
Coin Tossing Explains the Activity of Opposing Microtubule ...
Upload
independentCategory
view
1download
0
Neuropeptide FF Receptors Have Opposing ModulatoryEffects on Nociception
Jelveh Lameh, Fabio Bertozzi, Nicholas Kelly, Paula M. Jacobi, Derek Nguyen,Abhishek Bajpai, Gilles Gaubert, Roger Olsson, and Luis R. GardellACADIA Pharmaceuticals, Inc., San Diego, California
Received December 8, 2009; accepted March 25, 2010
ABSTRACTThe role of neuropeptide FF (NPFF) and its analogs in painmodulation is ambiguous. Although NPFF was first character-ized as an antiopioid peptide, both antinociceptive and prono-ciceptive effects have been reported, depending on the route ofadministration. Currently, two NPFF receptors, termed FF1 andFF2, have been identified and cloned, but their roles in painmodulation remain elusive because of the lack of availability ofselective compounds suitable for systemic administration in invivo models. Ligand-binding studies confirm ubiquitous ex-pression of both subtypes in brain, whereas only FF2 receptorsare expressed spinally. This disparity in localization has servedas the foundation of the hypothesis that FF1 receptors mediatethe pronociceptive actions of NPFF. We have identified novelsmall molecule NPFF receptor agonists and antagonists withvarying degrees of FF2/FF1 functional selectivity. Using these
pharmacological tools in vivo has allowed us to define the rolesof NPFF receptor subtypes as pertains to the modulation ofnociception. We demonstrate that selective FF2 agonism doesnot modulate acute pain but instead ameliorates inflammatoryand neuropathic pains. Treatment with a nonselective FF1/FF2agonist potentiates allodynia in neuropathic rats and increasessensitivity to noxious thermal and to non-noxious mechanicalstimuli in normal rats in an FF1 antagonist-reversible manner.Treatment with FF1 antagonists reversed established mechan-ical allodynia, indicating the possibility of increased NPFF tonethrough FF1 receptors. In conclusion, we provide evidence forthe opposing roles of NPFF receptors and highlight selectiveFF2 agonism and/or selective FF1 antagonism as potentialtargets warranting further investigation.
The role of neuropeptide FF (NPFF) and its analogs in painmodulation is ambiguous. Although “classically” NPFF ischaracterized as an antiopioid peptide, it has been shown toalso elicit robust antinociceptive effects depending on thedose and route of administration used (for review, see Yanget al., 2008). For example, intrathecal administration ofNPFF or of its stable analog [D-Tyr1,(NMe)Phe3]NPFF(1DMe) either elicits antinociception or potentiates the an-tinociceptive effect of morphine (Kontinen and Kalso, 1995;Gouarderes et al., 1996; Xu et al., 1999). In contrast, intra-cerebroventricular administration of these agents antago-nizes morphine-induced antinociception (Gicquel et al., 1992;Oberling et al., 1993; Dupouy and Zajac, 1995).
To date, two NPFF receptor subtypes, termed FF1 andFF2, have been identified and subsequently cloned (Bonini et
al., 2000; Elshourbagy et al., 2000). At present, the specificrole of each NPFF receptor subtype, with respect to painmodulation, has not been adequately described. The lack ofselective pharmacological tools suitable for systemic admin-istration has made elucidating the in vivo pharmacology ofthese receptors challenging. Based on results from ligand-binding studies in rodents, it is clear that both NPFF recep-tors are widely expressed in brain tissue, whereas only theFF2 receptor is expressed spinally (Bonini et al., 2000; Liu etal., 2001; Yang and Iadarola, 2006). The lack of spinal FF1receptors has led to the hypothesis that the antinociceptiveactions of NPFF are mediated via FF2 receptors, whereas thepronociceptive actions of NPFF are mediated via FF1 recep-tors (Liu et al., 2001). We have identified several novel smallmolecule, nonpeptidic, ligands with varying degrees of func-tional selectivity for the NPFF receptor subtypes (Gaubert etal., 2009). The goal of this investigation was to use thesepharmacological tools to define the roles of the NPFF recep-tors as they pertain to pain modulation.
This study was supported by ACADIA Pharmaceuticals, Inc.Article, publication date, and citation information can be found at
http://jpet.aspetjournals.org.doi:10.1124/jpet.109.164384.
ABBREVIATIONS: NPFF, neuropeptide FF; 1DMe, [D-Tyr1,(NMe)Phe3]NPFF; SNL, spinal nerve ligation; NPAF, neuropeptide AF; BIBP-3226,(R)-N(2)-(diphenylacetyl)-N-[(4-hydroxyphenyl)-methyl]-argininamide; dPQR, dansyl-Pro-Gln-Arg-NH2; %MPE, percent maximum possible effect;PWT, paw withdrawal threshold; ANOVA, analysis of variance; CI, confidence interval; R-SAT, receptor selection and amplification technology; h,human; CCK, cholecystokinin.
0022-3565/10/3341-244–254$20.00THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS Vol. 334, No. 1Copyright © 2010 by The American Society for Pharmacology and Experimental Therapeutics 164384/3594465JPET 334:244–254, 2010 Printed in U.S.A.
244
at ASPE
T Journals on Septem
ber 28, 2016jpet.aspetjournals.org
Dow
nloaded from

In this study, we present the in vitro and in vivo profilesfor a representative set of ligands that have allowed us tounmask the putative roles of the NPFF receptor subtypes.Based on our profiling we have identified compounds that,according to in vitro functional assays, are 1) selectiveagonists for FF2 receptors (i.e., AC-263093), 2) nonselec-tive agonists for NPFF receptors (i.e., AC-262616), and 3)selective FF1 receptor antagonists (i.e., AC-262620 andAC-262970). Systemic administration of a nonselectiveFF1/FF2 receptor agonist resulted in a pronociceptive phe-notype as indicated by increased sensitivity to noxiousthermal as well as to non-noxious mechanical stimuli. Instark contrast, systemic administration of a selective FF2receptor agonist resulted in an antinociceptive phenotypeas indicated by 1) attenuation of phase II of the formalintest, 2) reversal of carrageenan-induced thermal hyperal-gesia, and 3) reversal of mechanical hypersensitivity in-duced by L5/L6 spinal nerve ligation (SNL). Similar re-sults were obtained with FF1 receptor antagonists.
Taken together, these data suggest that the antinocicep-tive actions of NPFF are driven by activation of FF2 recep-tors, whereas the pronociceptive and by extension the antio-pioid actions of NPFF are mediated via activation of FF1receptors. Therefore, selective FF2 receptor agonists andselective FF1 receptor antagonists, alone or in combina-tion, may hold promise for the treatment of various typesof chronic pains whether inflammatory or neuropathic inorigin.
Materials and MethodsCompounds and Dosing Solutions. The novel small molecule,
nonpeptidic NPFF ligands AC-262616, AC-263093, AC-262620, andAC-262970 were synthesized by ACADIA Pharmaceuticals. Furtherdetails pertaining to the chemical properties and structures of thesecompounds have been disclosed elsewhere (Gaubert et al., 2009).NPFF (FLFQPQRF-NH2) and NPAF (AGEGLNSQFWSLAAPQRF-NH2) were purchased from American Peptide Co., Inc. (Sunnyvale, CA)and Bachem Biosciences (King of Prussia, PA), respectively. (R)-N(2)-(diphenylacetyl)-N-[(4-hydroxyphenyl)-methyl]-argininamide (BIBP-3226)was purchased from Sigma-Aldrich (St. Louis, MO). The N-terminallymodified tripeptide amide and putative NPFF antagonist, dansyl-PQR-NH2 (dPQR), was synthesized by Phoenix Pharmaceuticals (Burlingame,CA). Morphine sulfate was purchased from Sigma-Aldrich. For intraperi-toneal administration compounds were dissolved in dimethyl sulfoxide andbrought up to volume in sterile 0.9% saline and administered in a finalvolume of 1 ml/kg b.wt. Doses are expressed in terms of freebase content.
Receptor Selection and Amplification Technology. NIH-3T3cells were cotransfected with a mixture of plasmid DNAs containingreceptor of interest and �-galactosidase. The plates were incubatedat 37°C in a humidified CO2 incubator for 18 to 24 h, the transfectionmedium was removed, and cells were harvested and frozen for futureuse. On the day of the assay, cells were plated onto 96-well tissueculture plates containing various concentrations of ligands. For ag-onist assays, only agonist was added to the wells. For antagonistassays, antagonist was added to the wells, and the cells were spikedwith a fixed concentration of agonist (NPFF and NPAF for FF2 andFF1 receptors, respectively) before addition to the wells. After 5 daysof incubation, the cells were assayed for production of �-galactosi-dase by adding ortho-nitrophenyl-�-galactoside in a 0.5% NonidetP40 solution to the wells and measuring the absorbance at 420 nm(Burstein et al., 1997).
cAMP Inhibition. HEK-293T cells were transiently cotrans-fected with DNA for either human FF1 or FF2 receptors and theDNA for EP2 receptor in 10-cm2 tissue culture plates. After 48 h,
cells were harvested and plated onto 96-well TC plates. Inhibition ofcAMP production was measured in the presence of varying concen-trations of the agonist of interest after stimulation of cAMP produc-tion by prostaglandin E2 (agonist of the EP2 receptor). For antago-nist assays, the ability of each compound to reverse the inhibitoryeffect of NPFF on cAMP production stimulated by prostaglandin E2
was measured. cAMP levels were measured using a DiscoveRx HitHunter cAMP CL kit following the manufacturer’s instructions.Dose-response curves were fitted to a nonlinear regression modelwith an equation for one-site competition using Prism (GraphPadSoftware Inc., La Jolla, CA).
Binding Assays. HEK-T cells were transfected with either FF1 orFF2 receptors. Cells were harvested 48 h after transfection, and cellhomogenates were prepared. The Kd of 125I-NPFF was determined tobe 50 and 100 pM for FF2 and FF1 receptors, respectively. Compe-tition binding assays were performed in the presence of 70 pM125I-NPFF (GE Healthcare, Little Chalfont, Buckinghamshire, UK)and various concentration of the ligand in 96-well plates as describedpreviously (Mollereau et al., 2002). Incubations were terminated byfiltration onto GF/B filters followed by three washes with ice-coldbuffer. Radioactivity retained on the filters was quantitated by scin-tillation counting in a Top Count counter (PerkinElmer Life andAnalytical Sciences, Waltham, MA). Data were fitted to a nonlinearregression model with an equation for one-site competition usingPrism (GraphPad Software Inc.).
Animals. Male, Sprague-Dawley rats (150–300 g; Harlan, India-napolis, IN) were housed in groups of three in a climate-controlledroom on a 12-h light/dark cycle (lights on at 7:00 AM), with food andwater available ad libitum. All testing was performed in accordancewith the policies and recommendations of the International Associ-ation for the Study of Pain and the National Institutes of Healthguidelines for the handling and use of laboratory animals and re-ceived approval from the Institutional Animal Care and Use Com-mittee of ACADIA Pharmaceuticals.
Formalin Test. Naive rats were injected with 50 �l of a 5.0%formalin solution into the dorsal surface of a hind paw and thenplaced in individual plastic cages for observation. The number ofnociceptive responses (i.e., paw flinches/licks/bites) was counted for aperiod of 60 min after formalin injection. Vehicle or test compoundswere administered 15 min before formalin injection.
Carrageenan-Induced Thermal Hyperalgesia. Naive ratswere assessed for their responsiveness to a noxious thermal stimu-lus. Response latencies were measured using the hot-plate test.Inflammatory pain was produced by injecting 0.1 ml of 2% �-carra-geenan (Sigma-Aldrich) into the left hind paw. Three hours aftercarrageenan injection, hot-plate latencies were again obtained. Asignificant reduction in the hot-plate latency was interpreted as thepresence of thermal hyperalgesia. Rats were injected with vehicle ortest compound and then tested at various time points after admin-istration. To generate dose-response curves, raw data were convertedto percent maximum possible effect (%MPE) by the formula, %MPE �[(experimental score � postcarrageenan score)/(naive score � post-carrageenan score)] � 100, where the experimental score is thehot-plate latency obtained after compound administration, the post-carrageenan score is the hot-plate latency obtained 3 h after carra-geenan treatment, and the naive score is the hot-plate latency ob-tained before any treatments. Each dose was assessed in separategroups of rats.
Assessment of Thermal Hyperalgesia. This test was per-formed by placing rats in a Plexiglas enclosure on a thermostaticallycontrolled metal plate maintained at 52°C. The time elapsed untilthe rat showed an obvious nociceptive response (i.e., licking/stomp-ing/elevating hind paw) was measured. Animals were tested beforeand at various time points after compound administration. A cutofftime of 40 s was used to prevent tissue damage.
L5/L6 Spinal Nerve Ligation Surgery. SNL injury was pro-duced as described previously (Kim and Chung, 1992) in six separatecohorts of rats, five of which had n � 24 and the sixth had n � 30
NPFF Receptors Have Opposing Roles 245
at ASPE
T Journals on Septem
ber 28, 2016jpet.aspetjournals.org
Dow
nloaded from
subjects. Anesthesia was induced with 2% isoflurane in O2 at 2 l/minand maintained with 1.5% isoflurane in O2. The dorsal vertebralcolumn from L4 to S2 was exposed, and the L5 and L6 spinal nerveswere identified and carefully isolated. The L5 and L6 spinal nerveswere tightly ligated distal to the dorsal root ganglion with 6-0 silksuture. The incision was closed, and the animals were observed foruneventful recoveries. Rats were allowed a period of at least 2 weeksbefore test compound evaluation.
Assessment of Mechanical Hypersensitivity. Paw withdrawalthresholds (PWTs) of the left hind paws (i.e., ipsilateral to SNL) ofthe rats were determined in response to probing with eight cali-brated von Frey filaments (Stoelting Inc., Wood Dale, IL) in logarith-mically spaced increments ranging from 0.41 to 15 g (4–150 mN).Each filament was applied perpendicularly to the plantar surface ofthe paw of rats kept in suspended wire-mesh cages. PWT was deter-mined in grams by sequentially increasing and decreasing the stim-ulus intensity and estimated using a Dixon nonparametric test. Asignificant reduction in PWT from the pre-SNL baseline value indi-cated the presence of mechanical hypersensitivity. Rats were allowedto recover for a minimum of 2 weeks before test compound assess-ment. Any rat that exhibited motor deficiency or a lack of subsequentincreased sensitivity to innocuous mechanical stimulation (PWT�4.3 g) was excluded from additional testing (12 of a possible 186).To generate dose-response curves, raw data were converted to%MPE by the formula, %MPE � [(PWT after drug � PWT for vehiclecontrol)/(15 � PWT for vehicle-control)] � 100. Each dose was as-sessed in separate groups of rats.
Statistical Analyses. Statistical analyses were conducted usingPrism 5 (GraphPad Software Inc.). The effects of test compoundswere analyzed by performing two-way ANOVAs. Planned compari-sons of means (each group versus vehicle) were performed using aBonferroni post hoc test, provided a significant main effect wasdetected. Significance was set at the level of p � 0.05. The dose thatelicits 50% efficacy (ED50) and the corresponding 95% confidenceinterval (95% CI) were determined using linear regression analysis.These calculations were performed with the pharmacological statis-
tics package FlashCalc (Dr. Michael H. Ossipov, University of Ari-zona, Tucson, AZ).
ResultsIn Vitro Characterization of Nonpeptidic Small Mol-
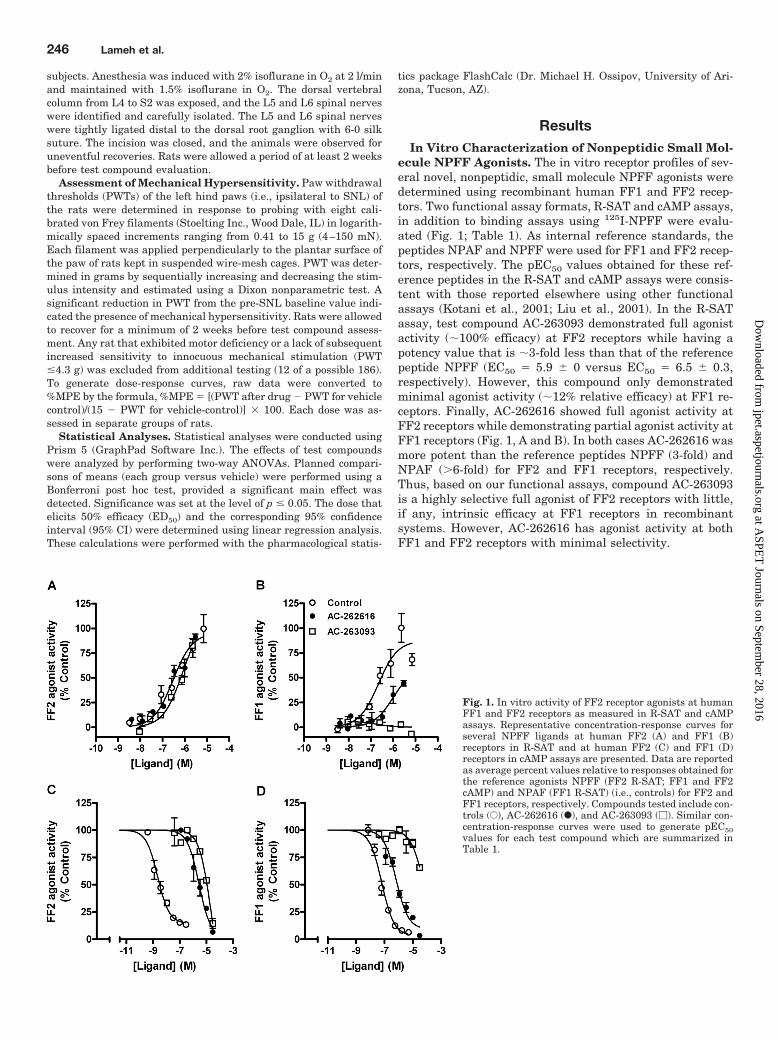
ecule NPFF Agonists. The in vitro receptor profiles of sev-eral novel, nonpeptidic, small molecule NPFF agonists weredetermined using recombinant human FF1 and FF2 recep-tors. Two functional assay formats, R-SAT and cAMP assays,in addition to binding assays using 125I-NPFF were evalu-ated (Fig. 1; Table 1). As internal reference standards, thepeptides NPAF and NPFF were used for FF1 and FF2 recep-tors, respectively. The pEC50 values obtained for these ref-erence peptides in the R-SAT and cAMP assays were consis-tent with those reported elsewhere using other functionalassays (Kotani et al., 2001; Liu et al., 2001). In the R-SATassay, test compound AC-263093 demonstrated full agonistactivity (�100% efficacy) at FF2 receptors while having apotency value that is �3-fold less than that of the referencepeptide NPFF (EC50 � 5.9 � 0 versus EC50 � 6.5 � 0.3,respectively). However, this compound only demonstratedminimal agonist activity (�12% relative efficacy) at FF1 re-ceptors. Finally, AC-262616 showed full agonist activity atFF2 receptors while demonstrating partial agonist activity atFF1 receptors (Fig. 1, A and B). In both cases AC-262616 wasmore potent than the reference peptides NPFF (3-fold) andNPAF (6-fold) for FF2 and FF1 receptors, respectively.Thus, based on our functional assays, compound AC-263093is a highly selective full agonist of FF2 receptors with little,if any, intrinsic efficacy at FF1 receptors in recombinantsystems. However, AC-262616 has agonist activity at bothFF1 and FF2 receptors with minimal selectivity.
Fig. 1. In vitro activity of FF2 receptor agonists at humanFF1 and FF2 receptors as measured in R-SAT and cAMPassays. Representative concentration-response curves forseveral NPFF ligands at human FF2 (A) and FF1 (B)receptors in R-SAT and at human FF2 (C) and FF1 (D)receptors in cAMP assays are presented. Data are reportedas average percent values relative to responses obtained forthe reference agonists NPFF (FF2 R-SAT; FF1 and FF2cAMP) and NPAF (FF1 R-SAT) (i.e., controls) for FF2 andFF1 receptors, respectively. Compounds tested include con-trols (E), AC-262616 (F), and AC-263093 (�). Similar con-centration-response curves were used to generate pEC50values for each test compound which are summarized inTable 1.
246 Lameh et al.
at ASPE
T Journals on Septem
ber 28, 2016jpet.aspetjournals.org
Dow
nloaded from

The agonist activity and functional selectivity of the testcompounds were confirmed in the cAMP assay. The selectiveFF2 agonist, AC-263093, was fully efficacious at FF2 recep-tors and only showed weak agonist activity at FF1 receptors(�23% relative efficacy), confirming that this compound isfunctionally selective for FF2 over FF1 receptors. AC-262616,which displayed agonist activity at both NPFF receptors inthe R-SAT assay, behaved similarly in the cAMP assay.Moreover, as in R-SAT, AC-262616 demonstrated slightlyhigher potency at FF1 over FF2 receptors in the cAMP assay(Fig. 1, C and D).
The selective FF2 agonist, AC-263093, retained its selec-tivity in the binding assay (�2 fold for FF2 over FF1),whereas AC-262616 displayed equal or increased affinity forthe FF1 receptor (Table 1). Furthermore, AC-263093 showedlittle to no binding across a broad screening panel (n � 50) of
other receptors, channels, and enzymes (IC50 � 10 �M;CEREP, Poitiers, France).
In Vitro Characterization of Nonpeptidic SmallMolecule FF1 Antagonists. The in vitro receptor profilesof several novel, nonpeptidic, small molecule NPFF antag-onists were determined using recombinant human FF1and FF2 receptors. Two functional assay formats, R-SATand cAMP assays, as well as binding assays using 125I-NPFF were used for this characterization (Fig. 2; Table 2).As internal reference standards, the peptides BIBP-3226and dPQR were included for comparison in both functionalassays (Table 2). The pKi values obtained for BIBP-3226 inthe R-SAT and cAMP assays were consistent with thosereported elsewhere (Mollereau et al., 2002). The lack ofstability of dPQR precluded us from characterizing it inR-SAT; however, the pKi value obtained in the cAMP for-
TABLE 1In vitro activity of NPFF receptor agonists at human FF1 and FF2 receptors as measured by R-SAT, cAMP, and binding assaysEfficacy values are expressed relative to the internal reference standards, NPAF and NPFF, for FF1 and FF2 receptors, respectively (R-SAT) and NPFF (cAMP).
Compound
R-SAT cAMP Binding Ki
hFF2 hFF1 hFF2 hFF1hFF2 hFF1
%Eff pEC50 %Eff pEC50 %Eff pEC50 %Eff pEC50
nM
NPAF 105 � 16 8.7 � 0.6 100 6.6 � 0.4 8.5 � 0.6 100 7.2 � 0.3 3.2 � 4.0 9.8 � 0NPFF 100 6.5 � 0.3 N.T. 100 8.2 � 0.4 7.3 � 0.4 0.98 � 1.1 0.9 � 0AC-262616 73 � 13 7.0 � 0.4 52 � 3a 7.4 114 � 8 5.5 � 0 93 � 9 5.9 � 0.1 230 � 11 172 � 160AC-263093 90 � 15 5.9 � 0 12 � 0a N.D. 78 � 24 5.2 � 0.3 23 � 9a N.D. 1296 � 942 3320 � 2890
%Eff, percent efficacy; N.D., could not be determined; N.T., not tested.a %Eff values reported are those obtained at the highest testable concentration of compound (6 �M for RSAT and 30 �M for cAMP assays). Because of the toxicity of the
compound, higher concentrations could not be tested for these compounds. Because proper dose-response could not be constructed, potency of these compounds has beenestimated.
Fig. 2. In vitro activity of selective FF1 receptor antagonists at human FF1 and FF2 receptors as measured in R-SAT, cAMP, and binding assays.Representative concentration-response curves for AC-262970 and AC-262620 at human FF1 receptors in R-SAT (A and D) and cAMP (B and E) assaysare presented. Data are reported as average percentage of response obtained relative to the reference FF1 antagonist BIBP-3226 (E). Radioligandbinding assays for AC-262970 (C) and AC-262620 (F) were also conducted in membranes isolated from cells expressing either human FF1 (E) or FF2(F) receptors. Similar concentration-response curves were used to generate pEC50 and pKi values for each test compound, which are summarized inTable 2.
NPFF Receptors Have Opposing Roles 247
at ASPE
T Journals on Septem
ber 28, 2016jpet.aspetjournals.org
Dow
nloaded from

mat was concordant with previous binding studies (Prokaiet al., 2001).
Test compounds AC-262970 and AC-262620 demonstratedpotent antagonist activity at FF1 receptors in both R-SATand cAMP assay formats. Although showing similar bindingaffinity for FF2 and FF1 receptors (Ki values of 17.3 � 20.5and 14.3 � 3.0 nM, respectively) (Fig. 2C), AC-262970 was aselective antagonist of FF1 receptors. AC-262970 antago-nized the effect of NPAF (R-SAT) and NPFF (cAMP) at FF1receptors, achieving pKi values of 8.2 � 0.3 and 7.4 � 0.4,respectively (Fig. 2, A and B). AC-262970 behaved as a fullagonist at FF2 receptors with a pEC50 value of 6.7 � 0.3,representing a 30-fold lower activity at FF2 receptors basedon functional activity in the R-SAT assay (Table 2). Likewise,AC-262620 showed functional selectivity as an antagonist atthe FF1 receptor. This compound antagonized the effect ofNPAF (R-SAT) and NPFF (cAMP) at FF1 receptors, achiev-ing pKi values of 8.1 � 0.3 and 7.7 � 0.4, respectively (Fig. 2,D and E). AC-262620 behaved as a full agonist at FF2 recep-tors with a pEC50 value of 5.9 � 0.5, representing 150-foldlower activity at FF2 receptors based on functional activity inthe R-SAT assay (Table 2). However, unlike AC-262970, se-lectivity of AC-262620 for FF1 receptors was further demon-strated by 20-fold higher binding affinity for FF1 receptorscompared with FF2 receptors, having Ki values of 16.4 � 10.1and 320.3 � 84.8 nM, respectively (Fig. 2F). Furthermore,AC-262970 showed little to no binding to a broad screeningpanel (n � 50) of other receptors, channels, and enzymes(IC50 � 10 �M).
Effects of the Selective FF2 Receptor Agonist AC-263093 in the Formalin Test. Given that antinociceptiveeffects of NPFF and 1DMe were unremarkable in the tail-flick test after spinal administration (data not shown), weprofiled AC-263093 (a selective FF2 agonist) in the formalinmodel head-to-head with morphine. The two-way ANOVAyielded F2,15 � 217.6, p 0.0001 and F1,15 � 391.9, p 0.0001 for factors associated with treatment and phase, re-spectively. Furthermore, the treatment � phase interactionwas statistically significant (F2,15 � 170.2, p 0.0001). For-malin administration produced the characteristic biphasicflinching response in vehicle-treated rats: 68.3 � 4.7 and562.8 � 17.6 flinches across phase 1 (i.e., 0–10 min) andphase 2 (i.e., 10.1–60 min), respectively (Fig. 3A). Consistentwith the pharmacology of �-opioid agonists, 10 mg/kg mor-phine significantly decreased flinching in both phases of theformalin test; values obtained were 18.0 � 6.0 and 27.7 � 9.1for phase 1 (i.e., 0–10 min) and phase 2 (i.e., 10.1–60 min),respectively (p 0.05, Bonferroni post-tests). In contrastwith the effect of morphine, intraperitoneal administration of
10 mg/kg AC-263093 selectively inhibited flinching behaviorin phase 2 (i.e., 11–60 min) of the formalin test; the valueobtained for phase 2 was 213.3 � 26.0 (p 0.001, Bonferronipost-test).
Effect of the Selective FF2 Receptor Agonist AC-263093 on Thermal Hyperalgesia Produced by Carra-geenan. At 3 h after intraplantar carrageenan administra-tion, rats demonstrated significant reductions in hot-platelatency; values obtained before carrageenan and after carra-geenan were 10.7 � 0.1 and 6.8 � 0.1 s, respectively (p �0.05, paired t test). Administration of AC-263093 intraperi-toneally produced a significant dose- and time-dependentattenuation of thermal hypersensitivity produced by carra-geenan treatment (Fig. 3B). The two-way ANOVA yieldedF3,120 � 28.4, p 0.0001 and F6,120 � 55.5, p 0.0001 forfactors associated with dose and time, respectively. Further-more, the dose � time interaction was statistically signifi-cant, F18,120 � 5.4, p 0.0001. AC-263093 at a dose of 1mg/kg was without effect. In contrast, both the 3 and 10mg/kg doses of AC-263093 produced significant attenuationof carrageenan-induced thermal hypersensitivity, and thelevels of efficacy were maintained for 2 to 3 h (p 0.05,Bonferroni post-tests). The effect of 10 mg/kg AC-263093 wasabsent by 3 h as indicated by a return of hot-plate latency topretreatment levels (p 0.05, Bonferroni post-tests). Thecalculated ED50 value for intraperitoneal AC-263093 was 2.4mg/kg [1.6–3.6 (95% CI)] after 30 min post-treatment. Nosignificant effects of 10 mg/kg AC-263093 were noted in non-inflamed rats (p 0.05, Bonferroni post-tests), suggestingthat systemic administration of a selective FF2 agonist isunlikely to alter basal sensory thresholds (Fig. 3C).
Effects of the Selective FF2 Receptor Agonist AC-263093 on SNL-Induced Mechanical Hypersensitiv-ity. After L5/L6 SNL rats show significant reductions inPWTs, values obtained before SNL and after SNL were15 � 0 and 2.7 � 0.2 g, respectively (p � 0.05, paired ttest). AC-263093 produced a dose-dependent reversal ofSNL-induced mechanical hypersensitivity after intraperi-toneal administration (Fig. 4A). A two-way repeated-mea-sures ANOVA of these data revealed significant main ef-fects for both dose and time, F4,150 � 26.4, p 0.0001 andF6,150 � 267.3, p 0.0001, respectively, as well as, asignificant dose � time interaction, F24,150 � 14.3, p 0.0001. AC-263093 at doses of 1 and 3 mg/kg producedsmall increases in PWTs, which peaked at 30 min andsteadily declined with time; this effect was not signifi-cantly different from those values obtained for the vehicle-treated controls (p 0.05, Bonferroni post-tests). In con-trast, both the 10 and 30 mg/kg doses of AC-263093
TABLE 2In vitro activity of NPFF receptor antagonists at human FF1 and FF2 receptors as measured by R-SAT, cAMP, and binding assaysNPAF and NPFF were used as the FF1 agonists in the R-SAT and cAMP formats, respectively. pKi (i.e., �log Ki) values are expressed relative to the internal referencestandard BIBP-3226. Ki values were calculated using the Cheng-Prusoff equation: Ki � IC50/(1 � �agonist /EC50 agonist).
CompoundhFF1 hFF2
R-SAT Antagonist pKi cAMP Antagonist pKi Binding Ki (nM) R-SAT Agonist pEC50 (%Eff) Binding Ki (nM)
BIBP-3226 7.6 � 0.6 7.5 � 0.4 N.T. 7.2 � 0 (66 � 0) N.T.dPQR N.D.a 4.4 � 0.4 N.T. N.D.a N.T.AC-262620 8.1 � 0.3 7.7 � 0.4 16.4 � 10.1 5.9 � 0.5 (92 � 25) 320.3 � 84.8AC-262970 8.2 � 0.3 7.4 � 0.4 14.3 � 3.0 6.7 � 0.3 (96 � 38) 17.3 � 0.20.5
N.T., not tested.a dPQR is not stable enough for R-SAT assays and thus was not determined (N.D.).
248 Lameh et al.
at ASPE
T Journals on Septem
ber 28, 2016jpet.aspetjournals.org
Dow
nloaded from
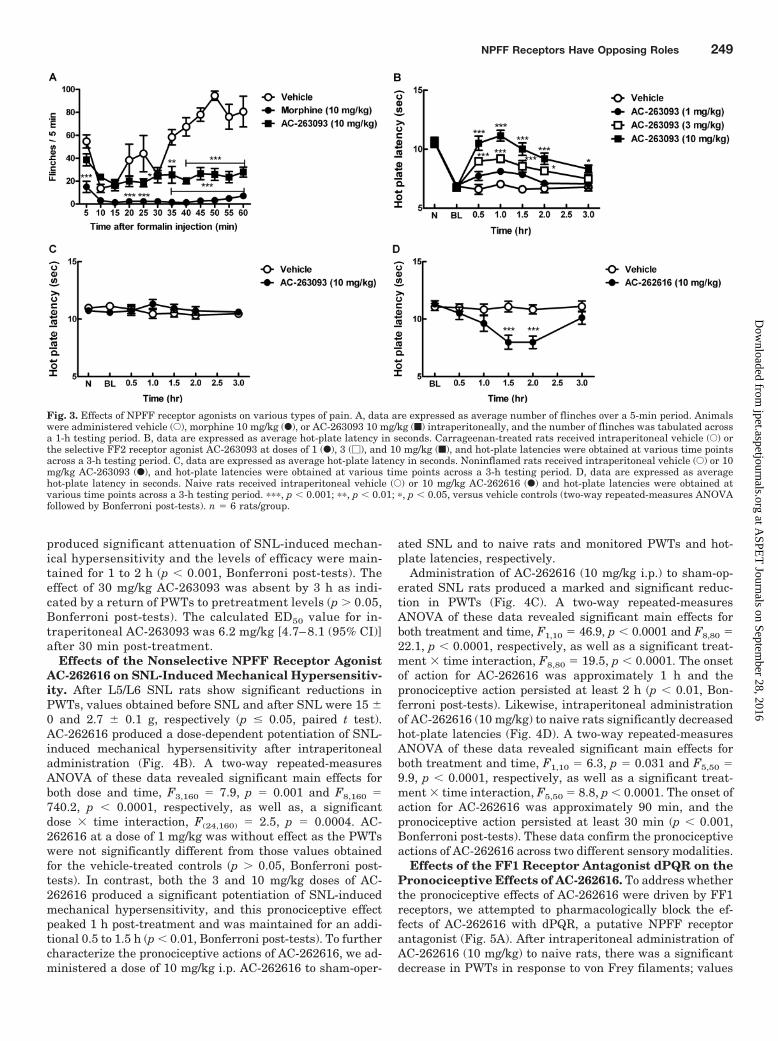
produced significant attenuation of SNL-induced mechan-ical hypersensitivity and the levels of efficacy were main-tained for 1 to 2 h (p 0.001, Bonferroni post-tests). Theeffect of 30 mg/kg AC-263093 was absent by 3 h as indi-cated by a return of PWTs to pretreatment levels (p 0.05,Bonferroni post-tests). The calculated ED50 value for in-traperitoneal AC-263093 was 6.2 mg/kg [4.7– 8.1 (95% CI)]after 30 min post-treatment.
Effects of the Nonselective NPFF Receptor AgonistAC-262616 on SNL-Induced Mechanical Hypersensitiv-ity. After L5/L6 SNL rats show significant reductions inPWTs, values obtained before SNL and after SNL were 15 �0 and 2.7 � 0.1 g, respectively (p � 0.05, paired t test).AC-262616 produced a dose-dependent potentiation of SNL-induced mechanical hypersensitivity after intraperitonealadministration (Fig. 4B). A two-way repeated-measuresANOVA of these data revealed significant main effects forboth dose and time, F3,160 � 7.9, p � 0.001 and F8,160 �740.2, p 0.0001, respectively, as well as, a significantdose � time interaction, F(24,160) � 2.5, p � 0.0004. AC-262616 at a dose of 1 mg/kg was without effect as the PWTswere not significantly different from those values obtainedfor the vehicle-treated controls (p 0.05, Bonferroni post-tests). In contrast, both the 3 and 10 mg/kg doses of AC-262616 produced a significant potentiation of SNL-inducedmechanical hypersensitivity, and this pronociceptive effectpeaked 1 h post-treatment and was maintained for an addi-tional 0.5 to 1.5 h (p 0.01, Bonferroni post-tests). To furthercharacterize the pronociceptive actions of AC-262616, we ad-ministered a dose of 10 mg/kg i.p. AC-262616 to sham-oper-
ated SNL and to naive rats and monitored PWTs and hot-plate latencies, respectively.
Administration of AC-262616 (10 mg/kg i.p.) to sham-op-erated SNL rats produced a marked and significant reduc-tion in PWTs (Fig. 4C). A two-way repeated-measuresANOVA of these data revealed significant main effects forboth treatment and time, F1,10 � 46.9, p 0.0001 and F8,80 �22.1, p 0.0001, respectively, as well as a significant treat-ment � time interaction, F8,80 � 19.5, p 0.0001. The onsetof action for AC-262616 was approximately 1 h and thepronociceptive action persisted at least 2 h (p 0.01, Bon-ferroni post-tests). Likewise, intraperitoneal administrationof AC-262616 (10 mg/kg) to naive rats significantly decreasedhot-plate latencies (Fig. 4D). A two-way repeated-measuresANOVA of these data revealed significant main effects forboth treatment and time, F1,10 � 6.3, p � 0.031 and F5,50 �9.9, p 0.0001, respectively, as well as a significant treat-ment � time interaction, F5,50 � 8.8, p 0.0001. The onset ofaction for AC-262616 was approximately 90 min, and thepronociceptive action persisted at least 30 min (p 0.001,Bonferroni post-tests). These data confirm the pronociceptiveactions of AC-262616 across two different sensory modalities.
Effects of the FF1 Receptor Antagonist dPQR on thePronociceptive Effects of AC-262616. To address whetherthe pronociceptive effects of AC-262616 were driven by FF1receptors, we attempted to pharmacologically block the ef-fects of AC-262616 with dPQR, a putative NPFF receptorantagonist (Fig. 5A). After intraperitoneal administration ofAC-262616 (10 mg/kg) to naive rats, there was a significantdecrease in PWTs in response to von Frey filaments; values
Fig. 3. Effects of NPFF receptor agonists on various types of pain. A, data are expressed as average number of flinches over a 5-min period. Animalswere administered vehicle (E), morphine 10 mg/kg (F), or AC-263093 10 mg/kg (f) intraperitoneally, and the number of flinches was tabulated acrossa 1-h testing period. B, data are expressed as average hot-plate latency in seconds. Carrageenan-treated rats received intraperitoneal vehicle (E) orthe selective FF2 receptor agonist AC-263093 at doses of 1 (F), 3 (�), and 10 mg/kg (f), and hot-plate latencies were obtained at various time pointsacross a 3-h testing period. C, data are expressed as average hot-plate latency in seconds. Noninflamed rats received intraperitoneal vehicle (E) or 10mg/kg AC-263093 (F), and hot-plate latencies were obtained at various time points across a 3-h testing period. D, data are expressed as averagehot-plate latency in seconds. Naive rats received intraperitoneal vehicle (E) or 10 mg/kg AC-262616 (F) and hot-plate latencies were obtained atvarious time points across a 3-h testing period. ���, p 0.001; ��, p 0.01; �, p 0.05, versus vehicle controls (two-way repeated-measures ANOVAfollowed by Bonferroni post-tests). n � 6 rats/group.
NPFF Receptors Have Opposing Roles 249
at ASPE
T Journals on Septem
ber 28, 2016jpet.aspetjournals.org
Dow
nloaded from

obtained before treatment (i.e., naive) and after treatment(i.e., baseline) were 14.7 � 0.2 and 4.4 � 0.4 g, respectively(p � 0.05, paired t test). Immediately after baseline testing,rats received either vehicle or dPQR (30 mg/kg) intraperito-neally and then were tested across a 2.5-h period. A two-wayrepeated-measures ANOVA of these data revealed signifi-cant main effects for both treatment and time, F2,120 � 64.7,p 0.0001 and F8,120 � 51.1, p 0.0001, respectively, as wellas, a significant treatment � time interaction, F16,120 � 23.9,p 0.0001. AC-262616-treated rats that received intraperi-toneal vehicle treatment continued to demonstrate signifi-cantly lower PWTs relative to vehicle-treated controls (p 0.001, Bonferroni post-tests). In contrast, AC-262616-treatedrats that received 30 mg/kg i.p. dPQR showed a significantreversal of mechanical hypersensitivity. The effect of 30mg/kg dPQR was almost complete, but short-lived as thereversal was maintained for approximately 0.5 h (p 0.001).
Effects of the FF1 Receptor Antagonist dPQR onSNL-Induced Mechanical Hypersensitivity. After L5/L6SNL rats show significant reductions in PWTs; values ob-tained before SNL and after SNL were 15 � 0 and 2.7 � 0.1 g,respectively (p � 0.05, paired t test). Administration of in-traperitoneal dPQR produced a dose-dependent reversal ofSNL-induced mechanical hypersensitivity (Fig. 5B). A two-way repeated-measures ANOVA of these data revealed sig-nificant main effects for both dose and time, F3,160 � 17.4,p 0.0001 and F8,160 � 141.6, p 0.0001, respectively, aswell as a significant dose � time interaction, F24,160 � 6.5,p 0.0001. A dose of 3 mg/kg dPQR produced a small in-
crease in PWTs, which peaked at 15 min and declined overtime; this effect was not significantly different from thosevalues obtained for the vehicle-treated controls (p 0.05,Bonferroni post-tests). In contrast, both the 10 and 30 mg/kgdoses of dPQR produced significant attenuation of SNL-in-duced mechanical hypersensitivity, and the levels of efficacywere maintained for 1 h (p 0.05, Bonferroni post-tests).The effects of 10 and 30 mg/kg dPQR were absent by 1 h asindicated by a return of PWTs to pretreatment levels (p 0.05, Bonferroni post-tests). The short-lived effects ofdPQR are consistent with its published pharmacokineticparameters (Prokai et al., 2000). The calculated ED50
value for dPQR administered intraperitoneally was 9.5mg/kg [7.0 –12.7 (95% CI)] after 15 min post-treatment.
Effects of the NPFF Receptor Antagonists AC-262970and AC-262620 on SNL-Induced Mechanical Hypersen-sitivity. After L5/L6 SNL rats show significant reductions inPWTs; values obtained before SNL and after SNL were 15 �0 and 2.4 � 0.2 g, respectively (p � 0.05, paired t test).AC-262970 produced a dose-dependent reversal of SNL-in-duced mechanical hypersensitivity after intraperitonealadministration (Fig. 6A). A two-way repeated-measuresANOVA of these data revealed significant main effects forboth dose and time, F3,90 � 12.8, p � 0.0001 and F5,90 �203.3, p 0.0001, respectively, as well as a significant dose �time interaction, F15,90 � 6.4, p 0.0001. AC-262970 at adose of 1 mg/kg produced a small increase in PWTs, whichpeaked at 30 min and declined over time; this effect was notsignificantly different from those values obtained for the
Fig. 4. Effects of AC-263093 and AC-262616 on L5/L6 SNL-induced mechanical hypersensitivity after intraperitoneal administration. Data areexpressed as average PWT of the injured paw in SNL rats. A, SNL rats were given intraperitoneal vehicle (E) or the selective FF2 receptor agonistAC-263093 at doses of 1 (F), 3 (�), 10 (f), and 30 mg/kg (‚), and PWTs were obtained at various time points across a 3-h testing period. B, SNL ratsreceived vehicle (E) or the nonselective FF1/FF2 receptor agonist AC-262616 at doses of 1 (F), 3 (�), and 10 mg/kg (f) and PWTs were obtained atvarious time points across a 3.5-h testing period. C, sham-operated SNL rats were given either vehicle (E) or 10 mg/kg i.p. of the nonselective FF1/FF2receptor agonist AC-262616 (F), and PWTs were obtained at various time points across a 3.5-h testing period. N, presurgical response thresholds; BL,baseline response before test compound administration. ���, p 0.001; ��, p 0.01; �, p 0.05, versus vehicle controls (two-way repeated-measuresANOVA followed by Bonferroni post-tests). n � 6 rats/group.
250 Lameh et al.
at ASPE
T Journals on Septem
ber 28, 2016jpet.aspetjournals.org
Dow
nloaded from

vehicle-treated controls (p 0.05, Bonferroni post-tests). Incontrast, both the 3 and 10 mg/kg doses of AC-262970 pro-duced significant attenuation of SNL-induced mechanical hy-persensitivity, and the levels of efficacy were maintained forup to 1 h (p 0.05, Bonferroni post-tests). The effect of 3 and10 mg/kg AC-262970 were absent by 1 h as indicated by areturn of PWTs to pretreatment levels (p 0.05, Bonferronipost-tests). The calculated ED50 value for intraperitonealAC-262970 was 3.5 mg/kg [2.1–5.7 (95% CI)] after 30 minpost-treatment. As described previously, the FF1 antagonistsused herein also showed full agonist activity at FF2 receptorswith potency values similar to that obtained for AC-263093.To ascertain whether the efficacy in the SNL model is drivenprimarily via FF1 receptor antagonism, we next evaluatedAC-262620. AC-262620 is a potent FF1 antagonist with 20-and 150-fold selectivity for FF1 receptors in binding andR-SAT assays, respectively.
Rats show significant reductions in PWTs after SNL; val-ues obtained before SNL and after SNL were 15 � 0 and2.5 � 0.3 g, respectively (p � 0.05, paired t test). AC-262620at a dose of 10 mg/kg produced a time-dependent reversal ofSNL-induced mechanical hypersensitivity after intraperito-neal administration (Fig. 6B). A two-way repeated-measuresANOVA of these data revealed significant main effects for
both drug and time, F1,54 � 21.1, p � 0.0013 and F6,54 � 38.1,p 0.0001, respectively, as well as a significant drug � timeinteraction, F6,54 � 8.7, p 0.0001. AC-262620 at a dose of 10mg/kg produced a significant attenuation of SNL-inducedmechanical hypersensitivity, and the levels of efficacy weremaintained for up to 1.5 h (p 0.01, Bonferroni post-tests).The effect of 10 mg/kg AC-262620 was absent by 2 h asindicated by a return of PWTs to pretreatment levels (p 0.05, Bonferroni post-tests).
DiscussionIn the present investigation, we report on the activity of a
set of novel, small molecule, NPFF receptor ligands that areexemplified by 1) AC-263093, a selective FF2 receptor ago-nist, which is fully efficacious in vitro in both R-SAT andcAMP functional assays at hFF2 receptors (relative to NPFF)while virtually devoid of intrinsic efficacy at hFF1 receptors(relative to NPAF), 2) AC-262616, a pan FF1/FF2 receptoragonist, which is fully efficacious at both NPFF receptorsubtypes in an cAMP assay, and 3) AC-262970 and AC-262620, selective FF1 antagonists, which bind hFF1 recep-tors with low nanomolar affinity and display functional se-lectivity values of 30- and 150-fold, respectively, for FF1 overFF2 receptors. Using these molecules we have, for the firsttime, been able to elucidate the pharmacology of NPFF re-ceptors after systemic administration. Our results providethe first direct in vivo evidence for the opposing roles ofNPFF receptor subtypes as pertains to the modulation ofnociception. Using these compounds, we have been able todirectly show that FF1 receptor activation is pronociceptive,whereas FF2 receptor activation is antinociceptive.
Intrathecal administration of NPFF elicits acute antinoci-ception and enhances the efficacy of spinal opioids, whereasintracerebroventricular administration of NPFF antagonizesthe effects of opioids and results in pronociceptive effects [forreview, see Yang et al., (2008)]. Receptor localization studieshave established that whereas NPFF receptors are expressedubiquitously in the brain, only the FF2 receptor has beendetected at the level of the spinal cord (Bonini et al., 2000;Liu et al., 2001; Yang and Iadarola, 2006). The apparent lackof FF1 receptor expression spinally has led to the idea thatthe pronociceptive actions of NPFF are mediated via FF1receptors (Liu et al., 2001). However, this hypothesis hasnever been adequately addressed in vivo because of a lack ofhighly selective nonpeptidic NPFF ligands suitable for sys-temic administration. Using our novel small molecule, non-peptidic NPFF receptor ligands as pharmacological tools, wehave clearly demonstrated that NPFF receptors act in anopposing manner to modulate nociceptive input.
We were unable to detect any significant antinociceptiveeffects of AC-263093, a selective FF2 selective agonist, afterintraperitoneal administration in the hot-plate test. Al-though it remains plausible that NPFF agonists can poten-tiate the antinociceptive actions of opioids (Kontinen andKalso, 1995; Gouarderes et al., 1996; Xu et al., 1999), depend-ing on the route of administration, this possibility was be-yond the scope of the present investigation. Given the lack ofactivity of AC-263093 on acute sensory thresholds, we alsoprofiled this compound in the formalin and carrageenan mod-els. Like morphine, AC-263093 markedly attenuated flinch-ing behavior, although this effect was specific to the second
Fig. 5. Effects of the putative NPFF receptor antagonist dPQR on me-chanical hypersensitivity after intraperitoneal administration. A, dataare expressed as average PWT of the left hind paw. After predrug PWTs(N) were obtained, rats were immediately administered intraperitonealvehicle (E) or 10 mg/kg AC-262616, and 75 min later PWTs were againobtained (BL). Immediately after testing, rats received either intraperi-toneal vehicle (F) or 30 mg/kg dPQR (�), and PWTs were obtained atvarious time points across a 2.5-h testing period. B, data are expressed asaverage PWT of the injured paw in rats. Animals were administeredintraperitoneal vehicle (E) or dPQR at doses of 3 (F), 10 (�), and 30 mg/kg(f), and PWTs were obtained at various time points across a 3-h testingperiod. ���, p 0.001; �, p 0.05, versus vehicle controls (two-wayrepeated-measures ANOVA followed by Bonferroni post-tests). n � 6rats/group.
NPFF Receptors Have Opposing Roles 251
at ASPE
T Journals on Septem
ber 28, 2016jpet.aspetjournals.org
Dow
nloaded from

phase, suggesting that AC-263093 may be active in models ofspinal sensitization. Consistent with this interpretation, AC-263093 dose dependently alleviated thermal hyperalgesiaproduced by intraplantar carrageenan, an effect similar tothat previously reported for 1DMe after intrathecal admin-istration (Xu et al., 1999). Increases in NPFF immunoreac-tivity have been reported in the spinal cord dorsal horn aftercarrageenan treatment (Kontinen et al., 1997). Morphinepretreatment completely blocks the increase in spinal NPFFimmunoreactivity (Kontinen et al., 1997), suggesting thatenhanced release of NPFF may promote thermal hyperalge-sia. NPFF binding sites are not significantly altered by eitherneonatal capsaicin treatment or by dorsal rhizotomy recep-tors, suggesting that these receptors are almost exclusivelyexpressed postsynaptically in the spinal cord (Lombard et al.,1995). It is possible that spinal FF2 receptors may act asautoreceptors to negatively regulate NPFF release. A reduc-tion in endogenous NPFF release could drive the antihyper-algesic effects of selective FF2 agonists as well as provide amechanism by which the antinociceptive activity of intrathe-cal morphine may be enhanced by these agents.
In addition, systemic administration of AC-263093 dosedependently attenuated mechanical hypersensitivity afterSNL, without altering the response thresholds of sham-oper-ated rats, consistent with previous studies using microinjec-tions in discrete brainstem regions (Xu et al., 1999; Altier etal., 2000; Wei et al., 2001). In stark contrast to the effectsobserved with AC-263093, intraperitoneal administration ofthe nonselective FF1/FF2 receptor agonist AC-262616 poten-tiated the severity of the SNL-induced hypersensitivity. Fur-thermore, intraperitoneal administration of AC-262616 pro-
duced marked increases in sensitivity to both noxiousthermal and to non-noxious mechanical stimuli in both naiveand sham-operated rats, respectively. Given that efficacyobserved in the SNL model is correlated with the degree ofFF2 functional selectivity, we hypothesized that the prono-ciceptive actions were a direct result of supraspinal FF1receptor activation.
To date, the only well characterized and commerciallyavailable NPFF receptor antagonist is BIBP-3226 (Mollereauet al., 2002; Fang et al., 2006). Although intracerebroventric-ular administration of BIBP-3226 has been shown to blockthe effects produced by NPFF and NPVF (Fang et al., 2006),BIBP-3226 remains a very potent and selective neuropeptideY1 receptor antagonist (Doods et al., 1995). As a result,efficacy measured in vivo would be confounded as NPY1antagonists effectively reverse SNL-induced mechanical hy-persensitivity in their own right (Ossipov et al., 2002). There-fore, we opted for dPQR, a putative NPFF receptor antago-nist (Prokai et al., 2001), which crosses the blood-brainbarrier (Prokai et al., 2000), for our initial in vivo assess-ments. Systemic administration of dPQR reversed the prono-ciceptive actions of AC-262616, confirming biological activityof this FF1 receptor antagonist.
To determine whether endogenous NPFF systems play arole in the maintenance of mechanical hypersensitivity afterSNL, we administered dPQR to neuropathic rats. Systemicadministration of dPQR dose dependently alleviated SNL-induced allodynia with a duration of action consistent withits published pharmacokinetic profile (Prokai et al., 2000).These initial proof-of-concept data provided the impetus forthe discovery of FF1 receptor antagonists. Like dPQR, both
Fig. 6. Effects of the FF1 receptor antagonists AC-262970and AC-262620 on L5/L6 SNL-induced mechanical hyper-sensitivity after intraperitoneal administration. Data areexpressed as average PWT of the injured paw in rats.A, animals were administered intraperitoneal vehicle (E)or the potent FF1 receptor agonist AC-262970 at doses of 1(F), 3 (�), and 10 mg/kg (f), and PWTs were obtained atvarious time points across a 2-h testing period. B, animalswere administered intraperitoneal vehicle (E) or the potentand selective FF1 receptor agonist AC-262620 at a dose of10 mg/kg (F), and PWTs were obtained at various timepoints across a 3-h testing period. N, PWTs before SNL;BL, PWTs after SNL. ���, p 0.001; ��, p 0.01; �, p 0.05, versus vehicle controls (two-way repeated-measuresANOVA followed by Bonferroni post-tests). n � 6 rats/group.
252 Lameh et al.
at ASPE
T Journals on Septem
ber 28, 2016jpet.aspetjournals.org
Dow
nloaded from

AC-262970 and AC-262620, two highly potent FF1 antago-nists, with low nanomolar affinity for FF1 receptors, atten-uated SNL-induced mechanical hypersensitivity. It should benoted that these antagonists also elicited full agonist activityat FF2 receptors with potencies in the range of AC-263093.Therefore, one possible interpretation is that the FF2 agonistactivity of AC-262970 and AC-262620 is sufficient to atten-uate SNL-induced hypersensitivity. However, this possibilityis unlikely given that 1) administration of AC-262616 pro-duced mechanical and thermal hypersensitivity in naive rats,2) dPQR is devoid of FF2 agonist activity, and 3) AC-262620is 150-fold selective in the functional R-SAT assay and is20-fold selective for FF1 receptors in binding assays. Takentogether, these data support the concept that after peripheralnerve injury, FF1 receptor tone may be enhanced.
As mentioned previously, we are unaware of any reportsconfirming the presence of FF1 receptors in the spinal cord ofrats; therefore, the pronociceptive actions of FF1 receptoractivation would most likely originate in the brain. BecauseNPVF preferentially activates FF1 receptors, this endoge-nous peptide may be the source of pronociceptive drive, as invitro studies have confirmed that it possesses antiopioid ac-tivity (Liu et al., 2001; Kersante et al., 2006). The ability ofAC-262616 to significantly decrease baseline sensory thresh-olds suggests the possibility that FF1 receptor activation, inaddition to promoting morphine antinociceptive tolerance,may also contribute to opioid-induced paradoxical pain. Thisidea is supported by the observation that intracerebroven-tricular administration of an anti-NPFF antiserum com-pletely restores the efficacy of intracerebroventricular mor-phine in tolerant rats (Lake et al., 1991). In addition,administration of a pan FF1/FF2 receptor antagonist, RF9,prevents heroin-induced hyperalgesia and effectively blocksthe development of antinociceptive tolerance after repeatedheroin treatment (Simonin et al., 2006).
Mechanistically, opioid-induced paradoxical pain and neu-ropathic pain share common features, including the activa-tion of descending pain facilitatory pathways (Porreca et al.,2001; Burgess et al., 2002; Gardell et al., 2002). The activa-tion of descending pain facilitation by antiopioid peptides isnot without precedence. For example, microinjection of cho-lecystokinin (CCK) into the rostral ventromedial medullaproduces mechanical hypersensitivity in naive rats (Xie etal., 2005), an effect similar to that observed after adminis-tration of AC-262616. Furthermore, CCK2 receptor antago-nists restore the antinociceptive potency of morphine in tol-erant rats, reverse opioid-induced paradoxical pain (Xie etal., 2005), and diminish mechanical allodynia in SNL rats(Kovelowski et al., 2000). Given the similarities between theaforementioned effects with CCK and those reported hereinwith our FF1 receptor ligands, together with those reportedelsewhere (Simonin et al., 2006), there is a possibility thatsupraspinal FF1 receptor activation may drive descendingpain facilitatory pathways, although further work in thisarea is warranted.
In addition to the activation of descending pain facilitatorysystems, opioid-induced paradoxical pain and neuropathicpain share another common feature, the up-regulation ofspinal dynorphin (Gardell et al., 2002, 2003). Althoughdynorphin is an endogenous � opioid receptor agonist, en-hanced levels of this peptide promote neuropathic pain(Wang et al., 2001; Burgess et al., 2002), as well as opioid
antinociceptive tolerance and opioid-induced paradoxicalpain (Gardell et al., 2002). Critically, manipulations thatinhibit the pronociceptive actions of spinal dynorphin allevi-ate neuropathic pain (Wang et al., 2001; Burgess et al., 2002;Gardell et al., 2003, 2004) and reverse antinociceptive toler-ance and the accompanying abnormal pain (Vanderah et al.,2000; Gardell et al., 2002). This result is highly relevantbecause a recent study has shown that 1DMe suppressesspinal dynorphin release in anesthetized rats (Ballet et al.,2002). Because FF1 receptors are not localized spinally, theeffects of 1DMe must therefore be driven by FF2 receptorsand thus represents a mechanism by which selective FF2agonists may alleviate neuropathic pain and restore the an-tinociceptive potency of opioids. Moreover, the suppression ofdynorphin release also would have beneficial effects inchronic inflammatory pain as this type of pain is associatedwith both descending pain facilitation as well as spinaldynorphin up-regulation (Dubner and Ruda, 1992). Ofcourse, this proposed mechanism of selective FF2 agonismdoes not preclude potential supraspinal effects, although fur-ther work in this area is warranted.
In conclusion, we have demonstrated the opposing roles ofFF1 and FF2 receptors as they pertain to the modulation ofnociception. We have shown that FF2 agonism is efficaciousin various pain models, whereas FF1 agonism is pronocicep-tive. Selective FF2 agonists, FF1 antagonists, or bifunctionalligands may represent novel approaches for the treatment ofchronic pain whether of inflammatory or neuropathic in ori-gin. However, identification of the selective tools necessary tovalidate these targets is only the first step.
Acknowledgments
We thank Dr. Douglas Bonhaus for his critical review of thismanuscript. We also thank William Keefe, Norman Nash, andDr. Audra L. Scully for their work related to the enablement of theR-SAT functional assays.
ReferencesAltier N, Dray A, Menard D, and Henry JL (2000) Neuropeptide FF attenuates
allodynia in models of chronic inflammation and neuropathy following intrathecalor intracerebroventricular administration. Eur J Pharmacol 407:245–255.
Ballet S, Braz J, Mauborgne A, Bourgoin S, Zajac JM, Hamon M, and Cesselin F(2002) The neuropeptide FF analogue, 1DMe, reduces in vivo dynorphin releasefrom the rat spinal cord. J Neurochem 81:659–662.
Bonini JA, Jones KA, Adham N, Forray C, Artymyshyn R, Durkin MM, Smith KE,Tamm JA, Boteju LW, Lakhlani PP, et al. (2000) Identification and characteriza-tion of two G protein-coupled receptors for neuropeptide FF. J Biol Chem 275:39324–39331.
Burgess SE, Gardell LR, Ossipov MH, Malan TP Jr, Vanderah TW, Lai J, andPorreca F (2002) Time-dependent descending facilitation from the rostral ventro-medial medulla maintains, but does not initiate, neuropathic pain. J Neurosci22:5129–5136.
Burstein ES, Spalding TA, and Brann MR (1997) Pharmacology of muscarinicreceptor subtypes constitutively activated by G proteins. Mol Pharmacol 51:312–319.
Doods HN, Wienen W, Entzeroth M, Rudolf K, Eberlein W, Engel W, and WielandHA (1995) Pharmacological characterization of the selective nonpeptide neuropep-tide Y Y1 receptor antagonist BIBP 3226. J Pharmacol Exp Ther 275:136–142.
Dubner R and Ruda MA (1992) Activity-dependent neuronal plasticity followingtissue injury and inflammation. Trends Neurosci 15:96–103.
Dupouy V and Zajac JM (1995) Effects of neuropeptide FF analogs on morphineanalgesia in the nucleus raphe dorsalis. Regul Pept 59:349–356.
Elshourbagy NA, Ames RS, Fitzgerald LR, Foley JJ, Chambers JK, Szekeres PG,Evans NA, Schmidt DB, Buckley PT, Dytko GM, et al. (2000) Receptor for the painmodulatory neuropeptides FF and AF is an orphan G protein-coupled receptor.J Biol Chem 275:25965–25971.
Fang Q, Guo J, He F, Peng YL, Chang M, and Wang R (2006) In vivo inhibition ofneuropeptide FF agonism by BIBP3226, an NPY Y1 receptor antagonist. Peptides27:2207–2213.
Gardell LR, Ibrahim M, Wang R, Wang Z, Ossipov MH, Malan TP Jr, Porreca F, andLai J (2004) Mouse strains that lack spinal dynorphin upregulation after periph-eral nerve injury do not develop neuropathic pain. Neuroscience 123:43–52.
Gardell LR, Vanderah TW, Gardell SE, Wang R, Ossipov MH, Lai J, and Porreca F
NPFF Receptors Have Opposing Roles 253
at ASPE
T Journals on Septem
ber 28, 2016jpet.aspetjournals.org
Dow
nloaded from

(2003) Enhanced evoked excitatory transmitter release in experimental neuropa-thy requires descending facilitation. J Neurosci 23:8370–8379.
Gardell LR, Wang R, Burgess SE, Ossipov MH, Vanderah TW, Malan TP Jr, Lai J,and Porreca F (2002) Sustained morphine exposure induces a spinal dynorphin-dependent enhancement of excitatory transmitter release from primary afferentfibers. J Neurosci 22:6747–6755.
Gaubert G, Bertozzi F, Kelly NM, Pawlas J, Scully AL, Nash NR, Gardell LR, LamehJ, and Olsson R (2009) Discovery of selective nonpeptidergic neuropeptide FF2receptor agonists. J Med Chem 12:6511–6514.
Gicquel S, Mazarguil H, Allard M, Simonnet G, and Zajac JM (1992) Analogues ofF8Famide resistant to degradation, with high affinity and in vivo effects. EurJ Pharmacol 222:61–67.
Gouarderes C, Jhamandas K, Sutak M, and Zajac JM (1996) Role of opioid receptorsin the spinal antinociceptive effects of neuropeptide FF analogues. Br J Pharmacol117:493–501.
Kersante F, Mollereau C, Zajac JM, and Roumy M (2006) Anti-opioid activities ofNPFF1 receptors in a SH-SY5Y model. Peptides 27:980–989.
Kim SH and Chung JM (1992) An experimental model for peripheral neuropathyproduced by segmental spinal nerve ligation in the rat. Pain 50:355–363.
Kontinen VK, Aarnisalo AA, Idanpaan-Heikkila JJ, Panula P, and Kalso E (1997)Neuropeptide FF in the rat spinal cord during carrageenan inflammation. Peptides18:287–292.
Kontinen VK and Kalso EA (1995) Differential modulation of �2-adrenergic and�-opioid spinal antinociception by neuropeptide FF. Peptides 16:973–977.
Kotani M, Mollereau C, Detheux M, Le Poul E, Brezillon S, Vakili J, Mazarguil H,Vassart G, Zajac JM, and Parmentier M (2001) Functional characterization of ahuman receptor for neuropeptide FF and related peptides. Br J Pharmacol 133:138–144.
Kovelowski CJ, Ossipov MH, Sun H, Lai J, Malan TP, and Porreca F (2000) Su-praspinal cholecystokinin may drive tonic descending facilitation mechanisms tomaintain neuropathic pain in the rat. Pain 87:265–273.
Lake JR, Hammond MV, Shaddox RC, Hunsicker LM, Yang HY, and Malin DH(1991) IgG from neuropeptide FF antiserum reverses morphine tolerance in therat. Neurosci Lett 132:29–32.
Liu Q, Guan XM, Martin WJ, McDonald TP, Clements MK, Jiang Q, Zeng Z,Jacobson M, Williams DL Jr, Yu H, et al. (2001) Identification and characteriza-tion of novel mammalian neuropeptide FF-like peptides that attenuate morphine-induced antinociception. J Biol Chem 276:36961–36969.
Lombard MC, Simonnet G, Zajac JM, Besson JM, and Allard M (1995) Distributionof neuropeptide FF (FLFQPQRFamide) receptors in the adult rat spinal cord:effects of dorsal rhizotomy and neonatal capsaicin. Neuroscience 68:1229–1235.
Mollereau C, Mazarguil H, Marcus D, Quelven I, Kotani M, Lannoy V, Dumont Y,Quirion R, Detheux M, Parmentier M, et al. (2002) Pharmacological characteriza-tion of human NPFF1 and NPFF2 receptors expressed in CHO cells by using NPYY1 receptor antagonists. Eur J Pharmacol 451:245–256.
Oberling P, Stinus L, Le Moal M, and Simonnet G (1993) Biphasic effect on nocicep-tion and antiopiate activity of the neuropeptide FF (FLFQPQRFamide) in the rat.Peptides 14:919–924.
Ossipov MH, Zhang ET, Carvajal C, Gardell L, Quirion R, Dumont Y, Lai J, andPorreca F (2002) Selective mediation of nerve injury-induced tactile hypersensi-tivity by neuropeptide Y. J Neurosci 22:9858–9867.
Porreca F, Burgess SE, Gardell LR, Vanderah TW, Malan TP Jr, Ossipov MH, LappiDA, and Lai J (2001) Inhibition of neuropathic pain by selective ablation ofbrainstem medullary cells expressing the �-opioid receptor. J Neurosci 21:5281–5288.
Prokai L, Prokai-Tatrai K, Zharikova A, Li X, and Rocca JR (2001) Combinatoriallead optimization of a neuropeptide FF antagonist. J Med Chem 44:1623–1626.
Prokai L, Zharikova AD, Janaky T, and Prokai-Tatrai K (2000) Exploratory phar-macokinetics and brain distribution study of a neuropeptide FF antagonist byliquid chromatography/atmospheric pressure ionization tandem mass spectrome-try. Rapid Commun Mass Spectrom 14:2412–2418.
Simonin F, Schmitt M, Laulin JP, Laboureyras E, Jhamandas JH, MacTavish D,Matifas A, Mollereau C, Laurent P, Parmentier M, et al. (2006) RF9, a potent andselective neuropeptide FF receptor antagonist, prevents opioid-induced toleranceassociated with hyperalgesia. Proc Natl Acad Sci USA 103:466–471.
Vanderah TW, Gardell LR, Burgess SE, Ibrahim M, Dogrul A, Zhong CM, Zhang ET,Malan TP Jr, Ossipov MH, Lai J, et al. (2000) Dynorphin promotes abnormal painand spinal opioid antinociceptive tolerance. J Neurosci 20:7074–7079.
Wang Z, Gardell LR, Ossipov MH, Vanderah TW, Brennan MB, Hochgeschwender U,Hruby VJ, Malan TP Jr, Lai J, and Porreca F (2001) Pronociceptive actions ofdynorphin maintain chronic neuropathic pain. J Neurosci 21:1779–1786.
Wei H, Panula P, and Pertovaara A (2001) Modulation of pain by [1DMe]NPYF, astable analogue of neuropeptide FF, in neuropathic rats. Brain Res 900:234–243.
Xie JY, Herman DS, Stiller CO, Gardell LR, Ossipov MH, Lai J, Porreca F, andVanderah TW (2005) Cholecystokinin in the rostral ventromedial medulla medi-ates opioid-induced hyperalgesia and antinociceptive tolerance. J Neurosci 25:409–416.
Xu M, Kontinen VK, Panula P, and Kalso E (1999) Effects of (1DMe)NPYF, asynthetic neuropeptide FF analogue, in different pain models. Peptides 20:1071–1077.
Yang HY and Iadarola MJ (2006) Modulatory roles of the NPFF system in painmechanisms at the spinal level. Peptides 27:943–952.
Yang HY, Tao T, and Iadarola MJ (2008) Modulatory role of neuropeptide FF systemin nociception and opiate analgesia. Neuropeptides 42:1–18.
Address correspondence to: Dr. Luis R. Gardell, Cara Therapeutics, Inc.,One Parrott Dr., Shelton, CT 06484. E-mail: [email protected]
254 Lameh et al.
at ASPE
T Journals on Septem
ber 28, 2016jpet.aspetjournals.org
Dow
nloaded from
Copyright © 2022 FDOKUMEN




















