
Opposing effects of hMOF and SIRT1 on H4K16 acetylation and the sensitivity to the topoisomerase II...
13
ORIGINAL ARTICLE Opposing effects of hMOF and SIRT1 on H4K16 acetylation and the sensitivity to the topoisomerase II inhibitor etoposide N Hajji 1,3 , K Wallenborg 2 , P Vlachos 1 , J Fu¨llgrabe 1 , O Hermanson 2 and B Joseph 1 1 Department of Oncology Pathology, Cancer Centrum Karolinska, Karolinska Institutet, Stockholm, Sweden and 2 Department of Neuroscience, CoE in Developmental Biology (CEDB/DBRM), Karolinska Institutet, Stockholm, Sweden Various inhibitors of histone deacetylase (HDAC) activity can sensitize drug resistant cancer cells to chemothera- peutic agents. However, the mechanisms underlying such effects of distinct HDAC inhibitors (HDACi) remain poorly understood. Here we show that both the HDACi trichostatin A and valproic acid induced a sensitization of multidrug-resistant cancer cells to the topoisomerase II inhibitor etoposide/VP16. This effect was associated with increased acetylation of certain lysines on histones H3 and H4, including lysine 16 on histone H4 (H4K16). Over- expression of the histone acetyltransferase hMOF, known to target H4K16, was sufficient to mimic HDACi treatment on sensitization and H4K16 acetylation, and importantly, small-interfering RNA (siRNA)-mediated knockdown of hMOF abolished the HDACi-mediated sensitizing effects as well as the increase in H4K16 ace- tylation. Conversely, siRNA-mediated knockdown of the H4K16 deacetylase SIRT1 mimicked HDACi treatment whereas overexpression of SIRT1 abolished H4K16 acetylation and significantly reduced the sensitizing effects of HDACi. Interestingly, the effects of hMOF on H4K16 acetylation and sensitization to the topoisomerase II inhi- bitor could be directly counteracted by exogenous expres- sion of increasing amounts of SIRT1 and vice versa. Our study results suggest that hMOF and SIRT1 activities are critical parameters in HDACi-mediated sensitization of multidrug-resistant cancer cells to topoisomerase II inhibitor and increased H4K16 acetylation. Oncogene (2010) 29, 2192–2204; doi:10.1038/onc.2009.505; published online 1 February 2010 Keywords: cell death; topoisomerase inhibitor; HDAC inhibitor; hMOF; SIRT1; H4K16 acetylation Introduction Histone lysine modifications are directly influencing activation and repression of transcription, and are also essential for proper elongation and higher-order chro- matin structure depending on which residue is modified and at what position in the gene such residue is located (Schneider and Grosschedl, 2007). Histone deacetylases (HDACs) and histone acetyltransferases (HAT) control the acetylation state of lysine residues, including those situated in the N-terminal ‘tails’ of the histones. In addition, posttranslational modification by acetylation/ deacetylation by these classes of enzymes of non-histone proteins, including transcription factors and co-regula- tors, has emerged as a regulatory mechanism controlling the function, activity and stability of these proteins (Hermanson et al., 2002; Glozak et al., 2005; Liu et al., 2009). In promoters, the acetylation of such histone lysines, including lysine 9 on histone H3 (H3K9), is believed to directly influence the recruitment of activator and repressor complexes to the chromatin (Schneider and Grosschedl, 2007). In addition, modifications of H3K9 and lysine 16 on histone H4 (H4K16) in exons are associated with elongation control, and the acetylation state of lysine residues thus regulates both the local chromatin dynamics and the higher-order chromatin structure (Shogren-Knaak et al., 2006). HDAC inhibitors (HDACi) constitute a relatively new class of chemotherapeutic drugs. These inhibitors exert multiple antineoplastic effects both in vitro and in vivo. In a short period of time, HDACi have shown promising results in preclinical and clinical trials with low toxicity in certain solid tumors and hematological malignancies (Bolden et al., 2006). In addition to their intrinsic anticancer properties, HDACi can modulate cellular responses to other cytotoxic modalities includ- ing ionizing and ultraviolet radiation and chemother- apeutic drugs (Karagiannis and El-Osta, 2006; Hajji et al., 2008). Many mechanisms have been proposed to explain the action of various HDACi, yet little is understood regarding how distinct HDACi can yield similar cellular effects seemingly regardless of structure and specificity. Interestingly, loss of total H4K16 acetylation has been suggested to represent one common hallmark of cancer cells (Fraga et al., 2005). The H4K16 residue is of particular interest as acetylation of this lysine residue is sufficient to induce reorganization of higher-order chromatin structure (Shogren-Knaak et al., 2006). The aim of this study was to investigate putative common mechanisms underlying sensitization by differ- ent HDACi of drug-resistant cancer cells to the Received 23 May 2009; revised 12 October 2009; accepted 27 November 2009; published online 1 February 2010 Correspondence: Dr B Joseph, Department of Oncology Pathology, Cancer Centrum Karolinska, Karolinska Institutet, R8:03, Stockholm S-171 76, Sweden. E-mail: [email protected] 3 Current address: Department of Experimental Medicine and Toxico- logy, Division of Investigative Science, Imperial College London, UK. Oncogene (2010) 29, 2192–2204 & 2010 Macmillan Publishers Limited All rights reserved 0950-9232/10 $32.00 www.nature.com/onc
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Opposing effects of hMOF and SIRT1 on H4K16 acetylation and the sensitivity to the topoisomerase II...

ORIGINAL ARTICLE
Opposing effects of hMOF and SIRT1 on H4K16 acetylation and the
sensitivity to the topoisomerase II inhibitor etoposide
N Hajji1,3, K Wallenborg2, P Vlachos1, J Fullgrabe1, O Hermanson2 and B Joseph1
1Department of Oncology Pathology, Cancer Centrum Karolinska, Karolinska Institutet, Stockholm, Sweden and2Department of Neuroscience, CoE in Developmental Biology (CEDB/DBRM), Karolinska Institutet, Stockholm, Sweden
Various inhibitors of histone deacetylase (HDAC) activitycan sensitize drug resistant cancer cells to chemothera-peutic agents. However, the mechanisms underlying sucheffects of distinct HDAC inhibitors (HDACi) remainpoorly understood. Here we show that both the HDACitrichostatin A and valproic acid induced a sensitization ofmultidrug-resistant cancer cells to the topoisomerase IIinhibitor etoposide/VP16. This effect was associated withincreased acetylation of certain lysines on histones H3 andH4, including lysine 16 on histone H4 (H4K16). Over-expression of the histone acetyltransferase hMOF, knownto target H4K16, was sufficient to mimic HDACitreatment on sensitization and H4K16 acetylation,and importantly, small-interfering RNA (siRNA)-mediatedknockdown of hMOF abolished the HDACi-mediatedsensitizing effects as well as the increase in H4K16 ace-tylation. Conversely, siRNA-mediated knockdown of theH4K16 deacetylase SIRT1 mimicked HDACi treatmentwhereas overexpression of SIRT1 abolished H4K16acetylation and significantly reduced the sensitizing effectsof HDACi. Interestingly, the effects of hMOF on H4K16acetylation and sensitization to the topoisomerase II inhi-bitor could be directly counteracted by exogenous expres-sion of increasing amounts of SIRT1 and vice versa. Ourstudy results suggest that hMOF and SIRT1 activities arecritical parameters in HDACi-mediated sensitizationof multidrug-resistant cancer cells to topoisomerase IIinhibitor and increased H4K16 acetylation.Oncogene (2010) 29, 2192–2204; doi:10.1038/onc.2009.505;published online 1 February 2010
Keywords: cell death; topoisomerase inhibitor; HDACinhibitor; hMOF; SIRT1; H4K16 acetylation
Introduction
Histone lysine modifications are directly influencingactivation and repression of transcription, and are also
essential for proper elongation and higher-order chro-matin structure depending on which residue is modifiedand at what position in the gene such residue is located(Schneider and Grosschedl, 2007). Histone deacetylases(HDACs) and histone acetyltransferases (HAT) controlthe acetylation state of lysine residues, including thosesituated in the N-terminal ‘tails’ of the histones. Inaddition, posttranslational modification by acetylation/deacetylation by these classes of enzymes of non-histoneproteins, including transcription factors and co-regula-tors, has emerged as a regulatory mechanism controllingthe function, activity and stability of these proteins(Hermanson et al., 2002; Glozak et al., 2005; Liu et al.,2009). In promoters, the acetylation of such histonelysines, including lysine 9 on histone H3 (H3K9), isbelieved to directly influence the recruitment of activatorand repressor complexes to the chromatin (Schneiderand Grosschedl, 2007). In addition, modifications ofH3K9 and lysine 16 on histone H4 (H4K16) in exons areassociated with elongation control, and the acetylationstate of lysine residues thus regulates both the localchromatin dynamics and the higher-order chromatinstructure (Shogren-Knaak et al., 2006).
HDAC inhibitors (HDACi) constitute a relativelynew class of chemotherapeutic drugs. These inhibitorsexert multiple antineoplastic effects both in vitro andin vivo. In a short period of time, HDACi have shownpromising results in preclinical and clinical trials withlow toxicity in certain solid tumors and hematologicalmalignancies (Bolden et al., 2006). In addition to theirintrinsic anticancer properties, HDACi can modulatecellular responses to other cytotoxic modalities includ-ing ionizing and ultraviolet radiation and chemother-apeutic drugs (Karagiannis and El-Osta, 2006; Hajjiet al., 2008). Many mechanisms have been proposed toexplain the action of various HDACi, yet little isunderstood regarding how distinct HDACi can yieldsimilar cellular effects seemingly regardless of structureand specificity. Interestingly, loss of total H4K16acetylation has been suggested to represent one commonhallmark of cancer cells (Fraga et al., 2005). The H4K16residue is of particular interest as acetylation of thislysine residue is sufficient to induce reorganization ofhigher-order chromatin structure (Shogren-Knaak et al.,2006).
The aim of this study was to investigate putativecommon mechanisms underlying sensitization by differ-ent HDACi of drug-resistant cancer cells to the
Received 23 May 2009; revised 12 October 2009; accepted 27 November2009; published online 1 February 2010
Correspondence: Dr B Joseph, Department of Oncology Pathology,Cancer Centrum Karolinska, Karolinska Institutet, R8:03, StockholmS-171 76, Sweden.E-mail: [email protected] address: Department of Experimental Medicine and Toxico-logy, Division of Investigative Science, Imperial College London, UK.
Oncogene (2010) 29, 2192–2204& 2010 Macmillan Publishers Limited All rights reserved 0950-9232/10 $32.00
www.nature.com/onc

topoisomerase II inhibitor etoposide/VP16. Our studyresults suggest that HDACi-induced sensitization wasassociated with increased histone acetylation, includingthat of H4K16, and that the HAT hMOF and theHDAC SIRT1 influenced not only H4K16 acetylationlevels but also the HDACi-induced sensitization of thedrug-resistant cells to etoposide.
Results
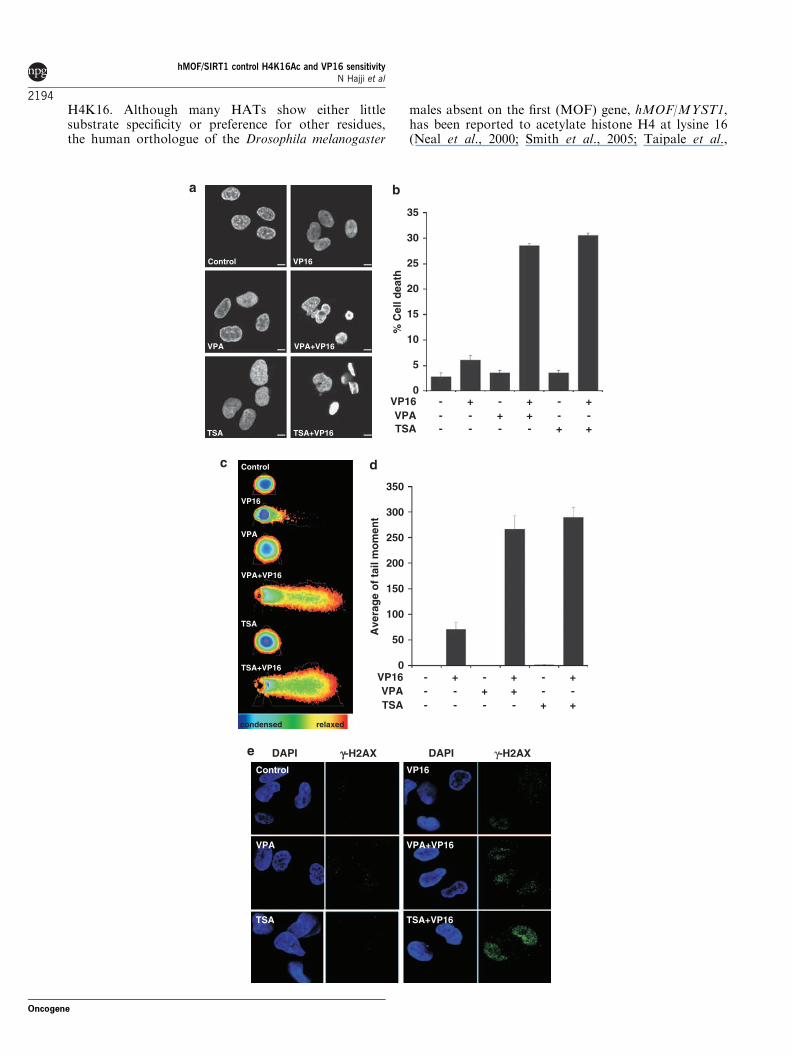
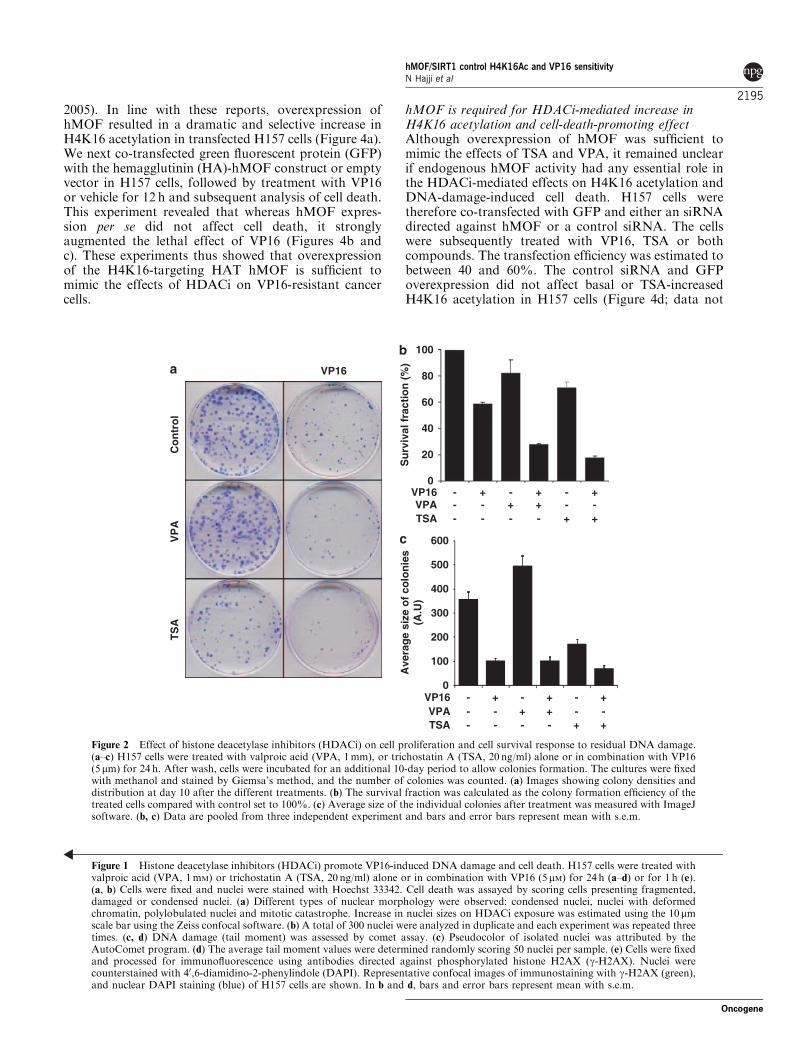
HDAC inhibitors reverse NSCLC cell resistanceto VP16-induced DNA damage and cell deathNon-small-cell lung carcinoma (NSCLC) H157 cellswere initially selected for this study based on theirknown resistance to g-ionizing radiation as well asetoposide/VP16, a well-characterized and specific topo-isomerase II inhibitor used clinically in chemotherapyand known to be lethal to normal cell types (Hajji et al.,2008). We first treated cells with structurally andfunctionally divergent inhibitors (HDACi) valproic acid(VPA, 1mM) or trichostatin A (TSA, 20 ng/ml; 66 nM)alone or in combination with VP16 (5 mM). Treatmentwith either TSA or VPA alone did not induce anyapparent increase in cell death, whereas both HDACisensitized H157 cells to VP16-induced cell death(Figures 1a and b). We have recently shown thatexposure to TSA but not VPA induces decreased BclxLexpression and thereby initiation of a caspase-mediatedAIF-dependent apoptotic cell death (Hajji et al., 2008).Whereas TSA promoted VP16-induced apoptotic celldeath, VPA/VP16 co-treatment induced nuclear mor-phology resembling mitotic catastrophe (Figure 1b), anevent associated with block in mitosis, mitotic spindledisorganization, failed chromosome segregation andformation of multinucleated cells (Castedo et al.,2004). Later on these cells died by necrosis-like lysis(Vakifahmetoglu et al., 2008). The promoting effect ofTSA and VPA treatment on VP16-induced cell deathwas further investigated and confirmed by clonogenicassay at 10 days (Figures 2a–c). Colonies from VPA-only treated cells presented an apparent increase in size,indicating that VPA treatment stimulated cell prolifera-tion (Figure 2c), which could argue in favor of mitoticcell death upon DNA damages.
Because the mechanisms downstream of the VP16-induced cell death promoted by TSA versus VPAappeared distinct, these results raised the centralquestion of how TSA and VPA yet can be as efficientin inducing sensitization to VP16-mediated cell death.We hypothesized that the sensitizing effect could dependon an increased ability of the topoisomerase II inhibitorto provoke DNA double-strand breaks. We thereforeassessed DNA damage by the ‘comet assay’. VP16, TSAor VPA alone produced only limited DNA damagein H157 cells as assessed by average tail moment(Figures 1c and d). In contrast, cells co-treated withTSA/VP16 or VPA/VP16 exhibited a dramatic increasein DNA damage (Figures 1c and d). Analysis of thephosphorylation of H2AX, one of the earliest events
following the induction of double-strand breaks, con-firmed these results (Figure 1e).
TSA or VPA trigger an increase in histone H4 lysine 16acetylation in cancer cellsTo investigate putative effects of the HDACi on histoneacetylation, we next analyzed the steady-state level oftotal acetylation at specific single lysine residues onhistone H3 and H4. Using specific antibodies againstacetylated lysine 9 on histone H3 (H3K9), H3K14,H4K5, H4K8, H4K12 and H4K16, followed byconfocal imaging analysis and immunoblotting ofhistone-enriched cell extracts, we detected an increasedacetylation of a subset of lysine residues after treatmentof H157 cells with VPA or TSA alone or in combinationwith VP16 (Figures 3a and b). The most pronouncedincrease in acetylation was detected at H4K16 asassessed by immunocytochemistry and immunoblotting(Figures 3a–c). H4K16 acetylation is known to influencehistone–histone interactions and higher-order chroma-tin structure, and has previously been shown to displaylow levels of total acetylation in cancer cells (Fragaet al., 2005).
To elucidate whether the effects of HDACi on H4K16acetylation were cell specific or general in nature, weexamined two additional cancer cell lines and a primarystem cell culture: NSCLC H1299 cell line, neuroblasto-ma SK-N-BE(2) cell line and primary fibroblastgrowth factor (FGF2)-expanded embryonic neural stemcells prepared from rat cortices (Jepsen et al., 2007;Andersson et al., 2009). SK-N-BE(2) is a highlymalignant cell line derived from chemotherapy-resistantneuroblastoma, which presents an intrinsic resistance tomultiple drugs including several DNA-damaging agents(Keshelava et al., 1998). Interestingly, all drug-resistantcancer cells analyzed exhibited a lower level of H4K16acetylation compared with the drug-sensitive neuralstem cells (Figure 3c) in part consistent with previousreports (Fraga et al., 2005). Treatment with TSA orVPA induced a clear increase in H4K16 acetylation inthe cancer cells, whereas the primary neural stem cellsshowed high basal total H4K16 acetylation levels andthis acetylation was in contrast unaffected by HDACitreatment. The level of H4K16 acetylation may thusreflect differences in chromatin acetylation machi-nery between stem cells and cancer cells (Figure 3c).Interestingly, the increase in H4K16 acetylation wasaccompanied by the sensitization of these cancer celllines to VP16-induced cell death (Figure 3d), whereasthe neural stem cells, displaying high basal H4K16acetylation levels, showed massive cell death in responseto VP16, without need of sensitization by HDACi. TotalH4K16 acetylation levels thus correlates with thesensitivity to DNA-damaging agents in neural stemand drug-resistant cancer cells.
Overexpression of hMOF is sufficient to increase H4K16acetylation and promote DNA damage induced cell deathWe next aimed at identifying the potential HAT thatcould mediate the effect of HDACi on acetylation of
hMOF/SIRT1 control H4K16Ac and VP16 sensitivityN Hajji et al
2193
Oncogene
H4K16. Although many HATs show either littlesubstrate specificity or preference for other residues,the human orthologue of the Drosophila melanogaster
males absent on the first (MOF) gene, hMOF/MYST1,has been reported to acetylate histone H4 at lysine 16(Neal et al., 2000; Smith et al., 2005; Taipale et al.,
Control
TSA+VP16
VPA+VP16
VPA
VP16
TSA
VP16%
Cel
l dea
th
- -- -+ +- -- - + +
+- - -+ +
TSAVPA
Control
VPA+VP16VPA
VP16
TSA+VP16TSA
35
30
25
20
15
10
5
0
350
300
250
200
150
100
50
0
condensed relaxed
VP16
Ave
rag
e o
f ta
il m
om
ent
- -- -+ +- -- - + +
+- - -+ +
TSAVPA
Control
TSA+VP16
VPA+VP16VPA
VP16
TSA
γγ-H2AXDAPI DAPI γ-H2AX
hMOF/SIRT1 control H4K16Ac and VP16 sensitivityN Hajji et al
2194
Oncogene
2005). In line with these reports, overexpression ofhMOF resulted in a dramatic and selective increase inH4K16 acetylation in transfected H157 cells (Figure 4a).We next co-transfected green fluorescent protein (GFP)with the hemagglutinin (HA)-hMOF construct or emptyvector in H157 cells, followed by treatment with VP16or vehicle for 12 h and subsequent analysis of cell death.This experiment revealed that whereas hMOF expres-sion per se did not affect cell death, it stronglyaugmented the lethal effect of VP16 (Figures 4b andc). These experiments thus showed that overexpressionof the H4K16-targeting HAT hMOF is sufficient tomimic the effects of HDACi on VP16-resistant cancercells.
hMOF is required for HDACi-mediated increase inH4K16 acetylation and cell-death-promoting effectAlthough overexpression of hMOF was sufficient tomimic the effects of TSA and VPA, it remained unclearif endogenous hMOF activity had any essential role inthe HDACi-mediated effects on H4K16 acetylation andDNA-damage-induced cell death. H157 cells weretherefore co-transfected with GFP and either an siRNAdirected against hMOF or a control siRNA. The cellswere subsequently treated with VP16, TSA or bothcompounds. The transfection efficiency was estimated tobetween 40 and 60%. The control siRNA and GFPoverexpression did not affect basal or TSA-increasedH4K16 acetylation in H157 cells (Figure 4d; data not
VP16
Su
rviv
al f
ract
ion
(%
)
100
80
60
40
20
0VP16
- - -+ + -- - +- - +
- + -- + +
TSAVPA
VP16- - -+ + -- - +- - +
- + -- + +
TSAVPA
Ave
rag
e si
ze o
f co
lon
ies
(A.U
)
0
100
200
300
400
500
600
TS
AV
PA
Co
ntr
ol
Figure 2 Effect of histone deacetylase inhibitors (HDACi) on cell proliferation and cell survival response to residual DNA damage.(a–c) H157 cells were treated with valproic acid (VPA, 1mm), or trichostatin A (TSA, 20 ng/ml) alone or in combination with VP16(5mm) for 24 h. After wash, cells were incubated for an additional 10-day period to allow colonies formation. The cultures were fixedwith methanol and stained by Giemsa’s method, and the number of colonies was counted. (a) Images showing colony densities anddistribution at day 10 after the different treatments. (b) The survival fraction was calculated as the colony formation efficiency of thetreated cells compared with control set to 100%. (c) Average size of the individual colonies after treatment was measured with ImageJsoftware. (b, c) Data are pooled from three independent experiment and bars and error bars represent mean with s.e.m.
Figure 1 Histone deacetylase inhibitors (HDACi) promote VP16-induced DNA damage and cell death. H157 cells were treated withvalproic acid (VPA, 1mM) or trichostatin A (TSA, 20 ng/ml) alone or in combination with VP16 (5 mM) for 24 h (a–d) or for 1 h (e).(a, b) Cells were fixed and nuclei were stained with Hoechst 33342. Cell death was assayed by scoring cells presenting fragmented,damaged or condensed nuclei. (a) Different types of nuclear morphology were observed: condensed nuclei, nuclei with deformedchromatin, polylobulated nuclei and mitotic catastrophe. Increase in nuclei sizes on HDACi exposure was estimated using the 10mmscale bar using the Zeiss confocal software. (b) A total of 300 nuclei were analyzed in duplicate and each experiment was repeated threetimes. (c, d) DNA damage (tail moment) was assessed by comet assay. (c) Pseudocolor of isolated nuclei was attributed by theAutoComet program. (d) The average tail moment values were determined randomly scoring 50 nuclei per sample. (e) Cells were fixedand processed for immunofluorescence using antibodies directed against phosphorylated histone H2AX (g-H2AX). Nuclei werecounterstained with 40,6-diamidino-2-phenylindole (DAPI). Representative confocal images of immunostaining with g-H2AX (green),and nuclear DAPI staining (blue) of H157 cells are shown. In b and d, bars and error bars represent mean with s.e.m.
hMOF/SIRT1 control H4K16Ac and VP16 sensitivityN Hajji et al
2195
Oncogene
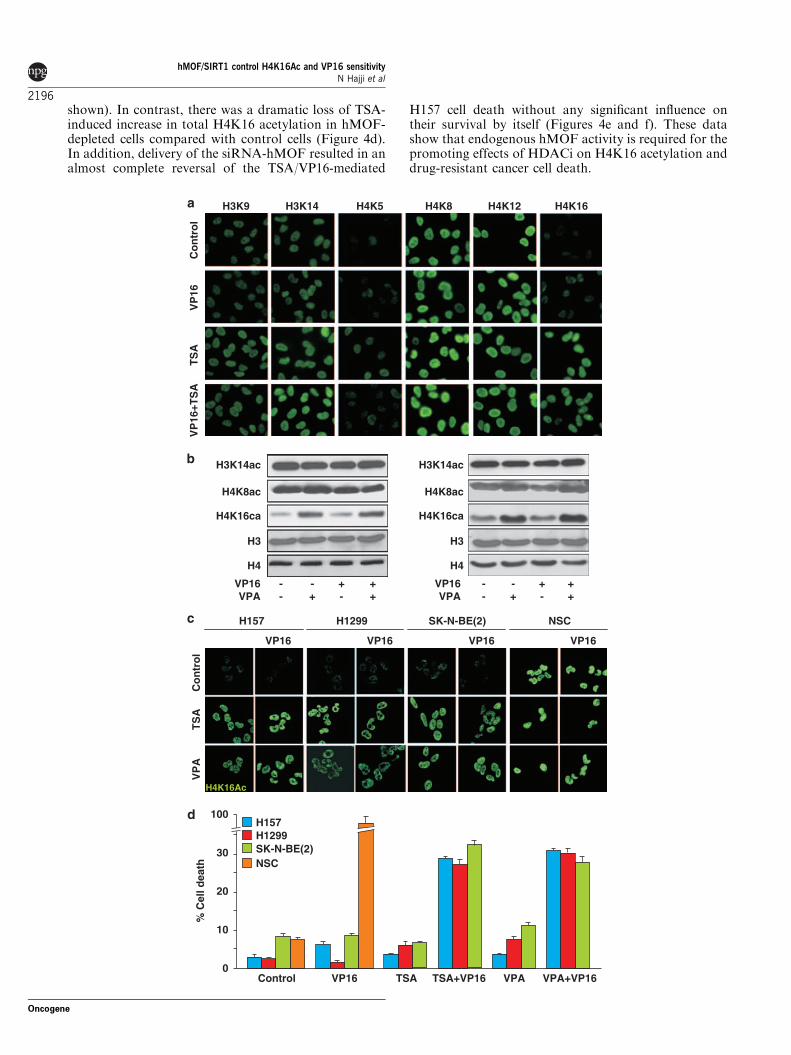
shown). In contrast, there was a dramatic loss of TSA-induced increase in total H4K16 acetylation in hMOF-depleted cells compared with control cells (Figure 4d).In addition, delivery of the siRNA-hMOF resulted in analmost complete reversal of the TSA/VP16-mediated
H157 cell death without any significant influence ontheir survival by itself (Figures 4e and f). These datashow that endogenous hMOF activity is required for thepromoting effects of HDACi on H4K16 acetylation anddrug-resistant cancer cell death.
H4K8 H4K12 H4K16H3K14 H4K5H3K9
H1299 SK-N-BE(2)
VP16
NSC
VP16 VP16VP16
H157
H4K16Ac
H157
% C
ell d
eath
100
30
20
10
0
H1299SK-N-BE(2)
VP
16+T
SA
TS
AV
P16
Co
ntr
ol
H3
H4K16ca
H4K8ac
H3K14ac
VP16VPA -
-+-
-+ +
+VP16VPA -
-+-
-+ +
+
H4
H3
H4K16ca
H4K8ac
H3K14ac
H4
NSC
TSA TSA+VP16 VPA+VP16VPAControl VP16
VP
AT
SA
Co
ntr
ol
hMOF/SIRT1 control H4K16Ac and VP16 sensitivityN Hajji et al
2196
Oncogene

SIRT1 inhibition mediates increase in H4K16 acetylationand promotes cell deathWe next asked which HDAC could counteract the effectof hMOF by deacetylating H4K16Ac and contribute tothe resistance to VP16-induced cell death. Whereas classI HDACs show little substrate specificity and the directHDAC activity of class II HDACs has been debated, theclass III HDAC SIRT1 has been reported to showpreference for deacetylation of H4K16 (Vaquero et al.,2004, 2007; Pruitt et al., 2006). Further, HDACs 1–8were ubiquitously expressed in H157 cells as assessed byimmunoblotting, and did not show any change inexpression after VPA or TSA exposure (data notshown). VPA and TSA indeed bind directly to andinhibit class I and class II HDACs, but importantlythese molecules have also been shown to affect class IIIsirtuin deacetylases indirectly, including SIRT1, inmammalian cells by regulating their gene expression(Kyrylenko et al., 2003; KW and OH, unpublishedobservations).
We therefore examined the effects of TSA and VPAexposure on SIRT1 protein levels (Figure 5a). Inaddition, we analyzed the protein levels of hMOF andSIRT2, a cytoplasmic HDAC that has been shown todeacetylate H4K16Ac during mitosis (Vaquero et al.,2006, 2007). SIRT1, SIRT2 and hMOF proteins were allexpressed in untreated cells. However, whereas theexpression levels of both hMOF and SIRT2 remainedunaltered on HDACi treatment, both TSA and VPAstimulation led to a significant decrease in SIRT1protein levels within 3 h (Figure 5a).
H157 cells were next co-transfected with DsRed-Mito and either an siRNA directed against humanSIRT1 or a control siRNA, followed by subsequenttreatment with VP16. Confocal imaging revealed a clearincrease in H4K16 acetylation in SIRT1-depleted H157cells compared to neighboring nontransfected cells(Figure 5b). H157 cells co-transfected with GFP/SIRT1siRNA or GFP/control siRNA were then treated withVP16 or vehicle for 12 h and the cell death was assessed(Figure 5c). In contrast to hMOF overexpression,incubation of H157 cells with siRNA directed againstSIRT1 was sufficient to reduce their survival, indicatingthat SIRT1 may have multiple roles in cell death andsurvival as previously reported (Figure 5c; Deng, 2009;Liu et al., 2009). Importantly however, SIRT1 silencingresulted in a significant increase in VP16-induced celldeath. Similar results regarding sensitization andH4K16 acetylation were obtained using EX-527, aselective inhibitor of SIRT1 that does not inhibit HDACor other sirtuin deacetylase family members (Napper
et al., 2005; Figures 5d and e). These data indicate thatinhibition of SIRT1 activity resulted in the sensitizationto cell death induced by topoisomerase II inhibitor, andthat this effect was associated with an increase in totalH4K16 acetylation.
SIRT1 downregulation is required for TSA-mediatedincrease in H4K16 acetylation and death-promoting effectWe next assessed whether SIRT1 downregulation wasrequired for the HDACi promotion of cancer cell death.We therefore transiently overexpressed Flag-taggedSIRT1 in H157 cells and treated them with TSA for3 h. Overexpression of SIRT1 in H157 cells resulted inan abolishment of the TSA-induced increase in H4K16acetylation as assessed by single-cell analysis (Figure 5f).We next co-transfected GFP with the Flag-SIRT1construct or empty vector in H157 cells, followed bytreatment with vehicle, TSA, VP16 or the combinedtreatment TSA/VP16 for 12 h and assessed the celldeath. This experiment revealed that whereas SIRT1overexpression by itself did not affect the rate of celldeath, it drastically decreased the killing effect of TSA/VP16 (Figures 5g and h). These experiments thussuggest that overexpression of SIRT1 is sufficient toinhibit the sensitizing effects of TSA on drug-resistantNSCLC cells.
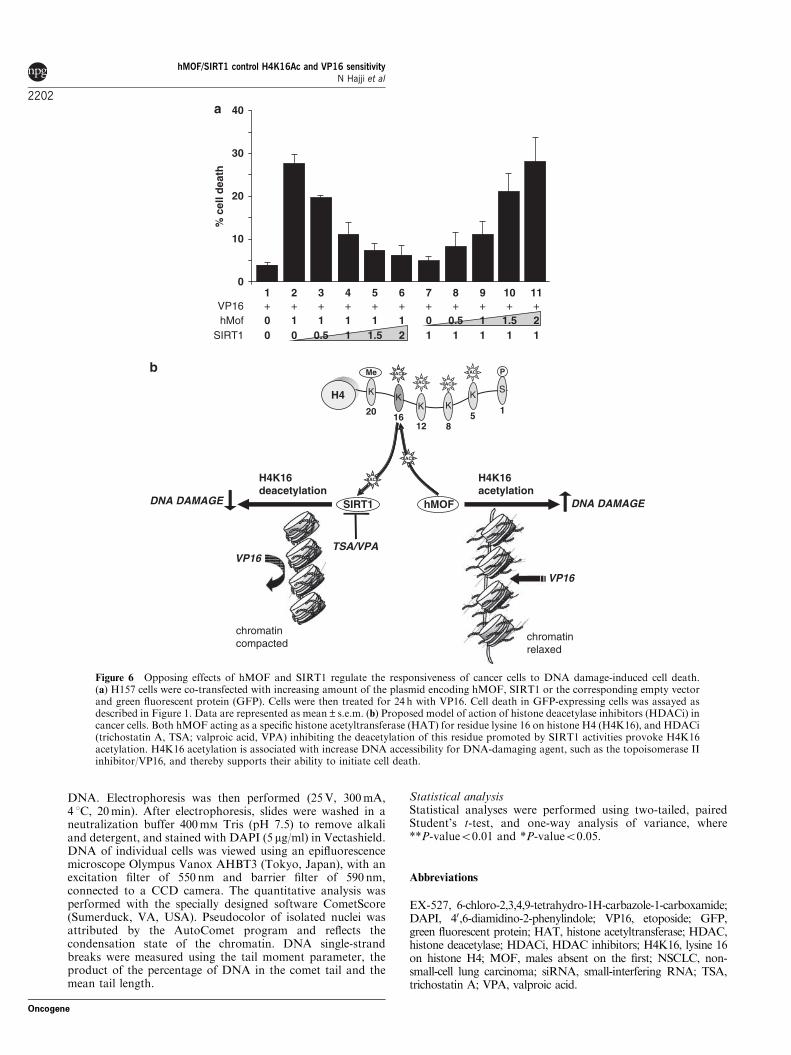
Opposing effects of hMOF and SIRT1 regulate theresponse of cancer cells to topoisomerase II inhibitionOn the basis of these results, we hypothesized that adirect balance between the opposing effects of hMOFand SIRT1 may control the responsiveness of cancercells to the topoisomerase II inhibitor VP16. To test thishypothesis, we co-transfected H157 cells with differentratios of SIRT1 and hMOF (1:0; 1:0.5; 1:1; 1:1.5 and1:2), treated with VP16 for 24 h and subsequentlyexamined for cell death (Figure 6a). Intriguingly,transfecting increasing levels of SIRT1 inhibited thestimulation by hMOF on VP16-induced cell death in adose-dependent manner, whereas increasing levels ofhMOF expression conversely inhibited the survivaleffect of SIRT1 expression after exposure to thetopoisomerase II inhibitor.
Discussion
A number of studies have previously shown thattreatment with diverse HDACi triggers or promotes celldeath in different types of cancer cells, resulting in a
Figure 3 Histone deacetylase inhibitors (HDACi)-induced lysine 16 on histone H4 (H4K16) acetylation in cancer cells associates withtheir sensitization to VP16 killing effect. H157 non-small-cell lung carcinoma (NSCLC) cells (a–d), H1299 NSCLC cells, SK-N-BE(2)neuroblastoma cells and neural stem cells (c, d) were treated with valproic acid (VPA, 1mM), or trichostatin A (TSA, 20 ng/ml) alone orin combination with VP16 (5mM) for 3 h (a–c) or 24 h (d). (a) H157 cells were then fixed and processed for immunofluorescence usingantibodies directed against monoacetylated lysine K9 or K14 on histone H3 and lysine K5, K8, K12 or K16 on histone H4. (b) Acid-soluble nuclear proteins were extracted and analyzed by immunoblot for acetylation at residues H3K14, H4K8 and H4K16. Theamount of histones loaded was controlled by staining the membrane for histones H3 and H4. (c) Cells were fixed and processed forimmunofluorescence using antibodies directed against H4K16Ac. (d) Cells were fixed and nuclei were stained with Hoechst 33342. Celldeath was assayed as described in Figure 1. A total of 300 nuclei were analyzed in duplicate and each experiment was repeated threetimes. Data are represented as mean±s.e.m.
hMOF/SIRT1 control H4K16Ac and VP16 sensitivityN Hajji et al
2197
Oncogene

series of clinical trials involving HDACi treatment(Villar-Garea and Esteller, 2004; Drummond et al.,2005; Bolden et al., 2006; Minucci and Pelicci, 2006).
Although several mechanisms have been suggested,a common mechanism underlying similar effects ofHDACi with distinct structure and function has
**
VP16Control
% c
ell d
eath
100
80
60
40
20
0
siRNA CFP+CFP
siRNA hMOF+GFP
% c
ell d
eath
100
80
60
40
20
0VP16TSA - - + +
- + - +
**
MergehMOF
Co
ntr
ol
DAPI
siRNA CFP+GFP +TSA
siR
NA
CF
P+G
FP
siRNA hMOF+GFP +TSA
TSA+VP16
siR
NA
hM
OF
+GF
P
GFP hMOF MergepcDNA+GFPhMOF+GFP
Co
ntr
ol
VP
16
DA
PI
GF
P
GFP DAPI Merge
acH
4K16
Mer
ge
DAPI H4K16ac
Figure 4 Human males absent on the first (hMOF) provokes lysine 16 on histone H4 (H4K16) acetylation and sensitizes H157 cells toVP16-induced cell death. (a) H157 cells were transfected with a plasmid encoding HA-tagged hMOF or the corresponding emptyvector. Cells were fixed and immunofluorescence detection was performed using antibodies directed against the HA tag and H4K16Ac.Nuclei were counterstained with 40,6-diamidino-2-phenylindole (DAPI). Confocal images of immunostaining with HA-hMOF (red),H4K16Ac (green) antibodies and nuclear DAPI staining (blue) of H157 cells on hMOF overexpression are depicted. (b, c) H157 cellswere co-transfected with a plasmid encoding HA-tagged hMOF or the corresponding empty vector and green fluorescent protein(GFP) and then treated with VP16 (5mM) for 24 h (b). Cells were fixed and immunofluorescence detection was performed usingantibodies directed against the HA tag of hMOF. Nuclei were counterstained with DAPI. Confocal images of GFP (green),immunostaining with HA-hMOF antibodies (red) and nuclear DAPI staining (blue) of H157 cells on hMOF overexpression andtreatment are depicted. (c) Cell death was assayed as described in Figure 1. A total of 300 nuclei from GFP-expressing cells wereanalyzed in duplicate and each experiment was repeated three times. Data are represented as mean±s.e.m. (d–f) H157 cells weretransiently co-transfected with plasmid encoding GFP together with small-interfering RNAs (siRNAs) pool designed to interferespecifically with the expression of human MOF. As control, an siRNA directed against enhanced cyan fluorescent protein (ECFP) wasused. At 24 h after transfection, cells were treated with VP16, trichostatin A (TSA) or a combination of both, and incubated foradditional 24 h. (d) Confocal analysis of H4K16 acetylation performed by immunofluorescence using a polyclonal antibody againstH4K16Ac revealed by an Alexa Fluor 594-conjugated secondary antibody. Nuclei were counterstained with DAPI. GFP was used astransfection maker. Confocal images of GFP (green), immunostaining with H4K16Ac (red) antibodies and nuclear DAPI staining(blue) of H157 cells after TSA treatment are depicted. (e) Confocal images of GFP (green), nuclear DAPI staining (blue) of H157 cellsafter TSA/VP16 treatment are depicted. Asterisks in panel indicate condensed/fragmented cell nuclei. These experiments wereperformed three times with similar results. (f) Cell death was assayed by scoring of GFP-expressing cells presenting condensed/fragmented nuclei. A total of 300 nuclei were analyzed in duplicate and each experiment was repeated three times. Bars and error barsrepresent mean with s.e.m.
hMOF/SIRT1 control H4K16Ac and VP16 sensitivityN Hajji et al
2198
Oncogene

remained unclear. Our study results reveal a novel andunexpected mechanism underlying HDACi-inducedeffects on multidrug-resistant cells to topoisomerase IIinhibitor involving hMOF and SIRT1. We speculatethat the effects could depend on the opposing activitiesof hMOF and SIRT1 on H4K16 acetylation and thusincreased chromatin accessibility (Shogren-Knaak et al.,2006; Figure 6b).
Importantly, this single histone modification influ-ences both higher-order chromatin structure andfunctional interactions between histones as well as anon-histone protein and the chromatin fiber (Shogren-Knaak et al., 2006). It has been shown that reduction inH4K16 acetylation correlate with tumor progression(Fraga et al., 2005). In addition, the enzymaticcomponents of the regulatory system described in thisstudy have been reported to be altered in cancer disease,although it should noted that both hMOF and SIRT1have been associated with both survival and death-promoting mechanisms. For example, hMOF expres-sion is frequently downregulated in primary breastcarcinoma and medulloblastoma and constitutes abiomarker for clinical outcome in medulloblastoma(Pfister et al., 2008). There is however additionalevidence that hMOF, similar to the related HATTip60, is involved in mechanisms leading to DNArepair as well as in death signaling pathways (Ikuraet al., 2000). The cell fate may depend on the amount ofDNA damage, exemplified by the fact that Tip60appears to exert its effect primarily at the initial phaseof DNA damage (Ikura et al., 2000).
The function of SIRT1 in different cell contexts seemsto be even more complex. SIRT1 expression issignificantly elevated in prostate and colon cancers(Huffman et al., 2007; Stunkel et al., 2007), and HIC1(hypermethylated in cancer 1), an inhibitor of SIRT1transcription, is epigenetically silenced in many tumors(Chen et al., 2005). SIRT1 has however in addition toH4K16 been shown to be a protein deacetylase, andplural effects of SIRT1 on cell survival have beenreported. SIRT1 has been shown to promote cellsurvival by deacetylating the DNA-damage-protectingprotein NBS1 (Yuan et al., 2007). SIRT1 has also beenshown to regulate acetylation levels of p53, but theimplications of this regulation are less clear as it hasbeen reported on one hand that SIRT1 deacetylationinfluences p53-dependent DNA damage response, buton the other that the SIRT1-mediated p53 acetylationhas a minor biological role (Vaziri et al., 2001; Mottaet al., 2004). It is therefore worth noting that none of thecancer cell lines used in this study expresses functionalp53. Furthermore, SIRT1 does not exert protectiveeffect after DNA damage in all cell types (Solomonet al., 2006), and cytoplasm-localized SIRT1 has evenbeen reported to enhance apoptosis (Jin et al., 2007).Thus the subcellular localization, expression and activitylevels of SIRT1 are essential for its effect on cell survival(Deng, 2009; Liu et al., 2009).
Altogether, our observations support a model wherethe hMOF/SIRT1 enzymatic system regulates H4K16acetylation levels and where its deregulation is asso-
ciated with cancer occurrence and sensitivity to topo-isomerase inhibitor (Figure 6b). The results of our studymay further provide a mechanism justifying ongoingclinical trials on neuroectodermal tumors using com-bined treatment with valproate and etoposide (www.clinicaltrials.gov).
Materials and methods
ReagentsEtoposide (Vepeside, VP16, 20mg/ml in dextran) was fromBristol-Myers (New York, NY, USA). 6-Chloro-2,3,4,9-tetrahydro-1H-carbazole-1-carboxamide (EX-527; 50mm inethanol; Tocris Bioscience, Ellisville, MO, USA), TSA (1mmin dimethyl sulfoxide; Sigma) and 2-propylpentanoic acid(VPA, 1M in ethanol; Sigma) were stored at �20 1C in dark.Subsequent dilutions were performed in culture medium.
Cell culture and treatmentsHuman non-small-cell lung carcinoma cell line NCL-H157(CRL-5802; ATCC, Manassas, VA, USA) and NCI-H1299(CRL-5803; ATCC) and human neuroblastoma SK-N-BE(2)cell line (CRL-2271; ATCC) were used in this study (Josephet al., 2001, 2002). None of the cell lines express functionalp53: one is p53 null, one has a frameshift in p53 and the thirdhas a p53 hot stop mutation (p53 mutation databases at http://www-p53.iarc.fr and http://p53.free.fr). Cells were cultured at37 1C, 5% CO2 in RPMI 1640 medium (H157 and H1299), ormodified Eagle’s medium mixed with F-12 Nutrient Mixture(Ham) at ratio 1:1 (SK-N-BE(2)), all supplemented with 10%heat-inactivated fetal calf serum, 2mm l-glutamine, penicillin(100U/ml) and streptomycin (100mg/ml).Isolation and culture of multipotent neural progenitors,
referred to as neural stem cells, were performed essentially asdescribed elsewhere (Jepsen et al., 2007; Andersson et al.,2009). In brief, cortices from rats at embryonic day 15 weredissected and mechanically dispersed in a modified serum-free,4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)-free N2-supplemented Dulbecco’s modified Eagle’s medium/F12 medium. The primary cells were plated on 35mm dishescoated with poly-l-ornithine and fibronectin (Sigma). The cellswere treated with human recombinant basic FGF2 at 10 ng/mlevery 24 h and the N2 medium was replaced every 48 h. Alltreatments were performed after the first passage when 499%of the cells showed nestin expression and typical neural stemcell morphology and o1% of the cells expressed neuronal andglial differentiation markers.At 24 h after setting in culture dishes with fresh medium, we
treated cells with 5 mm VP16, 1mm VPA, 20 ng/ml TSA aloneor in combination. The cell density was kept at levels allowingexponential growth.
Detection of cell deathAfter treatment, cells were fixed in 4% paraformaldehyde andsubsequently DNA was stained with Hoechst 33342 (0.1 mg/ml;Molecular Probes, Paisley, UK). The number of dying cellswas measured quantitatively by assessing the percentage ofcells with fragmented, damaged or condensed nuclei.
Colony formation assayCell survival after treatment was measured by clonogenic assayin monolayer (Gallego et al., 2004). Single-cell suspensions wereplated out at 500 cells per 100mm dish in triplicate. Cells were
hMOF/SIRT1 control H4K16Ac and VP16 sensitivityN Hajji et al
2199
Oncogene

then exposed for 24h to the HDACi alone or in combinationwith VP16. After washes, cells were incubated in completeculture medium at 37 1C for an additional 10-day period.Eventually, the medium was aspirated and dishes were fixed inmethanol and stained with 3% Giemsa for 30min, then rinsedand air-dried. Surviving colonies made up of more than 50 cellsper colony were counted. The size of the individual colonies aftertreatment was measured with ImageJ software (US NationalInstitutes of Health, Bethesda, MD, USA). The survival datawere normalized to the cloning efficiencies of untreated controls.Three separate experiments were carried out for each treatment.
Extraction of acid-soluble nuclear proteins, western blotTo detect single acetylated lysine residue on histone H4 andH3 tails, we extracted acid-soluble nuclear proteins as follows:
cells were spun down, washed with phosphate-buffered saline,and resuspended in an histone lysis buffer containing 10mM
Tris (pH 6.5), 50mM sodium bisulfate, 10mM MgCl2, 8.6%sucrose, 1% Triton X-100 and 0.2 M sulfuric acid, andincubated on ice for 1 h. Samples were then centrifuged at11 000 g for 30min at 4 1C and the supernatant wereprecipitated with acetone overnight, and centrifuged at11 000 g for 1 h at 4 1C. Pellets were dried out at roomtemperature. Acid-extracted histones were obtained by cen-trifugation at 11 000 g for 1 h at 4 1C, and the concentrations ofhistones were measured by the Bradford method. Afterseparation by SDS–polyacrylamide gel electrophoresis (15%gel), proteins were transferred on to nitrocellulose membrane(Amersham, Uppsala, Sweden). Membranes were blocked for2 h with 5% non-fat milk in phosphate-buffered saline at roomtemperature and subsequently probed overnight with primary
% c
ell d
eath
% c
ell d
eath
VP16EX-527
--
+-
-+ +
+
40
30
20
10
0
% c
ell d
eath
80
60
40
20
0
100siRNACFP+GFP
siRNASIRT1+GFP
VP16Control
vector+GFPSIRT1+GFP
100
80
60
40
20
0VP16TSA - - + +
- + - +
60kDa
43kDa
30kDa
hMOF
SIRT2
SIRT1DAPI
DAPI
Merge
Merge
Co
ntr
ol
EX
-527
H4K16Ac
H4K16Ac
H4K16AcSIRT1DAPI
TS
Ave
cto
r +G
FP
SIR
T1
+GF
P
siR
NA
SIR
T1
+DsR
edm
ito
DsRedmito
GAPDH
120kDa
VPATSA
--
+-
-+
Fo
ld o
ver
con
rol SIRT1
SIRT2
VPATSA
--
+-
-+
1.0
2.0
0
0.5
1.5 hMOF
**
**
**
TSA+VP16
MergeDAPIGFP
hMOF/SIRT1 control H4K16Ac and VP16 sensitivityN Hajji et al
2200
Oncogene

antibodies raised against histone H3 and H4 (Abcam,Cambridge, UK) or acetylated histone lysine H3K9, H3K14,H4K5, H4K8, H4K12 and H4K16 (Upstate Biotechnology,Billerica, MA, USA). Binding of primary antibodies wasdetected with a horseradish-peroxidase-conjugated goat anti-mouse or goat anti-rabbit IgGs (Pierce, Rockford, IL, USA).After repeated washing in phosphate-buffered saline, bandswere visualized by enhanced chemiluminescence (ECL Plus)following the manufacturer’s instructions (Amersham).
TransfectionPlasmids encoding HA-human MOF and Flag-human SIRT1were generous gifts from Robert G Roeder (The RockefellerUniversity) and Leonard Guarente (Massachusetts Institute ofTechnology), respectively. EGFP and DsRed-Mito plasmidused as control were from Clontech (Saint-Germain-en-Laye,France). At 24 h after setting in culture dishes with freshmedium, cells were transfected using Lipofectamine 2000(Invitrogen, Paisley, UK) according to the manufacturer’sprotocol. Transfection efficiency was monitored by fluores-cence using EGFP plasmid. Small-interfering RNA (siRNA)sequence for ECFP: 50-GGCCUUGAUGCCGUUCUUC99-30
(Eurogentech, Seraing, Belgium). siRNA MOF (human,MYST1 NM_032188), siGenome ON-TARGET plus SMART-pool (catalog no L-014800), siRNA SIRT1 (human, SIR2L1NM_012238), siGenome ON-TARGET plus SMARTpool (catno L-003540), both were from Dharmacon (Chicago, IL, USA).siRNA knockdown efficiency was confirmed by immunoblot-ting (Supplementary Figure S1).
Immunofluorescence and confocal microscopyFor confocal microscope analysis, the adherent cells weregrown on coverslips. Anti-HA (Roche, Indianapolis, IN,USA), phosphorylated g-H2AX (Upstate Biotechnology) andacetylated H3K9, H3K14, H4K5, H4K8, H4K12, H4K16(Upstate Biotechnology) antibodies were used in immuno-detection. Alexa Fluor 488- or 594-conjugated anti-IgGs wereused as secondary antibodies (Molecular Probes). Paraformal-dehyde-fixed cells were blocked in HEPES/3% bovine serumalbumin/0.3% Triton X-100 and incubated with primary (4 1C,overnight) and secondary antibodies (room temperature, 1 h).
Cell nuclei were counterstained with 40,6-diamidino-2-phenyl-indole (DAPI; 1mg/ml). Samples were mounted with Vecta-shield (Vector Laboratories, Burlingame, CA, USA) andanalyzed with Zeiss 510 META confocal laser scanningmicroscopy (Zeiss, Jena, Germany) equipped with an invertedZeiss Axiovert 200 microscope with an a � 63 oil immersionobjective. Mix dyes were acquired by sequential multiplechannel fluorescence scanning to avoid bleeds through.Routinely, 0.15–0.20mM thick focal planes were scanned.Chats were quantified and images were processed using theZeiss software (LSM 510 Meta).
Immunoblotting analysisTotal protein extracts were carried out as reported (Nymanet al., 2009). For immunoblot analysis, protein extracts wereresolved on SDS–polyacrylamide gel electrophoresis andblotted; protein pieces were detected with antibodies raisedagainst SIRT1, SIRT2 (both from Cell Signaling, Beverely,MA, USA), and hMOF (Santa Cruz Biotechnology, SantaCruz, CA, USA). Immunoblot with anti-G3PDH (Trevigen,Gaithersburg, MD, USA) was used for standardization ofprotein loading.
Comet assayComet assay essentially measures the degree of fragmentationof DNA within the cell. The comet assay was performed asdescribed previously (Singh et al., 1988). Briefly, 10 000 cellswere mixed with 85ml low-melting agarose (0.7% in phos-phate-buffered saline) kept at 37 1C and the cell–agarosemixture was placed on 1% normal-melting agarose-coatedglass slides and covered with a coverslip. The cell–agarosemixtures were allowed to solidify for 10min at 4 1C. Thecoverslip was removed and further 100 ml low-melting agarosewas added. After solidification at 4 1C, the samples wereincubated for 60min in a chilled lysis buffer containing 2.5MNaCl, 100mM Na2EDTA, 10mM Tris, 1% sodium sarcosinate,1% Triton X-100, and 10% dimethyl sulfoxide (pH 10).Subsequently, the slides were drained and placed in ahorizontal gel electrophoresis unit. The chamber was filledwith chilled electrophoresis buffer for alkaline comet assay(1mM Na2EDTA, 300mM NaOH (pH 12.8)) and the sampleswere left in this solution for 20min to allow unwinding of the
Figure 5 Trichostatin A (TSA)-induced SIRT1 down-regulation triggers lysine 16 on histone H4 (H4K16) acetylation and sensitizesH157 cells to VP16-induced cell death. (a) H157 cells were treated with valproic acid (VPA, 1mm) or TSA (20 ng/ml) for 3 h. Total cellextracts were prepared and subjected to immunoblotting with anti-SIRT1, SIRT2 and hMOF antibodies. G3PDH was used asstandard for equal loading of protein. (b, c) H157 cells were transiently co-transfected with plasmid encoding green fluorescent protein(GFP) together with small-interfering RNAs (siRNAs) pool designed to interfere specifically with the expression of human SIRT1. Ascontrol, an siRNA directed against enhanced cyan fluorescent protein (ECFP) was used. (b) Confocal analysis of H4K16 acetylationwas performed by immunofluorescence using a polyclonal antibody against H4K16Ac revealed by an Alexa Fluor 488-conjugatedsecondary antibody. Nuclei were counterstained with 40,6-diamidino-2-phenylindole (DAPI). DsRed-Mito was used as transfectionmaker. Confocal images of DsRed-Mito (red), immunostaining with H4K16Ac (green) antibodies and nuclear DAPI staining (blue) ofH157 cells after TSA treatment are depicted. (c) At 24 h after transfection, cells were treated with VP16, and incubated for additional12 h. Cell death was assayed by scoring of GFP-expressing cells presenting condensed/fragmented nuclei. A total of 300 nuclei wereanalyzed in duplicate and each experiment was repeated three times. Bars and error bars represent mean with s.e.m.(d) H157 cells were treated with EX-527 (1mm) for 3 h. Cells were fixed and immunofluorescence detection was performed usingantibodies directed against H4K16Ac. Nuclei were counterstained with DAPI. (e) H157 cells were treated with EX-527 (1mm) alone orin combination with VP16 (5mm) for 24 h. Cell death was assayed as in Figure 1. (f) H157 cells were transfected with a plasmidencoding Flag-tagged SIRT1 or the corresponding empty vector. At 24 h after transfection, cells were treated with TSA, and incubatedfor additional 3 h. Cells were fixed and immunofluorescence detection was performed using antibodies directed against H4K16Ac.Nuclei were counterstained with DAPI. Confocal images of immunostaining with Flag (red), H4K16Ac (green) antibodies and nuclearDAPI staining (blue) of H157 cells on SIRT1 overexpression are depicted. (g, h) H157 cells were transiently co-transfected withplasmid encoding GFP together with Flag-tagged SIRT1 or the corresponding empty vector. At 24 h after transfection, cells weretreated with VP16, TSA or a combination of both, and incubated for additional 12 h. (g) Confocal images of GFP (green), nuclearDAPI staining (blue) of H157 cells after TSA/VP16 treatment are depicted. Asterisks in the panel indicate condensed/fragmented cellnuclei. These experiments were performed three times with similar results. (h) Cell death was assayed by scoring of GFP-expressingcells presenting condensed/fragmented nuclei. A total of 300 nuclei were analyzed in duplicate and each experiment was repeated threetimes. Bars and error bars represent mean with s.e.m.
hMOF/SIRT1 control H4K16Ac and VP16 sensitivityN Hajji et al
2201
Oncogene
DNA. Electrophoresis was then performed (25V, 300mA,4 1C, 20min). After electrophoresis, slides were washed in aneutralization buffer 400mM Tris (pH 7.5) to remove alkaliand detergent, and stained with DAPI (5 mg/ml) in Vectashield.DNA of individual cells was viewed using an epifluorescencemicroscope Olympus Vanox AHBT3 (Tokyo, Japan), with anexcitation filter of 550 nm and barrier filter of 590 nm,connected to a CCD camera. The quantitative analysis wasperformed with the specially designed software CometScore(Sumerduck, VA, USA). Pseudocolor of isolated nuclei wasattributed by the AutoComet program and reflects thecondensation state of the chromatin. DNA single-strandbreaks were measured using the tail moment parameter, theproduct of the percentage of DNA in the comet tail and themean tail length.
Statistical analysisStatistical analyses were performed using two-tailed, pairedStudent’s t-test, and one-way analysis of variance, where**P-valueo0.01 and *P-valueo0.05.
Abbreviations
EX-527, 6-chloro-2,3,4,9-tetrahydro-1H-carbazole-1-carboxamide;DAPI, 40,6-diamidino-2-phenylindole; VP16, etoposide; GFP,green fluorescent protein; HAT, histone acetyltransferase; HDAC,histone deacetylase; HDACi, HDAC inhibitors; H4K16, lysine 16on histone H4; MOF, males absent on the first; NSCLC, non-small-cell lung carcinoma; siRNA, small-interfering RNA; TSA,trichostatin A; VPA, valproic acid.
VP16hMof
SIRT1
H4 KK
KKK
S
PMe
2016
125
8
1
SIRT1 hMOF
AC
ACAC
AC
H4K16acetylation
H4K16deacetylation
TSA/VPA
DNA DAMAGE DNA DAMAGE
VP16
chromatincompacted
chromatinrelaxed
VP16
% c
ell d
eath
40
30
20
10
0
10 0 0.5 1 1.51 1 11 200 0.5 1 1 1 1 121.5 1
+ + + + + + + + ++ +1 2 3 4 6 7 8 9 105 11
AC
AC
Figure 6 Opposing effects of hMOF and SIRT1 regulate the responsiveness of cancer cells to DNA damage-induced cell death.(a) H157 cells were co-transfected with increasing amount of the plasmid encoding hMOF, SIRT1 or the corresponding empty vectorand green fluorescent protein (GFP). Cells were then treated for 24 h with VP16. Cell death in GFP-expressing cells was assayed asdescribed in Figure 1. Data are represented as mean±s.e.m. (b) Proposed model of action of histone deacetylase inhibitors (HDACi) incancer cells. Both hMOF acting as a specific histone acetyltransferase (HAT) for residue lysine 16 on histone H4 (H4K16), and HDACi(trichostatin A, TSA; valproic acid, VPA) inhibiting the deacetylation of this residue promoted by SIRT1 activities provoke H4K16acetylation. H4K16 acetylation is associated with increase DNA accessibility for DNA-damaging agent, such as the topoisomerase IIinhibitor/VP16, and thereby supports their ability to initiate cell death.
hMOF/SIRT1 control H4K16Ac and VP16 sensitivityN Hajji et al
2202
Oncogene

Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
We thank RG Roeder (The Rockefeller University), L Guarente(Massachusetts Institute of Technology) and M Henriksson
(Karolinska Institutet) for providing us with hMOF DNAconstruct, SIRT1 DNA construct and SK-N-BE(2) cell line,respectively, and S Orrenius, T Perlmann and members ofHermanson and Joseph’s labs for helpful comments. This workwas supported by the Swedish Research Council, the SwedishCancer Society, the Ake Wiberg Foundation, the SwedishMedical Society, the Karolinska Institutet Foundations (KICancer) (BJ and OH), SSF (CEDB & OBOE) and the SwedishChildren’s Cancer Foundation (OH).
References
Andersson T, Sodersten E, Duckworth JK, Cascante A, Fritz N,Sacchetti P et al. (2009). CXXC5 is a novel BMP4-regulated modulatorof Wnt signaling in neural stem cells. J Biol Chem 284: 3672–3681.
Bolden JE, Peart MJ, Johnstone RW. (2006). Anticancer activities ofhistone deacetylase inhibitors. Nat Rev Drug Discov 5: 769–784.
Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R,Kroemer G. (2004). Cell death by mitotic catastrophe: a moleculardefinition. Oncogene 23: 2825–2837.
Chen WY, Wang DH, Yen RC, Luo J, Gu W, Baylin SB. (2005).Tumor suppressor HIC1 directly regulates SIRT1 to modulatep53-dependent DNA-damage responses. Cell 123: 437–448.
Deng CX. (2009). SIRT1, is it a tumor promoter or tumor suppressor?Int J Biol Sci 5: 147–152.
Drummond DC, Noble CO, Kirpotin DB, Guo Z, Scott GK, BenzCC. (2005). Clinical development of histone deacetylase inhibitorsas anticancer agents. Annu Rev Pharmacol Toxicol 45: 495–528.
Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J,Schotta G et al. (2005). Loss of acetylation at Lys16 andtrimethylation at Lys20 of histone H4 is a common hallmark ofhuman cancer. Nat Genet 37: 391–400.
Gallego MA, Joseph B, Hemstrom TH, Tamiji S, Mortier L,Kroemer G et al. (2004). Apoptosis-inducing factor determines thechemoresistance of non-small-cell lung carcinomas. Oncogene 23:6282–6291.
Glozak MA, Sengupta N, Zhang X, Seto E. (2005). Acetylation anddeacetylation of non-histone proteins. Gene 363: 15–23.
Hajji N, Wallenborg K, Vlachos P, Nyman U, Hermanson O, JosephB. (2008). Combinatorial action of the HDAC inhibitor trichostatinA and etoposide induces caspase-mediated AIF-dependent apopto-tic cell death in non-small cell lung carcinoma cells. Oncogene 27:3134–3144.
Hermanson O, Glass CK, Rosenfeld MG. (2002). Nuclear receptorcoregulators: multiple modes of modification. Trends EndocrinolMetab 13: 55–60.
Huffman DM, Grizzle WE, Bamman MM, Kim JS, Eltoum IA,Elgavish A et al. (2007). SIRT1 is significantly elevated in mouseand human prostate cancer. Cancer Res 67: 6612–6618.
Ikura T, Ogryzko VV, Grigoriev M, Groisman R, Wang J, HorikoshiM et al. (2000). Involvement of the TIP60 histone acetylase complexin DNA repair and apoptosis. Cell 102: 463–473.
Jepsen K, Solum D, Zhou T, McEvilly RJ, Kim HJ, Glass CK et al.(2007). SMRT-mediated repression of an H3K27 demethylase inprogression from neural stem cell to neuron. Nature 450: 415–419.
Jin Q, Yan T, Ge X, Sun C, Shi X, Zhai Q. (2007). Cytoplasm-localized SIRT1 enhances apoptosis. J Cell Physiol 213: 88–97.
Joseph B, Ekedahl J, Lewensohn R, Marchetti P, Formstecher P,Zhivotovsky B. (2001). Defective caspase-3 relocalization in non-small cell lung carcinoma. Oncogene 20: 2877–2888.
Joseph B, Marchetti P, Formstecher P, Kroemer G, Lewensohn R,Zhivotovsky B. (2002). Mitochondrial dysfunction is an essentialstep for killing of non-small cell lung carcinomas resistant toconventional treatment. Oncogene 21: 65–77.
Karagiannis TC, El-Osta A. (2006). Modulation of cellular radiationresponses by histone deacetylase inhibitors. Oncogene 25:3885–3893.
Keshelava N, Seeger RC, Groshen S, Reynolds CP. (1998). Drugresistance patterns of human neuroblastoma cell lines derived frompatients at different phases of therapy. Cancer Res 58: 5396–5405.
Kyrylenko S, Kyrylenko O, Suuronen T, Salminen A. (2003).Differential regulation of the Sir2 histone deacetylase gene familyby inhibitors of class I and II histone deacetylases. Cell Mol Life Sci
60: 1990–1997.Liu T, Liu PY, Marshall GM. (2009). The critical role of the class III
histone deacetylase SIRT1 in cancer. Cancer Res 69: 1702–1705.Minucci S, Pelicci PG. (2006). Histone deacetylase inhibitors and the
promise of epigenetic (and more) treatments for cancer. Nat Rev
Cancer 6: 38–51.Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W et al.
(2004). Mammalian SIRT1 represses forkhead transcription factors.Cell 116: 551–563.
Napper AD, Hixon J, McDonagh T, Keavey K, Pons JF, Barker Jet al. (2005). Discovery of indoles as potent and selective inhibitorsof the deacetylase SIRT1. J Med Chem 48: 8045–8054.
Neal KC, Pannuti A, Smith ER, Lucchesi JC. (2000). A new humanmember of the MYST family of histone acetyl transferases with highsequence similarity to DrosophilaMOF. Biochim Biophys Acta 1490:170–174.
Nyman U, Vlachos P, Cascante A, Hermanson O, Zhivotovsky B,Joseph B. (2009). Protein kinase C-dependent phosphorylationregulates the cell cycle-inhibitory function of the p73 carboxyterminus transactivation domain. Mol Cell Biol 29: 1814–1825.
Pfister S, Rea S, Taipale M, Mendrzyk F, Straub B, Ittrich C et al.(2008). The histone acetyltransferase hMOF is frequently down-regulated in primary breast carcinoma and medulloblastoma andconstitutes a biomarker for clinical outcome in medulloblastoma.Int J Cancer 122: 1207–1213.
Pruitt K, Zinn RL, Ohm JE, McGarvey KM, Kang SH, Watkins DNet al. (2006). Inhibition of SIRT1 reactivates silenced cancergenes without loss of promoter DNA hypermethylation. PLoS
Genet 2: e40.Schneider R, Grosschedl R. (2007). Dynamics and interplay of nuclear
architecture, genome organization, and gene expression. Genes Dev
21: 3027–3043.Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson
CL. (2006). Histone H4-K16 acetylation controls chromatinstructure and protein interactions. Science 311: 844–847.
Singh NP, McCoy MT, Tice RR, Schneider EL. (1988). A simpletechnique for quantitation of low levels of DNA damage inindividual cells. Exp Cell Res 175: 184–191.
Smith ER, Cayrou C, Huang R, Lane WS, Cote J, Lucchesi JC. (2005).A human protein complex homologous to the Drosophila MSLcomplex is responsible for the majority of histone H4 acetylation atlysine 16. Mol Cell Biol 25: 9175–9188.
Solomon JM, Pasupuleti R, Xu L, McDonagh T, Curtis R, DiStefanoPS et al. (2006). Inhibition of SIRT1 catalytic activity increases p53acetylation but does not alter cell survival following DNA damage.Mol Cell Biol 26: 28–38.
Stunkel W, Peh BK, Tan YC, Nayagam VM, Wang X, Salto-Tellez Met al. (2007). Function of the SIRT1 protein deacetylase in cancer.Biotechnol J 2: 1360–1368.
hMOF/SIRT1 control H4K16Ac and VP16 sensitivityN Hajji et al
2203
Oncogene

Taipale M, Rea S, Richter K, Vilar A, Lichter P, Imhof A et al. (2005).hMOF histone acetyltransferase is required for histone H4 lysine 16acetylation in mammalian cells. Mol Cell Biol 25: 6798–6810.
Vakifahmetoglu H, Olsson M, Tamm C, Heidari N, Orrenius S,Zhivotovsky B. (2008). DNA damage induces two distinct modes ofcell death in ovarian carcinomas. Cell Death Differ 15: 555–566.
Vaquero A, ScherM, Lee D, Erdjument-Bromage H, Tempst P, ReinbergD. (2004). Human SirT1 interacts with histone H1 and promotesformation of facultative heterochromatin. Mol Cell 16: 93–105.
Vaquero A, Scher MB, Lee DH, Sutton A, Cheng HL, Alt FW et al.(2006). SirT2 is a histone deacetylase with preference for histone H4Lys 16 during mitosis. Genes Dev 20: 1256–1261.
Vaquero A, Sternglanz R, Reinberg D. (2007). NAD+-dependentdeacetylation of H4 lysine 16 by class III HDACs. Oncogene 26:5505–5520.
Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TKet al. (2001). hSIR2(SIRT1) functions as an NAD-dependent p53deacetylase. Cell 107: 149–159.
Villar-Garea A, Esteller M. (2004). Histone deacetylase inhibitors:understanding a new wave of anticancer agents. Int J Cancer 112:171–178.
Yuan Z, Zhang X, Sengupta N, Lane WS, Seto E. (2007). SIRT1regulates the function of the Nijmegen breakage syndrome protein.Mol Cell 27: 149–162.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
hMOF/SIRT1 control H4K16Ac and VP16 sensitivityN Hajji et al
2204
Oncogene




















