
Elucidating the Conformational Dependence of Calculated p K a Values
Upload
uapfilialayacuchoCategory
view
0download
0
doi:10.1016/j.jmb.2009.11.038 J. Mol. Biol. (2010) 396, 280–292
Available online at www.sciencedirect.com
A Single Mutation at the Sheet Switch RegionResults in Conformational Changes Favoring λ6Light-Chain Fibrillogenesis
Alejandra Hernández-Santoyo1⁎†, Luis del Pozo Yauner2,3†,Deyanira Fuentes-Silva1, Ernesto Ortiz2, Enrique Rudiño-Piñera2,Rosana Sánchez-López2, Eduardo Horjales4, Baltazar Becerril2†and Adela Rodríguez-Romero1⁎†
1Instituto de Química,Universidad NacionalAutónoma de México, CircuitoExterior, Ciudad Universitaria,Mexico D.F. 04510, Mexico2Instituto de Biotecnología,Universidad NacionalAutónoma de México, ApartadoPostal 510-3, Cuernavaca,Morelos 62250, Mexico3Instituto Nacional de MedicinaGenómica, Periférico Sur 4124,Álvaro Obregón, Mexico D.F.01900, Mexico4Instituto de Física de SãoCarlos, Universidade de SãoPaulo, Avenida do TrabalhadorSão-Carlense 400, São Carlos,São Paulo 13560-970, BrazilReceived 1 August 2009;received in revised form5 November 2009;accepted 15 November 2009Available online24 November 2009
*Corresponding authors. E-mail ad† A.H.-S., L.d.P.Y., B.B., and A.R.Abbreviations used: ASU, asymm
framework region; LC, light chain; Pvariable region of the light chain; W
0022-2836/$ - see front matter © 2009 E
Systemic amyloid light-chain (LC) amyloidosis is a disease processcharacterized by the pathological deposition of monoclonal LCs in tissue.All LC subtypes are capable of fibril formation although λ chains,particularly those belonging to the λ6 type, are overrepresented. Here, wereport the thermodynamic and in vitro fibrillogenic properties of severalmutants of the λ6 protein 6aJL2 in which Pro7 and/or His8 was substitutedby Ser or Pro. The H8P and H8S mutants were almost as stable as the wild-type protein and were poorly fibrillogenic. In contrast, the P7S mutationdecreased the thermodynamic stability of 6aJL2 and greatly enhanced itscapacity to form amyloid-like fibrils in vitro. The crystal structure of the P7Smutant showed that the substitution induced both local and long-distanceeffects, such as the rearrangement of the VL (variable region of the lightchain)–VL interface. This mutant crystallized in two orthorhombicpolymorphs, P212121 and C2221. In the latter, a monomer that was notarranged in the typical Bence–Jones dimer was observed for the first time.Crystal-packing analysis of the C2221 lattice showed the establishment ofintermolecular β–β interactions that involved the N-terminus and β-strandB and that these could be relevant in the mechanism of LC fibril formation.Our results strongly suggest that Pro7 is a key residue in the conformationof the N-terminal sheet switch motif and, through long-distance interac-tions, is also critically involved in the contacts that stabilized the VLinterface in λ6 LCs.
© 2009 Elsevier Ltd. All rights reserved.
Keywords: AL amyloidosis; crystal structure; fibrillogenesis; λ6 subgroup;thermodynamic stability
Edited by S. Radforddresses: [email protected]; [email protected]. made equal contributions to this work.etric unit; BJ, Bence–Jones; CDR, complementarity-determining region; FR,BS, phosphate-buffered saline; PCR, polymerase chain reaction; ThT, thioflavin T; VL,T, wild type.
lsevier Ltd. All rights reserved.

281Molecular Mechanism of Light–Chain Fibrillogenesis
Introduction
Amyloidosis derived from amyloid light chain(LC) is a systemic disease characterized by theextracellular deposition of a fibrillar proteinaceousmaterial made of immunoglobulin LC fragments,comprising the variable domain (VL) either alone orwith a portion of the constant domain (CL).
1–4 Themechanisms involved in fibril formation remainunclear. Several lines of evidence point to thermo-dynamic stability as one of the major factors thatinfluence the amyloidogenic behavior of the LCs.5–9
Point mutations that destabilize the folding of theLC, extreme values of pH and temperature, and thepresence of chemical denaturants induce the forma-tion of amyloid-like fibril in vitro.10–16 These datasuggest that misfolded species play a key role in theaggregation route.6,11,14,17 Although ample datasupport this model, little is known about the natureof the structural changes leading to the amyloidaggregation of LC.The VL adopts the typical immunoglobulin fold
characterized by the topological organization of ninestrands (A, B, C, C′, C″, D, E, F, and G) as a Greekkey motif.18 As in other β-sheet proteins, the LCsmake use of “negative design” features to preventthe edge strands from forming promiscuous βinteractions, thereby preventing aggregation of thenative state. The VL makes use of several edge-protection strategies, including the favorable place-ment of prolines and charged residues, a β-bulge onedge strand G (C-terminal region), and a loopcomprising residues 40–60 that caps edge strandsC″ and λ LCs, N-terminal strand A (residues 1–14)adopts a characteristic conformation known as the“sheet switch,” which has been proposed to protectthe two edge strands B and G from developingspurious intermolecular interactions that lead toaggregation.19
Consistent with the misfolding hypothesis ofprotein aggregation, it could be rationally predictedthat the VL amyloidogenesis proceeds via confor-mational intermediates that compromise the effec-
Table 1. Thermodynamic and kinetic parameters for the chemproteins
Protein
Thermal unfolding GdnHCl unfo
ΔG25 ″Cb
(kcal/mol)Tm
c
(°C)ΔHm
c
(kcal/mol)ΔGH2O
(kcal/mol)−m
(kcal/m
6aJL2 5.2 49.9 86.2 5.1 3.6P7S 3.7 43.0 79.5 2.5 3.0H8P 4.4 50.1 84.8 4.5 3.0H8S 5.0 49.5 84.4 4.5 3.5P7S–H8S 3.8 43.7 79.4 N.D. N.D
N.D. means not determined.a Cumulative free energy change relative to 6aJL2: ΔΔGunf= (m
6aJL
b Calculated as described by Pace et al.39c Calculated from the plot of ΔG versus T.d Calculated by extrapolating the linear region of the hyperbolic phe Asymptotic ThT fluorescence value of the elongation reaction, nof Pseudo-first-order growth rate constant fits from the data shown
tiveness of these protective motifs (e.g., the N-terminal sheet switch). Consequently, aggregation-prone regions, such as the edge strands that arenormally buried in the native state, may partiallyaccess the molecular surface. If this assumption iscorrect, the substitution of residues at position 7and/or position 8 will disturb the interactionnetwork that stabilizes the N-terminal sheet switchin λ LC, thereby favoring its aggregation intoamyloid-like fibrils.To confirm this hypothesis, we used the 6aJL2
protein as a model. This VL contains the sequencesencoded by the λ6 germ-line genes.21 In this work,we report the thermodynamic and in vitro fibrillo-genic properties of 6aJL2 mutants in which Sersubstitutes Pro7 and/or His8. Serine is present inother VLλ gene segments in both positions. Theeffect of substituting His8, a residue characteristic ofthe λ6 proteins, by Pro was also studied. Weobserved that the P7S mutation decreased thethermodynamic stability of 6aJL2 and enhanced itsfibrillogenic potential in vitro. In contrast, H8P andH8S mutants were almost as stable as the wild-type(WT) protein and were poorly fibrillogenic. Thecrystal structures of the native 6aJL2 and the pointmutant P7S were determined to aid in the interpre-tation of the biophysical data. Our structural dataclearly indicate that Pro7 is a key residue that affectsthe stability of the sheet switch at the N-terminus,thus supporting its protective function. Additional-ly, this residue is involved in long-distance interac-tions that stabilize the whole domain, in particular,the conformation of the VL–VL interface.
Results
Thermodynamic stability of mutants and thekinetics of amyloid fibril formation
The thermodynamic parameters of the differentmutants (P7S, H8P, H8S, and P7S–H8S), as deter-mined from thermal and chemical equilibrium
ical and thermal unfolding and the aggregation of the λ6
lding
ΔΔGa
(kcal/mol)
In vitro fibrillogenesis
ol)Cm(M)
Without seed With seed
tlag (min)d AThTe Kf (s−1)
1.41 — 900 1 5.3×10−5
0.84 2.1 50 1.6 1.1×10−4
1.48 −0.3 1000 0.5 6.1×10−5
1.28 0.5 500 0.3 4.8×10−5
. N.D. N.D. 130 2.0 1.2×10−4
2×Cmmut)− (m6aJL2×Cm
6aJL2).
ase back to the abscissa.rmalized to 6aJL2.in Fig. 1d.
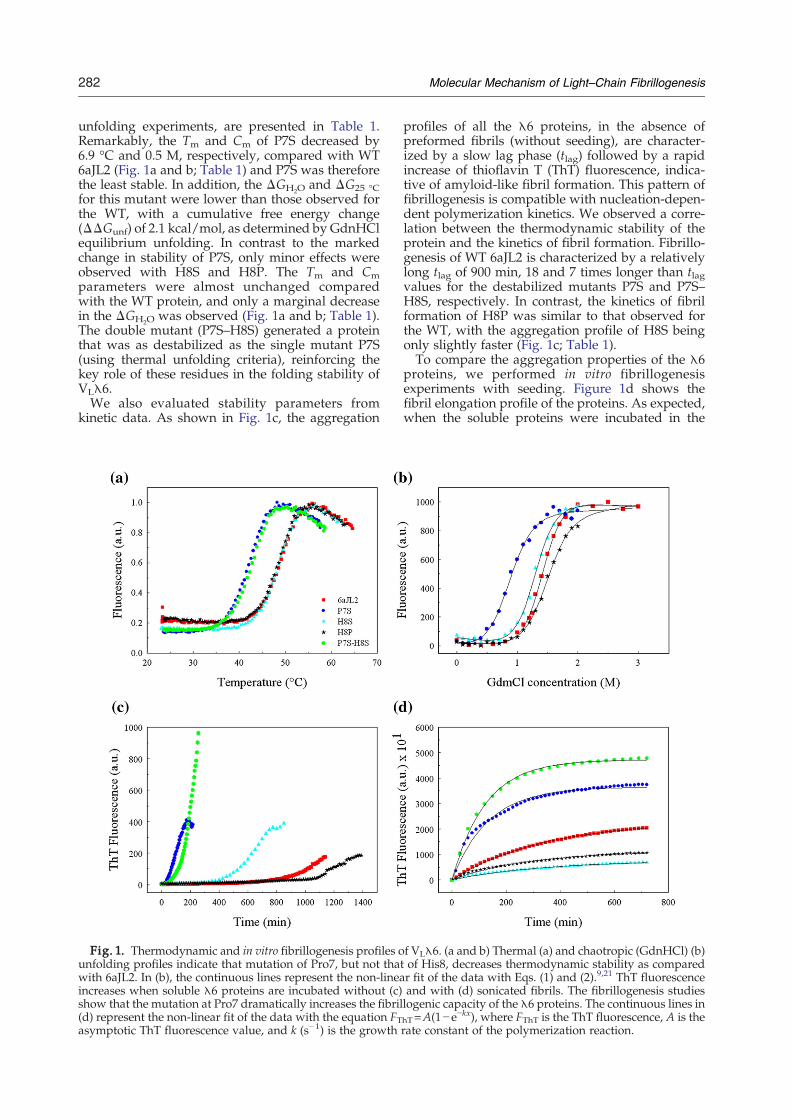
282 Molecular Mechanism of Light–Chain Fibrillogenesis
unfolding experiments, are presented in Table 1.Remarkably, the Tm and Cm of P7S decreased by6.9 °C and 0.5 M, respectively, compared with WT6aJL2 (Fig. 1a and b; Table 1) and P7S was thereforethe least stable. In addition, the ΔGH2O and ΔG25 °Cfor this mutant were lower than those observed forthe WT, with a cumulative free energy change(ΔΔGunf) of 2.1 kcal/mol, as determined by GdnHClequilibrium unfolding. In contrast to the markedchange in stability of P7S, only minor effects wereobserved with H8S and H8P. The Tm and Cmparameters were almost unchanged comparedwith the WT protein, and only a marginal decreasein the ΔGH2O was observed (Fig. 1a and b; Table 1).The double mutant (P7S–H8S) generated a proteinthat was as destabilized as the single mutant P7S(using thermal unfolding criteria), reinforcing thekey role of these residues in the folding stability ofVLλ6.We also evaluated stability parameters from
kinetic data. As shown in Fig. 1c, the aggregation
Fig. 1. Thermodynamic and in vitro fibrillogenesis profiles ounfolding profiles indicate that mutation of Pro7, but not thatwith 6aJL2. In (b), the continuous lines represent the non-lineaincreases when soluble λ6 proteins are incubated without (c)show that the mutation at Pro7 dramatically increases the fibril(d) represent the non-linear fit of the data with the equation FTasymptotic ThT fluorescence value, and k (s−1) is the growth
profiles of all the λ6 proteins, in the absence ofpreformed fibrils (without seeding), are character-ized by a slow lag phase (tlag) followed by a rapidincrease of thioflavin T (ThT) fluorescence, indica-tive of amyloid-like fibril formation. This pattern offibrillogenesis is compatible with nucleation-depen-dent polymerization kinetics. We observed a corre-lation between the thermodynamic stability of theprotein and the kinetics of fibril formation. Fibrillo-genesis of WT 6aJL2 is characterized by a relativelylong tlag of 900 min, 18 and 7 times longer than tlagvalues for the destabilized mutants P7S and P7S–H8S, respectively. In contrast, the kinetics of fibrilformation of H8P was similar to that observed forthe WT, with the aggregation profile of H8S beingonly slightly faster (Fig. 1c; Table 1).To compare the aggregation properties of the λ6
proteins, we performed in vitro fibrillogenesisexperiments with seeding. Figure 1d shows thefibril elongation profile of the proteins. As expected,when the soluble proteins were incubated in the
f VLλ6. (a and b) Thermal (a) and chaotropic (GdnHCl) (b)of His8, decreases thermodynamic stability as comparedr fit of the data with Eqs. (1) and (2).9,21 ThT fluorescenceand with (d) sonicated fibrils. The fibrillogenesis studieslogenic capacity of the λ6 proteins. The continuous lines inhT=A(1−e−kx), where FThT is the ThT fluorescence, A is therate constant of the polymerization reaction.

283Molecular Mechanism of Light–Chain Fibrillogenesis
presence of fragmented fibrils (seeds), the ThTfluorescence increased without a lag phase andreached a plateau. Even when similar time-coursecurves and kinetics were obtained for all proteins,the parameters describing the kinetics of theelongation phase differed from one protein toanother (Table 1). The asymptotic trace of ThTfluorescence (parameter A), which is indicative ofthe amount of fibrils accumulated at a position ofequilibrium between the fibril and the monomer,was 1.6-fold higher for the mutant P7S and was2-fold higher for the double mutant P7S–H8S thanfor the WT protein. Similarly, the pseudo-first-ordergrowth rate constant (k s−1) is nearly twice as highfor the Pro7 and P7–H8mutants. In contrast, theHis8mutants elongated their fibrils by 30% to 50%compared with the WT protein, although thepseudo-first-order growth rate constants were quitesimilar.
Fig. 2. Electron microscopic images of the fibrillar aggreg(b) P7S. (c) H8S. (d) H8P. (e) P7S–H8S. Scale bars for all ca
Electron microscopy
We confirmed that all the proteins formedcharacteristic thin unbranched fibrils (Fig. 2). How-ever, there were differences in morphology andlength. The mutant P7S–H8S formed rod-like fibersthat appeared shorter and rigid in structure com-pared with the long and slender fibers of the WTprotein. The fibrils of the mutants H8P and H8S tendto be as long as those of the WT. It is interesting tonote the twisted-ribbon appearance of the P7Sfibrils.
Structural analysis
The structures described here correspond to thenative 6aJL2 and the P7S mutant. The formercrystallized in the space group P4122 [Protein DataBank (PDB) ID 2W0K], whereas the latter (the most
ates formed by the WT 6aJL2 and its mutants. (a) 6aJL2.ses represent 100 nm.

284 Molecular Mechanism of Light–Chain Fibrillogenesis
fibrillogenic) crystallized in two space groups,P212121 (PDB ID 3B5G) and C2221 (PDB ID 3BDX),respectively. Both polymorphs contained a Bence–
Fig. 3. Structural analysis of native 6aJL2 and its mutant P7P212121 and C2221 structures of the P7S mutant. Monomer c (gmonomers (C2221 in orange, P212121 blue, and native in olive) texception of the N-termini that show a conformational changeand monomers a and b of the native 6aJL2 are similar. (d) Supeof approximately 13 deg between the orientation of the forme
Jones (BJ) dimer in the asymmetric unit (ASU) (Fig.3a and b); however, the C2221 crystal showed anadditional monomer that has not been previously
S. (a and b) Ribbon diagrams and molecular surfaces of thereen) never forms the BJ assembly. (c) Superposition of allhat reveals a high level of similarity among them, with thein monomers a and b of the P7S mutant (inset). Monomer crposition of the WT and the P7S dimers showing a rotationr and that of the mutant (C2221).

285Molecular Mechanism of Light–Chain Fibrillogenesis
observed (Fig. 3b). This monomer did not establishcontacts with any symmetry-related molecules toconfigure a BJ arrangement. Crystallographic dataand refinement statistics for the three structures arepresented in Table 2. We have used continuousnumbering for the amino acid residues in all of theproteins included in this article.The native 6aJL2 and the P7S structures had
classic LC immunoglobulin folds (Fig. 3a and b).The final 2Fo−Fc electron density maps showedgood quality and overall agreement with thestructures in each complementarity-determiningregion (CDR) and framework region (FR). Thisallowed the backbone and side-chain conforma-tions to be modeled with high accuracy. A super-position of all monomers showed the threestructures to be highly similar (Fig. 3c), with thebackbone RMSD varying from 0.14 to 0.49 Å. Themost significant differences among the structuresinvolved the residues in β-bulge 1 (residues 7–9),FR-3, and CDR-1. Regardless of the differencesobserved, the CDR-1 is folded, in all the proteins,in a characteristic canonical structure denominated“curly.”22
The interfaces of the three crystallographic struc-tures were analyzed using the PISA server.23 We
Table 2. Data collection and structure refinementstatistics
Crystal2W0K(WT) 3B5G (P7S) 3BDX (P7S)
Data collectionSpace group P4122 P212121 C2221Cell dimensions (Å)a 71.27 40.77 56.11b 71.27 68.59 106.12c 95.45 83.85 133.36Unique reflectionsa 8166 (1297) 19,189 (2713) 16,902 (2544Resolution limit (Å) 50.2–2.45 53.07–1.90 29.96–2.3Rmerge
b 0.051 (0.26) 0.0098 (0.361) 0.084 (0.227)Multiplicity 3.0 (2.9) 11.6 (9.9) 2.3 (2.2)Completeness (%) 87.9 (90.8) 99.9 (99.5) 93.9 (97.8)I/σ(I) 9.8 (2.8) 21.5 (6.1) 8.7 (3.9)
Refinement statisticsR/Rfree
c (%) 20.8/25.3 16.7/20.9 20.5/24.6No. of atomsProtein/solvent/acetate
1680/45/0 1780/161/2 2517/100/5
Glycerol/Na/Mes 7/1/0 4/0/1Average B-value (Å2) 41.2 17.5 22.0RMSD from idealityBond length (Å) 0.01 0.013 0.007Bond angle (°) 1.24 1.44 1.3Ramachandran plot (%)Allowed regions 99.1 99.0 98.9Disallowed regions 0.9 1.0 1.1a Values in parentheses correspond to the last resolution shellb Rmerge=∑j∑h(|Ij,h− ⟨Ih⟩|)/∑j∑h(⟨Ih⟩), where h is the unique
reflection index, Ij,h is the intensity of the symmetry-relatedreflection, and ⟨Ih⟩ is the mean intensity.
c R=∑h||Fo|h−|Fc|h||/∑h|Fo|h for all reflections, where Foand Fc are the observed and calculated structure factorsrespectively, and h defines unique reflections. Rfree is calculatedanalogously for the test reflections, randomly selected, andexcluded from the refinement.
)
.
,
manually checked all contact salt bridges andhydrogen bonds provided by this program. The BJdimers contained in the ASU of the native proteinand of the P7S mutant (P212121) were slightlydifferent at the interface, which showed a differenceof about 5 deg. Each of these proteins showed thesame set of interactions between monomers a and b,including six hydrogen bonds established amongthe residues located in FR-2 (residues 35–49), CDR-3(residues 89–96), and FR-4 (residues 97–111). Nota-bly, for the C2221 BJ dimer, the number of interfaceinteractions increased to seven hydrogen bonds andtwo salt bridges, now including CDR-2 (residues 50–56). This demonstrated some differences in thearrangement of its monomers. It is also importantto note that for the C2221 structure (Fig. 3b), theinteractions of monomer c were not involved in theclassic BJ arrangement with any other symmetry-related molecule. Instead, their FR-2, FR-4, andCDR-3, which are normally implicated in thisinterface, interact mainly with FR-1 of monomer a.The contacts between monomers a and c includeonly five hydrogen bonds.Additionally, we analyzed the P-value using the
PISA server, which is a criterion of the significanceand specificity of the interactions detected. AP-valueN0.5 means that the interface is less hydrophobicthan expected and is therefore likely to be an artifactof crystal packing. On the other hand, Pb0.5 indi-cates that the interface is interaction specific. TheP-values are very similar for the interfaces betweenmonomers a and b of all structures and range from0.22 to 0.29, indicating that the interactions arespecific. Nonetheless, the orientation between thesetwo monomers in the C2221 dimer is different byabout 13 deg (Fig. 3d), implying that the interfaceinteractions are made highly flexible by the rear-rangement of other regions in the molecule. Asexpected, monomer c presents a P-value of 0.40,indicating that its interaction with monomer a is lessspecific.
Molecular environment at the N-terminal region
The topology of the N-terminal region was similarin all of the structures and included a β-bulgebetween two segments of β-strand A; nonetheless,different contacts were observed with neighboringregions, such as the C-terminus and β-strand B.Interestingly, the size of the β-bulge in monomers aand b of the P7S mutant (C2221) was increased byone residue, causing the second part of β-strand Ato be shorter (Fig. 4a and b). The main-chainconformations at residues 7 and 8 of the WT andin monomer c of the C2221 structure are the same. Itis worth mentioning that when this region wascompared with that of monomers a and b of the P7Sstructures, we noted that they had undergone aconformational change (Fig. 3c, inset) that hadstrengthened the interaction with strand B andweakened the interaction with the C-terminus.When this change occurred (Fig. 4a, inset), threeadditional hydrogen bonds were established with

286 Molecular Mechanism of Light–Chain Fibrillogenesis
strand B (OG-Ser7···N-Ser21, OG-Ser7···O-Ser21, andN-Ser7···O-Ser21); therefore, three hydrogen bondswere lost at the C-terminal region. Overall, these
Fig. 4. Structural analysis of the N-terminal region of thdiagrams of monomer a or monomer b and monomer c, respterminus (A) are shown in orange. The size of the second part2Fo−Fc map showing the conformational change that allowbetween Ser7 and Ser21 (OG-Ser7···N-Ser21, OG-Ser7···O-Ser3.03 Å, respectively). These are not established in monomer c (ihydrogen bonds are increased to 3.9, 4.0 and 3.8 Å, respective
contacts generated the conformational change that isassociated with the rotations of approximately 13and 5 deg at the interface for the C2221 and the
e P7S mutant. (a and b) Molecular surfaces and ribbonectively. The strands (B and G) that interact with the N-of strand A is longer in monomer c (C2221). Inset in (a) iss the establishment of three additional hydrogen bonds21, and N-Ser7···O-Ser21, with distances of 2.52, 3.5 andnset b), where the distances between the atoms involved inly.

287Molecular Mechanism of Light–Chain Fibrillogenesis
P212121 cells, respectively, as described above(Fig. 3d).Analysis of the intermolecular packing contacts
for the three structures revealed different interac-tions. The most relevant contacts were observed inthe P7S (C2221) molecule, where antiparallel β
Fig. 5. Crystal-packing interactions. (a) Interactions of therun near perpendicular to the longitudinal axes of a putativeprotein does not show this β–β motif.
interactions between monomer b and a symmetry-related molecule bsym and between monomer c anda symmetry molecule csym were established. Thestrands involved in this interaction, B and the N-terminus, generate a periodic stack with strands thatare near perpendicular to the direction of the
C2221 structure where the β–β motifs (b–bsym and c–csym)filament. Insets show these motifs in detail. (b) The native

288 Molecular Mechanism of Light–Chain Fibrillogenesis
longitudinal array (Fig. 5a). This organization wasnot observed for the other structures as shown inFig. 5b for the native protein.
Discussion
Although we are only beginning to understandthe subtleties involved in the conversion of nativestates into amyloid fibrils, it has been proposed thatnon-native species play a central role in VLfibrillogenesis.6–8,11,12,14,17 Nonetheless, little infor-mation is available about the regions that areinvolved in the conformational changes that pro-mote aggregation. Several reports indicate thatproteins have acquired sequence and structuraladaptations that enable them to avoid undesiredprotein aggregation and the formation of amyloid-like fibrils. Three anti-aggregation motifs have beendescribed in the VL, two β-bulges corresponding tothe N- and C-terminal regions (strands A and G)and the loop formed by residues 40–60.19,20 In thiswork, we chose to study the N-terminus to evaluateits protective role against fibril formation, therebydemonstrating the importance of residue Pro7 in theconformation and stability of the VLλ6 domain.P7S and the double mutant (P7S–H8S) were less
thermodynamically stable than WT 6aJL2, whereasthe mutations H8S and H8P had only a minor effecton stability (Table 1). The thermodynamic conse-quence of the P7S mutation supports the postulatethat the N-terminus is involved in the network ofinteractions that cooperatively stabilize the VLdomain, as we previously suggested.21 In additionto the interactions with β-strands B and G from theβ-sandwich, the N-terminus is involved in contactswith the hydrophobic core of the domain. Here, wehave demonstrated that the P7S mutation modifiesthe network of H-bonds between the N-terminusand β-strands B and G (see the inset in Fig. 4a andb). In line with our findings, it has previously beendemonstrated that proteolytic removal of the N-terminal segment in a λ-type VL protein results inthe destabilization of the domain, in associationwith local and long-distance structural effects.24
As previously established, we have observed acorrelation between the thermodynamic stabilityand the kinetics of the in vitro formation of fibrilsfrom the soluble precursors.5,7–9,12,21 The tlag valuesof the destabilized mutants P7S and P7S–H8S were18 and 7 times shorter, respectively, than the tlag of6aJL2. In contrast, the kinetics of fibril formation byH8Pwas similar to that observed for theWT protein,whereas it was slightly faster for H8S (Fig. 1c; Table1). Additionally, the P7S substitution increased theability of the protein to elongate the fibril seeds. Thelarger amount of P7S fibril that accumulated at theequilibrium point, as determined by ThT fluores-cence, is indicative of a lower critical concentration(Cr) of the soluble monomers, reflecting an aggre-gation reaction that is thermodynamically morefavorable than that for the WT.25
The mutants H8P and H8S elongate the fibrilspoorly, suggesting that His8 plays some role instabilizing the structure of the fibril (Fig. 1; Table 1).In contrast, the P7S mutation increased the rateconstant for the polymerization of the fibrils. Theanti-aggregation role that has been proposed for theN-terminal segment20,26 suggests that some sort ofstructural reorganization, in which this region playsa critical role, is a key component of the aggregationmechanism. This is supported by the conformationalrearrangements observed at the N-terminus of theP7S mutant that reflect a main chain that is moreflexible near position 7 (Fig. 3c, inset). Recently, ithas been found that the elongation rate of 6aJL2fibrils increases significantly with increasingGdnHCl concentration, reaching a maximum thatcoincides approximately with the midpoint ofunfolding (Cm) of the native monomer and thenstarts to decrease.27 These findings indicate that thecompetent species that elongate the fibril are moreflexible and undergo conformational changes.It is interesting to note that the P7S mutant
crystallized in two orthorhombic space groups,P212121 and C2221. The first polymorph thatcontained a dimer in the ASU grew in a few daysas long, thin needles, whereas the C2221 crystalswere obtained in about 2 months, allowing theestablishment of interactions that involved mono-mers and dimers. In this regard, it is noted thatfibrillogenesis closely resembles the crystallizationprocess due to similarities in the nucleation-depen-dent kinetics of fibril formation in vitro and therequirement of a critical protein concentration,below which polymerization cannot occur.28 Asalient feature of our results is the presence of amonomer in the C2221 structure, which suggests alower affinity in the VL–VL interactions for the P7Smutant. Moreover, the conformational change at thesheet switch in P7S promoted the establishment ofthree additional hydrogen bonds between Ser7 ofthe N-terminus and Ser21 of β-strand B, therebyincreasing the intramolecular interactions with thisstrand and decreasing the interaction with the C-terminal region (Fig. 4a, inset). All these changesgenerated a BJ dimer, which was characterized byan interface rotation of approximately 13 deg (Fig.3d). These findings strongly suggest that Pro7 helpsstabilize the sheet switch locally and that it iscritically involved in the contacts that stabilize theVL interface through long-distance interactions. Theincreased fibrillogenic potential of the P7S and P7S–H8S mutants could also be explained by thedestabilization of the VL–VL dimer interface, inconsonance with data presented by others.5,16,29
Crystal-packing analysis of the three structuresshowed that only for the C2221 polymorph did therearrangement of the interface enable the establish-ment of two antiparallel motifs between β-strandsAand B of one molecule with the same regions in asymmetrically related partner. These are reminis-cent of the β–β interactions postulated for the cross-β structure in several amyloid fibers.30–34 Throughthese interactions, the presence of a putative

289Molecular Mechanism of Light–Chain Fibrillogenesis
filament was evinced, wherein the β–β motifs runnear perpendicular to its longitudinal axis, as shownin Fig. 5a. Nevertheless, it should be noted that suchsignificant intermolecular head-to-head interactionsare not complemented by tail-to-tail interactions (asare typical of amyloid fibers), limiting the periodic-ity of the β–β interactions. A noteworthy feature ofthe data is that these β–βmotifs were only observedbetween monomers b–bsym and c–csym of the C2221structure. This arrangement, which clearly does notsatisfy the periodicity predicted for the fibril cross-βstructure, suggests that VL fibrillogenesis proceedsvia a more complex conformational adjustment thatinvolves other regions of the domain as well. One ofthe implications of our results is that the modificationof the first β-bulge to deprotect strand B allows theestablishment of intermolecular interactions that mayrelate to the mechanism of LC fibril formation. Basedon our findings, we suggest that additionalmolecularevents that expose one or both of theβ edge strandsCandD are required tomake theVL domain competentfor amyloid aggregation.
Materials and Methods
Site-directed mutagenesis
The expression vector containing the sequence of theVLλ6 6aJL2 gene was generated as described previously.21
The 6aJL2-P7S, 6aJL2-H8S, 6aJL2-H8P, and 6aJL2-P7S–H8S genes were generated by polymerase chain reaction(PCR) using a two-step strategy: First, a mutagenic PCRwas performed using the 6aJL2 gene as template and theproper combination of mutagenic forward primers andthe reverse primer 6a-NotI. In the second step, theresultant product was used in conjunction with theprimers 6a-SfiI (forward) and 6a-NotI (reverse) to assem-ble the entire mutated gene by PCR.
Cloning, expression, and purification
The mutant genes were cloned as described previously.21
Briefly, the PCR product was purified, digested with SfiIand NotI restriction enzymes (New England Biolabs,Beverly, MA), and ligated into the SfiI/NotI-digestedpSyn1 expression vector.35 The vector was electroporatedinto Escherichia coli electrocompetent cells, independentclones were selected, and the construct was verified byDNA sequencing.36 WT 6aJL2 and its mutants wereexpressed and purified as described previously.21 Theprotein purity was verified using SDS-PAGE electrophore-sis, and the identity of the purified protein was establishedby electrospray ionization ion-trapmass spectrometry in anLCQmass spectrometer equipped with a nanospray source(Thermo Fisher Scientific, Waltham, MA) (results notshown). The concentration of the purified proteins wasdetermined spectrophotometrically at 280 nm in 6.5 MGdnHCl and 20 mM sodium phosphate buffer, pH 7.5,using molar extinction coefficients of 13,494 M−1 cm−1, ascalculated from the amino acid sequence using the softwareProtParam, which is available at the ExPASy Web site‡.
‡http://ca.expasy.org
GdnHCl and thermally induced equilibrium unfolding
Chemical unfolding studies of 6aJL2 mutants wereperformed as follows: Protein samples (4.1 μM) wereincubated at 25 °C overnight in the presence of increasingconcentrations (0–3 M) of GdnHCl buffered with 20 mMsodium phosphate at pH 7.5. Fluorescence emissionspectra were determined at 25 °C from 310 to 410 nmusing an excitation wavelength of 290 nm and an LS50Bspectrofluorimeter (Perkin Elmer, Norwalk, CT) equippedwith a thermostated cell compartment. Thermodynamicparameters were calculated from fluorescence data usingnon-linear least-squares fitting to the following equationconsidering a two-state model between the native state(N) and the unfolded state (U), N↔U:
yobs =yN + mN D½ �ð Þ + yU + mU D½ �ð Þe−
�DG0
H2Oþm=RT
�
1 + e−�DG0
H2Oþm=RT
� ð1Þ
where yobs is themeasured fluorescence intensity at 350 nm,yN is the fluorescence intensity of the native protein in thepretransition range, and yU is the fluorescence intensity ofthe unfolded protein at high concentrations of GdnHCl.DG0
H2O is the free energy of unfolding in the absence ofdenaturant. D is the concentration of GdnHCl, and m isδΔG/δ[D], the free energy change permole of denaturant.RandT are the ideal gas constant and absolute temperature inKelvin, respectively. The pretransition and posttransitionbaselines are described by the linear slopes mN and mU,respectively.37,38 Cm, the denaturant concentration whereΔG=0, was calculated from:
Cm = − DGH20 =m
Thermal unfolding experiments were performed as de-scribed previously,9,21 with samples containing 50 μgprotein/ml in phosphate-buffered saline (PBS). The datafrom temperature-induced unfolding were analyzed asdescribed by Pace et al.39 The equilibrium constant (K) wascalculated using:
K = yn − yð Þ y − ydð Þ ð2Þwhere y is the measured tryptophan fluorescence intensityand yn and yd are the y-intercepts for the native anddenatured forms of the protein, respectively. The freeenergy,ΔG, at each temperaturewas calculatedbyapplyingthe following equation:
DG = − RTlnK ð3ÞThe midpoint temperature (Tm) and the enthalpy (ΔHm)and entropy (ΔSm) changes at theTmwere determined froma plot ofΔG versus T, because at Tm,ΔG=0 andΔHm=(Tm)(DSm). The value of ΔG at room temperature (25 °C) wascalculated as described by Pace et al.39
DG Tð Þ = DHm 1 − T=Tmð Þ − DCp Tm − Tð Þ + Tln T=Tmð Þ½ �ð4Þ
where T is 298 K and ΔCp is the heat capacity changeassociatedwith unfolding. The latter is calculated accordingto the method of Milardi et al.40
In vitro fibril formation
In vitro fibrillogenesis from the soluble precursor wasperformed using a modification of a method described

290 Molecular Mechanism of Light–Chain Fibrillogenesis
elsewhere.7 An appropriate volume of 8.3 μM proteinsolution in PBS, pH 7.4, containing 20 μM ThT (Sigma-Aldrich) and 0.05% sodium azide was added to a 4.5-mlcell and incubated at 37 °C for 5 min. The fluorescenceemission was recorded at 482 nm every 15–30 s using anexcitation wavelength of 450 nm (the excitation andemission slit widths were 2.5 and 5.0 nm, respectively).
Seeding assay
Amyloid fibrils were prepared at 37 °C by incubating 1to 2 mg/ml of protein solutions in PBS, pH 7.4, containing0.05% sodium azide with continuous stirring. After thecompletion of fibrillogenesis (48 h), the aggregates wereharvested by centrifugation. The fibril pellet was washedtwice with PBS, pH 7.4, and the concentration wasdetermined using the Micro BCA™ Protein AssayReagent Kit (Pierce, Rockford, IL). The seeds wereprepared by sonication of a 240-μg/ml dilution of astock fibril suspension using a Branson Sonifier 450(Branson Ultrasonics Co., Danbury, CT). The fibrilsuspension was kept on ice throughout the fragmentationprocedure.Mixtures containing 18 μM soluble monomer in PBS,
21.6 μg/ml of seeds, and 33 μM ThT were prepared on iceat 4 °C. One hundred microliters of each mixture wasapplied to each well of a 96-well, flat-bottom Ultra-LowAttachment Polystyrene Plate (Ultra-Low Cluster, Corn-ing Incorporated, Corning, NY), sealed with Mylar platesealers (Dynex), and loaded into the fluorescence platereader at 37 °C. The fluorescence data were measured at15-min intervals using a multiplate reader. The excitationand emission wavelength filters were 450 and 485 nm,respectively.
Electron microscopy
After completion of the fibrillogenesis assays, weapplied 10 μl from each sample to a Formvar-coated(200-mesh) copper grid, stabilized with evaporated carbonfilm (Electron Microscopy Science, Fort Washington, PA),for 1–3 min. The excess liquid was removed with a smallfilter paper strip, and the grids were negatively stained byfloating them onto a drop of freshly prepared 4% uranylacetate for 1–2 min. Electron microscopy was performed at80 kV on a Zeiss EM900 Transmission Electron Micro-scope. Images were recorded by means of a CCDDualVision 300W camera at a resolution of 1030×1300pixels (Gatan, Pleasanton, CA).
Crystallization of the recombinant proteins
Native 6aJL2 and the P7S mutant were crystallizedusing hanging-drop vapor-diffusion techniques at 18 °C.For the native protein, the best results were obtained indroplets composed by mixing equal volumes of 10 mg/mlof protein solution in PBS with 100 mM Mes, pH 6.5, and1.6–2.0 M sodium acetate. Crystals of the P7S mutant wereobtained under two conditions using a protein concentra-tion of 7 mg/ml. Under the first condition, 0.1 M sodiumcacodylate and 1.4 M sodium acetate, pH 6.5, were used asthe precipitant. These crystals were long, thin needles withan orthorhombic P212121 space group and a BJ dimer inthe ASU. Larger crystals with a different morphology, butthat were also orthorhombic (C2221), with three moleculesin the ASU, were grown using 0.1 M Mes, pH 6.5, and2.0 M sodium acetate.
Data collection and processing
Diffraction data of the P7S crystal (C2221) weremeasured in-house using an R-Axis II-C image-platedetector mounted on a Rigaku rotating-anode generator(Cu–Kα, λ=1.5416 Å) at 100 K. Diffraction images wereprocessed and scaled using MOSFLM41 and SCALA in theCCP4 suite.42 Native 6aJL2 and P7S (P212121) crystals weretaken to the National Synchrotron Light Source (Upton,NY), and data were collected at 100 K using a Quantum210 CCD detector (Area Detector System Corporation,Poway, CA) at beamline X6A. The data for these crystalswere finally processed and scaled using the programsindicated above. The three crystals were cryoprotected byincreasing the concentration of glycerol to 30%.
Structural determination and refinement
The phases for the three crystals were determined bymolecular replacement. For native 6aJL2 and P7S(P212121), we used the coordinates of a monomer of thehuman λ6 WIL VL (PDB ID 2CD0)42 and the programPHASER43 from CCP4.44 The coordinates of a monomer ofthe refined P7S mutant (P212121) were used to solve thestructure of the P7S mutant (C2221) with PHASER. In therefinement of the native 6aJL2 structure, we used bothrestrained non-crystallographic symmetry and transla-tion–libration–screw-rotation tensor refinement, as imple-mented in PHENIX.45 For the P7S mutant (P212121), werefined the structure using REFMAC46 and CNS47 for thepolymorph C2221. The crystallographic refinement statis-tics are shown in Table 2.Complete models were built using the program
COOT.48 Water molecules were added near the end ofthe refinement by a search procedure based on peaksobserved in the difference maps and hydrogen-bondcriteria. The stereochemistry and the agreement betweenthe model and the X-ray data were verified using COOT48
and PROCHECK.49 Molecular comparisons were per-formed with ALIGN,50 and the figures were preparedwith PyMOL51 and Chimera.52 The interfacial areas on thedimers and different arrangements were calculated usingPISA23 and CONTACT in CNS.47 The distance calcula-tions considered residues within 4.0 Å of the proteinmolecules.
Accession numbers
The atomic coordinates and structure factors of the finalmodels have been deposited in the Research Collabora-tory for Structural Bioinformatics PDB with accessionnumber 2W0K for native 6aJL2 and accession numbers3B5G and 3BDX for the P212121 and C2221 polymorphs ofthe P7S mutant, respectively.
Acknowledgements
This work was supported in part by grants fromDirección General de Asuntos del Personal Acadé-mico-Universidad Nacional Autónoma de México(UNAM) (IN 209506-3 to A.R.-R. and IN220707-3 toB.B.) and from Consejo Nacional de Ciencia yTecnología (82947 to A.R.-R. and D44122Q to B.B.).

291Molecular Mechanism of Light–Chain Fibrillogenesis
Experimental data corresponding to PDB ID 3BDXwere collected at LUEP-IQ-UNAM. Other data weremeasured at beamline X6A of the National Syn-chrotron Light Source. We thank Vivian Stojanoffand Jean Jankovick for their help during datacollections at line X6A-NSLS. The X6A beamline isfunded by the National Institute of General MedicalSciences under agreement GM#0080. The NationalSynchrotron Light Source is supported by the U.S.Department of Energy through contract no. DE-AC02-98CH10886. Finally, we thank LeopoldoGuereca, BSc, Rosalba Sánchez, and Oscar Villafrom IBT-UNAM for expression and purification ofthe variable domains used in this work and formass spectrometry analysis of the proteins.
References
1. Buxbaum, J. (1992). Mechanisms of disease: monoclo-nal immunoglobulin deposition, amyloidosis, lightchain deposition disease, and light and heavy chaindeposition disease. Hematol. Oncol. Clin. North Am. 6,323–346.
2. Cohen, A. S. (1993). Primary (AL) amyloidosis. RenalFailure, 15, 429–433.
3. Skinner, M. (2000). AL amyloidosis: the last 30 years.Amyloid: Int. J. Exp. Clin. Invest. 7, 13–14.
4. Solomon, A. (1986). Clinical implications ofmonoclonallight chains. Semin. Oncol. 13, 341–349.
5. Baden, E. M., Randles, E. G., Aboagye, A. K.,Thompson, J. R. & Ramirez-Alvarado, M. (2008).Structural insights into the role of mutations inamyloidosis. J. Biol. Chem. 283, 30950–30956.
6. Dealwis, C. & Wall, J. (2004). Towards understandingthe structure–function relationship of human amyloiddisease. Curr. Drug Targets, 5, 159–171.
7. Hurle, M. R., Helms, L. R., Li, L., Chan, W. & Wetzel,R. (1994). A role for destabilizing amino acid replace-ments in light chain amyloidosis. Proc. Natl Acad. Sci.USA, 91, 5446–5450.
8. Raffen, R., Dieckman, L. J., Szpunar, M., Wunschl, C.,Pokkuluri, P. R., Dave, P. et al. (1999). Physicochemicalconsequences of amino acid variations that contributeto fibril formation by immunoglobulin light chain.Protein Sci. 8, 509–517.
9. Wall, J., Schell, M., Murphy, C., Hrncic, R., Stevens,F. J. & Solomon, A. (1999). Thermodynamic insta-bility of human λ6 light chain: correlation withfibrillogenicity. Biochemistry, 38, 14101–14108.
10. Idicula-Thomas, S. & Balaji, P. V. (2005). Understand-ing the relationship between the primary structure ofproteins and their amyloidogenic propensity: cluesfrom inclusion body formation. Protein Eng. Des. Sel.18, 175–180.
11. Khurana, R., Gillespie, J. R., Talapatra, A., Minert, L.J., Ionescu-Zanetti, C., Millett, I. & Fink, A. L. (2001).Partially folded intermediates as critical precursors oflight chain amyloid fibrils and amorphous aggregates.Biochemistry, 40, 3525–3535.
12. Kim, Y. S., Cape, S. P., Chi, E., Raffen, R., Wilkins-Stevens, P., Stevens, F. J. et al. (2001). Counteractingeffects of renal solutes on amyloid fibril formation byimmunoglobulin light chains. J. Biol. Chem. 276,1626–1633.
13. Klafki, H. W., Pick, A. I., Pardowitz, I., Cole, T., Awni,L. A., Barnikol, H. U. et al. (1993). Reduction of
disulfide bonds in an amyloidogenic Bence–Jonesprotein leads to formation of “amyloid-like” fibrils invitro. Biol. Chem. Hoppe-Seyler, 374, 1117–1122.
14. Qin, Z., Hu, D., Zhu, M. & Fink, A. L. (2007).Structural characterization of the partially foldedintermediates of an immunoglobulin light chainleading to amyloid fibrillation and amorphous aggre-gation. Biochemistry, 46, 3521–3531.
15. Rostagno, A., Vidal, R., Kaplan, B., Chuba, J., Kumar,A., Elliott, J. I. et al. (1999). pH-dependent fibrillogen-esis of a VκIII Bence–Jones protein. Br. J. Haematol. 107,835–843.
16. Souillac, P. O., Uversky, V. N., Millett, I. S., Khurana,R., Doniach, S. & Fink, A. L. (2002). Effect ofassociation state and conformation stability on thekinetics of immunoglobulin light chain amyloid fibrilformation at physiological pH. J. Biol. Chem. 277,12657–12665.
17. Uversky, V. N. & Fink, A. L. (2004). Conformationalconstraints for amyloid fibrillation: the importance ofbeing unfolded. Biochim. Biophys. Acta, 1698, 131–153.
18. Schiffer, M. (1996). Molecular anatomy and thepathological expression of antibody light chains.Am. J. Pathol. 148, 1397–1406.
19. Richardson, J. S. & Richardson, D. C. (2002). Naturalβ-sheet proteins use negative design to avoid edge-to-edge aggregation. Proc. Natl Acad. Sci. USA, 99,2754–2759.
20. Nowak, M. (2004). Immunoglobulin kappa light chainand its amyloidogenic mutants: a molecular dynamicsstudy. Proteins, 55, 11–21.
21. del Pozo Yauner, L., Ortiz, E., Sanchez, R., Sanchez-Lopez, R., Guereca, L., Murphy, C. L. et al. (2008).Influence of the germline sequence on the thermody-namic stability and fibrillogenicity of human lambda 6light chains. Proteins, 72, 684–692.
22. del Pozo Yauner, L., Ortiz, E. & Becerril, B. (2006). TheCDR1 of the human lambdaVI light chains adopts anew canonical structure. Proteins, 62, 122–129.
23. Krissinel, E. & Henrick, K. (2007). Inference ofmacromolecular assemblies from crystalline state. J.Mol. Biol. 372, 774–797.
24. Krol, M., Roterman, I., Piekarska, B., Konieczny, L.,Rybarska, J. & Stopa, B. (2003). Local and long-rangestructural effects caused by the removal of the N-terminal polypeptide fragment from immunoglobulinL chain k. Biopolymers, 69, 189–200.
25. Wetzel, R. (2006). Kinetics and thermodynamics ofamyloid fibril assembly. Acc. Chem. Res. 39, 671–679.
26. O'Nuallain, B., Allen, A., Kennel, S. J., Weiss, D. T.,Solomon, A. & Wall, J. S. (2007). Localization of aconformational epitope common to non-native andfibrillar immunoglobulin light chains. Biochemistry, 46,1240–1247.
27. Blancas-Mejia, L. M., Tellez, L. A., del Pozo-Yauner,L., Becerril, B., Sanchez-Ruiz, J. M. & Fernandez-Velasco, D. A. (2009). Thermodynamic and kineticcharacterization of a germ line human lambda6 light-chain protein: the relation between unfolding andfibrillogenesis. J. Mol. Biol. 386, 1153–1166.
28. Makino, D. L., Henschen-Edman, A. H., Larson, S. B.& McPherson, A. (2007). Bence–Jones KWR proteinstructures determined by X-ray crystallography. ActaCrystallogr., Sect. D: Biol. Crystallogr. 63, 780–792.
29. Baden, E. M., Owen, B. A., Peterson, F. C., Volkman,B. F., Ramirez-Alvarado, M. & Thompson, J. R.(2008). Altered dimer interface decreases stabilityin an amyloidogenic protein. J. Biol. Chem. 283,15853–15860.

292 Molecular Mechanism of Light–Chain Fibrillogenesis
30. Nelson, R., Sawaya, M. R., Balbirnie, M., Madsen,A. O., Riekel, C., Grothe, R. & Eisenberg, D. (2005).Structure of the cross-beta spine of amyloid-likefibrils. Nature, 435, 773–778.
31. Sawaya, M. R., Sambashivan, S., Nelson, R., Ivanova,M. I., Sievers, S. A., Apostol, M. I. et al. (2007). Atomicstructures of amyloid cross-beta spines reveal variedsteric zippers. Nature, 447, 453–457.
32. Wiltzius, J. J., Sievers, S. A., Sawaya, M. R., Cascio, D.,Popov, D., Riekel, C. & Eisenberg, D. (2008). Atomicstructure of the cross-beta spine of islet amyloidpolypeptide (amylin). Protein Sci. 17, 1467–1474.
33. Janowski, R., Kozak, M., Abrahamson, M., Grubb, A.& Jaskolski, M. (2005). 3D domain-swapped humancystatin C with amyloid-like intermolecular beta-sheets. Proteins, 61, 570–578.
34. Nelson, R. & Eisenberg, D. (2006). Recent atomicmodels of amyloid fibril structure. Curr. Opin. Struct.Biol. 16, 260–265.
35. Schier, R., Marks, J. D., Wolf, E. J., Apell, G., Wong, C.,McCartney, J. E. et al. (1995). In vitro and in vivocharacterization of a human anti-c-erbB-2 single-chainFv isolated from a filamentous phage antibodylibrary. Immunotechnology, 1, 73–81.
36. Sanger, F., Nicklen, S. & Coulson, A. R. (1977). DNAsequencing with chain-terminating inhibitors. Proc.Natl Acad. Sci. USA, 74, 5463–5467.
37. Bolen, D. W. & Santoro, M. M. (1988). Unfolding freeenergy changes determined by the linear extrapola-tion method: 2. Incorporation of delta G degrees N–Uvalues in a thermodynamic cycle. Biochemistry, 27,8069–8074.
38. Santoro, M. M. & Bolen, D. W. (1988). Unfolding freeenergy changes determined by the linear extrapola-tion method: 1. Unfolding of phenylmethanesulfonylalpha-chymotrypsin using different denaturants. Bio-chemistry, 27, 8063–8068.
39. Pace, C. N., Shirley, B. A. & Thomson, J. A. (1989).Measuring the conformational stability of a protein. InProtein structure: a practical approach. Te, C., (ed), IRLPress, Oxford, UK.
40. Milardi, D., la Rosa, C., Fasone, S. & Grasso, D. (1997).An alternative approach in the structure-based pre-dictions of the thermodynamics of protein unfolding.Biophys. Chem. 69, 43–51.
41. Leslie, A. G. (1999). Integration of macromoleculardiffraction data.ActaCrystallogr., Sect. D: Biol. Crystallogr.55, 1696–1702.
42. Pokkuluri, P. R., Solomon,A.,Weiss, D. T., Stevens, F. J.& Schiffer, M. (1999). Tertiary structure of human λ6light chains.Amyloid: Int. J. Exp. Clin. Invest. 6, 165–171.
43. McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D.,Winn, M. D., Storoni, L. C. & Read, R. J. (2007). Phasercrystallographic software. J. Appl. Crystallogr. 40,658–674.
44. Collaborative Computational Project 4. (1994). TheCCP4 suite: programs for protein crystallography.Acta Crystallogr., Sect. D: Biol. Crystallogr. 50, 760–763.
45. Adams, P. D., Grosse-Kunstleve, R. W., Hung, L.-W.,Ioerger, T. R.,McCoy,A. J.,Moriarty, N.W. et al. (2002).PHENIX: building new software for automated crys-tallographic structure determination. Acta Crystallogr.,Sect. D: Biol. Crystallogr. 58, 1948–1954.
46. Winn, M. D., Isupov, M. N. & Murshudov, G. N.(2001). Use of TLS parameters to model anisotropicdisplacements in macromolecular refinement. ActaCrystallogr., Sect. D: Biol. Crystallogr. 57, 122–133.
47. Brunger, A. T., Adams, P. D., Clore, G. M., DeLano,W. L., Gros, P., Grosse-Kunstleve, R. W. et al. (1998).Crystallography and NMR System: a new softwaresuite for macromolecular structure determination.Acta Crystallogr., Sect. D: Biol. Crystallogr. 54, 905–921.
48. Emsley, P. & Cowtan, K. (2004). Coot: model-buildingtools for molecular graphics. Acta Crystallogr., Sect. D:Biol. Crystallogr. 60, 2126–2132.
49. Laskowski, R. A., MacArthur, M. W., Moss, D. S. &Thornton, J. M. (1993). PROCHECK: a program tocheck the stereochemical quality of protein structures.J. Appl. Crystallogr. 26, 283–291.
50. Cohen, G. H. (1997). ALIGN: a program to superim-pose protein coordinates, accounting for insertionsand deletions. J. Appl. Crystallogr. 30, 1160–1161.
51. DeLano, W. L. (2002). The PyMOL Molecular GraphicsSystem.DeLano Scientific, San Carlos, CA; http://pymol.sourceforge.net/.
52. Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch,G. S., Greenblatt, D. M., Meng, E. C. & Ferrin, T. E.(2004). UCSF Chimera—a visualization system forexploratory research and analysis. J. Comput. Chem. 25,1605–1612.
Copyright © 2022 FDOKUMEN




















