
The novel histone deacetylase inhibitor BML-210 exerts growth inhibitory, proapoptotic and...
10
The novel histone deacetylase inhibitor BML-210 exerts growth inhibitory, proapoptotic and differentiation stimulating effects on the human leukemia cell lines Jurate Savickiene a , Veronika-Viktorija Borutinskaite a,b , Grazina Treigyte a , Karl-Eric Magnusson b , Ruta Navakauskiene a, ⁎ a Department of Developmental Biology, Institute of Biochemistry, LT-08662 Vilnius, Lithuania b Division of Medical Microbiology, Department of Molecular and Clinical Medicine, SE-583 53 Linkoping, Sweden Received 30 March 2006; received in revised form 25 July 2006; accepted 2 August 2006 Available online 15 August 2006 Abstract Histone deacetylase inhibitors have a potent role in the strategy for the treatment of leukemias. BML-210 (N-(2-Aminophenyl)-N′ phenyloctanol diamine) is the novel histone deacetylase inhibitor, and its mechanism of action has not been characterized. In this study, we examined the in vitro effects of BML-210 on the human leukemia cell lines (NB4, HL-60, THP-1, and K562). We found that BML-210 inhibits the growth of all cell lines and promotes apoptosis in a dose- and time-dependent manner. BML-210 alone induces HL-60 and K562 cell differentiation (up to 30%) to granulocytes and erythrocytes, respectively, and in combination with differentiation agents — all-trans retinoic acid and hemin, markedly potentates it. Those treatments cause G1 arrest and histone H4 acetylation, affects transcription factor NF-κB and Sp1 binding activity to their consensus sequences, the p21 or the FasL promoters, and influences expression of Sp1, NF-κB, p21 and FasL. These findings suggest that BML-210 could be a promising antileukemic agent to induce apoptosis and to modulate differentiation through the modulation of histone acetylation and gene expression. © 2006 Elsevier B.V. All rights reserved. Keywords: Apoptosis; Differentiation; Histone deacetylase inhibitor; Leukemia; Transcription factors 1. Introduction Histone deacetylase (HDAC) inhibitors comprise a diverse group of compounds that block histone deacetylation resulting in hyperacetylation of core histones, transcription factors and proteins involved in transcription (Yoshida et al., 2001). Transient acetylation of core histones is a prerequisite for chromatin remodeling and gene transcription (Strahl and Allis, 2000). This modification leads to loosening of histone-DNA contacts and facilitates the accessibility of a variety of factors to DNA (Wolffe et al., 1997). A number of transcriptional repressors and co- repressors have been shown to recruit the HDAC complex to the promoter regions. In acute promyelocytic leukemia (APL) cells, the promyelocytic leukemia–retinoic acid receptor alpha (PML– RARα) fusion protein, generated by chromosomal translocation, represses the gene expression required for myeloid leukemia by recruiting many HDAC molecules and promoting the develop- ment of leukemia (Grignani et al., 1999). Pharmacological doses of all-trans retinoic acid (ATRA) can bind to the fusion protein with a loss of HDAC activity and thereby induce reinitiating of differentiation (Lin et al., 1999). APL cells containing promye- locytic leukemia zinc finger–retinoic acid receptor alpha (PLZF– RARα) are resistant to ATRA, but transcriptional repression mediated by HDAC can be released with HDAC inhibitors (Redner et al., 1999). The clinical benefits of HDAC inhibitors and their implications for differentiation therapy currently are under active investigation. The addition of HDAC inhibitors has been shown to inhibit proliferation and to stimulate differentiation and apoptosis in transformed cells both in vitro and in vivo (Yoshida et al., 2003). Growth inhibitory effects have been documented in virtually all transformed cell types, including cell lines that arise from hematological (leukemia's, lymphoma's and mycelia's) and European Journal of Pharmacology 549 (2006) 9 – 18 www.elsevier.com/locate/ejphar ⁎ Corresponding author. Tel.: +370 5 2729187; fax: +370 5 2729196. E-mail address: [email protected] (R. Navakauskiene). 0014-2999/$ - see front matter © 2006 Elsevier B.V. All rights reserved. doi:10.1016/j.ejphar.2006.08.010
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of The novel histone deacetylase inhibitor BML-210 exerts growth inhibitory, proapoptotic and...

logy 549 (2006) 9–18www.elsevier.com/locate/ejphar
European Journal of Pharmaco
The novel histone deacetylase inhibitor BML-210 exerts growth inhibitory,proapoptotic and differentiation stimulating effects
on the human leukemia cell lines
Jurate Savickiene a, Veronika-Viktorija Borutinskaite a,b, Grazina Treigyte a,Karl-Eric Magnusson b, Ruta Navakauskiene a,⁎
a Department of Developmental Biology, Institute of Biochemistry, LT-08662 Vilnius, Lithuaniab Division of Medical Microbiology, Department of Molecular and Clinical Medicine, SE-583 53 Linkoping, Sweden
Received 30 March 2006; received in revised form 25 July 2006; accepted 2 August 2006Available online 15 August 2006
Abstract
Histone deacetylase inhibitors have a potent role in the strategy for the treatment of leukemias. BML-210 (N-(2-Aminophenyl)-N′phenyloctanol diamine) is the novel histone deacetylase inhibitor, and its mechanism of action has not been characterized. In this study, weexamined the in vitro effects of BML-210 on the human leukemia cell lines (NB4, HL-60, THP-1, and K562). We found that BML-210 inhibitsthe growth of all cell lines and promotes apoptosis in a dose- and time-dependent manner. BML-210 alone induces HL-60 and K562 celldifferentiation (up to 30%) to granulocytes and erythrocytes, respectively, and in combination with differentiation agents — all-trans retinoic acidand hemin, markedly potentates it. Those treatments cause G1 arrest and histone H4 acetylation, affects transcription factor NF-κB and Sp1binding activity to their consensus sequences, the p21 or the FasL promoters, and influences expression of Sp1, NF-κB, p21 and FasL. Thesefindings suggest that BML-210 could be a promising antileukemic agent to induce apoptosis and to modulate differentiation through themodulation of histone acetylation and gene expression.© 2006 Elsevier B.V. All rights reserved.
Keywords: Apoptosis; Differentiation; Histone deacetylase inhibitor; Leukemia; Transcription factors
1. Introduction
Histone deacetylase (HDAC) inhibitors comprise a diversegroup of compounds that block histone deacetylation resulting inhyperacetylation of core histones, transcription factors andproteins involved in transcription (Yoshida et al., 2001). Transientacetylation of core histones is a prerequisite for chromatinremodeling and gene transcription (Strahl and Allis, 2000). Thismodification leads to loosening of histone-DNA contacts andfacilitates the accessibility of a variety of factors to DNA (Wolffeet al., 1997). A number of transcriptional repressors and co-repressors have been shown to recruit the HDAC complex to thepromoter regions. In acute promyelocytic leukemia (APL) cells,the promyelocytic leukemia–retinoic acid receptor alpha (PML–RARα) fusion protein, generated by chromosomal translocation,
⁎ Corresponding author. Tel.: +370 5 2729187; fax: +370 5 2729196.E-mail address: [email protected] (R. Navakauskiene).
0014-2999/$ - see front matter © 2006 Elsevier B.V. All rights reserved.doi:10.1016/j.ejphar.2006.08.010
represses the gene expression required for myeloid leukemia byrecruiting many HDAC molecules and promoting the develop-ment of leukemia (Grignani et al., 1999). Pharmacological dosesof all-trans retinoic acid (ATRA) can bind to the fusion proteinwith a loss of HDAC activity and thereby induce reinitiating ofdifferentiation (Lin et al., 1999). APL cells containing promye-locytic leukemia zinc finger–retinoic acid receptor alpha (PLZF–RARα) are resistant to ATRA, but transcriptional repressionmediated by HDAC can be released with HDAC inhibitors(Redner et al., 1999). The clinical benefits of HDAC inhibitorsand their implications for differentiation therapy currently areunder active investigation. The addition of HDAC inhibitors hasbeen shown to inhibit proliferation and to stimulate differentiationand apoptosis in transformed cells both in vitro and in vivo(Yoshida et al., 2003).
Growth inhibitory effects have been documented in virtually alltransformed cell types, including cell lines that arise fromhematological (leukemia's, lymphoma's and mycelia's) and

10 J. Savickiene et al. / European Journal of Pharmacology 549 (2006) 9–18
epithelial (breast, bladder, ovarian, prostate, lung) tumors (Markset al., 2001a). This effect leads to the activation of transcription of afinite number of genes (about 2–8%) via histone acetylation(Chambers et al., 2003). HDAC inhibitors invariably inhibitproliferation of transformed cells in culture (Marks et al., 2001b). Itis, however, not yet understood which genes are targeted byhistone acetylation and how their expression is modulated by theinhibitors. Themechanism of gene expression selectivity currentlyis also under intensive study.
Currently, a number of structurally different HDAC inhibitorsare being evaluated as a potential new class of therapeutic agentsbut little is known about the molecular events that control theireffectiveness (Marks et al., 2001b; Minucci et al., 2001; Vigushinand Combers, 2002). Short-chain fatty acid compounds, likebutyrates, represent a class that is currently approved for the use inthe clinic (Newmark et al., 1994), inducing cell differentiationand/or apoptosis programs in a wide variety of neoplastic cells.The limitations of its clinical use because of rapid metabolism anda very short half-life in the blood may be overcome by butyratederivates or prodrugs of butyric acid (Gozzini et al., 2003). Inleukemia cells, HDAC inhibitors, such as butyrate derivates andsuberoylanilide hydroxamic acid (SAHA) induce differentiationor apoptosis (Richon et al., 1998; Rivero and Adunyah, 1998;Vrana et al., 1999). Several HDAC inhibitors are now beingevaluated clinically: SAHA is currently in a phase II clinical studyagainst T-cell lymphomas and metastatic squamous cell carcino-mas of the head and neck, MS-275 is in trial for advanced solidtumors or lymphomas and for poor-risk hematological malignan-cy; CI-994 is used for treatment of leukemia (Kelly et al., 2003;Marks et al., 2001a). The depsipeptide FK228 is a particularlypotent agent with broad clinical potential in chronic leukemia,small lymphoblastic lymphoma, acute lymphoblastic leukemia,cutaneous T-cell lymphoma or small cell non-small cell lungcancer (Yoshida et al., 2003). Recently, other classes ofcompounds, such as novel hybrid compounds of trapoxin andtrichostatin A, called CHAPs derivates (Yoshida et al., 2001),have been proposed as a promising candidate for antitumor drugswith strong HDAC-inhibiting activity. In some cases, HDACinhibitors can overcome drug resistance in other chemotherapeu-tical treatments (Redner et al., 1999). However, the use of HDACinhibitors is limited by progressive constitutional symptomsobserved in many patients.
In our study, we have investigated the biological effects of thenovel HDAC inhibitor, BML-210, on leukemia cell proliferation,viability, differentiation and transcriptional regulation of cellcycle– and apoptosis–regulating genes by transcription factorsSp1, NF-κB and p53. Based on our findings, we suggest thatBML-210 could be a promising antileukemic agent to induceapoptosis and to regulate differentiation through the modulationof histone acetylation and gene expression.
2. Materials and methods
2.1. Materials
BML-210 was obtained from Biomol Research Laboratories(USA). ATRA and hemin chloride were purchased from Sigma
Chemical Co. (St. Louis, MO), phenyl butyrate from Calbio-chem (Germany). The stock solutions of BML-210 (5 mM indimethyl sulphoxide (DMSO)), ATRA (500 μM in ethanol) andhemin chloride (25 mg/ml in 1.4 M ammonium hydroxide) werestored at −20 °C.
2.2. Cell cultures
The human leukemia cell lines (promyelocytic HL-60, acutepromyelocytic NB4, chronic myeloid K562 and acute mono-cytic THP-1) were maintained in RPMI 1640 mediumsupplemented with 10% fetal bovine serum, 100 U/ml penicillinand 100 μg/ml streptomycin (Gibco, Grand Island, NY). Cellswere grown at 37 °C in a humidified 5% CO2 atmosphere andused for assays during exponential phase of growth. Cell via-bility was assayed by exclusion of 0.2% Trypan blue. Cellnumber was determined by counting cells in suspension in ahaemocytometer. Granulocytic differentiation of HL-60 andNB4 cells was determined by nitro blue tetrazolium (NBT)reduction (Collins, 1987). Hemoglobin production duringerythroid differentiation of K562 cells was established bybenzidine staining (Jeannesson et al., 1984).
2.3. Cell cycle analysis
Untreated and HDAC inhibitor-treated cells were collectedby centrifugation, suspended in PBS and fixed in ice cold 70%ethanol (ratio 1:10) for 24 h at −20 °C. After centrifugation at500 ×g for 5 min, cells were suspended in phosphate-bufferedsaline (PBS) containing propidium iodide (PI) (50 μg/ml) andRNase (0.2 mg/ml) and incubated at room temperature for30 min. The tubes were then taken at 4 °C in the dark untilanalysis by flow cytometer (Becton-Dickinson FACS Calibur,USA). The percentage of cells in G0/G1, S and G2/M wasevaluated with CellQuest software. Apoptotic cells werequantified on PI histogram as a hypodiploid peak and the datawere registered on a logarithmic scale.
2.4. Assessment of CD11b
NB4 or HL-60 cells (5×105 cells/sample) were washedtwice in PBS, pH 7.4, then treated with monoclonal anti-humanCD11b, C3bi receptor/RPE (DakoCytomation, Denmark) in thedark at 4 °C for 30 min. Cells were washed with PBS containing2% bovine serum albumin (BSA), fixed in 4% paraformalde-hyde for 15–30 min on ice and pellet was resuspended in PBS.Eight thousand events were analyzed for each sample byimmunofluorescence using flow cytometry. Proliferating cellswith and without CD11b antibodies were used as a control.
2.5. Preparation of cytosolic and nuclear proteins
The cells (5×106 to 107) were harvested by centrifugation at500 ×g for 6 min, washed twice in ice cold PBS and suspendedin Nuclei EZ lysis buffer (Sigma, St. Louis, MO) for 5 min onice. The cell homogenates were then centrifuged at 1500 ×g for5 min. Supernatant, corresponding to the cytosolic fraction, was

Fig. 1. Dose-dependent effects of BML-210 on the growth of leukemia cell linesNB4, HL-60, THP-1 and K562. Cells were exposed to different concentrationsof BML-210 for 5 days. Aliquots of the cultures were subjected to countingfollowing staining with 0.2% Trypan blue for the determination of total numberof viable cells. Results are given as mean±S.E.M. (n=3).
11J. Savickiene et al. / European Journal of Pharmacology 549 (2006) 9–18
clarified by centrifuging at 15,000 ×g for 15 min and then it wasfrozen at −76 °C or immediately used for sodium dodecylsulfate/polyacrylamide gel electrophoresis (SDS/PAGE). Nu-clei were washed in the same cold Nuclei EZ buffer, vortexedbriefly and set on ice for 5 min. They were pelleted at 500 ×g for5 min, then completely suspended in Nuclei EZ storage buffer(Sigma, St. Louis, MO) and frozen at −76 °C.
Nuclear protein extracts for electrophoretic mobility shiftassay (EMSA) were prepared by lysis of nuclei in buffer(20 mM Tris-HCl, pH 8.0, 200 μM EDTA, 2 mM EGTA, 20%glycerol, 400 mM NaCl) containing 3 mM dithiothreitol (DTT),1 mM phenylmethylsulfonyl fluoride (PMSF) and 1× proteaseinhibitor cocktail Complete (Sigma, St. Louis, MO). Afterincubation for 1 h on ice, the extracts were centrifuged at18,000 ×g for 20 min, and used immediately. Proteinconcentrations were determined by RCDC protein assay asrecommended by the manufacturer (Bio Rad Lab., CA).
For SDS-PAGE analysis of total nuclear proteins, nuclei wereresuspended (approx. 5×107/ml) in two volumes (v/v) of 2× lysissolution (100 mMTris, pH 7.4, 5 mMMgCl2, 200 mMDTTand4% SDS), and thereafter 3 volumes of 1× lysis solution andbenzonase (Pure Grade, Merck, Germany) was added to give afinal concentration of 2.5 units/ml. The lysates were incubated for1 h at 0 °C and then centrifuged at 15,000 ×g for 30 min. Thesupernatants were immediately subjected to electrophoresis orfrozen at −76 °C.
2.6. Electrophoretic mobility shift assay (EMSA)
The probes used were synthetic oligonucleotides (MWG-Biotech AG, Denmark) representing binding sites: (5′-AGTTGAGGGGACTTTCCCAGGC-3′) consensus NF-κB; (5′-AAGCCTGGGCAACATAGAAAGTCCCCATCTGTACAAAA-3′) NF-κB from the FasL promoter; (5′-ATTCGATCGGGGCGGGGCGAGC-3′) consensus Sp1; (5′-ATTCGATCGGTTCGGGGCGAGC-3′) mutated consensus Sp1; (5′-GGCCGAGCGCGGGT CCCGCCTCCTTGAGGCGGG-3′)Sp1-3 (element 3) from the p21 promoter; (5′-ATCAGAAAATTGTGGGCGGAAACTTCCAGG-3′) Sp1 from theFasL promoter; (5′-CTGCGCGGGGCGGGCGCCGC-3′) Sp1from the p65 promoter; (5′-CTGCGCGGGTTGGGCGCCGC-3′)mutated Sp1from the p65 promoter; (5′-ATCAGGAACATGTCCCAA CATGTTGAGCTCT-3′) p53 from the p21 promoter; (5′-ATCAGGAATCGCTCCCAA CATGTTGAGCTCT-3′) mutatedp53 from the p21 promoter; (5′-GCCCTGTGCCAGGGGAGAGGAAGTGGAGGG-3′) PU.1 from the human neutrophil elastasepromoter.
Standard DNA reactions were performed with 10 μg nuclearextracts in a 20 μl of reaction buffer (10 mM HEPES pH 7.9,3 mM MgCl2, 0.1 mM EDTA, 40 mM NaCl, 10% glycerol)containing 2 μg BSA, 1 μg poly(dI-dC), 1 pM labeledoligonucleotide for 30 min at room temperature. When desired,unlabeled competitor oligonucleotide was added to proteinextracts at 50 or 100 foldmolar excess for a 15min preincubation.DNA-protein complexes were resolved on 5% polyacrylamidegel containing 1× Tris-borate buffer. After electrophoresis, gelswere dried and then exposed to X-ray films.
2.7. Isolation and fractionation of histones
Histones were extracted as described previously (Treigyteand Gineitis, 1979). Shortly, histones were extracted fromnuclei twice by 0.4 N H2SO4 and precipitated by adding 5 vol ofethanol at −20 °C overnight. Histone electrophoresis wascarried out essentially as described (Hurley, 1977). Firstly,histones (5 μg) were dissolved in a buffer, containing 0.9 Macetic acid, 10% glycerol, 6.25 M urea and 5% β-mercap-toethanol, then resolved on 15% polyacrylamide gels contain-ing 6 M urea and 0.9 M acetic acid, and transferred toImmobilon™ polyvinylidene difluoride (PVDF) membrane(Millipore, Bedford, MA). The membranes were probed withanti-hyperacetylated histone H4 (Penta) antibodies (Upstate,Lake Placid, NY).
2.8. Gel electrophoresis and immunoblot analysis
Cytosolic and nuclear proteins were resolved on 7–15%polyacrylamide gradient gels (Invitrogen, CA), then transferredto Immobilon™ PVDF membranes and probed with antibodiesagainst p21, p53, Sp1, FasL, NF-κB p50 or NF-κB p65 (SantaCruz, Biotechnology, Inc., CA) at a concentration of 1 μg/ml inPBS containing 0.18% Tween-20, 0.35 M NaCl, and 1% BSA.The membranes were subsequently washed with PBS-Tweenand then incubated with horseradish peroxidase-conjugatedsecondary antibody (DAKO, A/S, Denmark) for 1 h at roomtemperature. Immunoreactive bands were detected by enhancedchemiluminescence using ECL™ Western blotting detectionreagents (Amersham Pharmacia, Sweden) according to theinstructions of the manufacturer.

Fig. 3. Influence of BML-210 on expression of CD11b in HL-60 and NB4 cells.Control cells and treated with 5 μMBML-210 for 24 and 48 h were analyzed forthe early myeloid cell surface differentiation marker CD11b by flow cytometry.Control cells — full grey diagram, treated cells — empty diagram.
12 J. Savickiene et al. / European Journal of Pharmacology 549 (2006) 9–18
3. Results
3.1. Growth inhibition of leukemia cell lines by BML-210
In order to estimate the effects of different doses of BML-210,the leukemia cell lines NB4, HL-60, THP-1 and K562 weretreated with HDAC inhibitor alone for 5–6 days. In all cell linestested, BML-210, as a single agent, inhibited cell growth in adose- and time-dependent manner (Fig. 1). The optimal concen-tration of BML-210 for the cells was 5 μM. At a concentration of10 μM, HL-60 and THP-1 cells were more sensitive for cellgrowth inhibition and survival. The maximum growth inhibitionwas achieved with 20–30 μM BML-210. HDAC inhibitortreatment at those doses for 72 h of exposure produced a loss ofviability in all cell lines tested. In the apoptosis-resistant cell lineK562, the cytotoxic effect was delayed. A combination of BML-210 and ATRA exerted additive growth inhibitory effects in NB4and HL-60 cells (data not shown). 24 h exposition to lethal doses(20–30 μM) BML-210 caused NB4 or THP-1 cell growthrestoration after 6 or 9 days, respectively (data not shown).
3.2. BML-210 enhances leukemia cell differentiation inducedby different agents
To examine the differentiating activity of BML-210,leukemia cell lines HL-60, NB4 and K562 were treated with5–10 μM BML-210. Interestingly, BML-210 alone induced a
Fig. 2. Dose-dependent effects of BML-210 on leukemia cell differentiation.(A) HL-60 and NB4 cells were treated with 1 μM ATRA or pretreated with 5 μMBML-210 for 24 h before induction of differentiation with 1 μMATRA alone (BMLpretr-ATRA), or cotreated with 5 μM BML-210 and 1 μM ATRA (BML+ATRA).(B) HL-60 cells were treated with different doses of BML-210 alone. (C) K562 cellswere treatedwith 7μMBML-210 or 10μMhemin alone, and in combination.During5 days in culture, HL-60 and NB4 cell differentiation toward granulocytes wasdetermined by the ability of cells to reduceNBT, and hemoglobin-positiveK562 cellswere established by benzidine staining. Results are given as mean±S.E.M. (n=3).
dose-dependent differentiation of HL-60 cells to granulocytes,and K562 cells to erythrocytes up to 30% on day 4 (Fig. 2B, C).By contrast, BML-210 did not induce NB4 cell differentiation.NBT-positivity (Fig. 2B) and expression of the early differen-tiation marker CD11b at 24 h (Fig. 3) was noticed in HL-60 cellsonly. The in vitro differentiation experiments with inducers ofdifferentiation were performed at a lower concentration of BML-210 (5 μM) to avoid cytotoxicity (see Fig. 1). Indeed, BML-210in combination with 1 μM ATRA accelerated and noticeablyenhanced NB4 and HL-60 cell granulocytic differentiationduring 4 days, with a stronger effect on HL-60 cells (Fig. 2A).Pretreatment with 5 μM BML-210 for 24 h before subsequenttreatment with ATRA alone resulted in greater differentiationeffects in comparison with ATRA treatment (Fig. 2A). K562 cellerythroid differentiation by hemin alone was delayed till day 5,while combined treatment with 10 μM hemin and 7 μM BML-210 accelerated the cell maturation to erythrocytes (Fig. 2C).
We compared BML-210 effects on NB4 cell growth anddifferentiation with those obtained using another well knownHDAC inhibitor, phenyl butyrate. This agent at concentrationsof 0.5–4 mM inhibited cell growth in a dose-dependent manner,and in combination with ATRA (but not alone) increased theNBT-positivity (data not shown).
3.3. Influence of BML-210 on NB4 and HL-60 cell cycleprogression
NB4 and HL-60 cells were incubated with BML-210 (5 μM)alone and in combination with ATRA (1 μM), or pretreated withHDAC inhibitor for 24 h and then transferred to fresh medium
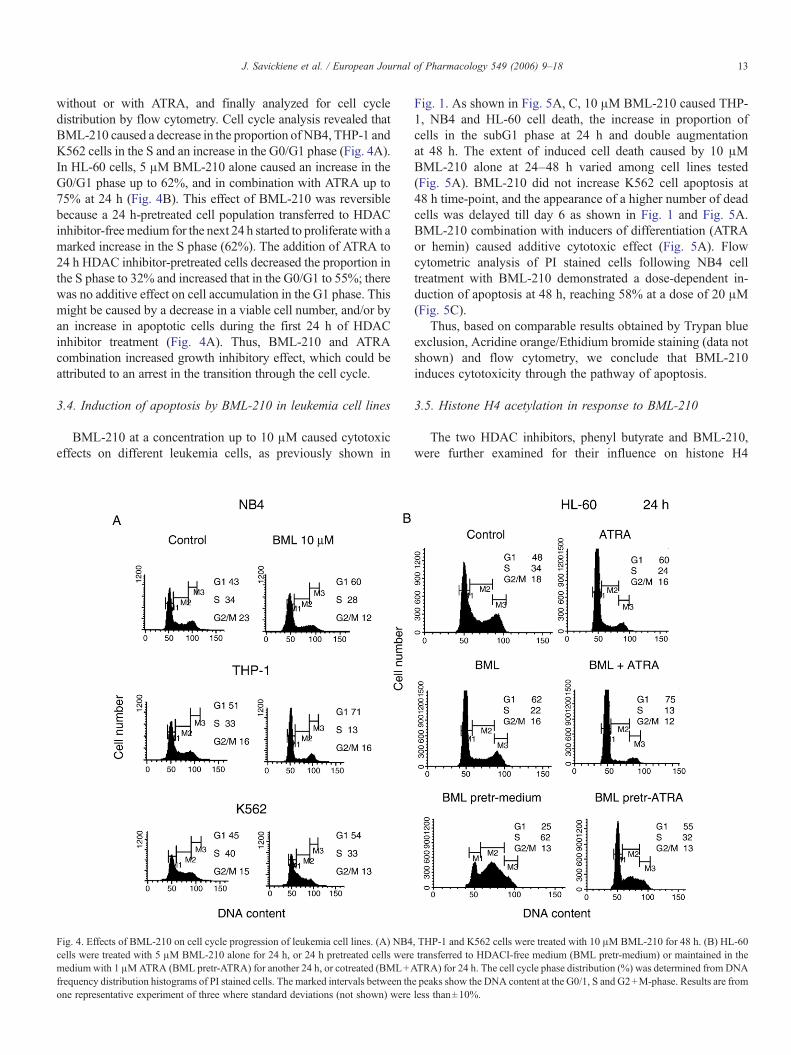
13J. Savickiene et al. / European Journal of Pharmacology 549 (2006) 9–18
without or with ATRA, and finally analyzed for cell cycledistribution by flow cytometry. Cell cycle analysis revealed thatBML-210 caused a decrease in the proportion of NB4, THP-1 andK562 cells in the S and an increase in the G0/G1 phase (Fig. 4A).In HL-60 cells, 5 μM BML-210 alone caused an increase in theG0/G1 phase up to 62%, and in combination with ATRA up to75% at 24 h (Fig. 4B). This effect of BML-210 was reversiblebecause a 24 h-pretreated cell population transferred to HDACinhibitor-freemedium for the next 24 h started to proliferate with amarked increase in the S phase (62%). The addition of ATRA to24 h HDAC inhibitor-pretreated cells decreased the proportion inthe S phase to 32% and increased that in the G0/G1 to 55%; therewas no additive effect on cell accumulation in the G1 phase. Thismight be caused by a decrease in a viable cell number, and/or byan increase in apoptotic cells during the first 24 h of HDACinhibitor treatment (Fig. 4A). Thus, BML-210 and ATRAcombination increased growth inhibitory effect, which could beattributed to an arrest in the transition through the cell cycle.
3.4. Induction of apoptosis by BML-210 in leukemia cell lines
BML-210 at a concentration up to 10 μM caused cytotoxiceffects on different leukemia cells, as previously shown in
Fig. 4. Effects of BML-210 on cell cycle progression of leukemia cell lines. (A) NB4cells were treated with 5 μM BML-210 alone for 24 h, or 24 h pretreated cells weremedium with 1 μMATRA (BML pretr-ATRA) for another 24 h, or cotreated (BML+Afrequency distribution histograms of PI stained cells. The marked intervals between thone representative experiment of three where standard deviations (not shown) were
Fig. 1. As shown in Fig. 5A, C, 10 μM BML-210 caused THP-1, NB4 and HL-60 cell death, the increase in proportion ofcells in the subG1 phase at 24 h and double augmentationat 48 h. The extent of induced cell death caused by 10 μMBML-210 alone at 24–48 h varied among cell lines tested(Fig. 5A). BML-210 did not increase K562 cell apoptosis at48 h time-point, and the appearance of a higher number of deadcells was delayed till day 6 as shown in Fig. 1 and Fig. 5A.BML-210 combination with inducers of differentiation (ATRAor hemin) caused additive cytotoxic effect (Fig. 5A). Flowcytometric analysis of PI stained cells following NB4 celltreatment with BML-210 demonstrated a dose-dependent in-duction of apoptosis at 48 h, reaching 58% at a dose of 20 μM(Fig. 5C).
Thus, based on comparable results obtained by Trypan blueexclusion, Acridine orange/Ethidium bromide staining (data notshown) and flow cytometry, we conclude that BML-210induces cytotoxicity through the pathway of apoptosis.
3.5. Histone H4 acetylation in response to BML-210
The two HDAC inhibitors, phenyl butyrate and BML-210,were further examined for their influence on histone H4
, THP-1 and K562 cells were treated with 10 μM BML-210 for 48 h. (B) HL-60transferred to HDACI-free medium (BML pretr-medium) or maintained in theTRA) for 24 h. The cell cycle phase distribution (%) was determined from DNAe peaks show the DNA content at the G0/1, S and G2+M-phase. Results are fromless than±10%.
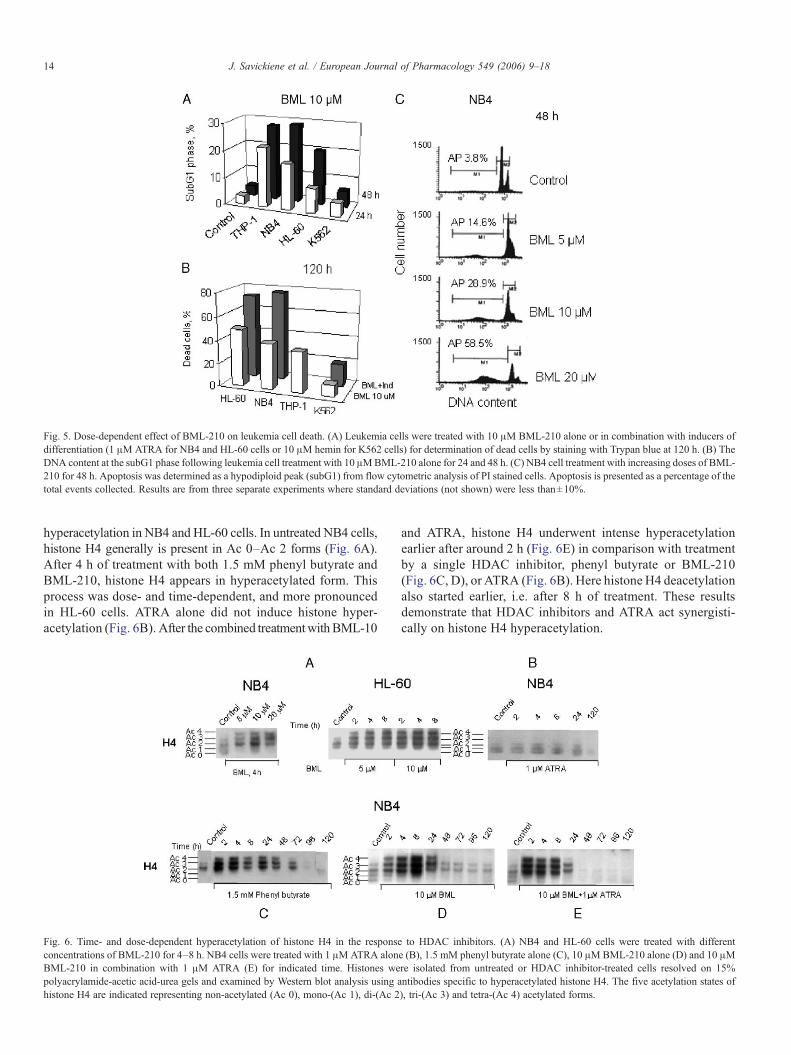
Fig. 5. Dose-dependent effect of BML-210 on leukemia cell death. (A) Leukemia cells were treated with 10 μM BML-210 alone or in combination with inducers ofdifferentiation (1 μM ATRA for NB4 and HL-60 cells or 10 μM hemin for K562 cells) for determination of dead cells by staining with Trypan blue at 120 h. (B) TheDNA content at the subG1 phase following leukemia cell treatment with 10 μMBML-210 alone for 24 and 48 h. (C) NB4 cell treatment with increasing doses of BML-210 for 48 h. Apoptosis was determined as a hypodiploid peak (subG1) from flow cytometric analysis of PI stained cells. Apoptosis is presented as a percentage of thetotal events collected. Results are from three separate experiments where standard deviations (not shown) were less than±10%.
14 J. Savickiene et al. / European Journal of Pharmacology 549 (2006) 9–18
hyperacetylation in NB4 and HL-60 cells. In untreated NB4 cells,histone H4 generally is present in Ac 0–Ac 2 forms (Fig. 6A).After 4 h of treatment with both 1.5 mM phenyl butyrate andBML-210, histone H4 appears in hyperacetylated form. Thisprocess was dose- and time-dependent, and more pronouncedin HL-60 cells. ATRA alone did not induce histone hyper-acetylation (Fig. 6B). After the combined treatmentwithBML-10
Fig. 6. Time- and dose-dependent hyperacetylation of histone H4 in the responseconcentrations of BML-210 for 4–8 h. NB4 cells were treated with 1 μMATRA alonBML-210 in combination with 1 μM ATRA (E) for indicated time. Histones wepolyacrylamide-acetic acid-urea gels and examined by Western blot analysis using ahistone H4 are indicated representing non-acetylated (Ac 0), mono-(Ac 1), di-(Ac 2
and ATRA, histone H4 underwent intense hyperacetylationearlier after around 2 h (Fig. 6E) in comparison with treatmentby a single HDAC inhibitor, phenyl butyrate or BML-210(Fig. 6C, D), or ATRA (Fig. 6B). Here histone H4 deacetylationalso started earlier, i.e. after 8 h of treatment. These resultsdemonstrate that HDAC inhibitors and ATRA act synergisti-cally on histone H4 hyperacetylation.
to HDAC inhibitors. (A) NB4 and HL-60 cells were treated with differente (B), 1.5 mM phenyl butyrate alone (C), 10 μMBML-210 alone (D) and 10 μMre isolated from untreated or HDAC inhibitor-treated cells resolved on 15%ntibodies specific to hyperacetylated histone H4. The five acetylation states of), tri-(Ac 3) and tetra-(Ac 4) acetylated forms.

Fig. 7. Sp1, p53, PU.1 and NFκ-B binding to the promoters of myeloid origin.EMSA was performed using nuclear extracts from 8 h BML-210-treated NB4cells and a labeled oligonucleotide representing: (A) Sp1 consensus motif, (B)Sp1 binding sites from the p21 promoter-element 3, (C) the FasL promoter, (D)the NF-κB component-p65, promoter, (E) NF-κB consensus motif, (F) NF-κBbinding site from the FasL promoter, (G) PU.1 binding site from the HNEpromoter, (H) p53 binding site from the p21 promoter. Sp1 and NF-κB bindingwas eliminated competitively by addition of a 50–100 fold molar excess ofunlabeled competitor (Cold) or using a probe containing mutated binding site(Mut) from the target promoter. Arrows indicate DNA complexes with Sp1, NF-κB, PU.1 and p53 proteins.
15J. Savickiene et al. / European Journal of Pharmacology 549 (2006) 9–18
3.6. Modulation of transcription factors binding activity byBML-210
To study the binding activity of transcription factors Sp1,NF-κB, p53 and PU.1, we performed EMSA with radiolabeledoligonucleotides and nuclear extracts from untreated NB4 cells,and cells treated with BML-210 alone or in combination withATRA. Previously, we showed a specific Sp1 binding to theconsensus sequence and the binding site 3 (Sp1-3) of the p21promoter, p53 binding affinity to the p21 promoter or NF-κB tothe consensus sequence and to the FasL promoter, or PU.1 to thehuman neutrophil elastase (HNE) promoter (Fig. 7). Thebinding of those transcription factors to the promoters wasefficiently competed by a 50 or 100 M excess of the unlabeledoligonucleotide (cold), or by a mutated oligonucleotide.
Fig. 8. Transcription factors binding activity in response to BML-210 alone or in com10 μMBML-210 for 2–8 h. (B) NB4 and HL-60 cells were treated with 10 μMBMLwith 1 μM ATRA during 4 days. EMSAwas performed using a total 10 μg protein findicated promoters. Arrows indicate DNA complexes with Sp1, NF-κB, PU.1 and
After that we investigated whether treatment with differentHDAC inhibitors, BML-210 and phenyl butyrate, affected thebinding activity of transcription factors in NB4 and HL-60 cells.By EMSA, both HDAC inhibitors induced NF-κB or Sp1binding to their consensus sequences and Sp1 to the p65 pro-moter, or p53 to the p21 promoter during the first 8 h (Fig. 8A).Fig. 8B demonstrates different NF-κB binding activity toconsensus sequence in HL-60 and NB4 cells during 4 days oftreatment with 10 μM BML-210. This was associated withdifferentiation induction in HL-60 cells only leading toenhanced complex formation the first two days. NF-κB bindingto the FasL promoter increased in both cell lines. In NB4 cells,binding activity of Sp1 to the FasL or Sp1-3 to the p21 promoterwas pronounced on days 3-4. In HL-60 cells, we did not observeSp1 binding, because the Sp1 protein exists in a truncated form,which is not able to bind to the GC-rich elements in DNA (Raoet al., 1999; Savickiene et al., 2005).
Combined treatment with BML-210 and ATRA causedelevated binding capacity of many transcription factors (NF-κB,PU.1, Sp1–3, p53) to the promoters tested during first two days andsubsequent decrease on day 3–4 (Fig. 8C). In contrast, Sp1 bindingto the FasL promoter markedly increased on day 3–4, which islikely associated with the induction of apoptosis at this time-point.
3.7. Distribution of some regulatory proteins in the cytoplasmand the nucleus of NB4 cells during treatment with BML-210and ATRA alone or in combination
The total cytoplasmic and nuclear levels of p21, p53, Sp1,FasL and NF-κB components, i.e. p50 and p65, were estimatedby Western blot (Fig. 9). ATRA (1 μM) treatment for 96 h hadno influence on either the cytoplasmic or the nuclear levels ofp21 and p53 expression, but increased the cytoplasmic level ofp50, p65 and FasL. At the same time Sp1 protein expressionslightly decreased in the cytoplasm. 5 μM BML-210 decreased
bination with ATRA. (A) NB4 cells were treated with 1.5 mM phenyl butyrate or-210 for 4 days. (C) NB4 cells were treated with 5 μMBML-210 in combinationrom each nuclear extract and oligonucleotides containing binding sites from thep53 proteins.

Fig. 9. Expression of cytoplasmic and nuclear proteins in response to BML-210. NB4 cells were treated with 10 μM BML-210 or with 1 μM ATRA alone or in itscombination for indicated time-points. Cytosolic and nuclear proteins were fractionated on 7–15% SDS-PAGE and analyzed by Western blotting with antibodiesagainst p21, p53, Sp1, NF-κB and FasL.
16 J. Savickiene et al. / European Journal of Pharmacology 549 (2006) 9–18
both the cytoplasmic and the nuclear levels of p21, Sp1, p50 andp65 and only p53 cytoplasmic level after 24 h of treatment.Expression of FasL in the nucleus only slightly increased during96 h of HDAC inhibitor treatment. Cotreatment with BML-210and ATRA for 24–96 h had no significant changes in thecytoplasmic level of transcription factors examined, butdecreased expression of p21, Sp1 and p65 in the nucleusduring cell maturation. These results are in agreement with thedata obtained in EMSA and demonstrate that investigatedregulatory proteins are involved in the processes caused byBML-210 alone or together with differentiation inducer.
4. Discussion
BML-210 is a novel histone deacetylase inhibitorwith putativeeffects on leukemia cell growth, differentiation and apoptosis.Wefound here that BML-210 is comparable with a well knownHDAC inhibitor, phenyl butyrate, in its ability to cause cytotoxiceffects dependent on exposure time and dose. The growth of fourdifferent leukemia cell lines (NB4, HL-60, K562, and THP-1)was inhibited and the apoptotic process was promoted by BML-210. These cellular responses were associated with the increasedaccumulation of the cells in G1 and subG1 phases of the cell cycle(Fig. 4). It is known that inhibitor of cyclin-dependent kinases,p21 (Waf1/Cip1), plays an important role in G1 cell cycle arrest(Harper et al., 1993). It has also been reported that HDACinhibitors, like sodium butyrate, FK228, trichostatin A (TSA),MS-27-275, induce the expression of p21 (Kosugi et al., 1999;Richon et al., 2000; Sasakawa et al., 2002). Our resultsdemonstrate clearly that BML-210 caused p21 expression duringthe first 24 h-treatment, probably via acetylation of histone H3and H4 in the p21 promoter region (Kwon et al., 2002). Thiscoincides with transcription activation through Sp1 sites of thep21 promoter, which contains six Sp1 binding sites and plays amajor role in the regulation of p21 transcription (Gartel and Tyner,1999; Sowa et al., 1997). Cell arrest in the G1 was necessary for
the induction of apoptosis by higher doses of BML-210. ManyHDAC inhibitors cause cell growth arrest and induction ofdifferentiation or apoptosis, although the mechanisms of theinduction of apoptosis are cell type (Bernhard et al., 1999;Bernhard et al., 2001) or HDAC inhibitor-specific (Bernhardet al., 2001; Kwon et al., 2002; Peart et al., 2003). Our data showthat BML-210 alone or in combination with the differentiationinducer ATRA markedly increased Sp1 binding to the FasLpromoter during 4 days of treatment in both HL-60 and NB4 cells(Fig. 8). These results are consistent with an increased number ofapoptotic cells and up-regulation of FasL expression in thecytoplasm (Fig. 9) supporting the data about the involvement ofFas/FasL apoptotic pathway (Bernhard et al., 2001; Glick et al.,1999; Kwon et al., 2002).
We found that BML-210 alone induces differentiation in K562and HL-60 cell lines. We detected the early myeloid differenti-ation marker CD11b expression and NBT-reduction in HL-60 butnot NB4 cells. This difference was associated with a morepronounced histone acetylation in HL-60 cells (Fig. 6) or withdirect/indirect alterations of the transcriptional profile (Fig. 8).However, BML-210 accelerated and markedly enhanced HL-60and NB4 cell granulocytic or K562 cell erythroid differentiation,when used in combination with inducers of differentiation. Thedata is supported by our morphological and immunochemicalobservations and increased PU.1 binding to the HNE promoter(Fig. 8); the expression of the latter is normally up-regulatedduring the early phases of granulocytic/monocytic differentiation(Scott et al., 1994). This biological effect was accompanied byhistone H4 hyperacetylation and more rapid appearance of thismodification during 24 h of cotreatment with BML-210 andATRA (Fig. 6). It is known that histone modifications play acrucial role in recruitment and the function of pre-initiationcomplex components on promoters during the activation ofcertain genes (Fischle et al., 2003; Kouzarides, 2002, 2003). Infact, we have demonstrated that BML-210 alone and incombination with ATRA affects the binding activity of

17J. Savickiene et al. / European Journal of Pharmacology 549 (2006) 9–18
transcription factors NF-κB, p53, Sp1 to the p21, FasL and othersmyeloid promoters with the following changes in the expressionlevels of several regulatory proteins.
NF-κB represents a family of inducible transcription factorsparticipating in the regulation of various cellular processes, suchas cell growth, differentiation, apoptosis, inflammation andimmunity (Glosh, 1999). NF-κB-mediated gene induction wasshown to be enhanced by HDAC inhibitors, like TSA or sodiumbutyrate (Adam et al., 2003; Chen and Greene, 2003; Quivy andVan Lint, 2004). The most abundant form of NF-κB is a p50/p65heterodimer, in which p65 contains the transcriptional activationdomain. Both the subunits are acetylated upon activation withinvolvement of p300/CBP (CREB binding protein) (Chen andGreene, 2003; Gerritsen et al., 1997). p65 binds to CBP and itshomolog p300 (Roth et al., 2001), while p50 fails to recruit thetranscriptional co-activators. This resulted in transcriptionalactivation, presumably via associated histone-directed acetylaseactivity, which includes localized chromatin (Kiernan et al., 2003).Our results demonstrate that BML-210 enhances NF-κB bindingto its consensus sequence during the first hours of treatment. Thisearly effect was most pronounced in HL-60 cells and coincidedwith the induction of cell differentiation. In NB4 cells, NF-κBactivity on days 3–4 represents the survival of proliferating cells(Fig. 8B). During apoptosis, the decrease in NF-κB bindingactivity to consensus site and to the FasL promoter on day 4 ran inparallel during our experiments with BML-210 alone or incombination with ATRA (Fig. 8). Alternatively, it has beendemonstrated that an important functional role of NF-κB in p21induction, G1 arrest and cell maturation by HDAC inhibitor,sodium butyrate, and the disruption of NF-κB pathway caused thecells to engage an alternative, apoptotic program (Dai et al., 2003).
Sp1 sites are often found in NF-κB-regulated genes and this canprovide a number of combinatorial regulations. The interaction ofSp1 with DNA may raise the expression level of NF-κB targetgenes even in the absence of activated NF-κB (Hirano et al., 1998).On the other hand, we detected elevated Sp1 binding activity to thep65 promoter and NF-κB to the consensus sequence during 2–4 hof BML-210 treatment. This correlated temporally with theincreased p65 protein level in NB4 cell nucleus, which couldaccount for the transcriptional activation ofNF-κB-activable genes.There are two classes of such genes: those containing constitutivelyand immediately accessible NF-κB binding sites in their promoterand those that need the hyperacetylation to become accesible toNF-κB (Saccani et al., 2001). HDAC inhibitors would thus increaseNF-κB-dependent transcription by blocking the HDAC activityassociated with p65, thereby resulting in histone hyperacetylationand subsequently a higher level of gene expression.
p53 protein conformation for DNA binding to the promotersmay play a role in modulating the rate of its acetylation andfurther recruitment of chromatin remodeling factors (Dornan etal., 2003; Vogelstein et al., 2000). Recently, Lagger et al. (2003)have indeed demonstrated that p53 cooperates with Sp1 in theactivation of the p21 promoter. It is known that HDACinhibitors, such as sodium butyrate, TSA, apicidin, SAHA andothers, are able to induce p21 (Calonghi et al., 2005; Joseph etal., 2005). Our results show elevated binding activity of bothSp1–3 and p53 to the p21 promoter during 24 h-exposure to
BML-210 alone or together with ATRA in NB4 cells. Aftersuch treatment, p21 protein level increased in both thecytoplasm and the nucleus immediately.
Taken together, our results demonstrate that the novel HDACinhibitor BML-210 could be a promising agent for leukemiatherapy to induce apoptosis and to modulate differentiation viahistone acetylation and gene expression.
Acknowledgements
This research was supported by the Swedish Institute (VisbyProgram No. 01361/2006), the Swedish Research Council,Lithuanian State Science and Studies Foundation (No.V-73/2006 and No.T-37/06) and EU Marie Curie Programme for V.-V. Borutinskaite. Authors are grateful to Dr. Almantas Šiaurysfor technical assistance in flow cytometric analysis.
References
Adam, E., Quivy, V., Bex, F., Chariot, A., Collette, Y., Vanhulle, C.,Schoonbroodt, S., Goffin, V., Nguyen, T.L., Gloire, G., Carrard, G., Friguet,B., De Launoit, Y., Burny, A., Bours, V., Piette, J., Van Lint, C., 2003.Potentiation of tumor necrosis factor-induced NF-kappa B activation bydeacetylase inhibitors is associated with a delayed cytoplasmic reappearanceof I kappa B alpha. Mol. Cell. Biol. 23, 6200–6209.
Bernhard, D., Ausserlechner, M.J., Tonko, M., Loffler, M., Hartmann, B.L.,Csordas, A., Kofler, R., 1999. Apoptosis induced by the histone deacetylaseinhibitor sodium butyrate in human leukemic lymphoblasts. FASEB J. 13,1991–2001.
Bernhard, D., Skvortsov, S., Tinhofer, I., Hubl, H., Greil, R., Csordas, A.,Kofler, R., 2001. Inhibition of histone deacetylase activity enhances Fasreceptor-mediated apoptosis in leukemic lymphoblasts. Cell Death Differ. 8,1014–1021.
Calonghi, N., Cappadone, C., Pagnotta, E., Boga, C., Bertucci, C., Fiori, J.,Tasco, G., Casadio, R., Masotti, L., 2005. Histone deacetylase 1: a target of9-hydroxystearic acid in the inhibition of cell growth in human colon cancer.J. Lipid Res. 46, 1596–1603.
Collins, S., 1987. The HL-60 promyelocytic leukemia cell line: proliferation,differentiation and cellular oncogene expression. Blood 70, 1233–1244.
Chambers, A.E., Banerjee, S., Chaplin, T., Dunne, J., Debernardi, S., Joel, S.P.,Young, B.D., 2003. Histone acetylation-mediated regulation of genes inleukaemic cells. Eur. J. Cancer 39, 1165–1175.
Chen, L.F., Greene, W.C., 2003. Regulation of distinct biological activities ofthe NF-kappa B transcription factor complex by acetylation. J. Mol. Med.81, 549–557.
Dai, Y., Rahmani, M., Grant, S., 2003. An intact NF-kappa B pathway isrequired for histone deacetylase inhibitor-induced G1 arrest and maturationin U937 human myeloid leukemia cells. Cell Cycle 2, 462–472.
Dornan, D., Shimizu, H., Burch, L., Smith, A.J., Hupp, T.R., 2003. The prolinerepeat domain of p53 binds directly to the transcriptional coactivator p300and allosterically controls DNA-dependent acetylation of p53. Mol. Cell.Biol. 23, 8846–8861.
Fischle, W., Wang, Y., Allis, C.D., 2003. Histone and chromatin cross-talk. Curr.Opin. Cell. Biol. 15, 172–183.
Gartel, A.L., Tyner, A.L., 1999. Transcription regulation of the p21 (WAF1/CIP1)
gene. Exp. Cell Res. 246, 280–289.Gerritsen, M.E., Williams, A.J., Neish, A.S., Moore, S., Shi, Y., Collins, T.,
1997. CREB-binding protein/p300 are transcriptional coactivators of p65.Proc. Natl. Acad. Sci. U. S. A. 94, 2927–2932.
Glick, R.D., Swendeman, A.L., Coffey, D.C., Rifkind, R.A., Marks, P.A.,Richon, V.M., La Quaglia, M.P., 1999. Hybrid polar histone deacetylaseinhibitor induces apoptosis and CD95/CD95 ligand expression in humanneuroblastoma. Cancer Res. 59, 4392–4399.
Glosh, S., 1999. Regulation of inducible gene expression by the transcriptionfactor NF-κB. Immunol. Res. 19, 183–189.

18 J. Savickiene et al. / European Journal of Pharmacology 549 (2006) 9–18
Gozzini, A., Rovida, E., Sbarba, P., Galimbert, S., Santini, V., 2003. Butyrates,as a single drug, induce histone acetylation and granulocytic maturation:possible selectivity on core binding factor— acute myeloid leukemia blasts.Cancer Res. 63, 8955–8961.
Grignani, F., Gelmetti, V., Fanelli, M., Rogaia, D., De Matteis, S., Ferrara, F.F.,Bonci, D., Grignani, F., Nervi, C., Pelicci, P.G., 1999. Formation of PML/RAR alpha high molecular weight nuclear complexes through the PMLcoiled-coil region is essential for the PML/RAR alpha-mediated retinoic acidresponse. Oncogene 18, 6313–6321.
Harper, J.W., Adami, G.R., Wei, N., Keymarsi, K., Elledge, S.J., 1993. The p21Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependentkinases. Cell 75, 806–816.
Hirano, F., Tanaka, H., Hirano, Y., Hiramoto, M., Handa, H., Makino, I.,Schneidereit, C., 1998. Functional interference of Sp1 and NF-kappa Bthrough the same DNA binding site. Mol. Cell. Biol. 18, 1266–1274.
Hurley, C.K., 1977. Electrophoresis of histones: a modified Panyim andChalkley system for slab gels. Anal. Biochem. 80, 624–626.
Jeannesson, P., Ginot, L., Manfait, M., Jardillier, J.C., 1984. Induction ofhemoglobin synthesis in human leukemic K 562 cells by adriamycin.Anticancer Res. 4, 47–51.
Joseph, J., Wajapeyee, N., Somasundaram, K., 2005. Role of p53 status inchemosensitivity determination of cancer cells against histone deacetylaseinhibitor sodium butyrate. Int. J. Cancer 115, 11–18.
Kelly, W.K., Richon, V.M., O‘Conor, O., Curley, T., MacGregore-Curtelli, B.,Tong, W., Klang, M., Schwartz, L., Richardson, S., Rosa, E., Drobnjak, M.,Cordon-Cordo, C., Chiao, J.H., Rifkind, R., Marks, P.A., Scher, H., 2003.Phase I clinical trial of histone deacetylase inhibitor: suberoylanilidehydroxamic acid administered intravenously. Clin. Cancer Res. 9, 3578–3588.
Kiernan, R., Bres, V., Ng, R.W., Coudart, M.P., El Messaoudi, S., Sardet, C., Jin,D.Y., Emiliani, S., Benkirane,M., 2003. Post-activation turn-off of NF-kappaB-dependent transcription is regulated by acetylation of p65. J. Biol. Chem.278, 2758–2766.
Kosugi, H., Towatari, M., Hatano, S., Kitamura, K., Kiyoi, H., Kinoshita, T.,Tanimoto, M., Murate, T., Kawashima, K., Saito, H., Naoe, T., 1999.Histone deacetylase inhibitors are potent inducer/enhancer of differentiationin acute myeloid leukemia: a new approach to anti-leukemia therapy.Leukemia 13, 1316–1324.
Kouzarides, T., 2002. Histone methylation in transcriptional control. Curr. Opin.Genet. Dev. 12, 198–209.
Kouzarides, T., 2003. Wellcome Trust Award Lecture. Chromatin-modifyingenzymes in transcription and cancer. Biochem. Soc. Trans. 31, 741–743.
Kwon, S.H., Ahn, S.H., Kim, Y.K., Bae, G.U., Yoon, J.W., Hong, S., Lee, H.Y.,Lee, Y.W., Han, J.W., 2002. Apicidin, a histone deacetylase inhibitor,induces apoptosis and Fas/Fas ligand expression in human acutepromyelocytic leukemia cells. J. Biol. Chem. 277, 2073–2080.
Lagger, G., Doetzlhofer, A., Schuettengruber, B., Haidweger, E., Simboeck, E.,Tischler, J., Chiocca, S., Suske, G., Rotheneder, H., Wintersberger, E.,Seiser, C., 2003. The tumor suppressor p53 and histone deacetylase 1 areantagonistic regulators of the cyclin-dependent kinase inhibitor p21/WAF1/CIP1 gene. Mol. Cell. Biol. 23, 2669–2679.
Lin, R.J., Egan, D.A., Evans, R.M., 1999. Molecular genetics of acutepromyelocytic leukemia. Trends Genet. 15, 197–202.
Marks, P.A., Richon, V.M., Breslow, R., Rifkind, R.A., 2001a. Histonedeacetylase inhibitors as new cancer drugs. Curr. Opin. Oncol. 13, 477–483.
Marks, P.A., Rifkind, R.A., Richon, V.M., Breslow, R., Miller, T., Kelly, W.K.,2001b. Histone deacetylases and cancer: causes and therapies. Nat. Rev.Cancer 1, 194–202.
Minucci, S., Nervi, C., Lo Coco, F., Pelicci, P.G., 2001. Histone deacetylase: acommon molecular target for differentiation treatment of acute myeloidleukemias. Oncogene 20, 3110–3115.
Newmark, H.L., Lupton, J.R., Young, C.W., 1994. Butyrate as a differentiatingagent: pharmacokinetics, analogues and current status. Cancer Lett. 78, 1–5.
Quivy, V., Van Lint, C., 2004. Regulation at multiple levels of NF-κB-mediatedtransactivation by protein acetylation. Biochem. Pharmacol. 68, 1221–1229.
Peart,M.J., Tainton,K.M., Ruefli, A.A., Dear, A.E., Sedelies,K.A., O’Reilly, L.A.,Waterhouse, N.J., Trapani, J.A., Johnstone, R.W., 2003. Novel mechanisms ofapoptosis induced by histone deacetylase inhibitors. Cancer Res. 63,4460–4471.
Redner, R.L., Wang, J., Liu, J.M., 1999. Chromatin remodeling and leukemia:new therapeutic paradigms. Blood 94, 793–802.
Rao, J., Zhang, F., Donnelly, R.J., Spector, N.L., Studzinski, G.P., 1999.Truncation of Sp1 transcription factor by myeloblastin in undifferentiatedHL-60 cells. J. Cell. Physiol. 175, 121–128.
Richon, V.M., Emiliani, S., Verdin, E., Webb, Y., Breslow, R., Rifkind, R.A.,Marks, P.A., 1998. A class of hybrid polar inducers of transformed celldifferentiation inhibits histone deacetylases. Proc. Natl. Acad. Sci. U. S. A. 95,3003–3007.
Richon, V.M., Sandhoff, T.W., Rifkind, R.A., Marks, P.A., 2000. Histonedeacetylase inhibitor selectivity induces p21 WAF1 expression and generatedhistone acetylation. Proc. Natl. Acad. Sci. U. S. A. 97, 10014–10169.
Rivero, J.A., Adunyah, S.E., 1998. Sodium butyrate stimulates PKC activationand induces differential expression of certain PKC isoforms during erythroiddifferentiation. Biochem. Biophys. Res. Commun. 248, 664–668.
Roth, S.Y., Denu, J.M., Allis, C.D., 2001. Histone acetyltransferase. Annu. Rev.Biochem. 70, 81–120.
Saccani, S., Pantano, S., Natoli, G., 2001. Two waves of nuclear factor κBrecruitment to target promoters. J. Exp. Med. 193, 1351–1359.
Sasakawa, Y., Naoe, Y., Inoue, T., Sasakawa, T., Matsuo, M., Manda, T., Mutoh,S., 2002. Effects of FK228, a novel histone deacetylase inhibitor, on humanlymphoma U-937 cells in vitro and in vivo. Biochem. Pharmacol. 64,1079–1090.
Savickiene, J., Treigyte, G., Magnusson, K.-E., Navakauskiene, R., 2005. p21(Waf1/Cip1) and FasL gene activation via Sp1 and NFκB is required forleukemia cell survival but not for cell death induced by diverse stimuli. Int.J. Biochem. Cell Biol. 37, 784–796.
Scott, E.W., Simon, M.C., Anastasi, J., Singh, H., 1994. Requirement oftranscription factor PU.1 in the development of multiple haematopoieticlineages. Science 265, 1573–1577.
Sowa, Y., Orita, T., Minamikawa, S., Nakano, K., Mizuno, T., Nomura, H., Sakai,T., 1997.Histone deacetylase inhibitor activates theWAF1/Cip1gene promoterthrough the Sp1 sites. Biochem. Biophys. Res. Commun. 241, 142–150.
Strahl, B.D., Allis, C.D., 2000. The language of covalent histone modifications.Nature 403, 41–45.
Treigyte, G., Gineitis, A., 1979. Specific changes in the biosynthesis andacetylation of nucleosomal histones in the early stages of embryogenesis ofsea urchin. Exp. Cell Res. 121, 127–134.
Vigushin, D.M., Combers, R.C., 2002. Histone deacetylase inhibitors in cancertreatment. Anticancer Drugs 1, 1–13.
Vogelstein, B., Lane, D., Levine, A.J., 2000. Surfing the p53 network. Nature408, 307–310.
Wolffe, A.P., Wong, J., Pruss, D., 1997. Activators and repressors: making useof chromatin to regulate transcription. Genes Cells 2, 291–302.
Vrana, J.A., Decker, R.H., Johnson, C.R., Wang, Z., Jarvis, W.D., Richon, V.M.,Eginger, M., Fisher, P.B., Grant, S., 1999. Induction of apoptosis in U937human leukemia cells by suberoylanilide hydroxamic acid (SAHA)proceeds through pathways that are regulated by Bcl-2/Bcl-XL, c-Jun, andindependent of p53. Oncogene 18, 7016–7025.
Yoshida,M., Furumai, R., Nishiyama,M.,Komatsu,Y.,Nishino,N.,Horinouchi, S.,2001. Histone deacetylase as a new target for cancer chemotherapy. CancerChemother. Pharmacol. Suppl: 1, S20–S26.
Yoshida, M., Shimazu, T., Matuyama, A., 2003. Protein deacetylases: enzymeswith functional diversity as novel therapeutic targets. Prog. Cell Cycle Res.5, 269–278.




















