
SIRT1 is a redox-sensitive deacetylase that is post-translationally modified by oxidants and...
15
The FASEB Journal • Research Communication SIRT1 is a redox-sensitive deacetylase that is post-translationally modified by oxidants and carbonyl stress Samuel Caito,* Saravanan Rajendrasozhan,* Suzanne Cook,* Sangwoon Chung,* Hongwei Yao,* Alan E. Friedman,* Paul S. Brookes, † and Irfan Rahman* ,1 *Department of Environmental Medicine, Lung Biology and Disease Program; and † Department of Anesthesiology, University of Rochester Medical Center, Rochester, New York, USA ABSTRACT Sirtuin1 (SIRT1) deacetylase levels are decreased in chronic inflammatory conditions and ag- ing where oxidative stress occurs. We determined the mechanism of SIRT1 redox post-translational modifi- cations leading to its degradation. Human lung epithe- lial cells exposed to hydrogen peroxide (150 –250 M), aldehyde-acrolein (10 –30 M), and cigarette smoke extract (CSE; 0.1–1.5%) in the presence of intracellular glutathione-modulating agents at 1–24 h, and oxidative post-translational modifications were assayed in cells, as well as in lungs of mice lacking and overexpressing glutaredoxin-1 (Glrx1), and wild-type (WT) mice in response to cigarette smoke (CS). CSE and aldehydes dose and time dependently decreased SIRT1 protein levels, with EC 50 of 1% for CSE and 30 M for acrolein at 6 h, and >80% inhibition at 24 h with CSE, which was regulated by modulation of intracellular thiol status of the cells. CS decreased the lung levels of SIRT1 in WT mice, which was enhanced by deficiency of Glrx1 and prevented by overexpression of Glrx1. Oxidants, alde- hydes, and CS induced carbonyl modifications on SIRT1 on cysteine residues concomitant with decreased SIRT1 activity. Proteomics studies revealed alkylation of cysteine residue on SIRT1. Our data suggest that oxidants/aldehydes covalently modify SIRT1, decreas- ing enzymatic activity and marking the protein for proteasomal degradation, which has implications in inflammatory conditions.—Caito, S., Rajendrasozhan, S., Cook, S., Chung, S., Yao, H., Friedman, A. E., Brookes, P. S., Rahman, I. SIRT1 is a redox-sensitive deacetylase that is post-translationally modified by oxi- dants and carbonyl stress. FASEB J. 24, 3145–3159 (2010). www.fasebj.org Key Words: sirtuins aldehydes epithelium cigarette smoke inflammation Sirtuins, class III histone deacetylases (HDACs), seem to have evolved to respond to a variety of stresses and are emerging as key antiaging molecules and regulators in many diseases, including type II diabetes, cardiovascular disease, Alzheimer’s disease, and chronic obstructive pulmonary disease (COPD), as well as aging (1–7). SIRT1, which is homologous to yeast (Saccharo- myces cerevisiae) silent information regulator 2 (Sir2), is the most extensively studied sirtuin, has been shown to be the key molecule responsible for extending life span in response to caloric restriction (8) and is involved in silencing the chromatin of the mating locus (2, 9). SIRT1 can deacetylate histones, but it can also suppress inflammation through the deacetylation of the RelA/ p65 subunit of NF-B (10), suppress apoptosis through the deacetylation of p53, Ku70, and FOXO3a (3, 11, 12), and inhibit senescence through the deacetylation of FOXO3a, FOXO4, and p53 (2, 13), which are hallmarks of several chronic inflammatory diseases. Oxidative stress has been shown to contribute to a variety of chronic inflammatory diseases (14, 15). Dur- ing oxidative stress, reversible and irreversible modifi- cations on proteins are formed as a result of direct oxidation of amino acid side chains or addition of reactive intermediates from the oxidation of other cellular components on cysteine, histidine, and lysine residues. Modifications, such as formation of disulfide bonds, S-nitrosylation, and S-glutathionylation can eas- ily be reversed by thioredoxin and glutaredoxin (Glrx) enzymes (16). However, irreversible covalent modifica- tions, such as carbonylation and tyrosine nitration, can lead to loss of protein function, protein aggregation folding, or degradation (16). The biochemical regulation of SIRT1 seems to be complex (17). SIRT1 has been shown to reduce cellular oxidative stress burden through deacetylation of FOXO3a, leading to an up-regulation of catalase and manganese superoxide dismutase (SOD) (1) SIRT1 also regulates aging and oxidative stress in the heart (18), and oxidative stress leads to a redistribution of SIRT1 on chromatin (19). We have recently shown that the levels of SIRT1 are decreased in vitro in macro- phages in response to cigarette smoke extract (CSE), as well as in lungs of patients with COPD (20, 21). 1 Correspondence: Department of Environmental Medicine, University of Rochester Medical Center, 601 Elmwood Ave., Box 850, Rochester, NY 14642, USA. E-mail: irfan_rahman@urmc. rochester.edu doi: 10.1096/fj.09-151308 3145 0892-6638/10/0024-3145 © FASEB
Transcript of SIRT1 is a redox-sensitive deacetylase that is post-translationally modified by oxidants and...

The FASEB Journal • Research Communication
SIRT1 is a redox-sensitive deacetylase that ispost-translationally modified by oxidants andcarbonyl stress
Samuel Caito,* Saravanan Rajendrasozhan,* Suzanne Cook,* Sangwoon Chung,*Hongwei Yao,* Alan E. Friedman,* Paul S. Brookes,† and Irfan Rahman*,1
*Department of Environmental Medicine, Lung Biology and Disease Program; and †Department ofAnesthesiology, University of Rochester Medical Center, Rochester, New York, USA
ABSTRACT Sirtuin1 (SIRT1) deacetylase levels aredecreased in chronic inflammatory conditions and ag-ing where oxidative stress occurs. We determined themechanism of SIRT1 redox post-translational modifi-cations leading to its degradation. Human lung epithe-lial cells exposed to hydrogen peroxide (150–250 �M),aldehyde-acrolein (10–30 �M), and cigarette smokeextract (CSE; 0.1–1.5%) in the presence of intracellularglutathione-modulating agents at 1–24 h, and oxidativepost-translational modifications were assayed in cells,as well as in lungs of mice lacking and overexpressingglutaredoxin-1 (Glrx1), and wild-type (WT) mice inresponse to cigarette smoke (CS). CSE and aldehydesdose and time dependently decreased SIRT1 proteinlevels, with EC50 of 1% for CSE and 30 �M for acroleinat 6 h, and >80% inhibition at 24 h with CSE, which wasregulated by modulation of intracellular thiol status ofthe cells. CS decreased the lung levels of SIRT1 in WTmice, which was enhanced by deficiency of Glrx1 andprevented by overexpression of Glrx1. Oxidants, alde-hydes, and CS induced carbonyl modifications onSIRT1 on cysteine residues concomitant with decreasedSIRT1 activity. Proteomics studies revealed alkylationof cysteine residue on SIRT1. Our data suggest thatoxidants/aldehydes covalently modify SIRT1, decreas-ing enzymatic activity and marking the protein forproteasomal degradation, which has implications ininflammatory conditions.—Caito, S., Rajendrasozhan,S., Cook, S., Chung, S., Yao, H., Friedman, A. E.,Brookes, P. S., Rahman, I. SIRT1 is a redox-sensitivedeacetylase that is post-translationally modified by oxi-dants and carbonyl stress. FASEB J. 24, 3145–3159(2010). www.fasebj.org
Key Words: sirtuins � aldehydes � epithelium � cigarette smoke� inflammation
Sirtuins, class III histone deacetylases (HDACs),seem to have evolved to respond to a variety of stressesand are emerging as key antiaging molecules andregulators in many diseases, including type II diabetes,cardiovascular disease, Alzheimer’s disease, and chronicobstructive pulmonary disease (COPD), as well as aging
(1–7). SIRT1, which is homologous to yeast (Saccharo-myces cerevisiae) silent information regulator 2 (Sir2), isthe most extensively studied sirtuin, has been shown tobe the key molecule responsible for extending life spanin response to caloric restriction (8) and is involved insilencing the chromatin of the mating locus (2, 9).SIRT1 can deacetylate histones, but it can also suppressinflammation through the deacetylation of the RelA/p65 subunit of NF-�B (10), suppress apoptosis throughthe deacetylation of p53, Ku70, and FOXO3a (3, 11,12), and inhibit senescence through the deacetylationof FOXO3a, FOXO4, and p53 (2, 13), which arehallmarks of several chronic inflammatory diseases.
Oxidative stress has been shown to contribute to avariety of chronic inflammatory diseases (14, 15). Dur-ing oxidative stress, reversible and irreversible modifi-cations on proteins are formed as a result of directoxidation of amino acid side chains or addition ofreactive intermediates from the oxidation of othercellular components on cysteine, histidine, and lysineresidues. Modifications, such as formation of disulfidebonds, S-nitrosylation, and S-glutathionylation can eas-ily be reversed by thioredoxin and glutaredoxin (Glrx)enzymes (16). However, irreversible covalent modifica-tions, such as carbonylation and tyrosine nitration, canlead to loss of protein function, protein aggregationfolding, or degradation (16).
The biochemical regulation of SIRT1 seems to becomplex (17). SIRT1 has been shown to reduce cellularoxidative stress burden through deacetylation ofFOXO3a, leading to an up-regulation of catalase andmanganese superoxide dismutase (SOD) (1) SIRT1also regulates aging and oxidative stress in the heart(18), and oxidative stress leads to a redistribution ofSIRT1 on chromatin (19). We have recently shown thatthe levels of SIRT1 are decreased in vitro in macro-phages in response to cigarette smoke extract (CSE), aswell as in lungs of patients with COPD (20, 21).
1 Correspondence: Department of Environmental Medicine,University of Rochester Medical Center, 601 Elmwood Ave., Box850, Rochester, NY 14642, USA. E-mail: [email protected]
doi: 10.1096/fj.09-151308
31450892-6638/10/0024-3145 © FASEB

However, the mechanism of this down-regulation byCSE and its reactive components (oxidants and alde-hydes) in lung cells is not known. On the basis of theabove observations, we hypothesized that cigarettesmoke (CS)-mediated oxidative stress decreases SIRT1protein level and activity by a redox-dependent mech-anism; causing irreversible oxidative post-translationalmodifications on SIRT1 leading to inactivation anddecreased protein level in lung epithelial cells in vitroand in mouse lungs in vivo. We tested this hypothesis byaltering the redox status of human bronchial andairway epithelial cells and in lungs of mice exposed toCS. We further studied the posttranslational modifica-tions of SIRT1 by carbonyls in lung epithelial cells, aswell as in mice-deficient and overexpressing Glrx1, akey enzyme, which reverses protein S-glutathionylationand is involved in maintaining the redox status ofproteins.
MATERIALS AND METHODS
Reagents
Unless otherwise stated, all biochemical reagents used in thisstudy were purchased from Sigma Chemicals (St. Louis, MO,USA). Antibodies used to detect SIRT1 include mouse-spe-cific SIRT1 (catalog no. 2028; Cell Signaling, Danvers, MA,USA), human SIRT1 (2310; Cell Signaling) for Western blotanalysis, and human SIRT1 (7343; Abcam, Cambridge, MA,USA) for immunocytochemistry and immunoprecipitation.
Mouse exposure
Adult male C57BL/6J mice (Jackson Laboratory, Bar Harbor,ME, USA) and Glrx1-knockout (Glrx1-KO) and Glrx1-trans-genic (Glrx1-Tg) mice overexpressing Glrx1 only in lungalveolar epithelial cells (obtained from Dr. Ye-Shih Ho,Wayne State University, Detroit, MI, USA; ref. 22) werehoused in the Inhalation Core Facility at the University ofRochester for 1 wk of acclimatization before CS exposure. Allanimal procedures were approved by the Committee onAnimal Research of the University of Rochester. Eight- to10-wk-old mice (6–8 mice/group) were used for CS exposurefor 3 d. Briefly, mice were housed in an individual wire cagecompartment, which was placed inside an aerated plastic box,which was connected to the smoke source. Research-gradecigarettes (2R4F; University of Kentucky, Lexington, KY; 9.7mg tar and 0.85 mg nicotine/cigarette) were used to generatesmoke, and mice were exposed according to the U.S. FederalTrade Commission protocol (1 puff/min of 2 s duration and35 ml volume) using an automatic cigarette-smoking machine(Baumgartner-Jaeger CSM2072i; CH Technologies, West-wood, NJ, USA). Mainstream CS was diluted with filtered airand smoke exposure [300 mg/m3 total particulate matter(TPM) of air in chamber corresponding to human consump-tion of 1–1.5 packs/d] was monitored in real time with anaerosol monitor (MicroDust Pro, Casella CEL, Bedford, UK),verified daily by gravimetric sampling (21, 23, 24). Chamberatmosphere was also monitored for TPM by adjustment of theflow rate of the diluted medical air (24). The exposureregimen consisted of two 1-h CS exposures 1 h apart for 3consecutive days, with the mice sacrificed 24 h after the lastexposure.
Protein extraction from lung tissue
Mice were injected with pentobarbiturate (100 mg/kg bodyweight, i.p.; Abbott Laboratories, Chevy Chase, MD, USA)and sacrificed by exsanguination. The lungs were removed enbloc, and the left lungs were lavaged 3 times with 0.5 ml of0.9% NaCl, while the right lung lobe was frozen for immu-noblot analysis. One hundred milligrams of right lung lobewas mechanically homogenized in 0.5 ml of ice-cold RIPAbuffer supplemented with a protease inhibitor cocktail (leu-peptin, aprotinin, pepstatin, and PMSF), and then placed onice for 45 min to allow for total cell lysis to occur. Thehomogenate was centrifuged at 13,000 rpm in a bench-topcentrifuge for 25 min at 4°C for removal of cellular debris.The supernatant was then transferred to a fresh 1.7-mlEppendorf tube and used as whole lysate.
Cell culture
The human bronchial epithelial cell line BEAS-2B was grownin DMEM-Ham’s F12 50:50 mixture (DMEM-F12; Mediatech,Manassas, VA, USA) supplemented with 5% FBS, 15 mMHEPES, 100 �g/ml penicillin, and 100 U/ml streptomycin.Human small airway epithelial cells (SAECs) derived from asingle healthy donor were purchased from Lonza (formerlyCambrex, Walkersville, MD, USA) along with growth medium[Small Airway Epithelial Cell Growth Medium (SAGM)]bullet kit supplemented with 52 �g bovine pituitary extract,0.5 ng/ml human recombinant epidermal growth factor(EGF), 0.5 �g/ml epinephrine, 10 �g/ml transferrin, 5�g/ml insulin, 0.1 ng/ml retinoic acid (RA), 6.5 ng/mltriiodothyronine, 50 �g/ml gentamicin/amphotericin-B (GA-1000), and 50 �g/ml fatty acid-free BSA. Cells were cultured at37°C in a humidified atmosphere containing 7.5% CO2.
Preparation of aqueous CSE
Ten percent CSE was prepared by bubbling smoke from one2R4F research-grade cigarette into 10 ml of culture mediumat a rate of 1 cigarette/2 min, as described previously (21,25–27), using a modification of the method described byCarp and Janoff (28). The CSE was adjusted to pH 7.4 and wassterile-filtered through a 0.45-�m filter (25-mm Acrodisc; PallCorporation, Ann Arbor, MI, USA). The CSE preparation wasstandardized by monitoring the absorbance at 320 nm(OD�0.75�0.05). The spectral variations observed betweendifferent CSE preparations at �320 were minimal. CSE wasfreshly prepared for each experiment and diluted with cul-ture medium containing 1% FBS immediately before use.Control medium was prepared by bubbling air through 10 mlof culture medium supplemented with 1% FBS, adjusting pHto 7.4, and sterile filtering as described for 10% CSE.
Treatments
BEAS-2B cells were seeded at a density of 1 � 106 cells/well(final volume 2 ml with medium) and grown to �80–90%confluency in 6-well plates containing DMEM-F12 mediumwith 1% FBS. The cells were treated with CSE (0.1–1.5%) orH2O2 (150, 250, or 500 �M) for 6 or 24 h in the presence orabsence of buthionine sulfoximine (BSO; 100 �M) pretreat-ment for 18 h or with N-acetyl-l-cysteine (NAC; 2 mM) for 2 hat 37°C with 7.5% CO2. Cells were also treated with polyeth-ylene glycol (PEG)-conjugated SOD (200 U/ml), catalase(200 U/ml), or a mixture of both (200 U/ml each) for 30min prior to treatment with CSE for 6 h. Cells were treated for1 or 6 h with KO2 (1 �M), a superoxide anion generator,prepared in dry dimethyl sulfoxide (DMSO), as described previ-
3146 Vol. 24 September 2010 CAITO ET AL.The FASEB Journal � www.fasebj.org

ously (29). At the end of treatment, the cells were washed withcold sterile Ca2�/Mg2�-free PBS and lysed using RIPA buffersupplemented with a protease inhibitor cocktail (leupeptin,aprotinin, pepstatin, and PMSF).
Immunoblot analysis
Protein levels were measured using the bicinchoninic acid kit(Pierce, Rockford, IL, USA). Linear regression was used todetermine the protein concentration of the samples. Twentymicrograms of BEAS-2B whole-cell extract, prepared as de-scribed above, was subjected to electrophoresis on 7.5%PAGE gels and transferred onto nitrocellulose membranes(Amersham, Arlington Heights, IL, USA). The nitrocellulosemembrane was blocked with 5% BSA and subsequently incu-bated with rabbit polyclonal anti-human SIRT1 antibody(1:1000 dilution; Cell Signaling). After 3 washing steps (10min each), the levels of protein were detected using goatanti-rabbit antibody (1:20,000 dilution) linked to horseradishperoxidase (Dako, Santa Barbara, CA, USA), and boundcomplexes were detected using enhanced chemilumines-cence method (ECL; Jackson Immunology Research, WestGrove, PA, USA).
Covalent modification of SIRT1 by carbonylation
SIRT1 was immunoprecipitated using whole-cell extracts;SIRT1 antibody (1:80 dilution; Abcam) was added to 100 �gof protein in a final volume of 400 �l and incubated for 1 h.Protein-A/G agarose beads (20 �l) (Santa Cruz Biotechnol-ogy, Santa Cruz, CA, USA) were added to each sample andleft overnight at 4°C on a rotator (Barnstead Thermolyne,Dubuque, IA, USA). The samples were then centrifuged at13,000 rpm at 4°C for 5 min. The supernatant was discarded,and the beads were washed 3 times and then resuspended in50 �l lysis buffer. For immunoblots, 100 �g of the immuno-precipitated SIRT1 agarose bead suspension was added to 5�sample loading buffer, boiled, and resolved using SDS-PAGEas described above. Agarose beads alone were used as nega-tive control. To determine the carbonylation of SIRT1, blotswere probed first with anti-SIRT1 antibody (Abcam). Afterstripping, membranes were equilibrated with 20% (v/v)methanol, 80% Tris-buffered saline for 5 min, then incubatedwith 0.5 mM 2,4-dinitrophenylhydrazine (DNP) for 30 min atroom temperature. The membranes were washed and thenincubated overnight in anti-DNP antibody, as described pre-viously (30).
Biotin-switch assay
Modifications on cysteine residues were measured by maleim-ide-PEO2-biotin labeling, as described previously (31). Afterimmunoprecipitation of SIRT1, SIRT1 conjugated to theagarose beads were diluted in PBS and 1 �l of maleimide-PEO2-biotin (Pierce) was added for 2 h rocking at roomtemperature in the dark. Samples were then boiled andelectrophoresed on a polyacrylamide gel. After transferringthe gel, the nitrocellulose membrane was blocked with 5%BSA, streptavidin conjugated to horseradish peroxidase wasadded for 1 h, and visualized using ECL.
MALDI-TOF/TOF MS spectra of SIRT1 modified byreactive aldehydes
Recombinant human SIRT1 (30 �g) (Enzo Life Sciences,Plymouth Meeting, PA, USA; cat no. SE-239, 82 kDa, 747 aa,which was produced from human cDNA and expressed in E.
coli and sequence identical to that at GenBank accession no.NM012238) was modified with 4-hydroxy-2-nonenal (4-HNE;30 �M) for 18 h at 37°C, which was then digested with trypsingold solution (Promega, Madison, WI, USA) in ammoniumbicarbonate (50 mM) in 10% acetonitrile (pH 8) for 18 h at37°C. The protein was not reduced during the digestion; nodithiothreitol was used. Peptides were extracted using 50%acetonitrile, 5% formic acid, and analyzed by MALDI TOF/TOF mass spectrometry (AutoflexIII TOF/TOF MALDI massspectrometer; Bruker Daltonics, Billerica, MA, USA). Datawere analyzed using Mascot 2.1.04 (Matrix Science, London,UK).
SIRT1 activity assay
SIRT1 activity was assayed using a deacetylase colorimetricactivity assay kit (Enzo Life Sciences), according to themanufacturer’s instructions. Briefly, SIRT1 was immunopre-cipitated as described above from whole-cell extracts treatedwith CSE (0.1%–1.5%) or H2O2 (150 �M) for 6 or 24 h. Cellswere also treated with resveratrol (5 �M) or sirtinol (10 �M),a well-characterized activator and inhibitor of SIRT1, respec-tively. After the final washing, Color de Lys substrate reagentand NAD� was added to the SIRT1-conjugated beads andincubated for 80 min at 37°C. The substrate-SIRT1 mixturewas then placed on a 96-well plate, and the Color de Lysdeveloper reagent was added to the wells for 20 min at 37°C.The plate was then read at 405 nm using a spectrophotometer(Model 680 microplate reader, Bio-Rad, Hercules, CA, USA).
Immunofluorescence
BEAS-2B cells were grown on 8-well chamber slides (1�104
cells/well), treated with H2O2 (150 �M) or CSE (0.5–1.5%)in the presence or absence of NAC (2 mM), and then fixed in4% paraformaldehyde for 10 min. The cells were thenpermeabilized for 10 min in 0.3% Triton X-100 in PBS, andblocked for 1 h using 10% normal goat serum in PBS.Samples were incubated with antibodies specific for SIRT1 ina humidified chamber overnight. The primary antibody wasdetected with Alexa Fluor 594 goat anti-rabbit secondaryantibody (Invitrogen, Carlsbad, CA, USA). Nuclei werestained with 1 �g/ml Hoechst 33342 for 1 min. Sampleswithout primary antibodies were used as negative controls.The coverslips were mounted onto the slides using Vecta-Shield (Vector Laboratories, Burlingame, CA, USA) andviewed under a Nikon TE2000-E microscope (Nikon, Tokyo,Japan).
Statistical analysis
Results are shown as means � se. Statistical significance wascalculated using 1-way ANOVA by StatView; values of P 0.05were considered nonsignificant.
RESULTS
SIRT1 protein levels and activity are dose and timedependently decreased by CS-mediated oxidativestress and aldehyde in both primary and transformedcells
CS is shown to decrease SIRT1 protein levels andactivity in lungs of rats and humans (20, 21, 32) and inmacrophages in vitro (20, 21), but the mechanism of
3147REDOX REGULATION OF SIRT1

CS-induced oxidative modifications of SIRT1 is notknown. We investigated the mechanism of reduction ofSIRT1 by CS in both primary and transformed epithe-lial cell lines. Whole-cell extracts of BEAS-2B cellsshowed reduced SIRT1 levels in response to increasingconcentrations of CSE from 6 to 24 h. Prior to 6 h,there was no reduction in SIRT1 levels (data notshown). As a control for oxidative stress, KO2, whichgenerates superoxide anion, and H2O2 were used.H2O2 (150 �M) did not significantly decrease SIRT1levels at either 6 or 24 h (Fig. 1A). Treatment withhigher concentrations of H2O2 (250 and 500 �M) for6 h resulted in significant loss of SIRT1; however, therewas an increasing level of cytotoxicity associated withthese treatments (data not shown). Cells treated withKO2 (1 �M) showed a time-dependent loss of SIRT1levels (Fig. 1A). As cigarettes contain between 1.92 mgand 3.14 mg of reactive carbonyl compounds, SIRT1levels were examined in response to the reactive alde-hyde acrolein, a component of CS (394 �M of acroleinand up to 500 �g acrolein/cigarette, which is �39.4�M in 1% of CSE) (33–36). Whole-cell extracts ofBEAS-2B cells treated with acrolein (10 or 30 �M)showed a dose-dependent loss of SIRT1 protein, withEC50 of 30 �M at 6 h, with 80% inhibition at 24 h(Fig. 1B), suggesting that aldehyde/carbonyl stress maylead to decreased SIRT1 levels. To compare the find-ings of the transformed cell lines, human primaryepithelial cells were examined for SIRT1 levels inresponse to CS. Treatment of human SAECs with CSE(0.5% and 1%) for 4 h resulted in a dose-dependent
decrease in SIRT1 levels in whole-cell extracts (Fig. 1C).CSE (0.1% to 1.5%) was equivalent to �5 to 50 �M ofaldehydes present in CS (34–36).
SIRT1 activity was assayed to determine whether CSEreduced SIRT1 deacetylase activity in lung epithelialcells. SIRT1 activity was reduced in a dose-dependentmanner at 6 and 24 h by CSE (Fig. 2A, B). H2O2 (150�M) caused a reduction in SIRT1 activity without areduction in protein level at both 6 and 24 h (Fig. 2A,B). Acrolein with EC50 of 30 �M also decreased SIRT1activity significantly at 6 h treatment (Fig. 2A, B). As asecond measure of SIRT1 activity, levels of acetylatedp53, a target of SIRT1, was measured in BEAS-2B cellstreated with CSE, H2O2, acrolein, resveratrol, or sirtinolfor 6 h. CSE, H2O2, acrolein, and sirtinol increasedacetylation on lysine 382 residue on p53, whereasuntreated cells and resveratrol treated cells had lowlevels of acetylated p53 (Fig. 2C). These data confirmthat SIRT1 activity is inhibited by carbonyl and oxida-tive stress.
SIRT1 is degraded by the proteasome in response toCSE
The proteasome is the major route of protein degrada-tion in the cell; therefore, it was examined whether CSdecreased SIRT1 levels through proteasomal degrada-tion. BEAS-2B cells were pretreated with the protea-some inhibitors N-Acetyl-Leu-Leu-Nle-CHO (ALLN; 5�M) and MG-132 (10 �M) for 30 min, washed withPBS, and treated for 6 h with CSE (1 and 1.5%).
Figure 1. Aldehyde and CS lower SIRT1 levels in human lung epithelial cells. A) BEAS-2Bcells were treated with varying concentrations of CSE (0.1–1.5%) or H2O2 (150, 250, or 500�M) for 6 or 24 h. Cells were also treated for 1 or 6 h with either KO2 (1 �M) or dry DMSOvehicle (Veh). Ctrl, untreated control. B) BEAS-2B cells were treated with acrolein (10 or30 �M) for 6 h. C) SAECs were treated with CSE (0.5 and 1%) for 4 h. Whole-cell extractsfrom treated cells were used for immunoblot analysis for SIRT1. -Actin was used as ahousekeeping loading control. Blots are representative of �3 separate experiments (n�3).*P � 0.05, **P � 0.01, ***P � 0.001 vs. controls.
3148 Vol. 24 September 2010 CAITO ET AL.The FASEB Journal � www.fasebj.org
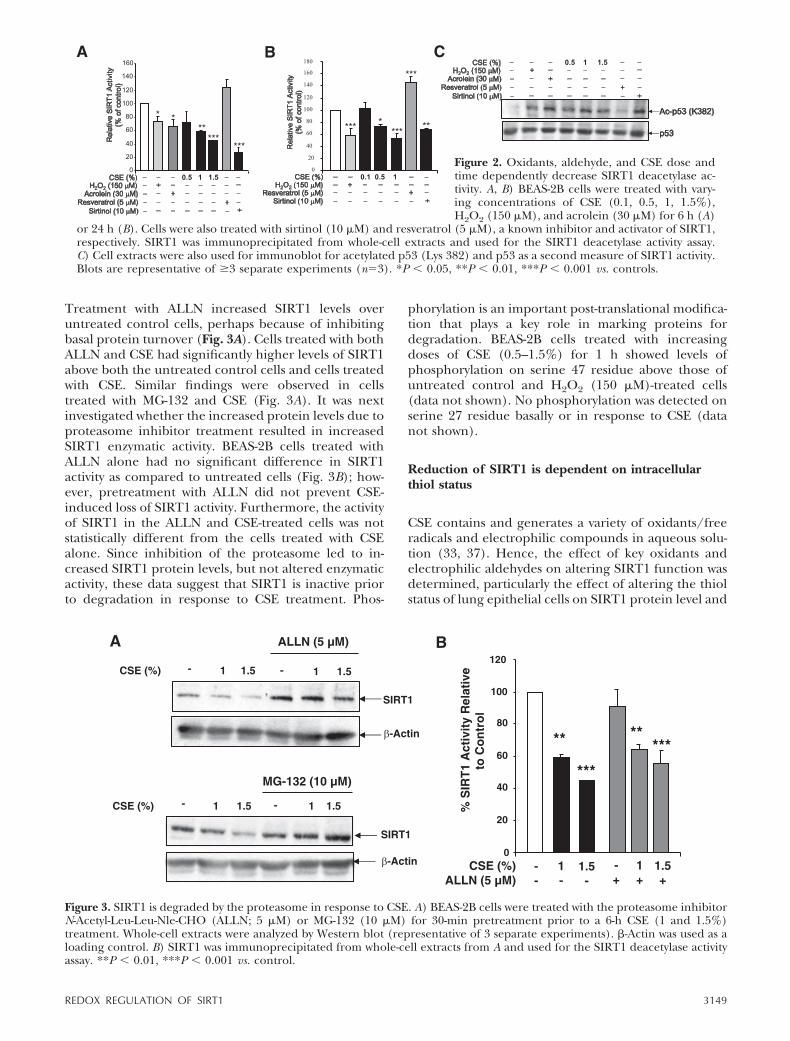
Treatment with ALLN increased SIRT1 levels overuntreated control cells, perhaps because of inhibitingbasal protein turnover (Fig. 3A). Cells treated with bothALLN and CSE had significantly higher levels of SIRT1above both the untreated control cells and cells treatedwith CSE. Similar findings were observed in cellstreated with MG-132 and CSE (Fig. 3A). It was nextinvestigated whether the increased protein levels due toproteasome inhibitor treatment resulted in increasedSIRT1 enzymatic activity. BEAS-2B cells treated withALLN alone had no significant difference in SIRT1activity as compared to untreated cells (Fig. 3B); how-ever, pretreatment with ALLN did not prevent CSE-induced loss of SIRT1 activity. Furthermore, the activityof SIRT1 in the ALLN and CSE-treated cells was notstatistically different from the cells treated with CSEalone. Since inhibition of the proteasome led to in-creased SIRT1 protein levels, but not altered enzymaticactivity, these data suggest that SIRT1 is inactive priorto degradation in response to CSE treatment. Phos-
phorylation is an important post-translational modifica-tion that plays a key role in marking proteins fordegradation. BEAS-2B cells treated with increasingdoses of CSE (0.5–1.5%) for 1 h showed levels ofphosphorylation on serine 47 residue above those ofuntreated control and H2O2 (150 �M)-treated cells(data not shown). No phosphorylation was detected onserine 27 residue basally or in response to CSE (datanot shown).
Reduction of SIRT1 is dependent on intracellularthiol status
CSE contains and generates a variety of oxidants/freeradicals and electrophilic compounds in aqueous solu-tion (33, 37). Hence, the effect of key oxidants andelectrophilic aldehydes on altering SIRT1 function wasdetermined, particularly the effect of altering the thiolstatus of lung epithelial cells on SIRT1 protein level and
Figure 2. Oxidants, aldehyde, and CSE dose andtime dependently decrease SIRT1 deacetylase ac-tivity. A, B) BEAS-2B cells were treated with vary-ing concentrations of CSE (0.1, 0.5, 1, 1.5%),H2O2 (150 �M), and acrolein (30 �M) for 6 h (A)
or 24 h (B). Cells were also treated with sirtinol (10 �M) and resveratrol (5 �M), a known inhibitor and activator of SIRT1,respectively. SIRT1 was immunoprecipitated from whole-cell extracts and used for the SIRT1 deacetylase activity assay.C) Cell extracts were also used for immunoblot for acetylated p53 (Lys 382) and p53 as a second measure of SIRT1 activity.Blots are representative of �3 separate experiments (n�3). *P � 0.05, **P � 0.01, ***P � 0.001 vs. controls.
Figure 3. SIRT1 is degraded by the proteasome in response to CSE. A) BEAS-2B cells were treated with the proteasome inhibitorN-Acetyl-Leu-Leu-Nle-CHO (ALLN; 5 �M) or MG-132 (10 �M) for 30-min pretreatment prior to a 6-h CSE (1 and 1.5%)treatment. Whole-cell extracts were analyzed by Western blot (representative of 3 separate experiments). -Actin was used as aloading control. B) SIRT1 was immunoprecipitated from whole-cell extracts from A and used for the SIRT1 deacetylase activityassay. **P � 0.01, ***P � 0.001 vs. control.
3149REDOX REGULATION OF SIRT1

activity in response to CSE. First, the BEAS-2B cells’glutathione (GSH) levels were depleted by the additionof BSO (100 �M) 18 h prior to a 24-h treatment withCSE (0.5 and 1%) or H2O2 (150 �M). Pretreatmentwith BSO resulted in further loss of SIRT1 in responseto CSE treatment; SIRT1 levels were significantly lowerin cells treated with BSO and CSE (0.5%) than in cellstreated with CSE (0.5%) alone (P�0.01) (Fig. 4A). Itwas interesting to note that BSO-treated cells did nothave significantly lower SIRT1 levels than untreatedcontrol cells. These data suggest that an oxidativeenvironment may prime cells for SIRT1 loss, however,does not lead to SIRT1 loss in the absence of anenvironmental insult.
It was next determined whether increasing the GSHlevels with NAC (2 mM), and thus increasing the lungcells’ thiol levels (38), could prevent CSE-mediated SIRT1loss. BEAS-2B cells pretreated with NAC for 2 h before a24-h treatment with CSE (0.5 and 1%) had significantlyhigher levels of SIRT1 than cells that were treated withCSE alone (P�0.001) (Fig. 4B). Levels of SIRT1 in cellstreated with both NAC and CSE were also significantlyhigher than control levels, suggesting that intracellularthiols may be able to prevent CSE-dependent loss ofSIRT1. BEAS-2B cells and SAEC were also treated withglutathione monoethyl ester (GSHmee; 2 mM), a cell-permeable form of GSH, for 2 h prior to CSE (1%)treatment for 6 h to determine whether directly increas-ing GSH levels could restore SIRT1 protein. Pretreatmentwith GSHmee also prevented CSE-induced SIRT1 loss atthis shorter time point (Fig. 4C, D). Altogether, these datasuggest that intracellular thiol levels play an importantrole in down-regulation of SIRT1 protein level in re-sponse to CS.
To determine whether other ROS scavengers couldalso prevent CSE-mediated SIRT1 loss, BEAS-2B cellspretreated with either PEG-conjugated SOD (200U/ml), catalase (200 U/ml), or a combination of both(200 U/ml of each) for 30 min prior to a 6-h treatmentwith 1% CSE. Surprisingly, the combination of theseantioxidant enzymes with CSE did not restore SIRT1levels as NAC had done (Fig. 4E). These data furthersuggest that the thiol status of the cell is important formaintaining SIRT1 protein levels in response to CS.
It was next determined whether these changes onSIRT1 protein level had any effect on its enzymaticactivity. BEAS-2B cells treated with CSE (0.5%) for 24 hin the presence or absence of an 18-h pretreatmentwith BSO or a 2-h pretreatment with NAC were assayedfor SIRT1 activity. BSO treatment alone lowered SIRT1activity (P�0.001), whereas NAC had no effect onSIRT1 activity (Fig. 5). H2O2 reduced SIRT1 activity(P�0.001), which was prevented by NAC pretreatment(P�0.01). BSO pretreatment had no significant effecton SIRT1 activity in combination with H2O2. Similarly,CSE significantly lowered SIRT1 activity (P�0.05),which was prevented by pretreatment with NAC(P�0.05). Interestingly, BSO in combination with CSEdid not further decrease SIRT1 activity compared toCSE alone. These data suggest that the thiol status ofthe cell is important not only for maintaining SIRT1protein level in response to smoke, but also its activity.
Oxidative stress causes nucleocytoplasmic shuttling ofSIRT1 by a redox-dependent mechanism
It is known that in response to antimycin A, a mitochon-drial complex III inhibitor, SIRT1, is shuttled out of the
Figure 4. Modulation of cellular thiol level regu-lates SIRT1 protein level. A) BEAS-2B cells weretreated with BSO (100 �M) to deplete GSH levels.BSO was pretreated 18 h before a 24-h treatmentwith CSE (0.5 or 1%) or H2O2 (150 �M).B) BEAS-2B cells were treated with NAC (2 mM) for2 h prior to a 24-h treatment with CSE (0.5 or 1%)or H2O2 (150 �M). C, D) BEAS-2B cells (C) and
SAECs (D) were treated with glutathione monoethyl ester (GSHmee; 2 mM) for 2 h, washed off with sterile PBS, and treatedwith CSE (1%) for 6 h. E) PEG-conjugated SOD (200 U/ml) and catalase (200 U/ml) were given to BEAS-2B cells aloneor in combination for 30 min prior to being washed off with PBS, followed by a 6-h treatment with CSE (1%). Whole-cellextracts were analyzed by immunoblot. Gels are representative of 3 separate experiments (n�3). -Actin was used as aloading control.
3150 Vol. 24 September 2010 CAITO ET AL.The FASEB Journal � www.fasebj.org
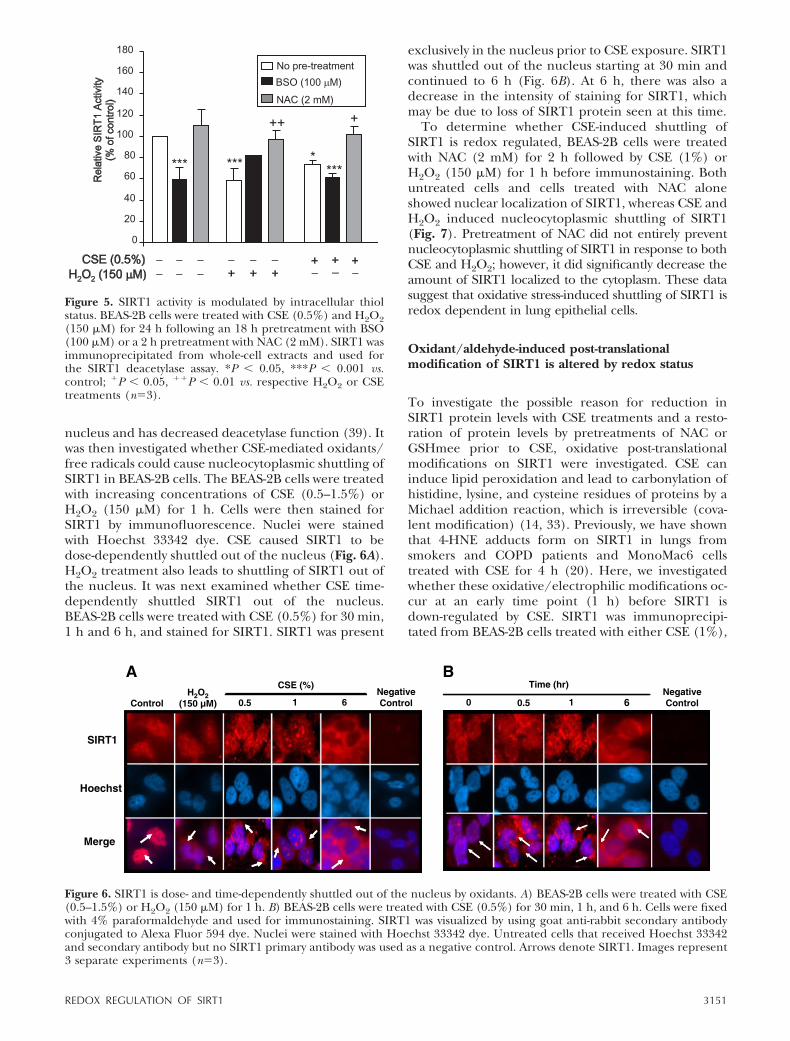
nucleus and has decreased deacetylase function (39). Itwas then investigated whether CSE-mediated oxidants/free radicals could cause nucleocytoplasmic shuttling ofSIRT1 in BEAS-2B cells. The BEAS-2B cells were treatedwith increasing concentrations of CSE (0.5–1.5%) orH2O2 (150 �M) for 1 h. Cells were then stained forSIRT1 by immunofluorescence. Nuclei were stainedwith Hoechst 33342 dye. CSE caused SIRT1 to bedose-dependently shuttled out of the nucleus (Fig. 6A).H2O2 treatment also leads to shuttling of SIRT1 out ofthe nucleus. It was next examined whether CSE time-dependently shuttled SIRT1 out of the nucleus.BEAS-2B cells were treated with CSE (0.5%) for 30 min,1 h and 6 h, and stained for SIRT1. SIRT1 was present
exclusively in the nucleus prior to CSE exposure. SIRT1was shuttled out of the nucleus starting at 30 min andcontinued to 6 h (Fig. 6B). At 6 h, there was also adecrease in the intensity of staining for SIRT1, whichmay be due to loss of SIRT1 protein seen at this time.
To determine whether CSE-induced shuttling ofSIRT1 is redox regulated, BEAS-2B cells were treatedwith NAC (2 mM) for 2 h followed by CSE (1%) orH2O2 (150 �M) for 1 h before immunostaining. Bothuntreated cells and cells treated with NAC aloneshowed nuclear localization of SIRT1, whereas CSE andH2O2 induced nucleocytoplasmic shuttling of SIRT1(Fig. 7). Pretreatment of NAC did not entirely preventnucleocytoplasmic shuttling of SIRT1 in response to bothCSE and H2O2; however, it did significantly decrease theamount of SIRT1 localized to the cytoplasm. These datasuggest that oxidative stress-induced shuttling of SIRT1 isredox dependent in lung epithelial cells.
Oxidant/aldehyde-induced post-translationalmodification of SIRT1 is altered by redox status
To investigate the possible reason for reduction inSIRT1 protein levels with CSE treatments and a resto-ration of protein levels by pretreatments of NAC orGSHmee prior to CSE, oxidative post-translationalmodifications on SIRT1 were investigated. CSE caninduce lipid peroxidation and lead to carbonylation ofhistidine, lysine, and cysteine residues of proteins by aMichael addition reaction, which is irreversible (cova-lent modification) (14, 33). Previously, we have shownthat 4-HNE adducts form on SIRT1 in lungs fromsmokers and COPD patients and MonoMac6 cellstreated with CSE for 4 h (20). Here, we investigatedwhether these oxidative/electrophilic modifications oc-cur at an early time point (1 h) before SIRT1 isdown-regulated by CSE. SIRT1 was immunoprecipi-tated from BEAS-2B cells treated with either CSE (1%),
Figure 5. SIRT1 activity is modulated by intracellular thiolstatus. BEAS-2B cells were treated with CSE (0.5%) and H2O2(150 �M) for 24 h following an 18 h pretreatment with BSO(100 �M) or a 2 h pretreatment with NAC (2 mM). SIRT1 wasimmunoprecipitated from whole-cell extracts and used forthe SIRT1 deacetylase assay. *P � 0.05, ***P � 0.001 vs.control; �P � 0.05, ��P � 0.01 vs. respective H2O2 or CSEtreatments (n�3).
6 6
Figure 6. SIRT1 is dose- and time-dependently shuttled out of the nucleus by oxidants. A) BEAS-2B cells were treated with CSE(0.5–1.5%) or H2O2 (150 �M) for 1 h. B) BEAS-2B cells were treated with CSE (0.5%) for 30 min, 1 h, and 6 h. Cells were fixedwith 4% paraformaldehyde and used for immunostaining. SIRT1 was visualized by using goat anti-rabbit secondary antibodyconjugated to Alexa Fluor 594 dye. Nuclei were stained with Hoechst 33342 dye. Untreated cells that received Hoechst 33342and secondary antibody but no SIRT1 primary antibody was used as a negative control. Arrows denote SIRT1. Images represent3 separate experiments (n�3).
3151REDOX REGULATION OF SIRT1

H2O2 (150 �M), or acrolein (30 �M) for 1 h. Totalcarbonyl modification on SIRT1 was measured by deriv-itizing the immunoprecipitated SIRT1 with 2,4-dinitrophe-nyl hydrazine (DNPH). There were relatively low levelsof carbonylation on SIRT1 in untreated cells; however,H2O2, acrolein and CSE lead to increased carbonyla-tion (Fig. 8). Pretreatment with NAC led to a significantreduction in levels of carbonylation adducts in CSE-treated cells (P�0.01). This suggests that carbonylationof SIRT1 in response to CSE occurs early (1 h) and isfollowed by the reduction in SIRT1 protein levels orenzymatic activity (6 to 24 h).
It was also investigated whether there was increasedtotal carbonylation in vivo in lungs of mice exposed to CS.SIRT1 was significantly decreased in mouse whole lungextracts from mice exposed to CS (P�0.05) (Fig. 9A).Mice exposed to CS had increased carbonylation onSIRT1 as compared to air-exposed mice, suggestingcarbonylation is an important post-translational modi-fication to SIRT1 that occurs in vivo (Fig. 9B). Tofurther investigate the role of carbonyl modificationson SIRT1 levels, mice lacking Glrx1 were exposed toCS. These mice are unable to repair/reverse S-gluta-thionylation, a reversible modification on proteins andthus are susceptible to oxidative stress. The level ofSIRT1 was dramatically reduced in response to CS inthe lungs of Glrx1-KO mice as compared to air-exposedGlrx1-KO mice (Fig. 9A). In contrast, mice overexpress-ing Glrx1 (Glrx1-Tg) showed no reduction in SIRT1levels in response to CS compared to WT mice (Fig.9A). SIRT1 was then immunoprecipitated from WT,Glrx1-KO, and Glrx1-Tg mice, and protein carbonyla-tion and glutathionylation levels were measured. CSincreased S-glutathionylation on SIRT1, which was aug-mented in the Glrx1-KO mice exposed to CS (Fig. 9B).There was also increased S-glutathionylation on SIRT1
in lungs of air-exposed Glrx1-KO mice. S-glutathionyla-tion was not seen in lungs of Glrx1-Tg mice exposed toeither air or CS. Carbonylation on SIRT1 was increasedin both air- and CS-exposed Glrx1-KO mice comparedto their counterpart WT mice. Glrx1-Tg mice exposed to
Figure 7. Nucleocytoplasmic shuttling of SIRT1 by oxidative stress is reversed by NAC. BEAS-2B cells were treated with NAC (2mM) for 2 h prior to a 1-h treatment with either CSE (1%) or H2O2 (150 �M). Cells were fixed with 4% paraformaldehyde andused for immunostaining. SIRT1 was visualized by using goat anti-rabbit secondary antibody conjugated to Alexa Fluor 594 dye.Nuclei were stained with Hoechst 33342 dye. Untreated cells that received Hoechst 33342 and secondary antibody but no SIRT1primary antibody was used as a negative control. Arrows denote SIRT1. Images represent 3 separate experiments (n�3).
Figure 8. Oxidative stress and CSE induce carbonylation onSIRT1. SIRT1 was immunoprecipitated (IP) from whole-cellextracts of BEAS-2B cells treated for 1 h with acrolein (30�M), H2O2 (150 �M), and CSE (1%) in the presence orabsence of a 2-h pretreatment with NAC (2 mM). Equalamount (100 �g) of immunoprecipitated SIRT1 protein wasused for immunoblotting (IB). Carbonylation was detected byfirst derivitizing the samples with DNPH and immunoblottingwith an anti-DNP antibody. Blot is representative of �3separate experiments **P � 0.01, ***P � 0.001 vs. control;��P � 0.01 vs. CSE.
3152 Vol. 24 September 2010 CAITO ET AL.The FASEB Journal � www.fasebj.org

air had no carbonylation present on SIRT1 in the lung,particularly lung epithelial cells, as these mice expressGlrx1 only in airway epithelial cells, while CS-exposedtransgenic mice had carbonylation on SIRT1 similar tothe WT CS-exposed mice (Fig. 9B). Taken together, these
data suggest that covalent modifications on SIRT1 affectthe SIRT1 protein level in response to CS.
It was next determined whether the CSE-inducedcarbonylation was on cysteine residues on SIRT1 inBEAS-2B cells and mouse lung and whether there wasan effect on altering the intracellular thiol levels on thecysteine modifications. SIRT1 was immunoprecipitatedfrom whole-cell extracts of BEAS-2B cells exposed toCSE for 6 h in the presence or absence of a 2-hpretreatment with NAC. Free cysteine residues werelabeled with maleimide-PEO2-biotin. Treatment withCSE to BEAS-2B cells led to decreased maleimide-PEO2-biotin labeling of cysteine residues on SIRT1compared to control (Fig. 10A). Treatment with eitherNAC alone did not affect the maleimide-PEO2-biotinlabeling; however, the combination of NAC and CSEresulted in labeling that was similar to control cells,suggesting that NAC pretreatment prevented CSE frommodifying free cysteine residues on SIRT1. Immuno-precipitation of SIRT1 from WT mouse lung extractsalso revealed that there were decreased free cysteineresidues on SIRT1 with CS exposure, as compared toair-exposed mice (Fig. 10B).
It was next determined whether inducing alkylationof free cysteines on SIRT1 would lead to decreasedactivity or protein levels. BEAS-2B cells were treatedwith the alkylating agent N-ethylmaleimide (NEM; 50or 100 �M) for 30 min or 3 h, and then examined forSIRT1 levels and activity. Alkylation of SIRT1 by NEMled to decreased enzymatic activity at both time pointsand concentrations (Fig. 11A). Interestingly, 50 �M
d
d
Figure 10. Altering cell thiol status affects post-translational modifica-tions on cysteine residues of SIRT1 in response to aldehydes andoxidants. SIRT1 was immunoprecipitated (IP) from whole-cell ex-tracts of BEAS-2B cells treated for 6 h with acrolein (30 �M), H2O2
(150 �M), and CSE (1%) in the presence or absence of a 2-h pretreatment with NAC (2 mM) (A) or wild typeC57BL/6J mice exposed to air or CS (B). Free cysteine residues were labeled using maleimide-PEO2-biotin andvisualized using immunoblot with streptavidin conjugated to horseradish peroxidase. Blot is representative of threeseparate experiments (n�3). **P � 0.01, ***P � 0.001 vs. controls; �P � 0.05 vs. CSE (1%).
Figure 9. Reversible covalent modification on SIRT1 by CSdoes not affect total SIRT1 levels. A) Whole-lung extractswere blotted for SIRT1 levels from WT C57BL/6J, Glrx1-KO,and Glrx1-Tg mice exposed to air or CS (300 mg/m3 TPM)for 3 d and sacrificed 24 h after last exposure, as described inMaterials and Methods. B) Whole lung cell extract (100 �g)was immunoprecipitated and S-glutathionylated, and totalcarbonylated SIRT1 levels were detected by immunoblot withanti-GSH and anti-DNP antibodies. Blots are representative of3 separate experiments (n�3).
3153REDOX REGULATION OF SIRT1

NEM only slightly decreased SIRT1 levels only at 3 h,whereas 100 �M NEM treatment decreased SIRT1levels at both 30 min and 3 h (Fig. 11B). To determinewhether there was increased labeling of SIRT1 with thelonger incubations with NEM, SIRT1 was immunopre-cipitated from BEAS-2B cells and labeled with maleimide-PEO2-biotin (Fig. 11C). As the treatment time of NEMincreased, more cysteines were alkylated, giving a faintsignal at 3 h compared to untreated cells. These datasuggest that alkylation of SIRT1 on cysteine residues candirectly decrease SIRT1 activity and protein levels.
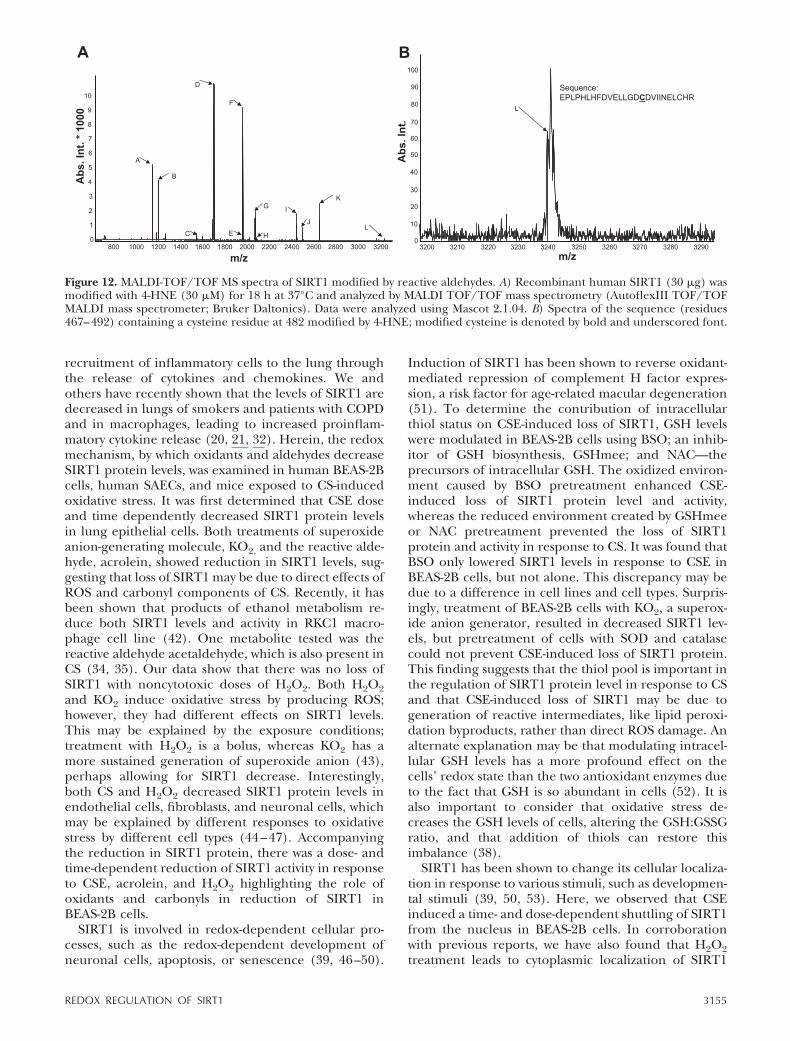
MALDI-TOF/TOF MS spectra of SIRT1 modified byreactive aldehydes
To further confirm carbonylation/alkylation on SIRT1 inresponse to carbonyl stress, recombinant human SIRT1was modified with 4-HNE and analyzed by MALDI TOF/
TOF mass spectrometry. MS/MS spectra were collected bythe lift method in positive mode to fragment parentpeaks, and fragment sequences were compared to humanSIRT1 using MASCOT version 2.1.04 (40, 41). As thepeptides were extracted under acidic conditions, the onlypossible modification sites observed would be on cysteineresidues since modifications on arginines, lysines, andhistidines are acid labile. We found carbonylation/alkyla-tion of cysteine residues between aa 467 and 492 onSIRT1 with 4-HNE (Table 1 and Fig. 12), suggesting thatenvironmental reactive aldehydes are able to directlymodify SIRT1 by post-translational modification.
DISCUSSION
Epithelial cells are important cells in initiating andperpetuating the inflammatory response to CS by the
d
β
Figure 11. Cysteine alkylation de-creases SIRT1 protein levels andenzymatic activity. A) BEAS-2Bcells were treated with NEM (50 or100 �M) for 30 min or 3 h, andSIRT1 was immunoprecipitatedfrom whole-cell extracts for mea-surement of deacetylase activity.B) Extracts were also used for im-munoblot for SIRT1 protein levels.-Actin was used as a loading con-trol. C) Nonalkylated/free cysteine
residues on immunoprecipitated SIRT1 were labeled using maleimide-PEO2-biotin and visualized using immunoblot withstreptavidin conjugated to horseradish peroxidase. Blots are representative of 3 separate experiments (n�3). *P � 0.05,**P � 0.01, ***P � 0.001 vs. controls; �P � 0.05 vs. NEM (50 �M) at 3 h.
TABLE 1. Peptides identified by MALDI TOF/TOF-MS from SIRT1 modified By 4-HNE
Mass (Da) Sequence position Sequence Peak
1144.817 385–394 GDIFNQVVPR A1200.815 66–77 GCPGAAAAALWR 2:Carbamidom ethyl (C) B1547.042 637–649 LDGNQYLFLPPNR C1703.201 636–649 RLDGNQYLFLPPNR D1961.387 256–274 IIVLTGAGVSVSCGIPDFR 13:Carbamidom ethyl (C) E1963.323 342–358 NYTQNIDTLEQVAGIQR F2073.405 182–199 IGPYTFVQQHLMIGTDPR G2089.480 255–274 KIIVLTGAGVSVSCGGIPDFR 14:Carbamidom ethyl (C) H2448.817 445–466 VRPVALIPSSIPHEVPQILINR I2499.581 283–303 LAVDFPDLPDPQAMFDIEYFR J2653.705 520–543 ELAYLSELPPTPLHVSEDSSSPER K3239.535 467–492 EPLPHLHFDVELLGDCDVIINELCHR 4-HNE (C), Carbamidom
ethyl (C), on either residue 16 or 24L
3154 Vol. 24 September 2010 CAITO ET AL.The FASEB Journal � www.fasebj.org
recruitment of inflammatory cells to the lung throughthe release of cytokines and chemokines. We andothers have recently shown that the levels of SIRT1 aredecreased in lungs of smokers and patients with COPDand in macrophages, leading to increased proinflam-matory cytokine release (20, 21, 32). Herein, the redoxmechanism, by which oxidants and aldehydes decreaseSIRT1 protein levels, was examined in human BEAS-2Bcells, human SAECs, and mice exposed to CS-inducedoxidative stress. It was first determined that CSE doseand time dependently decreased SIRT1 protein levelsin lung epithelial cells. Both treatments of superoxideanion-generating molecule, KO2, and the reactive alde-hyde, acrolein, showed reduction in SIRT1 levels, sug-gesting that loss of SIRT1 may be due to direct effects ofROS and carbonyl components of CS. Recently, it hasbeen shown that products of ethanol metabolism re-duce both SIRT1 levels and activity in RKC1 macro-phage cell line (42). One metabolite tested was thereactive aldehyde acetaldehyde, which is also present inCS (34, 35). Our data show that there was no loss ofSIRT1 with noncytotoxic doses of H2O2. Both H2O2and KO2 induce oxidative stress by producing ROS;however, they had different effects on SIRT1 levels.This may be explained by the exposure conditions;treatment with H2O2 is a bolus, whereas KO2 has amore sustained generation of superoxide anion (43),perhaps allowing for SIRT1 decrease. Interestingly,both CS and H2O2 decreased SIRT1 protein levels inendothelial cells, fibroblasts, and neuronal cells, whichmay be explained by different responses to oxidativestress by different cell types (44–47). Accompanyingthe reduction in SIRT1 protein, there was a dose- andtime-dependent reduction of SIRT1 activity in responseto CSE, acrolein, and H2O2 highlighting the role ofoxidants and carbonyls in reduction of SIRT1 inBEAS-2B cells.
SIRT1 is involved in redox-dependent cellular pro-cesses, such as the redox-dependent development ofneuronal cells, apoptosis, or senescence (39, 46–50).
Induction of SIRT1 has been shown to reverse oxidant-mediated repression of complement H factor expres-sion, a risk factor for age-related macular degeneration(51). To determine the contribution of intracellularthiol status on CSE-induced loss of SIRT1, GSH levelswere modulated in BEAS-2B cells using BSO; an inhib-itor of GSH biosynthesis, GSHmee; and NAC—theprecursors of intracellular GSH. The oxidized environ-ment caused by BSO pretreatment enhanced CSE-induced loss of SIRT1 protein level and activity,whereas the reduced environment created by GSHmeeor NAC pretreatment prevented the loss of SIRT1protein and activity in response to CS. It was found thatBSO only lowered SIRT1 levels in response to CSE inBEAS-2B cells, but not alone. This discrepancy may bedue to a difference in cell lines and cell types. Surpris-ingly, treatment of BEAS-2B cells with KO2, a superox-ide anion generator, resulted in decreased SIRT1 lev-els, but pretreatment of cells with SOD and catalasecould not prevent CSE-induced loss of SIRT1 protein.This finding suggests that the thiol pool is important inthe regulation of SIRT1 protein level in response to CSand that CSE-induced loss of SIRT1 may be due togeneration of reactive intermediates, like lipid peroxi-dation byproducts, rather than direct ROS damage. Analternate explanation may be that modulating intracel-lular GSH levels has a more profound effect on thecells’ redox state than the two antioxidant enzymes dueto the fact that GSH is so abundant in cells (52). It isalso important to consider that oxidative stress de-creases the GSH levels of cells, altering the GSH:GSSGratio, and that addition of thiols can restore thisimbalance (38).
SIRT1 has been shown to change its cellular localiza-tion in response to various stimuli, such as developmen-tal stimuli (39, 50, 53). Here, we observed that CSEinduced a time- and dose-dependent shuttling of SIRT1from the nucleus in BEAS-2B cells. In corroborationwith previous reports, we have also found that H2O2treatment leads to cytoplasmic localization of SIRT1
Figure 12. MALDI-TOF/TOF MS spectra of SIRT1 modified by reactive aldehydes. A) Recombinant human SIRT1 (30 �g) wasmodified with 4-HNE (30 �M) for 18 h at 37°C and analyzed by MALDI TOF/TOF mass spectrometry (AutoflexIII TOF/TOFMALDI mass spectrometer; Bruker Daltonics). Data were analyzed using Mascot 2.1.04. B) Spectra of the sequence (residues467–492) containing a cysteine residue at 482 modified by 4-HNE; modified cysteine is denoted by bold and underscored font.
3155REDOX REGULATION OF SIRT1

(50). Since CSE induces loss of SIRT1, our data suggestthat nucleocytoplasmic shuttling of SIRT1 may facili-tate degradation in the proteasome in the cytoplasm,although H2O2 also induces shuttling of SIRT1 butdoes not lead to SIRT1 protein loss. Recent studies haveestablished that cytoplasmic localized SIRT1 occursduring apoptosis; however, no cell death was observedunder the CSE and H2O2 (150 �M) treatment condi-tions performed in this study. Whether cytoplasmiclocalization of SIRT1 contributes to senescence, both ofwhich can be induced by CS and H2O2 (47, 54, 55),remains to be determined. It was also observed thatshuttling of SIRT1 by oxidative stress could be reducedby pretreatment of cells with NAC, suggesting thatnucleocytoplasmic shuttling is mediated by a redox-sensitive mechanism. Phosphorylation by a redox-sen-sitive kinase may explain this observation. Oxidativestress has been shown to activate redox-sensitive ki-nases, such as I-�B kinase (IKK), phosphoinositide3-kinase (PI-3K), and extracellular signal-related kinase1/2 (ERK1/2) (56, 57). PI-3K has been shown tobe involved in the nuclear localization of SIRT1, as theuse of PI-3K inhibitors leads to cytoplasmic localizationof SIRT1 (39), while phosphorylation by CK2 increasesSIRT1 activity (58, 59). Phosphorylation is a key deter-miner of cellular localization for several proteins, suchas FOXO3 transcription factor (60, 61), and sinceSIRT1 has several phosphorylation sites of unknownfunction (58, 62), it is speculated that phosphorylationmay contribute to CS-induced nucleocytoplasmic shut-tling of SIRT1.
Oxidants present in CS are known to cause peroxi-dation of sensitive biomolecules, such as DNA, lipids,and proteins. Carbonylation of proteins results fromreactive �--unsaturated aldehydes reacting with cys-teine, histidine, and lysine residues by Michael addi-tion. It was found that CS, H2O2, and acrolein allincreased carbonylation on SIRT1 both in vitro inBEAS-2B cells and in vivo in mouse lung. Furthermore,these modifications occurred before the loss of SIRT1protein level and activity in BEAS-2B cells. These car-bonyl adducts were decreased in cells pretreated withNAC prior to CSE exposure, which is in agreement withprevious reports that NAC can prevent lipid peroxida-tion (63). Although carbonylation occurs on specificamino acid residues, several proteins may be affected byoxidative stress/reactive carbonyls. Whether there areprotein targets of CS-induced carbonylation that areactivated or inhibited and subsequently affect SIRT1remains to be determined.
Oxidative stress can induce sulfenic acid formationon cysteine residues of proteins, which can oxidize to amore stable acid or react with thiols to produce disul-fides or GSH to form adducts, termed glutathionylation(64). Glutathionylation can be reversed by the enzymeGlrx1. It was investigated whether this reversible oxida-tive modification occurred on SIRT1 in response to CS.Genetic ablation of Glrx1 leads to increased SIRT1 loss,while overexpressing Glrx1 was protective. Glrx1 is acytosolic protein (64), which implies that its action on
SIRT1, a predominantly nuclear protein, may occurpost-translationally before SIRT1 is shuttled to thenucleus or once SIRT1 is shuttled out of the nucleus inresponse to oxidative stress. Further investigation isneeded to explore this relationship. Levels of glutathio-nylation and carbonylation on SIRT1 were increasedin lungs of air- and CS-exposed Glrx1-KO mice, whiletransgenic mice showed the same amount of carbony-lation as wild-type mice, but no effect on glutathio-nylation, suggesting that carbonylation and gluta-thionylation on SIRT1 occur independently.Currently, it is not known whether glutathionylationor carbonylation occurs first and if these two modifica-tions have similar effects on SIRT1.
Since cysteine, lysine, and histidine residues arepotential targets for oxidative adduct formation, it wasinvestigated whether cysteines on SIRT1 were the tar-gets of CSE-induced modifications. Free cysteines wereevaluated using a biotin switch assay; free cysteines werestrongly labeled in untreated cells using maleimide-PEO2-biotin; however, acrolein, H2O2, and CSE greatlydecreased this labeling, suggesting that it had modifiedthese residues. NAC given before CSE could preventthe modification of cysteines. The modification ofcysteine residues was also seen in vivo in whole-cellextracts from mouse lungs exposed to CS. Cysteines donot play a role in the enzymatic functioning of SIRT1,however, there are key cysteine residues located in thesirtuin fold domain, which are responsible for thecoordination of a structural Zn atom (17, 65). Thisdomain along with a Rossmann fold domain, are be-lieved to form a cleft for NAD� and substrate binding(17). It is possible that CS modifies cysteine residues,causing a loss of SIRT1’s function by altering thisbinding cleft.
Oxidative post-translational modifications to proteinsare deleterious: often resulting in inactivation, loss offunction, and/or degradation (15, 66). It was evidentthat carbonyl modifications were increased on SIRT1 inresponse to acrolein, H2O2, and CSE, and that onlyacrolein and CSE decreased SIRT1 protein levels, thequestion remained what was the functional conse-quence of the carbonylation on cysteine residues. Todetermine whether cysteine alkylation resulted in lossof SIRT1 protein level and/or activity, BEAS-2B cellswere treated with NEM. NEM caused a dose-dependentloss of SIRT1 activity, most likely because of the alkyla-tion of the critical cysteines in the NAD� bindingpocket. There was also a dose- and time-dependent lossof SIRT1 protein in response to NEM, suggesting thatloss of SIRT1 can occur by modifying cysteine residues.MALDI TOF/TOF mass spectrometry further con-firmed that cysteine residues are able to be alkylated/carbonylated by reactive aldehydes on a SIRT1, findinga cysteine residue between aa 467 and 492 that wascarbonylated. The possibility of carbonylation of otherelectrophilic target amino acids, such as histidine andlysine, cannot be ruled out as the MALDI TOF/TOFanalysis was performed under acid-labile conditions,where any modifications on lysine and histidine would
3156 Vol. 24 September 2010 CAITO ET AL.The FASEB Journal � www.fasebj.org

be removed. In addition, the endogenous alkylation orcarbonylation target of SIRT1 in response to oxidative/carbonyl stress in vivo may vary since in our study, weused recombinant SIRT1 for proteomic studies.
Oxidatively modified proteins may be degraded bythe proteasome by ubiquitin-independent or -depen-dent mechanisms (14). Indeed, proteasomal degrada-tion of SIRT1 in response to CS was observed in thisstudy. Furthermore, SIRT1, which was accumulatedwhen the proteasome was inhibited had lower activitythan untreated cells, suggesting SIRT1 is inactivated byCS before degradation. Oxidative and carbonyl stressesand thiol modulation may cause a multitude of biomo-lecular/bioenergetic effects contributing to loss ofSIRT1 in response to CSE, and whether any specificpathway is involved has yet to be determined. There aremany signals for protein degradation (67). It is hypoth-esized that oxidative modifications, such as carbonyla-tion, lead to an unstable hydrophobic conformationand directly lead to degradation by the 20S proteasome(68). This may be the case with NEM treatments, aslevels of phosphorylated or ubiquitinated SIRT1 werenot investigated. Recently, ubiquitin-independent pro-teasome degradation of SIRT1 has been observed inresponse to ionizing radiation in articular chondrocytes(48). However, in CS-induced loss of SIRT1, the possi-ble role of phosphorylation-ubiquitination pathwaycannot be ignored as oxidatively modified proteins likealcohol dehydrogenase are degraded by the ubiquitin-dependent 26S proteasome (69). Ubiquitin-dependentproteasomal degradation has been observed in thediaphragm of patients with COPD (70), and our labo-ratory has shown increased ubiquitin-dependent degra-dation of HDAC2 in response to CSE (71), suggesting apossible role for ubiquitin-dependent proteasomal deg-radation of SIRT1 in response to CS. Recent researchhas identified several sites of phosphorylation on SIRT1(58, 62, 72, 73) and shown that phosphorylation ofserine 27 residue of SIRT1 is important in proteinstability in cancer (74). We have observed phosphory-lation on serine 47 residue; however, whether thisresidue is involved in proteasomal degradation ofSIRT1 by CS remains to be determined. Phosphoryla-tion is a reversible modification, but oxidative/covalentcarbonyl modifications are irreversible; therefore, it willbe important to determine the order of events thatoccur prior to SIRT1 degradation in response to CS.We propose that electrophilic compounds present inCS/CSE would covalently modify SIRT1 along withphosphorylation, leading to its degradation, as seen inthe present study and reported earlier in lungs ofsmokers and patients with COPD (20, 31).
Overall, our data show that SIRT1 is oxidativelydown-regulated by CS/aldehydes, leading to post-trans-lational modification, inactivation, and protein loss viathe proteasome. Further inducing oxidative stress withthe combination of GSH depletion and CSE resulted ingreater loss of SIRT1 protein. Thiol-replenishing agentscould prevent CSE-induced modification of SIRT1,leading to increased SIRT1 protein levels when com-
pared to CSE-treated cells; however, the antioxidantenzymes SOD and catalase could not prevent SIRT1protein loss. CS is a complex mixture of free radicals,electrophilic compounds, and oxidants (33), and theoxidative/carbonyl stress it induced had different ef-fects on SIRT1 than that of H2O2; which decreasedSIRT1 activity but had no effect on protein level inBEAS-2B cells. This suggests that there are multiplepathways and events occurring during CS exposure thataffect SIRT1 functioning. SIRT1 was shown to becarbonylated and glutathionylated in vivo in mice ex-posed to CS and that these modifications could beattenuated by increasing intracellular thiols by NAC invitro and in vivo in mice overexpressing Glrx1, anenzyme that repairs glutathionylated proteins. Ourfindings suggest that oxidant/carbonyl stress-mediatedreduction of SIRT1 will lead to loss of its control ofacetylation of its target proteins p53, RelA/p65, andFOXO1/3, and hence this will enhance inflammatory,senescence and apoptosis responses (hallmarks of var-ious chronic inflammatory diseases). There has beenrecent development of SIRT1 activators for the poten-tial use in the treatment of Type 2 diabetes and otherchronic inflammatory diseases (75–77). However, itremains to be determined whether these agents arebeneficial in chronic inflammatory conditions or in treat-ing diseases, like COPD, where there is a decrease inSIRT1 protein level. Our findings advance the emergingfield of research on SIRT1 regulation by suggesting that asimple activation of SIRT1 by pharmacological agents maynot be effective since SIRT1 is covalently modified inoxidative/inflammatory conditions. We propose that re-versal of oxidative post-translationally modified SIRT1may be an avenue before effective therapeutic strategiescan be designed for chronic inflammatory diseases. Nev-ertheless, our data not only demonstrate the basic under-standing of oxidant/carbonyl-mediated reduction ofSIRT1 and redox regulation/modifications of SIRT1 butalso have widespread implications in understanding thepathogenesis of various chronic inflammatory diseaseswhere oxidative/carbonyl stress occur, leading to inflam-mation-aging (inflammaging).
This work was supported by U.S. National Institutes of Healthgrants 1R01-HL092842, R01-HL085613, and 1R01HL097751-01; National Institute of Environmental Health SciencesCenter grant ES01247; and Toxicology Training Programgrant T32-ES07026. The authors thank Dr. Se-Ran Yang forher technical assistance. The authors also thank Dr. Ye-Shih Ho (Institute of Environmental Health Sciences andDepartment of Biochemistry and Molecular Biology,Wayne State University, Detroit, MI, USA) for providingGlrx1-KO and Glrx1-Tg mice. None of the authors have anyethical conflicts of interest for publication of this manu-script.
REFERENCES
1. Elliott, P. J., and Jirousek, M. (2008) Sirtuins: novel targets formetabolic disease. Curr. Opin. Investig. Drugs 9, 371–378
3157REDOX REGULATION OF SIRT1

2. Michan, S., and Sinclair, D. (2007) Sirtuins in mammals: in-sights into their biological function. Biochem. J. 404, 1–13
3. Westphal, C. H., Dipp, M. A., and Guarente, L. (2007) Atherapeutic role for sirtuins in diseases of aging? Trends. Biochem.Sci. 32, 555–560
4. Chen, J., Zhou, Y., Mueller-Steiner, S., Chen, L. F., Kwon, H., Yi,S., Mucke, L., and Gan, L. (2005) SIRT1 protects againstmicroglia-dependent amyloid-beta toxicity through inhibitingNF-�B signaling. J. Biol. Chem. 280, 40364–40374
5. Vogelmeier, C., and Bals, R. (2007) Chronic obstructive pulmo-nary disease and premature aging. Am. J. Respir. Crit. Care Med.175, 1217–1218
6. Tuder, R. M. (2006) Aging and cigarette smoke: fueling the fire.Am. J. Respir. Crit. Care. Med. 174, 490–491
7. Karrasch, S., Holz, O., and Jorres, R. A. (2008) Aging andinduced senescence as factors in the pathogenesis of lungemphysema. Respir. Med. 102, 1215–1230
8. Cohen, H. Y., Miller, C., Bitterman, K. J., Wall, N. R., Hekking,B., Kessler, B., Howitz, K. T., Gorospe, M., de Cabo, R., andSinclair, D. A. (2004) Calorie restriction promotes mammaliancell survival by inducing the SIRT1 deacetylase. Science 305,390–392
9. Zhang, T., and Kraus, W. L. (2009) SIRT1-dependent regulation ofchromatin and transcription: Linking NAD(�) metabolism andsignaling to the control of cellular functions. [E-pub ahead ofprint] Biochim. Biophys. Acta doi: 10.1016/j.bbapap.2009.10.022
10. Yeung, F., Hoberg, J. E., Ramsey, C. S., Keller, M. D., Jones,D. R., Frye, R. A., and Mayo, M. W. (2004) Modulation ofNF-�B-dependent transcription and cell survival by the SIRT1deacetylase. EMBO J. 23, 2369–2380
11. Brunet, A., Sweeney, L. B., Sturgill, J. F., Chua, K. F., Greer,P. L., Lin, Y., Tran, H., Ross, S. E., Mostoslavsky, R., Cohen,H. Y., Hu, L. S., Cheng, H. L., Jedrychowski, M. P., Gygi, S. P.,Sinclair, D. A., Alt, F. W., and Greenberg, M. E. (2004) Stress-dependent regulation of FOXO transcription factors by theSIRT1 deacetylase. Science 303, 2011–2015
12. Luo, J., Nikolaev, A. Y., Imai, S., Chen, D., Su, F., Shiloh, A.,Guarente, L., and Gu, W. (2001) Negative control of p53 bySir2alpha promotes cell survival under stress. Cell 107, 137–148
13. Salminen, A., Ojala, J., Huuskonen, J., Kauppinen, A., Suu-ronen, T., and Kaarniranta, K. (2008) Interaction of aging-associated signaling cascades: inhibition of NF-kappaB signalingby longevity factors FoxOs and SIRT1. Cell. Mol. Life Sci. 65,1049–1058
14. Grimsrud, P. A., Xie, H., Griffin, T. J., and Bernlohr, D. A.(2008) Oxidative stress and covalent modification of proteinwith bioactive aldehydes. J. Biol. Chem. 283, 21837–21841
15. Nystrom, T. (2005) Role of oxidative carbonylation in proteinquality control and senescence. EMBO J. 24, 1311–1317
16. Ghezzi, P., and Bonetto, V. (2003) Redox proteomics: identifi-cation of oxidatively modified proteins. Proteomics 3, 1145–1153
17. Sauve, A. A., Wolberger, C., Schramm, V. L., and Boeke, J. D.(2006) The biochemistry of sirtuins. Annu. Rev. Biochem. 75,435–465
18. Alcendor, R. R., Gao, S., Zhai, P., Zablocki, D., Holle, E., Yu, X.,Tian, B., Wagner, T., Vatner, S. F., and Sadoshima, J. (2007)Sirt1 regulates aging and resistance to oxidative stress in theheart. Circ. Res. 100, 1512–1521
19. Oberdoerffer, P., Michan, S., McVay, M., Mostoslavsky, R., Vann,J., Park, S. K., Hartlerode, A., Stegmuller, J., Hafner, A., Loerch,P., Wright, S. M., Mills, K. D., Bonni, A., Yankner, B. A., Scully,R., Prolla, T. A., Alt, F. W., and Sinclair, D. A. (2008) SIRT1redistribution on chromatin promotes genomic stability butalters gene expression during aging. Cell 135, 907–918
20. Rajendrasozhan, S., Yang, S. R., Kinnula, V. L., and Rahman, I.(2008) SIRT1, an antiinflammatory and antiaging protein, isdecreased in lungs of patients with chronic obstructive pulmo-nary disease. Am. J. Respir. Crit. Care Med. 177, 861–870
21. Yang, S. R., Wright, J., Bauter, M., Seweryniak, K., Kode, A., andRahman, I. (2007) Sirtuin regulates cigarette smoke-inducedproinflammatory mediator release via RelA/p65 NF-�B in mac-rophages in vitro and in rat lungs in vivo: implications forchronic inflammation and aging. Am. J. Physiol. Lung Cell. Mol.Physiol. 292, L567–L576
22. Ho, Y. S., Xiong, Y., Ho, D. S., Gao, J., Chua, B. H., Pai, H., andMieyal, J. J. (2007) Targeted disruption of the glutaredoxin 1gene does not sensitize adult mice to tissue injury induced by
ischemia/reperfusion and hyperoxia. Free Radic. Biol. Med. 43,1299–1312
23. Thatcher, T. H., Maggirwar, S. B., Baglole, C. J., Lakatos, H. F.,Gasiewicz, T. A., Phipps, R. P., and Sime, P. J. (2007) Arylhydrocarbon receptor-deficient mice develop heightened in-flammatory responses to cigarette smoke and endotoxin associ-ated with rapid loss of the nuclear factor-�B component RelB.Am. J. Pathol. 170, 855–864
24. Yao, H., Edirisinghe, I., Rajendrasozhan, S., Yang, S. R., Caito,S., Adenuga, D., and Rahman, I. (2008) Cigarette smoke-mediated inflammatory and oxidative responses are strain-dependent in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 294,L1174–L1186
25. Caito, S., Yang, S. R., Kode, A., Edirisinghe, I., Rajendrasozhan,S., Phipps, R. P., and Rahman, I. (2008) Rosiglitazone and15-deoxy-Delta12,14-prostaglandin J2, PPAR agonists, differen-tially regulate cigarette smoke-mediated pro-inflammatory cyto-kine release in monocytes/macrophages. Antioxid. Redox Signal10, 253–260
26. Kode, A., Yang, S. R., and Rahman, I. (2006) Differential effectsof cigarette smoke on oxidative stress and proinflammatorycytokine release in primary human airway epithelial cells and ina variety of transformed alveolar epithelial cells. Respir. Res. 7,132–151
27. Edirisinghe, I., Yang, S. R., Yao, H., Rajendrasozhan, S., Caito,S., Adenuga, D., Wong, C., Rahman, A., Phipps, R. P., Jin, Z. G.,and Rahman, I. (2008) VEGFR-2 inhibition augments cigarettesmoke-induced oxidative stress and inflammatory responsesleading to endothelial dysfunction. FASEB J. 22, 2297–2310
28. Carp, H., and Janoff, A. (1978) Possible mechanisms of emphy-sema in smokers. In vitro suppression of serum elastase-inhibi-tory capacity by fresh cigarette smoke and its prevention byantioxidants. Am. Rev. Respir. Dis. 118, 617–621
29. Reiter, C. D., Teng, R. J., and Beckman, J. S. (2000) Superoxidereacts with nitric oxide to nitrate tyrosine at physiological pH viaperoxynitrite. J. Biol. Chem. 275, 32460–32466
30. Murtaza, I., Wang, H. X., Feng, X., Alenina, N., Bader, M.,Prabhakar, B. S., and Li, P. F. (2008) Down-regulation ofcatalase and oxidative modification of protein kinase CK2 leadto the failure of apoptosis repressor with caspase recruitmentdomain to inhibit cardiomyocyte hypertrophy. J. Biol. Chem. 283,5996–6004
31. Ying, J., Clavreul, N., Sethuraman, M., Adachi, T., and Cohen,R. A. (2007) Thiol oxidation in signaling and response to stress:detection and quantification of physiological and pathophysio-logical thiol modifications. Free Radic. Biol. Med. 43, 1099–1108
32. Nakamaru, Y., Vuppusetty, C., Wada, H., Milne, J. C., Ito, M.,Rossios, C., Elliot, M., Hogg, J., Kharitonov, S., Goto, H., Bemis,J. E., Elliott, P., Barnes, P. J., and Ito, K. (2009) A proteindeacetylase SIRT1 is a negative regulator of metalloproteinase-9.FASEB J. 23, 2810–2819
33. Church, D. F., and Pryor, W. A. (1985) Free-radical chemistry ofcigarette smoke and its toxicological implications. Environ.Health Perspect. 64, 111–126
34. Eiserich, J. P., van der Vliet, A., Handelman, G. J., Halliwell, B.,and Cross, C. E. (1995) Dietary antioxidants and cigarettesmoke-induced biomolecular damage: a complex interaction.Am. J. Clin. Nutr. 62, 1490S–1500S
35. Fujioka, K., and Shibamoto, T. (2006) Determination of toxiccarbonyl compounds in cigarette smoke. Environ. Toxicol. 21,47–54
36. Smith, C. J., and Hansch, C. (2000) The relative toxicity ofcompounds in mainstream cigarette smoke condensate. FoodChem. Toxicol. 38, 637–646
37. Kodama, M., Kaneko, M., Aida, M., Inoue, F., Nakayama, T., andAkimoto, H. (1997) Free radical chemistry of cigarette smokeand its implication in human cancer. Anticancer Res. 17, 433–437
38. Mulier, B., Rahman, I., Watchorn, T., Donaldson, K., MacNee,W., and Jeffery, P. K. (1998) Hydrogen peroxide-induced epi-thelial injury: the protective role of intracellular nonproteinthiols (NPSH). Eur. Respir. J. 11, 384–391
39. Tanno, M., Sakamoto, J., Miura, T., Shimamoto, K., and Horio,Y. (2007) Nucleocytoplasmic shuttling of the NAD�-dependenthistone deacetylase SIRT1. J. Biol. Chem. 282, 6823–6832
40. Sayre, L. M., Lin, D., Yuan, Q., Zhu, X., and Tang, X. (2006)Protein adducts generated from products of lipid oxidation:focus on HNE and one. Drug Metab. Rev. 38, 651–675
3158 Vol. 24 September 2010 CAITO ET AL.The FASEB Journal � www.fasebj.org

41. Shevchenko, A., Tomas, H., Havlis, J., Olsen, J. V., and Mann, M.(2006) In-gel digestion for mass spectrometric characterizationof proteins and proteomes. Nat. Protoc. 1, 2856–2860
42. Shen, Z., Ajmo, J. M., Rogers, C. Q., Liang, X., Le, L., Murr,M. M., Peng, Y., and You, M. (2009) Role of SIRT1 in regulationof LPS- or two ethanol metabolites-induced TNF� production incultured macrophage cell lines. Am. J. Physiol. Gastrointest. LiverPhysiol. 296, G1047–G1053
43. Marklund, S. (1976) Spectrophotometric study of spontaneousdisproportionation of superoxide anion radical and sensitivedirect assay for superoxide dismutase. J. Biol. Chem. 251, 7504–7507
44. Csiszar, A., Labinskyy, N., Podlutsky, A., Kaminski, P. M., Wolin,M. S., Zhang, C., Mukhopadhyay, P., Pacher, P., Hu, F., de Cabo,R., Ballabh, P., and Ungvari, Z. (2008) Vasoprotective effects ofresveratrol and SIRT1: attenuation of cigarette smoke-inducedoxidative stress and proinflammatory phenotypic alterations.Am. J. Physiol. Heart Circ. Physiol. 294, H2721–H2735
45. Ota, H., Eto, M., Kano, M. R., Ogawa, S., Iijima, K., Akishita, M.,and Ouchi, Y. (2008) Cilostazol inhibits oxidative stress-inducedpremature senescence via upregulation of Sirt1 in humanendothelial cells. Arterioscler. Thromb. Vasc. Biol. 28, 1634–1639
46. Prozorovski, T., Schulze-Topphoff, U., Glumm, R., Baumgart, J.,Schroter, F., Ninnemann, O., Siegert, E., Bendix, I., Brustle, O.,Nitsch, R., Zipp, F., and Aktas, O. (2008) Sirt1 contributescritically to the redox-dependent fate of neural progenitors.Nat. Cell Biol. 10, 385–394
47. Furukawa, A., Tada-Oikawa, S., Kawanishi, S., and Oikawa, S.(2007) H2O2 accelerates cellular senescence by accumulation ofacetylated p53 via decrease in the function of SIRT1 by NAD�depletion. Cell. Physiol. Biochem. 20, 45–54
48. Hong, E. H., Lee, S. J., Kim, J. S., Lee, K. H., Um, H. D., Kim,J. H., Kim, S. J., Kim, J. I., and Hwang, S. G. (2010) Ionizingradiation induces cellular senescence of articular chondrocytesvia negative regulation of SIRT1 by p38 kinase. J. Biol. Chem. 285,1283–1295
49. Fratelli, M., Goodwin, L. O., Orom, U. A., Lombardi, S., Tonelli,R., Mengozzi, M., and Ghezzi, P. (2005) Gene expressionprofiling reveals a signaling role of glutathione in redox regu-lation. Proc. Natl. Acad. Sci. U. S. A. 102, 13998–14003
50. Jin, Q., Yan, T., Ge, X., Sun, C., Shi, X., and Zhai, Q. (2007)Cytoplasm-localized SIRT1 enhances apoptosis. J. Cell. Physiol.213, 88–97
51. Wu, Z., Lauer, T. W., Sick, A., Hackett, S. F., and Campochiaro,P. A. (2007) Oxidative stress modulates complement factor Hexpression in retinal pigmented epithelial cells by acetylation ofFOXO3. J. Biol. Chem. 282, 22414–22425
52. Rahman, I., and MacNee, W. (1999) Lung glutathione andoxidative stress: implications in cigarette smoke-induced airwaydisease. Am. J. Physiol. Lung Physiol. 277, L1067–L1088
53. Hisahara, S., Chiba, S., Matsumoto, H., Tanno, M., Yagi, H.,Shimohama, S., Sato, M., and Horio, Y. (2008) Histone deacety-lase SIRT1 modulates neuronal differentiation by its nucleartranslocation. Proc. Natl. Acad. Sci. U. S. A. 105, 15599–15604
54. Tsuji, T., Aoshiba, K., and Nagai, A. (2006) Alveolar cellsenescence in patients with pulmonary emphysema. Am. J.Respir. Crit. Care Med. 174, 886–893
55. Tsuji, T., Aoshiba, K., and Nagai, A. (2004) Cigarette smokeinduces senescence in alveolar epithelial cells. Am. J. Respir. Cell.Mol. Biol. 31, 643–649
56. Mercer, B. A., Kolesnikova, N., Sonett, J., and D’Armiento, J.(2004) Extracellular regulated kinase/mitogen activated pro-tein kinase is up-regulated in pulmonary emphysema andmediates matrix metalloproteinase-1 induction by cigarettesmoke. J. Biol. Chem. 279, 17690–17696
57. Pantano, C., Reynaert, N. L., van der Vliet, A., and Janssen-Heininger, Y. M. (2006) Redox-sensitive kinases of the nuclearfactor-�B signaling pathway. Antioxid. Redox Signal 8, 1791–1806
58. Zschoernig, B., and Mahlknecht, U. (2009) Carboxy-terminalphosphorylation of SIRT1 by protein kinase CK2. Biochem.Biophys. Res. Commun. 381, 372–377
59. Kang, H., Jung, J. W., Kim, M. K., and Chung, J. H. (2009) CK2is the regulator of SIRT1 substrate-binding affinity, deacetylase
activity and cellular response to DNA-damage. PLoS One 4,e6611
60. Van Der Heide, L. P., Hoekman, M. F., and Smidt, M. P. (2004)The ins and outs of FoxO shuttling: mechanisms of FoxOtranslocation and transcriptional regulation. Biochem. J. 380,297–309
61. Poon, I. K., and Jans, D. A. (2005) Regulation of nucleartransport: central role in development and transformation?Traffic 6, 173–186
62. Sasaki, T., Maier, B., Koclega, K. D., Chruszcz, M., Gluba, W.,Stukenberg, P. T., Minor, W., and Scrable, H. (2008) Phosphor-ylation regulates SIRT1 function. PLoS ONE 3, e4020
63. Whitekus, M. J., Li, N., Zhang, M., Wang, M., Horwitz, M. A.,Nelson, S. K., Horwitz, L. D., Brechun, N., Diaz-Sanchez, D., andNel, A. E. (2002) Thiol antioxidants inhibit the adjuvant effectsof aerosolized diesel exhaust particles in a murine model forovalbumin sensitization. J. Immunol. 168, 2560–2567
64. Gallogly, M. M., and Mieyal, J. J. (2007) Mechanisms of revers-ible protein glutathionylation in redox signaling and oxidativestress. Curr. Opin. Pharmacol. 7, 381–391
65. Sanders, B. D., Jackson, B., and Marmorstein, R. (2010) Struc-tural basis for sirtuin function: What we know and what wedon’t. [E-pub ahead of print] Biochim. Biophys. Acta doi: 10.1016/j.bbapap.2009.09.009
66. Petersen, D. R., and Doorn, J. A. (2004) Reactions of 4-hy-droxynonenal with proteins and cellular targets. Free Radic. Biol.Med. 37, 937–945
67. Ravid, T., and Hochstrasser, M. (2008) Diversity of degradationsignals in the ubiquitin-proteasome system. Nat. Rev. Mol. Cell.Biol. 9, 679–690
68. Grune, T., Merker, K., Sandig, G., and Davies, K. J. (2003)Selective degradation of oxidatively modified protein substratesby the proteasome. Biochem. Biophys. Res. Commun. 305, 709–718
69. Carbone, D. L., Doorn, J. A., and Petersen, D. R. (2004)4-Hydroxynonenal regulates 26S proteasomal degradation ofalcohol dehydrogenase. Free Radic. Biol. Med. 37, 1430–1439
70. Ottenheijm, C. A., Heunks, L. M., Li, Y. P., Jin, B., Minnaard, R.,van Hees, H. W., and Dekhuijzen, P. N. (2006) Activation of theubiquitin-proteasome pathway in the diaphragm in chronicobstructive pulmonary disease. Am. J. Respir. Crit. Care. Med. 174,997–1002
71. Adenuga, D., Yao, H., March, T. H., Seagrave, J., and Rahman,I. (2009) Histone deacetylase 2 is phosphorylated, ubiquiti-nated, and degraded by cigarette smoke. Am. J. Respir. Cell. Mol.Biol. 40, 464–473
72. Beausoleil, S. A., Jedrychowski, M., Schwartz, D., Elias, J. E.,Villen, J., Li, J., Cohn, M. A., Cantley, L. C., and Gygi, S. P.(2004) Large-scale characterization of HeLa cell nuclear phos-phoproteins. Proc. Natl. Acad. Sci. U. S. A. 101, 12130–12135
73. Olsen, J. V., Blagoev, B., Gnad, F., Macek, B., Kumar, C.,Mortensen, P., and Mann, M. (2006) Global, in vivo, andsite-specific phosphorylation dynamics in signaling networks.Cell 127, 635–648
74. Ford, J., Ahmed, S., Allison, S., Jiang, M., and Milner, J. (2008)JNK2-dependent regulation of SIRT1 protein stability. Cell Cycle7, 3091–3097
75. Milne, J. C., Lambert, P. D., Schenk, S., Carney, D. P., Smith,J. J., Gagne, D. J., Jin, L., Boss, O., Perni, R. B., Vu, C. B., Bemis,J. E., Xie, R., Disch, J. S., Ng, P. Y., Nunes, J. J., Lynch, A. V.,Yang, H., Galonek, H., Israelian, K., Choy, W., Iffland, A., Lavu,S., Medvedik, O., Sinclair, D. A., Olefsky, J. M., Jirousek, M. R.,Elliott, P. J., and Westphal, C. H. (2007) Small moleculeactivators of SIRT1 as therapeutics for the treatment of type 2diabetes. Nature 450, 712–716
76. Baur, J. A. (2009) Biochemical effects of SIRT1 activators.[E-pub ahead of print] Biochim. Biophys. Acta doi: 10.1016/j.bbapap.2009.10.025
77. Lavu, S., Boss, O., Elliott, P. J., and Lambert, P. D. (2008)Sirtuins–novel therapeutic targets to treat age-associated dis-eases. Nat. Rev. Drug Discov. 7, 841–853
Received for publication November 21, 2009.Accepted for publication March 18, 2010.
3159REDOX REGULATION OF SIRT1




















