Symmetry: 181 A 182 A 183 A 184 A 185 A Eigenvalue: 23.636857 23.732598 24.629441 24.774262 24.860531 Symmetry: 186 A 187 A 188 A 189 A 190 A Eigenvalue: 25.049703 25.611353 25.658137 30.163290 30.321176 Symmetry: 191 A 192 A Eigenvalue: 31.537206 31.644838
Lampiran 4. Log File Energi HOMO-LUMO Senyawa Kompleks bis-4-heksiloksibenzilamin tembaga(I)
EIGENVALUES(eV)
Symmetry: 1 A 2 A 3 A 4 A 5 A
Eigenvalue: -8869.646671-1103.314886 -981.254159 -980.375999 -980.299916
Symmetry: 6 A 7 A 8 A 9 A 10 A
Eigenvalue: -554.713694 -554.295505 -424.995825 -424.835401 -306.465354
Symmetry: 11 A 12 A 13 A 14 A 15 A
Eigenvalue: -306.327021 -304.322292 -304.281731 -304.277591 -304.257754
Symmetry: 16 A 17 A 18 A 19 A 20 A
Eigenvalue: -303.952035 -303.552823 -303.421521 -303.410382 -303.387698
Symmetry: 21 A 22 A 23 A 24 A 25 A
Eigenvalue: -303.381597 -303.380947 -303.349292 -302.600270 -302.545057
Symmetry: 26 A 27 A 28 A 29 A 30 A
Eigenvalue: -302.507945 -302.323219 -302.311364 -302.057108 -302.003335
Symmetry: 31 A 32 A 33 A 34 A 35 A
Eigenvalue: -301.907021 -301.776874 -301.692025 -301.570439 -301.379677
Symmetry: 36 A 37 A 38 A 39 A 40 A
Eigenvalue: -144.688953 -102.525859 -97.826244 -97.352043 -38.370000
Symmetry: 41 A 42 A 43 A 44 A 45 A
Eigenvalue: -38.186254 -37.347316 -37.043061 -33.387032 -32.915331
Symmetry: 46 A 47 A 48 A 49 A 50 A
Eigenvalue: -30.596260 -30.154784 -30.138984 -29.681182 -29.617593
Symmetry: 51 A 52 A 53 A 54 A 55 A
Eigenvalue: -29.325142 -28.841481 -28.626102 -27.511266 -27.414169
Symmetry: 56 A 57 A 58 A 59 A 60 A
Eigenvalue: -26.689522 -26.365050 -24.949580 -24.614447 -24.390428
Symmetry: 61 A 62 A 63 A 64 A 65 A
Eigenvalue: -23.954731 -23.441742 -23.192445 -23.035911 -22.917896
Symmetry: 66 A 67 A 68 A 69 A 70 A
Eigenvalue: -22.709033 -22.556859 -22.278640 -22.058955 -21.651467
Symmetry: 71 A 72 A 73 A 74 A 75 A
Eigenvalue: -21.189758 -20.796976 -19.978118 -19.730054 -19.633623
Symmetry: 76 A 77 A 78 A 79 A 80 A
Eigenvalue: -19.352207 -19.297817 -19.166383 -19.047434 -18.826047
Symmetry: 81 A 82 A 83 A 84 A 85 A
Eigenvalue: -18.505229 -18.219291 -18.186700 -18.053416 -17.969999
Symmetry: 86 A 87 A 88 A 89 A 90 A
Eigenvalue: -17.866377 -17.665741 -17.546834 -17.285920 -16.919759
Symmetry: 91 A 92 A 93 A 94 A 95 A
75
Eigenvalue: -16.825471 -16.794605 -16.744064 -16.519689 -16.175533
Symmetry: 96 A 97 A 98 A 99 A 100 A
Eigenvalue: -16.075462 -15.908465 -15.767832 -15.739890 -15.688386
Symmetry: 101 A 102 A 103 A 104 A 105 A
Eigenvalue: -15.517805 -15.406017 -15.241046 -15.095578 -15.043134
Symmetry: 106 A 107 A 108 A 109 A 110 A
Eigenvalue: -14.904268 -14.890769 -14.623710 -14.383970 -14.368151
Symmetry: 111 A 112 A 113 A 114 A 115 A
Eigenvalue: -14.115698 -14.012973 -13.865321 -13.513732 -13.387927
Symmetry: 116 A 117 A 118 A 119 A 120 A
Eigenvalue: -13.363934 -13.337063 -13.211857 -13.143160 -13.065767
Symmetry: 121 A 122 A 123 A 124 A 125 A
Eigenvalue: -12.805009 -12.510332 -12.284468 -12.176459 -11.914186
Symmetry: 126 A 127 A 128 A 129 A 130 A
Eigenvalue: -11.085292 -9.598812 ----0.953190 2.1826400.953190 2.1826400.953190 2.1826400.953190 2.182640 4.582261
Symmetry: 131 A 132 A 133 A 134 A 135 A
Eigenvalue: 4.829649 5.005436 5.149244 5.484207 6.946505
Symmetry: 136 A 137 A 138 A 139 A 140 A
Eigenvalue: 9.874535 10.459069 10.897132 11.219603 11.342538
Symmetry: 141 A 142 A 143 A 144 A 145 A
Eigenvalue: 11.499012 11.577951 11.713926 12.093128 12.167197
Symmetry: 146 A 147 A 148 A 149 A 150 A
Eigenvalue: 12.751347 12.880884 13.565883 14.126948 14.229733
Symmetry: 151 A 152 A 153 A 154 A 155 A
Eigenvalue: 14.319420 14.402075 14.453314 14.691873 14.951579
Symmetry: 156 A 157 A 158 A 159 A 160 A
Eigenvalue: 15.260448 15.578195 15.615922 15.627808 15.746054
Symmetry: 161 A 162 A 163 A 164 A 165 A
Eigenvalue: 15.826296 15.875667 15.985379 16.036278 16.070868
Symmetry: 166 A 167 A 168 A 169 A 170 A
Eigenvalue: 16.167615 16.348417 16.441313 16.506306 16.814109
Symmetry: 171 A 172 A 173 A 174 A 175 A
Eigenvalue: 16.847422 17.083440 17.152121 17.357849 17.388959
Symmetry: 176 A 177 A 178 A 179 A 180 A
Eigenvalue: 17.780701 17.940896 18.291821 18.575451 18.622825
Symmetry: 181 A 182 A 183 A 184 A 185 A
Eigenvalue: 18.708538 18.811656 18.839521 18.906618 18.995897
Symmetry: 186 A 187 A 188 A 189 A 190 A
Eigenvalue: 19.105339 19.455463 19.662727 19.735645 19.885489
Symmetry: 191 A 192 A 193 A 194 A 195 A
Eigenvalue: 19.997321 20.044583 20.063197 20.172233 20.299797
Symmetry: 196 A 197 A 198 A 199 A 200 A
Eigenvalue: 20.329888 20.507645 20.559571 21.186389 21.224651
Symmetry: 201 A 202 A 203 A 204 A 205 A
Eigenvalue: 21.426639 21.494465 21.649906 21.761106 22.318551
Symmetry: 206 A 207 A 208 A 209 A 210 A
Eigenvalue: 22.645892 26.567779 27.072581 28.048334 28.464323

76
Lampiran 5. Spektrum Transisi Elektronik Berupa Panjang Gelombang dan Intensitas Serapan Senyawa Bis-4-heksiloksibenzilamin
Lampiran 6. Spektrum Transisi Elektronik Berupa Panjang Gelombang dan Intensitas Serapan Senyawa Kompleks bis-4-heksiloksibenzilamin tembaga(I)

77
Lampiran 7. Input Frekuensi Senyawa Bis-4-heksiloksibenzilamin dalam Gaussian 98 for Windows
---------------------------------------------------------- # freq=raman rhf/sto-3g guess=mix scf=qc geom=connectivity ---------------------------------------------------------- Charge = 0 Multiplicity = 1
Coordinates (Angstroms) X Y Z
C 4.949004 1.363963 -0.214993 C 4.409941 1.486048 -1.495182 C 3.080220 1.788341 -1.680009 C 2.237263 1.976591 -0.576755 C 2.766391 1.862643 0.703677 C 4.110597 1.561793 0.868727 C 6.431943 1.054881 -0.023513 N 7.230574 2.315065 -0.000400 O 0.911697 2.278209 -0.893901 C 0.014087 2.314082 0.225357 C -1.405962 2.554266 -0.333706 C -2.474988 2.546134 0.776984 C -3.890725 2.797582 0.218664 C -4.975743 2.738429 1.314020 C -6.385437 2.999627 0.754517 C 3.090344 -2.722984 0.299750 C 2.787149 -2.489187 -1.039766 C 4.532769 -2.978229 0.743290 N 4.783058 -2.430642 2.103123 C 1.488494 -2.277161 -1.445626 C 0.444657 -2.291137 -0.512298 C 0.738705 -2.521823 0.826408 C 2.052307 -2.738355 1.216542 O -0.826623 -2.057481 -1.045547
C -1.894996 -2.093731 -0.089795 C -3.207774 -1.811994 -0.851386 C -4.430379 -1.822499 0.088136 C -5.742090 -1.511059 -0.661556 C -6.966843 -1.517632 0.276578 C -8.275441 -1.196207 -0.466212 H 5.040447 1.342351 -2.347657 H 2.688028 1.880096 -2.671304 H 2.139317 2.006423 1.558666 H 4.509291 1.480634 1.858351 H 6.744564 0.511763 -0.890803 H 6.528745 0.614830 0.946995 H 8.192579 2.097070 0.163989 H 7.140349 2.783050 -0.879519 H 0.020157 1.340082 0.668288 H 0.272997 3.170384 0.812394 H -1.624145 1.718077 -0.964645 H -1.403948 3.544398 -0.739335 H -2.483119 1.553774 1.177053 H -2.252151 3.377457 1.412709 H -4.096150 1.985831 -0.447490 H -3.891878 3.803159 -0.147004 H -4.980285 1.731119 1.674862 H -4.766843 3.545891 1.984293 H -6.324422 3.172204 -0.299710 H -7.006612 2.148647 0.941264 H -6.805378 3.859630 1.232998 H 3.574878 -2.474156 -1.763753 H 4.641791 -4.039869 0.820321 H 5.157156 -2.423419 0.074540 H 5.740822 -2.577331 2.350453 H 4.193298 -2.895575 2.763441 H 1.275923 -2.100842 -2.479369 H -0.045788 -2.532378 1.553979 H 2.268269 -2.921262 2.248436 H -1.953397 -3.095455 0.281738 H -1.741275 -1.279944 0.587715 H -3.342941 -2.629595 -1.528259 H -3.127571 -0.813563 -1.227689 H -4.522610 -2.826279 0.447052 H -4.283117 -1.018736 0.778917 H -5.890028 -2.316081 -1.350725

78
H -5.648345 -0.508159 -1.022534 H -7.064862 -2.522407 0.631148 H -6.814392 -0.717799 0.970787 H -8.065950 -1.030285 -1.502303 H -8.953266 -2.018108 -0.366546 H -8.717120 -0.316708 -0.046332
Lampiran 8. Input Frekuensi Senyawa Kompleks bis-4-heksiloksibenzilamin
tembaga(I) Gaussian 98 for Windows -------------------------------------------------------------- # opt freq=raman rhf/sto-3g guess=mix scf=qc geom=connectivity -------------------------------------------------------------- Charge = 1 Multiplicity = 1
Coordinates (Angstroms)
X Y Z C 4.691044 1.757135 0.208463 C 4.170610 1.970818 1.488422 C 2.842624 2.264271 1.665433 C 1.978125 2.362085 0.558793 C 2.496885 2.167781 -0.720358 C 3.844226 1.870972 -0.880750 C 6.170356 1.453946 0.033895 O 0.661620 2.647769 0.877800 N 6.513379 0.037790 0.501243 C -0.257897 2.679107 -0.228676 C -1.664573 2.924003 0.358066 C -2.758701 2.907622 -0.729317 C -4.159359 3.147850 -0.131993 C -5.268526 3.115071 -1.203790 C -6.663783 3.370863 -0.607205 C 1.741969 -1.913393 -0.290955 C 1.356958 -1.404267 0.948601 C 0.070745 -1.570944 1.406106 C -0.883525 -2.233178 0.619398 C -0.494791 -2.762868 -0.609330 C 0.809207 -2.602246 -1.048154 C 3.123920 -1.641947 -0.878394 N 4.260536 -1.807803 0.118312 O -2.158087 -2.301623 1.163324 C -3.204442 -2.559849 0.207890 C -4.548900 -2.191100 0.870121 C -5.698549 -2.165820 -0.157007 C -7.033126 -1.722030 0.474692 C -8.155432 -1.588303 -0.574472 C -9.485338 -1.129599 0.048299 Cu 6.076904 -1.471975 -0.760351 H 4.815696 1.904655 2.339531 H 2.460596 2.420126 2.652684 H 1.859194 2.247031 -1.575910 H 4.235603 1.727389 -1.866198 H 6.693272 2.121137 0.686823 H 6.360079 1.477526 -1.018887 H 7.483391 -0.141567 0.337210 H 6.319362 -0.046853 1.478583 H -0.009100 3.532105 -0.824826 H -0.261629 1.702911 -0.666773 H -1.653538 3.917307 0.755715 H -1.869186 2.092909 1.000184 H -2.558267 3.741104 -1.369657 H -2.769153 1.915516 -1.129961 H -4.150288 4.144736 0.256639 H -4.350776 2.321221 0.519886 H -5.071018 3.934929 -1.862367 H -5.285794 2.115221 -1.584451 H -6.578015 3.521988 0.448591 H -7.090496 4.242048 -1.058705 H -7.292989 2.526412 -0.796684 H 2.069158 -0.878720 1.549826 H -0.203995 -1.192940 2.368671 H -1.202077 -3.292601 -1.212678 H 1.101266 -3.018172 -1.989752 H 3.125927 -0.608005 -1.153823

79
H 3.277149 -2.394399 -1.623536 H 5.133962 -1.652805 -0.343319 H 4.153140 -1.145303 0.859634 H -3.059434 -1.883995 -0.608869 H -3.218178 -3.615356 0.032918 H -4.442991 -1.188252 1.227861 H -4.774186 -2.982235 1.554410 H -5.442457 -1.413318 -0.873286 H -5.839612 -3.178565 -0.472203 H -6.867770 -0.738530 0.862357 H -7.330531 -2.512865 1.131222 H -7.851466 -0.805749 -1.237882 H -8.328119 -2.573956 -0.953376 H -10.229440 -1.052099 -0.716689 H -9.351061 -0.175294 0.513242 H -9.800454 -1.842376 0.781477








![Reactions of bis[1,2-bis(dialkylphosphino)ethane]-(dihydrogen)hydridoiron(1+) with alkynes](https://static.fdokumen.com/doc/165x107/63146d10c32ab5e46f0ce1ad/reactions-of-bis12-bisdialkylphosphinoethane-dihydrogenhydridoiron1-with.jpg)




![Bis[(1 S *,2 S *)- trans -1,2-bis(diphenylphosphinoxy)cyclohexane]chloridoruthenium(II) trifluoromethanesulfonate dichloromethane disolvate](https://static.fdokumen.com/doc/165x107/63360a7bcd4bf2402c0b5520/bis1-s-2-s-trans-12-bisdiphenylphosphinoxycyclohexanechloridorutheniumii.jpg)






