
Nicotine withdrawal alleviates acetic acid-induced gastric injury in rats

ORIGINAL PAPER
Alpha Lipoic Acid Alleviates Oxidative Stress and PreservesBlood Brain Permeability in Rats with Subarachnoid Hemorrhage
Mehmet Ersahin • Hale Z. Toklu • Sule Cetinel •
Meral Yuksel • Can Erzik • M. Zafer Berkman •
Berrak C. Yegen • Goksel Sener
Accepted: 22 September 2009 / Published online: 13 October 2009
� Springer Science+Business Media, LLC 2009
Abstract The neuroprotective effect of alpha lipoic acid
(ALA; 100 mg/kg, po), a dithiol antioxidant, on experi-
mentally induced subarachnoid hemorrhage (SAH) was
assessed in Wistar albino rats. Neurological examination
scores recorded at the 48th h of SAH induction were
increased in SAH groups, which were accompanied with
significant increases in the formation of reactive oxygen
species, DNA fragmentation ratios, malondialdehyde levels
and myeloperoxidase activity, while significant decreases
in the brain glutathione content and Na?, K?-ATPase
activity were observed. On the other hand, ALA treatment
reversed all these biochemical indices as well as SAH-
induced histopathological alterations. Increased brain edema,
impaired blood-brain-barrier permeability and neurological
scores were also improved by ALA treatment. The results
demonstrate that ALA exerts neuroprotective effects via
the enhancement of endogenous antioxidant enzyme activity,
the inhibition of neutrophil accumulation and free radical
generation, suggesting a therapeutic potential in reducing
secondary injury after SAH in patients.
Keywords Alpha lipoic acid � Antioxidant �Cerebral vasospasm � Subarachnoid hemorrhage �Lipid peroxidation
Introduction
Despite sophisticated medical management and neurosur-
gical techniques, subarachnoid hemorrhage (SAH) from
ruptured intracranial aneurysm is associated with high
morbidity and mortality rates. Experimental and clinical
studies have shown that the acute symptoms of SAH are
attributed to increased intracranial pressure, decreased
cerebral perfusion pressure and the resulting ischemia [1,
2]. In the clinical setting, most deaths occur within hours
after onset of SAH [3], and those patients who survive the
initial hemorrhage and overcome vasospasms frequently
suffer from persistent cognitive deficits, psychosocial
impairments, and a decreased quality of life because of
acute brain injury [4].
A growing body of evidence suggests that the potential
vasoconstrictors released from activated platelets, as well as
from mechanically damaged erythrocytes, participate in the
acute vasospastic/ischemic response following SAH [5].
Experimental studies indicate that the generation of reactive
oxygen species (ROS) is one of the mechanisms playing a
crucial role in the acute brain injury which leads to the
delayed cerebral vasospasm following SAH [6–8] and that
M. Ersahin � M. Z. Berkman
Department of Neurosurgery, Haydarpasa Numune Education
and Research Hospital, Istanbul, Turkey
H. Z. Toklu � G. Sener (&)
Department of Pharmacology, School of Pharmacy, Marmara
University, Tıbbiye Cad., 34668 Istanbul, Turkey
e-mail: [email protected]; [email protected]
S. Cetinel
Department of Histology & Embryology, School of Medicine,
Marmara University, Istanbul, Turkey
M. Yuksel
Vocational School of Health Related Professions, Marmara
University, Istanbul, Turkey
C. Erzik
Department of Medical Biology, School of Medicine, Marmara
University, Istanbul, Turkey
B. C. Yegen
Department of Physiology, School of Medicine, Marmara
University, Istanbul, Turkey
123
Neurochem Res (2010) 35:418–428
DOI 10.1007/s11064-009-0072-z

these radicals are casually related to the expression of cere-
bral ischemia [9, 10]. Thus, the use of free radical scavengers
in animal models of SAH [11, 12] and some clinical trials
[13, 14] have demonstrated beneficial effects of these agents
on ischemic neurological deficits due to SAH-induced
vasospasms. We have recently shown that melatonin, which
is a potent free radical scavenger, reversed SAH-induced
histopathological and biochemical alterations [15].
Alpha lipoic (ALA) acid, a dithiol antioxidant, is an
important co-factor in pyruvate dehydrogenase and
a-ketoglutarate dehydrogenase in the mitochondria [16].
The metabolic antioxidant lipoic acid is a low molecular
weight substance that is absorbed from the diet and crosses
the blood-brain barrier. Although the metabolic role of
ALA has been known for more than 50 years, its effects
when given exogenously have become known recently.
Several studies have provided evidence that ALA supple-
mentation decreases oxidative stress and restores reduced
levels of other antioxidants in vivo [17]. ALA influences a
number of cell processes, including direct radical scav-
enging, recycling of other antioxidants, accelerating glu-
tathione (GSH) synthesis, and modulating transcription
factor activity, especially that of nuclear factor (NF)-kappa
B, which may account for its dramatic effects in oxidative
stress conditions [18].
Several studies have shown that ALA exerts multiple
pharmacological actions that prevent nerve degeneration in
experimental in vitro models of diabetes [19], Parkinson
[20], and Alzheimer diseases [21]. Similarly, ALA has
been shown to exert a neuroprotective effect in several in
vivo models of neurologic injury, such as traumatic brain
injury [22], cerebral ischemia [23], seizures [24] and
autoimmune encephalomyelitis [25].
In the light of these findings, the aim of the current study
is to determine the effects of alpha lipoic acid on SAH-
induced oxidative brain injury and neurological symptoms
using biochemical, neurological and histopathological
approaches.
Materials and Methods
Animals and Experimental Design
All experimental protocols were approved by the Marmara
University Animal Care and Use Committee. Male Wistar
albino rats (300–350 g) were housed in a temperature-
controlled room (22 ± 2�C) with standardized light/dark
(12/12 h) cycles, where the relative humidity (65–70%)
was kept constant. Rats were fed with standard rat pellets
and tap water ad libitum.
Rats were divided into three groups as: (1) sham-
operated and vehicle-treated control group (n = 18), (2)
vehicle-treated SAH group (n = 18), (3) ALA (100 mg/kg/
day, po)-treated SAH group (n = 18). The active ingredi-
ent ALA (99% purity, (R)-a-lipoic acid) was supplied by
Mikrogen Pharmaceuticals (Istanbul, Turkey) and it was
dissolved in 10% DMSO (v/v in corn oil). Either ALA or
DMSO was administered through an orogastric tube
immediately after the induction of SAH and was continued
daily for 2 days.
At the 48th h of surgery, neurological examinations
were performed in all groups. Then, subgroups of the rats
in each group were decapitated to obtain brain tissue
samples for the biochemical analyses (n = 6), for Evans
blue assay and edema evaluation (n = 6). The remaining
rats (n = 6) in each group were given the fixative perfusion
for histological preparation and analysis.
Induction of SAH
In the present study, the rat basilar artery vasospasm model
was used according as previously described [26]. In the
anesthetized rats injected intraperitoneally (ip) with keta-
mine (100 mg/kg) and chlorpromazine (2 mg/kg), a small
incision was made in the area of the occipitocervical
junction to expose the atlanto-occipital membrane. After
the animals were placed on the stereotaxic frame, the cis-
terna magna was tapped with a 27-gauge needle, and
0.3 ml of cerebral spinal fluid was gently aspirated. Freshly
drawn blood (0.3 ml) taken from the femoral artery was
then injected aseptically into the cisterna magna within a
2-min period. Immediately after the injection of blood, the
puncture site was sealed with glue to prevent the formation
of a fistula. To permit blood distribution around the basal
arteries, each rat was tilted at an angle of 20� for 30 min
with its head lowered.
Neurological Examination
Since functional scoring is important in testing neuropro-
tective drugs, a simple set of commonly used neurological
tests were used to assess the normal and abnormal function
following SAH in rats. The neurological examination scores
were conducted according to Bederson’s modified neuro-
logical examination test [22, 27] by a ‘blinded’ investigator.
A twenty-point neuroscore was used to assess motor and
behavioral deficits. The sequence of testing animals by a
given task was randomized for the animals. Briefly, the
consciousness, performance in a smooth climbing platform,
extremity tonus, walking and postural reflexes, circling and
response to the nociceptive stimuli were assessed. For
walking and posture, rats were allowed to move about freely
on the floor, while they were observed. In the circling test,
the rats were held gently from the tail, suspended one meter
above the floor, and observed for forelimb flexion, where
Neurochem Res (2010) 35:418–428 419
123

normal rats are expected to extend both forelimbs toward
the floor. The rotation degree and time were measured.
Finally, the responses to the nociceptive stimuli were
assessed by tail-immersion test in 56�C water.
Evans Blue Assay for the Evaluation of Blood Brain
Barrier Permeability
To evaluate the blood brain barrier (BBB) integrity, Evans
blue dye (EB) was used as a marker of albumin extrava-
sation [22]. Briefly, EB (2% in saline, 4 ml/kg) was
injected via the jugular vein at the 48th h of the SAH
induction and it was allowed to remain in circulation for
30 min. Then, chests were opened and the rats were per-
fused transcardially with 250 ml of saline at a pressure of
110 mm Hg for approximately 15 min. After decapitation,
the brain was removed and dissected into cerebral cortex
and cerebellum, which were then weighed separately for
the quantitative measurement of EB-albumin extravasation.
Brain samples were homogenized in 2.5 ml phosphate-
buffered saline and mixed by vortexing for 2 min after the
addition of 2.5 ml of 60% trichloroacetic acid to precipitate
the protein. Samples were cooled and then centrifuged for
30 min at 1000g. The supernatant was measured at 620 nm
for the absorbance of EB using a spectrophotometer
(Shimadzu UV1208, Japan). EB was expressed as lg/mg of
brain tissue against a standard curve.
Evaluation of the Brain Edema
Brain edema was evaluated by the gravimetric method
based on the measurement of the water content of brain
[22]. The whole brain was weighed and then dried for
48 h at 100�C, afterwards re-weighed. The percentage of
water was calculated according to the following formula:
% H2O = [(wet weight - dry weight)/wet weight] 9 100.
Chemiluminescence (CL) Assay
To assess the role of reactive oxygen species in SAH-
induced brain damage, luminol and lucigenin chemilumi-
nescences were measured as indicators of radical forma-
tion. Lucigenin (bis-N-methylacridiniumnitrate) and
luminol (5-amino-2,3-dihydro-1,4-phthalazinedione) were
obtained from Sigma (St Louis, MO, USA). Measurements
were made at room temperature using Junior LB 9509
luminometer (EG&G Berthold, Germany). Tissue samples
cut into small pieces were put into vials containing PBS-
HEPES buffer (0.5 M PBS containing 20 mM HEPES, pH
7.2). ROM were quantitated after the addition of the
enhancers, lucigenin or luminol, for a final concentration
of 0.2 mM. Luminol detects a group of reactive species,
such as •OH, H2O2, HOCl radicals, while lucigenin is
selective for O2-. Counts were obtained at 1 min intervals
and the results were given as the area under curve (AUC)
for a counting period of 5 min. Counts was corrected for
wet tissue weight and expressed as relative light units (rlu)
per milligram of tissue [28].
DNA Fragmentation Assay
The percentage of DNA fragmentation in the brain tissue
was determined as an indicator of cell death, including
apoptosis. Brain tissue samples were homogenized in 10
volumes of a lysis buffer (5 mM Tris–HCL, 20 mM ethyl-
ene diamine tetraacetic acid [EDTA], 0.5% (v/v) t-octyl-
phenoxypolyethoxyethanol [Triton-X 100]; pH = 8.0).
Two separate samples of 1 ml were taken from the mucosal
samples and centrifuged at 25,000g for 30 min to separate
the intact chromatin in the pellet from the fragmented DNA
in the supernatant [29]. The supernatant was taken out to be
saved and the pellet was re-suspended in 1 ml of Tri-EDTA
buffer (pH = 8.0), 10 mM:1 mM, respectively. Both the
supernatant and the re-suspended pellet were assayed then
for the DNA content by diphenylamine reaction described
by Burton [30].
Malondialdehyde and Glutathione Assays
Brain samples were homogenized with ice-cold 150 mM
KCl for the determination of malondialdehyde (MDA) and
glutathione (GSH) levels, indicating lipid peroxidation and
intracellular antioxidant status, respectively. The MDA
levels were assayed for the products of lipid peroxidation
and results are expressed as nmol MDA/g tissue [31]. GSH
was determined by a spectrophotometric method based on
the use of Ellman’s reagent and results are expressed as
lmol GSH/g tissue [32].
Measurement of Myeloperoxidase Activity
Myeloperoxidase (MPO) activity, which is accepted as an
indicator of neutrophil infiltration in tissues, was measured
by a procedure similar to that described by Hillegas et al.
[33]. Brain tissue samples were homogenized in 50 mM
potassium phosphate buffer with a pH of 6.0, and centrifuged
at 41,400g for 10 min. The pellets were then suspended in
50 mM PB containing 0.5% hexadecyltrimethylammonium
bromide (HETAB). After three freeze and thaw-cycles, with
sonication between cycles, the samples were centrifuged at
41,400g for 10 min. Aliquots (0.3 ml) were added to 2.3 ml
of reaction mixture containing 50 mM PB, o-dianisidine,
and 20 mM H2O2 solution. One unit of enzyme activity was
defined as the amount of MPO present that caused a change
in absorbance, measured at 460 nm for 3 min. MPO activity
was expressed as U/g tissue.
420 Neurochem Res (2010) 35:418–428
123

Na?, K?-ATPase Activity
Since the activity of Na?, K?-ATPase, a membrane-
bound enzyme required for cellular transport, is very
sensitive to free radical reactions and lipid peroxidation,
reductions in this activity can indicate membrane damage
indirectly. Measurement of Na?, K?-ATPase activity is
based on the measurement of inorganic phosphate
released by ATP hydrolysis during incubation of brain
homogenates with an appropriate medium containing
3 mM ATP as a substrate. The total ATPase activity was
determined in the presence of 100 mM NaCl, 5 mM KCl,
6 mM MgCl2, 0.1 mM EDTA, 30 mM Tris–HCl (pH
7.4), while the Mg2?-ATPase activity was determined in
the presence of 1 mM ouabain. The difference between
the total and the Mg2?-ATPase activities was taken as a
measure of the Na?, K?-ATPase activity [34]. The
reaction was initiated with the addition of the homogenate
(0.1 ml) and a 5-min preincubation period at 37�C was
allowed. Following the addition of Na2ATP and a 10-min
re-incubation period, the reaction was terminated by the
addition of ice-cold 6% perchloric acid. The mixture was
then centrifuged at 3500g, and Pi in the supernatant
fraction was determined by the method of Fiske and
Subarrow [35]. The specific activity of the enzyme was
expressed as nmol Pi mg-1 protein h-1. The protein
concentration of the supernatant was measured by the
Lowry method [36].
Histopathological Preparation and Analysis
Anesthetized (ip ketamine and chlorpromazine) rats were
perfused transcardially with a solution of 2.5% glutaral-
dehyde in 0.1 M PBS (pH 7.4). To obtain basilar artery
sections, anterior midline of the brainstem was removed.
For light microscopic analysis, perfused tissue specimens
were fixed in 10% formaldehyde, dehydrated in alcohol
series, clearing in toluene end embedding in paraffin.
Paraffin sections (5 lm) were stained with hematoxylin
and eosin (H&E) and examined under a photomicroscope
(Olympus BH 2, Tokyo, Japan).
For the electron microscopic evaluation, tissues
obtained from perfused rats were post-fixed with 1% OsO4,
dehydrated in routine alcohol series and embedded in
Epon-812 resin. Semi-thin (1 lm) sections, stained with
toluidine blue, and thin (60 nm) sections, stained with 1%
lead citrate and uranyl acetate, were examined under a
JEOL 5200 JSM (Japan) electron microscope.
Statistical Analysis
Statistical analysis was done using a GraphPad Prism 3.0
(GraphPad Software, San Diego; CA; USA). All data are
expressed as means ± SEM. Groups of data were com-
pared with an analysis of variance (ANOVA) followed by
Tukey’s multiple comparison tests. Values of P \ 0.05
were considered as significant.
Results
Neurological examination score recorded at the 48th h of
surgery was significantly higher in the vehicle-treated SAH
group when compared with the control group (P \ 0.001),
while the score was significantly reduced in the ALA-
treated SAH group (P \ 0.05; Table 1). Similarly, brain
water content and EB content were also significantly higher
than the control group, indicating brain edema and BBB
permeability (P \ 0.05 and P \ 0.001). On the other hand,
ALA treatment reduced both parameters.
Luminol and lucigenin CL in the brain tissues of the
vehicle-treated SAH group were significantly higher than
those of the control group (P \ 0.01 and P \ 0.05), indi-
cating enhanced generation of ROS in the tissue while
these increases were significantly suppressed by ALA
treatment (P \ 0.05; Fig. 1).
In the vehicle-treated SAH group, as compared with the
control group, the MDA level in the brain samples showed
a marked increase, indicating enhanced lipid peroxidation
(P \ 0.001), while ALA administration abolished the ele-
vation in the brain MDA level (Fig. 2a). In accordance
with that, SAH caused a significant depletion in brain GSH
Table 1 Neurological examination scores, brain water content, Evans blue (EB) content, luminol and lucigenin chemiluminescence (CL) values
in the brain tissues of rats in the vehicle- or ALA-treated sham-operated control (C) and subarachnoid hemorrhage (SAH) groups
C SAH
Vehicle-treated ALA-treated Vehicle-treated ALA-treated
Neurological score 1.1 ± 0.9 1.3 ± 0.3 8.33 ± 3.5*** 3.50 ± 2.8?
Brain water content (%) 77.5 ± 0.4 75.9 ± 0.3 81.0 ± 1.4* 78.7 ± 0.5
EB content (mg/g tissue) 2.0 ± 0.1 2.1 ± 0.2 3.0 ± 0.2*** 2.5 ± 0.1?
Each group consists of six rats
* P \ 0.05, *** P \ 0.001, compared to control group; ? P \ 0.05, compared to vehicle-treated SAH group
Neurochem Res (2010) 35:418–428 421
123

content when compared to control group (P \ 0.01), while
in the ALA-treated SAH group, brain GSH content was
found to be preserved (Fig. 2b).
In the brain samples of the vehicle-treated SAH group,
DNA fragmentation was significantly increased as com-
pared to the control group (P \ 0.001; Fig. 2c), while ALA
treatment abolished the SAH-induced increase in DNA
fragmentation.
Myeloperoxidase activity was significantly elevated in
the brain tissues of the vehicle-treated SAH group as
compared to that of the control group (P \ 0.001; Fig. 3a).
However, ALA treatment reversed the SAH induced
increase in brain MPO level to normal. On the other hand,
in the vehicle-treated SAH group Na?, K?-ATPase activity
was significantly reduced, implicating an impaired mem-
brane transport function, while the measured Na?, K?-
ATPase activity in the brain tissue of the ALA-treated SAH
group was not different than that of the control rats
(Fig. 3b).
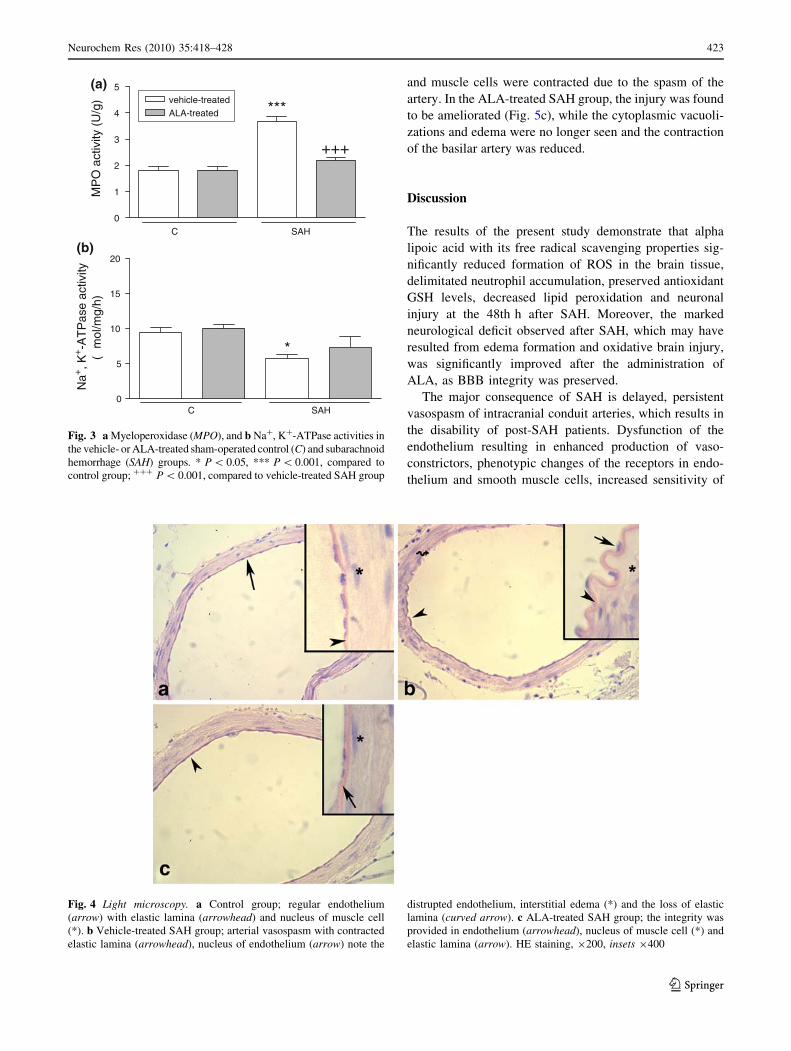
Light microscopic evaluation revealed that basilar
arteries obtained from sham-operated control rats had a
regular alignment with endothelial cells and elastic lamina
(Fig. 4a), while in the vehicle-treated SAH group a
prominent vasospasm of the basilar arteries was clearly
demonstrated, and the continuity of both endothelium and
elastic lamina was distrupted, indicating a severe degen-
eration (Fig. 4b). In the basilar arteries of the rats treated
with ALA, arterial vasospasm was decreased providing the
integrity of the endothelium (Fig. 4c).
On transmission electron microscopic analysis, compared
to control (Fig. 5a) group, SAH injury in the vehicle-treated
animals (Fig. 5b) was observed as severe degeneration in
the endothelium, elastic lamina and connective tissue of the
basilar artery with intense edema and vacuolizations in the
cytoplasm of both endothelial and muscle cells. Elastic
lamina showed detachments and has lost its continuity in
most of the observed areas. The nuclei of both endothelial
0
10
20
30
**
+
C SAH
ALA-treated
vehicle-treated(a)
Lum
inol
CL
(rlu
/mg)
0
5
10
15
20
25 *(b)
C SAH
+
Luci
geni
n C
L (r
lu/m
g)
Fig. 1 a Luminol and b lucigenin chemiluminescence (CL) levels in
the vehicle- or ALA-treated sham-operated control (C) and subarach-
noid hemorrhage (SAH) groups. Each group consists of six rats.
Values are represented as mean ± SEM. * P \ 0.05, ** P \ 0.01,
compared to control group; ? P \ 0.05, compared to vehicle-treated
SAH group
0
10
20
30
40
50
60 ***
+++
(a)
C SAH
ALA-treated
vehicle-treated
MD
A (
nmol
/g)
0.0
0.5
1.0
1.5
2.0
**+
(b)
GS
H (
µmol
/g)
0.0
0.1
0.2 ***
++
(c)
DN
A fr
agm
enta
tion
%C SAH
C SAH
Fig. 2 a Malondialdehyde (MDA), b glutathione (GSH) levels, and
c DNA fragmentation ratio (%) in the vehicle- or ALA-treated sham-
operated control (C) and subarachnoid hemorrhage (SAH) groups.
Each group consists of six rats. Values are represented as mean ±
SEM. ** P \ 0.01, *** P \ 0.001, compared to control group;? P \ 0.05, ?? P \ 0.01, ??? P \ 0.001, compared to vehicle-
treated SAH group
422 Neurochem Res (2010) 35:418–428
123
and muscle cells were contracted due to the spasm of the
artery. In the ALA-treated SAH group, the injury was found
to be ameliorated (Fig. 5c), while the cytoplasmic vacuoli-
zations and edema were no longer seen and the contraction
of the basilar artery was reduced.
Discussion
The results of the present study demonstrate that alpha
lipoic acid with its free radical scavenging properties sig-
nificantly reduced formation of ROS in the brain tissue,
delimitated neutrophil accumulation, preserved antioxidant
GSH levels, decreased lipid peroxidation and neuronal
injury at the 48th h after SAH. Moreover, the marked
neurological deficit observed after SAH, which may have
resulted from edema formation and oxidative brain injury,
was significantly improved after the administration of
ALA, as BBB integrity was preserved.
The major consequence of SAH is delayed, persistent
vasospasm of intracranial conduit arteries, which results in
the disability of post-SAH patients. Dysfunction of the
endothelium resulting in enhanced production of vaso-
constrictors, phenotypic changes of the receptors in endo-
thelium and smooth muscle cells, increased sensitivity of
0
1
2
3
4
5
***
+++
(a)
ALA-treatedvehicle-treated
C SAH
MP
O a
ctiv
ity (
U/g
)
0
5
10
15
20(b)
*
C SAH
Na+
, K+-A
TP
ase
activ
ity( µ
mol
/mg/
h)
Fig. 3 a Myeloperoxidase (MPO), and b Na?, K?-ATPase activities in
the vehicle- or ALA-treated sham-operated control (C) and subarachnoid
hemorrhage (SAH) groups. * P \ 0.05, *** P \ 0.001, compared to
control group; ??? P \ 0.001, compared to vehicle-treated SAH group
Fig. 4 Light microscopy. a Control group; regular endothelium
(arrow) with elastic lamina (arrowhead) and nucleus of muscle cell
(*). b Vehicle-treated SAH group; arterial vasospasm with contracted
elastic lamina (arrowhead), nucleus of endothelium (arrow) note the
distrupted endothelium, interstitial edema (*) and the loss of elastic
lamina (curved arrow). c ALA-treated SAH group; the integrity was
provided in endothelium (arrowhead), nucleus of muscle cell (*) and
elastic lamina (arrow). HE staining, 9200, insets 9400
Neurochem Res (2010) 35:418–428 423
123

vascular smooth muscle cells to vasoconstrictors, release of
spasmogens from lysed blood clot and inflammatory
response of the vascular wall have been proposed as
pathological mechanisms participating in the development
of post-SAH spasm [1]. Although the causative factors
underlying the development of cerebral vasospasm are
poorly understood, it is well known that cerebrovascular
effects of ROS contribute to disease progression following
SAH [37]. Since SAH-related cerebral ischemia still has a
substantial impact on mortality while the prognosis of the
surviving patients remains poor, extensive experimental
and human studies are currently performed to aid in
designing advanced therapeutic strategies for better pro-
tection of the cerebral tissues. ALA and its reduced form,
dihydrolipoic acid (DHLA) are capable of quenching ROS
such as hydroxyl radicals, peroxyl radicals, superoxide,
hypochlorous acid and peroxynitrite and they prevent sin-
glet oxygen-induced DNA damage, exhibit chelating
activity, reduce lipid peroxidation and increase intracellu-
lar glutathione levels [17]. Therefore, ALA and DHLA
have the potency to improve SAH-induced oxidative
injury. Previously, Panigrafi et al. [38] have shown that the
natural thiol antioxidant ALA is effective in improving
survival and protecting the rat against reperfusion injury
following cerebral ischemia.
Although the major mechanism of action of ALA
appears to be due to its ability to substitute for glutathione,
ALA was shown to increase NO biosynthesis in cultured
endothelial cells and improve endothelial NO-dependent
vasomotor function in a variety of conditions [39, 40] via
amelioration of endothelial GSH status [41]. Moreover,
ALA was also shown to reduce plasma levels of IL-6 and
plasminogen activator inhibitor-1, suggesting that mecha-
nisms other than antioxidant effects may be involved [42].
ALA down-regulates the expression of redox-sensitive pro-
inflammatory proteins including TNF and inducible nitric
oxide synthase. As a naturally occurring cofactor for the
mitochondrial enzymes pyruvate dehydrogenase and alpha-
ketoglutarate dehydrogenase, ALA has been shown to have
a variety of properties which can interfere with the path-
ogenesis or progression of neurodegenerative diseases [43].
It was found that ALA inhibits alpha-ketoglutarate dehy-
drogenase-mediated generation of superoxide and hydro-
gen peroxide [44].
Various experimental studies demonstrated that disrup-
tion of the BBB is the primary cause of SAH-induced brain
edema [45–47]. Doczi et al. [48] have reported that early
cerebral edema, one of the major determinants of mortality
following SAH, develops due to BBB breakdown, while
increased BBB permeability has been correlated with
delayed cerebral ischemia and poor clinical outcome.
Ischemia of the brain tissue causes destruction of the
functional and structural integrity of the BBB, which is
responsible for regulating the transport of fluids and soluble
Fig. 5 Transmission electronmicroscopy: a1 Control group;
endothelial cell (arrowhead)
and elastic lamina (arrow) and
the nuclei of muscle cells (n).
b1 Vehicle-treated SAH group;
detachment of elastic lamina
(arrow), severe edema of
interstitium (white doubleheaded arrow) contracted nuclei
of muscle (**) and degeneration
of endothelial cell
(arrowheads). c1 ALA-treated
SAH group; reduction of edema
and vacuolizations and
maintaining the regular
morphology of endothelial cell
(arrowhead), sarcoplasm (s) and
elastic lamina (arrows)
424 Neurochem Res (2010) 35:418–428
123

substances [46]. In this sense, Na?, K?-ATPase, which is
responsible for the maintenance of neuronal excitability and
the control of cellular volume in the central nervous system
[49], is an important parameter to investigate SAH-induced
brain damage. Inhibition of the activity of this crucial
enzyme is associated with various neuropathological con-
ditions, including cerebral ischemia, neurodegenerative
disorders and spinal cord edema, which also provide evi-
dence for the vulnerability of Na?, K?-ATPase to free
radical attacks [50–52]. The decrease in Na? and K? gra-
dients under ischemic conditions, resulting in reduced
uptake and thereby increased extracellular concentration of
different neurotransmitters, may further exacerbate the
ischemic conditions [53]. Similarly, in the present study,
decreased Na?, K?-ATPase activity due to SAH, indicating
membrane damage and deterioration of membrane fluidity,
is in accordance with impaired BBB, increased lipid per-
oxidation and increased brain water content. Decreased
plasma membrane transporter in the brain of animals sub-
jected to SAH may also be due to overall cell death with the
consequent reduction in Na?, K?-ATPase activity, which is
further supported by increased DNA fragmentation. On the
other hand, ALA treatment, via its powerful antioxidative
properties, decreased brain edema and lipid peroxidation,
preserved BBB, increased Na?, K?-ATPase activity and
improved SAH-induced injury. Our results are in agreement
with the studies which have shown that lipoic acid preserves
the integrity of the BBB, while alleviating the oxidative
stress-induced changes in different models of brain injury
[22, 54].
It is generally accepted that cerebral vasospasm after
SAH is produced by the compounds released from the
subarachnoid clot, which decrease blood flow to the
affected brain regions, leading to ischemia and generation
of ROS, causing a secondary injury and brain edema that
result in neurological deficits [55]. It has been shown that
SAH causes an imbalance between vasoconstrictors and
vasodilators, which is mainly responsible for the neuro-
logical deficits, is closely related to the generation of ROS
[56]. Several experimental and human studies have shown
that inhibition of ROS by several antioxidant compounds
decreased SAH-induced cerebral vasospasm and ischemia
[14, 15] In the present study, luminol Cl levels, which
detect •OH, H2O2, HOCl radicals, as well as lucigenin CL
levels detecting O2- were significantly increased, impli-
cating that SAH causes increased ROS production in the
cerebral tissue and ALA treatment significantly inhibited
SAH-induced ROS production. The CL results of the cur-
rent study are in agreement with a number of studies which
show that ALA and its reduced form DHLA directly
scavenge reactive oxygen and nitrogen species [16–18].
It has been demonstrated that SAH-induced free radical
generation leads to neuronal damage by promoting lipid
peroxidation, protein breakdown and DNA damage, which
in turn lead to cellular apoptosis, endothelial injury and
increases BBB permeability [57]. Thus, lipid peroxidation
plays an important role in the genesis of chronic vasospasm
and associated neurological outcomes in SAH. In the
present study, lipid peroxidation is increased significantly
while the antioxidant GSH level is decreased in the cere-
bral tissues, indicating oxidative damage in the brain of
SAH-induced rats. It is well known that GSH, a major
intracellular antioxidant molecule, and GSH-related anti-
oxidant enzymes protect tissues from the damaging effects
of free radicals and constitute an important mechanism
against oxidative stress [58]. In the current study, as ALA
reduced the oxidative injury of cellular structures, the level
of the intracellular antioxidant GSH, which is otherwise
oxidized when inactivating free radicals, was not changed.
Thus, it appears that the anti-oxidative effect of ALA on
lipid peroxidation does not directly involve the expenditure
of tissue GSH stores, but the antioxidant pool is further
supported by the action of ALA. Several other studies have
demonstrated that ALA scavenges ROS and reactive
nitrogen species, prevents DNA damage, exhibits chelating
activity, reduces lipid peroxidation and increases intracel-
lular GSH levels [17]. Besides its antioxidant properties
through scavenging the ROS, the specific stimulatory effect
of ALA on the intracellular GSH levels [59, 60] further
suggests that it may be an important component of the
treatment regimen of the cerebrovascular ischemic
diseases.
It has been demonstrated that SAH damages the BBB,
and then the blood cells such as neutrophils and macro-
phages accumulate in the brain and further sustain the
cerebral inflammatory cascade [46]. Neutrophils could
greatly exacerbate the pathological sequelae of brain
trauma by altering vascular permeability, contributing to
oxidative damage, inducing further neuronal damage via
the secretion of lysosomal enzymes and cytokines or by
altering cerebral vascular blood flow [61, 62]. Activity of
myeloperoxidase (MPO) enzyme, which is extensively
used for the quantitative indication of inflammation, was
increased in the brain tissue at the 48th h of subarachnoid
hemorrhage, implicating that SAH-induced cerebral
ischemic damage is a neutrophil-dependent inflammatory
condition. Similarly, polymorphonuclear leukocyte
recruitment has been correlated with increased cerebral
edema [63] and neutrophil depletion has been reported to
attenuate increases in brain water content and to decrease
infarct size following ischemic brain injury [64]. On the
other hand, ALA treatment in the current study offered
neuronal protection by a neutrophil-dependent mechanism,
as it was previously shown to exert mucosal protection
through the inhibition of neutrophil recruitment in the
gastric and colonic tissues [65, 66]. The current results are
Neurochem Res (2010) 35:418–428 425
123

in agreement with similar studies, which have shown that
increased MPO activity plays a role in SAH-induced
ischemic brain damage and the antioxidant drugs prevent
the damage by inhibiting neutrophil infiltration [15, 67].
Alpha lipoic acid treatment, in the present study, sup-
pressed the high percentage of DNA fragmentation in the
brain, while oxidative brain injury was improved. Simi-
larly, it was demonstrated that application of the antioxi-
dant ALA in animal and cell culture models decreases
oxidative stress and supports the endogenous antioxidant
systems potently- and apoptosis-related cell death in tissues
exposed to oxidant injury [68, 69].
Despite sophisticated medical management and neuro-
surgical techniques, subarachnoid hemorrhage still results
in a high mortality rate. Considering that oxidative stress
plays important roles in the pathogenesis of acute brain
injury after SAH [70, 71], reducing oxidative stress,
thereby activating the survival pathway could be a thera-
peutic target for acute brain injury after SAH in a clinical
situation. The results of the current study demonstrate that
ALA has a significant neuroprotective effect mediated via
enhancing the activity of endogenous antioxidant enzymes,
inhibiting neutrophil accumulation and free radical gener-
ation. Since ALA is known to have various metabolic
antioxidant properties, can cross the blood-brain barrier,
and does not exhibit any serious side effects, it is possible
to speculate that ALA has the potential to be a novel
therapeutic agent for the reduction of secondary injury after
SAH in patients.
References
1. Kozniewska E, Michalık R, Rafałowska J et al (2006) Mecha-
nisms of vascular dysfunction after subarachnoid hemorrhage. J
Physiol Pharmacol 57(Suppl 11):145–160
2. Takao A (1999) Oxyhemoglobin as the principal cause of cere-
bral vasospasm: a holistic view of its actions. Crit Rev Neurosurg
9:303–318
3. Broderick JP, Brott TG, Duldner JE et al (1994) Initial and
recurrent bleeding are the major causes of death following sub-
arachnoid hemorrhage. Stroke 25:1342–1347
4. Hutter BO, Kreitschmann-Andermahr I, Gilsbach JM (2001)
Health-related quality of life after aneurysmal subarachnoid
hemorrhage: impacts of bleeding severity, computerized tomog-
raphy findings, surgery, vasospasm, and neurological grade. J
Neurosurg 94:241–251
5. Peyrot F, Ducrocq C (2008) Potential role of tryptophan deriva-
tives in stress responses characterized by the generation of
reactive oxygen and nitrogen species. J Pineal Res 45:235–246
6. Nishizawa S, Yamamoto S, Yokoyama T et al (1997) Dysfunc-
tion of nitric oxide induces protein kinase C activation resulting
in vasospasm after subarachnoid hemorrhage. Neurol Res
19:558–562
7. Imperatore C, Germano A, d’Avella D et al (2000) Effects of the
radical scavenger AVS on behavioral and BBB changes after
experimental subarachnoid hemorrhage. Life Sci 66:779–790
8. Ostrowski RP, Tang J, Zhang JH (2006) Hyperbaric oxygen
suppresses NADPH oxidase in a rat subarachnoid hemorrhage
model. Stroke 37:1314–1318
9. Asano T, Takakura K, Sano K et al (1996) Effects of a hydroxyl
radical scavenger on delayed ischemic neurological deficits fol-
lowing aneurysmal subarachnoid hemorrhage: results of a mul-
ticenter, placebo-controlled double-blind trial. J Neurosurg
84:792–803
10. Haley EC Jr, Kassell NF, Apperson-Hansen C et al (1997) A
randomized, double-blind, vehicle-controlled trial of tirilazad
mesylate in patients with aneurysmal subarachnoid hemorrhage: a
cooperative study in North America. J Neurosurg 86:467–474
11. Karaoglan A, Akdemir O, Barut S et al (2008) The effects of
resveratrol on vasospasm after experimental subarachnoidal
hemorrhage in rats. Surg Neurol 70:337–343
12. Aladag MA, Turkoz Y, Ozcan C et al (2006) Caffeic acid
phenethyl ester (CAPE) attenuates cerebral vasospasm after
experimental subarachnoidal haemorrhage by increasing brain
nitric oxide levels. Int J Dev Neurosci 24:9–14
13. Green AR, Ashwood T (2005) Free radical trapping as a thera-
peutic approach to neuroprotection in stroke: experimental and
clinical studies with NXY-059 and free radical scavengers. Curr
Drug Targets CNS Neurol Disord 4:109–118
14. Munakata A, Ohkuma H, Nakano T et al (2009) Effect of a free
radical scavenger, edaravone, in the treatment of patients with
aneurysmal subarachnoid hemorrhage. Neurosurgery 64:423–428
(discussion 428-9)
15. Ersahin M, Toklu HZ, Cetinel S et al (2009) Melatonin reduces
experimental subarachnoid hemorrhage-induced oxidative brain
damage and neurological symptoms. J Pineal Res 46:324–332
16. Packer L, Witt EH, Tritschler HJ (1995) a-Lipoic acid as a bio-
logical antioxidant. Free Radic Biol Med 19:227–250
17. Moini H, Packer L, Saris NE (2002) Antioxidant and prooxidant
activities of alpha-lipoic acid and dihydrolipoic acid. Toxicol
Appl Pharmacol 182:84–90
18. Packer L (1998) Alpha-lipoic acid: a metabolic antioxidant which
regulates NF-kappa B signal transduction and protects against
oxidative injury. Drug Metab Rev 30:245–275
19. Vincent AM, Stevens MJ, Backus C et al (2005) Cell culture
modeling to test therapies against hyperglycemia-mediated
oxidative stress and injury. Antioxid Redox Signal 7:1494–
1506
20. Bharat S, Cochran BC, Hsu M et al (2002) Pre-treatment with R-
lipoic acid alleviates the effects of GSH depletion in PC12 cells:
implications for Parkinson’s disease therapy. Neurotoxicology
23:479–486
21. Abdul HM, Butterfield DA (2007) Involvement of PI3K/PKG/
ERK1/2 signaling pathways in cortical neurons to trigger pro-
tection by cotreatment of acetyl-Lcarnitine and alpha-lipoic acid
against HNE-mediated oxidative stress and neurotoxicity:
implications for Alzheimer’s disease. Free Radic Biol Med
42:371–384
22. Toklu HZ, Hakan T, Biber N et al (2009) The protective effect of
alpha lipoic acid against traumatic brain injury in rats. Free Radic
Res 25:1–10
23. do Vale OC, Fonteles DS, Cabral FR et al (2003) A dual action of
alpha-lipoic acid in the brain: an electrophysiological evaluation.
Arq Neuropsiquiatr 61(3B):738–745
24. Freitas RM (2009) The evaluation of effects of lipoic acid on the
lipid peroxidation, nitrite formation and antioxidant enzymes in
the hippocampus of rats after pilocarpine-induced seizures.
Neurosci Lett 455:140–144
25. Jones RE, Moes N, Zwickey H et al (2008) Treatment of
experimental autoimmune encephalomyelitis with alpha lipoic
acid and associative conditioning. Brain Behav Immun 22:538–
543
426 Neurochem Res (2010) 35:418–428
123

26. Delgado TJ, Brismar J, Svendgaard NA (1985) Subarachnoid
haemorrhage in the rat: angiography and fluorescence microscopy
of the major cerebral arteries. Stroke 16:595–602
27. Bederson JB, Pitts LH, Tsuji M et al (1986) Rat middle cerebral
artery occlusion: evaluation of the model and development of a
neurological examination. Stroke 17:472–476
28. Haklar G, Yuksel M, Yalcın AS (1998) Chemiluminescence in
the measurement of free radicals: theory and application on a
tissue injury model. Marmara Med J 11:56–60
29. Wyllie H (1980) Glucocorticoid induced thymocyte apoptosis is
associated with endogenous endonuclease activation. Nature
284:555–556
30. Burton K (1956) A study of the conditions and mechanism of the
diphenylamine reaction for the colorimetric estimation of
deoxyribonucleic acid. Biochem J 62:315–323
31. Beuge JA, Aust SD (1978) Microsomal lipid peroxidation.
Methods Enzymol 53:302–311
32. Beutler E, Duron O, Kelly BM (1963) Improved method for the
determination of blood glutathione. J Lab Clin Med 61:882–888
33. Hillegas LM, Griswold DE, Brickson B et al (1990) Assessment
of myeloperoxidase activity in whole rat kidney. J Pharmacol
Methods 24:285–295
34. Reading HW, Isbir T (1980) The role of cation activated ATPase
in transmitter release from the rat iris. Q J Exp Physiol 65:105–
116
35. Fiske CH, Subbarow Y (1925) The colorimetric determination of
phosphorus. J Biol Chem 66:375–400
36. Lowry OH, Rosenbrough NJ, Farr AL et al (1951) Protein
measurements with the folin phenol reagent. J Biol Chem
193:265–275
37. Paravicini TM, Sobey CG (2003) Cerebral vascular effects of
reactive oxygen species: recent evidence for a role of NADPH-
oxidase. Clin Exp Pharmacol Physiol 30:855–859
38. Panigrahi M, Sadguna Y, Shivakumar BR et al (1996) Alpha-
lipoic acid protects against reperfusion injury following cerebral
ischemia in rats. Brain Res 717:184–188
39. Lee WJ, Lee IK, Kim HS, Kim YM et al (2005) Alpha-lipoic acid
prevents endothelial dysfunction in obese rats via activation of
AMP-activated protein kinase. Arterioscler Thromb Vasc Biol
25:2488–2494
40. Sena CM, Nunes E, Louro T (2007) Effects of alpha-lipoic acid
on endothelial function in aged diabetic and high-fat fed rats. Br J
Pharmacol. doi:10.1038/sj.bjp.0707474 (e-pub ahead of print 1
October 2007)
41. Smith AR, Hagen TM (2003) Vascular endothelial dysfunction in
aging: loss of Akt-dependent endothelial nitric oxide synthase
phosphorylation and partial restoration by (R)-alpha-lipoic acid.
Biochem Soc Trans 31:1447–1449
42. Sola S, Mir MQ, Cheema FA et al (2005) Irbesartan and lipoic
acid improve endothelial function and reduce markers of
inflammation in the metabolic syndrome: results of the Irbesartan
and Lipoic Acid in Endothelial Dysfunction (ISLAND) study.
Circulation 111:343–348
43. Maczurek A, Hager K, Kenklies M et al (2008) Lipoic acid as an
anti-inflammatory and neuroprotective treatment for Alzheimer’s
disease. Adv Drug Deliv Rev 60:1463–1470
44. Ambrus A, Tretter L, Adam-Vizi V (2009) Inhibition of the
alpha-ketoglutarate dehydrogenase-mediated reactive oxygen
species generation by lipoic acid. J Neurochem 109(Suppl
1):222–229
45. Germano A, d’Avella D, Cicciarello R et al (1992) Blood-brain
barrier permeability changes after experimental subarachnoid
hemorrhage. Neurosurgery 30:882–886
46. Del Zoppo GJ, Hallenbeck JM (2000) Advances in the vascular
pathophysiology of ischemic stroke. Thromb Res 98:73–81
47. Doczi TP, Joo F, Adam G et al (1986) Blood-brain barrier
damage during the acute stage of subarachnoid hemorrhage, as
exemplified by a new animal model. Neurosurgery 18:733–739
48. Doczi TP, Joo F, Balas I (1995) Atrial natriuretic peptide (ANP)
attenuates brain oedema accompanying experimental subarach-
noid haemorrhage. Acta Neurochir (Wien) 132:87–91
49. Streck EL, Feier G, Burigo M et al (2006) Effects of electro-
convulsive seizures on Na(?), K(?)-ATPase activity in the rat
hippocampus. Neurosci Lett 404:254–257
50. Grisar T (1984) Glial and neuronal Na?–K? pump in epilepsy.
Ann Neurol 16(Suppl):S128–S134
51. de Souza Wyse AT, Streck EL, Worm P et al (2000) Precondi-
tioning prevents the inhibition of Na?, K?-ATPase activity after
brain ischemia. Neurochem Res 25:971–975
52. Yang YB, Piao YJ (2003) Effects of resveratrol on secondary
damages after acute spinal cord injury in rats. Acta Pharmacol Sin
24:703–710
53. Stys PK (1998) Anoxic and ischemic injury of myelinated axons
in CNS white matter: from mechanistic concepts to therapeutics.
J Cereb Blood Flow Metab 18:2–25
54. Morini M, Roccatagliata L, Dell’Eva R et al (2004) Alpha-lipoic
acid is effective in prevention and treatment of experimental
autoimmune encephalomyelitis. J Neuroimmunol 148:146–153
55. Kolias AG, Sen J, Belli A (2009) Pathogenesis of cerebral
vasospasm following aneurysmal subarachnoid hemorrhage:
putative mechanisms and novel approaches. J Neurosci Res
87:1–11
56. Ayer RE, Zhang JH (2008) Oxidative stress in subarachnoid
haemorrhage: significance in acute brain injury and vasospasm.
Acta Neurochir Suppl 104:33–41
57. Lewen A, Matz P, Chan PH (2000) Free radical pathways in CNS
injury. J Neurotrauma 17:871–890
58. Reiter RJ, Tan DX, Acuna-Castroviejo D et al (2000) Melatonin:
mechanisms and actions as an antioxidant. Curr Top Biophys
24:171–183
59. Busse E, Zimmer G, Schopohl B et al (1992) Influence of alpha-
lipoic acid on intracellular glutathione in vitro and in vivo.
Arzneimittelforschung 42:829–831
60. Han D, Tritschler HJ, Packer L (1995) Alpha-lipoic acid
increases intracellular glutathione in a human T-lymphocyte
Jurkat cell line. Biochem Biophys Res 207:258–264
61. Wedmore CV, Williams JT (1981) Control of vascular perme-
ability by polymorphonuclear leukocytes in inflammation. Nature
289:646–650
62. Tonnesen MG, Worthen GS, Johnston RB (1988) Neutrophil
emigration, activation, and tissue damage. In: Clark RAE, Hensen
PM (eds) The molecular and cellular biology of wound repair.
Plenum, New York, pp 149–184
63. Schoettle RJ, Kochanek PM, Magargee JM et al (1990) Early
polymorphonuclear leukocyte accumulation correlates with the
development of post-traumatic cerebral edema in rats. J Neuro-
trauma 7:207–217
64. Matsuo Y, Onodera H, Shiga Y et al (1994) Correlation between
myeloperoxidase-quantified neutrophil accumulation and ische-
mic brain injury in the rat: effects of neutrophil depletion. Stroke
25:1469–1475
65. Kolgazi M, Jahovic N, Yuksel M et al (2007) Alpha-lipoic acid
modulates gut inflammation induced by trinitrobenzene sulfonic
acid in rats. J Gastroenterol Hepatol 22:1859–1865
66. Sehirli O, Tatlidede E, Yuksel M et al (2008) Antioxidant effect
of alpha-lipoic acid against ethanol-induced gastric mucosal
erosion in rats. Pharmacology 81:173–180
67. Wilson JX, Gelb AW (2002) Free radicals, antioxidants, and
neurologic injury: possible relationship to cerebral protection by
anesthetics. J Neurosurg Anesthesiol 14:66–79
Neurochem Res (2010) 35:418–428 427
123

68. Dincer Y, Telci A, Kayali R et al (2002) Effect of alpha-
lipoic acid on lipid peroxidation and anti-oxidant enzyme
activities in diabetic rats. Clin Exp Pharmacol Physiol 29:
281–284
69. Vincent AM, McLean LL, Backus C et al (2005) Short-term
hyperglycemia produces oxidative damage and apoptosis in
neurons. FASEB J 19:638–640
70. Matz PG, Copin J-C, Chan PH (2000) Cell death after exposure to
subarachnoid hemolysate correlates inversely with expression of
CuZn-superoxide dismutase. Stroke 31:2450–2458
71. Matz PG, Fujimura M, Lewen A et al (2001) Increased cyto-
chrome c-mediated DNA fragmentation and cell death in man-
ganese-superoxide dismutase-deficient mice after exposure to
subarachnoid hemolysate. Stroke 32:506–515
428 Neurochem Res (2010) 35:418–428
123
Copyright © 2022 FDOKUMEN




















