
Zinc-α2-Glycoprotein Exerts Antifibrotic Effects in Kidney and Heart
10
BASIC RESEARCH www.jasn.org Zinc-a2-Glycoprotein Exerts Antifibrotic Effects in Kidney and Heart Inga Sörensen-Zender,* Sagar Bhayana,* Nathan Susnik,* Veronique Rolli, † Sandor Batkai, ‡ Baisantry Arpita,* § Siamak Bahram, † Payel Sen,* Beina Teng,* Robert Lindner, | Mario Schiffer,* Thomas Thum, ‡¶ Anette Melk, § Hermann Haller,* and Roland Schmitt* Departments of *Nephrology and Hypertension, § Pediatric Kidney, Liver, and Metabolic Diseases, and | Cell Biology and ‡ Institute of Molecular and Translational Therapeutic Strategies, Integriertes Forschungs- und Behandlungszentrum Transplantation, Hannover Medical School, Hannover, Germany; † Immunogénétique Moléculaire Humaine, Centre de Recherche d’Immunologie et d’Hématologie, Faculté de Médecine, Hôpitaux Universitaires de Strasbourg, Strasbourg, France; and ¶ National Heart and Lung Institute, Imperial College, London, United Kingdom ABSTRACT Zinc-a2-glycoprotein (AZGP1) is a secreted protein synthesized by epithelial cells and adipocytes that has roles in lipid metabolism, cell cycling, and cancer progression. Our previous findings in AKI indicated a new role for AZGP1 in the regulation of fibrosis, which is a unifying feature of CKD. Using two models of chronic kidney injury, we now show that mice with genetic AZGP1 deletion develop significantly more kidney fibrosis. This destructive phenotype was rescued by injection of recombinant AZGP1. Exposure of AZGP1- deficient mice to cardiac stress by thoracic aortic constriction revealed that antifibrotic effects were not restricted to the kidney but were cardioprotective. In vitro, recombinant AZGP1 inhibited kidney epithelial dedifferentiation and antagonized fibroblast activation by negatively regulating TGF-b signaling. Patient sera with high levels of AZGP1 similarly attenuated TGF-b signaling in fibroblasts. Taken together, these findings indicate a novel role for AZGP1 as a negative regulator of fibrosis progression, suggesting that recombinant AZGP1 may have translational effect for treating fibrotic disease. J Am Soc Nephrol 26: ccc–ccc, 2015. doi: 10.1681/ASN.2014050485 Fibrosis of the kidney is present in almost all forms of chronic renal disease and has been identified as one of the most crucial determinants of progressive loss of renal function. 1 Studying age-dependent dif- ferences in renal repair, we observed that mice treated with small interfering RNA (siRNA) against zinc-a2-glycoprotein (AZGP1) were unexpectedly susceptible to kidney fibrosis, 2 suggesting a poten- tial link between AZGP1 and the development of fibrosis. AZGP1 is a secreted glycoprotein that is synthe- sized by adipocytes and epithelial cells of many organs. First isolated from human plasma, it was later found in other body fluids, such as saliva, sweat, milk, and urine. 3,4 The biologic function of AZGP1 is still poorly understood, and although the molecular structure of AZGP1 resembles a secre- tory form of MHC-I, the protein seems to be devoid of immunologic functions. 5 AZGP1 plays a role in lipid metabolism and potentially, cancer cachexia, where it has been described as a lipid-mobilizing fac- tor. 6 In agreement, AZGP1 mobilized fat in cultured adipocytes through stimulation of b3-adrenoceptors. 7 Furthermore, rodents that were administered exoge- nous AZGP1 showed increased fat mobilization, 8,9 whereas genetic deletion of AZGP1 was associated with increased body fat. 5 With the molecular mass of 41 kD, AZGP1 is freely filtered in the glomerulus and subsequently Received May 18, 2014. Accepted January 5, 2015. Published online ahead of print. Publication date available at www.jasn.org. Correspondence: Dr. Roland Schmitt, Hannover Medical School, Carl-Neuberg-Strasse 1, 30625 Hannover, Germany. Email: [email protected] Copyright © 2015 by the American Society of Nephrology J Am Soc Nephrol 26: ccc–ccc, 2015 ISSN : 1046-6673/2611-ccc 1
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Zinc-α2-Glycoprotein Exerts Antifibrotic Effects in Kidney and Heart

BASIC RESEARCH www.jasn.org
Zinc-a2-Glycoprotein Exerts Antifibrotic Effects inKidney and Heart
Inga Sörensen-Zender,* Sagar Bhayana,* Nathan Susnik,* Veronique Rolli,† Sandor Batkai,‡
Baisantry Arpita,*§ Siamak Bahram,† Payel Sen,* Beina Teng,* Robert Lindner,|
Mario Schiffer,* Thomas Thum,‡¶ Anette Melk,§ Hermann Haller,* and Roland Schmitt*
Departments of *Nephrology and Hypertension, §Pediatric Kidney, Liver, and Metabolic Diseases, and |Cell Biologyand ‡Institute of Molecular and Translational Therapeutic Strategies, Integriertes Forschungs- und BehandlungszentrumTransplantation, Hannover Medical School, Hannover, Germany; †Immunogénétique Moléculaire Humaine, Centrede Recherche d’Immunologie et d’Hématologie, Faculté de Médecine, Hôpitaux Universitaires de Strasbourg,Strasbourg, France; and ¶National Heart and Lung Institute, Imperial College, London, United Kingdom
ABSTRACTZinc-a2-glycoprotein (AZGP1) is a secreted protein synthesized by epithelial cells and adipocytes that hasroles in lipidmetabolism, cell cycling, and cancer progression. Our previous findings in AKI indicated a newrole for AZGP1 in the regulation of fibrosis, which is a unifying feature of CKD. Using twomodels of chronickidney injury, we now show that mice with genetic AZGP1 deletion develop significantly more kidneyfibrosis. This destructive phenotypewas rescued by injection of recombinant AZGP1. Exposure of AZGP1-deficient mice to cardiac stress by thoracic aortic constriction revealed that antifibrotic effects were notrestricted to the kidney butwere cardioprotective. In vitro, recombinant AZGP1 inhibited kidney epithelialdedifferentiation and antagonized fibroblast activation by negatively regulating TGF-b signaling. Patientsera with high levels of AZGP1 similarly attenuated TGF-b signaling in fibroblasts. Taken together, thesefindings indicate a novel role for AZGP1 as a negative regulator of fibrosis progression, suggesting thatrecombinant AZGP1 may have translational effect for treating fibrotic disease.
J Am Soc Nephrol 26: ccc–ccc, 2015. doi: 10.1681/ASN.2014050485
Fibrosis of the kidney is present in almost all formsof chronic renal disease and has been identified asone of the most crucial determinants of progressiveloss of renal function.1 Studying age-dependent dif-ferences in renal repair, we observed that micetreated with small interfering RNA (siRNA) againstzinc-a2-glycoprotein (AZGP1) were unexpectedlysusceptible to kidney fibrosis,2 suggesting a poten-tial link between AZGP1 and the development offibrosis.
AZGP1 is a secreted glycoprotein that is synthe-sized by adipocytes and epithelial cells of manyorgans. First isolated from human plasma, it waslater found in other body fluids, such as saliva,sweat, milk, and urine.3,4 The biologic function ofAZGP1 is still poorly understood, and although themolecular structure of AZGP1 resembles a secre-tory form ofMHC-I, the protein seems to be devoidof immunologic functions.5 AZGP1 plays a role in
lipid metabolism and potentially, cancer cachexia,where it has been described as a lipid-mobilizing fac-tor.6 In agreement, AZGP1 mobilized fat in culturedadipocytes through stimulation ofb3-adrenoceptors.7
Furthermore, rodents that were administered exoge-nous AZGP1 showed increased fat mobilization,8,9
whereas genetic deletion of AZGP1 was associatedwith increased body fat.5
With the molecular mass of 41 kD, AZGP1 isfreely filtered in the glomerulus and subsequently
Received May 18, 2014. Accepted January 5, 2015.
Published online ahead of print. Publication date available atwww.jasn.org.
Correspondence: Dr. Roland Schmitt, Hannover Medical School,Carl-Neuberg-Strasse 1, 30625 Hannover, Germany. Email:[email protected]
Copyright © 2015 by the American Society of Nephrology
J Am Soc Nephrol 26: ccc–ccc, 2015 ISSN : 1046-6673/2611-ccc 1

cleared by kidney tubular cells through reabsorption andlysosomal degradation.10 Circulating AZGP1 significantly ac-cumulates when renal clearance is disrupted in patients withacute and chronic renal failure.11–13 Stimulated by a uremicenvironment, adipocytes might secrete more AZGP1, furtherincreasing protein levels.14 However, the functional effect ofelevated AZGP1 levels in renal disease remains unclear. Be-cause AZGP1 was inversely associated with proatherogenicfactors and oxidative stress in patients undergoing hemodial-ysis, Leal et al.15 suggested that it might act as a cardiovascularprotective factor in this patient population.
Similar to other organs, kidney fibrosis is characterized by adedifferentiation of epithelial cells and an activation of in-terstitial fibroblasts. Like in other organs, the single mostimportant mediator of kidney fibrosis is TGF-b.16 The keyrole of TGF-b in progressive renal disease was underlined bystudies of TGF-b blockade and transgenic TGF-b overex-pression.17,18 These studies showed that TGF-b can potentlydisrupt renal cell homeostasis by activating resting fibroblastsinto myofibroblasts, which produce excessive amounts ofcollagen and other extracellular matrix components.1,16 Inparallel, sustained TGF-b signaling pro-motes fibrosis progression by inducingthe dedifferentiation of parenchymal epi-thelial cells.19 Interestingly, AZGP1 hasbeen shown to promote epithelial celldifferentiation and stabilization duringcarcinogenesis.20–23 Along these lines,AZGP1 was linked to inhibition of TGF-b–mediated dedifferentiation in pancre-atic cancer cells.24 On the basis of thesedata, we tested the hypothesis thatAZGP1 negatively regulates the develop-ment of organ fibrosis.
RESULTS
AZGP1 Deficiency ExacerbatesExperimental Kidney FibrosisOn the basis of our previous data,2 we hy-pothesized that AZGP1 inhibits profibroticprocesses in the kidney. To test this hy-pothesis, we induced experimental renalfibrosis in AZGP1-deficient mice. So far,the only phenotypical difference found inthese mice was a moderately increasedbody weight combined with a reduced li-polytic activity.5 To minimize potentiallyconfounding effects of this phenotypicaldifference, we used heterozygous litter-mates (AZGP1+/2) as controls instead ofwild-type (AZGP1+/+) mice in most ex-periments. AZGP1+/2 mice did not differsignificantly fromAZGP12/2mice in body
weight, kidney function, and triglyceride levels (SupplementalFigure 1, A–D). AZGP1+/2 showed global AZGP1 expressionat a level of approximately 50% of AZGP1+/+ mice (Supple-mental Figure 1, E and F), and there were no discernable dif-ferences in renal expression levels of E-cadherin and vimentin,suggesting a similar tubulointerstitial composition betweenunstressed AZGP12/2 and AZGP1+/2 kidneys (SupplementalFigure 1, G and H).
Mice were subjected to two well established experimentalmodels of renal tubulointerstitial fibrosis: aristolochic acidnephropathy (AAN) and unilateral ureteral obstruction(UUO). In both models, kidneys of AZGP12/2 mice hadmore tubular damage and increased tubulointerstitial matrixexpansion compared with AZGP1+/2 kidneys (Figure 1, A andC). In UUO, AZGP1 deletion was associated with a more se-vere loss of intact tubular brush border membrane, a sign ofepithelial damage and dedifferentiation, as shown by reducedLotus Tetragonolobus Lectin (LTL) staining (Figure 1, B andE). In both models, AZGP12/2 kidneys had enhanced depo-sition of interstitial collagen as reflected by Picrosirius Redstaining (Figure 1, C, D, and F).
Figure 1. Genetic deficiency of AZGP1 exacerbates progression of renal fibrosis inUUO and AAN. (A) Representative images of hematoxylin/eosin staining of AZGP1+/2
and AZGP12/2 kidney sections after 2 weeks of UUO. (B) Representative imagesshowing LTL staining as a marker of intact proximal tubular brush border in UUOkidneys. Representative images of Picrosirius Red staining in (C) bright-field and (D)polarized light at 4 weeks of AAN. (E) Quantification of the LTL-positive area in theouter medulla region of UUO and AAN kidneys. (F) Quantification of birefringentcollagen fibers in Picrosirius Red–stained kidney sections after UUO and AAN. UUOwas evaluated for all data at 2 weeks, and AAN was evaluated for all data at 4 weeks.Values are given as means6SEMs (n=8 for E and F). Original magnifications, 3200 inA, C, and D; 3100 in B. *P,0.05.
2 Journal of the American Society of Nephrology J Am Soc Nephrol 26: ccc–ccc, 2015
BASIC RESEARCH www.jasn.org

In addition, AZGP12/2 kidneys showed significantly in-creased interstitial a-Smooth Muscle Actin (aSMA), a markerof fibroblast activation, and enhanced expression of profi-brotic Collagen I and vimentin in both models (Figure 2,A–E). No changes were found in the overall numbers ofCD45-positive leukocytes and F4/80-positive macrophages.Renal expression of proinflammatory monocyte chemotacticprotein-1 (MCP1) was not different, but mRNA levels ofIL-1b were significantly higher in AZGP12/2 UUO kid-neys (Figure 2, E and F). The expressions of tubular damagemarkers kidney injury molecule-1 (Kim-1) and neutrophilgelatinase–associated lipocalin (NGAL) were also markedlystronger in AZGP12/2 kidneys (Figure 2, C–E). Additionally,in UUO, there was a significant reduction of expression of theepithelial differentiation marker E-cadherin (Figure 2, B andD). For additional genotype-phenotype correlation, UUOwasalso performed in wild-type littermates. In the renal analysis,wild-type kidneys showed a mild trend for additional protec-tion compared with kidneys from AZGP1+/2 mice, but noneof the differences reached statistical significance (Supplemen-tal Figure 2).
Deletion of AZGP1 Exacerbates Experimental CardiacFibrosisTo determine whether the observed antifibrotic effects ofAZGP1 were also present in other organs, we induced cardiachypertrophy and fibrosis in AZGP1+/2 and AZGP12/2 miceby a thoracic aortic constriction (TAC) model that leads toleft ventricular pressure overload. After 4 weeks of TAC,AZGP1+/2 and AZGP12/2 mice had significantly increasedventricular mass compared with sham-operated mice (13969and 14265 mg versus 91612 mg; each P,0.05), but there wasno significant difference between the genotypes. Despite sim-ilar degrees of hypertrophy, AZGP12/2 hearts had more ex-tracellular matrix deposition, which was indicated by Massontrichrome staining (Figure 3A), and an increased accumulationof collagen, whichwas evidenced by quantification of PicrosiriusRed staining (Figure 3, B–D). This was accompanied by a sig-nificantly higher abundance ofaSMA, Collagen I, and fibronec-tin in cardiac tissue of AZGP12/2 mice (Figure 3, E and F).
Systemic Treatment with AZGP1 Rescues theProfibrotic Phenotype of AZGP12/2 Mice in UUO
To test whether exogenous application ofAZGP1 would rescue the profibrotic pheno-type seen in AZGP12/2 mice, recombinantmurine AZGP1was injected into AZGP12/2
animals during UUO. Systemic treatmentwith 200 mg AZGP1 intravenously (ap-proximately 8 mg/kg) on 5 consecutivedays during UUO significantly attenuatedprofibrotic changes in AZGP12/2 kidneys,preserving the integrity of tubular brushborder membranes (Figure 4A), suppress-ing the expression of profibrotic aSMAand Collagen I (Figure 4B), and reducingexpression of tubular damage markersKim-1 and NGAL to levels only slightlyhigher than in AZGP1+/2 controls (Figure4B).
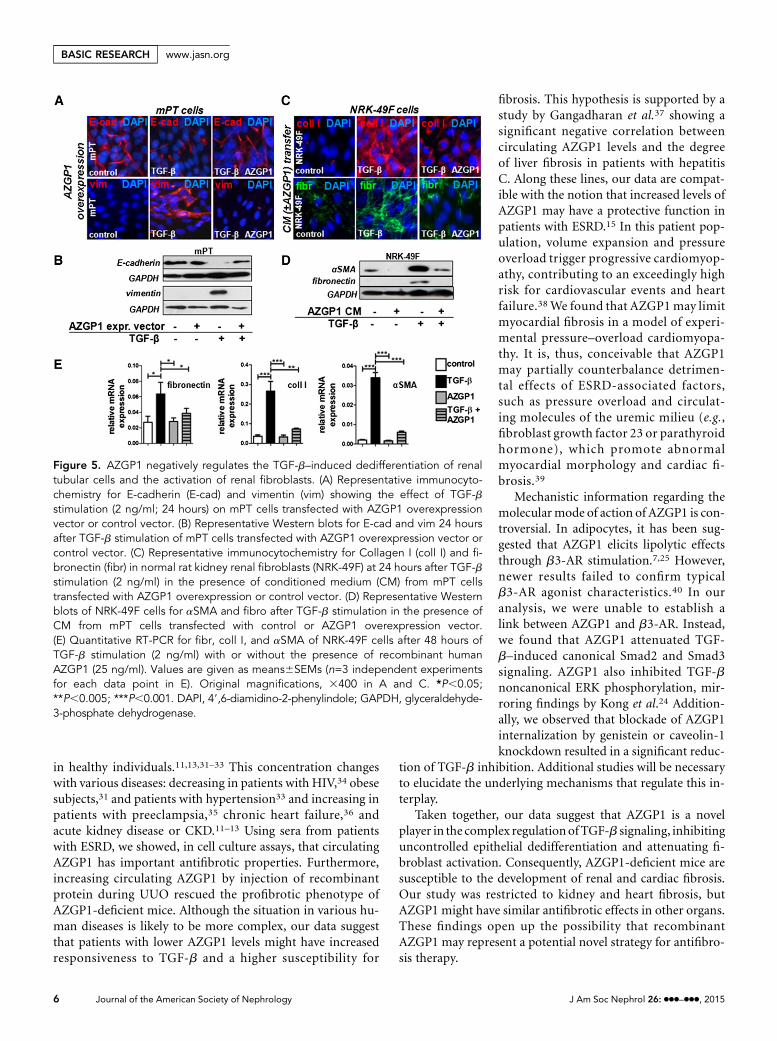
AZGP1 Attenuates TGF-b–InducedEffects in Renal Tubular Cells andRenal FibroblastsTGF-b is the single most important media-tor offibrosis in the kidney and other organs,exerting its detrimental effect by inducingepithelial dedifferentiation and activatinglocal fibroblasts.16 To test whether the an-tifibrotic effects of AZGP1 might be ex-plained through TGF-b antagonism,AZGP1 was overexpressed in cultured mu-rine proximal tubular (mPT) cells thatwere challenged with TGF-b. On additionof TGF-b, AZGP1-overexpressing cellsshowed sustained E-cadherin expressionand did not express vimentin, indicating
Figure 2. Genetic deficiency of AZGP1 is associated with enhanced epithelial de-differentiation and extracellular matrix accumulation. (A) Representative images ofimmunofluorescence staining of aSMA in AZGP1+/2 and AZGP12/2 kidney sections4 weeks after AAN. (B) Representative Western blots for aSMA and E-cadherin at2 weeks of UUO. (C) Representative Western blots for aSMA and Kim-1 at 4 weeks ofAAN. (D and E) Quantitative RT-PCR for fibrosis genes, tubular damage markers, andinflammatory genes in UUO and AAN kidneys. (F and G) Quantification of cells pos-itive for CD45 or F4/80 immunofluorescence in UUO and AAN kidney sections. Valuesare given as means6SEMs (n=8 for D–G). Original magnification, 3400 in A.*P,0.05;**P,0.005. DAPI, 49,6-diamidino-2-phenylindole; GAPDH, glyceraldehyde-3-phosphatedehydrogenase.
J Am Soc Nephrol 26: ccc–ccc, 2015 AZGP1 Inhibits Fibrosis 3
www.jasn.org BASIC RESEARCH

an inhibition of TGF-b–induced epithelial dedifferentiation(Figure 5, A and B).
Fibroblasts do not express AZGP1 but are exposed to itthrough local and systemic secretions.2 To mimic this situation,conditioned medium from AZGP1–overexpressing mPT cellswas transferred to normal rat kidney fibroblasts (NRK-49Fs).When fibroblasts were subsequently stimulated with TGF-b, thetypical profibrotic cell activation as mirrored by upregulation ofCollagen I, fibronectin, and aSMA was significantly inhibited(Figure 5, C andD). The same effect was found when fibroblastswere exposed to purified recombinant human (Figure 5E) orrecombinant murine AZGP1 (Supplemental Figure 3A).
AZGP1 Inhibits TGF-b Signaling by anEndocytosis-Dependent MechanismTo elucidate the mechanism behind AZGP1 antagonism of theTGF-b pathway, we performed luciferase assays of the Smadpromoter, revealing less TGF-b–induced transcription activ-ity in the presence of AZGP1 (Figure 6A). Consistently, therewas reduced TGF-b–induced phosphorylation of Smad2 andSmad3, whereas the mild increase in Smad1/Smad5/Smad9
phosphorylation was not significantly altered (Figure 6B).We also found that AZGP1 suppressed TGF-b–dependent ex-tracellular signal–regulated kinase (ERK) phosphorylation,whereas it had no effect on basal ERK activation (Figure6C). To test the possibility that AZGP1 antagonizes TGF-bby direct binding or competitive blocking of its receptor,coimmunoprecipitation studies were performed but failed toshow any protein-protein interaction (Supplemental Figure 3,B and C). Similarly, propranolol blockade of the only de-scribed putative receptor of AZGP1, the b3-adrenergic recep-tor (b3-AR),25 had no significant effect on AZGP1-inducedchanges in fibroblasts (Supplemental Figure 3, D and E). Con-focal microscopy revealed that fluochrome-labeled AZGP1was readily internalized into renal fibroblasts (Figure 6D). In-ternalization was significantly diminished after preincubationwith genistein, a tyrosine kinase inhibitor, which is often usedto block caveolin-dependent endocytosis (Figure 6, E and F).More specific inhibition using caveolin-1 siRNA also resultedin a reduction in AZGP1 uptake (Figure 6, G–I). Diminishedinternalization of AZGP1 after genistein treatment andcaveolin-1 knockdown was paralleled by an attenuated TGF-b
Figure 3. Genetic deficiency of AZGP1 exacerbates progression of cardiac fibrosis in the TAC model. (A) Representative images ofMasson trichrome–stained heart sections from AZGP1+/2 and AZGP12/2 mice after 4 weeks of TAC. Representative images of Pic-rosirius Red staining in (B) bright-field and (C) polarized light microscopy. (D) Quantification of Picrosirius Red birefringent signal.(E) Representative Western blots of aSMA and Collagen I in cardiac tissue of AZGP1+/2 and AZGP12/2 mice at 4 weeks of TAC.(F) Quantitative RT-PCR for fibrosis genes at 4 weeks of TAC. Values are given as means6SEMs (n=8 for each data point in D andF). Original magnifications, 3200 in A; 3400 in B and C. *P,0.05; **P,0.005. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
4 Journal of the American Society of Nephrology J Am Soc Nephrol 26: ccc–ccc, 2015
BASIC RESEARCH www.jasn.org

inhibitory effect of AZGP1 (Figure 6, J–M). These resultssuggest a link between endocytosis and AZGP1-dependent an-tagonism of the TGF-b pathway.
AZGP1 in Patient Sera Exerts TGF-b Inhibitory EffectsTo test the hypothesis that serum AZGP1 might confer anti-fibroticeffects,weusedsera frompatientswithESRD,whichhaveparticularly high levels of circulating AZGP1.11–13 Fibroblaststhat were exposed to medium supplemented with ESRD serumshowed a markedly lower TGF-b response (Figure 7A). WhenAZGP1 levels were reduced by antibody-mediated depletion(Figure 7B), the inhibitory effect of ESRD serum was signifi-cantly attenuated (Figure 7A). These findings suggest that circu-lating AZGP1 in patient sera may act as a negative regulator oftheTGF-b pathway,which suggests that individual AZGP1 levelscould change the susceptibility to profibrotic disease processes.
DISCUSSION
In this study, we characterized AZGP1 as a negative regulatorof TGF-b–induced effects in renal epithelial cells and renal
fibroblasts. We found that genetic deletion of AZGP1 aggra-vated kidney and heart fibrosis in experimental injury models.This destructive phenotype was rescued by exogenous admin-istration of recombinant AZGP1. Cell culture assays revealedthat TGF-b antagonistic effects of AZGP1 were linked to en-docytosis and also found in patient sera. Together, these find-ings introduce AZGP1 as a novel player in the balance of theepithelial-interstitial homeostasis.
One of the main observations of this study was that AZGP1attenuated TGF-b–induced epithelial dedifferentiation and fi-broblast activation, which are common features of organ fi-brosis and cancer progression.26 Enhanced tumor progressionhas been shown to correlate with lower expression of epithelialAZGP1.20–23 Kong et al.24 found that AZGP1 expression canpreserve epithelial integrity by promoting mesenchymal-to-epithelial transition in pancreatic cancer cells. Kong et al.24
attributed the prodifferentiation effects of AZGP1 to TGF-bantagonistic properties. We observed similar anti–TGF-b ef-fects of AZGP1 in renal epithelial cells. Additionally, we foundthat AZGP1 attenuated TGF-b–induced activation offibroblasts. These cell culture findings were congruent to ourfindings in knockout mice. Genetic AZGP1 deletion was as-sociated with a more pronounced loss of epithelial differenti-ation and enhanced activation of fibroblasts in kidneysstressed by AAN or UUO. Both AAN and UUO importantlyinvolve the TGF-b pathway.27,28 AZGP1-deficient mice werealso more severely affected by TAC-induced cardiac fibrosis,which is also driven by TGF-b.29 Together with our cell cultureresults, these findings suggest that AZGP1 acquires its antifi-brotic properties through inhibition of pathologic TGF-bpathway activation. In accordance with this, we found thatphysiologically low levels of TGF-b were associated with aninconspicuous baseline phenotype in unstressed AZGP1–deficient mice. Importantly, there was no significant differencebetween UUO kidneys from wild-type and AZGP1+/2 mice,suggesting that a 50% reduction of normal AZGP1 is stillcompatible with an appropriate renal response to fibroticstress.
Agrowingnumberof clinical studies suggesting thatAZGP1plays a role inmany different cancers20–22 implies that our datamight be relevant to the field of oncology. If it is correct thatAZGP1 exerts antitumor effects through inhibition of TGF-b,24 AZGP1-deficient mice would be expected to have a highersusceptibility for tumor progression. The fact that we did notobserve pathologic tumor formation argues against a pivotalrole in cancer regulation. However, it is possible that antitu-mor effects of AZGP1 only become relevant in a tumor mi-croenvironment containing increased TGF-b levels. In thiscontext, it is important to keep in mind that TGF-b can alsoact as a tumor suppressor, possibly repressing carcinogenesisat early stages.30 Therefore, the TGF-b inhibitory effect ofAZGP1 might be heterogeneous in cancer development,which should be explored in experimental studies.
AZGP1 is a glycoprotein secreted by most epithelial tissues,reaching a considerable serum concentration of 40–70 mg/ml
Figure 4. Administration of recombinant AZGP1 rescues profibroticphenotype of AZGP12/2 kidneys. (A) Quantification of LTL-positivestaining in 1-week UUO kidneys from AZGP1+/2 and AZGP12/2
mice compared with AZGP12/2 mice after systemic injection ofrecombinant AZGP1 (200 mg intravenously on 5 consecutivedays). (B) Quantitative RT-PCR for fibrosis and tubular damagemarkers. Values are given as means6SEMs (n=8 for each datapoint). *P,0.05; **P,0.005.
J Am Soc Nephrol 26: ccc–ccc, 2015 AZGP1 Inhibits Fibrosis 5
www.jasn.org BASIC RESEARCH
in healthy individuals.11,13,31–33 This concentration changeswith various diseases: decreasing in patients with HIV,34 obesesubjects,31 and patients with hypertension33 and increasing inpatients with preeclampsia,35 chronic heart failure,36 andacute kidney disease or CKD.11–13 Using sera from patientswith ESRD, we showed, in cell culture assays, that circulatingAZGP1 has important antifibrotic properties. Furthermore,increasing circulating AZGP1 by injection of recombinantprotein during UUO rescued the profibrotic phenotype ofAZGP1-deficient mice. Although the situation in various hu-man diseases is likely to be more complex, our data suggestthat patients with lower AZGP1 levels might have increasedresponsiveness to TGF-b and a higher susceptibility for
fibrosis. This hypothesis is supported by astudy by Gangadharan et al.37 showing asignificant negative correlation betweencirculating AZGP1 levels and the degreeof liver fibrosis in patients with hepatitisC. Along these lines, our data are compat-ible with the notion that increased levels ofAZGP1 may have a protective function inpatients with ESRD.15 In this patient pop-ulation, volume expansion and pressureoverload trigger progressive cardiomyop-athy, contributing to an exceedingly highrisk for cardiovascular events and heartfailure.38We found that AZGP1may limitmyocardial fibrosis in a model of experi-mental pressure–overload cardiomyopa-thy. It is, thus, conceivable that AZGP1may partially counterbalance detrimen-tal effects of ESRD-associated factors,such as pressure overload and circulat-ing molecules of the uremic milieu (e.g.,fibroblast growth factor 23 or parathyroidhormone), which promote abnormalmyocardial morphology and cardiac fi-brosis.39
Mechanistic information regarding themolecularmode of action of AZGP1 is con-troversial. In adipocytes, it has been sug-gested that AZGP1 elicits lipolytic effectsthrough b3-AR stimulation.7,25 However,newer results failed to confirm typicalb3-AR agonist characteristics.40 In ouranalysis, we were unable to establish alink between AZGP1 and b3-AR. Instead,we found that AZGP1 attenuated TGF-b–induced canonical Smad2 and Smad3signaling. AZGP1 also inhibited TGF-bnoncanonical ERK phosphorylation, mir-roring findings by Kong et al.24 Addition-ally, we observed that blockade of AZGP1internalization by genistein or caveolin-1knockdown resulted in a significant reduc-
tion of TGF-b inhibition. Additional studies will be necessaryto elucidate the underlying mechanisms that regulate this in-terplay.
Taken together, our data suggest that AZGP1 is a novelplayer in the complex regulationofTGF-b signaling, inhibitinguncontrolled epithelial dedifferentiation and attenuating fi-broblast activation. Consequently, AZGP1-deficient mice aresusceptible to the development of renal and cardiac fibrosis.Our study was restricted to kidney and heart fibrosis, butAZGP1 might have similar antifibrotic effects in other organs.These findings open up the possibility that recombinantAZGP1 may represent a potential novel strategy for antifibro-sis therapy.
Figure 5. AZGP1 negatively regulates the TGF-b–induced dedifferentiation of renaltubular cells and the activation of renal fibroblasts. (A) Representative immunocyto-chemistry for E-cadherin (E-cad) and vimentin (vim) showing the effect of TGF-bstimulation (2 ng/ml; 24 hours) on mPT cells transfected with AZGP1 overexpressionvector or control vector. (B) Representative Western blots for E-cad and vim 24 hoursafter TGF-b stimulation of mPT cells transfected with AZGP1 overexpression vector orcontrol vector. (C) Representative immunocytochemistry for Collagen I (coll I) and fi-bronectin (fibr) in normal rat kidney renal fibroblasts (NRK-49F) at 24 hours after TGF-bstimulation (2 ng/ml) in the presence of conditioned medium (CM) from mPT cellstransfected with AZGP1 overexpression or control vector. (D) Representative Westernblots of NRK-49F cells for aSMA and fibro after TGF-b stimulation in the presence ofCM from mPT cells transfected with control or AZGP1 overexpression vector.(E) Quantitative RT-PCR for fibr, coll I, and aSMA of NRK-49F cells after 48 hours ofTGF-b stimulation (2 ng/ml) with or without the presence of recombinant humanAZGP1 (25 ng/ml). Values are given as means6SEMs (n=3 independent experimentsfor each data point in E). Original magnifications, 3400 in A and C. *P,0.05;**P,0.005; ***P,0.001. DAPI, 49,6-diamidino-2-phenylindole; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
6 Journal of the American Society of Nephrology J Am Soc Nephrol 26: ccc–ccc, 2015
BASIC RESEARCH www.jasn.org

CONCISE METHODS
UUOAZGP1-deficient mice were generated and genotyped as previously
described.5 Male wild-type, AZGP1-deficient, and heterozygote lit-
termates (10–12 weeks old) were subjected to UUO as previously
reported (n=8 for each condition and group).41 Mice were anesthe-
tized and underwent left proximal ureteral ligation through amidline
abdominal incision. Mice were euthanized 7 or 14 days after UUO,
and kidneys were analyzed. Representative slices of each kidney
(including cortex andmedulla) were frozen in liq-
uid nitrogen or fixed in 4% paraformaldehyde
(PFA). For the rescue experiment, AZGP1-
deficientmicewere treatedwith systemic injections
of AZGP1 (200mg intravenously; approximately
8 mg/kg) on 5 consecutive days. For functional
studies, blood samples were taken, and renal
function was estimated by serum creatinine
and urea measurements as well as measurement
of triglycerides using an automated method
(Beckman Analyzer; Beckman Instruments
GmbH, Munich, Germany). All experimental
procedures were in agreement with institutional
and legislator regulations and approved by the
local authorities.
AANFor AAN, 10 mg/kg aristolochic acid (Sigma-
Aldrich, St. Louis, MO) was injected intraper-
itoneally into male 10- to 12-week-old mice as
previously described (n=8 for each condition
and group).42 The control group was injected
with the vehicle solution comprising of
DMSO. Mice were euthanized 28 days after
injection. Representative slices of each kidney
(including cortex and medulla) were frozen in
liquid nitrogen or fixed in 4% PFA.
Cardiac Fibrosis (TAC Model)A cardiac fibrotic response was induced by a
model of cardiac pressure overload caused by
TAC as previously described (n=8 for each con-
dition and group).43 Male AZGP1–deficient
mice and heterozygote littermates (10–12 weeks
old) were used for this purpose. Sham-operated
animals served as controls. Hearts were explan-
ted after 4 weeks of TAC and prepared for his-
tologic and biochemical analysis.
Histologic and ImmunohistochemicalStainingObstructed and contralateral kidneys were dis-
sected, fixed in 4% PFA, and embedded in par-
affin. Four-micrometer sections were used for
hematoxylin/eosin staining, Picrosirius Red
staining, and immunohistochemistry. Immunohistochemistry was
performed using the following primary antibodies: rat anti-mouse
F4/80 (Serotec, Oxford, UK), mouse anti-aSMA, clone 1A4 (Sigma-
Aldrich), rat anti-mouse CD45 (BD Bioscience), and anti-Collagen I
(Calbiochem, Darmstadt, Germany). Biotinylated LTL was pur-
chased from Vector Laboratories (Burlingame, CA). Deparaffinized
kidney sections were boiled for 20 minutes in citrate buffer for an-
tigen-retrieval, blocked with 5%milk, and incubated overnight with
primary antibodies. Staining was visualized with Alexa 488/Alexa
547 secondary antibodies (Molecular Probes/Invitrogen, Carlsbad,
Figure 6. AZGP1 inhibits TGF-b–induced Smad and ERK phosphorylation, which islinked to its internalization by endocytosis. (A) Smad reporter luciferase assay inNRK-49F cells after 24 hours of TGF-b stimulation (2 ng/ml) in the presence or ab-sence of recombinant AZGP1 (25 ng/ml). (B and C) Representative Western blots forphosphorylated Smad2, Smad3, Smad1/Smad5/Smad9, ERK, and their respectivetotal protein after 24 hours of TGF-b stimulation (2 ng/ml) in the presence or absenceof recombinant AZGP1 (25 ng/ml). (D) Representative confocal microscopy imageshowing NRK-49F cells at 2 hours after administration of FITC–labeled recombinanthuman AZGP1. Cellular uptake of labeled AZGP1 is significantly reduced by (E and F)pretreatment with genistein (200 mM) and (G and H) after siRNA-mediated knockdownof caveolin-1. (I) Quantification of cytoplasmic FITC–positive pixels after treatmentwith control or anticaveolin-1 siRNA. Quantitative RT-PCR shows change in AZGP1effect on aSMA expression (J and K) after pretreatment with genistein (200 mM) and(L and M) after siRNA-mediated knockdown of caveolin-1. Values are given as means6SEMs (n=3 independent experiments for each data point in A, I, and J–M.) Originalmagnifications, 363 in D–H. *P,0.05; **P,0.005; ***P,0.001.
J Am Soc Nephrol 26: ccc–ccc, 2015 AZGP1 Inhibits Fibrosis 7
www.jasn.org BASIC RESEARCH

CA) or the ABC Vectastain Kit (Vector Laboratories). For Picrosirius
Red staining, deparaffinized kidney sections were stained in Picro-
sirius Red staining solution (Sigma-Aldrich) for 1 hour, washed in
acidified water, and mounted. The staining was analyzed under po-
larized light, considering birefringence as a positive signal. Quanti-
fication of F4/80- and CD45-expressing cells was done by counting
the positive cells in 10 randomly chosen, nonoverlapping fields
(3200 magnification) in the outer medulla. aSMA, LTL, and Picro-
sirius Red staining was quantified using Photoshop-based image
analysis (Adobe Photoshop software; Adobe Systems, San Jose, CA).
Western Blot and CoimmunoprecipitationWestern analysis was performed as previously described.2 In brief, a
representative part of each kidney (kidney slices containing cortical
and medullary tissues) was frozen immediately after harvesting in
liquid nitrogen. Tissue was later homogenized, and after protein elec-
trophoresis, proteins were transferred to polyvinylidene difluoride
membranes, blocked with 5% milk in phosphate buffered saline/
tween, and probed overnight at 4°C with primary antibodies: anti-
aSMA (Sigma-Aldrich), rabbit anti-human KIM-1 (Novus Bio-
logicals, Littleton, CO), goat anti-mouse Lipocalin2/NGAL (R&D
Systems, Minneapolis, MN), rabbit anti-human E-Cadherin (Santa
Cruz Biotechnology, Santa Cruz, CA), rabbit anti-human Vimentin
(Santa Cruz Biotechnology), goat anti-human Fibronectin (Santa Cruz
Biotechnology), rabbit anti–rat-ERK1/2 (p44/42 mitogen-activated
protein kinase), rabbit anti-human phospho-ERK1/2 (phospho-
p44/42 mitogen-activated protein kinase; Cell Signaling Technology,
Danvers, MA), rabbit anti-Smad2 (Cell Signaling Technology), rabbit
antiphospho-Smad2 (Cell Signaling Technology), rabbit anti-Smad3
(Cell Signaling Technology), rabbit antiphospho-Smad3 (Cell Sig-
naling Technology), rabbit anti-Smad5 (Cell Signaling Technology),
rabbit antiphospho-Smad1/5/9 (Cell Signaling Technology), goat anti-
mouse AZGP1 (Zag [E-20]; Santa Cruz Biotech-
nology), and mouse anti-human AZGP1 (Zag
[1D4]; Santa Cruz Biotechnology). horseradish
peroxidase–conjugated secondary antibodies
were purchased from Santa Cruz Biotechnology.
Antibody binding was visualized by chemilumi-
nescence (SuperSignal West Pico Chemilumines-
cent; Thermo Fisher Scientific, Rockford, IL).
Rabbit anti-mouse glyceraldehyde-3-phosphate
dehydrogenase (Sigma-Aldrich) was used as an
internal loading control and normalization of
protein quantification. For coimmunoprecipita-
tion, protein lysate was incubated overnight with
anti-AZGP1 (H-123 and E-20), anti–TGF-b, and
anti–TGF-b receptor-II antibody (Santa Cruz
Biotechnology) followed by the addition of Pro-
tein G PLUS-Agarose beads (Santa Cruz Biotech-
nology) for 4 hours at 4°C. Beads were washed
and boiled in 43 gel-loading dye and analyzed by
immunoblotting for anti-AZGP1, anti–TGF-b,
and anti–TGF-b receptor-II antibody (same anti-
bodies as for immunoprecipitation).
Cell CultureNormal rat fibroblasts (NRK-49F; ATCC) as well as mPT epithelial
cells (provided by Lloyd G. Cantley, Yale School of Medicine, New
Haven, CT) were cultured in DMEM/F12 with 10% FCS. Cells were
treatedwithhuman recombinantTGF-b (2 ng/ml; R&DSystems) and
human recombinant AZGP1 (25 ng/ml; R&D Systems) for 6, 24, or
48 hours. Before treatment, cells were starved in DMEM/F12 without
FCS for 16 hours. Fibroblasts and epithelial cells were transfected
with expression or reporter plasmids using TransIT-LT1 transfection
reagent (Mirus Bio LLC) according to the manufacturer’s instruc-
tions. For blockage of endocytosis, NRK-49F cells were transfected
with 10 nM caveolin-1 siRNA (Qiagen, Hilden, Germany) using Lip-
ofectamine RNAiMAX transfection reagent (Life Technologies,
Darmstadt, Germany) for 48 hours before FITC–labeled recombi-
nant AZGP1 was added (knockdown efficiency was confirmed by
quantitative PCR showing a 70%–80% reduction of caveolin-1
mRNA) or preincubated with Genistein (200 mM; Sigma-Aldrich) for
30 minutes. Cells were fixed after 15 minutes or 2 hours of exposure.
Uptake of labeled AZGP1 was quantified by analyzing cytoplasmic
fluorescent pixels using confocal laser–scanning microphotographs
and National Institutes of Health ImageJ software. For inhibition of
b-adrenoreceptor, activation cells were preincubated with 10 mM
propanolol (Sigma-Aldrich) for 30 minutes and harvested after
24 hours. Sera from patients with ESRD (n=11) were diluted 1:20 in
culture medium.
Luciferase AssayNRK-49F cells were transfected with Smad luciferase reporter
construct SBE-Luc vector 16527 from Addgene (Cambridge, MA)
and treated with TGF-b and AZGP1 as described above. After
24 hours, luciferase activity was measured using the Dual-Luciferase
Reporter Assay (Promega,Madison,WI) according to themanufacturer’s
Figure 7. Sera from patients with ESRD negatively regulate TGF-b–mediated renalfibroblast activation in an AZGP1-dependent manner. (A) Quantitative RT-PCR foraSMA expression in NRK-49F cells with or without TGF-b stimulation (2 ng/ml) in thepresence or absence of 5% ESRD patient sera. (B) AZGP1 levels in ESRD patient serawere reduced by antibody treatment as shown by ELISA quantification before andafter depletion treatment. Values are given as means6SEMs (n=11 for each data pointusing sera from 11 patients with ESRD) in A and B. *P,0.05; ***P,0.001.
8 Journal of the American Society of Nephrology J Am Soc Nephrol 26: ccc–ccc, 2015
BASIC RESEARCH www.jasn.org

instructions. Firefly luciferase activity was normalized to cotransfected
Renilla luciferase activity (pRL-TK; Promega).
Production and Purification of AZGP1HEK293-F cells were seeded in FreeStyle Medium (Gibco; Life
Technologies, Carlsbad, CA) and transfected with expression vector
containing mouse AZGP1. Harvesting and purification of recombi-
nant AZGP1 was done as previously described by Russell and Tisdale9
using centrifugal concentrating filters (Amicon, Billerica, MA) with a
10-kD cutoff. Concentrated medium was mixed 1 g per 10 ml
diethylaminoethyl cellulose (Sigma-Aldrich) in 10 mM Tris (pH 8.8)
to allow binding of negatively charged AZGP1. Diethylaminoethyl cel-
lulose was subsequently centrifuged, and AZGP1 was eluted by add-
ing 10mMTris/0.3MNaCl. The supernatant was concentrated using
Amicon centrifugal filters with a cutoff of 30 kD. Protein concentra-
tion was measured by Bradford Assay. For in vivo injection, the pro-
tein was diluted in 0.9% NaCl. For fluorescent labeling of AZGP1,
recombinant mouse AZGP1 was labeled with the Atto 488 Protein
Labeling Kit (Sigma-Aldrich) according to the manufacturer’s in-
structions.
Quantitative Real–Time PCRRNA was isolated from NRK-49F cells and frozen kidney tissue
(kidney slices containing cortical and medullary tissue) using the
RNeasy Mini Kit (Qiagen) according to the manufacturer’s instruc-
tions. Reverse transcription was performed with M-MLV Reverse
Transcription (Promega). cDNA was used as a template for quanti-
tative PCR. The levels ofmRNAexpressionwere determined by quan-
titative real-time PCR using a Roche Lightcycler 480 System with
SYBR green master mix and specific primers: NGAL forward: TGA
AGGAACGTT TCACCCGCT TTG,NGAL reverse: ACAGGAAAG
ATG GAG TGG CAG ACA; KIM-1 forward: AAA CCA GAG ATT
CCC ACA CG, KIM-1 reverse: GTC GTG GGT CTT CCT GTA GC;
aSMA forward: GTG CTA TGT CGC TCT GGA CTT TGA; aSMA
reverse: ATG AAA GAT GGC TGG AAG AGG GTC; Collagen I for-
ward: TGT CCC AAC CCC CAA AGA C, Collagen I reverse: CCC
TCG ACT CCTACATCT GA; Fibronectin forward: CGG TAG GAC
CTT CTA TTC CT, Fibronectin reverse: GAT ACA TGA CCC CTT
CAT TG; E-cadherin forward: AGT CCC GGC TTC AGT TCC,
E-cadherin reverse: CTG TGA TGG TGC CGT CTG T; Vimentin
forward: CAC ACG CAC CTA CAG TCT G, Vimentin reverse: GTC
CACCGAGTCTTGAAGC; IL-1b forward: AGGTCCACGGGAAA-
GACACAGG, IL-1b reverse: GGGCTGCTTCCAAACCTTTGAC;
MCP1 forward: TCATGCTTCTGGGCCTGC, MCP1 reverse:
CAGCCTACTCATTGGGATCATC; and Actin forward: AGC CAT
GTA CGT AGC CAT CC, Actin reverse: CTC TCA GCT GTG GTA
A. Melting curves were examined to verify that a single product was
amplified. For quantitative analysis, all samples were normalized to
Actin gene expression using the DDCT (cycle threshold) value
method.
Serum DepletionSera from 11 patients on maintenance hemodialysis were collected
after the long weekly interval as previously described.13 All patients
consented in writing to donating blood for scientific evaluation. The
protocol was approved by the Institutional Review Board atHannover
Medical School. AZGP1 was depleted from serum using a standard
immunoprecipitation protocol. In brief, serum was incubated with
200mg/ml AZGP1 antibody (Zag [H123]; Santa Cruz Biotechnology)
at 4°C with rotation. Protein A/G Plus beads (Santa Cruz Biotech-
nology) were added and incubated overnight at 4°C with rotation.
Beads were precipitated, and the supernatant was used for the treat-
ment of NRK-49F cells. AZGP1 concentration was determined by
ELISA (Zinc-Alpha2 Glycoprotein Human ELISA; Biovendor
GmbH, Heidelberg, Germany).
Statistical AnalysesResults are expressed as means6SEMs. Statistical significance among
multiple groups was determined by one-way ANOVA tests, with a post
hoc Bonferroni test to determine significance between groups.
To determine significance for comparisons between two groups, a
t test was used. P,0.05 was considered to be statistically significant.
Prism 4.0 (GraphPad Software, San Diego, CA) was used to perform
statistical tests.
ACKNOWLEDGMENTS
We thank H. Ricci and B. Gewecke for technical assistance.
This work was supported by Deutsche Forschungsgemeinschaft
Grant SCHM 2146/3-1 and the 7th Framework Program of the Eu-
ropean Union (FIBROTARGET).
Parts of this work have been presented at the American Society of
Nephrology Renal Week, November 16–21, 2010 in Denver, CO.
DISCLOSURESNone.
REFERENCES
1. Farris AB, Colvin RB: Renal interstitial fibrosis: Mechanisms and evalu-ation. Curr Opin Nephrol Hypertens 21: 289–300, 2012
2. Schmitt R, Marlier A, Cantley LG: Zag expression during aging sup-presses proliferation after kidney injury. J Am Soc Nephrol 19: 2375–2383, 2008
3. BurgiW, SchmidK: Preparation andproperties of Zn-alpha 2-glycoproteinof normal human plasma. J Biol Chem 236: 1066–1074, 1961
4. Hassan MI, Waheed A, Yadav S, Singh TP, Ahmad F: Zinc alpha2-glycoprotein: A multidisciplinary protein. Mol Cancer Res 6: 892–906, 2008
5. Rolli V, Radosavljevic M, Astier V, Macquin C, Castan-Laurell I, VisentinV, Guigné C, Carpéné C, Valet P, Gilfillan S, Bahram S: Lipolysis is al-tered in MHC class I zinc-alpha(2)-glycoprotein deficient mice. FEBSLett 581: 394–400, 2007
6. Hirai K, Hussey HJ, Barber MD, Price SA, Tisdale MJ: Biological eval-uation of a lipid-mobilizing factor isolated from the urine of cancerpatients. Cancer Res 58: 2359–2365, 1998
7. Russell ST, Tisdale MJ: Role of b-adrenergic receptors in the anti-obesity and anti-diabetic effects of zinc-a2-glycoprotien (ZAG). Bio-chim Biophys Acta 1821: 590–599, 2012
8. Bing C, Bao Y, Jenkins J, Sanders P, Manieri M, Cinti S, Tisdale MJ,Trayhurn P: Zinc-alpha2-glycoprotein, a lipid mobilizing factor, is
J Am Soc Nephrol 26: ccc–ccc, 2015 AZGP1 Inhibits Fibrosis 9
www.jasn.org BASIC RESEARCH

expressed in adipocytes and is up-regulated in mice with cancer ca-chexia. Proc Natl Acad Sci U S A 101: 2500–2505, 2004
9. Russell ST, Tisdale MJ: Mechanism of attenuation of skeletal muscle at-rophyby zinc-alpha2-glycoprotein.Endocrinology151:4696–4704, 2010
10. Ekman R, Johansson BG, Ravnskov U: Renal handling of Zn-alpha2-glycoprotein as compared with that of albumin and the retinol-bindingprotein. J Clin Invest 57: 945–954, 1976
11. Leal VO, Lobo JC, Stockler-Pinto MB, Farage NE, Velarde GC, FouqueD, Leite M Jr., Mafra D: Zinc-a2-glycoprotein: Is there association be-tween this new adipokine and body composition in hemodialysis pa-tients? Ren Fail 34: 1062–1067, 2012
12. Philipp A, Kralisch S, Bachmann A, Lossner U, Kratzsch J, Blüher M,Stumvoll M, Fasshauer M: Serum levels of the adipokine zinc-a2-glycoprotein are increased in chronic hemodialysis. Metabolism 60:669–672, 2011
13. Sörensen-Zender I, Beneke J, Schmidt BM, Menne J, Haller H, SchmittR: Zinc-alpha2-glycoprotein in patients with acute and chronic kidneydisease. BMC Nephrol 14: 145, 2013
14. Pelletier CC, Koppe L, Croze ML, Kalbacher E, Vella RE, Guebre-Egziabher F,GéloënA, Badet L, FouqueD, SoulageCO:White adiposetissue overproduces the lipid-mobilizing factor zinc a2-glycoprotein inchronic kidney disease. Kidney Int 83: 878–886, 2013
15. Leal VO, Lobo JC, Stockler-Pinto MB, Farage NE, Abdalla DS, Leite MJr., Mafra D: Is zinc-a2-glycoprotein a cardiovascular protective factorfor patients undergoing hemodialysis? Clin Chim Acta 413: 616–619,2012
16. Zeisberg M, Kalluri R: Cellular mechanisms of tissue fibrosis. 1. Com-mon and organ-specificmechanisms associatedwith tissue fibrosis.AmJ Physiol Cell Physiol 304: C216–C225, 2013
17. Lan HY: Diverse roles of TGF-b/Smads in renal fibrosis and in-flammation. Int J Biol Sci 7: 1056–1067, 2011
18. Koesters R, Kaissling B, Lehir M, Picard N, Theilig F, Gebhardt R, GlickAB, Hähnel B, Hosser H, Gröne HJ, Kriz W: Tubular overexpression oftransforming growth factor-beta1 induces autophagy and fibrosis butnot mesenchymal transition of renal epithelial cells. Am J Pathol 177:632–643, 2010
19. Ishibe S, Cantley LG: Epithelial-mesenchymal-epithelial cycling in kid-ney repair. Curr Opin Nephrol Hypertens 17: 379–385, 2008
20. Lapointe J, Li C, Higgins JP, van de Rijn M, Bair E, Montgomery K,Ferrari M, Egevad L, RayfordW, Bergerheim U, Ekman P, DeMarzo AM,Tibshirani R, Botstein D, Brown PO, Brooks JD, Pollack JR: Gene ex-pression profiling identifies clinically relevant subtypes of prostatecancer. Proc Natl Acad Sci U S A 101: 811–816, 2004
21. Huang CY, Zhao JJ, Lv L, Chen YB, Li YF, Jiang SS, Wang W, Pan K,Zheng Y, Zhao BW, Wang DD, Chen YM, Yang L, Zhou ZW, Xia JC:Decreased expression of AZGP1 is associated with poor prognosis inprimary gastric cancer. PLoS ONE 8: e69155, 2013
22. Parris TZ, Kovács A, Aziz L, Hajizadeh S, Nemes S, Semaan M, Forssell-Aronsson E, Karlsson P, Helou K: Additive effect of the AZGP1, PIP,S100A8 and UBE2C molecular biomarkers improves outcome pre-diction in breast carcinoma. Int J Cancer 134: 1617–1629, 2014
23. Brysk MM, Lei G, Adler-Storthz K, Chen Z, Brysk H, Tyring SK, Arany I:Zinc-alpha2-glycoprotein expression as a marker of differentiation inhuman oral tumors. Cancer Lett 137: 117–120, 1999
24. Kong B, Michalski CW, Hong X, Valkovskaya N, Rieder S, Abiatari I,Streit S, Erkan M, Esposito I, Friess H, Kleeff J: AZGP1 is a tumor sup-pressor in pancreatic cancer inducing mesenchymal-to-epithelialtransdifferentiation by inhibiting TGF-b-mediated ERK signaling. On-cogene 29: 5146–5158, 2010
25. Russell ST, Tisdale MJ: Role of b-adrenergic receptors in the oral activityof zinc-a2-glycoprotein (ZAG). Endocrinology 153: 4696–4704, 2012
26. De Wever O, Mareel M: Role of tissue stroma in cancer cell invasion.J Pathol 200: 429–447, 2003
27. Bascands JL, Schanstra JP: Obstructive nephropathy: Insights fromgenetically engineered animals. Kidney Int 68: 925–937, 2005
28. Zhou L, Fu P, Huang XR, Liu F, ChungAC, Lai KN, LanHY:Mechanismofchronic aristolochic acid nephropathy: Role of Smad3. Am J PhysiolRenal Physiol 298: F1006–F1017, 2010
29. Xia Y, Lee K, Li N, Corbett D, Mendoza L, Frangogiannis NG: Charac-terization of the inflammatory and fibrotic response in a mouse modelof cardiac pressure overload. Histochem Cell Biol 131: 471–481, 2009
30. Buijs JT, Henriquez NV, van Overveld PG, van der Horst G, ten Dijke P,van der Pluijm G: TGF-beta and BMP7 interactions in tumour pro-gression and bone metastasis. Clin Exp Metastasis 24: 609–617, 2007
31. Selva DM, Lecube A, Hernández C, Baena JA, Fort JM, Simó R: Lowerzinc-alpha2-glycoprotein production by adipose tissue and liver inobese patients unrelated to insulin resistance. J Clin Endocrinol Metab94: 4499–4507, 2009
32. Gong FY, Zhang SJ, Deng JY, Zhu HJ, Pan H, Li NS, Shi YF: Zinc-alpha2-glycoprotein is involved in regulation of body weight through inhibition oflipogenicenzymes inadipose tissue. Int JObes (Lond)33:1023–1030, 2009
33. Zhu HJ, Wang XQ, Pan H, Gong FY, Zhang DX, Li NS, Wang LJ, YangHB: Serum levels of the adipokine zinc- a 2-glycoprotein are decreasedin patients with hypertension. ISRN Endocrinol 2014: 374090, 2014
34. Ceperuelo-Mallafré V, Escoté X, Viladés C, Peraire J, Domingo P,Solano E, Sirvent JJ, Pastor R, Tinahones F, LealM, Richart C, Vendrell J,Vidal F, Alba V, Aguilar A, Auguet T, Chacón MR, López-Dupla M,Megia A, Miranda M, Olona M, Saurí A, Vargas M, Velasco I, Veloso S,Fontanet A, Gutiérrez M, Mateo G, Muñoz J, Sambeat MA; HIV-1 Lip-odystrophy Study Group: Zinc alpha-2 glycoprotein is implicated indyslipidaemia in HIV-1-infected patients treated with antiretroviraldrugs. HIV Med 13: 297–303, 2012
35. Stepan H, Philipp A, Roth I, Kralisch S, Jank A, Schaarschmidt W,Lössner U, Kratzsch J, BlüherM, Stumvoll M, FasshauerM: Serum levelsof the adipokine zinc-a2-glycoprotein are increased in preeclampsia.J Endocrinol Invest 35: 562–565, 2012
36. Tedeschi S, Pilotti E, Parenti E, Vicini V, Coghi P, Montanari A, RegolistiG, Fiaccadori E, Cabassi A: Serum adipokine zinc a2-glycoprotein andlipolysis in cachectic and noncachectic heart failure patients: Re-lationship with neurohormonal and inflammatory biomarkers. Metab-olism 61: 37–42, 2012
37. Gangadharan B, Antrobus R, Dwek RA, Zitzmann N: Novel serum bio-marker candidates for liver fibrosis in hepatitis C patients. Clin Chem53: 1792–1799, 2007
38. McIntyre CW, Odudu A: Hemodialysis-associated cardiomyopathy:A newly defined disease entity. Semin Dial 27: 87–97, 2014
39. Silver J, Rodriguez M, Slatopolsky E: FGF23 and PTH—double agentsat the heart of CKD. Nephrol Dial Transplant 27: 1715–1720, 2012
40. Wargent ET, O’Dowd JF, Zaibi MS, Gao D, Bing C, Trayhurn P,Cawthorne MA, Arch JR, Stocker CJ: Contrasts between the effects ofzinc-a2-glycoprotein, a putative b3/2-adrenoceptor agonist and theb3/2-adrenoceptor agonist BRL35135 in C57Bl/6 (ob/ob) mice. J En-docrinol 216: 157–168, 2013
41. Sörensen I, Susnik N, Inhester T, Degen JL, Melk A, Haller H, Schmitt R:Fibrinogen acting as a mitogen for tubulointerstitial fibroblasts, pro-motes renal fibrosis. Kidney Int 80: 1035–1044, 2011
42. Susnik N, Sörensen-Zender I, Rong S, von Vietinghoff S, Lu X, Rubera I,TaucM, Falk CS, AlexanderWS,Melk A, Haller H, Schmitt R: Ablation ofproximal tubular suppressor of cytokine signaling 3 enhances tubularcell cycling and modifies macrophage phenotype during acute kidneyinjury. Kidney Int 85: 1357–1368, 2014
43. RockmanHA, Ross RS, Harris AN, Knowlton KU, SteinhelperME, Field LJ,Ross J Jr., Chien KR: Segregation of atrial-specific and inducible ex-pressionof an atrial natriuretic factor transgene in an in vivomurinemodelof cardiac hypertrophy. Proc Natl Acad Sci U S A 88: 8277–8281, 1991
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2014050485/-/DCSupplemental.
10 Journal of the American Society of Nephrology J Am Soc Nephrol 26: ccc–ccc, 2015
BASIC RESEARCH www.jasn.org




















