
Presynaptic α2-receptors regulate reverse Na+/Ca2+-exchange and transmitter release in Na+-loaded...
13
Neurochemistry International 45 (2004) 699–711 Presynaptic 2 -receptors regulate reverse Na + /Ca 2+ -exchange and transmitter release in Na + -loaded peripheral sympathetic nerves Tamás L. Török a,∗ , Zsolt Nagykáldi a , Zsuzsanna Sáska a , Timur Kovács a , Somaia A. Nada a , Stefan Zilliikens a , Kálmán Magyar a , E. Sylvester Vizi b,c a Department of Pharmacodynamics, Semmelweis University, Nagyvárad-tér 4, P.O. Box 370, H-1445 Budapest, Hungary b Department of Pharmacology and Pharmacotherapy, Semmelweis University, Nagyvárad-tér 4, P.O. Box 370, H-1445 Budapest, Hungary c Institute of Experimental Medicine, Hungarian Academy of Sciences, P.O. Box 67, H-1450 Budapest, Hungary Received 10 February 2004; accepted 3 March 2004 Available online 8 May 2004 Abstract Electrical depolarisation-(2 Hz, 1 ms)-induced [ 3 H]noradrenaline ([ 3 H]NA) release has been measured from the isolated main pulmonary artery of the rabbit in the presence of uptake blockers (cocaine, 3 × 10 −5 M; corticosterone, 5 × 10 −5 M). Substitution of most of the exter- nal Na + by Li + (113 mM; [Na + ] 0 : 25 mM) slightly potentiated the axonal stimulation-evoked release of [ 3 H]NA in a tetrodotoxin (TTX, 10 −7 M) sensitive manner. The reverse Na + /Ca 2+ -exchange inhibitor KB-R7943 (3 × 10 −5 M) failed to inhibit the stimulation-evoked release of [ 3 H]NA, but increased the resting outflow of neurotransmitter. The ‘N-type’ voltage-sensitive Ca 2+ -channel (VSCC) blocker -conotoxin (-CgTx) GVIA (10 −8 M) significantly and irreversibly inhibited the release of [ 3 H]NA on stimulation (∼60–70%). The ‘residual release’ of NA was abolished either by TTX or by reducing external Ca 2+ from 2.5 to 0.25 mM. The ‘residual release’ of NA was also blocked by the non-selective VSCC-blocker neomycin (3 × 10 −3 M). Correlation was obtained between the extent of VSCC-inhibition and the transmitter release-enhancing effect of presynaptic 2 -receptor blocker yohimbine (3 × 10 −7 M). When the release of [ 3 H]NA was blocked by -CgTx GVIA plus neomycin, yohimbine was ineffective. Inhibition of the Na + -pump by removal of K + from the external medium increased both the resting and the axonal stimulation-evoked release of [ 3 H]NA in the absence of functioning VSCCs (i.e., in the presence of neomycin and after -CgTx treatment). Under these conditions the stimulation-evoked release of NA was abolished either by TTX or by external Ca 2+ -removal (+1mM EGTA). Similarly, external Li + (113 mM) or the reverse Na + /Ca 2+ exchange blocker KB-R7943 (3 × 10 −5 M) significantly inhibited the stimulation-induced transmitter release in ‘K + -free’ solution. KB-R7943 decreased the resting outflow of NA as well. Under conditions in which the Na + -pump was inhibited in the absence of functioning VSCCs, yohim- bine (3 × 10 −7 M) further enhanced the release of neurotransmitter, while l-noradrenaline (l-NA, 10 −6 M), an agonist of presynaptic 2 -receptors, inhibited it. The yohimbine-induced enhancement of NA-release was abolished by Li + -substitution and significantly inhib- ited by KB-R7943 application. It is concluded that after blockade of VSCCs brief depolarising pulses may reverse Na + /Ca 2+ -exchange and release neurotransmitter in Na + -loaded sympathetic nerves. Further, similar to that of VSCCs, the reverse Na + /Ca 2+ -exchange may also be regulated by presynaptic 2 -receptors. © 2004 Elsevier Ltd. All rights reserved. Keywords: Rabbit pulmonary artery; Reverse Na + /Ca 2+ -exchange; [ 3 H]noradrenaline release; 2 -Receptor modulation 1. Introduction During ‘excitation–secretion coupling’ the influx of Ca 2+ through voltage-sensitive Ca 2+ -channels (VSCCs) trig- gers neurotransmitter release (Llinás et al., 1992; Stanley, 1997). The other pathway that may be involved in in- crease of internal Ca 2+ is the Ca 2+ -influx mode of the Na + /Ca 2+ -exchanger (for reviews, see Blaustein and Lederer, 1999; Philipson and Nicoll, 2000). The Na + /Ca 2+ - ∗ Corresponding author. Tel.: +36-1-210-4411; fax: +36-1-210-4411. E-mail address: [email protected] (T.L. Török). exchange protein is expressed in high concentration at presynaptic nerve terminals (Luther et al., 1992; Reuter and Porzig, 1995) and may play an important role in Ca 2+ -extrusion after excitation (Blaustein and Lederer, 1999). However, it localises some distance from the release site (Juhaszova et al., 2000). Leblanc and Hume (1990) were the first to suggest in cardiac muscle that the influx of Ca 2+ through the exchanger (reverse mode; outward Na + /Ca 2+ -exchange current) can trigger SR Ca 2+ -release. In nerve terminals however, the action potential (AP) duration is less than a millisecond (Llinás et al., 1992), thus much shorter than 0197-0186/$ – see front matter © 2004 Elsevier Ltd. All rights reserved. doi:10.1016/j.neuint.2004.03.002
Transcript of Presynaptic α2-receptors regulate reverse Na+/Ca2+-exchange and transmitter release in Na+-loaded...

Neurochemistry International 45 (2004) 699–711
Presynaptic�2-receptors regulate reverse Na+/Ca2+-exchange andtransmitter release in Na+-loaded peripheral sympathetic nerves
Tamás L. Töröka,∗, Zsolt Nagykáldia, Zsuzsanna Sáskaa, Timur Kovácsa, Somaia A. Nadaa,Stefan Zilliikensa, Kálmán Magyara, E. Sylvester Vizib,c
a Department of Pharmacodynamics, Semmelweis University, Nagyvárad-tér 4, P.O. Box 370, H-1445 Budapest, Hungaryb Department of Pharmacology and Pharmacotherapy, Semmelweis University, Nagyvárad-tér 4, P.O. Box 370, H-1445 Budapest, Hungary
c Institute of Experimental Medicine, Hungarian Academy of Sciences, P.O. Box 67, H-1450 Budapest, Hungary
Received 10 February 2004; accepted 3 March 2004
Available online 8 May 2004
Abstract
Electrical depolarisation-(2 Hz, 1 ms)-induced [3H]noradrenaline ([3H]NA) release has been measured from the isolated main pulmonaryartery of the rabbit in the presence of uptake blockers (cocaine, 3×10−5 M; corticosterone, 5×10−5 M). Substitution of most of the exter-nal Na+ by Li+ (113 mM; [Na+]0: 25 mM) slightly potentiated the axonal stimulation-evoked release of [3H]NA in a tetrodotoxin (TTX,10−7 M) sensitive manner. The reverse Na+/Ca2+-exchange inhibitor KB-R7943 (3× 10−5 M) failed to inhibit the stimulation-evokedrelease of [3H]NA, but increased the resting outflow of neurotransmitter. The ‘N-type’ voltage-sensitive Ca2+-channel (VSCC) blocker�-conotoxin (�-CgTx) GVIA (10−8 M) significantly and irreversibly inhibited the release of [3H]NA on stimulation (∼60–70%). The‘residual release’ of NA was abolished either by TTX or by reducing external Ca2+ from 2.5 to 0.25 mM. The ‘residual release’ of NA wasalso blocked by the non-selective VSCC-blocker neomycin (3×10−3 M). Correlation was obtained between the extent of VSCC-inhibitionand the transmitter release-enhancing effect of presynaptic�2-receptor blocker yohimbine (3×10−7 M). When the release of [3H]NA wasblocked by�-CgTx GVIA plus neomycin, yohimbine was ineffective. Inhibition of the Na+-pump by removal of K+ from the externalmedium increased both the resting and the axonal stimulation-evoked release of [3H]NA in the absence of functioning VSCCs (i.e., in thepresence of neomycin and after�-CgTx treatment). Under these conditions the stimulation-evoked release of NA was abolished eitherby TTX or by external Ca2+-removal (+1 mM EGTA). Similarly, external Li+ (113 mM) or the reverse Na+/Ca2+ exchange blockerKB-R7943 (3× 10−5 M) significantly inhibited the stimulation-induced transmitter release in ‘K+-free’ solution. KB-R7943 decreasedthe resting outflow of NA as well. Under conditions in which the Na+-pump was inhibited in the absence of functioning VSCCs, yohim-bine (3× 10−7 M) further enhanced the release of neurotransmitter, while l-noradrenaline (l-NA, 10−6 M), an agonist of presynaptic�2-receptors, inhibited it. The yohimbine-induced enhancement of NA-release was abolished by Li+-substitution and significantly inhib-ited by KB-R7943 application. It is concluded that after blockade of VSCCs brief depolarising pulses may reverse Na+/Ca2+-exchangeand release neurotransmitter in Na+-loaded sympathetic nerves. Further, similar to that of VSCCs, the reverse Na+/Ca2+-exchange mayalso be regulated by presynaptic�2-receptors.© 2004 Elsevier Ltd. All rights reserved.
Keywords: Rabbit pulmonary artery; Reverse Na+/Ca2+-exchange; [3H]noradrenaline release;�2-Receptor modulation
1. Introduction
During ‘excitation–secretion coupling’ the influx of Ca2+through voltage-sensitive Ca2+-channels (VSCCs) trig-gers neurotransmitter release (Llinás et al., 1992; Stanley,1997). The other pathway that may be involved in in-crease of internal Ca2+ is the Ca2+-influx mode of theNa+/Ca2+-exchanger (for reviews, seeBlaustein andLederer, 1999; Philipson and Nicoll, 2000). The Na+/Ca2+-
∗ Corresponding author. Tel.:+36-1-210-4411; fax:+36-1-210-4411.E-mail address: [email protected] (T.L. Török).
exchange protein is expressed in high concentration atpresynaptic nerve terminals (Luther et al., 1992; Reuterand Porzig, 1995) and may play an important role inCa2+-extrusion after excitation (Blaustein and Lederer,1999). However, it localises some distance from the releasesite (Juhaszova et al., 2000).
Leblanc and Hume (1990)were the first to suggestin cardiac muscle that the influx of Ca2+ through theexchanger (reverse mode; outward Na+/Ca2+-exchangecurrent) can trigger SR Ca2+-release. In nerve terminalshowever, the action potential (AP) duration is less than amillisecond (Llinás et al., 1992), thus much shorter than
0197-0186/$ – see front matter © 2004 Elsevier Ltd. All rights reserved.doi:10.1016/j.neuint.2004.03.002

700 T.L. Török et al. / Neurochemistry International 45 (2004) 699–711
in heart muscle. In addition, the turnover rate of a singleopen VSCC is much higher (107 Ca2+/s; Hille, 1992) thanthat of a single exchanger (∼5 × 103 Ca2+/s; Hilgemann,1996). Therefore, the VSCCs are activated primarily ondepolarisation (cf.Blaustein and Lederer, 1999). Further,when internal Ca2+concentration is elevated as a resultof VSCC-activation, the exchanger may will be switchedback to the Ca2+-efflux mode (normal mode operation ofNa+/Ca2+-exchanger; cf.Philipson and Nicoll, 2000).
NeverthelessGleason et al. (1994, 1995)have shown incultured retinal amacrine cells that after closure of VSCCs,prolonged depolarisation produces a second rise in [Ca2+]iand transmitter release. This [Ca2+]i-elevation was accom-panied by an outward current, possibly mediated by reverseNa+/Ca2+-exchange.
In the present study we measured neurotransmitter re-lease from peripheral sympathetic nerves in response tophysiological depolarising stimuli (2 Hz, 1 ms). After phar-macological blockade of VSCCs, brief pulses could elicittransmitter release, but only if the nerves were Na+-loaded(Na+-pump inhibition). The release of neurotransmitter isprobably reflects reverse Na+/Ca2+-exchange activation,since it was blocked by TTX or by Ca2+-removal from theexternal medium (+1 mM EGTA). The release was alsosignificantly inhibited either when the fast Na+-current wascarried by Li+, or when the reverse Na+/Ca2+-exchangeblocker KB-R7943 was applied. The neurotransmitter re-lease, obtained under these conditions, was regulated bypresynaptic�2-receptors, since it was further enhancedby yohimbine and inhibited by l-noradrenaline. Theyohimbine-induced enhancement of NA-release was alsosignificantly inhibited either by Li+ or by KB-R7943.
2. Methods
2.1. Rabbit main pulmonary artery
The experiments have been carried out in the isolatedmain pulmonary artery of the rabbit (Starke et al., 1974;Török et al., 1992). Rabbits of either sex (2–3 kg) werekilled by cervical dislocation and exsanguination throughthe carotid artery. The main pulmonary artery (weight,40–80 mg) was dissected and placed into normal Krebssolution which contained the monoamine oxidase inhibitorpargyline (1.2 × 10−4 M) and which was fully equilibratedwith carbogene (5% CO2 in O2; pH 7.4). Then the arterywas opened longitudinally, fixed by two threads in such away that the circular muscle fibres were running betweenthe threads, and placed into a loading bath (volume, 1.5 ml)for 30 min at 37◦C. The composition of normal Krebs so-lution was (mM): Na+, 138.0; K+, 5.9; Ca2+, 2.5; Mg2+,1.2; Cl−, 122.7; HCO3
−, 25.0; H2PO4−, 1.2; SO4
2−, 1.2;glucose, 11.1. In ‘K+-free’ solution, KCl (4.7 mM) andKH2PO4 (1.2 mM) were omitted with no substitution forKCl but substitution for KH2PO4 with an equimolar con-
centration of NaH2PO4. In Ca2+-deficient solutions, CaCl2(2.5 mM) was reduced to 1.25, 0.75 and 0.25 mM, respec-tively, without ionic substitution. In Ca2+-free solution,CaCl2 was omitted and 1 mM EGTA was added. Low Na+solution contained 25.0 or 26.2 mM Na+ (in K+-containing-or in ‘K+-free’-solution), with Na+ being replaced by Li+(113.0 mM). Glucose (11.1 mM) was always maintained inthe solution, because its withdrawal dramatically increasedthe [Ca2+]0-independent release (Milusheva et al., 1992;Milusheva and Baranyi, 2003; Brassai et al., 2002).
2.2. Measurement of [3H]noradrenaline release
The method had been described previously (Borowskiet al., 1977; Török et al., 1992). Briefly, after the arteryhad been placed in Krebs solution, 25�l [ 3H]noradrenaline([3H]NA; specific activity, 12.0 Ci mmol−1) was added to theincubation solution (final concentration of [3H]NA, 1.39×10−6 M) for 45 min (pargyline and the NA-stabilising agentsascorbic acid, 3×10−4 M and disodium edetate (Na2EDTA),3×10−5 M, were present). Subsequently the artery was sus-pended in an organ bath (capacity, 2 ml) and perfused at arate of 8 ml/min with 800 ml medium, containing the neu-ronal uptake blocker cocaine (3× 10−5 M) instead of par-gyline. At the end of the washing period the flow rate waschanged to 4 ml/min and the extraneuronal uptake blockercorticosterone (5×10−5 M) was also added to the Krebs so-lution for 30 min. In order to induce neurotransmitter release,field stimulation (2 Hz, 1 ms, 60 V) was used for 3 min (360pulses) by means of two platinum electrodes fixed verticallyon opposite sides of the tissue. Tetrodotoxin (TTX, 10−7 M)abolished the field stimulation-induced [3H]NA-release andthe post-synaptic contractile response indicating the nervousorigin of liberated NA. Previously it was shown that in thepresence of uptake blockers 86% of nerve-evoked releaseof NA is unmetabolised (Endo et al., 1977). Since pargy-line was present at the beginning of our experiments theresting outflow of NA is probably unmetabolised too. Onthe basis of this assumption and knowing the specific ac-tivity of [ 3H]NA, we calculated the release of labelled NAin picomoles per 3 min according to the method ofEndoet al. (1977). The stimulation-evoked [3H]NA-release wascalculated by subtraction of the resting outflow, determinedimmediately before stimulation, from the release obtainedand up to 6 min after stimulation. Two control stimulationperiods (S1, S2) were performed and then modified Krebssolution was used or drugs were added 3, 6 or 18 min be-fore the third stimulation period (S3) as required (see in-dividual experiments). In most of the experiments�-CgTxGVIA was pre-perfused 18 min before S3, and was presentduring stimulation. Subsequently the toxin was washed outand a stimulation period was performed (S4). Neomycin wasadded to the perfusion solution 18 min before the fifth stim-ulation period (S5) and was present during S5 and the sub-sequent stimulation periods (S6–S10). External K+ was re-moved 18 min before S6, and was readmitted 6 min before

T.L. Török et al. / Neurochemistry International 45 (2004) 699–711 701
S10 (120 min ‘K+-free’ perfusion). Li+ was added to Krebssolution 3 min before stimulation (S3 or S7), since longerperiods increased the resting outflow of [3H]NA. KB-R7943was pre-perfused 6 min before stimulation (S3 or S7). Longerperiod (18 min) markedly accelerated the resting outflow oflabelled NA. Reduction of external Ca2+ from 2.5 to 1.25,0.75 or 0.25 mM was carried out 18 min before the stimu-lation periods (S5, S7 and S9). Meantime (S6 and S8) andafterwards (S10) normal Ca2+ (2.5 mM) was readmitted tothe external medium. Ca2+-omission (+1 mM EGTA)- orTTX-, yohimbine-, l-noradrenaline (l-NA)- and ryanodine-addition to ‘K+-free’ solution were carried out 18 min beforeS7. The effect of drugs or external ionic modifications wereexpressed as the ratio between the release of3H ([3H]NAin pmol) evoked by stimulation-3, -5, -7 and the overflowevoked by stimulation-2 (S3,5,7/S2). In control experimentsthe ratio between S3 and S2 was 0.98 ± 0.01 (n = 19).The perfusate was collected in 3 or 6 min samples. At theend of the experiments the preparations were dissolved in1 ml Soluene-350 (Packard) and the radioactivity of per-fusate samples and tissues were determined using a liquidscintillation counter (Beckman, LS-5000 TA). The total ini-tial [3H]NA content of the arteries was 462.5 ± 28.7 pmol(means± S.E.M. of 45 tissues; weight 56.2 ± 2.1 mg).
2.3. Stimulation technique
Square-wave pulses of 1 ms duration were delivered froma Grass S44 stimulator. Two platinum wire electrodes wereused for stimulation of nerves. The electrodes were fixedvertically on opposite sides of the tissue at the top and thebottom of the organ bath. The distance between the tops ofthe electrodes was 20 mm.
2.4. Drugs and statistics
The following drugs were used: l-[7,83H]noradrenaline(specific activity, 12.0 Ci/mmol; Radiochemical Centre,Amersham, UK); pargyline hydrochloride (Serva); co-caine hydrochloride (Merck); corticosterone (Fluka); ascor-bic acid (EGA); disodium ethylenediaminetetraacetate(Na2EDTA, Aldrich-Europe); tetrodotoxin (TTX, Cal-biochem);�-conotoxin-(�-CgTx)-GVIA (Sigma, AlamoneLabs Ltd.); KB-R7943 (2-[2-[4-(4-nitrobenzyloxy)phenyl]ethyl]isothiourea methanesulphonate, Nippon OrganonK.K., Osaka, Japan); ryanodine (Calbiochem); ethylene-glycol-bis-�-aminoethyl-ether)N,N′-tetraacetic acid (EGTA,Sigma); neomycin sulphate (Sigma); yohimbine hydrochlo-ride (Serva); l-noradrenaline hydrochloride (l-NA, Fluka).The drugs were dissolved in Krebs solution. Corticosteronewas dissolved in propylene glycol (final concentration,0.05%, v/v). KB-R7943 was dissolved in dimethyl sulphox-ide (DMSO, final concentration, 0.3%, v/v). These solvents,separately or together, did not affect neurotransmitter re-lease. All of the chemicals used for preparing Krebs solutionwere of analytical grade.
Means± S.E.M. are given. Data were compared usingStudent’s paired or unpairedt-test and a one-way analysisof variance. Differences between means were consideredsignificant ifP < 0.05.
3. Results
3.1. The effects of external Li+ and KB-R7943 on[3H]noradrenaline release from pulmonary arteries
Substitution of most of the external Na+ by Li+(113 mM; [Na+]0: 25.0 mM) failed to increase the releaseof [3H]NA in response to nerve-stimulation. The ratio ofstimulation-evoked release increased from 0.98 ± 0.03 to1.13 ± 0.06 (n = 4), however the difference was not sig-nificant (P > 0.05). Short pre-perfusion period was usedwith Li+-containing solution (3 min), since a longer pe-riod (18 min) markedly increased the resting outflow of[3H]NA (not shown). This effect is probably due to inhibi-tion of [Na+]0/[Ca2+]i-exchange (Reuter and Seitz, 1968)or activation of [Na+]i/[Ca2+]0-exchange (Baker et al.,1969). The [3H]NA-release obtained in Li+-solution wasreversibly blocked by the voltage-sensitive Na+-channelblocker tetrodotoxin (TTX, 10−7 M). These results sug-gest that Li+-influx through fast Na+-channel (Hille, 1992)may depolarise the membrane and open voltage-sensitiveCa2+-channels. The influx of Ca2+ may cause exocytoticrelease of neurotransmitter.
The reverse Na+/Ca2+-exchange inhibitor KB-R7943(Watano et al., 1996; Iwamoto et al., 1996), in large con-centration (3× 10−5 M), failed to affect the peak releaseof [3H]NA in response to nerve-stimulation (Fig. 1a).However, it prolonged the recovery of transmitter releaseafter stimulation and increased the resting outflow of la-belled NA immediately after its application (6 min; from0.45± 0.06 to 0.59± 0.07 pmol/3 min,n = 7). If a longerpre-perfusion period was used with KB-R7943 (18 min),the resting outflow of [3H]NA markedly increased (from0.82 ± 0.10 to 4.09 ± 0.78 pmol/3 min,n = 4; Fig. 1b).This effect was completely reversible, since after washingout of KB-R7943 the resting outflow and the nerve-evokedrelease of labelled NA returned to the control values within60–80 min (Fig. 1b). We had to use this large concentrationof KB-R7943, since 10−5 M did not inhibit the release of[3H]NA when external Ca2+ was readmitted to Na+-loadednerves (i.e., after 300 min perfusion with Ca2+/K+-free so-lution; Kovács and Török, unpublished observation). Thissuggests that the exchanger of sympathetic nerves is lesssensitive to this blocker.
3.2. Cessation of [3H]noradrenaline release byvoltage-sensitive Ca2+-channel blockers and the lack oftransmitter-releasing effect of yohimbine
The selective and irreversible ‘N-type’ VSCC-blocker�-conotoxin-(�-CgTx)-GVIA in low concentration (10−8 M)

702 T.L. Török et al. / Neurochemistry International 45 (2004) 699–711
Fig. 1. The effect of Na+/Ca2+-exchange inhibitor KB-R7943 (3×10−5 M) on spontaneous and nerve-stimulation induced [3H]NA-release from pulmonaryarteries (cocaine, 3× 10−5 M and corticosterone, 5× 10−5 M are present).Ordinate: [3H]NA-release (pmol (3 min)−1); abscissa: time (min). Horizontalbars above the axis represent the stimulation periods (3 min every 27 min). Dashed lines indicate the presence of KB-R7943 (3× 10−5 M). (a) Aftertwo control stimulation periods KB-R7943 was pre-perfused 6 min before the third stimulation period (S3; arrow). Note that KB-R7943 increased theresting outflow of [3H]NA but did not affect the peak release in response to stimulation (ratio (S3/S2): 1.35± 0.14). In addition, note that the recoveryof [3H]NA-release was delayed after cessation of stimulation, and the release of [3H]NA was slightly potentiated during the next two control stimulationperiods. Means± S.E.M. of seven identical experiments are given. (b) As in (a) except KB-R7943 was applied 18 min before the third stimulationperiod (S3; arrow). Note that KB-R7943 markedly increased the resting outflow of [3H]NA and delayed the recovery of3H release after wash-out. Inaddition, note that the resting outflow and the stimulation-induced3H release returned to the control values within 60–80 min on removal of KB-R7943.Means± S.E.M. of four identical experiments are given.

T.L. Török et al. / Neurochemistry International 45 (2004) 699–711 703
Fig. 2. [3H]NA-release in Na+-pump inhibited (‘K+-free’ solution) arteries in the presence of neomycin (3× 10−3 M) and after�-CgTx GVIA (10−8 M)pre-treatment.Ordinate: [3H]NA-release (pmol (3 min)−1); abscissa: time (min). Horizontal bars above the axis represent the stimulation periods. Dashedlines indicate the presence of�-CgTx and neomycin and the absence of external K+. Note that [K+]0 was removed for 120 min.�-CgTx significantlyinhibited the nerve-evoked release of [3H]NA (ratio (S3/S2): 0.39± 0.02), which further decreased on removal of�-CgTx. Neomycin, perfused 18 minbefore S5, abolished the release of3H (ratio (S5/S2): 0.03 ± 0.02). Subsequently, removal of K+, for four successive stimulation periods, increasedboth the resting and the nerve-evoked release of3H (neomycin was present). The peak release was obtained after 45 min of removal of K+ (secondstimulation in ‘K+-free’ solution; S7). Note that during the subsequent stimulation periods (S8, S9) the evoked-release of3H slightly decreased. After120 min ‘K+-free’ perfusion, external K+ (5.9 mM) was readmitted, 6 min before S10. Note that the resting outflow of3H was significantly reduced, andthe nerve-evoked release was abolished on K+-readmission. Means± S.E.M. of four identical experiments are given.
inhibited the release of [3H]NA by about 60–70% on nervestimulation (Table 1). The ‘residual release’ of [3H]NA wasblocked either by TTX (10−7 M) or by reducing externalCa2+ from 2.5 to 0.25 mM. These results suggest that depo-larisation induced Ca2+-influx is required for the ‘residualrelease’ observed. If a much larger concentration of�-CgTxGVIA was used (10−6 M; 60 min incubation period) the re-lease of [3H]NA was inhibited further (∼80%; not shown)but still some ‘residual release’ remained, suggesting thatother subtype(s) of VSCC(s) are probably activated.
The preferential�2-receptor antagonist yohimbine (3×10−7 M) increased the release of NA after�-CgTx GVIA(10−8 M) treatment (ratio of [3H]NA-release: 0.77± 0.05,n = 3); however its effectiveness in releasing NA was muchless than that of control (ratio: 4.11±0.57,n = 4; Table 1).
In intact arteries the non-selective Ca2+-channel inhibitorneomycin (Keith et al., 1992) inhibited the nerve-evoked re-lease of [3H]NA in a concentration-dependent manner (datanot shown). After 10−8 M �-CgTx GVIA pre-treatment3 × 10−3 M neomycin caused full inhibition of NA-release(ratio: 0.05±0.01,n = 3; Table 1). Under these conditions,yohimbine was ineffective in eliciting NA-release (ratio:0.07±0.01,n = 3;Table 1). During these experiments it was
also observed that the resting outflow of [3H]NA transientlyincreased on neomycin perfusion (see alsoFigs. 3 and 5).In GABA-ergic neuronesMartinez-Martos et al. (1997)have shown that neomycin activates fast, TTX-sensitiveNa+-channels. The neomycin-(3× 10−3 M)-caused fullinhibition of [3H]NA-release after 10−8 M �-CgTx GVIApre-treatment did not desensitise with time i.e., it was main-tained for four successive stimulation periods (not shown).
3.3. [3H]noradrenaline release without functioningVSCCs: activation of reverse Na+/Ca2+-exchange in‘K+-free’ solution, and the effects of TTX, Ca2+-removal,Li+-substitution or KB-R7943 application
In another series of experiments the Na+-pump wasinhibited by removal of K+ from the external medium.Previously the release of [3H]NA was blocked by�-CgTxGVIA (10−8 M) and neomycin (3× 10−3 M). K+-removalincreased both resting and the nerve-evoked release of[3H]NA (Fig. 2). The peak release was observed after45 min of K+-removal (second stimulation in ‘K+-free’solution; the ratio of [3H]NA-release: 1.36± 0.11, n = 4),
704 T.L. Török et al. / Neurochemistry International 45 (2004) 699–711
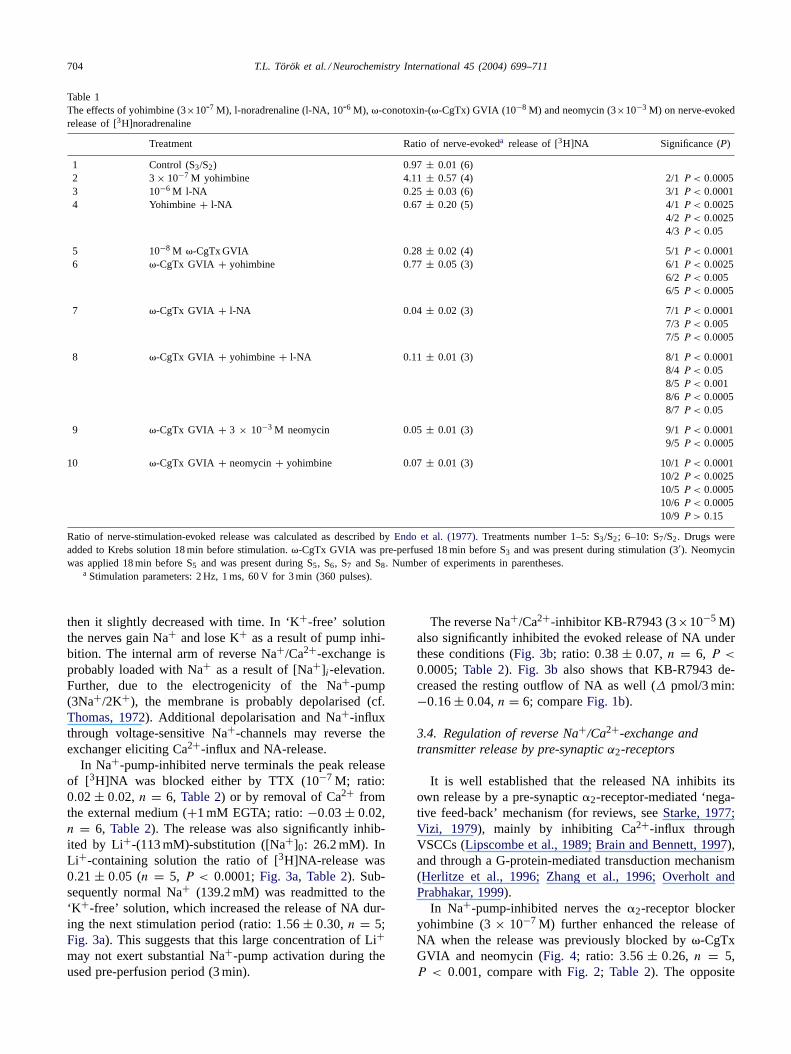
Table 1The effects of yohimbine (3×10-7 M), l-noradrenaline (l-NA, 10-6 M), �-conotoxin-(�-CgTx) GVIA (10−8 M) and neomycin (3×10−3 M) on nerve-evokedrelease of [3H]noradrenaline
Treatment Ratio of nerve-evokeda release of [3H]NA Significance (P)
1 Control (S3/S2) 0.97 ± 0.01 (6)2 3× 10−7 M yohimbine 4.11± 0.57 (4) 2/1P < 0.00053 10−6 M l-NA 0.25 ± 0.03 (6) 3/1P < 0.00014 Yohimbine+ l-NA 0.67 ± 0.20 (5) 4/1P < 0.0025
4/2 P < 0.00254/3 P < 0.05
5 10−8 M �-CgTx GVIA 0.28 ± 0.02 (4) 5/1P < 0.00016 �-CgTx GVIA + yohimbine 0.77± 0.05 (3) 6/1P < 0.0025
6/2 P < 0.0056/5 P < 0.0005
7 �-CgTx GVIA + l-NA 0.04 ± 0.02 (3) 7/1P < 0.00017/3 P < 0.0057/5 P < 0.0005
8 �-CgTx GVIA + yohimbine+ l-NA 0.11 ± 0.01 (3) 8/1P < 0.00018/4 P < 0.058/5 P < 0.0018/6 P < 0.00058/7 P < 0.05
9 �-CgTx GVIA + 3 × 10−3 M neomycin 0.05± 0.01 (3) 9/1P < 0.00019/5 P < 0.0005
10 �-CgTx GVIA + neomycin+ yohimbine 0.07± 0.01 (3) 10/1P < 0.000110/2 P < 0.002510/5 P < 0.000510/6 P < 0.000510/9 P > 0.15
Ratio of nerve-stimulation-evoked release was calculated as described byEndo et al. (1977). Treatments number 1–5: S3/S2; 6–10: S7/S2. Drugs wereadded to Krebs solution 18 min before stimulation.�-CgTx GVIA was pre-perfused 18 min before S3 and was present during stimulation (3′). Neomycinwas applied 18 min before S5 and was present during S5, S6, S7 and S8. Number of experiments in parentheses.
a Stimulation parameters: 2 Hz, 1 ms, 60 V for 3 min (360 pulses).
then it slightly decreased with time. In ‘K+-free’ solutionthe nerves gain Na+ and lose K+ as a result of pump inhi-bition. The internal arm of reverse Na+/Ca2+-exchange isprobably loaded with Na+ as a result of [Na+]i-elevation.Further, due to the electrogenicity of the Na+-pump(3Na+/2K+), the membrane is probably depolarised (cf.Thomas, 1972). Additional depolarisation and Na+-influxthrough voltage-sensitive Na+-channels may reverse theexchanger eliciting Ca2+-influx and NA-release.
In Na+-pump-inhibited nerve terminals the peak releaseof [3H]NA was blocked either by TTX (10−7 M; ratio:0.02± 0.02, n = 6, Table 2) or by removal of Ca2+ fromthe external medium (+1 mM EGTA; ratio:−0.03± 0.02,n = 6, Table 2). The release was also significantly inhib-ited by Li+-(113 mM)-substitution ([Na+]0: 26.2 mM). InLi+-containing solution the ratio of [3H]NA-release was0.21 ± 0.05 (n = 5, P < 0.0001; Fig. 3a, Table 2). Sub-sequently normal Na+ (139.2 mM) was readmitted to the‘K +-free’ solution, which increased the release of NA dur-ing the next stimulation period (ratio: 1.56± 0.30, n = 5;Fig. 3a). This suggests that this large concentration of Li+may not exert substantial Na+-pump activation during theused pre-perfusion period (3 min).
The reverse Na+/Ca2+-inhibitor KB-R7943 (3×10−5 M)also significantly inhibited the evoked release of NA underthese conditions (Fig. 3b; ratio: 0.38 ± 0.07, n = 6, P <
0.0005; Table 2). Fig. 3b also shows that KB-R7943 de-creased the resting outflow of NA as well (∆ pmol/3 min:−0.16± 0.04, n = 6; compareFig. 1b).
3.4. Regulation of reverse Na+/Ca2+-exchange andtransmitter release by pre-synaptic α2-receptors
It is well established that the released NA inhibits itsown release by a pre-synaptic�2-receptor-mediated ‘nega-tive feed-back’ mechanism (for reviews, seeStarke, 1977;Vizi, 1979), mainly by inhibiting Ca2+-influx throughVSCCs (Lipscombe et al., 1989; Brain and Bennett, 1997),and through a G-protein-mediated transduction mechanism(Herlitze et al., 1996; Zhang et al., 1996; Overholt andPrabhakar, 1999).
In Na+-pump-inhibited nerves the�2-receptor blockeryohimbine (3× 10−7 M) further enhanced the release ofNA when the release was previously blocked by�-CgTxGVIA and neomycin (Fig. 4; ratio: 3.56 ± 0.26, n = 5,P < 0.001, compare withFig. 2; Table 2). The opposite

T.L. Török et al. / Neurochemistry International 45 (2004) 699–711 705
Fig. 3. Inhibition of [3H]NA-release in Na+-pump inhibited arteries by external Li+ (113 mM; [Na+]0: 26.2 mM), or by KB-R7943-(3×10−5 M)-applicationin the presence of neomycin (3× 10−3 M) and after�-CgTx GVIA (10−8 M) pre-treatment. Horizontal bars above the axis represent the stimulationperiods.Ordinate: [3H]NA-release (pmol (3 min)−1); abscissa: time (min). (a)�-CgTx GVIA significantly reduced the evoked-release of [3H]NA, whichwas fully inhibited by neomycin, applied later (ratio (S5/S2): −0.01 ± 0.03). Note that neomycin transiently increased the resting outflow of3H.Subsequently, removal of K+, increased the release of3H. Li+ was pre-perfused 3 min before S7 (arrow), which abolished the nerve-evoked releaseof 3H in ‘K +-free’ solution. Note that during the subsequent stimulation period (S8) the release of3H was markedly increased. Means± S.E.M. offive identical experiments are given. (b) As in (a) except KB-R7943 was used instead of Li+. Neomycin abolished the evoked-release of [3H]NA after�-CgTx GVIA pre-treatment (ratio (S5/S2): −0.03± 0.03). KB-R7943, applied 6 min before S7 in ‘K +-free’ solution (arrow), significantly reduced theevoked-release of3H. Note that KB-R7943 also reduced the resting outflow of3H. In addition, note that after KB-R7943 removal, the evoked-releaseof 3H increased again (S8). Means± S.E.M. of six identical experiments are given.

706 T.L. Török et al. / Neurochemistry International 45 (2004) 699–711
Table 2[3H]noradrenaIine release induced by Na+-pump inhibition (‘K+-free’ solution) after blockade of VSCCs by�-CgTx GVIA (108 M) plus neomycin(3×10−3 M) and the effects of TTX (10−7 M), Ca2+-removal (+1 mM EGTA), yoblmbine (3×10−7 M), l-noradrenaline (10-6 M), external Li+ (113 mM)and KB-R7943 (3× 10−5 M)
Treatment Ratio of nerve-evokeda
release of [3H]NASignificance (P)
1 Control (S3/S2) 0.97 ± 0.01 (6)2 10-8 M �-CgTx GVIA + 3 × 10−3 M neomycin 0.02± 0.01 (6) 2/1P < 0.00013 ‘K+-free’ (45 min) in the presence of 3× 10−3 M neomycin and
after 10−8 M �-CgTx GVIA pre-treatment1.36 ± 0.11 (4) 3/1P < 0.005
3/2 P < 0.0001
4 ‘K+-free’ in the presence of neomycin and after�-CgTx GVIApre-treatment+ 10−7 M TTX
0.02 ± 0.02 (6) 4/3P < 0.0001
5 ‘K+-free’ in the presence of neomycin and after�-CgTx GVIApre-treatment+ Ca2+-free/EGTA sol.
-0.03 ± 0.02 (6) 5/3P < 0.0001
6 ‘K+-free’ in the presence of neomycin and after�-CgTx GVIApre-treatment+ 113 mM [Li+]0
b0.21 ± 0.05 (5) 6/1P < 0.0001
6/2 P < 0.0256/3 P < 0.0001
7 ‘K+-free’ in the presence of neomycin and after�-CgTx GVIApre-treatment+ 3 × 10−5 M KB-R7943c
0.38 ± 0.07 (6) 7/1P < 0.00017/2 P < 0.00257/3 P < 0.0005
8 ‘K+-free’ in the presence of neomycin and after�-CgTx GVIApre-treatment+ 3 × 10−7 M yohimbine
3.56 ± 0.26 (5) 8/1P < 0.00018/2 P < 0.00018/3 P < 0.001
9 ‘K+-free’ in the presence of neomycin and after�-CgTx GVIApre-treatment+ 10−6 M l-NA
0.55 ± 0.07 (6) 9/1P < 0.00059/2 P < 0.00059/3 P < 0.00059/8 P < 0.0005
10 ‘K+-free’ in the presence of neomycin and after�-CgTx GVIApre-treatment+ 3 × 10−7 M yohimbine+ 113 mM [Li+]0
b0.37 ± 0.03 (5) 10/1P < 0.0001
10/2 P < 0.000110/3 P < 0.000110/6 P < 0.0510/8 P < 0.0001
11 ‘K+-free’ in the presence of neomycin and after�-CgTx GVIApre-treatment+ 3×10−7 M yohimbine+ 3×10−5 M KB-R7943c
0.54 ± 0.07 (5) 11/1P < 0.000111/2 P < 0.000111/3 P < 0.000511/7 P > 0.1011/8 P < 0.0001
�-CgTx GVIA was pre-perfused 18 min before S3 and was present during stimulation (3′) Neomycin was applied 18 min before S5 and was presentthroughout. K+ was removed 18 min before S6.
a Stimulation parameters: 2 Hz, 1 ms, 60 V for 3 min (360 pulses) Treatments number 2–8: S7/S2.b Li+ was pre-perfused 3′ before S7 and was present during stimulation (3′).c KB-R7943 was pre-perfused 6′ before S7 and was present during stimulation (3′).
effect was obtained when the�2-receptors were activated byl-NA (10−6 M); i.e., the evoked-release of NA was inhibited(Fig. 5; ratio: 0.55± 0.07, n = 6, P < 0.0005;Table 2).
External Li+ (113 mM; [Na+]0: 26.2 mM) significantlyinhibited the release of NA which had been potentiated by�2-receptor inhibition. When the second stimulation periodwas carried out in yohimbine-containing ‘K+-free’ solutionto which Li+ was also added, the rate of transmitter re-lease was markedly inhibited (ratio: 0.37 ± 0.03, n = 5,P < 0.0001; Table 2). The yohimbine-induced enhance-ment of NA-release was similarly inhibited by KB-R7943(3 × 10−5 M; ratio: 0.54 ± 0.07, n = 5, P < 0.0001;Table 2).
From these experiments it can be concluded that similarto that of VSCCs the reverse Na+/Ca2+-exchange-mediated
NA-release may also be regulated by pre-synaptic�2-recep-tors. This regulation seems to be a kind of ‘short term’ reg-ulation since the Na+-pump was inhibited for only 45 min.
4. Discussion
The primary role of Na+/Ca2+-exchange is to extrudeCa2+ from the cells after ‘excitation–secretion/contraction’(cf. Blaustein and Lederer, 1999). Although the reversalpotential of the exchanger is around−40 mV, the maindifficulty to demonstrate reverse Na+/Ca2+-exchange con-tribution in transmitter release/muscle contraction is thatthe VSCCs are activated primarily on depolarisation (cf.Philipson and Nicoll, 2000). The reason is that the turnover

T.L. Török et al. / Neurochemistry International 45 (2004) 699–711 707
Fig. 4. [3H]NA-release-enhancing effect of yohimbine (3×10−7 M) in Na+-pump inhibited arteries, in which the evoked-release of [3H]NA was previouslyblocked by�-CgTx GVIA (10−8 M) and neomycin (3× 10−3 M). Ordinate: [3H]NA-release (pmol (3 min)−1); abscissa: time (min). Neomycin fullyinhibited the evoked-release of [3H]NA after �-CgTx GVIA pre-treatment (ratio (S5/S2): −0.01±0.04). Yohimbine, pre-perfused 18 min before the secondstimulation period in ‘K+-free’ solution (S7), markedly increased the release of3H on nerve-stimulation. Means± S.E.M. of five identical experimentsare given.
rate of Ca2+ by VSCC is much higher than that of theexchanger (for refs. see Introduction). Thus, if [Ca2+]i iselevated, as a result of VSCC(s) activation, this probablywill switch back the exchanger to the Ca2+-efflux mode(Philipson and Nicoll, 2000).
In the present study, electrical stimuli (2 Hz, 1 ms) wereused to elicit transmitter release from peripheral sym-pathetic nerves of rabbit pulmonary artery and Li+ wasused to carry the inward current through fast Na+-channel(Hille, 1992). Instead of inhibition, Li+ slightly poten-tiated the evoked-release of NA. Apart from the effectof Li+ on Na+/Ca2+-exchanger, it has also been shownthat Li+ exert an inhibitory effect on delayed rectifierK+-current (Kato et al., 1991), which is mainly responsiblefor AP-repolarisation of sympathetic nerves (Boehm andKubista, 2002).
The isothiourea-derivative Na+/Ca2+-exchange inhibitorKB-R7943 also failed to inhibit the evoked-release of NA athigh concentration (3× 10−5 M; Fig. 1a). KB-R7943 pref-erentially blocks reverse Na+/Ca2+-exchange in ventricularmyocytes (Watano et al., 1996; Iwamoto et al., 1996). How-ever, it also inhibits certain K+-channels (Matsuda et al.,2001), including the delayed rectifier (Tanaka et al., 2002).This latter may counteract to its inhibitory effect on reverseNa+/Ca2+-exchange. In pulmonary arteries KB-R7943 in-
creased the resting outflow of NA as well (Fig. 1), even if itwas used under reversed Ca2+-gradient (Ca2+-free+ 1 mMEGTA solution; unpublished observation). This raises thepossibility, that similar to that of cardiac and smooth mus-cle cells (Iwamoto et al., 1995), KB-R7943 may releaseCa2+ from internal stores of sympathetic nerves. On theother hand, KB-R7943 delayed the recovery of NA-releaseafter stimulation (Fig. 1a). Several effect may play a rolein this phenomena: including inhibition of normal mode ofoperation of Na+/Ca2+-exchanger (Iwamoto et al., 1996),Ca2+-releasing effect from internal stores (Iwamoto et al.,1995) or inhibition of delayed rectifier K+-current (Tanakaet al., 2002).
In pulmonary arteries, partial blockade of VSCCs sig-nificantly inhibited the evoked-release of NA. A lowconcentration of the selective and irreversible blocker of‘N-type’ VSCC �-CgTx GVIA (10−8 M) inhibited theevoked-release of NA by about 60–70% (Figs. 2–5). A muchlarger concentration of�-CgTx GVIA (10−6 M; 60 minpre-incubation) did not cause full inhibition (%-inhibition∼80; Zillikens and Török, unpublished observation). Thisis consistent with the observations of others (Kitagawaet al., 2003), namely, that in sympathetic neurons thedominant VSCC involved in transmitter release is the‘N-type’ (Lipscombe et al., 1989; Hille, 1994; Brain and

708 T.L. Török et al. / Neurochemistry International 45 (2004) 699–711
Fig. 5. Inhibition of [3H]NA-release by l-noradrenaline (l-NA, 10−6 M) in Na+-pump inhibited arteries in the presence of neomycin (3× 10−3 M)and after�-CgTx GVIA (10−8 M) pre-treatment.Ordinate: [3H]NA-release (pmol (3 min)−1); abscissa: time (min). Neomycin abolished the release of[3H]NA after �-CgTx GVIA application (ratio (S5/S2): 0.01± 0.02), and produced a transient increase of resting transmitter outflow. l-NA, pre-perfused18 min before the second stimulation period in 0K+-solution (S7), significantly inhibited the nerve-evoked release of3H. Means± S.E.M. of six identicalexperiments are given.
Bennett, 1997; for review, seeWaterman, 2000). The‘residual release’ of NA after�-CgTx-(10−8 M)-treatmentwas blocked by TTX or by reducing [Ca2+]0 from 2.5 to0.25 mM. The aminoglycoside antibiotic neomycin alsoinhibited, concentration-dependently, the ‘residual release’of NA in �-CgTx-treated arteries. Full inhibition was ob-served at a concentration of 3×10−3 M (Figs. 2–5). Keithet al. (1992)have shown that neomycin inhibits neuronal‘N-’, ‘L-’ and ‘non-N-’, ‘non-L-type’ high-voltage acti-vated Ca2+-channels. In peripheral sympathetic nervesSmith and Cunnane (1997)have shown that after�-CgTxGVIA treatment the ‘residual release’ of neurotransmit-ter can be blocked by the ‘P-/Q-type’ VSCC-antagonist�-agatoxin IVA or by the non-selective Ca2+-channelblocker �-grammotoxin SIA. In pulmonary arteries, rela-tively large concentration of�-agatoxin IVA (3× 10−7 M),which probably blocks both ‘P-/Q-type’ VSCCs, did notinhibit the ‘residual release’ after large concentration of�-CgTx-(10−6 M)-treatment (60 min incubation period; un-published observation). This suggests activation of othersubtype(s) of VSCC(s), or reverse Na+/Ca2+-exchange.This latter arises the possibility that neomycin may partiallyinhibit the exchanger as well.
In pulmonary arteries correlation was obtained be-tween the NA-release enhancing effect of the�2-receptorblocker yohimbine and the extent of previously released
NA. Namely, in normal solution, yohimbine markedly en-hanced the release of NA (Table 1). The release of NAwas much less in response to yohimbine after partial block-ade of ‘N-type’ VSCCs by�-CgTx GVIA, and finally,yohimbine was ineffective when the release was previ-ously blocked by�-CgTx GVIA plus neomycin (Table 1).This confirms the original suggestion of (Starke, 1977;Molderings et al., 2002), that NA-release is required forpre-synaptic �2-receptor-mediated ‘negative feed-back’mechanism.Brain and Bennett (1997)have succeeded inmeasuring [Ca2+]i-concentration in sympathetic varicosi-ties (�[Ca2+]v) of mouse vas deferens preparation. Theyshowed that yohimbine (10−5 M) increased the ampli-tude of�[Ca2+]v at 5 Hz stimulation frequency, while the�2-receptor agonist clonidine (10−6 M) had the oppositeeffect.
In order to demonstrate reverse Na+/Ca2+-exchangeactivation in NA-release and in response to brief (1 ms)depolarising stimuli, the sympathetic nerves were loadedwith Na+ by inhibiting the Na+-pump (Fig. 2) and trans-mitter release was previously blocked by�-CgTx GVIAand neomycin. In Na+-pump-inhibited axon terminals theinternal Na+-concentration is elevated and the membranemay be depolarised either due to switching off the elec-trogenic contribution of the Na+-pump (3Na+/2K+; cf.Thomas, 1972), or through reduction of the transmembrane

T.L. Török et al. / Neurochemistry International 45 (2004) 699–711 709
K+-gradient. As a result of [Na+]i-elevation, the internalarm of the exchanger is probably loaded with Na+. De-polarisation, by itself, activates the exchanger working inthe reverse mode. Additional depolarisation and Na+-influxthrough fast Na+-channel may activate the exchanger,producing Ca2+-influx and transmitter release. This wassupported by the findings that either TTX (10−7 M) orCa2+-removal (+1 mM EGTA) abolished the release ofNA observed (Table 2). Since the exchanger is electrogenic(3Na+/1Ca2+), the membrane is probably slightly hyper-polarised as internal Na+ is being exchanged for externalCa2+. Thus, depolarisation may not be absolutely requiredfor neurotransmitter release.
Theoretically it is possible that similar to that of cardiacmuscle (Leblanc and Hume, 1990), the exchanger medi-ated Ca2+-influx may release Ca2+ from internal stores. Inpulmonary arteries, large concentration of ryanodine (5×10−5 M) slightly, not significantly inhibited the release ofNA. The ratios of NA-release in the absence and in the pres-ence of ryanodine were: 1.10±0.19 (n = 5) and 0.82±0.15(n = 6), respectively, in two parallel series of experiments(not shown; %-inhibition∼25, P > 0.05). In normal so-lution ryanodine was ineffective (unpublished observation).It should be mentioned however, thatSmith and Cunnane(1996) could demonstrate ryanodine-effect in sympatheticnerves by using depolarisation (10–50 Hz) induced by highfrequency of electrical stimulation.
It is known that the reverse Na+/Ca2+-exchange re-quires internal Ca2+ for its activation (‘positive feed-back’mechanism;Baker and DiPolo, 1984; Kimura et al., 1986;Hilgemann, 1990; DiPolo and Beaugé, 1993), known as‘non-transported’ Ca2+. This Ca2+ may come from internalstores, possibly from the mitochondria by a [Na+]i-causedCa2+-release mechanism (Na+/Ca2+-exchange; Gunterand Pfeiffer, 1990; Brierley et al., 1994), and/or it is pro-vided by the plasma membrane exchanger (the Ca2+-effluxmode is inhibited and the Ca2+-influx mode is activated as[Na+]i is elevated). This latter suggestion was supported bythe finding that the reverse Na+/Ca2+-exchange inhibitorKB-R7943 decreased the resting outflow of NA in ‘K+-free’solution (compareFig. 3bwith Fig. 1).
External monovalent ions (K+, Rb+, Cs+, Li+) increasethe affinity of Ca2+ for the exchanger (Baker et al., 1969;Blaustein, 1977; Allen and Baker, 1986; Yasui and Kimura,1990), even if the Ca2+-binding is increased by reducingexternal Na+ (Török and Powis, 1990). These ions are nottransported and the requirement of the exchanger for alkalimetals is not absolute (cf.Blaustein and Lederer, 1999).Since we used ‘K+-free’ solution to load the nerves withNa+, the reverse exchanger mediated Ca2+-influx is prob-ably partly inhibited. An additional note, concerning K+ isas follows: although the depolarising pulse used was brief(1 ms), the duration of AP was probably longer. In ‘K+-free’solution the cells lose K+, since the gain of Na+ is coupledto K+-loss. Thus, less internal K+ is available to carry theoutward repolarising K+-current(s) through K+-channel(s).
The following experimental results may support theidea that brief depolarising pulses reverse the exchanger inNa+-loaded nerves in the absence of functioning VSCCs.
(i) The nerve-evoked release was fully blocked byTTX-application, or by removal of Ca2+ from the ex-ternal medium with parallel addition of 1 mM EGTA(Table 2).
(ii) The evoked release was markedly inhibited inLi+-containing solution (Fig. 3a, Table 2). Both fornormal and reverse Na+/Ca2+-exchange operation therequirement for Na+ is absolute, i.e., Li+0/i cannotsubstitute for Na+0/i in the exchange cycle (Bakeret al., 1969; Miura and Kimura, 1989; Reuter andPorzig, 1995; Doering et al., 1998). However, exter-nal Li+ can carry the inward current through fast,TTX-sensitive Na+-channels (Hille, 1992; Lipp andNiggli, 1994), thereby can depolarise the membraneand open VSCCs, but it does not ride through reverseNa+/Ca2+-exchange (Fig 3a). It should be mentionedhowever, that a large concentration of Li+ (113 mM)was used in our experiments. It is known that alkalimetals, such as Rb+, Cs+ and Li+ can reactivate theNa+-pump (cf. Thomas, 1972). However, this is un-likely in our experiments by using a short pre-perfusionperiod with Li+-containing solution (3 min), since thethird stimulation period in ‘K+-free’ solution signifi-cantly increased the release of NA (Fig. 3a; comparethe third and first stimulation periods in ‘K+-free’ solu-tion). Furthermore, when Li+ was added to ‘K+-free’solution in the presence of functioning VSCCs (i.e., inthe absence of�-CgTx and neomycin) the release of[3H]NA was maintained (ratio: 2.98 ± 0.55, n = 4;unpublished observation).
(iii) The reverse Na+/Ca2+-exchange-blocker KB-R7943(Watano et al., 1996; Shigekawa and Iwamoto, 2001)significantly inhibited the evoked-release of NA un-der these conditions (Fig. 3b, Table 2). A strikingdifference is that in 0K+-solution KB-R7943 de-creased the resting outflow of NA (compareFig. 3bwith Fig. 1). Thus, it seems that in ‘K+-free’ solutionthe plasma membrane reverse Na+/Ca2+-exchange isresponsible, at least partly, for the increased restingtransmitter-release observed. Further, the drug-causedinhibition of reverse Na+/Ca2+-exchange is prob-ably more pronounced than its Ca2+-releasing ef-fect from internal stores. KB-R7943 has little effecton Na+/K+-ATPase, Na+/H+-exchange, sarcolem-mal Ca2+-ATPase and SR Ca2+-ATPase (Iwamotoet al., 1996). However, it inhibits voltage-sensitiveNa+-channel, ‘L-type’ Ca2+-channel (Watano et al.,1996). In our experiments a substantial effect onfast Na+-channel is unlikely, since it did not re-duce the peak release of NA in normal solution(Fig. 1a). As for ‘L-type’ Ca2+-channels, neomycinis present, albeit this channel may not be involved in

710 T.L. Török et al. / Neurochemistry International 45 (2004) 699–711
NA-release of peripheral sympathetic nerves (Bennett,1999).
An important and new observation of the present studyis that in sympathetic nerves of rabbit pulmonary arterypre-synaptic�2-receptors may regulate NA-release thatis mediated by reverse Na+/Ca2+-exchange. Namely, inNa+-loaded arteries, in which VSCCs had been blockedpreviously, yohimbine further enhanced, while l-NA inhib-ited, the evoked-release of neurotransmitter (Figs. 4 and 5,Table 2). This modulation is effective in the ‘short-run’ sincethe duration time of Na+-loading period was only 45 min.The yohimbine-induced enhancement of NA-release wassignificantly inhibited either when the fast Na+-channelswere activated by Li+ (Table 2), or when KB-R7943 wassimultaneously present.
In conclusion, in peripheral sympathetic nerves particu-larly if there is a degree of Na+-loading (e.g., during hypoxicand hypoglycaemic conditions, cf.Milusheva et al., 1992)brief depolarising pulses may reverse Na+/Ca2+-exchangeby stimulating further Na+-influx, and thus activateCa2+-influx and transmitter release. This raises the pos-sibility that at least in peripheral sympathetic nerves theexchanger proteins and the vesicle-docking sites are closeto each other. Reverse Na+/Ca2+-exchange activation mayoccur physiologically especially at high frequency of nerveAPs, because of the small size of nerve terminals. Calcu-lations based on the reversal potential (ENa/Ca) and driv-ing force (�VNa/Ca) of the exchanger show that a smallincrease of internal Na+ (4–5 mM) is enough to reversethe exchanger. Further, similar to that of VSCCs, the re-verse Na+/Ca2+-exchange is probably also regulated bypre-synaptic�2-autoreceptors.
Acknowledgements
We thank Miss Mónika Juhász and Mrs. Ágnes Gáborházifor excellent technical assistance, Mr. Gyula Grézál for sta-tistical analysis and to Professor A.F. Brading for helpfulcomments and suggestions on the manuscript. CompoundKB-R7943 was kindly provided by Nippon Organon K.K.(Osaka, Japan). This work was supported, in part by OTKA(Grant Nos. 1020, T017749, TS040736), ETK-SOTE (GrantNo. G6), by ETT (Grant No. 237/96) and by a Grant fromThe Hungarian Academy of Sciences.
References
Allen, T.J.A., Baker, P.F., 1986. Comparison of the effects of potassiumand membrane potential on the calcium-dependent sodium efflux insquid axons. J. Physiol. 378, 53–76.
Baker, P.F., Blaustein, M.P., Hodgkin, A.L., Steinhardt, R.A., 1969. Theinfluence of calcium ions on sodium efflux in squid axons. J. Physiol.200, 431–458.
Baker, P.F., DiPolo, R., 1984. Axonal calcium and magnesiumhomeostasis. In: Baker, P.F. (Ed.), Current Topics in Membranes and
Transport, vol. 22, The Squid Axon. Academic Press, New York,London, pp. 195–247.
Bennett, M.R., 1999. The concept of a calcium sensor in transmitterrelease. Prog. Neurobiol. 59, 243–277.
Blaustein, M.P., 1977. Effects of external and internal cations and ATPon sodium–calcium exchange and calcium–calcium exchange in squidaxons. Biophys. J. 20, 79–111.
Blaustein, M.P., Lederer, W.J., 1999. Sodium/calcium exchange: itsphysiological implications. Physiol. Rev. 79, 763–854.
Boehm, S., Kubista, H., 2002. Fine tuning of sympathetic transmitterrelease via ionotropic and metabotropic presynaptic receptors.Pharmacol. Rev. 54, 43–99.
Borowski, E., Starke, K., Ehrl, H., Endo, T., 1977. A comparison of pre-and post-synaptic effects of alpha-adrenolytic drugs in the pulmonaryartery of the rabbit. Neuroscience 2, 285–296.
Brain, K.L., Bennett, M.R., 1997. Calcium in sympathetic varicositiesof mouse vas deferens during facilitation, augmentation andautoinhibition. J. Physiol. 502, 521–536.
Brassai, A., Mako, K., Domjanschitz, L., Sperlagh, B., 2002. Lack ofprejunctional modulation of noradrenaline release by endogenous nitricoxide in guinea pig pulmonary artery. Neurochem. Int. 41, 279–283.
Brierley, G.P., Baysal, K., Jung, D.W., 1994. Cation transport systemsin mitochondria: Na+ and K+ uniports and exchangers. J. Bioeng.Biomembr. 26, 519–526.
DiPolo, R., Beaugé, L., 1993. Ca2+ transport in nerve fibres. Biochim.Biophys. Acta 466, 481–499.
Doering, A.E., Nicoll, D.A., Lu, Y., Lu, L., Weiss, J.N., Philipson, K.D.,1998. Topology of a functionally important region of the cardiacNa+/Ca2+ exchanger. J. Biol. Chem. 273, 778–783.
Endo, T., Starke, K., Bangerter, A., Taube, H.D., 1977. Presynapticreceptor system on the noradrenergic neurones of the rabbit pulmonaryartery. Naunyn-Schmiedeberg’s Arch. Pharmacol. 296, 229–247.
Gleason, E., Borges, S., Wilson, M., 1994. Control of transmitter releasefrom retinal amacrine cells by Ca2+ influx and efflux. Neuron 13,1109–1117.
Gleason, E., Borges, S., Wilson, M., 1995. Electrogenic Na–Ca exchangeclears Ca2+ loads from retinal amacrine cells in culture. J. Neurosci.15, 3612–3621.
Gunter, T.E., Pfeiffer, D.R., 1990. Mechanisms by which mitochondriatransport calcium. Am. J. Physiol. 258, 755–786.
Herlitze, S., Garcia, D.E., Mackie, K., Hille, B., Scheuer, T., Catterall,W.A., 1996. Modulation of Ca2+ channels by G-protein�� subunits.Nature 380, 258–262.
Hilgemann, D.W., 1990. Regulation and deregulation of cardiac Na+,Ca2+ exchange in giant excised sarcolemmal membrane patches.Nature 344, 242–245.
Hilgemann, D.W., 1996. Unitary cardiac Na+, Ca2+ exchange currentmagnitudes determined from channel-like noise and charge movementsof ion transport. Biophys. J. 71, 759–768.
Hille, B., 1992. Ionic Channels of Excitable Membranes. SinauerAssociates Inc., Sunderland, MA, USA.
Hille, B., 1994. Modulation of ion-channel function by G-protein-coupledreceptors. Trends Neurosci. 17, 531–536.
Iwamoto, T., Wakabayashi, S., Shigekawa, M., 1995. Growth factor-induced phosphorylation and activation of aortic smooth muscleNa+/Ca2+ exchanger. J. Biol. Chem. 270, 8996–9001.
Iwamoto, T., Watano, T., Shigekawa, M., 1996. A novel isothioureaderivative selectively inhibits the reverse mode of Na+/Ca2+ exchangein cells expressing NCX1. J. Biol. Chem. 271, 22391–22397.
Juhaszova, M., Church, P., Blaustein, M.P., Stanley, E.F., 2000. Locationof calcium transporters at presynaptic terminals. Eur. J. Neurosci. 12,839–846.
Kato, M., Lledo, P.-M., Vincent, J.-D., 1991. Blockade by lithium ionsof potassium channels in rat anterior pituitary cells. Am. J. Physiol.261, (Cell. Physiol.) 30, C218–C223.
Keith, R.A., Margano, J.T., DeFeo, P.A., Horn, M.B., Salama, A.I.,1992. Actions of neomycin on neuronal L-, N-, and non-L/non-N-type

T.L. Török et al. / Neurochemistry International 45 (2004) 699–711 711
voltage-sensitive calcium channel responses. J. Mol. Neurosci. 3, 147–154.
Kimura, J., Noma, A., Irisawa, H., 1986. Na–Ca exchange current inmammalian heart cells. Nature 319, 596–597.
Kitagawa, H., Yamazaki, T., Akiyama, T., Mori, H., Sunagawa, K., 2003.Effects of ketamine on exocytotic and non-exocytotic noradrenalinerelease. Neurochem. Int. 42, 261–267.
Leblanc, N., Hume, J.R., 1990. Sodium-current induced release of calciumfrom cardiac sarcoplasmic reticulum. Science 248, 372–376.
Lipp, P., Niggli, E., 1994. Sodium-current induced calcium signals inisolated guinea-pig ventricular myocytes. J. Physiol. 474, 439–446.
Lipscombe, D., Kongsamut, S., Tsien, R.W., 1989.�-Adrenergic inhibitionof sympathetic neurotransmitter release mediated by modulation ofN-type calcium-channel gating. Nature 340, 639–642.
Llinás, R., Sugimori, M., Silver, R.B., 1992. Microdomains of highcalcium concentration in a presynaptic terminal. Science 256, 677–679.
Luther, P.W., Yip, R.K., Bloch, R.J., Ambesi, A., Lindenmayer, G.E.,Blaustein, M.P., 1992. Presynaptic localization of sodium/calciumexchangers in neuromuscular preparations. J. Neurosci. 12, 4898–4904.
Martinez-Martos, J.M., Iribar, M.C., Peinado, J.M., 1997. Evoked GABArelease is not mediated by N-type VDCC in the frontal cortex of awakerats: effects of neomycin. Brain Res. Bull. 43, 441–445.
Matsuda, T., Arakawa, N., Takuma, K., Kishida, Y., Kawasaki, Y.,Sakaue, M., Takahashi, K., Takahashi, T., Suzuki, T., Ota, T.,Hamano-Takahashi, A., Onishi, M., Tanaka, Y., Kameo, K., Baba, A.,2001. SEA0400, a novel and selective inhibitor of the Na+–Ca2+exchanger, attenuates reperfusion injury in the in vitro and in vivocerebral ischemic models. J. Pharmacol. Exp. Ther. 298, 249–256.
Milusheva, E.A., Baranyi, M., 2003. Implication of ionotropic glutamatereceptors in the release of noradrenaline in hippocampal CA1 and CA3subregions under oxygen and glucose deprivation. Neurochem. Int. 43,543–550.
Milusheva, E., Dóda, M., Pasztor, E., Lajtha, A., Sershen, H., Vizi,E.S., 1992. Regulatory interactions among axon terminals affecting therelease of different transmitters from rat striatal slices under hypoxicand hypoglycemic conditions. J. Neurochem. 59, 946–952.
Miura, Y., Kimura, J., 1989. Sodium–calcium exchange current:dependence on internal Ca and Na and competitive binding of externalNa and Ca. J. Gen. Physiol. 93, 1129–1145.
Molderings, G.J., Bonisch, H., Hammermann, R., Gothert, M., Bruss,M., 2002. Noradrenaline release-inhibiting receptors on PC12 cellsdevoid of alpha(2(-)) and CB(1) receptors: similarities to presynapticimidazoline and edg receptors. Neurochem. Int. 40, 157–167.
Overholt, J.L., Prabhakar, N.R., 1999. Norepinephrine inhibits a toxinresistant Ca2+ current in carotid body glomus cells: evidence for adirect G protein mechanism. J. Neurophysiol. 81, 225–233.
Philipson, K.D., Nicoll, D.A., 2000. Sodium-calcium exchange: amolecular perspective. Ann. Rev. Physiol. 62, 111–133.
Reuter, H., Porzig, H., 1995. Localization and functional significance ofthe Na+/Ca2+ exchanger in presynaptic boutons of hippocampal cellsin culture. Neuron 15, 1077–1084.
Reuter, H., Seitz, N., 1968. The dependence of calcium efflux from cardiacmuscle on temperature and external ion composition. J. Physiol. 195,451–470.
Shigekawa, M., Iwamoto, T., 2001. Cardiac Na+–Ca2+ exchange:molecular and pharmacological aspects. Circ. Res. 88, 864–876.
Smith, A.B., Cunnane, T.C., 1996. Ryanodine-sensitive calcium storesinvolved in neurotransmitter release from sympathetic nerve terminalsof the guinea-pig. J. Physiol. 497, 657–664.
Smith, A.B., Cunnane, T.C., 1997. Multiple calcium channels controlneurotransmitter release from rat postganglionic sympathetic nerveterminals. J. Physiol. 499, 341–349.
Stanley, E.F., 1997. The calcium channel and the organization of thepresynaptic transmitter release face. Trends Neurosci. 20, 404–409.
Starke, K., 1977. Regulation of noradrenaline release by presynapticreceptor systems. Rev. Physiol. Biochem. Pharmacol. 77, 1–124.
Starke, K., Montel, H., Gayk, W., Merker, R., 1974. Comparison ofthe effects of clonidine on pre- and post-synaptic adrenoceptors inthe rabbit pulmonary artery. Naunyn-Schmiedeberg’s Arch. Pharmacol.285, 133–138.
Tanaka, H., Nishimaru, K., Aikawa, T., Hirayama, W., Tanaka, Y.,Shigenobu, K., 2002. Effect of SEA0400, a novel inhibitor ofsodium-calcium exchanger, on myocardal ionic currents. Br. J.Pharmacol. 135, 1096–1100.
Thomas, R.C., 1972. Electrogenic sodium pump in nerve and musclecells. Physiol. Rev. 52, 563–594.
Török, T.L., Powis, D.A., 1990. Catecholamine release from bovinechromaffin cells: the role of sodium–calcium exchange inouabain-evoked release. Exp. Physiol. 75, 573–586.
Török, T.L., Tóth, P.T., Tóthfalusi, L., Azzidani, A.M., Magyar, K., 1992.Dependence of release of [3H]noradrenaline from rabbit pulmonaryartery on internal sodium. J. Physiol. 458, 11–25.
Vizi, E.S., 1979. Presynaptic modulation of neurochemical transmission.Prog. Neurobiol. 12, 181–290.
Watano, T., Kimura, J., Morita, T., Nakanishi, H., 1996. A novelantagonist, no. 7943, of the Na+/Ca2+ exchange current in guinea-pigcardiac ventricular cells. Br. J. Pharmacol. 119, 555–563.
Waterman, S.A., 2000. Voltage-gated calcium channels in autonomicneuroeffector transmission. Prog. Neurobiol. 60, 181–210.
Yasui, K., Kimura, J., 1990. Is potassium co-transported by thecardiac Na–Ca exchange? Pflügers Arch. Eur. J. Physiol. 415, 513–515.
Zhang, J.-F., Ellinor, P.T., Aldrich, R.W., Tsien, R.W., 1996. Multiplestructural elements in voltage-dependent Ca2+ channels support theirinhibition by G proteins. Neuron 17, 991–1003.




















