
A comparison of depression, anxiety, and health status in patients with progressive supranuclear...
20
Brief Reports A Comparison of Depression, Anxiety, and Health Status in Patients with Progressive Supranuclear Palsy and Multiple System Atrophy Anette Schrag, MD, PhD, 1 Shamira Sheikh, BSc, 1 Niall P. Quinn, MD, 1 Andrew J. Lees, MD, 1 Caroline Selai, PhD, 1 Chris Mathias, MD, 1,2 Irene Litvan, MD, 3 Anthony E. Lang, MD, 4 James H. Bower, MD, 5 David J. Burn, MD, 6 Philip Low, MD, 7 and Marjan Jahanshahi, PhD 1 * 1 Institute of Neurology, University College London, London; 2 Neurovascular Medicine Unit, Faculty of Medicine, Imperial College London, London, United Kingdom; 3 Movement Disorder Program, Department of Neurology, University of Louisville, Louisville, Kentucky, USA; 4 Division of Neurology, Toronto Western Hospital and the University of Toronto, Toronto, Ontario, Canada; 5 Department of Neurology, Mayo Clinic College of Medicine, Rochester, Minnesota, USA; 6 Institute for Ageing and Health, Newcastle University, Newcastle Upon Tyne, United Kingdom; 7 Department of Neurology, Mayo Clinic College of Medicine, Rochester, Minnesota Abstract: The objective of this study was to compare sub- jective health status and its correlates in progressive supranuclear palsy (PSP) and multiple system atrophy (MSA). One hundred eighty-eight patients with PSP and 286 patients with MSA completed EQ-5D and Hospital Depression and Anxiety Scale. The impact on mobility, usual activities, and self-care was similarly high in both groups after similar duration. Fifty-six percent of PSP and 43% of MSA had probable depression, and 37% of both groups had probable anxiety. Patients with PSP had signif- icantly higher depression scores, but groups did not differ in anxiety scores. Patients with MSA had significantly greater pain/discomfort than patients with PSP. The most important association with subjective health status was with depressive symptoms, which accounted for 38% and 29% of EQ-5D variance in patients with PSP and MSA, followed by disease severity and anxiety scores. We con- clude that depressive symptoms were common in both dis- orders, but more severe in PSP. Anxiety symptoms affected 37% of patients in both groups and contributed to impaired subjective health status. Pain was more problem- atic in MSA than PSP. Ó 2010 Movement Disorder Society Key words: health status; multiple system atrophy; pro- gressive supranuclear palsy; parkinsonism; depression; anxi- ety; quality of life Multiple system atrophy (MSA) and progressive supranuclear palsy (PSP) are two common types of atypical parkinsonism. Management is difficult but needs to be aimed at the most important clinical aspects that have an impact on the health status of patients. Few studies have assessed the impact of PSP and MSA on psychological well-being and health sta- tus, 1–3 and no direct comparison of health status between these disorders has been undertaken. We com- pared health status and its correlates in PSP and MSA to identify the most important targets for clinical man- agement. We hypothesized that after similar disease duration, health status would be severely and similarly affected in both groups, and that disease severity and disease duration would correlate negatively with health status in both groups. We predicted that depression and anxiety would occur but that the main association of poor health status would be with disease severity; and that depression and anxiety would contribute to a lesser extent to poor health status in both groups. PATIENTS AND METHODS We compared depressive and anxiety symptoms and health status in patients with PSP and patients with MSA from two surveys with similar methodology. 4,5 Patients were invited to participate either through the PSP (Europe) Association or the MSA Sarah Math- eson Trust (the UK) or recruited from neurology clinics *Correspondence to: Dr. Marjan Jahanshahi, Sobell Department of Motor Neuroscience and Movement Disorders, 33 Queen Square, London WC1N 3BG, United Kingdom. E-mail: m.jahanshahi@ion. ucl.ac.uk Potential conflict of interest: None reported. Received 26 January 2009; Revised 20 August 2009; Accepted 20 August 2009 Published online 13 April 2010 in Wiley InterScience (www. interscience.wiley.com). DOI: 10.1002/mds.22794 1077 Movement Disorders Vol. 25, No. 8, 2010, pp. 1077–1096 Ó 2010 Movement Disorder Society
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of A comparison of depression, anxiety, and health status in patients with progressive supranuclear...

Brief Reports
A Comparison of Depression,Anxiety, and Health Status inPatients with Progressive
Supranuclear Palsy and MultipleSystem Atrophy
Anette Schrag, MD, PhD,1 Shamira Sheikh, BSc,1
Niall P. Quinn, MD,1 Andrew J. Lees, MD,1
Caroline Selai, PhD,1
Chris Mathias, MD,1,2 Irene Litvan, MD,3
Anthony E. Lang, MD,4
James H. Bower, MD,5 David J. Burn, MD,6
Philip Low, MD,7
and Marjan Jahanshahi, PhD1*
1Institute of Neurology, University College London, London;2Neurovascular Medicine Unit, Faculty of Medicine, Imperial
College London, London, United Kingdom;3Movement Disorder Program, Department of Neurology,
University of Louisville, Louisville, Kentucky, USA;4Division of Neurology, Toronto Western Hospital and the
University of Toronto, Toronto, Ontario, Canada;5Department of Neurology, Mayo Clinic College of
Medicine, Rochester, Minnesota, USA; 6Institute for Ageingand Health, Newcastle University,
Newcastle Upon Tyne, United Kingdom; 7Department ofNeurology, Mayo Clinic College of Medicine,
Rochester, Minnesota
Abstract: The objective of this study was to compare sub-jective health status and its correlates in progressivesupranuclear palsy (PSP) and multiple system atrophy(MSA). One hundred eighty-eight patients with PSP and286 patients with MSA completed EQ-5D and HospitalDepression and Anxiety Scale. The impact on mobility,usual activities, and self-care was similarly high in bothgroups after similar duration. Fifty-six percent of PSP and43% of MSA had probable depression, and 37% of both
groups had probable anxiety. Patients with PSP had signif-icantly higher depression scores, but groups did not differin anxiety scores. Patients with MSA had significantlygreater pain/discomfort than patients with PSP. The mostimportant association with subjective health status waswith depressive symptoms, which accounted for 38% and29% of EQ-5D variance in patients with PSP and MSA,followed by disease severity and anxiety scores. We con-clude that depressive symptoms were common in both dis-orders, but more severe in PSP. Anxiety symptomsaffected 37% of patients in both groups and contributed toimpaired subjective health status. Pain was more problem-atic in MSA than PSP. � 2010 Movement Disorder Society
Key words: health status; multiple system atrophy; pro-gressive supranuclear palsy; parkinsonism; depression; anxi-ety; quality of life
Multiple system atrophy (MSA) and progressive
supranuclear palsy (PSP) are two common types of
atypical parkinsonism. Management is difficult but
needs to be aimed at the most important clinical
aspects that have an impact on the health status of
patients. Few studies have assessed the impact of PSP
and MSA on psychological well-being and health sta-
tus,1–3 and no direct comparison of health status
between these disorders has been undertaken. We com-
pared health status and its correlates in PSP and MSA
to identify the most important targets for clinical man-
agement. We hypothesized that after similar disease
duration, health status would be severely and similarly
affected in both groups, and that disease severity and
disease duration would correlate negatively with health
status in both groups. We predicted that depression and
anxiety would occur but that the main association of
poor health status would be with disease severity; and
that depression and anxiety would contribute to a
lesser extent to poor health status in both groups.
PATIENTS AND METHODS
We compared depressive and anxiety symptoms and
health status in patients with PSP and patients with MSA
from two surveys with similar methodology.4,5
Patients were invited to participate either through
the PSP (Europe) Association or the MSA Sarah Math-
eson Trust (the UK) or recruited from neurology clinics
*Correspondence to: Dr. Marjan Jahanshahi, Sobell Department ofMotor Neuroscience and Movement Disorders, 33 Queen Square,London WC1N 3BG, United Kingdom. E-mail: [email protected]
Potential conflict of interest: None reported.
Received 26 January 2009; Revised 20 August 2009; Accepted 20August 2009
Published online 13 April 2010 in Wiley InterScience (www.
interscience.wiley.com). DOI: 10.1002/mds.22794
1077
Movement DisordersVol. 25, No. 8, 2010, pp. 1077–1096� 2010 Movement Disorder Society

in United Kingdom and North-America. Details have
been described previously.4,5 All patients completed a
booklet of questionnaires which, in addition to demo-
graphic and clinical information, included the follow-
ing scales: The EQ-5D,6 a generic health status instru-
ment consisting of items on mobility, self care, usual
activities, pain/discomfort, and anxiety/depression,
from which a summary index can be calculated, and a
visual analogue scale (VAS) from 0 to 100, with 0
indicating the worst and 100 representing best health
status. The Hospital Anxiety and Depression Scale
(HADS)7 is a self-completed scale with an anxiety
(HADS-A) and a depression subscale (HADS-D).
Scores of less than 8 indicate no, 8 to 10 indicate pos-
sible, and 11 to 21 indicate probable anxiety or depres-
sion. Patients rated their MSA severity on a five-point
scale (no, some, moderate, severe problems with walk-
ing, balance, movement, coordination, bladder or sex-
ual function or dizziness, or wheelchair- or bed-bound)
or PSP severity on the six-point PSP staging system,8
modified for self completion. Except for the measures
of disease severity, the two studies used almost identi-
cal methodology with patient recruitment primarily
from patient organizations, use of the same generic
comparative scales and statistical analysis.
Statistical Analysis
Categorical data were analyzed using chi-square
tests. Means were compared with the Mann–Whitney
test. Spearman rank correlations were performed. Four
stepwise regression analyses were performed to iden-
tify the major associations of health status in patients
with PSP and patients with MSA with the EQ-5D
index and VAS as independent variables, entering age,
gender, disease duration, disease severity, HADS-D
and HADS-A subscores as dependent variables.
RESULTS
One hundred eighty-eight patients with PSP and
286 patients with MSA were included. Mean age was
68.8 years (SD 5 6.9) for the patients with PSP and
65.4 years (SD 5 9.8) for the patients with MSA (P 50.001). Mean disease duration was 5.7 years (SD 5 7.8)
for PSP and 6.3 years (SD 5 3.9; 0.3) for the MSA
group. Fifty-eight percent in both groups were men.
Depression and Anxiety
On the HADS-D scale, 55.6% of the patients with
PSP and 43.0% of the patients with MSA had probable
depression and an additional 19.8% of patients with
PSP and 28.1% of patients with MSA had possible
depression (P 5 0.03). The mean HADS-D scores
were 11.2 (SD 5 4.93) for the PSP and 10.1 (SD 54.06) for the MSA groups (P 5 0.01). On the HADS-
A scale, 37.3% of the patients with PSP and 37.3%
of the patients with MSA had probable anxiety and
an additional 14.5% of patients with PSP and 16.7%
of patients with MSA had possible anxiety (P 50.87). The mean HADS-A scores were 8.6 (SD 55.4) for the patients with PSP and 8.4 (SD 5 5.1) for
the patients with MSA (P 5 0.78). There were no
differences between the groups in anxiety items.
Patients with PSP significantly less often reported the
ability to enjoy a good book/radio/TV program (P <0.0001), and those with MSA significantly more often
reported they could still laugh and see the funny side
of things (P 5 0.009) and feel cheerful (P 5 0.001),
although they felt slowed down slightly more often
(P 5 0.05).
Health Status
The mean EQ-5D summary index score and VAS
scores are presented in Table 1. After a similar disease
duration, there was no difference in EQ-5D index
scores between the PSP and MSA groups. VAS scores
tended to be lower in MSA than in patients with PSP
with an average score of 39 (maximum 100) in MSA
and 42.8 in PSP (P 5 0.08). Responses to each of the
five items of the EQ-5D are presented in Table 2.
While similarly high numbers of patients had some or
severe problems in usual activities (96% in PSP and
98% in MSA), mobility (94 and 97%), self-care (88
and 87%), and anxiety/depression (65 and 68%),
patients with PSP tended to report problems with self-
care as severe more often (70 vs. 62%, P 5 0.07). In
contrast, pain was more frequent in MSA than PSP (56
vs. 78%, P < 0.001).
Correlates of Health Status in PSP and MSA
Age and disease duration did not correlate with
HADS-A or HADS-D or EQ-5D measures in either
disorder. Disease severity correlated significantly and
TABLE 1. EQ-5D mean scores (SD) in the PSP and theMSA groups
EQ5D PSP MSA P-value
n 188 286Summary index 0.23 (0.4) 0.20 (0.3) nsVAS 42.8 (24.6) 39.0 (20.3) 0.08
VAS, visual analogue scale.
1078 A. SCHRAG ET AL.
Movement Disorders, Vol. 25, No. 8, 2010

negatively with the EQ-5D summary index in patients
with PSP (r 5 20.58; P < 0.001) and patients with
MSA (r 5 20.51; P < 0.001). HADS-A and HADS-D
scores correlated negatively with the EQ-5D summary
index in the PSP (r 5 20.53 and r 5 20.60; P <0.001) and MSA groups (r 5 20.39 and r 5 20.55;
P < 0.001).
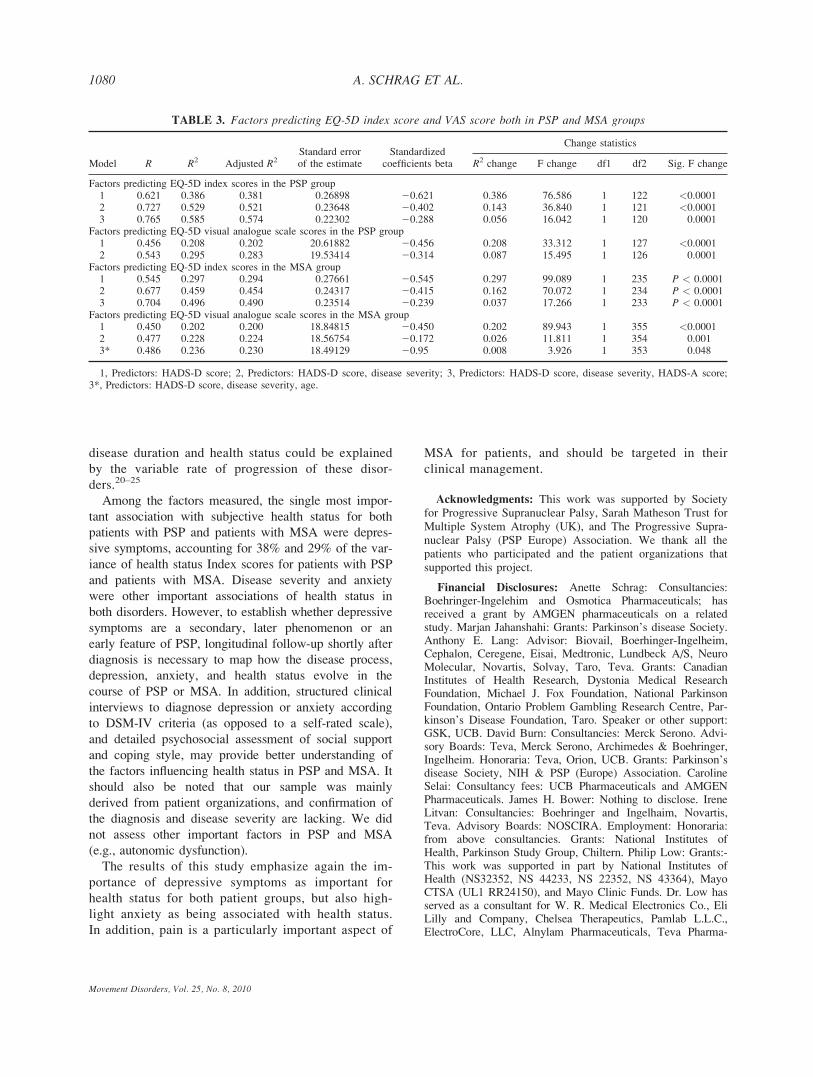
Using regression analysis, in the PSP group, 38% of
the variance of the EQ-5D Index Score was explained
by the HADS-D, 14% by disease severity and a further
5% by HADS-A (Table 3). For the EQ-5D VAS, 20%
of the variance was explained by HADS-D scores, and
a further 8% by disease severity (Table 3). In MSA,
29% of the variance of the EQ-5D Index Score was
explained by HADS-D, and a further 16% by disease
severity and 4% by HADS-A (Table 3). Twenty per-
cent of the variance of EQ-5D VAS was accounted
for by HADS-D and a further 2% by disease severity
(Table 3).
DISCUSSION
This is the first study to directly compare health sta-
tus in PSP and MSA. Both patients with PSP and
patients with MSA reported worse health status than
patients with Parkinson’s disease (PD) in a previous
study.9 Although both disorders had a similar impact
on the health status of patients, there were some qual-
itative differences. After a similar disease duration,
patients with PSP tended to have more difficulties
with self-care and had significantly more difficulties
with depressive symptoms. In contrast, patients with
MSA reported significantly greater pain/discomfort
than patients with PSP. The two groups did not differ
in terms of anxiety. Pain is an important aspect of
PD10–12 and MSA13,14 in which multiple mechanisms,
including hypoperfusion, rheumatic, sensory, dystonic
and off-period related aspects have been postulated.
In a previous study, MSA with predominant parkin-
sonism was associated with more pain than MSA with
predominant cerebellar ataxia.2 Difference between
pain in the MSA and PSP groups is likely to reflect
the different clinical presentation of PSP with rela-
tively less typical limb parkinsonism and more promi-
nent axial symptoms, less common response to levo-
dopa and fewer autonomic symptoms. However, we
did not explore the type and location of pain experi-
enced in MSA. Depressive symptoms, on the other
hand, were more problematic in PSP than MSA, and
this is agrees with studies reporting rates of depres-
sion in PSP from 40 to 70%,15–17 and in MSA from
16 to 39%.1,18,19 When comparing individual HADS
items, there were no differences in anxiety items, but
patients with PSP reported significantly more symp-
toms compatible with anhedonia and concentration/
apathy.
As expected, disease severity showed significant
negative correlations with the EQ-5D summary index
for both groups. In contrast to a previous finding in
PSP,3 disease duration did not correlate significantly
with health status for either the patients with PSP or
patients with MSA. This lack of association between
TABLE 2. Scores of EQ-5D dimensions in the PSP and MSA groups
PSP (%) MSA (%) P-value
EQ-5D No problems in walking about 6 3Some problems in walking about 75 79Confined to bed 19 18Total (some and severe problems) 94 97 ns
EQ-5D No problems with self-care 12 13Some problems washing or dressing myself 44 47Unable to wash or dress myself 44 40Total (some and severe problems) 88 87 ns
EQ-5D No problems with performing my usual activities 4 2Some problems with performing my usual 26 36Unable to perform my usual activities 70 62Total (some and severe problems) 96 98 0.07
EQ-5D No pain or discomfort 43 22Moderate pain or discomfort 46 61Extreme pain or discomfort 10 17Total (some and severe problems) 56 78 <0.001
EQ-5D Not anxious or depressed 35 32Moderately anxious or depressed 50 54Extremely anxious or depressed 15 14Total (some and severe problems) 65 68 ns
1079HEALTH-RELATED QUALITY OF LIFE IN MSA AND PSP
Movement Disorders, Vol. 25, No. 8, 2010
disease duration and health status could be explained
by the variable rate of progression of these disor-
ders.20–25
Among the factors measured, the single most impor-
tant association with subjective health status for both
patients with PSP and patients with MSA were depres-
sive symptoms, accounting for 38% and 29% of the var-
iance of health status Index scores for patients with PSP
and patients with MSA. Disease severity and anxiety
were other important associations of health status in
both disorders. However, to establish whether depressive
symptoms are a secondary, later phenomenon or an
early feature of PSP, longitudinal follow-up shortly after
diagnosis is necessary to map how the disease process,
depression, anxiety, and health status evolve in the
course of PSP or MSA. In addition, structured clinical
interviews to diagnose depression or anxiety according
to DSM-IV criteria (as opposed to a self-rated scale),
and detailed psychosocial assessment of social support
and coping style, may provide better understanding of
the factors influencing health status in PSP and MSA. It
should also be noted that our sample was mainly
derived from patient organizations, and confirmation of
the diagnosis and disease severity are lacking. We did
not assess other important factors in PSP and MSA
(e.g., autonomic dysfunction).
The results of this study emphasize again the im-
portance of depressive symptoms as important for
health status for both patient groups, but also high-
light anxiety as being associated with health status.
In addition, pain is a particularly important aspect of
MSA for patients, and should be targeted in their
clinical management.
Acknowledgments: This work was supported by Societyfor Progressive Supranuclear Palsy, Sarah Matheson Trust forMultiple System Atrophy (UK), and The Progressive Supra-nuclear Palsy (PSP Europe) Association. We thank all thepatients who participated and the patient organizations thatsupported this project.
Financial Disclosures: Anette Schrag: Consultancies:Boehringer-Ingelehim and Osmotica Pharmaceuticals; hasreceived a grant by AMGEN pharmaceuticals on a relatedstudy. Marjan Jahanshahi: Grants: Parkinson’s disease Society.Anthony E. Lang: Advisor: Biovail, Boerhinger-Ingelheim,Cephalon, Ceregene, Eisai, Medtronic, Lundbeck A/S, NeuroMolecular, Novartis, Solvay, Taro, Teva. Grants: CanadianInstitutes of Health Research, Dystonia Medical ResearchFoundation, Michael J. Fox Foundation, National ParkinsonFoundation, Ontario Problem Gambling Research Centre, Par-kinson’s Disease Foundation, Taro. Speaker or other support:GSK, UCB. David Burn: Consultancies: Merck Serono. Advi-sory Boards: Teva, Merck Serono, Archimedes & Boehringer,Ingelheim. Honoraria: Teva, Orion, UCB. Grants: Parkinson’sdisease Society, NIH & PSP (Europe) Association. CarolineSelai: Consultancy fees: UCB Pharmaceuticals and AMGENPharmaceuticals. James H. Bower: Nothing to disclose. IreneLitvan: Consultancies: Boehringer and Ingelhaim, Novartis,Teva. Advisory Boards: NOSCIRA. Employment: Honoraria:from above consultancies. Grants: National Institutes ofHealth, Parkinson Study Group, Chiltern. Philip Low: Grants:-This work was supported in part by National Institutes ofHealth (NS32352, NS 44233, NS 22352, NS 43364), MayoCTSA (UL1 RR24150), and Mayo Clinic Funds. Dr. Low hasserved as a consultant for W. R. Medical Electronics Co., EliLilly and Company, Chelsea Therapeutics, Pamlab L.L.C.,ElectroCore, LLC, Alnylam Pharmaceuticals, Teva Pharma-
TABLE 3. Factors predicting EQ-5D index score and VAS score both in PSP and MSA groups
Model R R2 Adjusted R2Standard errorof the estimate
Standardizedcoefficients beta
Change statistics
R2 change F change df1 df2 Sig. F change
Factors predicting EQ-5D index scores in the PSP group1 0.621 0.386 0.381 0.26898 20.621 0.386 76.586 1 122 <0.00012 0.727 0.529 0.521 0.23648 20.402 0.143 36.840 1 121 <0.00013 0.765 0.585 0.574 0.22302 20.288 0.056 16.042 1 120 0.0001
Factors predicting EQ-5D visual analogue scale scores in the PSP group1 0.456 0.208 0.202 20.61882 20.456 0.208 33.312 1 127 <0.00012 0.543 0.295 0.283 19.53414 20.314 0.087 15.495 1 126 0.0001
Factors predicting EQ-5D index scores in the MSA group1 0.545 0.297 0.294 0.27661 20.545 0.297 99.089 1 235 P < 0.00012 0.677 0.459 0.454 0.24317 20.415 0.162 70.072 1 234 P < 0.00013 0.704 0.496 0.490 0.23514 20.239 0.037 17.266 1 233 P < 0.0001
Factors predicting EQ-5D visual analogue scale scores in the MSA group1 0.450 0.202 0.200 18.84815 20.450 0.202 89.943 1 355 <0.00012 0.477 0.228 0.224 18.56754 20.172 0.026 11.811 1 354 0.0013* 0.486 0.236 0.230 18.49129 20.95 0.008 3.926 1 353 0.048
1, Predictors: HADS-D score; 2, Predictors: HADS-D score, disease severity; 3, Predictors: HADS-D score, disease severity, HADS-A score;3*, Predictors: HADS-D score, disease severity, age.
1080 A. SCHRAG ET AL.
Movement Disorders, Vol. 25, No. 8, 2010

ceutical Industries Ltd., AstraZeneca R&D Lund, FoldRxPharmaceuticals, Inc., Nordic Biotech Advisors Aps, SangamoBioSciences, Inc. Niall P. Quinn: Advisory board and Honora-ria: UCB. Honorarium: Orion Pharma. Chris Mathias: Consul-tancy: Chelsea Therapeutics. Andrew Lees: Consultancies: Ge-nus. Advisory Boards: Novartis, Teva, Meda, BoehringerIngelheim, GSK, Ipsen, Lundbeck, Allergan, Orion. Honoraria:Novartis, Teva, Meda, Boehringer Ingelheim, GSK, Ipsen,Lundbeck, Allergan, Orion. Grants: PSP Association, WestonTrust - The Reta Lila Howard Foundation. Employment: UCL/UCLH. Grant: Parkinson’s disease Society; Others: None.
Author Roles: Research Project: Conception: A. Schrag, M.Jahanshahi, C. Selai, N. Quinn; Organization: A. Schrag; Execu-tion: A. Schrag, N. Quinn, A. Lees, C. Mathias, I. Litvan, A.Lang, J. Bower, D. Burn, P. Low. Statistical analysis: Design:A. Schrag, M. Jahanshahi; Execution: A. Schrag; Review andcritique: A. Schrag, M. Jahanshahi. Manuscript: Writing of thefirst draft: S. Sheikh, M. Jahanshahi, A. Schrag; Review and cri-tique: M. Jahanshahi, A. Schrag, N. Quinn, A. Lees, C. Selai, C.Mathias, I. Litvan, A. Lang, J. Bower, D. Burn, P. Low.
REFERENCES
1. Benrud-Larson LM, Sandroni P, Schrag A, Low PA. Depressivesymptoms and life satisfaction in patients with multiple systematrophy. Mov Disord 2005;20:951–957.
2. SchragA, Geser F, Stampfer-KountchevM, et al. Health-related qualityof life inmultiple system atrophy.MovDisord 2006;21: 809–815.
3. Schrag A, Selai C, Davis J, Lees AJ, Jahanshahi M, Quinn N.Health-related quality of life in patients with progressive supra-nuclear palsy. Mov Disord 2003;18:1464–1469.
4. Schrag A, Selai C, Quinn N, et al. Measuring quality of life inPSP: the PSP-QoL. Neurology 2006;67:39–44.
5. Schrag A, Selai C, Mathias C, et al. Measuring health-related qualityof life in MSA: the MSA-QoL. Mov Disord 2007;22: 2332–2338.
6. The EuroQol Group. EuroQol—a new facility for the measurementof health-related quality of life. Health Policy 1990;16: 199–208.
7. Zigmond AS, Snaith RP. The hospital anxiety and depressionscale. Acta Psychiatr Scand 1983;67:361–370.
8. Golbe LI, Ohman-Strickland PA. A clinical rating scale for pro-gressive supranuclear palsy. Brain 2007;130:1552–1565.
9. Schrag A, Jahanshahi M, Quinn N. How does Parkinson’s dis-ease affect quality of life? A comparison with quality of life inthe general population. Mov Disord 2000;15:1112–1118.
10. Silva EG, Viana MA, Quagliato EM. Pain in Parkinson’s disease:analysis of 50 cases in a clinic of movement disorders. Arq Neu-ropsiquiatr 2008;66:26–29.
11. Defazio G, Berardelli A, Fabbrini G, et al. Pain as a nonmotorsymptom of Parkinson disease: evidence from a case-controlstudy. Arch Neurol 2008;65:1191–1194.
12. Mylius V, Engau I, Teepker M, et al. Pain sensitivity and de-scending inhibition of pain in Parkinson’s disease. J Neurol Neu-rosurg Psychiatry 2009;80:24–28.
13. Mathias CJ, Mallipeddi R, Bleasdale-Barr K. Symptoms associ-ated with orthostatic hypotension in pure autonomic failure andmultiple system atrophy. J Neurol 1999;246:893–898.
14. Tison F, Wenning GK, Volonte MA, Poewe WR, Henry P, QuinnNP. Pain in multiple system atrophy. J Neurol 1996;243: 153–156.
15. Esmonde T, Giles E, Gibson M, Hodges JR. Neuropsychologicalperformance, disease severity, and depression in progressivesupranuclear palsy. J Neurol 1996;243:638–643.
16. Borroni B, Turla M, Bertasi V, Agosti C, Gilberti N, PadovaniA. Cognitive and behavioral assessment in the early stages ofneurodegenerative extrapyramidal syndromes. Arch GerontolGeriatr 2008;47:53–61.
17. Menza MA, Cocchiola J, Golbe LI. Psychiatric symptoms in pro-gressive supranuclear palsy. Psychosomatics 1995;36:550–554.
18. Fetoni V, Soliveri P, Monza D, Testa D, Girotti F. Affectivesymptoms in multiple system atrophy and Parkinson’s disease:response to levodopa therapy. J Neurol Neurosurg Psychiatry 1999;66:541–544.
19. Gill CE, Khurana RK, Hibler RJ. Occurrence of depressivesymptoms in Shy-Drager syndrome. Clin Auton Res 1999;9:1–4.
20. Muller J, Wenning GK, Verny M, et al. Progression of dysarthriaand dysphagia in postmortem-confirmed parkinsonian disorders.Arch Neurol 2001;58:259–264.
21. Santacruz P, Uttl B, Litvan I, Grafman J. Progressive supranu-clear palsy: a survey of the disease course. Neurology 1998;50:1637–1647.
22. Nath U, Ben Shlomo Y, Thomson RG, Lees AJ, Burn DJ. Clini-
cal features and natural history of progressive supranuclear palsy:
a clinical cohort study. Neurology 2003;60:910–916.23. Schrag A, Wenning GK, Quinn N, Ben Shlomo Y. Survival in
multiple system atrophy. Mov Disord 2008;23:294–296.24. Papapetropoulos S, Gonzalez J, Mash DC. Natural history of pro-
gressive supranuclear palsy: a clinicopathologic study from a
population of brain donors. Eur Neurol 2005;54:1–9.25. Geser F, Wenning GK, Seppi K, et al. Progression of multiple
system atrophy (MSA): a prospective natural history study by theEuropean MSA Study Group (EMSA SG). Mov Disord 2006;21:179–186.
1081HEALTH-RELATED QUALITY OF LIFE IN MSA AND PSP
Movement Disorders, Vol. 25, No. 8, 2010

Impact of Belief inNeuroprotection on Therapeutic
Intervention in Parkinson’s Disease
Rodger J. Elble, MD, PhD,1*Oksana Suchowersky, MD,2
Stephanie Shaftman, MSc, MS,3,4
William J. Weiner, MD,5 Peng Huang, PhD,6 andBarbara Tilley, PhD7, On Behalf of the NINDS
NET-PD Investigators
1Department of Neurology, Southern Illinois UniversitySchool of Medicine, Springfield, Illinois, USA; 2Departmentof Medical Genetics, University of Calgary, Calgary, Alberta,Canada; 3Department of Clinical Neurosciences, Universityof Calgary, Calgary, Alberta, Canada; 4Department ofBiostatistics, Bioinformatics and Epidemiology, MedicalUniversity of South Carolina, Charleston, South Carolina,USA; 5Department of Neurology, University of Maryland
School of Medicine, Baltimore, Maryland, USA; 6Departmentof Oncology, Johns Hopkins University, Baltimore, Maryland,USA; 7Division of Biostatistics, University of Texas Health
Science Center School of Public Health, Houston,Texas, USA
Abstract: We explored the hypotheses that an investigator’sbelief in a putative neuroprotective agent might influencethe timing of symptomatic intervention and the assessmentof signs and symptoms of patients with Parkinson’s diseasewith the Unified Parkinson’s Disease Rating Scale(UPDRS). These hypotheses were tested with Cox and gen-eral linear modeling, using data from a previously pub-lished double-blind placebo-controlled futility trial of coen-zyme Q10 and GPI-1485. We found the investigators’ levelof confidence in these agents had no effect on the time tosymptomatic therapy or on the change in UPDRS during12 months of treatment. � 2010 Movement Disorder Society
Key words: Parkinson’s disease; bias; clinical trial; Uni-fied Parkinson’s Disease Rating Scale
The strong tendency for an investigator’s or patient’s
beliefs to influence the perceived response to a treat-
ment is well known. Consequently, patients are com-
monly randomly assigned to active agent or placebo,
and the patients and investigators are blinded to the
treatment.
In studies of putative neuroprotective agents,
patients are typically randomly assigned to the active
agent or placebo and then assessed periodically for
changes in their clinical condition. Investigators often
have the option of starting an established treatment if a
patient becomes sufficiently symptomatic. The time to
therapy should be greater in patients receiving an
effective neuroprotective drug. This study design has
been used extensively in Parkinson’s disease (PD)
since the DATATOP study of selegiline.1
The NINDS NET-PD investigators recently exam-
ined coenzyme Q10 (CoQ10, a putative neuroprotective
agent) and the neuroimmunophilin-ligand compound
GPI-1485 (a drug purported to promote neuronal repair
and regeneration) in a randomly assigned, double-
blind, placebo-controlled, calibrated futility trial of 213
untreated patients with early PD.2 At the beginning
and end of this trial, investigators were asked to rate
their confidence in these two drugs as neuroprotectants.
In this report, we examine whether the investigators’
level of confidence in CoQ10 and GPI-1485 at baseline
influenced the time to symptomatic therapy and the
change in the Unified Parkinson’s Disease Rating Scale
(UPDRS) and whether the investigators’ level of confi-
dence changed over time.
METHODS
The details and outcome of the original clinical trial
have been reported elsewhere.2 Briefly, 213 patients,
age 30 and over, with mild untreated PD not requiring
treatment, were randomly assigned 1:1:1 to receive one
of the following three treatment options for 12 months:
(1) 2,400 mg of CoQ10 and placebo for GPI-1485, (2)
placebo for CoQ10 and 4,000 mg of GPI-1485, or (3)
placebo for CoQ10 and placebo for GPI-1485. Partici-
pants were re-evaluated with the UPDRS and other
clinical scales at 1, 3, 6, 9, and 12 months (66 days)
after the baseline visit.
The primary, prespecified outcome measure was the
change in the total UPDRS score from baseline to the
time at which there was sufficient disability to warrant
symptomatic therapy for PD or from baseline to the end
of the 12-month treatment period if the patient did not
require symptomatic therapy. The decision to begin
symptomatic therapy was a clinical decision, made by
the investigator at each site. This decision was based on
a patient’s impairment in ambulation, activities of daily
living, and occupational status. The site investigators (N
5 48) were trained on this endpoint using case vignettes.
In the original study, a futility study design3 was
used to assess CoQ10 and GPI-1485 against a predeter-
Members of the ‘‘NET-PD Steering Committee’’ are listed as an
Appendix.
*Correspondence to: Dr. Rodger J. Elble, Department of Neuro-logy, Southern Illinois University School of Medicine, PO Box19643, Springfield, IL. E-mail: [email protected]
Potential conflict of interest: Nothing to report.Received 1 May 2009; Revised 8 October 2009; Accepted 11 Decem-
ber 2009Published online 3 February 2010 in Wiley InterScience (www.
interscience.wiley.com). DOI: 10.1002/mds.22997
Movement Disorders, Vol. 25, No. 8, 2010
1082 R.J. ELBLE ET AL.
mined change in UPDRS that is regarded as clinically
meaningful. Thus, the mean change in total UPDRS
for each treatment group was compared to a 30%
reduction in a historically derived change in total
UPDRS that was based on the placebo arm of the Dep-
renyl and Tocopherol Antioxidant Therapy of Parkin-
sonism trial.4 A placebo arm was included in the origi-
nal futility study to verify and update the historical
control assumptions used in sample size estimation.
The investigators’ confidence in CoQ10 and GPI-
1485 was assessed at the beginning and end of the
study using an 11-point rating scale in which confi-
dence in each agent was estimated to be 0, 10, 20, . . .,100%. For each study drug, investigators were asked
‘‘What is your level of confidence that the study drug
is an effective neuroprotectant for PD? The investiga-
tors were also asked (Yes/No) if their perceptions were
based on (1) preclinical data, (2) clinical information,
(3) steering committee recommendation, and (4)
‘‘nothing better to try.’’
We examined two post hoc hypotheses; hypothesis
1: investigator’s baseline belief influences the time to
symptomatic therapy and hypothesis 2: investigator’s
baseline belief influences the change from baseline in
the UPDRS. We also assessed change in beliefs over
time. The original study was placebo-controlled and
double-blind, and the two active drugs had no percepti-
ble symptomatic effect.2 Consequently, the data from
the three treatment arms were pooled in our statistical
analyses. However, treatment group was included as a
covariate.
All statistical analyses were performed with SAS/
STAT.5 Hypothesis 1 was tested statistically by model-
ing the time to symptomatic therapy (outcome mea-
sure) using a Cox model (SAS PHREG procedure)
with duration of disease, baseline UPDRS score, age at
baseline, and treatment group as covariates and using
confidence in CoQ10 and confidence in GPI-1485 as
independent variables. Hypothesis 2 was tested by con-
structing a general linear model using change in total
UPDRS from baseline to the 12 month visit as the out-
come, age, duration of disease, and treatment group as
covariates, and confidence in CoQ10 and confidence in
GPI-1485 as independent variables. We used Wilcoxon
signed rank tests to test the null hypothesis that change
from baseline to the end of the trial in investigator
confidence equal zero, separately for CoQ10 and GPI-
1485. Baseline data on the covariates were previously
published in the report on the trial.2
RESULTS
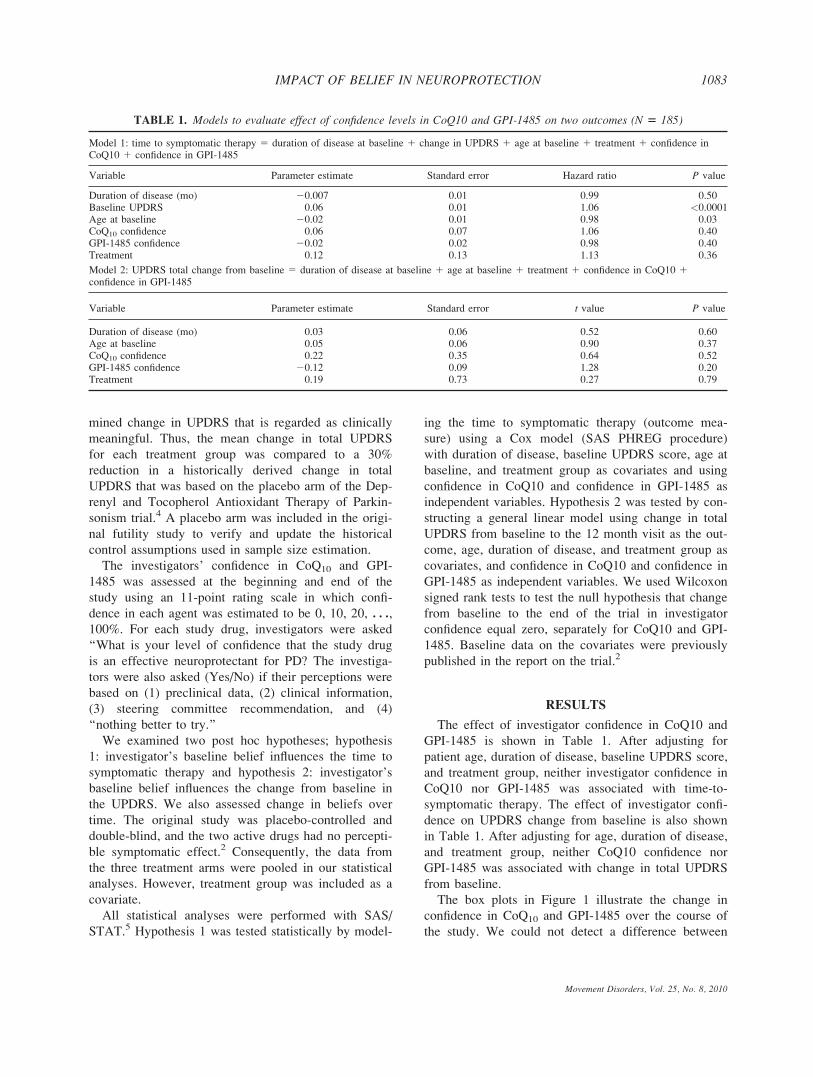
The effect of investigator confidence in CoQ10 and
GPI-1485 is shown in Table 1. After adjusting for
patient age, duration of disease, baseline UPDRS score,
and treatment group, neither investigator confidence in
CoQ10 nor GPI-1485 was associated with time-to-
symptomatic therapy. The effect of investigator confi-
dence on UPDRS change from baseline is also shown
in Table 1. After adjusting for age, duration of disease,
and treatment group, neither CoQ10 confidence nor
GPI-1485 was associated with change in total UPDRS
from baseline.
The box plots in Figure 1 illustrate the change in
confidence in CoQ10 and GPI-1485 over the course of
the study. We could not detect a difference between
TABLE 1. Models to evaluate effect of confidence levels in CoQ10 and GPI-1485 on two outcomes (N 5 185)
Model 1: time to symptomatic therapy 5 duration of disease at baseline 1 change in UPDRS 1 age at baseline 1 treatment 1 confidence inCoQ10 1 confidence in GPI-1485
Variable Parameter estimate Standard error Hazard ratio P value
Duration of disease (mo) 20.007 0.01 0.99 0.50Baseline UPDRS 0.06 0.01 1.06 <0.0001Age at baseline 20.02 0.01 0.98 0.03CoQ10 confidence 0.06 0.07 1.06 0.40GPI-1485 confidence 20.02 0.02 0.98 0.40Treatment 0.12 0.13 1.13 0.36
Model 2: UPDRS total change from baseline 5 duration of disease at baseline 1 age at baseline 1 treatment 1 confidence in CoQ10 1confidence in GPI-1485
Variable Parameter estimate Standard error t value P value
Duration of disease (mo) 0.03 0.06 0.52 0.60Age at baseline 0.05 0.06 0.90 0.37CoQ10 confidence 0.22 0.35 0.64 0.52GPI-1485 confidence 20.12 0.09 1.28 0.20Treatment 0.19 0.73 0.27 0.79
1083IMPACT OF BELIEF IN NEUROPROTECTION
Movement Disorders, Vol. 25, No. 8, 2010

baseline and final visit for either agent (P > 0.05)
using a Wilcoxon signed rank test. The mean (SD) for
level of confidence (0–100%) in CoQ10 at start and
end of study were 41.7 (17.8) and 42.0 (19.0), and the
mean (SD) for confidence in GPI-1485 at baseline and
end-of-study were 35.2 (18.8) and 34.6 (20.3). Thus,
confidence levels in CoQ10 and GPI-1485 were similar.
These analyses were conducted on the 46 of 48 investi-
gators who completed both confidence assessments.
The bases for investigator confidence at the start of
study were similar for CoQ10 and GPI-1485. An inves-
tigator could choose more than one reason. For CoQ10,
the percentage of investigators deriving confidence
from preclinical data was 73.3%, clinical information
95.5%, Steering Committee recommendations 20.5%,
and ‘‘nothing better to try’’ 15.9%. For GPI-1485, these
percentages were preclinical data 88.6%, clinical infor-
mation 50%, steering committee recommendations
29.6%, and ‘‘nothing better to try’’ 20.0%. Thus, the
investigators’ confidence stemmed mostly from preclin-
ical and clinical data.
DISCUSSION
Investigator bias is a well-known confounder in clin-
ical research. Consequently, our published clinical fu-
tility trial of CoQ10 and GPI-1485 was double-blinded
and placebo-controlled.2 Nevertheless, we were inter-
ested in knowing whether the investigators’ level of
confidence in these agents affected UPDRS scoring
and time to symptomatic therapy. Investigators with
greater confidence might influence or assess their
patients in such a way that the UPDRS changed more
slowly and the time-to-symptomatic therapy was
delayed. We found no evidence of either behavior. We
know of no other study of this type in PD.
The data in this study came from a published futility
study2 that included a placebo arm. Thus, the original
study was double-blind and placebo-controlled, and the
futility method of hypothesis testing in the original
study should have no effect on the results reported in
this report.
We suspect that our results would have been differ-
ent if our clinical trial had not been double-blind
and placebo-controlled. Because placebo associated
improvement is substantial in treatment trials for PD,6
the investigators had difficulty separating the placebo
effect from a treatment effect. Nevertheless, it is reas-
suring to know that investigator confidence had no
appreciable effect on UPDRS scoring and timing of
symptomatic therapy. It is important to note that the
investigators’ confidence in the two drugs did not sig-
nificantly change over time suggesting that clinical eq-
uipoise was maintained throughout the study.
Acknowledgments: Sponsored by the NIH (National Insti-tute of Neurological Disorders and Stroke), U01NS043127,U01NS 043128, and U10NS44415 through 4455.
Financial Disclosures: Rodger Elble is a consultant forJazz Pharmaceuticals, Ortho-McNeil and the Kinetics Foun-dation, and he receives grant support from the National Insti-tutes of Health (NINDS), GlaxoSmithKline, Pfizer, andKiwanis International (Illinois-Eastern Iowa District). Heserves on the medical advisory board of the InternationalEssential Tremor Foundation. Oksana Suchowersky is a con-sultant for Allergan and is an advisory board member forTeva. Stephanie Shaftman has no disclosures. WilliamWeiner is a consultant for the American Medical Associationand Santhera. He has received honoraria from Boehringer-Ingelheim and GlaxoSmithKline, and he receives royaltiesfrom Elsevier, Lippincott, and Johns Hopkins Press. PengHuang is a consultant for the National Institutes of Health(NIH) and American College of Physicians, and she receivesgrant support from the NIH, Maryland Cigarette RestitutionFund, Susan G. Komen Breast Cancer Foundation, and Avon.Barbara Tilley is a consultant for the National Institutes ofHealth and the Michael J. Fox Foundation. She receives grantsupport from the National Institutes of Health, Duke Endow-ment, Movement Disorder Society, and Michael J. Fox Foun-dation, and she received honoraria from the Michael J. FoxFoundation. The authors have no other stock ownership, con-sultancies, partnerships, intellectual property rights, experttestimony, contracts, employment, or royalties to report.
Author Roles: Rodger Elble and Stephanie Shaftmanwrote the first draft. Rodger Elble, Peng Huang, StephanieShaftman, Oksana Suchowersky, and William Weiner
FIG. 1. Box plots of investigator confidence at the beginning of thestudy and the change from baseline for CoQ10 and GPI-1485. Themedian (thick horizontal bars), 25th and 75th percentiles (box) andrange (whiskers) are shown.
1084 R.J. ELBLE ET AL.
Movement Disorders, Vol. 25, No. 8, 2010

designed and executed the study. All authors reviewed andcritiqued the final draft. Stephanie Shaftman, Peng Huang,and Barbara Tilley performed the statistical analyses. TheNET-PD investigators performed the original clinical trial,from which the data in this study were derived.
APPENDIX
NET-PD Steering Committee
Karl Kieburtz, MD, MPH (Principal Investigator,
Coordination Center): University of Rochester, Roches-ter, NY; Bernard Ravina, MD, MSCE*: National Insti-tutes of Health, Bethesda, MD; Wendy R. Galpern,
MD, PhD: National Institutes of Health, Bethesda,MD; Barbara Tilley, PhD (Principal Investigator, Sta-
tistical Center): Medical University of South Carolina,Charleston, SC; Kathleen Shannon, MD: Rush Univer-sity Medical Center, Chicago, IL; Caroline Tanner,
MD, PhD: The Parkinson’s Institute, Sunnyvale, CA;G. Frederick Wooten, MD: University of Virginia,Charlottesville, VA. *Current affiliation: University ofRochester, Rochester, NY.
Participating NET-PD Investigators
and Coordinators
Robert Hamill, MD: University of Vermont, Burling-ton, VT; Jacob I Sage, MD, Emily Kosa: UMDNJ Rob-ert Wood Johnson Medical School, New Brunswick,NJ; Ray L. Watts, MD, Natividad R. Stover, MD,
Rebecca McMurray, RN, MSN: University of Alabama,Birmingham, AL; Mark F. Lew, MD, Connie Kawai,
RN, BSN, CCRC: University of Southern California,Los Angeles, CA; David Coffey, MD, Pauline LeBlanc,
BS: Dartmouth Hitchcock Medical Center, Lebanon,NH; Julie Carter, RN, MN, ANP, Matthew Brodsky,
MD, Pamela Andrews: Oregon Health & Science Uni-versity, Portland, OR; Andrew Siderowf, MD, Sue
Reichwein, CCRC: University of Pennsylvania, Phila-delphia, PA; Lisa Shulman, MD, William J. Weiner,
MD, Katharine Pabst, CRNP, MS, MPH: University ofMaryland, Baltimore, MD; Kathleen Shannon, MD,
Jeana Jaglin, RN, CCRC: Rush University MedicalCenter, Chicago, IL; Robert Hauser, MD, Theresa
McClain, ARNP, Holly Delgado, RN: University ofSouth Florida, Tampa, FL; Oksana Suchowersky, MD,
Lorelei Derwent, RN, University of Calgary, Calgary,Canada; Jayaraman Rao, MD, Maureen Cook, RN,
BSN: Louisiana State University Health Sciences Cen-ter, New Orleans, LA; Michael J. Aminoff, MD, DSc,
Chad Christine, MD, Jessie Roth, RN: University ofCalifornia San Francisco, San Francisco; Maureen
Leehey, MD, Jacci Bainbridge, Pharm D: University ofColorado Health Sciences Center, Aurora, CO; G.
Webster Ross, MD, Stephanie Terashita, RN: PacificHealth Research Institute, Honolulu, HI; Carlos Singer,MD, Marian A. Perez, AA, Anita Blenke, PA-C, MS:
University of Miami, Miami, FL; Brad Racette, MD,
Patricia Deppen: Washington University School ofMedicine, St. Louis, MO; Rodger Elble, MD, PhD,
Charlene Young, RN, MSN, CFNP: Southern IllinoisUniversity, Springfield, IL; Caroline Tanner, MD,
Tracy Stewart, RN: The Parkinson’s Institute, Sunny-vale, CA; Kapil Sethi, MD, Buff Dill, BS, ED: MedicalCollege of Georgia, Augusta, GA; John Taylor, MD,
Peggy Roberge, RN: Hunter Holmes McGuire VeteransMedical Center, Richmond, VA; Richard B. Dewey, Jr.,
MD, Brigid Hayward: University of Texas Southwest-ern Medical School, Dallas, TX; Joseph Jankovic, MD,
Christine Hunter, RN, CCRC: Baylor College of Medi-cine, Houston, TX; Frederick Wooten, MD, Margaret
F. Keller, RN, MS, CCRC: University of Virginia,Charlottesville, VA; Danna Jennings, MD, Tammie
Kelsey, LPN: Institute for Neurodegenerative Disor-ders, New Haven, CT; Wayne Martin, MD, Germaine
McInnes, RN: University of Alberta Glenrose RehabHospital, Edmonton, AB, Canada; Joanne Wojcieszek,
MD, Joann Belden, RN: Indiana University School ofMedicine, Indianapolis, IN; Roger Albin, MD, Kristine
Wernette, RN, MS: University of Michigan, Ann Arbor,MI; Joseph Savitt, MD, PhD, Becky Dunlop, RN:
Johns Hopkins University, Baltimore, MD; Rajesh
Pahwa, MD, Kelly E. Lyons, PhD, Amy Parsons, RN,
BSN: University of Kansas Medical Center, KansasCity, KS; John Fang, MD, Dorothy Shearon, RN: Van-derbilt University Medical Center, Nashville, TN;Andrew Feigin, MD, Margaret Marie Cox, RN, BSN:
North Shore University Hospital, Manhasset, NY;Charles Adler, MD, PhD, Marlene Lind, RN: MayoClinic Scottsdale, Scottsdale, AZ; Burton Scott, MD,
Joanne Field, BSN, RN: Duke University, Durham,NC; Martha Nance, MD, Susan Peterson, RN: StruthersParkinson’s Center, Golden Valley, MN; Richard S.
Burns, MD, Lynn Marlor, RN: Barrow NeurologicalInstitute, Phoenix, AZ; Bodis-Wollner, MD, Elizabeth
Hayes, RN: State University of New York DownstateMedical Center, Brooklyn, NY; Jay Schneider, MD,
Stephanie Sendek: Thomas Jefferson University, Phila-delphia, PA; Stephen Gollomp, MD, Gwyn Vernon,
MSN, CRNP: Thomas Jefferson University/LankenauHospital, Wynnewood, PA; Peter LeWitt, MD, Maryan
DeAngelis, RN, CCRC: William Beaumont Hospital,Southfield, MI; Ivan G. David Simon, MD, Linda Paul,
RN, NP: Beth Israel Deaconess Medical Center,
1085IMPACT OF BELIEF IN NEUROPROTECTION
Movement Disorders, Vol. 25, No. 8, 2010

Boston, MA; Jay Gorell, MD (deceased), Shana Krstev-
ska, MD: Henry Ford Health System, Detroit, MI;Ryan Uitti, MD, Margaret Turk, RN: Mayo ClinicJacksonville, Jacksonville, FL; James Bower, MD,
Susan Torgrimson, RN Mayo Clinic Rochester, Roch-ester, MN; Marwan Sabbagh, MD, Zoran Obradov,
CRC: Sun Health Research Institute, Sun City, AZ.
REFERENCES
1. Parkinson Study Group. DATATOP: a multicenter controlledclinical trial in early Parkinson’s disease. Arch Neurol1989;46:1052–1060.
2. NINDS NET-PD Investigators. A randomized clinical trial ofcoenzyme Q10 and GPI-1485 in early Parkinson disease. Neurol-ogy 2007;68:20–28.
3. Tilley BC, Palesch YY, Kieburtz K, et al. Optimizing theongoing search for new treatments for Parkinson disease: usingfutility designs. Neurology 2006;66:628–633.
4. Parkinson Study Group. Effect of deprenyl on the progression ofdisability in early Parkinson’s disease. N Engl J Med1989;321:1364–1371.
5. SAS Institute Inc. SAS/STAT1 User’s Guide, Version 8. Cary,NC: SAS Institute Inc.; 1999.
6. Diederich NJ, Goetz CG. The placebo treatments in neuroscien-ces: new insights from clinical and neuroimaging studies. Neurol-ogy 2008;71:677–684.
Tyrosine Hydroxylase Deficiencyin Three Greek Patients with aCommon Ancestral Mutation
Roser Pons, MD,1* Mercedes Serrano, MD PhD,2,3
Aida Ormazabal, PhD,2,3 Claudio Toma, PhD,4
Angels Garcia-Cazorla, MD PhD,2,3 Estela Area, PhD,5
Marta Ribases, PhD,6 Emmanuel Kanavakis, MD,1
Kaliopi Drakaki, MD,1 Aristotelis Giannakopoulos, MD,1
Irene Orfanou, MD,1 Sotiris Youroukos, MD,1
Bru Cormand, PhD,4 and Rafael Artuch, MD, PhD2,3
1First Department of Pediatrics, Agia Sofia Hospital,University of Athens, Athens, Greece; 2Department of Neuro-pediatrics, Sant Joan de Deu Hospital, Center for BiomedicalResearch on Rare Diseases (CIBERER), Barcelona, Spain;3Department of Clinical Biochemistry, Sant Joan de Deu
Hospital, Center for Biomedical Research on Rare Diseases(CIBERER), Barcelona, Spain; 4Department of Genetics,
University of Barcelona, Institute of Biomedicine of the Universityof Barcelona (IBUB), Center for Biomedical Research on RareDiseases (CIBERER), Barcelona, Spain; 5Department of
Neurology, Columbia University, New York, New York, USA;6Department of Psychiatry, Vall d’Hebron University Hospital,
Barcelona, Spain
Abstract: We present the clinical, biochemical, and molec-ular findings of three Greek patients with tyrosinehydroxylase (TH) deficiency. All patients presented with asevere clinical phenotype characterized by prominentmotor delay, infantile parkinsonism, oculogyric crises,and signs of autonomic dysfunction. Cerebrospinal fluidanalysis disclosed reduced dopamine metabolites and nor-mal pterins. Response to levodopa was favorable thoughnot dramatic. All patients were homozygous for a previ-ously reported mutation (p.L236P). SNP haplotype analy-sis was consistent with a common ancestral mutation, thusindicating a founder effect in Greek patients with THdeficiency. � 2010 Movement Disorder Society
Key words: tyrosine hydroxylase; infantile parkinsonism;oculogyric crisis; autonomic dysfunction; founder effect
INTRODUCTION
Tyrosine hydroxylase (TH, EC 1.14.16.2) is the rate-
limiting step in the biosynthesis of catecholamines.1
TH deficiency (MIM#605407) is a rare metabolic dis-
order; it has been reported in �30 patients in the litera-
*Correspondence to: Dr. Roser Pons, Agia Sofia Hospital, Thivonand Levadias Street, Athens 11527, Greece.E-mail: [email protected]
Potential conflict of interest: Nothing to Report.
Received 18 September 2009; Revised 10 November 2009;Accepted 12 December 2009
Published online 2 March 2010 in Wiley InterScience (www.
interscience.wiley.com). DOI: 10.1002/mds.23002
1086 R. PONS ET AL.
Movement Disorders, Vol. 25, No. 8, 2010

ture.2–10 Clinical manifestations derive mainly from
chronic dopamine deficiency in the developing brain.9,11
Patients may present with a severe clinical phenotype
characterized by lack of motor development, parkinson-
ism, dystonia, and oculogyric crises associated with auto-
nomic and endocrine dysfunction.2–11 Intermediate phe-
notypes also occur in TH deficiency and some patients
may present with dopa responsive dystonia similar to
Segawa disease.12 Patients show a characteristic pattern
of biogenic amine metabolites in CSF, with decreased
homovanillic acid (HVA) and 3-methoxy-4-hydroxy-
phenylethyleneglycol (MHPG) and normal pterins.11
The treatment of choice in TH deficiency is levodopa
(L-dopa), but response is heterogenous.9,12
We report herein three unrelated patients from
Greece sharing a homozygous missense mutation in
the TH gene, and we analyze the possibility of a com-
mon ancestral origin for this particular mutation in the
Greek population.
PATIENTS AND METHODS
Patients
All patients were assessed clinically at Agia Sofia
Hospital in Athens. They belonged to three unrelated
families from different regions of Greece. Their parents
were healthy and nonconsanguineous. All pregnancies
and perinatal periods were uneventful. Their clinical
presentation and physical exams are depicted in Table 1.
All patients were treated with L-dopa (0.5–1 mg/kg/
day) and doses continue to be gradually titrated
upwards according to tolerability. They all showed
improvements in facial expression, symptoms of auto-
nomic dysfunction, and started gradually to make
motor progress (axial control and hand use). Drug-
induced dyskinesias were observed in Patients 1 and 3,
and they were managed with reduction of the L-dopa
dose and slow gradual increase of the medication.
Biochemical Analysis
CSF biogenic amines [3-ortho-methyldopa (3OMD),
MHPG, HVA, and 5-hydroxyindoleacetic acid (5-HIAA)]
and pterins (neopterin and biopterin) were analyzed by
HPLC in the Department of Clinical Biochemistry of the
Sant Joan de Deu Hospital in Barcelona.13
Samples were drawn in accordance with the Helsinki
declaration. The study was approved by the local
Ethics Committee and informed consent was obtained
from the patients’ parents.
TABLE 1. Clinical features and biogenic amines concentrations (nmol/L) in CSF
Patient 1 Patient 2 Patient 3
Age at onset (mo)/Age at diagnosis (mo) 3/24 5/5 3/5Symptoms at onsetNormal acquisition of head control 1 1 1Loss of head control followed by lack of motor acquisitions 1 1 1Tremor 1 1 1Oculogyric crises 1a 1 1a
Diurnal fluctuation/Sleep benefit 1 2 1Autonomic dysfunctionb 1 1 1Sleep disturbance 2 2 2
Examination at the time of diagnosisAlert and irritable 1 1 1Hypotonia 1 1 1Ptosis/Hypomimia 1 1 1Minimal spontaneous movements/Lack of axial control 1 1 1Tremor 2 1 1Dystoniac 1 1 1Babinski sign 1 1 2Hyperprolactinemia 1 1 1
CSF analysisHVA (normal range)d 50 (344–906) 31 (354–1328) 18.5 (354–1328)HIAA (normal range)d 197 (170–490) 270 (217–1142) 235 (217–1142)HVA/HIAA (normal range)d 0.25 (1.11–3.48) 0.11 (1.16–2.4) 0.08 (1.16–2.4)MHPG (normal range)d 20 (20–80) 1.6 (30–124) 1.4 (30–124)3OMD (normal range)d 10 (4–50) 4.6 (20–162) 10 (20–162)
aAssociated with prominent dystonic posturing of limbs and trunk.bExcessive sweating, increased upper respiratory secretions.cDystonic movements were observed when the infants were manipulated and stressed.dNormal range of metabolite concentrations is age dependent.HVA, homovanillic acid; HIAA, 5-hydroxyindoleacetic acid; MHPG, 3-methoxy-4-hydroxyphenylglycol; 3OMD,
3-ortho-methyldopa.
Movement Disorders, Vol. 25, No. 8, 2010
1087TYROSINE HYDROXYLASE DEFICIENCY
Genetic Study
Genomic DNA was isolated from peripheral blood.
Sequencing analysis of the TH gene was performed in
the Department of Clinical Biochemistry of the Sant
Joan de Deu Hospital in Barcelona. Further genetic
studies were performed at the Department of Genetics
of the University of Barcelona. We PCR-amplified and
sequenced (ABI Prism, Applied Biosystems) the cod-
ing region, splice sites, 114 bp preceding the initiation
codon, and 354 bp following the stop codon of the THgene using a set of 10 primer pairs, as previously
reported.14 Putative disease-causing mutations as well
as intronic and exonic polymorphisms were studied.
AluI (New England Biolabs, Ipswich, MA) restriction
analysis of a PCR product containing exons 5 and 6
(forward primer: 50-GTAGGGGAGGCTGCTTCAA-30;reverse primer: 50-CTGGTGACAAGATGGGTCCT-30)was performed to confirm the mutation identified in
the patients and to screen 250 healthy controls (200
Spanish and 50 Greek). The restriction analysis was
followed by agarose gel electrophoresis and ethidium
bromide staining. The mutation abolishes a restriction
site (normal pattern: 48 1 63 1 253 1 136 bp; mutant
pattern: 48 1 63 1 389 bp).
Haplotype analysis, including nine polymorphic sites
across the TH gene, was performed in the three
patients. Five of these sites were also genotyped in 50
unrelated control individuals of Greek origin by PCR
amplification and direct sequencing (Fig. 1). Three of
these five variations (rs12419447, rs6357, rs4074905)
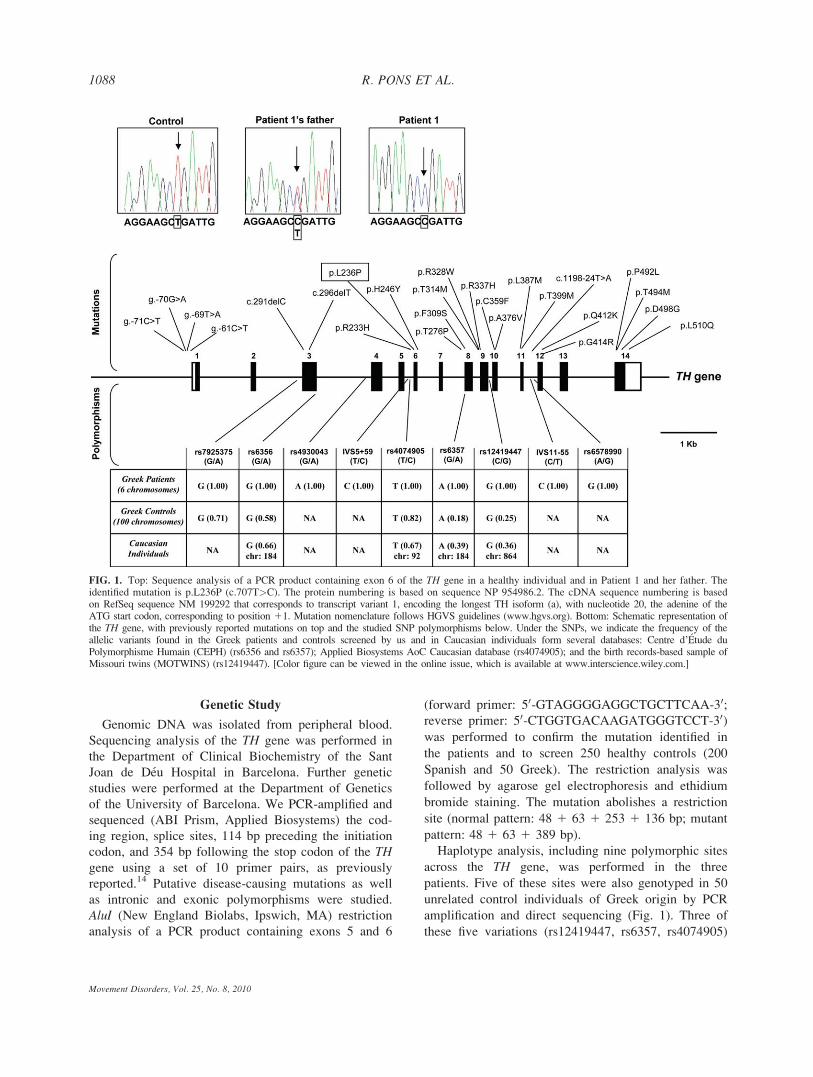
FIG. 1. Top: Sequence analysis of a PCR product containing exon 6 of the TH gene in a healthy individual and in Patient 1 and her father. Theidentified mutation is p.L236P (c.707T>C). The protein numbering is based on sequence NP 954986.2. The cDNA sequence numbering is basedon RefSeq sequence NM 199292 that corresponds to transcript variant 1, encoding the longest TH isoform (a), with nucleotide 20, the adenine of theATG start codon, corresponding to position 11. Mutation nomenclature follows HGVS guidelines (www.hgvs.org). Bottom: Schematic representation ofthe TH gene, with previously reported mutations on top and the studied SNP polymorphisms below. Under the SNPs, we indicate the frequency of theallelic variants found in the Greek patients and controls screened by us and in Caucasian individuals form several databases: Centre d’Etude duPolymorphisme Humain (CEPH) (rs6356 and rs6357); Applied Biosystems AoC Caucasian database (rs4074905); and the birth records-based sample ofMissouri twins (MOTWINS) (rs12419447). [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
1088 R. PONS ET AL.
Movement Disorders, Vol. 25, No. 8, 2010

were representative of a haplotype block spanning a
segment from introns 5 to 9, as defined by the Haplo-view v4.1 software,15 using the confidence intervals
method16 on the genotype data from the Greek con-
trols. Estimation of haplotype frequencies was also
performed by Haploview v4.1.
RESULTS
All patients showed decreased HVA and MHPG
concentrations, low HVA/5-HIAA ratio, (Table 1) and
normal pterin concentrations (data not shown). These
findings were consistent with TH deficiency.
Mutational analysis of the TH gene revealed a previ-
ously reported4 homozygous pathogenic mutation in
exon 6 (c.707T>C) in all three patients causing a substi-
tution of leucine for proline in residue 236 of the protein
(p.L236P), located in the a2 domain (Fig. 1). Parents
were heterozygous carriers of this mutation. Screening of
250 control subjects did not disclose the p.236P mutation.
We performed haplotype analysis of nine SNP poly-
morphisms along the gene revealing that all three
patients carried the same variants at homozygosis in all
the studied sites (Fig. 1). The scrutiny of five of these
nine SNPs in 100 chromosomes form 50 Greek control
individuals allowed estimation of haplotype frequencies
in the general population. Ten haplotypes were identified
with frequencies ranging from 0.0001 to 0.34, but the al-
lele combination found in the patients (rs7925375G-
rs6356G-rs4074905T-rs6357A-rs12419447G) was not
among them, indicating that it represents a rare haplo-
type. Under the assumption that the frequency of the
patients’ haplotype in the Greek population is <0.01, the
likelihood of independently encountering this haplotype
in six chromosomes can be estimated as <10 e 212,
which strongly supports the hypothesis of a common
origin for all the p.L236P alleles in opposition to a
recurrent mutational event.
DISCUSSION
In this report, we present three patients with TH
deficiency. All patients presented with a severe clinical
phenotype consisting of infantile parkinsonism (hypo-
kinesia and tremor), hypotonia, dystonia, and oculogy-
ric crises. Features of autonomic dysfunction included
ptosis, hyperhydrosis, and profuse nasal secretions;
whereas, hyperprolactinemia was the only endocrine
disturbance detected. The analysis of biogenic amine
metabolites in CSF showed reduced levels of catechol-
amine metabolites and normal pterins. Treatment with
L-dopa was followed by gradual improvement in motor
and autonomic function in all patients.
TH activity cannot be measured in easily accessible
tissues11 and confirmation of diagnosis is based on mo-
lecular analysis. Our patients were found to be homo-
zygous for the same missense mutation (p.L236P) that
has been previously reported as p.L205P.4 Expression
studies in different systems by Ludecke et al. revealed
that this mutation leads to normal TH RNA steady
state levels but reduced protein levels.4
Correlation between residual enzyme activity and
clinical severity has not been established in TH defi-
ciency. In contrast, it appears that the concentration of
HVA in CSF is indicative of phenotypic severity; HVA
levels range from undetectable to 30% of the lower limit
of the reference range in patients with severe pheno-
types4,7,8,17,18 and from 46 to 60% in patients with inter-
mediate phenotypes.10,19,20 HVA concentrations in our
patients (5.2–14.5%) fell into the levels suggested for
severe phenotypes of TH deficiency (Table 1). Interest-
ingly, the highest HVA concentration in CSF was found
in the eldest patients from our series (Patient 1), sug-
gesting compensatory mechanisms of dopamine turn-
over. This idea is supported by the finding of a higher
MHPG, the major product of norepinephrine and epi-
nephrine, in this patient (Table 1).
In our patients, the clinical response to L-dopa ther-
apy was favorable, but their motor progress is yet
insufficient and it may be too soon to reach conclu-
sions. Early initiation of therapy, gender, and tolerabil-
ity to treatment have been proposed as prognostic fac-
tors in these disorders.11,21–24 The latter is mainly rep-
resented by the occurrence of drug-induced dyskinesias
that may prevent reaching therapeutic doses of dopami-
nergic medications.24 In a TH knockout animal model,
hypersensitivity to L-dopa and dopamine receptor ago-
nists correlated with prominent locomotor hyperactiv-
ity.25 Chronic replacement with L-dopa relieved this
hypersensitivity. This phenomenon was also observed
in Patients 1 and 3 from our series, and it was also
reported in another patient with TH deficiency.17
Haplotype analysis across the TH gene in the six
p.L236P alleles identified in the three Greek patients
supported a common ancestral origin. In this regard,
the fact that the haplotype context of all the mutated
alleles is identical and extremely rare in the Greek
population indicates that the probability of a recurrent
mutational event is negligible. Supporting our findings,
the original patient in whom this mutation was first
reported was Greek.4 A second patient with the same
homozygous mutation has been reported, but his geo-
graphic origin was not stated.9
1089TYROSINE HYDROXYLASE DEFICIENCY
Movement Disorders, Vol. 25, No. 8, 2010

In summary, patients with TH deficiency harboring
the p.L236P mutation at homozygosity show a severe
phenotype with an early clinical presentation. A com-
mon origin for all the identified pL236P alleles in
Greek patients in this study is strongly supported by
our haplotype analysis.
Acknowledgments: This study was supported by ‘‘Agenciade Gestio d’Ajuts Universitaris i de Recerca AGAUR’’(2005SGR00848). Mercedes Serrano is a recipient of a Juande la Cierva grant from the Ministry of Science and Innova-tion (Spain).
Financial Disclosures: None.
Author Roles: Roser Pons, Mercedes Serrano, Bru Cor-mand and Rafael Artuch participated in the conception, orga-nization, and execution of the research project and in thewriting of the first draft and review and critique of the manu-script. Aida Ormazabal, Angels Garcia-Cazorla, Estela Area,Claudio Toma, Marta Ribases, Kaliopi Drakaki, AristotelisGiannakopoulos, Irene Orfanou, Emmanuel Kanavakis andSotiris Youroukos participated in the execution of theresearch project and the critical review of the manuscript.
REFERENCES
1. Nagatsu T, Levitt M, Udenfriend S. Tyrosine hydroxylase. Theinitial step in norepinephrine biosynthesis. J Biol Chem1964;239:2910–2917.
2. Knappskog PM, Flatmark T, Mallet J, Ludecke B, Bartholome K.Recessively inherited L-dopa-responsive dystonia caused by apoint mutation (Q381K) in the tyrosine hydroxylase gene. HumMol Genet 1995;4:1209–1212.
3. Ludecke B, Dworniczak B, Bartholome K. A point mutation inthe tyrosine hydroxylase gene associated with Segawa’s syn-drome. Hum Genet 1995;95:123–125.
4. Ludecke B, Knappskog M, Clayton T, et al. Recessively inheritedL-dopa-responsive parkinsonism in infancy caused by a pointmutation (L205P) in the tyrosine hydroxylase gene. Hum MolGenet 1996;5:1023–1028.
5. Steenbergen-Spanjers S, Janssen RJ, Wevers RA. A commonpoint mutation in the tyrosine hydroxylase gene in autosomalrecessive L-dopa-responsive dystonia in the Dutch population.Hum Genet 1998;102:644–646.
6. Brautigam C, Wevers RA, Jansen RJ, et al. Biochemical hall-marks of tyrosine hydroxylase deficiency. Clin Chem 1998;44:1897–1904.
7. De Lonlay P, Nassogne MC, van Gennip AH, et al. Tyrosinehydroxylase deficiency unresponsive to L-dopa treatment withunusual clinical and biochemical presentation. J Inherit MetabDis 2000;23:819–825.
8. Furukawa Y, Graf WD, Wong H, Shimadzu M, Kish SJ.Dopa-responsive dystonia simulating spastic paraplegia due to ty-rosine hydroxylase (TH) gene mutations. Neurology 2001;56:260–263.
9. Hoffmann GF, Assmann B, Brautigam C, et al. Tyrosine hydrox-ylase deficiency causes progressive encephalopathy and dopa-nonresponsive dystonia. Ann Neurol 2003;54:S56–S65.
10. Giovanello T, Leuzzi V, Carducci C, et al. Tyrosine hydroxylasedeficiency presenting with a biphasic clinical curse. Neuropediat-rics 2007;38:213–215.
11. Blau N, Thony B, Cotton RGH, Hyland K. Disorders of tetrahy-drobiopterin and related biogenic amines. In: Scriver CR, Beau-
det AL, Sly WS, Valle D, Childs B, Vogelstein B, editors. Themetabolic and molecular bases of inherited disease. New York:McGraw-Hill; 2001. p 1725–1776.
12. Furukawa Y, Kish SJ, Fahn S. Dopa-responsive dystonia due tomild tyrosine hydroxylase deficiency. Ann Neurol 2004;55:147–148.
13. Ormazabal A, Garcia-Cazorla A, Fernandez Y, Fernandez-Alvarez E, Campistol J, Artuch R. HPLC with electrochemicaland fluorescence detection procedures for the diagnosis of inbornerrors of biogenic amines and pterins. J Neurosci Methods2005;142:153–158.
14. Ribases M, Serrano M, Fernandez-Alvarez E, et al. A homozy-gous tyrosine hydroxylase gene promoter mutation in a patientwith dopa-responsive encephalopahty: clinical, biochemical andgenetic analysis. Mol Genet Metab 2007;92:274–277.
15. Barret JC, Fry B, Maller J, Daly MJ. Haploview: analysis of LDand haplotype maps. Bioinformatics 2005;21:263–265.
16. Gabriel SB, Schaffner SF, Nguyen H, et al. The structure of haplo-type blocks in the human genome. Science 2002;296:2225–2229.
17. Grattan-Smith PJ, Wevers RA, Steenbergen-Spanjers GC, FungVS, Earl J, Wilcken B. Tyrosine hydroxylase deficiency: clinicalmanifestations of catecholamine insufficiency in infancy. Mov Dis-ord 2002;17:354–359.
18. Brautigam C, Steenbergen-Spanjers GC, Hoffmann GF, et al. Bio-chemical and molecular genetic characteristics of the severe formof tyrosine hydroxylase deficiency. Clin Chem 1999;45: 2073–2078.
19. Diepold K, Schutz B, Rostasy K, et al. Levodopa-responsive infan-tile parkinsonism due to a novel mutation in the tyrosine hydroxy-lase gene and exacerbation by viral infections. Mov Disord2005;20:764–767.
20. Schiller A, Wevers RA, Steenbergen GC, Blau N, Jung HH.Long-term course of L-dopa-responsive dystonia caused by tyro-sine hydroxylase deficiency. Neurology 2004;63:1524–1526.
21. Sedel F, Ribeiro MJ, Remy P, Blau N, Saudubray JM, Agid Y.Dihydropteridine reductase deficiency: levodopa’s long-term effec-tiveness without dyskinesia. Neurology 2006;67:2243–2245.
22. Segawa M, Nomura Y, Nishiyama N. Autosomal dominant guano-sine triphosphate cyclohydrolase I deficiency (Segawa disease).Ann Neurol 2003;54:S32–S45.
23. Hyland K, Surtees RA, Rodeck C, Clayton PT. Aromatic L-aminoacid decarboxylase deficiency: clinical features, diagnosis, andtreatment of a new inborn error of neurotransmitter amine synthe-sis. Neurology 1992;42:1980–1988.
24. Pons R, Ford B, Chiriboga CA, et al. Aromatic L-amino acid de-carboxylase deficiency: clinical features, treatment, and prognosis.Neurology 2004;62:1058–1065.
25. Kim DS, Szczypka MS, Palmiter RD. Dopamine-deficient miceare hypersensitive to dopamine receptor agonists. J Neurosci2000;20:4405–4413.
1090 R. PONS ET AL.
Movement Disorders, Vol. 25, No. 8, 2010

Characterization of Lewy BodyPathology in 12- and 16-Year-OldIntrastriatal Mesencephalic Grafts
Surviving in a Patient WithParkinson’s Disease
Jia-Yi Li, MD, PhD,1* Elisabet Englund, MD,2
Hakan Widner, MD, PhD,3 Stig Rehncrona, MD,4
Anders Bjorklund, MD,5 Olle Lindvall, MD, PhD,3,6
Patrik Brundin, MD, PhD1
1Neuronal Survival Unit, Wallenberg Neuroscience Center,Lund University, Lund, Sweden; 2Department ofNeuropathology, Lund University, Lund, Sweden;
3Division of Neurology, Department of Clinical Sciences,Lund University, Lund, Sweden; 4Neurosurgery,Department of Clinical Sciences, Lund University,
Lund, Sweden; 5Neurobiology Unit,Wallenberg Neuroscience Center, Lund University, Lund,Sweden; 6Section of Restorative Neurology, WallenbergNeuroscience Center, Lund University, Lund, Sweden
Abstract: We previously reported the occurrence of Lewybodies in grafted human fetal mesencephalic neurons intwo patients with Parkinson’s disease. Here, we have usedimmunohistochemistry and electron microscopy to char-acterize the development of Lewy bodies in one of thesecases. This patient was operated in putamen on both sidesat 12 or 16 years before death, respectively. We demon-strate that 2% of the 12-year-old and 5% of the 16-year-old grafted, presumed dopaminergic neurons containedLewy bodies immunoreactive for a-synuclein. Based onmorphological analysis, two forms of a-synuclein-positiveaggregates were distinguished in the grafts, the first aclassical and compact Lewy body, the other a loose mesh-work aggregate. Lewy bodies in the grafts stainedpositively for ubiquitin and thioflavin-S, and containedcharacteristic a-synuclein immunoreactive electron densefibrillar structures on electron microscopy. Our data indi-cate that Lewy bodies develop gradually in transplanteddopaminergic neurons in a fashion similar to that in do-paminergic neurons in the host substantia nigra. � 2010Movement Disorder Society
Key words: neural transplantation; Lewy body; proteinaggregation; a-synuclein; transmissible neurological disease
In Parkinson’s disease (PD), neurodegeneration is
prominent in substantia nigra dopaminergic neurons.
Intraneuronal protein aggregates, rich in a-synucleinand called Lewy bodies/neurites (LBs/LNs), develop in
several brain regions and become widespread with
advancing disease. Patients with intrastriatal transplants
of human fetal mesencephalic tissue, rich in dopami-
nergic neurons, have displayed long-term clinical bene-
fits in open label trials (see Refs. 1 and 2). Brain imag-
ing studies provide evidence that the grafted neurons
survive and become functionally integrated in the host
brain.3 Postmortem studies on patients dying 18–52
months after grafting confirm that grafted dopaminer-
gic neurons survive and innervate the host striatum,
without any signs of pathology in the transplants.4–6
Recently, we reported that large numbers of dopami-
nergic neurons can survive up to 16 years after implan-
tation.7 We and others also demonstrated that a frac-
tion of the grafted cells developed PD pathology, i.e.,
LBs and LNs.7–9 Here we report a detailed analysis of
frequency, maturation and ultrastructural characteristics
of LBs in two separate grafts at 12 and 16 years after
implantation in the same PD patient.
MATERIALS AND METHODS
Postmortem Brain Preparation
This male patient, born in 1940, was transplanted
with human fetal mesencephalic tissue in 1989 (left
striatum) and 1993 (right striatum). Grafting procedure,
donor tissues, neurological outcome, and imaging data
have been reported previously.10,11 He died in 2005 of
acute aspiration coupled to advanced PD. The brain
was fixed in 6% buffered formaldehyde solution for 2
months. Basal ganglia and left mesencephalon were
cut into 10–15-mm thick blocks for frozen section
preparation. Remaining brain slices, including small
sections from basal ganglia and right mesencephalon,
were processed for paraffin embedding and sectioning
at 5 lm, followed by staining with antibodies against
a-synuclein and ubiquitin.
Immunohistochemistry
Paraffin sections were mounted on capillary glass
slides and treated in a microwave oven in citrate
buffer at pH 6.0 for 15 minutes at 800 W. The sec-
tions were washed three times with 0.1 M PB.
Thereafter, we incubated sections with a primary
antibody against a-synuclein (Clone, LB509, 1:600,
Zymed) and ubiquitin (1:200, Dako) overnight at
room temperature. After rinsing, we incubated with
The last two authors contributed equally to this work.*Correspondence to: Dr. Jia-Yi Li, Neuronal Survival Unit, Wal-
lenberg Neuroscience Center, Lund University, BMC A10, 221 84,Lund, Sweden. E-mail: [email protected] conflict of interest: Nothing to report.Received 11 September 2009; Revised 2 November 2009;
Accepted 15 December 2009
Published online 2 March 2010 in Wiley InterScience (www.
interscience.wiley.com). DOI: 10.1002/mds.23012
1091LEWY BODIES IN FETAL MESENCEPHALIC GRAFTS
Movement Disorders, Vol. 25, No. 8, 2010

the secondary antibodies (Biotinylated horse-anti-
mouse or goat-anti-rabbit, 1:200, Jackson Lab, West
Grove, PA), ABC-solution (Vector Lab) and finally
DAB. Immunoreactivity was assessed using a Nikon
microscope and images were processed with Adobe
Photoshop software. Grafts were easily distinguished
from the host brain. Since we observed decreased
TH immunoreactivity in grafted neurons, especially
in 16-year-old transplant, we used pigment granule-
containing neurons to estimate the proportion of do-
paminergic neurons within the grafts that contained
LBs.
Thioflavin-S Staining
Sections through grafts were mounted on glass
microscope slides, air-dried, treated in mixture of chlo-
roform and absolute ethanol (1:1) for 2 hours, and
hydrated through graded ethanol to distilled water. Sec-
tions were then placed in 0.1% thioflavin-S (Sigma)
for 10 minutes in the dark and developed in 80% etha-
nol for about 15 seconds. After rinsing in distilled
water, cover slip was placed over the sections, and
examined in a Nikon fluorescence microscope and a
Leica confocal microscope.
Electron Microscopic Analysis
Sections through the grafts (40 lm) were stained
with the a-synuclein antibody and fixed with 2.5% glu-
taraldehyde in PBS for 1 hour and 1% OsO4 in 0.1
PBS for 30 minutes. After rinsing in PBS, sections
were dehydrated in ethanol and propylene oxide and
flat-embedded in Epon 812 between two transparent
film leaves. After polymerization, sections were exam-
ined in a bright field microscope, and we identified
and cut small pieces (about 1 mm2) containing a-synu-clein immunoreactive grafted cells. These pieces were
mounted on plastic blocks for semithin and ultra-thin
sectioning. After contrasting the ultra-thin sections in
4% uranyl acetate, images were acquired in a Philips
CM-10 electron microscope.
RESULTS
Frequency of Lewy Bodies in Grafted Cells
In mesencephalic brain, 95% of dopaminergic neu-
rons in substantia nigra contain neuromelamin, the
amount of pigment progressively increasing with age.12
We found LBs in both the 12- and 16-year-old grafts as
evidenced by a-synuclein, ubiquitin, and thioflavin-S
staining (Figs. 1 and 3). In the 12-year-old grafts, a-syn-
uclein positive LBs were detected in 1.9% of the neuro-
melanin-containing, presumed dopaminergic neurons
(1,267 cells sampled). The frequency of a-synucleinpositive LBs had increased to 5.0% (1,196 cells
sampled) in the 16-year-old grafts (Fig. 1). We observed
that 2.1% of the pigmented neurons were ubiquitin-
immunopositive in the 12-year-old graft (905 cells
sampled), with only a slight change to 2.6% in the 16
year-old graft (966 cells counted). Interestingly, a-synu-clein positive LNs were sparsely distributed throughout
host striatum around the grafts (Fig. 2). In sections
stained with thioflavin-S, recognizing the beta sheet
structure of a-synuclein, we observed several positive
cells and neurite-like structures in the grafts (Fig. 3).
Different Forms of Lewy Bodies in Grafts
The a-synuclein positive aggregates in the grafts
exhibited different morphologies. Some of them were
classical compact LBs and LNs, whereas others were
fine granular aggregates, sometimes forming a loose
meshwork of a-synuclein positive material (Fig. 1).
The compact and granular forms of a-synuclein aggre-
gates were detected in both the 12- (Fig. 1A and B)
and 16- (Fig. 1C and D) year-old grafts, as well as in
the host substantia nigra (Fig. 1E and F). We also fre-
quently found LN structures in the transplants, presum-
ably belonging to grafted neurons (Fig. 1A–D).
Ultrastructural Appearance of Lewy Bodies
in Grafted Neurons
Electron microscopy showed electron dense material
in the soma and proximal dendrites of several pig-
mented neurons (Fig. 4A, asterisks). Some of them
contained fibrillar structures that were immunoreactive
for a-synuclein and formed presumed LBs (Fig. 4B)
and LNs (Fig. 4C).
DISCUSSION
Here we provide further evidence that the a-synu-clein-containing aggregates detected in neural grafts in
PD patients are LBs and LNs, and that a-synuclein is
present in a fibrillar form. Our data suggest that LBs in
grafted neurons exhibit different stages of maturation,
and that their formation is a slow, gradual process.
Lewy Body Formation in Grafts Resembles
That Seen in the Substantia Nigra
We and others previously reported that a-synucleinaccumulates in grafted dopaminergic neurons, and that
in some cases it misfolds and forms LBs beyond one
Movement Disorders, Vol. 25, No. 8, 2010
1092 J.-Y. LI ET AL.
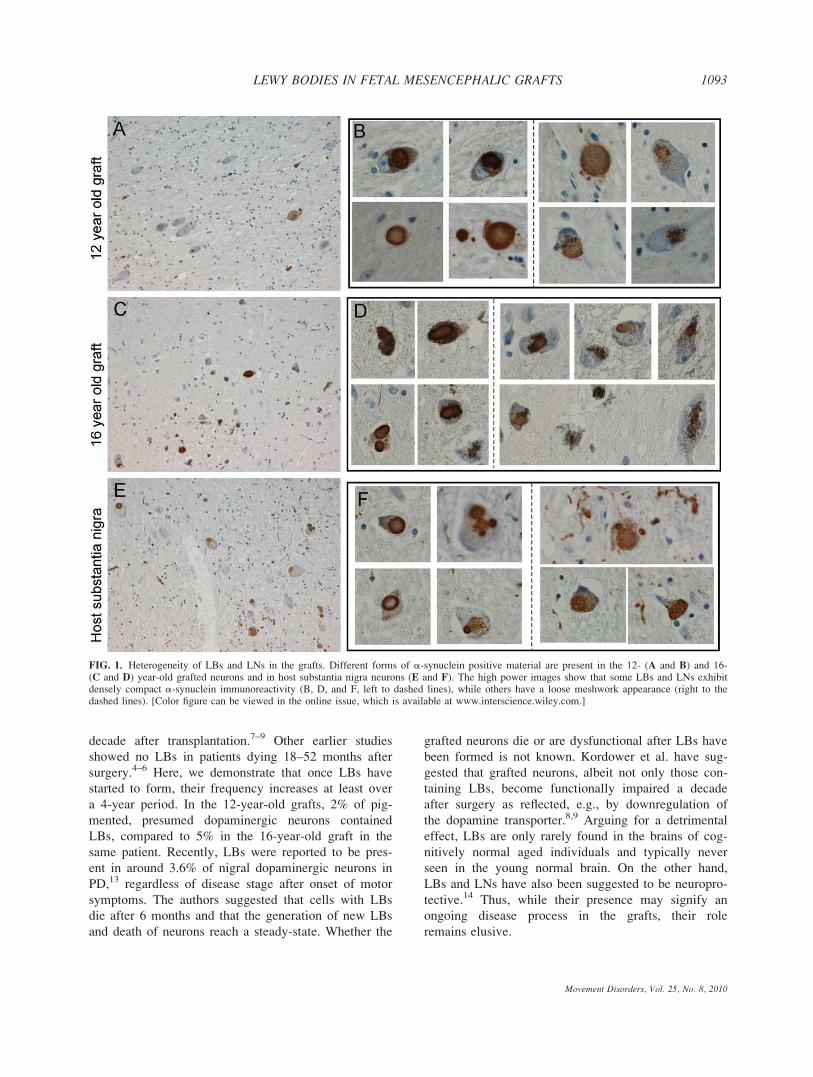
decade after transplantation.7–9 Other earlier studies
showed no LBs in patients dying 18–52 months after
surgery.4–6 Here, we demonstrate that once LBs have
started to form, their frequency increases at least over
a 4-year period. In the 12-year-old grafts, 2% of pig-
mented, presumed dopaminergic neurons contained
LBs, compared to 5% in the 16-year-old graft in the
same patient. Recently, LBs were reported to be pres-
ent in around 3.6% of nigral dopaminergic neurons in
PD,13 regardless of disease stage after onset of motor
symptoms. The authors suggested that cells with LBs
die after 6 months and that the generation of new LBs
and death of neurons reach a steady-state. Whether the
grafted neurons die or are dysfunctional after LBs have
been formed is not known. Kordower et al. have sug-
gested that grafted neurons, albeit not only those con-
taining LBs, become functionally impaired a decade
after surgery as reflected, e.g., by downregulation of
the dopamine transporter.8,9 Arguing for a detrimental
effect, LBs are only rarely found in the brains of cog-
nitively normal aged individuals and typically never
seen in the young normal brain. On the other hand,
LBs and LNs have also been suggested to be neuropro-
tective.14 Thus, while their presence may signify an
ongoing disease process in the grafts, their role
remains elusive.
FIG. 1. Heterogeneity of LBs and LNs in the grafts. Different forms of a-synuclein positive material are present in the 12- (A and B) and 16-(C and D) year-old grafted neurons and in host substantia nigra neurons (E and F). The high power images show that some LBs and LNs exhibitdensely compact a-synuclein immunoreactivity (B, D, and F, left to dashed lines), while others have a loose meshwork appearance (right to thedashed lines). [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
1093LEWY BODIES IN FETAL MESENCEPHALIC GRAFTS
Movement Disorders, Vol. 25, No. 8, 2010
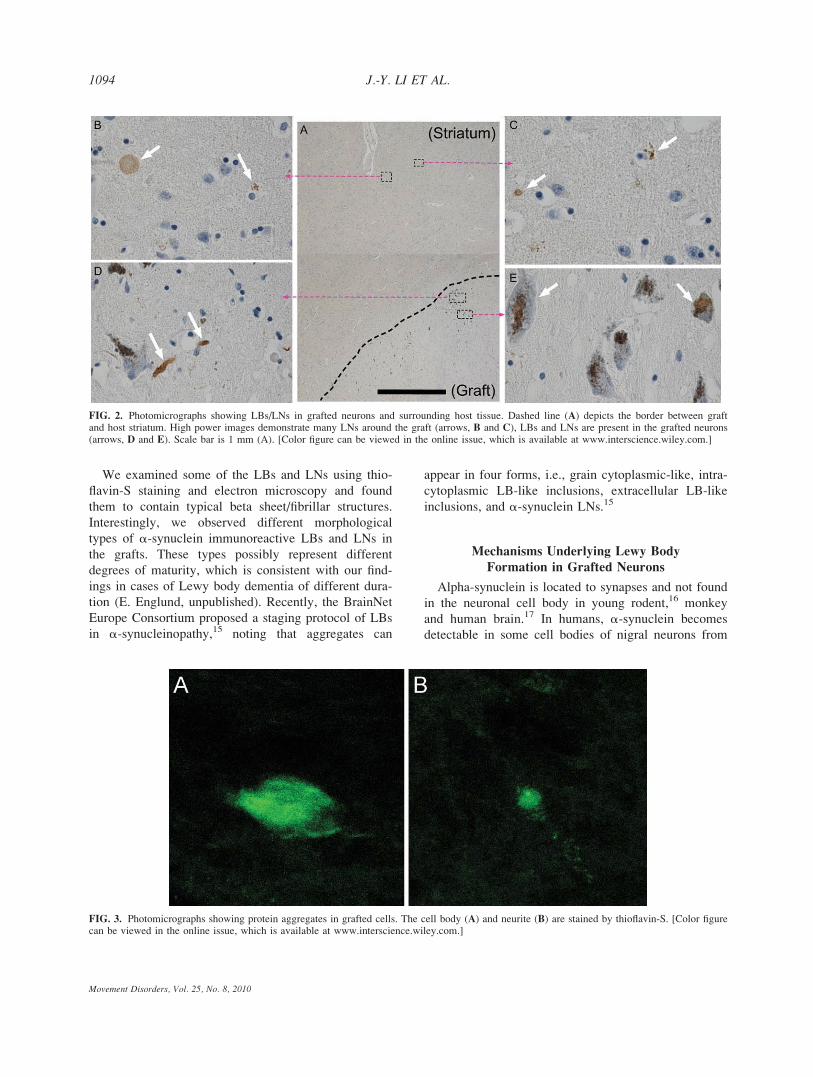
We examined some of the LBs and LNs using thio-
flavin-S staining and electron microscopy and found
them to contain typical beta sheet/fibrillar structures.
Interestingly, we observed different morphological
types of a-synuclein immunoreactive LBs and LNs in
the grafts. These types possibly represent different
degrees of maturity, which is consistent with our find-
ings in cases of Lewy body dementia of different dura-
tion (E. Englund, unpublished). Recently, the BrainNet
Europe Consortium proposed a staging protocol of LBs
in a-synucleinopathy,15 noting that aggregates can
appear in four forms, i.e., grain cytoplasmic-like, intra-
cytoplasmic LB-like inclusions, extracellular LB-like
inclusions, and a-synuclein LNs.15
Mechanisms Underlying Lewy Body
Formation in Grafted Neurons
Alpha-synuclein is located to synapses and not found
in the neuronal cell body in young rodent,16 monkey
and human brain.17 In humans, a-synuclein becomes
detectable in some cell bodies of nigral neurons from
FIG. 2. Photomicrographs showing LBs/LNs in grafted neurons and surrounding host tissue. Dashed line (A) depicts the border between graftand host striatum. High power images demonstrate many LNs around the graft (arrows, B and C), LBs and LNs are present in the grafted neurons(arrows, D and E). Scale bar is 1 mm (A). [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
FIG. 3. Photomicrographs showing protein aggregates in grafted cells. The cell body (A) and neurite (B) are stained by thioflavin-S. [Color figurecan be viewed in the online issue, which is available at www.interscience.wiley.com.]
1094 J.-Y. LI ET AL.
Movement Disorders, Vol. 25, No. 8, 2010

21 years of age, and cytoplasmic protein level then
increases with time.17 We previously reported that the
majority (80%) of the 16-year-old grafted neurons, but
only 40% of the cells in the 12-year-old graft in the
present patient exhibit high cytoplasmic levels of
a-synuclein.7 In contrast, we find here that only a mi-
nority of the grafted neurons contains LBs. These
observations suggest that the formation of LBs in
grafted neurons occurs in two steps, starting by
increased cytoplasmic levels of soluble a-synucleinfollowed by initiation of the aggregation process. The
mechanisms leading to increased cytoplasmic levels of
a-synuclein, followed by the generation of LBs and
LNs in grafted neurons are not known. We have specu-
lated that neuroinflammation, oxidative stress, excito-
toxicity or loss of trophic support could contribute.18
We also suggested that a-synuclein transfer from host
neurons to grafted cells may act as a seed for a-synu-clein aggregation, in a prion-like manner.7,18 In line
with this idea, we found LBs and LNs in the cortex7
and in the host striatum around the grafts in the present
patient. This hypothesis is further supported by a
recent study demonstrating that a-synuclein can move
from one cell to another in vitro and promote forma-
tion of aggregates in the new cells.19 Moreover, a-syn-uclein from host brain was found to enter neural
precursors grafted into transgenic mice overexpressing
a-synuclein.19 Transfer of aggregation-prone proteins
between cells is now suggested to play a role in many
proteinopathies.20 Protein containing expanded poly-
glutamine proteins and aggregates of mutant tau can
penetrate the outer membrane of cultured cells, act as
a seed and promote nucleation of endogenous proteins
expressing homologous sequences.21,22 Furthermore,
mutant human tau injected into mouse brains causes
aggregation of wild-type tau, and pathology can spread
from injection site to neighboring regions.23
Concluding Remarks
The findings of LBs in the grafted neurons provide
new insights into PD pathogenesis and may help to
explain how the pathology spreads within the patient’s
brain.18 Our observations are in line with the Braak
hypothesis24 concerning the propagation of Lewy pa-
thology according to stereotypic neuroanatomical pat-
terns, and suggest that transfer of misfolded protein
can be a key step of the process. The protracted devel-
opment of Lewy pathology indicates that there is a
large time window within which future therapies tar-
geting the transfer of proteins between cells can act.
Acknowledgments: This work was supported by grantsfrom the Swedish Research Council, Swedish ParkinsonFoundation, the Nordic Center of Excellence on Neurodegen-eration, the Strong Research Environment of the SwedishResearch Council (NeuroFortis) and Linne grant (BAGADI-LICO). We thank B.-M. Lindberg, A. Persson, and L. Geforsfor their excellent technical support.
Author Roles: J-YL designed and performed the detailedmorphological analysis of most of the materials. EE did au-topsy and routine neuropathology. AB provided expertise inneural transplantation. HW took care of the patient. SR oper-ated on the patient. PB dissected and prepared tissue for sur-
FIG. 4. Electron microscopic photomicrographs showing protein aggregates in grafted cells. Tissue sections were stained with an antibody againsthuman a-synuclein. (A) In the graft, three pigment-granule containing neurons (asterisks) did not exhibit any a-synuclein immunoreactive mate-rial, while one neuron was intensely positive for a-synuclein immunoreactivity. (B and C). The high power images (dashed squares) show detailsof LB (B) and LN (C). Fibril forms of a-synuclein positive structures are seen on zoomed up images (Insets). Scale bars 5 2 lm.
1095LEWY BODIES IN FETAL MESENCEPHALIC GRAFTS
Movement Disorders, Vol. 25, No. 8, 2010

geries. OL took care of the patient and headed the clinicaltransplantation program. J-YL wrote the manuscript. J-YL,EE, OL, and PB edited the manuscript. All authors gaveinput to the manuscript.
Financial Disclosure: Jia-Yi li: received grants fromSwedish Research Council, Swedish Parkinson Foundation,Segerfalk Foundation, Michael J. Fox Foundation, Lund Uni-versity. Elisabet Englund: received grants from Swedish Alz-heimer Foundation and Lund University. Hakan Widner:stock owership in medically related fields-NeuroVive AB;Advisory Boards-National Board for Health Evaluation (cur-rent); received grants from: Lund University; Region SkaneResearch Grants, Michael J. Fox Foundation, Swedish Par-kinson’s Disease Foundation; Ongoing studies with Neuro-Nova AB, Solvay Pharma AB, NeuroSearch A/S and Psycho-genics Inc, Medtronics AB. Stig Rehncrona: None. AndersBjorklund: Consultancies—NsGene A/S (Copenhagen, Den-mark), Amsterdam Molecular Therapeutics (Amsterdam);received grants from The Swedish Research Council, NEuro-StemCell (EU), Michael J Fox Foundation, Knut och AliceWallenbergs Stiftelse. Olle Lindvall: Consultancies—NsGene(Copenhagen, Denmark), Advisory Boards—Michael J. FoxFoundation for Parkinson’s Research, Merck Serono, Geneva,Switzerland; received grants from Swedish Research Council,Juvenile Diabetes Research Foundation; StemStroke (EUProject), The Soderberg, Crafoord, Segerfalk, and KockFoundations. Patrik Brundin: stock ownership in medicallyrelated fields—Lundbeck A/S; Medivir B, NeuroVive; Con-sultancies—NeuroNova AB (Stockholm, Sweden); AdvisoryBoards—H Lundbeck A/S (Copenhagen, Denmark), TEVAPharmaceutical Industries Ltd., Oxford Parkinson’s diseaseconsortium of the Department of Physiology, Member of theSAB of the Center for Molecular Physiology of the Brains,Gottingen, Germany, received grants from Swedish ResearchCouncil, NeuroNe, STEMS and AXREGEN (EU Projects),Human Frontier Science Foundation, Swedish Brain Founda-tion, MJ Fox Foundation, Soderberg Foundation.
REFERENCES
1. Hagell P, Brundin P. Cell survival and clinical outcome follow-ing intrastriatal transplantation in Parkinson disease. J Neuropa-thol Exp Neurol 2001;60:741–752.
2. Lindvall O, Bjorklund A. Cell therapy in Parkinson’s disease.NeuroRx 2004;1:382–393.
3. Piccini P, Brooks DJ, Bjorklund A, et al. Dopamine release fromnigral transplants visualized in vivo in a Parkinson’s patient. NatNeurosci 1999;2:1137–1140.
4. Mendez I, Sanchez-Pernaute R, Cooper O, et al. Cell type analy-sis of functional fetal dopamine cell suspension transplants in thestriatum and substantia nigra of patients with Parkinson’s disease.Brain 2005;128:1498–1510.
5. Kordower JH, Freeman TB, Chen EY, et al. Fetal nigral graftssurvive and mediate clinical benefit in a patient with Parkinson’sdisease. Mov Disord 1998;13:383–393.
6. Kordower JH, Freeman TB, Snow BJ, et al. Neuropathologicalevidence of graft survival and striatal reinnervation after thetransplantation of fetal mesencephalic tissue in a patient with Par-kinson’s disease. N Engl J Med 1995;332:1118–1124.
7. Li JY, Englund E, Holton JL, et al. Lewy bodies in grafted neu-rons in subjects with Parkinson’s disease suggest host-to-graftdisease propagation. Nat Med 2008;14:501–503.
8. Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW.Lewy body-like pathology in long-term embryonic nigral trans-plants in Parkinson’s disease. Nat Med 2008;14:504–506.
9. Kordower JH, Chu Y, Hauser RA, Olanow CW, Freeman TB.Transplanted dopaminergic neurons develop PD pathologicchanges: a second case report. Mov Disord 2008;23:2303–2306.
10. Hagell P, Schrag A, Piccini P, et al. Sequential bilateral trans-plantation in Parkinson’s disease: effects of the second graft.Brain 1999;122:1121–1132.
11. Lindvall O, Brundin P, Widner H, et al. Grafts of fetal dopamineneurons survive and improve motor function in Parkinson’sdisease. Science 1990;247:574–577.
12. Double KL, Dedov VN, Fedorow H, et al. The comparative biol-ogy of neuromelanin and lipofuscin in the human brain. Cell MolLife Sci 2008;65:1669–1682.
13. Greffard S, Verny M, Bonnet AM, Seilhean D, Hauw JJ, Duyck-aerts C. A stable proportion of Lewy body bearing neurons in thesubstantia nigra suggests a model in which the Lewy body causesneuronal death. Neurobiol Aging 2010;31:99–103.
14. Olanow CW, Perl DP, Demartino GN, Mcnaught KS. Lewy-bodyformation is an aggresome-related process: a hypothesis. LancetNeurol 2004;3:496–503.
15. Alafuzoff I, Ince PG, Arzberger T, et al. Staging/typing of Lewybody related alpha-synuclein pathology: a study of the BrainNetEurope Consortium. Acta Neuropathol 2009;117:635–652.
16. Li JY, Henning Jensen P, Dahlstrom A. Differential localizationof alpha-, beta- and gamma-synucleins in the rat CNS. Neuro-science 2002;113:463–478.
17. Chu Y, Kordower JH. Age-associated increases of alpha-synu-clein in monkeys and humans are associated with nigrostriataldopamine depletion: is this the target for Parkinson’s disease?Neurobiol Dis 2007;25:134–149.
18. Brundin P, Li JY, Holton JL, Lindvall O, Revesz T. Research inmotion: the enigma of Parkinson’s disease pathology spread. NatRev Neurosci 2008;9:741–745.
19. Desplats P, Lee HJ, Bae EJ, et al. Inclusion formation and neuro-nal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci USA 2009;106:13010–13015.
20. Aguzzi A. Cell biology: beyond the prion principle. Nature2009;459:924–925.
21. Frost B, Jacks RL, Diamond MI. Propagation of tau misfoldingfrom the outside to the inside of a cell. J Biol Chem 2009;284:12845–12852.
22. Ren PH, Lauckner JE, Kachirskaia I, Heuser JE, Melki R, KopitoRR. Cytoplasmic penetration and persistent infection of mammaliancells by polyglutamine aggregates. Nat Cell Biol 2009;11:219–225.
23. Clavaguera F, Bolmont T, Crowther RA, et al. Transmission andspreading of tauopathy in transgenic mouse brain. Nat Cell Biol2009;11:909–913.
24. Braak H, Del Tredici K, Rub U, De Vos RA, Jansen Steur EN, BraakE. Staging of brain pathology related to sporadic Parkinson’s dis-ease. Neurobiol Aging 2003;24:197–211.
1096 J.-Y. LI ET AL.
Movement Disorders, Vol. 25, No. 8, 2010




















