
Amyloid-β Membrane Binding and Permeabilization are Distinct Processes Influenced Separately by...
16
Amyloid-β Membrane Binding and Permeabilization are Distinct Processes Influenced Separately by Membrane Charge and Fluidity Pamela T. Wong 1 , Joseph A. Schauerte 1,2 , Kathleen C. Wisser 2 , Hao Ding 2 , Edgar L. Lee 1 , Duncan G. Steel 2 and Ari Gafni 1,2 , * 1 Department of Biological Chemistry, University of Michigan, 930 N. University, Ann Arbor, MI 48109, USA 2 Biophysics Research Division, University of Michigan, 930 N. University, Ann Arbor, MI 48109, USA Received 15 August 2008; received in revised form 25 November 2008; accepted 30 November 2008 Available online 16 December 2008 The 40 and 42 residue amyloid-β (Aβ) peptides are major components of the proteinaceous plaques prevalent in the Alzheimer's disease-afflicted brain and have been shown to have an important role in instigating neuronal degeneration. Whereas it was previously thought that Aβ becomes cyto- toxic upon forming large fibrillar aggregates, recent studies suggest that soluble intermediate-sized oligomeric species cause cell death through membrane permeabilization. The present study examines the interactions between Aβ40 and lipid membranes using liposomes as a model system to determine how changes in membrane composition influence the conversion of Aβ into these toxic species. Aβ40 membrane binding was monitored using fluorescence-based assays with a tryptophan-substituted peptide (Aβ40 [Y10W]). We extend previous observations that Aβ40 interacts pre- ferentially with negatively charged membranes, and show that binding of nonfibrillar, low molecular mass oligomers of Aβ40 to anionic, but not neutral, membranes involves insertion of the peptide into the bilayer, as well as sequential conformational changes corresponding to the degree of oligomerization induced. Significantly, while anionic membranes in the gel, liquid crystalline, and liquid ordered phases induce these conformational changes equally, membrane permeabilization is reduced dramatically as the fluidity of the membrane is decreased. These findings demonstrate that binding alone is not sufficient for membrane permeabilization, and that the latter is also highly dependent on the fluidity and phase of the membrane. We conclude that binding and pore formation are two distinct steps. The differences in Aβ behavior induced by membrane composition may have significant implications on the development and progression of AD as neuronal membrane composition is altered with age. © 2008 Elsevier Ltd. All rights reserved. Edited by S. Radford Keywords: amyloid-β; membrane; charge; fluidity; permeabilization *Corresponding author. Department of Biological Chemistry , University of Michigan, 930 N. University, Ann Arbor, MI 48109, USA. E-mail address: [email protected]. Abbreviations used: Aβ, amyloid-β;Aβ40 [Y10W], Aβ40 with a Tyr to Trp substitution at position 10; AD, Alzheimer's disease; AFM, atomic force microscopy; AMP, antimicrobial peptide; APP, amyloid precursor protein; CF, 5(6)-carboxyfluorescein; Dns-DPPE, N-(5-dimethylaminonaphthalene-1-sulfonyl)-1,2-dipalmitoyl-sn-glycero-3- phosphoethanolamine); DPPC, 1,2-dipalmitoyl-sn-glycero-3-phosphocholine; DPPC/PG, liposomes with equimolar DPPC and DPPG; DPPG, 1,2-dipalmitoyl-sn-glycero-3-[phospho-rac-(1-glycerol)]; DPPS, 1,2-dipalmitoyl-sn-glycero-3- [phospho-L-serine]; FRET, fluorescence resonance energy transfer; L:P, lipid to peptide molar ratio; LC, liquid-crystalline; LO, liquid-ordered; LUV, large unilamellar vesicle; NaP i , sodium phosphate; PFT, pore-forming toxin; POPC, 1-palmitoyl- 2-oleoyl-sn-glycero-3-phosphocholine; POPC/PG, liposomes with equimolar POPC and POPG; POPG, 1-palmitoyl-2- oleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)]; RT, room temperature; SUV, small unilamellar vesicle; WT, wild-type. doi:10.1016/j.jmb.2008.11.060 J. Mol. Biol. (2009) 386, 81–96 Available online at www.sciencedirect.com 0022-2836/$ - see front matter © 2008 Elsevier Ltd. All rights reserved.
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Amyloid-β Membrane Binding and Permeabilization are Distinct Processes Influenced Separately by...

doi:10.1016/j.jmb.2008.11.060 J. Mol. Biol. (2009) 386, 81–96
Available online at www.sciencedirect.com
Amyloid-β Membrane Binding and Permeabilization areDistinct Processes Influenced Separately by MembraneCharge and Fluidity
Pamela T. Wong1, Joseph A. Schauerte1,2, Kathleen C. Wisser2,Hao Ding2, Edgar L. Lee1, Duncan G. Steel2 and Ari Gafni1,2,*
1Department of BiologicalChemistry, University ofMichigan, 930 N. University,Ann Arbor, MI 48109, USA2Biophysics Research Division,University of Michigan,930 N. University, Ann Arbor,MI 48109, USA
Received 15 August 2008;received in revised form25 November 2008;accepted 30 November 2008Available online16 December 2008
*Corresponding author. Department48109, USA. E-mail address: arigafnAbbreviations used: Aβ, amyloid-
disease; AFM, atomic force microsco5(6)-carboxyfluorescein; Dns-DPPE,phosphoethanolamine); DPPC, 1,2-dDPPC and DPPG; DPPG, 1,2-dipalm[phospho-L-serine]; FRET, fluorescenLO, liquid-ordered; LUV, large unilam2-oleoyl-sn-glycero-3-phosphocholinoleoyl-sn-glycero-3-[phospho-rac-(1-
0022-2836/$ - see front matter © 2008 E
The 40 and 42 residue amyloid-β (Aβ) peptides are major components of theproteinaceous plaques prevalent in the Alzheimer's disease-afflicted brainand have been shown to have an important role in instigating neuronaldegeneration. Whereas it was previously thought that Aβ becomes cyto-toxic upon forming large fibrillar aggregates, recent studies suggest thatsoluble intermediate-sized oligomeric species cause cell death throughmembrane permeabilization. The present study examines the interactionsbetween Aβ40 and lipid membranes using liposomes as a model system todetermine how changes in membrane composition influence the conversionof Aβ into these toxic species. Aβ40 membrane binding was monitoredusing fluorescence-based assays with a tryptophan-substituted peptide(Aβ40 [Y10W]). We extend previous observations that Aβ40 interacts pre-ferentially with negatively charged membranes, and show that binding ofnonfibrillar, low molecular mass oligomers of Aβ40 to anionic, but notneutral, membranes involves insertion of the peptide into the bilayer, aswell as sequential conformational changes corresponding to the degree ofoligomerization induced. Significantly, while anionic membranes in the gel,liquid crystalline, and liquid ordered phases induce these conformationalchanges equally, membrane permeabilization is reduced dramatically as thefluidity of the membrane is decreased. These findings demonstrate thatbinding alone is not sufficient for membrane permeabilization, and that thelatter is also highly dependent on the fluidity and phase of the membrane.We conclude that binding and pore formation are two distinct steps. Thedifferences in Aβ behavior induced by membrane composition may havesignificant implications on the development and progression of AD asneuronal membrane composition is altered with age.
© 2008 Elsevier Ltd. All rights reserved.
Edited by S. Radford
Keywords: amyloid-β; membrane; charge; fluidity; permeabilizationof Biological Chemistry, University of Michigan, 930 N. University, Ann Arbor, [email protected].β; Aβ40 [Y10W], Aβ40 with a Tyr to Trp substitution at position 10; AD, Alzheimer'spy; AMP, antimicrobial peptide; APP, amyloid precursor protein; CF,N-(5-dimethylaminonaphthalene-1-sulfonyl)-1,2-dipalmitoyl-sn-glycero-3-ipalmitoyl-sn-glycero-3-phosphocholine; DPPC/PG, liposomes with equimolaritoyl-sn-glycero-3-[phospho-rac-(1-glycerol)]; DPPS, 1,2-dipalmitoyl-sn-glycero-3-ce resonance energy transfer; L:P, lipid to peptide molar ratio; LC, liquid-crystalline;ellar vesicle; NaPi, sodium phosphate; PFT, pore-forming toxin; POPC, 1-palmitoyl-
e; POPC/PG, liposomes with equimolar POPC and POPG; POPG, 1-palmitoyl-2-glycerol)]; RT, room temperature; SUV, small unilamellar vesicle; WT, wild-type.
lsevier Ltd. All rights reserved.

82 Aβ Membrane Interactions Depend on Lipid Composition
Introduction
Several age-related diseases are characterized byprotein misfolding and aggregation. Alzheimer'sdisease (AD) is a common form of progressive seniledementia caused by a loss of neurons in the cerebralcortex and hippocampal region of the brain.Pathologically, AD is characterized by the presenceof neurofibrillar tangles and extracellular senileplaques in the affected regions of the brain. Theseplaques are insoluble proteinaceous deposits com-posed primarily of aggregates of fibrils of the 40 and42 residue amyloid-β peptides (Aβ40 and Aβ42). Alarge body of work has implicated Aβ as a keyfactor in instigating neurodegeneration, althoughthe mechanism by which this occurs is still notentirely understood.1–4Aβ40 and Aβ42 are produced by cleavage of the
transmembrane protein, amyloid precursor proteinby several secretases.5 The Aβ portion of APP isreleased extracellularly as a soluble peptide, and isfound in the plasma and cerebrospinal fluid (CSF)under both normal and disease states. Developmentof AD cannot be explained by over-expression, sincetotal levels of Aβ found in the CSF are not con-sistently clearly higher in the diseased brain.6,7
While certain point mutations in APP found ininherited forms of early-onset AD lead to signifi-cantly increased levels of Aβ production, reports onAβ levels in late-onset “sporadic” AD brain areinconsistent, and do not show a clear elevation inAβ.6,8,9 Aβ40 is presumably cleaved as a monomerfrom APP, and forms a stable dimeric or trimericspecies.10–12 It subsequently oligomerizes to a highdegree, eventually forming large insoluble amyloidfibrils, which are characterized by a unique highlyordered cross β-sheet structure. In addition to fibrilsand monomers, lower-order oligomers and interme-diate order oligomers or protofibrils (N20-mer) canbe formed, and are present in neuronal tissue takenfrom AD patients.13–17 Whereas historically it wasassumed that Aβ becomes cytotoxic when it formslarge insoluble fibrillar aggregates in the brain,recent studies have found little correlation betweenamyloid plaque load and the clinical severity of thedisease.18,19 Furthermore, neuronal abnormalitiesand cognitive deficits begin to appear well beforeamyloid plaques are detected in transgenic animalsover-expressing APP.20–23 Moreover, recent studiesprovide evidence suggesting that the soluble inter-mediate-sized oligomeric species of Aβ effectivelycause cell death, potentially through membranepermeabilization. Indeed, a number of studies havedemonstrated that neurotoxicity is influenced by theinteraction of Aβ with membrane lipids.24–30 Sig-nificant increases in calcium influx into cells,31,32increased conductivity in membrane bilayers trea-ted with Aβ,25,33–36 and AFM images revealingannular Aβ structures bound to lipid membranes,35have provided evidence supporting a model inwhich Aβ forms channels or pores in the plasmamembrane, thereby permitting rapid influx ofcations such as calcium into the cell. Calcium influx
may subsequently trigger cell death directly or indi-rectly by stimulating apoptosis signaling path-ways.37 However, what specific factors promoteconversion of this normally expressed peptide into aneurotoxic entity remains elusive.An intriguing aspect of AD is that amyloid
plaques are found only in specific regions of thebrain, and are toxic to only certain types of neurons(cerebral cortex and hippocampal cells in particular)and not to other cells in the body.38,39 This points toan important role of the local environment, inparticular the plasma membrane, in promoting Aβtoxicity. Therefore, changes in the plasma mem-brane composition may be crucial in the develop-ment and progression of AD.A number of studies have shown that Aβ can
interact with synthetic lipids in vitro,35,40–42 and thatthe presence of Aβ induces changes in permeabilityand conductivity across lipid bilayers.25,33–36 In thepresent study, the specificity of Aβ membranebinding, and how peptide binding leads to per-meabilization, was further examined using lipo-somes as model membranes. Aβ40 membraneinteractions were characterized using the fluores-cence of a tryptophan-substituted peptide (Aβ40[Y10W]). The effects of lipid composition, inparticular the membrane charge and fluidity, onAβ40 binding, conformation, oligomerization state,and permeabilization activity were determined.Building on previous studies showing that Aβinteracts preferentially with anionic, as opposed toneutral, membranes,40–45 we present evidence forthe presence of at least two kinds of Aβ40 inter-actions with the membrane, which are highlydependent on the charge of the lipid. Significantly,we demonstrate that bilayer phase and fluidity asdetermined by phospholipid acyl chain saturationand cholesterol content, while not affecting Aβbinding, are critical in determining the permeabi-lization activity of the peptide.Aβ shares several characteristics with pore-
forming toxins (PFT) such as melittin, α-hemolysin,aerolysin and antimicrobial peptides.46,28 Like Aβ,these peptides exist in an unstructured randomcoiled state in solution, and undergo conforma-tional changes in the presence of membranes.47–49
These peptides are also capable of forming oligo-meric structures, and have been shown to alter thepermeability of their target membranes,50,51 Thus,greater insight into the mechanism of Aβ mem-brane interaction may be obtained by comparingits mechanism of action with that of AMPs andPFTs. Work with several AMPs (e.g., melittin,alamethicin, magainin, and protegrin) revealedthat in order to insert into a membrane and formchannels, a threshold ratio of lipid to peptide (L:P),the critical L:P, must be reached.52 The critical L:Pvalue is specific for the peptide and depends on themembrane it interacts with. When the L:P is abovethe critical value, the AMPs remain in an inactive,non-pore-forming state, with the axis of the helicalpeptide parallel with the plane of the mem-brane.49,52 On the other hand, below the critical
83Aβ Membrane Interactions Depend on Lipid Composition
L:P, the peptide undergoes a change in conforma-tion and orientation, allowing it to penetrate themembrane with the helical axis perpendicular tothe plane of the membrane, forming channels orpores. Here, we propose a similar mechanism forAβ, and compare the critical L:P values for dif-ferent membranes. Membrane binding and per-meabilization are pivotal steps in Aβ toxicity.Therefore, determining which factors promote orinhibit these processes is critical to understandingthe etiology of AD.
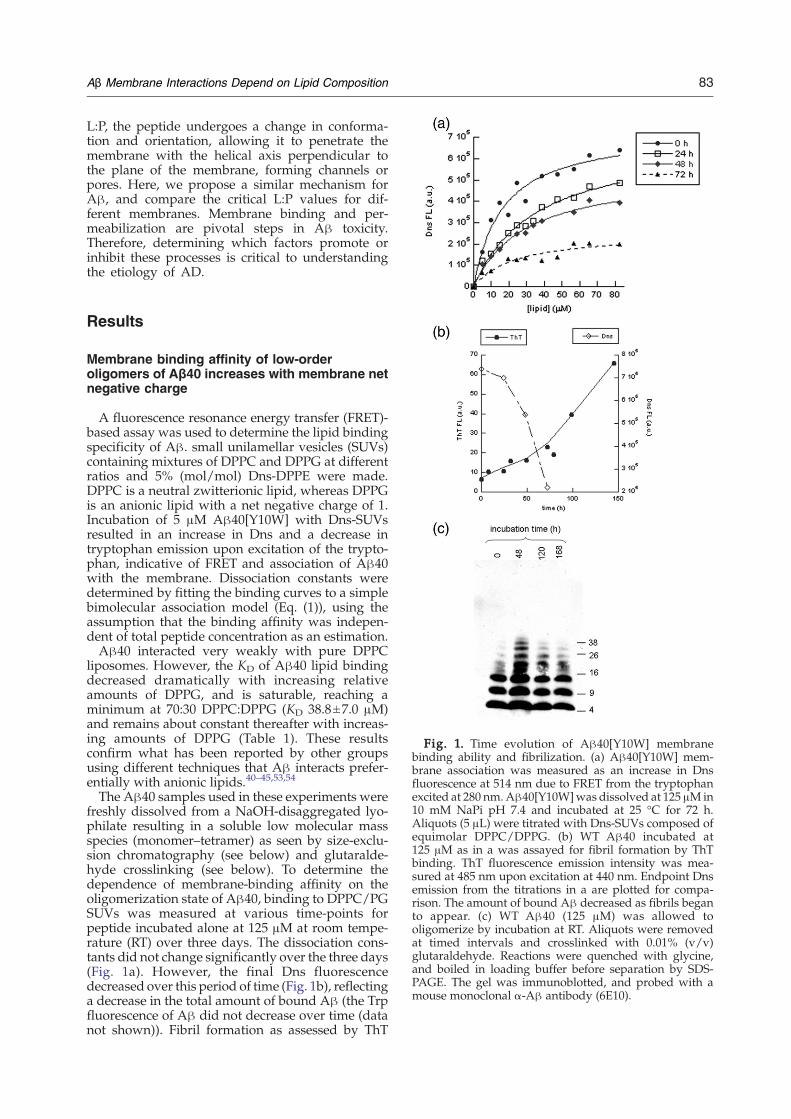
Fig. 1. Time evolution of Aβ40[Y10W] membranebinding ability and fibrilization. (a) Aβ40[Y10W] mem-brane association was measured as an increase in Dnsfluorescence at 514 nm due to FRET from the tryptophanexcited at 280 nm.Aβ40[Y10W]was dissolved at 125 μMin10 mM NaPi pH 7.4 and incubated at 25 °C for 72 h.Aliquots (5 μL) were titrated with Dns-SUVs composed ofequimolar DPPC/DPPG. (b) WT Aβ40 incubated at125 μM as in a was assayed for fibril formation by ThTbinding. ThT fluorescence emission intensity was mea-sured at 485 nm upon excitation at 440 nm. Endpoint Dnsemission from the titrations in a are plotted for compa-rison. The amount of bound Aβ decreased as fibrils beganto appear. (c) WT Aβ40 (125 μM) was allowed tooligomerize by incubation at RT. Aliquots were removedat timed intervals and crosslinked with 0.01% (v/v)glutaraldehyde. Reactions were quenched with glycine,and boiled in loading buffer before separation by SDS-PAGE. The gel was immunoblotted, and probed with amouse monoclonal α-Aβ antibody (6E10).
Results
Membrane binding affinity of low-orderoligomers of Aβ40 increases with membrane netnegative charge
A fluorescence resonance energy transfer (FRET)-based assay was used to determine the lipid bindingspecificity of Aβ. small unilamellar vesicles (SUVs)containing mixtures of DPPC and DPPG at differentratios and 5% (mol/mol) Dns-DPPE were made.DPPC is a neutral zwitterionic lipid, whereas DPPGis an anionic lipid with a net negative charge of 1.Incubation of 5 μM Aβ40[Y10W] with Dns-SUVsresulted in an increase in Dns and a decrease intryptophan emission upon excitation of the trypto-phan, indicative of FRET and association of Aβ40with the membrane. Dissociation constants weredetermined by fitting the binding curves to a simplebimolecular association model (Eq. (1)), using theassumption that the binding affinity was indepen-dent of total peptide concentration as an estimation.Aβ40 interacted very weakly with pure DPPC
liposomes. However, the KD of Aβ40 lipid bindingdecreased dramatically with increasing relativeamounts of DPPG, and is saturable, reaching aminimum at 70:30 DPPC:DPPG (KD 38.8±7.0 μM)and remains about constant thereafter with increas-ing amounts of DPPG (Table 1). These resultsconfirm what has been reported by other groupsusing different techniques that Aβ interacts prefer-entially with anionic lipids.40–45,53,54
The Aβ40 samples used in these experiments werefreshly dissolved from a NaOH-disaggregated lyo-philate resulting in a soluble low molecular massspecies (monomer–tetramer) as seen by size-exclu-sion chromatography (see below) and glutaralde-hyde crosslinking (see below). To determine thedependence of membrane-binding affinity on theoligomerization state of Aβ40, binding to DPPC/PGSUVs was measured at various time-points forpeptide incubated alone at 125 μM at room tempe-rature (RT) over three days. The dissociation cons-tants did not change significantly over the three days(Fig. 1a). However, the final Dns fluorescencedecreased over this period of time (Fig. 1b), reflectinga decrease in the total amount of bound Aβ (the Trpfluorescence of Aβ did not decrease over time (datanot shown)). Fibril formation as assessed by ThT
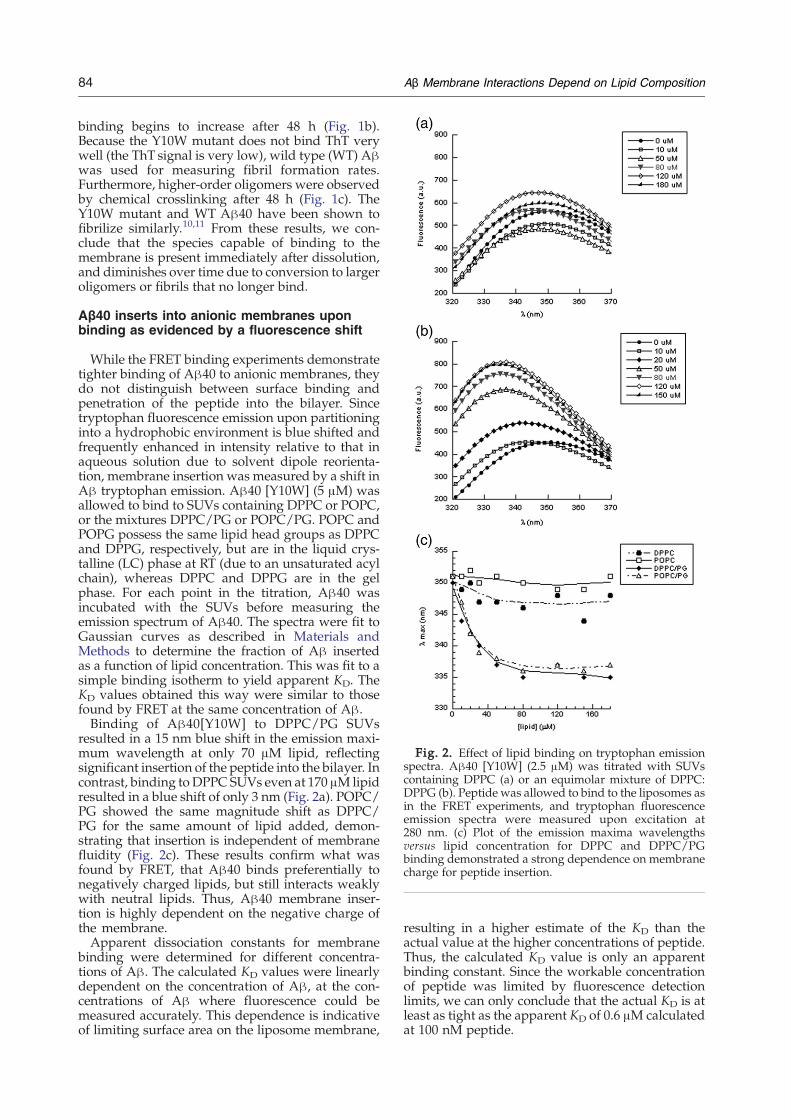
Fig. 2. Effect of lipid binding on tryptophan emissionspectra. Aβ40 [Y10W] (2.5 μM) was titrated with SUVscontaining DPPC (a) or an equimolar mixture of DPPC:DPPG (b). Peptide was allowed to bind to the liposomes asin the FRET experiments, and tryptophan fluorescenceemission spectra were measured upon excitation at280 nm. (c) Plot of the emission maxima wavelengthsversus lipid concentration for DPPC and DPPC/PGbinding demonstrated a strong dependence on membranecharge for peptide insertion.
84 Aβ Membrane Interactions Depend on Lipid Composition
binding begins to increase after 48 h (Fig. 1b).Because the Y10W mutant does not bind ThT verywell (the ThT signal is very low), wild type (WT) Aβwas used for measuring fibril formation rates.Furthermore, higher-order oligomers were observedby chemical crosslinking after 48 h (Fig. 1c). TheY10W mutant and WT Aβ40 have been shown tofibrilize similarly.10,11 From these results, we con-clude that the species capable of binding to themembrane is present immediately after dissolution,and diminishes over time due to conversion to largeroligomers or fibrils that no longer bind.
Aβ40 inserts into anionic membranes uponbinding as evidenced by a fluorescence shift
While the FRET binding experiments demonstratetighter binding of Aβ40 to anionic membranes, theydo not distinguish between surface binding andpenetration of the peptide into the bilayer. Sincetryptophan fluorescence emission upon partitioninginto a hydrophobic environment is blue shifted andfrequently enhanced in intensity relative to that inaqueous solution due to solvent dipole reorienta-tion, membrane insertion was measured by a shift inAβ tryptophan emission. Aβ40 [Y10W] (5 μM) wasallowed to bind to SUVs containing DPPC or POPC,or the mixtures DPPC/PG or POPC/PG. POPC andPOPG possess the same lipid head groups as DPPCand DPPG, respectively, but are in the liquid crys-talline (LC) phase at RT (due to an unsaturated acylchain), whereas DPPC and DPPG are in the gelphase. For each point in the titration, Aβ40 wasincubated with the SUVs before measuring theemission spectrum of Aβ40. The spectra were fit toGaussian curves as described in Materials andMethods to determine the fraction of Aβ insertedas a function of lipid concentration. This was fit to asimple binding isotherm to yield apparent KD. TheKD values obtained this way were similar to thosefound by FRET at the same concentration of Aβ.Binding of Aβ40[Y10W] to DPPC/PG SUVs
resulted in a 15 nm blue shift in the emission maxi-mum wavelength at only 70 μM lipid, reflectingsignificant insertion of the peptide into the bilayer. Incontrast, binding toDPPC SUVs even at 170 μM lipidresulted in a blue shift of only 3 nm (Fig. 2a). POPC/PG showed the same magnitude shift as DPPC/PG for the same amount of lipid added, demon-strating that insertion is independent of membranefluidity (Fig. 2c). These results confirm what wasfound by FRET, that Aβ40 binds preferentially tonegatively charged lipids, but still interacts weaklywith neutral lipids. Thus, Aβ40 membrane inser-tion is highly dependent on the negative charge ofthe membrane.Apparent dissociation constants for membrane
binding were determined for different concentra-tions of Aβ. The calculated KD values were linearlydependent on the concentration of Aβ, at the con-centrations of Aβ where fluorescence could bemeasured accurately. This dependence is indicativeof limiting surface area on the liposome membrane,
resulting in a higher estimate of the KD than theactual value at the higher concentrations of peptide.Thus, the calculated KD value is only an apparentbinding constant. Since the workable concentrationof peptide was limited by fluorescence detectionlimits, we can only conclude that the actual KD is atleast as tight as the apparent KD of 0.6 μM calculatedat 100 nM peptide.
85Aβ Membrane Interactions Depend on Lipid Composition
The tryptophan of Aβ40 is protected fromdynamic quenching upon binding to anionicliposomes
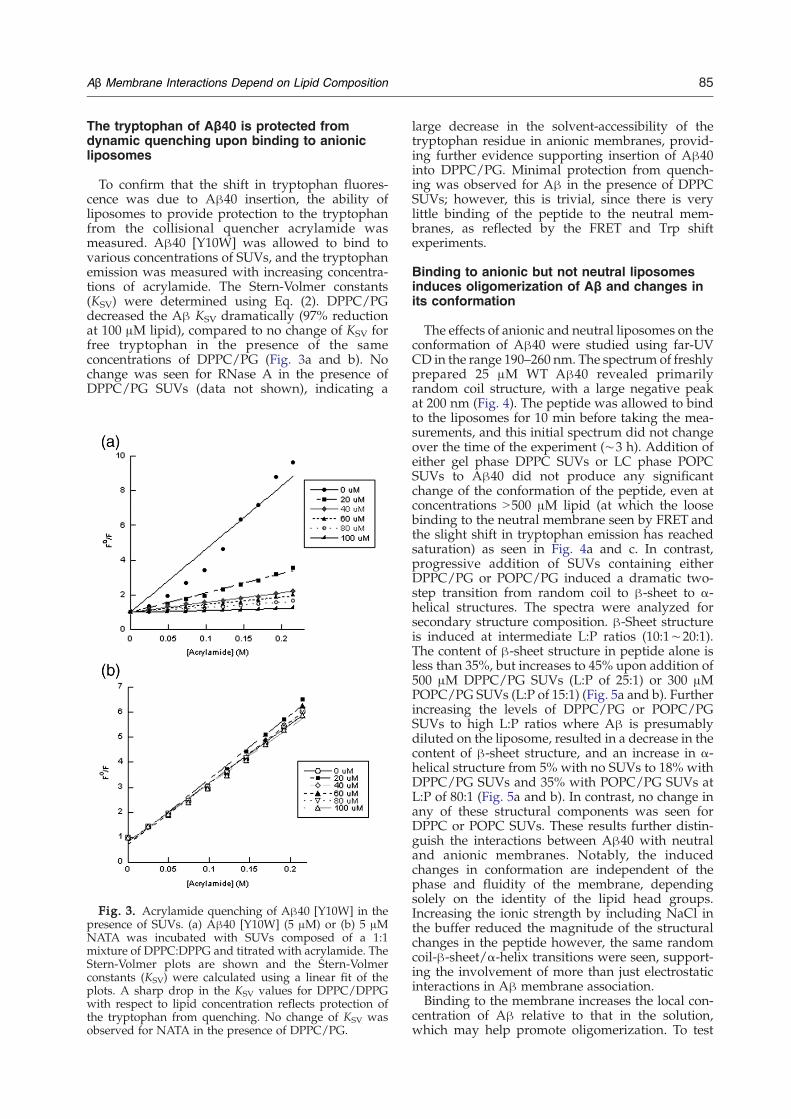
To confirm that the shift in tryptophan fluores-cence was due to Aβ40 insertion, the ability ofliposomes to provide protection to the tryptophanfrom the collisional quencher acrylamide wasmeasured. Aβ40 [Y10W] was allowed to bind tovarious concentrations of SUVs, and the tryptophanemission was measured with increasing concentra-tions of acrylamide. The Stern-Volmer constants(KSV) were determined using Eq. (2). DPPC/PGdecreased the Aβ KSV dramatically (97% reductionat 100 μM lipid), compared to no change of KSV forfree tryptophan in the presence of the sameconcentrations of DPPC/PG (Fig. 3a and b). Nochange was seen for RNase A in the presence ofDPPC/PG SUVs (data not shown), indicating a
Fig. 3. Acrylamide quenching of Aβ40 [Y10W] in thepresence of SUVs. (a) Aβ40 [Y10W] (5 μM) or (b) 5 μMNATA was incubated with SUVs composed of a 1:1mixture of DPPC:DPPG and titrated with acrylamide. TheStern-Volmer plots are shown and the Stern-Volmerconstants (KSV) were calculated using a linear fit of theplots. A sharp drop in the KSV values for DPPC/DPPGwith respect to lipid concentration reflects protection ofthe tryptophan from quenching. No change of KSV wasobserved for NATA in the presence of DPPC/PG.
large decrease in the solvent-accessibility of thetryptophan residue in anionic membranes, provid-ing further evidence supporting insertion of Aβ40into DPPC/PG. Minimal protection from quench-ing was observed for Aβ in the presence of DPPCSUVs; however, this is trivial, since there is verylittle binding of the peptide to the neutral mem-branes, as reflected by the FRET and Trp shiftexperiments.
Binding to anionic but not neutral liposomesinduces oligomerization of Aβ and changes inits conformation
The effects of anionic and neutral liposomes on theconformation of Aβ40 were studied using far-UVCD in the range 190–260 nm. The spectrum of freshlyprepared 25 μM WT Aβ40 revealed primarilyrandom coil structure, with a large negative peakat 200 nm (Fig. 4). The peptide was allowed to bindto the liposomes for 10 min before taking the mea-surements, and this initial spectrum did not changeover the time of the experiment (∼3 h). Addition ofeither gel phase DPPC SUVs or LC phase POPCSUVs to Aβ40 did not produce any significantchange of the conformation of the peptide, even atconcentrations N500 μM lipid (at which the loosebinding to the neutral membrane seen by FRET andthe slight shift in tryptophan emission has reachedsaturation) as seen in Fig. 4a and c. In contrast,progressive addition of SUVs containing eitherDPPC/PG or POPC/PG induced a dramatic two-step transition from random coil to β-sheet to α-helical structures. The spectra were analyzed forsecondary structure composition. β-Sheet structureis induced at intermediate L:P ratios (10:1∼20:1).The content of β-sheet structure in peptide alone isless than 35%, but increases to 45% upon addition of500 μM DPPC/PG SUVs (L:P of 25:1) or 300 μMPOPC/PG SUVs (L:P of 15:1) (Fig. 5a and b). Furtherincreasing the levels of DPPC/PG or POPC/PGSUVs to high L:P ratios where Aβ is presumablydiluted on the liposome, resulted in a decrease in thecontent of β-sheet structure, and an increase in α-helical structure from 5% with no SUVs to 18% withDPPC/PG SUVs and 35% with POPC/PG SUVs atL:P of 80:1 (Fig. 5a and b). In contrast, no change inany of these structural components was seen forDPPC or POPC SUVs. These results further distin-guish the interactions between Aβ40 with neutraland anionic membranes. Notably, the inducedchanges in conformation are independent of thephase and fluidity of the membrane, dependingsolely on the identity of the lipid head groups.Increasing the ionic strength by including NaCl inthe buffer reduced the magnitude of the structuralchanges in the peptide however, the same randomcoil-β-sheet/α-helix transitions were seen, support-ing the involvement of more than just electrostaticinteractions in Aβ membrane association.Binding to the membrane increases the local con-
centration of Aβ relative to that in the solution,which may help promote oligomerization. To test
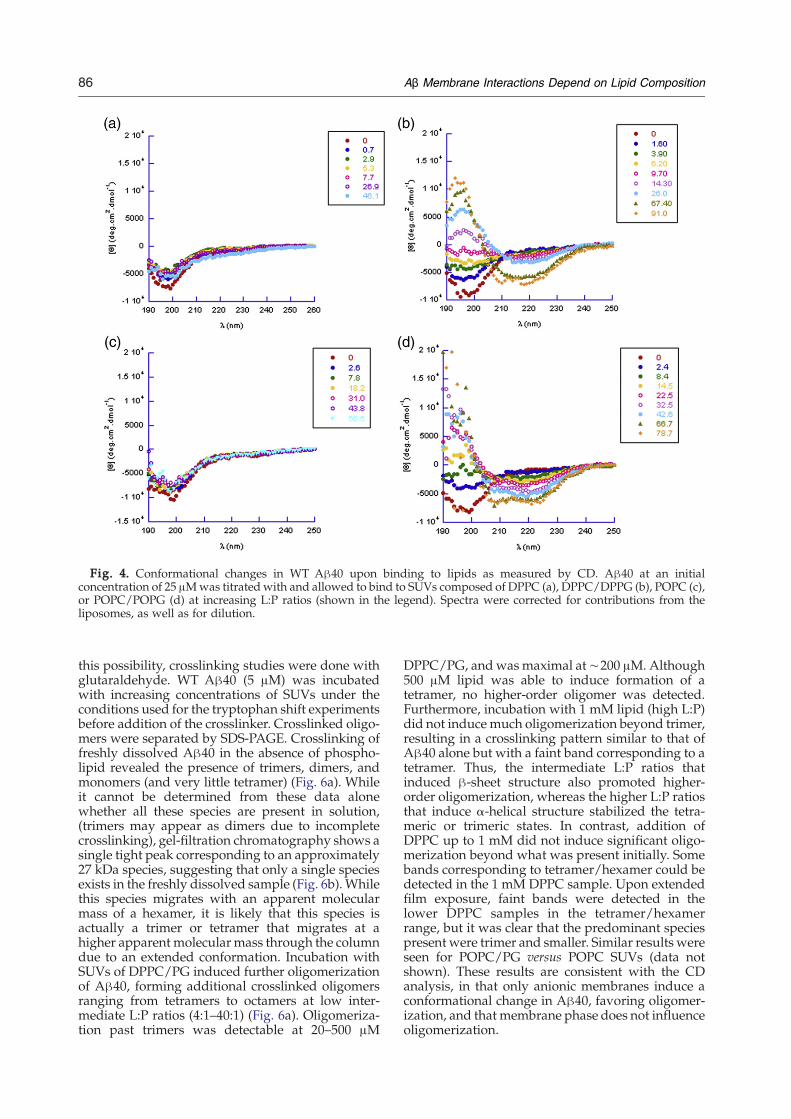
Fig. 4. Conformational changes in WT Aβ40 upon binding to lipids as measured by CD. Aβ40 at an initialconcentration of 25 μMwas titrated with and allowed to bind to SUVs composed of DPPC (a), DPPC/DPPG (b), POPC (c),or POPC/POPG (d) at increasing L:P ratios (shown in the legend). Spectra were corrected for contributions from theliposomes, as well as for dilution.
86 Aβ Membrane Interactions Depend on Lipid Composition
this possibility, crosslinking studies were done withglutaraldehyde. WT Aβ40 (5 μM) was incubatedwith increasing concentrations of SUVs under theconditions used for the tryptophan shift experimentsbefore addition of the crosslinker. Crosslinked oligo-mers were separated by SDS-PAGE. Crosslinking offreshly dissolved Aβ40 in the absence of phospho-lipid revealed the presence of trimers, dimers, andmonomers (and very little tetramer) (Fig. 6a). Whileit cannot be determined from these data alonewhether all these species are present in solution,(trimers may appear as dimers due to incompletecrosslinking), gel-filtration chromatography shows asingle tight peak corresponding to an approximately27 kDa species, suggesting that only a single speciesexists in the freshly dissolved sample (Fig. 6b).Whilethis species migrates with an apparent molecularmass of a hexamer, it is likely that this species isactually a trimer or tetramer that migrates at ahigher apparent molecular mass through the columndue to an extended conformation. Incubation withSUVs of DPPC/PG induced further oligomerizationof Aβ40, forming additional crosslinked oligomersranging from tetramers to octamers at low inter-mediate L:P ratios (4:1–40:1) (Fig. 6a). Oligomeriza-tion past trimers was detectable at 20–500 μM
DPPC/PG, and was maximal at∼200 μM. Although500 μM lipid was able to induce formation of atetramer, no higher-order oligomer was detected.Furthermore, incubation with 1 mM lipid (high L:P)did not induce much oligomerization beyond trimer,resulting in a crosslinking pattern similar to that ofAβ40 alone but with a faint band corresponding to atetramer. Thus, the intermediate L:P ratios thatinduced β-sheet structure also promoted higher-order oligomerization, whereas the higher L:P ratiosthat induce α-helical structure stabilized the tetra-meric or trimeric states. In contrast, addition ofDPPC up to 1 mM did not induce significant oligo-merization beyond what was present initially. Somebands corresponding to tetramer/hexamer could bedetected in the 1 mM DPPC sample. Upon extendedfilm exposure, faint bands were detected in thelower DPPC samples in the tetramer/hexamerrange, but it was clear that the predominant speciespresent were trimer and smaller. Similar results wereseen for POPC/PG versus POPC SUVs (data notshown). These results are consistent with the CDanalysis, in that only anionic membranes induce aconformational change in Aβ40, favoring oligomer-ization, and that membrane phase does not influenceoligomerization.
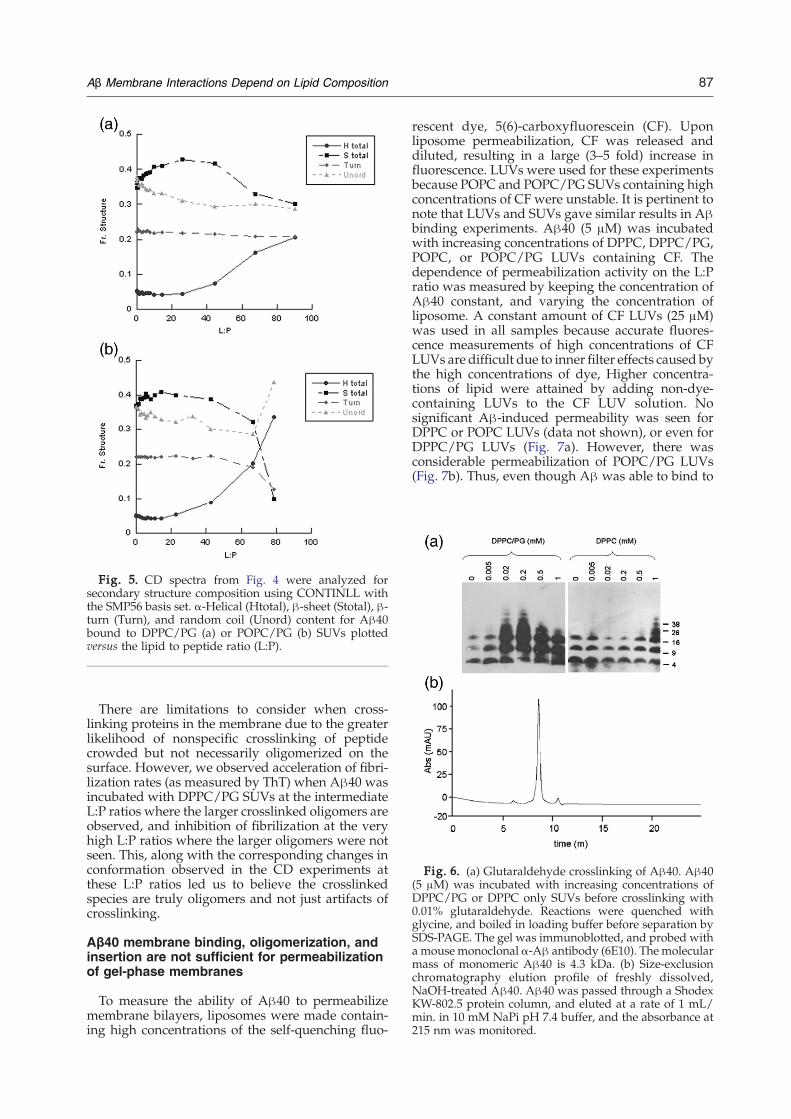
Fig. 5. CD spectra from Fig. 4 were analyzed forsecondary structure composition using CONTINLL withthe SMP56 basis set. α-Helical (Htotal), β-sheet (Stotal), β-turn (Turn), and random coil (Unord) content for Aβ40bound to DPPC/PG (a) or POPC/PG (b) SUVs plottedversus the lipid to peptide ratio (L:P).
Fig. 6. (a) Glutaraldehyde crosslinking of Aβ40. Aβ40(5 μM) was incubated with increasing concentrations ofDPPC/PG or DPPC only SUVs before crosslinking with0.01% glutaraldehyde. Reactions were quenched withglycine, and boiled in loading buffer before separation bySDS-PAGE. The gel was immunoblotted, and probed witha mouse monoclonal α-Aβ antibody (6E10). The molecularmass of monomeric Aβ40 is 4.3 kDa. (b) Size-exclusionchromatography elution profile of freshly dissolved,NaOH-treated Aβ40. Aβ40 was passed through a ShodexKW-802.5 protein column, and eluted at a rate of 1 mL/min. in 10 mM NaPi pH 7.4 buffer, and the absorbance at215 nm was monitored.
87Aβ Membrane Interactions Depend on Lipid Composition
There are limitations to consider when cross-linking proteins in the membrane due to the greaterlikelihood of nonspecific crosslinking of peptidecrowded but not necessarily oligomerized on thesurface. However, we observed acceleration of fibri-lization rates (as measured by ThT) when Aβ40 wasincubated with DPPC/PG SUVs at the intermediateL:P ratios where the larger crosslinked oligomers areobserved, and inhibition of fibrilization at the veryhigh L:P ratios where the larger oligomers were notseen. This, along with the corresponding changes inconformation observed in the CD experiments atthese L:P ratios led us to believe the crosslinkedspecies are truly oligomers and not just artifacts ofcrosslinking.
Aβ40 membrane binding, oligomerization, andinsertion are not sufficient for permeabilizationof gel-phase membranes
To measure the ability of Aβ40 to permeabilizemembrane bilayers, liposomes were made contain-ing high concentrations of the self-quenching fluo-
rescent dye, 5(6)-carboxyfluorescein (CF). Uponliposome permeabilization, CF was released anddiluted, resulting in a large (3–5 fold) increase influorescence. LUVs were used for these experimentsbecause POPC and POPC/PG SUVs containing highconcentrations of CF were unstable. It is pertinent tonote that LUVs and SUVs gave similar results in Aβbinding experiments. Aβ40 (5 μM) was incubatedwith increasing concentrations of DPPC, DPPC/PG,POPC, or POPC/PG LUVs containing CF. Thedependence of permeabilization activity on the L:Pratio was measured by keeping the concentration ofAβ40 constant, and varying the concentration ofliposome. A constant amount of CF LUVs (25 μM)was used in all samples because accurate fluores-cence measurements of high concentrations of CFLUVs are difficult due to inner filter effects caused bythe high concentrations of dye, Higher concentra-tions of lipid were attained by adding non-dye-containing LUVs to the CF LUV solution. Nosignificant Aβ-induced permeability was seen forDPPC or POPC LUVs (data not shown), or even forDPPC/PG LUVs (Fig. 7a). However, there wasconsiderable permeabilization of POPC/PG LUVs(Fig. 7b). Thus, even though Aβ was able to bind to
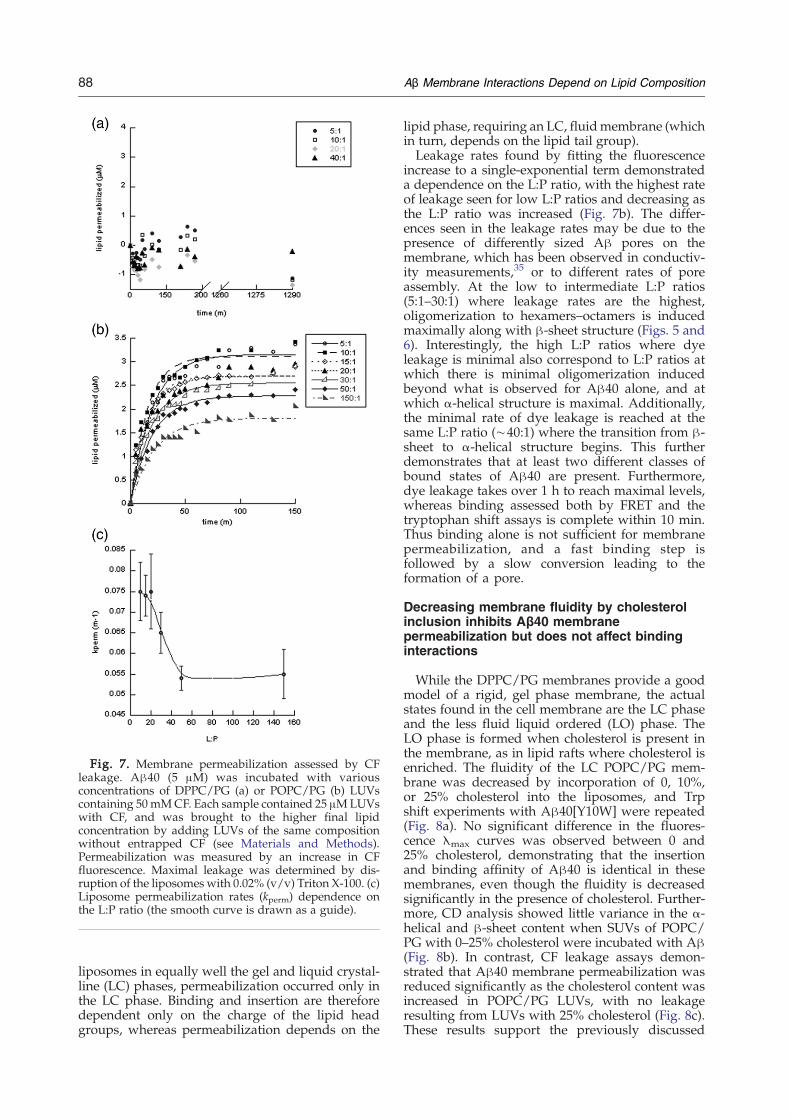
Fig. 7. Membrane permeabilization assessed by CFleakage. Aβ40 (5 μM) was incubated with variousconcentrations of DPPC/PG (a) or POPC/PG (b) LUVscontaining 50mMCF. Each sample contained 25 μMLUVswith CF, and was brought to the higher final lipidconcentration by adding LUVs of the same compositionwithout entrapped CF (see Materials and Methods).Permeabilization was measured by an increase in CFfluorescence. Maximal leakage was determined by dis-ruption of the liposomes with 0.02% (v/v) Triton X-100. (c)Liposome permeabilization rates (kperm) dependence onthe L:P ratio (the smooth curve is drawn as a guide).
88 Aβ Membrane Interactions Depend on Lipid Composition
liposomes in equally well the gel and liquid crystal-line (LC) phases, permeabilization occurred only inthe LC phase. Binding and insertion are thereforedependent only on the charge of the lipid headgroups, whereas permeabilization depends on the
lipid phase, requiring an LC, fluidmembrane (whichin turn, depends on the lipid tail group).Leakage rates found by fitting the fluorescence
increase to a single-exponential term demonstrateda dependence on the L:P ratio, with the highest rateof leakage seen for low L:P ratios and decreasing asthe L:P ratio was increased (Fig. 7b). The differ-ences seen in the leakage rates may be due to thepresence of differently sized Aβ pores on themembrane, which has been observed in conductiv-ity measurements,35 or to different rates of poreassembly. At the low to intermediate L:P ratios(5:1–30:1) where leakage rates are the highest,oligomerization to hexamers–octamers is inducedmaximally along with β-sheet structure (Figs. 5 and6). Interestingly, the high L:P ratios where dyeleakage is minimal also correspond to L:P ratios atwhich there is minimal oligomerization inducedbeyond what is observed for Aβ40 alone, and atwhich α-helical structure is maximal. Additionally,the minimal rate of dye leakage is reached at thesame L:P ratio (∼40:1) where the transition from β-sheet to α-helical structure begins. This furtherdemonstrates that at least two different classes ofbound states of Aβ40 are present. Furthermore,dye leakage takes over 1 h to reach maximal levels,whereas binding assessed both by FRET and thetryptophan shift assays is complete within 10 min.Thus binding alone is not sufficient for membranepermeabilization, and a fast binding step isfollowed by a slow conversion leading to theformation of a pore.
Decreasing membrane fluidity by cholesterolinclusion inhibits Aβ40 membranepermeabilization but does not affect bindinginteractions
While the DPPC/PG membranes provide a goodmodel of a rigid, gel phase membrane, the actualstates found in the cell membrane are the LC phaseand the less fluid liquid ordered (LO) phase. TheLO phase is formed when cholesterol is present inthe membrane, as in lipid rafts where cholesterol isenriched. The fluidity of the LC POPC/PG mem-brane was decreased by incorporation of 0, 10%,or 25% cholesterol into the liposomes, and Trpshift experiments with Aβ40[Y10W] were repeated(Fig. 8a). No significant difference in the fluores-cence λmax curves was observed between 0 and25% cholesterol, demonstrating that the insertionand binding affinity of Aβ40 is identical in thesemembranes, even though the fluidity is decreasedsignificantly in the presence of cholesterol. Further-more, CD analysis showed little variance in the α-helical and β-sheet content when SUVs of POPC/PG with 0–25% cholesterol were incubated with Aβ(Fig. 8b). In contrast, CF leakage assays demon-strated that Aβ40 membrane permeabilization wasreduced significantly as the cholesterol content wasincreased in POPC/PG LUVs, with no leakageresulting from LUVs with 25% cholesterol (Fig. 8c).These results support the previously discussed
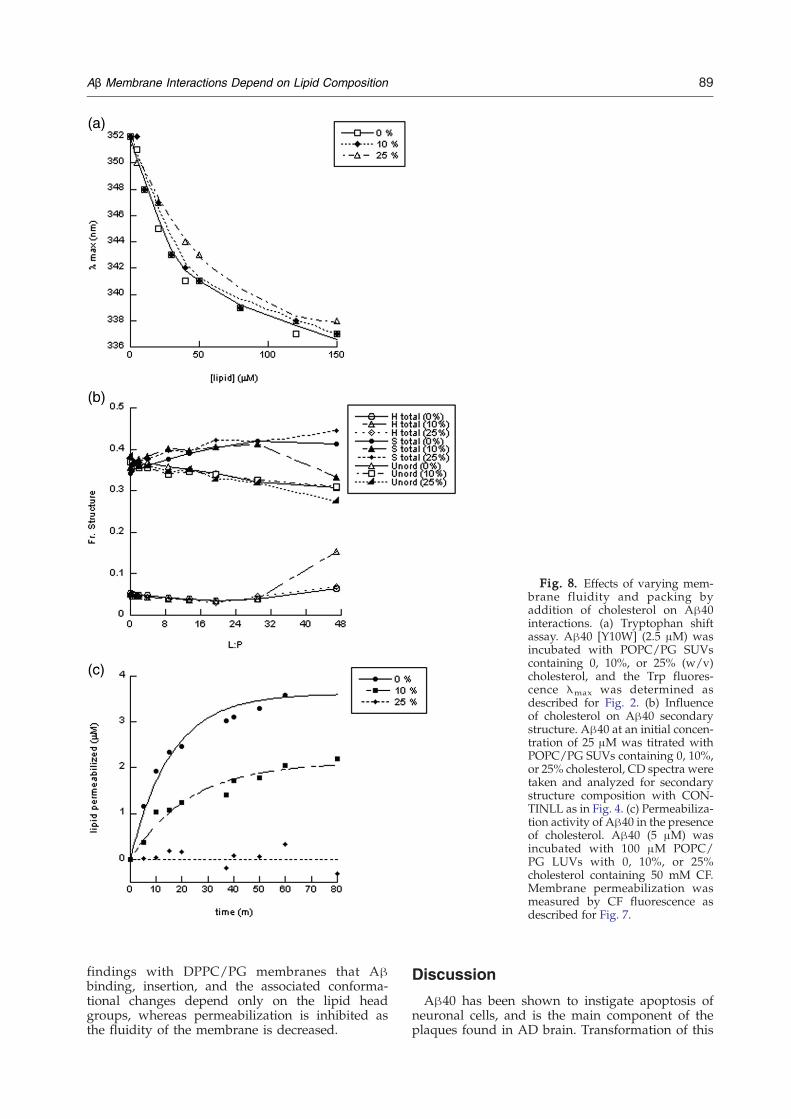
Fig. 8. Effects of varying mem-brane fluidity and packing byaddition of cholesterol on Aβ40interactions. (a) Tryptophan shiftassay. Aβ40 [Y10W] (2.5 μM) wasincubated with POPC/PG SUVscontaining 0, 10%, or 25% (w/v)cholesterol, and the Trp fluores-cence λmax was determined asdescribed for Fig. 2. (b) Influenceof cholesterol on Aβ40 secondarystructure. Aβ40 at an initial concen-tration of 25 μM was titrated withPOPC/PG SUVs containing 0, 10%,or 25% cholesterol, CD spectra weretaken and analyzed for secondarystructure composition with CON-TINLL as in Fig. 4. (c) Permeabiliza-tion activity of Aβ40 in the presenceof cholesterol. Aβ40 (5 μM) wasincubated with 100 μM POPC/PG LUVs with 0, 10%, or 25%cholesterol containing 50 mM CF.Membrane permeabilization wasmeasured by CF fluorescence asdescribed for Fig. 7.
89Aβ Membrane Interactions Depend on Lipid Composition
findings with DPPC/PG membranes that Aβbinding, insertion, and the associated conforma-tional changes depend only on the lipid headgroups, whereas permeabilization is inhibited asthe fluidity of the membrane is decreased.
Discussion
Aβ40 has been shown to instigate apoptosis ofneuronal cells, and is the main component of theplaques found in AD brain. Transformation of this

90 Aβ Membrane Interactions Depend on Lipid Composition
normally produced peptide into a neurotoxicspecies is a crucial step in the development andprogression of AD. Several studies have pointed tothe importance of membrane interactions in Aβtoxicity, revealing membrane-bound annular struc-tures of this peptide,35,55 and demonstrating thatmembrane permeability and conductivity are in-creased in the presence of Aβ.25,33–36,56 Previousstudies have shown that negatively charged lipidshave a role in mediating these interactions and ininfluencing the neurotoxic effects of Aβ.57 In thisstudy, we examined the mechanism of Aβ40membrane binding, and the effects that membraneinteractions have on the structure and permeabiliz-ing potency of the peptide. Liposomes containing avariety of phospholipids were used to determine theinfluence of lipid charge and phase on theseinteractions. Specifically, we found that (i) solublelow-order oligomers(b10mer) are the primary mem-brane binding species, (ii) Aβ40 binds more tightlyto and is inserted in negatively charged membranesindependent of phase or fluidity, (iii) interactionwith negatively charged membranes induces a tran-sition to either β-sheet or α-helical structure andincreases Aβ oligomerization in a way that dependson the L:P ratio but not on membrane fluidity, andmembrane binding, insertion, and conformationalchanges are not sufficient for permeabilization; thelatter is highly dependent on the phase and fluidityof themembrane, and is inhibited significantly as thefluidity of the membrane is decreased.
Low molecular mass species of Aβ40 bind andinsert preferentially into anionic membranesindependent of membrane phase
The preferential interaction of Aβ40 as well asother amyloid proteins and antimicrobial peptideswith anionic membranes has been reported inseveral studies.58,59 The FRET binding experimentsconfirmed these studies, demonstrating that Aβ40binding affinity becomes progressively higher asthe net negative charge (DPPG content of themembrane) is increased. It should be noted that thecalculated KD is not the actual binding affinity, andis only an apparent value, because binding islimited by the number of sites available on theliposome surface under these conditions, as dis-cussed below.While some studies found little or no interaction
of Aβ with neutral membranes,42,60 others havereported binding to PC membranes.61,62 Our fin-dings reveal loose binding to PC membranes. Fur-thermore, increasing the ionic strength of thesolution slightly reduced but did not abolish Aβ40binding to PC/PG membranes. This indicates thatthe Aβ–lipid interaction is not purely electrostatic,but also contains a hydrophobic component.Immediately after solvation in aqueous buffer Aβ
forms stable oligomeric species, mostly dimers andtrimers that are able to bind to the lipid membrane.However, we found that the binding decreased overtime as oligomerization of Aβ to larger species was
allowed to proceed, with very little bindingobserved for samples in which fibrils had formed.Therefore, once Aβ40 progresses past a certainoligomeric state it can no longer undergo the initialstep of binding to the membrane.Insertion of Aβ40 into the bilayer accompanies its
binding, as seen by a blue shift in the tryptophanfluorescence spectra of Aβ40[Y10W] bound toDPPC, POPC, DPPC/PG and POPC/PG SUVs.Much larger shifts were observed for anionic mem-branes, indicating peptide penetration into thebilayer not present for neutral membranes. Theinsertion of Aβ into the lipid bilayer was confirmedby the observed decrease in susceptibility of thetryptophan to dynamic quenching by acrylamideupon binding to anionic liposomes. These datasuggest that while Aβ40 is tightly bound andinserted in the bilayer for anionic membranes, alooser, more superficial surface binding exists forneutral membranes. While the Trp shift and protec-tion effect are dramatic, it is important to considerthe effects of the oligomerization of Aβ induced byinteraction with anionic liposomes. It is likely thatthe environment of the Trp residue becomes morehydrophobic and protected upon oligomerizationas well. Indeed, allowing Aβ40[Y10W] to formoligomers in solution in the absence of lipids resultsin an 8 nm blue shift in fluorescence. This issignificant, as interaction with anionic liposomesresults in a 15 nm shift. Thus, the observed effectsof lipid binding on Trp fluorescence are a combina-tion of membrane insertion and induced oligomer-ization. It is also hard to deduce information aboutpeptide insertion depth or orientation from theseexperiments. We cannot distinguish if the peptide isembedded in the lipid head region parallel WITHthe plane of the membrane bilayer, or fully insertedperpendicularly into the membrane. It would beideal to find conditions in which the processes ofbinding, oligomerization, and insertion could beseparated.The actual KD for Aβ binding to SUVs could not be
determined by the shift assay, and only an apparentKD of 600 nM at 100 nM Aβ could be determined.This value provides an upper limit for the actualdissociation constant, with the actual KD value likelybeing much smaller. Although PG is not found inmammalian cell membranes, it is commonly used asa model for anionic membranes, and repeating theseexperiments with PS lipids, an anionic lipid found inmammalian systems, showed similar results to whatwas seen with PG.
Induction of Aβ oligomerization and secondarystructure development by anionic membranesdepends on the L:P ratio, and is related topermeabilization activity
Immediately after solvation, Aβ40 in aqueousbuffer solution has a random coil structure, andremains unstructured in the presence of neutralSUVs. However, upon binding to anionic SUVseither in the gel phase (DPPC/PG) or in the liquid

91Aβ Membrane Interactions Depend on Lipid Composition
crystalline phase (POPC/PG), β-sheet structure israpidly induced at intermediate L:P ratios. As theconcentration of lipid is increased, the β-sheetcontent diminishes and the peptide is converted tomostly α-helical structure (Fig. 5). The selectivity foranionic membranes in terms of inducing structure issimilar to what is seen in several antimicrobial pep-tides, further relating Aβ with these peptides.50,59,63The two-step transition in secondary structureinduced upon interaction with anionic liposomes issimilar to what was observed by Terzi et al. in POPGliposomes.41 Here, we expanded this to examine therole of membrane phase in the structural transitions,and found no influence of membrane phase on theinduction of secondary structure. Therefore, only theinteractions between the lipid head groups and thepeptide influence conformational changes in Aβ.Interestingly, the induced structure correlates with
oligomerization, as detected by crosslinking experi-ments. Oligomerization was induced in the presenceof anionic liposomes only, with higher-order oligo-mers (up to octamers) present only at the inter-mediate L:P ratios where β-sheet structure wasprevalent. This order of oligomer is consistent withthe number of subunits observed in the annularmembrane-bound Aβ structures observed by Quistet al. by AFM.35 Higher L:P ratios that induced α-helical structure did not cause significant oligomer-ization beyond what was seen for Aβ in solution. Itappears that as Aβ binds to the membrane surface,the local concentration of peptide increases signifi-cantly, making it more conducive to the formation ofhigher-order oligomers. As more liposomes areadded, the density of Aβ on the membrane surfaceis lowered, reducing higher-order oligomerization.While Aβ40-induced permeabilization has been
seen in liposomes and bilayers before, many of thesestudies were done on membranes composed oflipids with mixed tail groups (i.e., phases) or LCphase membranes.34,43,64 In this study, we distin-guish between gel, LC, and LO phase membranes. Inassessing Aβ40 permeabilization activity by CFleakage from LUVs we found that, although Aβ40was able to bind equally well to anionic membranesboth in the gel and LC phases (with the samestructure and degree of oligomerization), this wasnot sufficient for permeabilization in gel phaseliposomes. Permeabilization was induced by Aβonly when bound to fluid liquid crystalline POPC/PG LUVs. The increased permeability in LC phasemembranes may be due to packing deficits in themembrane resulting from the presence of anunsaturated acyl chain. This would allow easierincorporation of the Aβ pore into the membrane incomparison to the DPPC/PG membrane, where theacyl chains are all saturated, resulting in tightpacking of the lipids in the bilayer. Thus bindingand permeabilization are distinct, separable steps,with Aβ pores assembling on the membrane ratherthan in solution.Since the cell membrane is typically composed of
either LC or LO phases, but not gel, LC membranefluidity was varied by the incorporation of 0–25%
cholesterol to form the less fluid LO phase (a healthyneuronal cell contains 25% cholesterol). Cholesterolis known to decrease the area per phospholipidmolecule in the membrane, thereby increasing thecohesiveness of lipids in the bilayer, making it morerigid.65 Binding affinity, peptide insertion, and theassociated conformational changes were unchangedas the fluidity of the membrane was decreased byaddition of cholesterol. However, permeabilizationwas decreased significantly at 10% cholesterol, andinhibited completely at 25% cholesterol. This sup-ports the result observed in the gel phase mem-branes, that permeabilization is highly dependenton the membrane phase. An intriguing possibilityfor future investigation is that the presence ofcholesterol in the membrane may still permit Aβ-induced perturbation of the membrane, but in a waythat does not allow CF to leak out of the liposome.For example, Aβ may form smaller pores, or formsmaller disruptions in the membrane that are notapparent by the methods used here.Interestingly, while Aβ binding and structural
changes were complete within 10 min, dye leakagereached a maximum only after 1–1.5 h. Peptidebinding is thus not the rate-limiting step in poreformation, which requires a slow conversion step onthe membrane to an active pore state. At low-intermediate L:P ratios, where oligomerization andβ-sheet structure are maximal, leakage rates are alsomaximal. In contrast, at the high L:P ratios wherethere is little induced oligomerization beyondtetramers and α-helical structure is maximal, leak-age rates were decreased but did not reach zero. Thissuggests that both the β-sheet and the α-helicalspecies can permeabilize the membrane, supportingthe presence of differently sized pores. Alternatively,this could be due to different rates of pore formation.Aβ40 membrane interactions may be described by
the two-state model proposed for antimicrobialpeptide–membrane interactions in which the L:Pratio determines the conformation of the peptidebound on the membrane.49,52 In this model, at veryhigh L:P ratios where the lipid is present at a largeexcess, the initially random-coiled peptide isinduced to develop α-helical structure, but binds tothe membrane parallel with the bilayer surface andremaining in an inactive state. Upon decreasing theL:P ratio, peptide density increases, driving thetransition to a pore with the helical axis perpendi-cular to the bilayer surface.52 As the L:P ratio isdecreased, pore formation becomes energeticallyfavored, possibly due to the oligomerization andconformational changes supported by the increasedlocal concentration of peptide on the membranesurface. On the other hand, if pore assembly occursin the membrane, the decrease in the rate of per-meabilization at high L:P ratios could be explainedby a slower rate of assembly due to a decrease indensity of the peptide on the liposome surface.The dependence of Aβ permeablization on the
membrane phase and fluidity resembles that of pore-forming toxins. For example, the pore-formingactivity of melittin has been shown to be highly

92 Aβ Membrane Interactions Depend on Lipid Composition
dependent on membrane fluidity, and is greatlyreduced as the concentration of cholesterol in themembrane is increased.66 There are several relatedfactors to consider when evaluating different mem-branes, such as lipid packing density, compressibilityand bending moduli, and membrane curvature,which are also likely to have an important role inthe formation of a pore. For instance, cholesterol isalso known to induce negative curvature in themembrane, which inhibits the formation of toroidalpores in particular, where the lipid bilayer must bendsuch that the lipid headgroups form part of the liningof the pore. The toroidal pore has been observed formelittin and, sinceAβ pore formation is also inhibitedby cholesterol, this may provide evidence supportingthe suggestion that Aβ forms toroidal pores.66The interaction of Aβ with lipid membranes is
clearly a complex process, and dependent on severalfactors. Here, we show that charge is important inthe initial membrane binding and oligomerizationsteps; however, permeabilization depends on mem-brane fluidity. These results may provide insightinto how Aβ becomes cytotoxic in vivo. Undernormal conditions, most of the negatively chargedlipids are found on the inner leaflet of the mem-brane.67 However, recent studies have shown thatdamage during aging or due to disease can alter themembrane composition, resulting in increasedcharge density on the outer leaflet.57,68 Differencesin cholesterol content and distribution in an agingbrain may also contribute to changes in Aβ mem-brane permeabilization activity due to alterations inthe fluidity of the membrane.69,70 The differenttypes of membrane interactions described here thatdepend on both charge and fluidity may help toexplain some of the differences in Aβ activity in thehealthy brain versus those in the AD brain.
Materials and Methods
Aβ40 monomerization and solubilization
Synthetic humanWTAβ40 peptide was purchased fromAnaspec Inc. (San Jose, CA). Mutant Aβ40 [Y10W] peptideis recombinant, and was purchased from rPeptide Inc.(Athens, GA). Peptides were dissolved at a concentrationof 1mg/mL in 50% (v/v) acetonitrile, sampleswere placedinto siliconized Eppendorf tubes, lyophilized overnight,and stored at –20 °C. The peptide was disaggregated bybase treatment as described.71,72 Briefly, the lyophilizedAβ40 was dissolved at a concentration of 0.5 mg/mL in2.5 mMNaOH, and the pH of the solution was checked toensure that it was above 10.5. Aβwas then sonicated in anFS20 bath sonicator (Fisher Scientific, Pittsburgh, PA) for1 min, lyophilized overnight and stored at –20 °C. Thisprocedure has been shown to disaggregate Aβ40.71,72
Immediately before each experiment, the peptide wassuspended in double-distilled water to a final concentra-tion of 1 mg/mL, and allowed to dissolve for 1 min beforemixing gently by pipetting. An equal volume of 20 mMsodium phosphate (NaPi), pH 7.4 was added, resulting ina 0.5 mg/mL solution of Aβ40 in 10 mMNaPi, pH 7.4. AllAβ used was treated and dissolved in this manner toensure homogeneity of the starting material.
Liposome formation
The synthetic lipids 1,2-dipalmitoyl-sn-glycero-3-phos-phocholine (DPPC), 1,2-dipalmitoyl-sn-glycero-3-[phos-pho-rac-(1-glycerol)] (DPPG), 1,2-dipalmitoyl-sn-glycero-3-[phospho-L-serine] (DPPS), 1-palmitoyl-2-oleoyl-sn-gly-cero-3-phosphocholine (POPC), and 1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (POPG) in chloro-form were purchased from Avanti Inc. (Alabaster, AL).Fluorescent N-(5-dimethylaminonaphthalene-1-sulfonyl)-1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (Dns-DPPE) was purchased from Invitrogen (Carlsbad, CA) asa powder, and was dissolved in chloroform/methanol(3:1, v/v). All lipids were stored at –20 °C. To formliposomes, lipids were mixed at the desired ratios, andthe chloroform/methanol was removed by evaporationin a glass test-tube under a gentle stream of nitrogen. Thedried samples were placed in a vacuum chamberovernight to remove any traces of solvent. Dried lipidfilms were rehydrated in 10 mM NaPi, pH 7.4 above thelipid transition temperature in a water-bath (for DPPCand mixtures of DPPC and DPPG (DPPC/PG) at 45 °C,for DPPC/PS at 50 °C, and for POPC and POPC/PGmixtures at 25 °C) for 2 h at a concentration of 2.5 mg/mL. Unless specified otherwise, the notation DPPC/PGrefers to liposomes composed of an equimolar mixture oftwo lipids with the same acyl chains but different headgroups — in this case, DPPC and DPPG. To form smallunilamellar vesicles (SUVs), the lipid suspension wasvortex mixed and then sonicated in an ultrasonic water-bath (Laboratory Supplies, Hicksville, NY) until thesolution became translucent (ca 5 min). Lipid concentra-tions were determined by the Stewart method.73 Allliposome concentrations are expressed as the concentra-tion of total lipid.
Fluorescence resonance energy transfer (FRET)binding assay
A 5 μM solution of freshly dissolved Aβ40 [Y10W] in10 mM NaPi, pH 7.4 was titrated with SUVs composed ofmixtures of DPPC and DPPG at different ratios andcontaining 5 mol % Dns-DPPE. Samples were incubatedfor 10 min at RT before taking measurements (nosignificant increase in fluorescence was seen after 10 min,confirming that the sample had reached equilibrium). Thefluorescence intensity was measured for both tryptophanat 352 nm and dansyl at 514 nm upon excitation at 280 nmusing an FP-6500 spectrofluorimeter (Jasco, Easton, MD).The fluorescence change due to FRET was evaluated bysubtracting the contribution of direct Dns excitation fromthe fluorescence signal of Dns-SUV with Aβ40 [Y10W].The tyrosine to tryptophan-substituted peptide Aβ40[Y10W] was used to maximize the spectral overlap andthus the FRET efficiency between the donor (Aβ) and theacceptor (Dns). It has been shown that this mutation doesnot alter the properties of Aβ40 significantly, and thatWT and Aβ40 [Y10W] can oligomerize and fibrilize in asimilar fashion.10,11 The change in fluorescence at 514 nmwas plotted versus lipid concentration, and fit to abinding equation:
DFL =FLf
1 + KDlipid½ �
� � ð1Þ
Where ΔFL is the fluorescence due to FRET, FLf is theendpoint fluorescence, KD is the dissociation constant, and[lipid] is the concentration of lipid added.

93Aβ Membrane Interactions Depend on Lipid Composition
To follow the effect of Aβ40 incubation time on itsmembrane binding ability, Aβ40[Y10W] was dissolved at125 μM in 10 mM NaPi as described. This sample wasincubated at RT for three days; samples were taken at 24 hintervals and their binding to Dns-SUVs was assayed.
Thioflavin T (ThT) binding assay for fibril formation
ThT binding to Aβ fibrils was determined asdescribed.74 Briefly, 6 μL samples were taken from a125 μMsolution ofWTAβ40 incubated in 10mMNaPi, pH7.4 in a 25°C water-bath for 150 h. The peptide wasincubated at 3.8 μM with 2.5 μM ThT in 50 mM glycine-NaOH, pH 8.5 for 3 min at RT. ThT fluorescence (excitationat 440 nm, emission at 485 nm) was measured.
Tryptophan emission shift
Freshly dissolved Aβ40 [Y10W] (2.5 μM in 10 mMNaPi, pH 7.4) was incubated with SUVs composed of anequimolar mixture of DPPC and DPPG (DPPC/PG) orPOPC and POPG (POPC/PG) at RT for 10 min beforemeasurements were taken. (Note that significant photo-bleaching of the Trp occurs upon excitation; thereforefluorescence was measured only once for each sample).Tryptophan emission spectra were recorded from 320 nmto 370 nm upon excitation at 280 nm. Spectra of lipo-somes alone at the same concentrations were subtracted.The KD for the wavelength shift was calculated with acorrection for the change in intensity for the 335 nm fullyblue-shifted fluorescence. The unshifted tryptophanemission at 350 nm and the final shifted emission at335 nm were fit individually to a single Gaussian curve(Gaussian width 3639273055448 for both peaks, and the335 nm peak had an intensity of 1.9 relative to the 350 nmpeak). All curves were fit as a linear combination of theinitial and final curves, and the relative proportion was fitby a simple binding isotherm.
Acrylamide quenching
Freshly dissolved Aβ40 [Y10W] (5 μM in 10 mM NaPi,pH 7.4) was incubated with various concentrations ofSUVs for 10min. at RTas described above. Acrylamidewastitrated up to 0.25M from a 5M stock solution inwater. Trpfluorescence was measured immediately (excitation at295 nm, emission at 352 nm). Samples were excited at295 nm instead of 280 nm to minimize acrylamide absor-bance. Stern-Volmer constants (KSV) were determined byEq. (2),
F0F
= 1 + Ksv Ac½ � ð2Þ
where F0 is the fluorescence without quencher, F is thefluorescence with quencher, and [Ac] is the concentrationof acrylamide added.
Circular dichroism
Circular dichroism was measured using a J-715 spec-tropolarimeter (Jasco, Easton, MD) at 22 °C using a 1 mmpathlength cuvette. Spectra of liposomes in buffer weresubtracted for each concentration of lipid used. Freshlydissolved WT Aβ40 (25 μM in 10 mM NaPi, pH 7.4) wastitrated with concentrated stocks of SUVs composed of
DPPC, POPC, DPPC/PG, or POPC/PG. After each addi-tion, the sample was allowed to equilibrate at 25 °C for10 min. before beginning the scans. Scans were taken at50 nm/min with a response time of 2 s. and a bandwithof 5 nm from 195 nm to 260 nm. Each spectrum was theresult of averaging five consecutive scans. The amountof lipid that could be added was limited, due to scat-tering effects. Secondary structure was evaluated fromthe spectra using CONTINLL algorithms within theCDPro analysis software.75 The reference basis set usedin the analysis was SMP56, which includes membraneproteins.
Size-exclusion chromatography
Aβ40 was passed through a Shodex KW-802.5 proteinsize-exclusion column (Shodex, Japan) on a Prostar 325HPLC (Varian, Palo Alto, CA) in 10 mM NaPi, pH 7.4 at aconstant flow rate of 1.0 mL/min. Absorbance wasmonitored at 215 nm. To determine molecular mass, thecolumn was calibrated with molecular mass standards(thyroglobulin, γ-globulin, ovalbumin, ribonuclease A,and N-acetyl tryptophanamide (NATA)).
Glutaraldehyde crosslinking
WT Aβ40 (5 μM) was incubated with increasingconcentrations of SUVs at RT for 10 min in 10 mM NaPi,pH 7.4. Peptide crosslinking was initiated by addition ofglutaraldehyde to 0.01%, and allowed to proceed for40 min at RT. Reactions were quenched with 100 mMglycine for 30 min at RT, then 4×SDS-PAGE loading buffer(250 mM Tris, pH 6.8, 8% (w/v) SDS, 40% (v/v) glycerol,4% (v/v) β-mercaptoethanol) was added, and sampleswere incubated at 100 °C for 5 min. Samples were sub-jected to electrophoresis in a 10%–20% (w/v) polyacry-lamide SDS Tris-Tricine Ready Gel (Bio-Rad, Hercules,CA) and immunoblotted onto a nitrocellulose membranewith a mouse monoclonal α-Aβ antibody (6E10) (AbcamInc., Cambridge, MA). Bound antibody was visualizedwith a horseradish peroxidase-conjugated α-mouseimmunoglobulin and ECL (Bio-Rad, Hercules, CA).
Liposome permeabilization
Liposomes containing 5(6)-carboxyfluorescein (CF)(Sigma-Aldrich, St. Louis, MO) were prepared by rehy-drating lipid films at a concentration of 5 mg/mL in10 mM NaPi, pH 7.4 containing 50 mM CF. Lipids wereincubated in a water-bath above the lipid meltingtemperature for 2 h. Rehydrated lipid suspensions werevortex mixed and subjected to at least eight freeze–thawcycles from liquid nitrogen to a 60 °C water-bath. To formLUVs, the lipid solution was extruded 21 times throughtwo stacked 100 nm pore-size polycarbonate Nucleporemembrane filters (Whatman, Florham Park, NJ). Free CFwas separated from liposomes containing CF by passageof the solution through a 10 mL G-50 Sephadex gel-filtration column (Sigma-Aldrich, St. Louis, MO) in 10 mMNaPi. For the permeabilization assay, freshly dissolved5 μM Aβ40 was incubated with increasing concentrationsof liposomes. In order to measure fluorescence accuratelyat high concentrations of liposome and avoid inner filtereffects due to high concentrations of CF, only 25 μM CFLUVs were included in each sample. Higher concentra-tions of lipid were attained by adding LUVs without CF.The CF fluorescence intensity (excitation at 490 nm,

94 Aβ Membrane Interactions Depend on Lipid Composition
emission at 514 nm) was measured at various time points.Dye leakage from liposomes alone was subtracted. Todetermine the fluorescence value for maximal dyeleakage, 0.2% Triton X-100 was added to the liposomesat the end of the time-course. Leakage is expressed as lipidpermeabilized as determined by dividing the fluorescencesignal by the fluorescence for 1 μM LUVs permeabilizedby treatment with Triton X-100.
Cholesterol incorporation
Cholesterol (Sigma-Aldrich, St. Louis, MO) dissolvedin chloroform was dried with POPC/PG at 0, 10%, or25% by mass. Liposomes were formed by rehydration at2.5 mg/mL at 25 °C in 10 mM NaPi, pH 7.4 and eithersonicated (SUVs) or extruded (LUVs) as described above.As cholesterol was increased, POPC/PG levels weredecreased in order to maintain constant total lipid(including cholesterol).
Acknowledgements
The authors thank Sudheendra Seetharamachar-aya for assistance with lipid methodology. Thiswork was supported by NIA grant R21 AG027370 toA.G., and NIH predoctoral fellowships T32GM008270 and T32 AG000114T32 to P.T.W.
References
1. Harper, J. D. & Lansbury, P. T., Jr (1997). Models ofamyloid seeding in Alzheimer's disease and scrapie:mechanistic truths and physiological consequences ofthe time-dependent solubility of amyloid proteins.Annu. Rev. Biochem. 66, 385–407.
2. Glenner, G. G. & Wong, C. W. (1984). Alzheimer'sdisease: initial report of the purification and cha-racterization of a novel cerebrovascular amyloidprotein. Biochem. Biophys. Res. Commun. 120,885–890.
3. Selkoe, D. J. (1991). The molecular pathology ofAlzheimer's disease. Neuron, 6, 487–498.
4. LaFerla, F. M. & Oddo, S. (2005). Alzheimer's disease:Abeta, tau and synaptic dysfunction. Trends Mol. Med.11, 170–176.
5. Lau, T. L., Ambroggio, E. E., Tew, D. J., Cappai,R., Masters, C. L., Fidelio, G. D. et al. (2006).Amyloid-beta peptide disruption of lipid mem-branes and the effect of metal ions. J. Mol. Biol.356, 759–770.
6. McLean, C. A., Cherny, R. A., Fraser, F. W., Fuller,S. J., Smith, M. J., Beyreuther, K. et al. (1999).Soluble pool of Abeta amyloid as a determinant ofseverity of neurodegeneration in Alzheimer's disease.Ann. Neurol. 46, 860–866.
7. Giedraitis, V., Sundelof, J., Irizarry, M. C., Garevik,N., Hyman, B. T., Wahlund, L. O. et al. (2007). Thenormal equilibrium between CSF and plasmaamyloid beta levels is disrupted in Alzheimer'sdisease. Neurosci. Lett. 427, 127–131.
8. Nilsberth, C., Westlind-Danielsson, A., Eckman, C.B.,Condron, M. M., Axelman, K., Forsell, C. et al. (2001).The ‘Arctic’ APP mutation (E693G) causes Alzhei-
mer's disease by enhanced Abeta protofibril forma-tion. Nature Neurosci. 4, 887–893.
9. Selkoe, D. J. (1999). Translating cell biology intotherapeutic advances in Alzheimer's disease. Nature,399, A23–A31.
10. Garzon-Rodriguez, W., Sepulveda-Becerra, M.,Milton, S. & Glabe, C. G. (1997). Soluble amyloidAbeta-(1-40) exists as a stable dimer at low con-centrations. J. Biol. Chem. 272, 21037–21044.
11. Garzon-Rodriguez, W., Vega, A., Sepulveda-Becerra,M., Milton, S., Johnson, D. A., Yatsimirsky, A. K. &Glabe, C. G. (2000). A conformation change in thecarboxyl terminus of Alzheimer's Abeta (1-40)accompanies the transition from dimer to fibril asrevealed by fluorescence quenching analysis. J. Biol.Chem. 275, 22645–22649.
12. Chen, Y. R. & Glabe, C. G. (2006). Distinct earlyfolding and aggregation properties of Alzheimeramyloid-β peptides Aβ40 and Aβ42: stable trimer ortetramer formation by Aβ42. J. Biol. Chem. 281,24414–24422.
13. Klein, W. L., Stine, W. B., Jr & Teplow, D. B. (2004).Small assemblies of unmodified amyloid beta-proteinare the proximate neurotoxin in Alzheimer's disease.Neurobiol. Aging, 25, 569–580.
14. Walsh, D. M., Hartley, D. M., Kusumoto, Y.,Fezoui, Y., Condron, M. M., Lomakin, A. et al.(1999). Amyloid beta-protein fibrillogenesis. Struc-ture and biological activity of protofibrillar inter-mediates. J. Biol. Chem, 274, 25945–25952.
15. Walsh, D. M., Lomakin, A., Benedek, G. B.,Condron, M. M. & Teplow, D. B. (1997).Amyloid beta-protein fibrillogenesis. Detection ofa protofibrillar intermediate. J. Biol. Chem. 272,22364–22372.
16. Walsh, D. M., Klyubin, I., Fadeeva, J. V., Cullen, W. K.,Anwyl, R., Wolfe, M. S. et al. (2002). Naturallysecreted oligomers of amyloid beta protein potentlyinhibit hippocampal long-term potentiation in vivo.Nature, 416, 535–539.
17. Gong, Y., Chang, L., Viola, K. L., Lacor, P. N.,Lambert, M. P., Finch, C. E. et al. (2003). Alzheimer'sdisease-affected brain: presence of oligomeric A betaligands (ADDLs) suggests a molecular basis forreversible memory loss. Proc. Natl Acad. Sci USA,100, 10417–10422.
18. Klein, W. L., Krafft, G. A. & Finch, C. E. (2001).Targeting small Abeta oligomers: the solution to anAlzheimer's disease conundrum? Trends Neurosci. 24,219; Proc. Natl Acad. Sci USA, 224.
19. Mucke, L., Masliah, E., Yu, G. Q., Mallory, M.,Rockenstein, E. M., Tatsuno, G. et al. (2000). High-level neuronal expression of abeta 1-42 in wild-typehuman amyloid protein precursor transgenic mice:synaptotoxicity without plaque formation. J. Neurosci.20, 4050–4058.
20. Lesne, S., Koh, M. T., Kotilinek, L., Kayed, R.,Glabe, C. G., Yang, A. et al. (2006). A specificamyloid-beta protein assembly in the brain impairsmemory. Nature, 440, 352–357.
21. Townsend, M., Shankar, G. M., Mehta, T., Walsh,D. M. & Selkoe, D. J. (2006). Effects of secretedoligomers of amyloid beta-protein on hippocam-pal synaptic plasticity: a potent role for trimers.J. Physiol. 572, 477–492.
22. Kawarabayashi, T., Shoji, M., Younkin, L. H.,Wen-Lang, L., Dickson, D. W., Murakami, T. et al.(2004). Dimeric amyloid beta protein rapidly accumu-lates in lipid rafts followed by apolipoprotein E and

95Aβ Membrane Interactions Depend on Lipid Composition
phosphorylated tau accumulation in the Tg2576mouse model of Alzheimer's disease. J. Neurosci. 24,3801–3809.
23. Haass, C. & Selkoe, D. J. (2007). Soluble proteinoligomers in neurodegeneration: lessons from theAlzheimer's amyloid beta-peptide. Nature Rev. Mol.Cell Biol. 8, 101–112.
24. Hertel, C., Terzi, E., Hauser, N., Jakob-Rotne, R.,Seelig, J. & Kemp, J. A. (1997). Inhibition of theelectrostatic interaction between beta-amyloid pep-tide and membranes prevents beta-amyloid-inducedtoxicity. Proc. Natl Acad. Sci. USA, 94, 9412–9416.
25. Kourie, J. I., Culverson, A. L., Farrelly, P. V., Henry,C. L. & Laohachai, K. N. (2002). Heterogeneousamyloid-formed ion channels as a common cyto-toxic mechanism: implications for therapeutic stra-tegies against amyloidosis. Cell Biochem. Biophys. 36,191–207.
26. Kayed, R., Sokolov, Y., Edmonds, B., McIntire, T. M.,Milton, S. C., Hall, J. E. & Glabe, C. G. (2004).Permeabilization of lipid bilayers is a commonconformation-dependent activity of soluble amyloidoligomers in protein misfolding diseases. J. Biol. Chem.279, 46363–46366.
27. Fagan, T., Kagan, B., Corbin, D., Hwang, W., Glabe,C., Nault, L. et al. (2006). Alzheimer Research ForumLive Discussion: Now you see them, now you don't:The amyloid channel hypothesis. J. Alzheimers Dis. 9,219–224.
28. Lashuel, H. A. & Lansbury, P. T., Jr (2006). Areamyloid diseases caused by protein aggregates thatmimic bacterial pore-forming toxins? Quart. Rev.Biophys. 39, 167–201.
29. Lin, H., Bhatia, R. & Lal, R. (2001). Amyloid β proteinforms ion channels: implications for Alzheimer'sdisease pathophysiology. FASEB J. 15, 2433–2444.
30. Simakova, O. & Arispe, N. J. (2006). Early and latecytotoxic effects of external application of the Alzhei-mer's Abeta result from the initial formation andfunction of Abeta ion channels. Biochemistry, 45,5907–5915.
31. Kawahara, M., Kuroda, Y., Arispe, N. & Rojas, E.(2000). Alzheimer's beta-amyloid, human isletamylin, and prion protein fragment evoke intra-cellular free calcium elevations by a commonmechanism in a hypothalamic GnRH neuronalcell line. J. Biol. Chem. 275, 14077–14083.
32. Demuro, A., Mina, E., Kayed, R., Milton, S. C.,Parker, I. & Glabe, C. G. (2005). Calcium dysregula-tion and membrane disruption as a ubiquitousneurotoxic mechanism of soluble amyloid oligomers.J. Biol. Chem. 280, 17294–17300.
33. Arispe, N., Rojas, E. & Pollard, H. B. (1993).Alzheimer disease amyloid beta protein forms cal-cium channels in bilayer membranes: blockade bytromethamine and aluminum. Proc. Natl Acad. Sci.USA, 90, 567–571.
34. Arispe, N., Pollard, H. B. & Rojas, E. (1993). Giantmultilevel cation channels formed by Alzheimerdisease amyloid beta-protein [A beta P-(1-40)] inbilayer membranes. Proc. Natl Acad. Sci. USA, 90,10573–10577.
35. Quist, A., Doudevski, I., Lin, H., Azimova, R., Ng,D., Frangione, B. et al. (2005). Amyloid ionchannels: a common structural link for protein-misfolding disease. Proc. Natl Acad. Sci. USA, 102,10427–10432.
36. Arispe, N., Pollard, H. B. & Rojas, E. (1996). Zn2+interaction with Alzheimer amyloid beta protein
calcium channels. Proc. Natl Acad. Sci. USA, 93,1710–1715.
37. Dong, Z., Saikumar, P., Weinberg, J. M. & Venkata-chalam, M. A. (2006). Calcium in cell injury and death.Annu. Rev. Pathol. 1, 405–434.
38. Selkoe, D. J. (2006). The ups and downs of Abeta.Nature Med. 12, 758–759.
39. Thal, D. R., Capetillo-Zarate, E., Del Tredici, K. &Braak, H. (2006). The development of amyloid betaprotein deposits in the aged brain. Sci. AgingKnowledgeEnviron. 2006, re1.
40. Terzi, E., Holzemann, G. & Seelig, J. (1995). Self-association of beta-amyloid peptide (1-40) in solu-tion and binding to lipid membranes. J. Mol. Biol.252, 633–642.
41. Terzi, E., Holzemann, G. & Seelig, J. (1997). Interac-tion of Alzheimer beta-amyloid peptide(1-40) withlipid membranes. Biochemistry, 36, 14845–14852.
42. Bokvist, M., Lindstrom, F., Watts, A. & Grobner, G.(2004). Two types of Alzheimer's beta-amyloid (1-40)peptide membrane interactions: aggregation prevent-ing transmembrane anchoring versus acceleratedsurface fibril formation. J. Mol. Biol. 335, 1039–1049.
43. McLaurin, J. & Chakrabartty, A. (1996). Membranedisruption by Alzheimer beta-amyloid peptidesmediated through specific binding to either phospho-lipids or gangliosides. Implications for neurotoxicity.J. Biol. Chem. 271, 26482–26489.
44. McLaurin, J. & Chakrabartty, A. (1997). Characteriza-tion of the interactions of Alzheimer beta-amyloidpeptides with phospholipid membranes. Eur. J.Biochem. 245, 355–363.
45. Alarcon, J. M., Brito, J. A., Hermosilla, T., Atwater, I.,Mears, D. & Rojas, E. (2006). Ion channel formation byAlzheimer's disease amyloid beta-peptide (Abeta40)in unilamellar liposomes is determined by anionicphospholipids. Peptides, 27, 95–104.
46. Rogaeva, E., Meng, Y., Lee, J. H., Gu, Y., Kawarai, T.,Zou, F. et al. (2007). The neuronal sortilin-relatedreceptor SORL1 is genetically associated with Alzhei-mer disease. Nature Genet. 39, 168–177.
47. Ladokhin, A. S. & White, S. H. (1999). Folding ofamphipathic alpha-helices on membranes: energeticsof helix formation by melittin. J. Mol. Biol. 285,1363–1369.
48. Vogel, H. (1981). Incorporation of melittin intophosphatidylcholine bilayers. Study of binding andconformational changes. FEBS Lett. 134, 37–42.
49. Huang, H. W. (2006). Molecular mechanism ofantimicrobial peptides: the origin of cooperativity.Biochim. Biophys. Acta, 1758, 1292–1302.
50. Wenk, M. R. & Seelig, J. (1998). Magainin 2 amideinteraction with lipid membranes: calorimetricdetection of peptide binding and pore formation.Biochemistry, 37, 3909–3916.
51. Zitzer, A., Palmer, M., Weller, U., Wassenaar, T.,Biermann, C., Tranum-Jensen, J. & Bhakdi, S. (1997).Mode of primary binding to target membranes andpore formation induced by Vibrio cholerae cytolysin(hemolysin). Eur. J. Biochem, 247, 209–216.
52. Huang, H.W. (2000). Action of antimicrobial peptides:two-state model. Biochemistry, 39, 8347–8352.
53. Terzi, E., Holzemann, G. & Seelig, J. (1994).Alzheimer beta-amyloid peptide 25-35: electro-static interactions with phospholipid membranes.Biochemistry, 33, 7434–7441.
54. Del Mar Martinez-Senac, M., Villalain, J. & Gomez-Fernandez, J. C. (1999). Structure of the Alzheimerbeta-amyloid peptide (25-35) and its interaction with

96 Aβ Membrane Interactions Depend on Lipid Composition
negatively charged phospholipid vesicles. Eur. J.Biochem. 265, 744–753.
55. Lal, R., Lin, H. & Quist, A. P. (2007). Amyloidbeta ion channel: 3D structure and relevance toamyloid channel paradigm. Biochim. Biophys. Acta,1768, 1966–1975.
56. Lin, M. C. & Kagan, B. L. (2002). Electrophysiologicproperties of channels induced by Abeta25-35 inplanar lipid bilayers. Peptides, 23, 1215–1228.
57. Simakova, O. & Arispe, N. J. (2007). The cell-selectiveneurotoxicity of the Alzheimer's Abeta peptide isdetermined by surface phosphatidylserine and cyto-solic ATP levels. Membrane binding is required forAbeta toxicity. J. Neurosci. 27, 13719–13729.
58. Breukink, E., van Kraaij, C., van Dalen, A., Demel,R. A., Siezen, R. J., de Kruijff, B. & Kuipers, O. P.(1998). The orientation of nisin in membranes.Biochemistry, 37, 8153–8162.
59. Blondelle, S. E., Lohner, K. & Aguilar, M. (1999).Lipid-induced conformation and lipid-binding pro-perties of cytolytic and antimicrobial peptides:determination and biological specificity. Biochim.Biophys. Acta, 1462, 89–108.
60. Choo-Smith, L. P., Garzon-Rodriguez, W., Glabe, C. G.& Surewicz, W. K. (1997). Acceleration of amyloidfibril formation by specific binding of Abeta-(1-40)peptide to ganglioside-containing membrane vesicles.J. Biol. Chem. 272, 22987–22990.
61. Ege, C. & Lee, K. Y. (2004). Insertion of Alzheimer's Abeta 40 peptide into lipid monolayers. Biophys. J. 87,1732–1740.
62. Devanathan, S., Salamon, Z., Lindblom, G., Grobner,G. & Tollin, G. (2006). Effects of sphingomyelin,cholesterol and zinc ions on the binding, insertionand aggregation of the amyloid Abeta(1-40) peptide insolid-supported lipid bilayers. FEBS J. 273, 1389–1402.
63. Zhao, H., Sood, R., Jutila, A., Bose, S., Fimland, G.,Nissen-Meyer, J. & Kinnunen, P. K. (2006). Interac-tion of the antimicrobial peptide pheromone Plan-taricin A with model membranes: implications for anovel mechanism of action. Biochim. Biophys. Acta,1758, 1461–1474.
64. Hirakura, Y., Lin, M. C. & Kagan, B. L. (1999).Alzheimer amyloid abeta1-42 channels: effects of
solvent, pH, and Congo Red. J. Neurosci. Res. 57,458–466.
65. Ohvo-Rekila, H., Ramstedt, B., Leppimaki, P. &Slotte, J. P. (2002). Cholesterol interactions withphospholipids in membranes. Progr. Lipid Res. 41,66–97.
66. Allende, D., Simon, S. A. & McIntosh, T. J. (2005).Melittin-induced bilayer leakage depends on lipidmaterial properties: evidence for toroidal pores.Biophys. J. 88, 1828–1837.
67. Sanderson, J. M. (2005). Peptide-lipid interactions:insights and perspectives. Org. Biomol. Chem. 3,201–212.
68. Balasubramanian, K., Mirnikjoo, B. & Schroit, A. J.(2007). Regulated externalization of phosphatidyl-serine at the cell surface: implications for apoptosis.J. Biol. Chem. 282, 18357–18364.
69. Thelen, K. M., Falkai, P., Bayer, T. A. & Lutjohann,D. (2006). Cholesterol synthesis rate in humanhippocampus declines with aging. Neurosci. Lett.403, 15–19.
70. Wood, W. G., Schroeder, F., Igbavboa, U., Avdulov,N.A. & Chochina, S. V. (2002). Brain membranecholesterol domains, aging and amyloid beta-pep-tides. Neurobiol. Aging, 23, 685–694.
71. Teplow, D. B. (2006). Preparation of amyloid beta-protein for structural and functional studies. MethodsEnzymol. 413, 20–33.
72. Fezoui, Y., Hartley, D. M., Harper, J. D., Khurana, R.,Walsh, D. M., Condron, M. M. et al. (2000). Animproved method of preparing the amyloid beta-protein for fibrillogenesis and neurotoxicity experi-ments. Amyloid, 7, 166–178.
73. Charles, J. & Stewart, M. (1980). Colorimetric deter-mination of phospholipids with ammonium ferrothio-cyanate. Anal. Biochem. 104, 10–14.
74. LeVine, H., III (1999). Quantification of beta-sheetamyloid fibril structures with thioflavin T. MethodsEnzymol. 309, 274–284.
75. Sreerama, N. & Woody, R. W. (2000). Estimation ofprotein secondary structure from circular dichroismspectra: comparison of CONTIN, SELCON, andCDSSTR methods with an expanded reference set.Anal. Biochem. 287, 252–260.




















