
Zebrafish embryo proteins induce apoptosis in human colon cancer cells (Caco2)
12
Apoptosis (2006) 11:1617–1628 DOI 10.1007/s10495-006-8895-4 Zebrafish embryo proteins induce apoptosis in human colon cancer cells (Caco2) Alessandra Cucina · Pier-Mario Biava · Fabrizio D’Anselmi · Pierpaolo Coluccia · Filippo Conti · Roberta di Clemente · Alfredo Miccheli · Luigi Frati · Alberto Gulino · Mariano Bizzarri Published online: 27 June 2006 C Springer Science + Business Media, LLC 2006 Abstract Previous studies have shown that proteins ex- tracted from Zebrafish embryo share some cytostatic charac- teristics in cancer cells. Our study was conducted to ascertain the biological properties of this protein network. Cancer cell growth and apoptosis were studied in Caco2 cells treated with embryonic extracts. Cell proliferation was significantly inhibited in a dose-dependent manner. Cell-cycle analysis in treated cells revealed a marked accumulation in the G 2 /M phase preceding induction of apoptosis. Embryo proteins in- duced a significant reduction in FLIP levels, and increased caspase-3 and caspase-8 activity as well as the apoptotic rate. Increased phosphorylated pRb values were obtained in treated Caco2 cells: the modified balance in pRb phospho- rylation was associated with an increase in E2F1 values and c-Myc over-expression. Our data support previous reports of an apoptotic enhancing effect displayed by embryo extracts, mainly through the pRb/E2F1 apoptotic pathway, which thus suggests that Zebrafish embryo proteins have complex anti- cancer properties. A. Cucina · F. D’Anselmi · P. Coluccia Department of Surgery “Pietro Valdoni”, Universit` a La Sapienza, Roma P.-M. Biava Foundation for Research into the Biological Therapy of Cancer, Milano F. Conti · R. d. Clemente · A. Miccheli Department of Chemistry, Universit` a La Sapienza, Roma L. Frati · A. Gulino · M. Bizzarri () Department of Experimental Medicine and Pathology, Universit` a La Sapienza, via Scarpa 14-16, 00161, Roma e-mail: [email protected] Keywords Apoptosis . Zebrafish embryo proteins . Caco2 . E2F1 . c-FLIP S Introduction Apoptosis plays an important role in living cells from the early stages of embryonic development. During organogen- esis, programmed cell death is as essential as cell differ- entiation and cell proliferation to properly regulate tissue and overall body shape and functions [1, 2] Both human and animal embryonic serum contain soluble stem cell dif- ferentiating and apoptosis-inducing proteins that selectively act on cancer cells, without interfering with normal prolif- erating tissues [3]. Generally, tumour development in em- bryo tissues, induced by several carcinogenic compounds, is strongly inhibited or almost totally suppressed when dif- ferentiating processes are in progress [4]. It is well known that carcinogens, before or during the main organogenetic period, cause a high rate of malformations and teratomas in offspring, though not tumour induction; once organogenesis is complete, the frequency of tumour induction rises with a concomitant decrease in the rate of malformations [5]. Moreover, teratocarcinoma, leukaemia and adrenal medulla carcinoma cells differentiate to normal tissues when placed in a normal embryonic environment [6]. Indeed, an increas- ing body of evidence from experimental studies suggests that human cancers may arise from the differentiation ar- rest of proliferating cancer stem cells. The maturation ar- rest of cancer stem cells can be overcome and the cancer stem cell induced to differentiate either by transplantation of the cancer cells into normal blastocysts [7] or by treat- ment with retinoids [8] or other differentiating factors [9]. This has led to the concept of “differentiation therapy” for cancer. Springer
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Zebrafish embryo proteins induce apoptosis in human colon cancer cells (Caco2)

Apoptosis (2006) 11:1617–1628DOI 10.1007/s10495-006-8895-4
Zebrafish embryo proteins induce apoptosis in human coloncancer cells (Caco2)Alessandra Cucina · Pier-Mario Biava ·Fabrizio D’Anselmi · Pierpaolo Coluccia ·Filippo Conti · Roberta di Clemente ·Alfredo Miccheli · Luigi Frati · Alberto Gulino ·Mariano Bizzarri
Published online: 27 June 2006C© Springer Science + Business Media, LLC 2006
Abstract Previous studies have shown that proteins ex-tracted from Zebrafish embryo share some cytostatic charac-teristics in cancer cells. Our study was conducted to ascertainthe biological properties of this protein network. Cancer cellgrowth and apoptosis were studied in Caco2 cells treatedwith embryonic extracts. Cell proliferation was significantlyinhibited in a dose-dependent manner. Cell-cycle analysis intreated cells revealed a marked accumulation in the G2/Mphase preceding induction of apoptosis. Embryo proteins in-duced a significant reduction in FLIP levels, and increasedcaspase-3 and caspase-8 activity as well as the apoptoticrate. Increased phosphorylated pRb values were obtained intreated Caco2 cells: the modified balance in pRb phospho-rylation was associated with an increase in E2F1 values andc-Myc over-expression. Our data support previous reports ofan apoptotic enhancing effect displayed by embryo extracts,mainly through the pRb/E2F1 apoptotic pathway, which thussuggests that Zebrafish embryo proteins have complex anti-cancer properties.
A. Cucina · F. D’Anselmi · P. ColucciaDepartment of Surgery “Pietro Valdoni”, Universita La Sapienza,Roma
P.-M. BiavaFoundation for Research into the Biological Therapy of Cancer,Milano
F. Conti · R. d. Clemente · A. MiccheliDepartment of Chemistry, Universita La Sapienza,Roma
L. Frati · A. Gulino · M. Bizzarri (�)Department of Experimental Medicine and Pathology, UniversitaLa Sapienza, via Scarpa 14-16, 00161,Romae-mail: [email protected]
Keywords Apoptosis . Zebrafish embryo proteins .
Caco2 . E2F1 . c-FLIPS
Introduction
Apoptosis plays an important role in living cells from theearly stages of embryonic development. During organogen-esis, programmed cell death is as essential as cell differ-entiation and cell proliferation to properly regulate tissueand overall body shape and functions [1, 2] Both humanand animal embryonic serum contain soluble stem cell dif-ferentiating and apoptosis-inducing proteins that selectivelyact on cancer cells, without interfering with normal prolif-erating tissues [3]. Generally, tumour development in em-bryo tissues, induced by several carcinogenic compounds,is strongly inhibited or almost totally suppressed when dif-ferentiating processes are in progress [4]. It is well knownthat carcinogens, before or during the main organogeneticperiod, cause a high rate of malformations and teratomas inoffspring, though not tumour induction; once organogenesisis complete, the frequency of tumour induction rises witha concomitant decrease in the rate of malformations [5].Moreover, teratocarcinoma, leukaemia and adrenal medullacarcinoma cells differentiate to normal tissues when placedin a normal embryonic environment [6]. Indeed, an increas-ing body of evidence from experimental studies suggeststhat human cancers may arise from the differentiation ar-rest of proliferating cancer stem cells. The maturation ar-rest of cancer stem cells can be overcome and the cancerstem cell induced to differentiate either by transplantationof the cancer cells into normal blastocysts [7] or by treat-ment with retinoids [8] or other differentiating factors [9].This has led to the concept of “differentiation therapy” forcancer.
Springer

1618 Apoptosis (2006) 11:1617–1628
These findings suggest that cancer development can, atleast in part, be controlled by embryonic factors sharingdifferentiating and/or apoptotic activity. It has been estab-lished that malignant cell transformation does not necessar-ily destroy the expression potential of differentiated char-acteristics, of apoptosis or of growth arrest [10]. Prelim-inary results have been obtained both from in vivo andin vitro studies with proteins extracted from Xenopus l.or Zebrafish during different embryonal stages of differ-entiation. Some clinical trials, performed using zebrafishembryo extract administered to advanced cancer patientswho no longer respond to conventional treatments, haveshown marked beneficial effects (improvement in perfor-mance status and significant increase in overall survival)[11, 12]. Likewise, in vitro studies have demonstrated thatembryo proteins synthesized in the pregnant uteri of mam-mals [13] or by totipotent embryonal zebrafish cells [14],activate an apoptotic response accompanied by a signifi-cant inhibition of the proliferation rate in several cancer celllines.
Caco2 colon cancer cells generally respond but mini-mally to cytostatic drugs and are particularly resistant toapoptosis-inducing factors, showing a highly malignant phe-notype characterised by the loss of the p-53 gene [15]. More-over, in some colon cancer cell lines, the pRb protein behavesparadoxically: the hypophosphorylated or unphosphorylatedpRb behaves as a tumour-suppressor gene, exerting a neg-ative control on cancer growth [16]. Inactivation of pRb byhyperphosphorylation in the late G1 phase causes the re-lease of E2F1, thereby allowing the transcription of genesrequired for DNA synthesis or induction of apoptosis [17,18] through both the intrinsic and the extrinsic apoptoticpathway. This process involves not only the overexpres-sion of caspase-3 (for review see Ginsberg [19]), but alsoa down-regulation of the cellular FLICE-inhibitory proteinshort [20].
The aim of this study was to shed light on the selectivebiochemical pathways involved in the inhibition of growthas well as in the regulation of apoptosis induced by in vitrotreatment of Caco2 cells with stem cell proteins obtainedfrom Zebrafish embryos.
Materials and methods
Embryo extracts
The embryos of Zebrafish (cultured under standard condi-tions as previously described [21]) at 50% of the epibolystage (ZEPEP) or at the gastrula stage (ZEPGA) were sepa-rately collected, washed in distilled water and dissolved witha turboemulsifier in cold PBS for 60 sec. The extracts wereprepared as 1 microgram/ml stocks in a solution of pure glyc-
erine and 30% ethylic alcohol at a ratio of 4:1. The extractswere then stored at 4◦C until use.
Cell culture
The human colorectal cancer cell line Caco2 was obtainedfrom the European Collection of Cell Cultures (ECACC).The cells were seeded in 25-cm2 flasks (Falcon; BectonDickinson Labware; Franklin Lakes NJ, USA) in Dulbeccomodified Eagle medium (DMEM) supplemented with 10%fetal calf serum (FCS), non essential aminoacids and antibi-otics (Penicillin 100 IU/ml, Streptomycin 100 µg/ml, Gen-tamycin 200 µg/ml) (standard medium). The cultures werekept at 37◦C in an atmosphere of 5% CO2 in air. The mediumwas changed every third day. At confluence, the cells weresubcultured after removal with 0.05% trypsin-0.01% EDTA.Caco2 cells were used for experiments from passages 30 to40. Cell viability was assessed by means of the Trypan Blue(Sigma Chemical Co., St. Louis MO, USA) dye exclusionmethod.
Proliferation assay
Caco2 cell proliferation was determined using a modifica-tion of the sulforhodamine B (SRB) assay, as previouslydescribed [22]. Briefly, assays were carried out in 96-wellculture plates (Falcon, Becton Dickinson Labware). Caco2cells were plated at a concentration of 5 × 103 cells/well in astandard medium and were allowed to attach overnight. Thefollowing day, the cells were refed with 200 µL of standardmedium containing ZEPEP or ZEPGA at concentrations rang-ing from 0.03 µg/ml to 3 µg/ml. The plates were incubatedfor 24, 48, 72, and 96 h at 37◦C in an atmosphere of 5% CO2
in air. Caco2 cells were fixed with cold 50% trichloroaceticacid (TCA) solution for 1 hr at 4◦C in the dark. The mediaand TCA were removed and the plates were rinsed with wa-ter five times and then air dried. The cells were stained byaddition of 50 µL of 0.4% SRB (Sigma Chemical Co.) in 1%acetic acid for 15 min. The stain was removed and the cellswere washed five times with 1% acetic acid and air dried;100 µl of 10 mmol/L unbuffered Tris was then added to eachwell to dissolve the dye. The plates were shaken for 15 minuntil the dye was uniformly distributed and then read on aMicrotiter Plate Reader (Opsys MR) at 490 nm. Standardmedium was used as the blank for these assays.
Cell count assay
Caco2 cells were seeded in 6-well culture plates (Falcon,Becton Dickinson Labware) at a concentration of 1 × 105
cells/well in a standard medium. The following day, thecells were refed with standard medium containing ZEPEP
or ZEPGA at concentrations ranging from 0.03 µg/ml to
Springer

Apoptosis (2006) 11:1617–1628 1619
3 µg/ml. The plates were incubated for 24, 48 and 72 hrs at37◦C in an atmosphere of 5% CO2 in air. Caco2 cells werethen detached from wells by trypsinization and centrifuged,and the cell pellets were resuspended in PBS. Cell count wasperformed by a particle count and size analyzer (BeckmanCoulter, Inc.).
Annexin V/7-AAD staining
Caco2 cells were cultured at confluence into 25-cm2 flasks(Falcon, Becton Dickinson Labware) in a standard medium.Caco2 cells were then treated with ZEPEP or ZEPGA at con-centrations of 0.3 µg/ml (ZEP1EP and ZEP1GA) and 3 µg/ml(ZEP2EP and ZEP2GA) for 24 and 72 hrs in a standardmedium, trypsinized and washed twice with PBS. Caco2cells were stained with FITC labelled Annexin V/7-AAD (7-aminoactinomycine-D) according to the manufacturer’s in-structions (Annexin V/7-AAD kit, Immunotech, Marseille,France). Briefly, a washed cell pellet was resuspended in1.0 ml binding buffer at a concentration of 105–106 cells/ml; 10 µl of Annexin V together with 20 µl 7-AAD wereadded to 400 µl cell suspension. Caco2 cells were incubatedfor 10 min on ice in the dark. The samples were analyzed byflow cytometry.
Flow cytometry
Flow cytometry was performed using an EPICS Coul-ter XL (Beckman-Coulter Inc. Fullerton, CA, USA). Thefluorescence of 10,000 events was measured. In general,this resulted in a situation in which viable, apoptotic anddead subpopulations could be adequately assessed (>200events/population). An excitation wavelength of 488 nm wasused in combination with standard filters to discriminate be-tween the FL1, FL2 and FL3 channels.
Cell cycle analysis
Caco2 cells were cultured at confluence into 25-cm2 flasks(Falcon, Becton Dickinson Labware) in a standard medium.Caco2 cells were then treated with ZEPEP or ZEPGA atconcentrations of 0.3 µg/ml and 3 µg/ml for 24 and72 hrs in a standard medium. Caco2 cells were harvested,washed and stained with aqueous staining solution contain-ing 0.75 µmol/L (0.5 µg ml−1) Propidium Iodide (PI), 4 µMsodium citrate, 1% Triton-X100 and 1% bovine serum albu-min (all from Sigma Chemical Co.) at 4◦C overnight. PI-stained cells were measured using the Coulter Epics XL andanalyzed by Modfit LT Software (Veruty software Inc. USA).
ELISA for Caco2 cell apoptosis detection
Caco2 cell apoptosis was measured by an ELISA based ondetection of histone-associated DNA fragments in the cy-
toplasm of apoptotic cells according to the manufacturer’sinstructions (Cell Death Detection ELISA Plus kit, Roche Di-agnostics). Caco2 cells, seeded at a concentration of 1.0 ×104 cells/well in a 96-well plate (Falcon, Becton DickinsonLabware), were cultured in a standard medium for 24 hrs.Caco2 cells were then treated with ZEPEP or ZEPGA at con-centrations of 0.3 µg/ml and 3 µg/ml for 24 and 72 hrs ina standard medium. Cells were then detached from wellsby trypsinization and centrifuged, and cell pellets were re-suspended in PBS. Cell count was performed by a particlecount and size analyzer (Beckman Coulter, Inc.). The cy-toplasmic nucleosome content, detected using the ELISAmethod, was determined by optical density measured at405 nm with a Microtiter Plate Reader (Opsys MR, DinexTechnologies, Inc. Chantilly, Virginia, USA). The enrich-ment of mono- and oligonucleosomes released into the cy-toplasm was calculated as the ratio of the absorbance of thesample cells to the absorbance of control cells. The enrich-ment factor was used as a parameter of apoptosis [23] andis shown on the y axis (Fig. 3(a)) as the mean ± standarddeviation from six separate experiments performed in tripli-cate. An enrichment factor of 1.0 represents background orspontaneous apoptosis (7% ± 0.8), as determined in par-allel by flow cytometry analysis after Annexin V/7-AADstaining.
Caspase activity assay
Caspase-3 and caspase-8 activity was measured using col-orimetric assay kits (Chemicon International, Inc. Temec-ula, CA, USA) according to the manufacturer’s instructions.The assay is based on spectrophotometric detection of thechromophore p-nitroaniline (pNA) after cleavage from thelabelled substrate that recognizes the optimal tetrapeptidesequence of individual activation sites. Briefly, Caco2 cells,treated with ZEPEP or ZEPGA at concentrations of 0.3 µg/ml(ZEP1EP and ZEP1GA) and 3 µg/ml (ZEP2EP or ZEP2GA) for24 hrs, were detached from culture flasks by trypsinizationand centrifuged; cell pellets were lysed in cell lysis buffer for10 min on ice and then centrifuged for 5 min at 10,000 × g.Lysed cells were incubated with 10 µl of substrate specificfor caspase-3 and caspase-8 in a 96-well plate for 2 hrs at37◦C. The specific substrate used was Ac-DEVD-pNA forcaspase-3 and Ac-IETD-pNA for caspase-8. The pNA lightemission was quantified using a Microtiter Plate Reader (Op-sys) at 405 nm. The optical density values obtained werecompared with a pNA standard titration curve to determinethe relative activities.
Immunocytochemical assay
Caco2 cells were seeded in a 60 mm diameter Petri dish(Falcon) in complete medium. At semiconfluence, cells weretreated with ZEPEP at concentrations of 0.3 µg/ml (ZEP1EP)
Springer

1620 Apoptosis (2006) 11:1617–1628
and 3 µg/ml (ZEP2EP) for 24 hrs at 37◦C in an atmosphere of5% CO2. Cells were washed twice with PBS, fixed in forma-lin for 30 min and then washed in PBS. Immunocytochem-istry was performed by a Labelled-[strept] Avidin-Biotin(LAB-SA) method using a commercial kit (Histostain-plusKit; Biømeda, Foster City, CA, USA) according to the man-ufacturer’s instructions. Cells were stained with a rabbitpolyclonal anti-caspase-3 (V10248, Biømeda) and with arabbit polyclonal anti-caspase-8 (V10251, Biømeda). Af-ter incubation, primary antibodies were removed and bi-otinilated secondary antibody was added for 30 min. Af-ter two washes in PBS, the avidin-biotin complex wasadded for 30 min at room temperature. Cells were washedin PBS and the peroxidase reaction was initiated using0.06% diaminobenzidine (DAB) and 0.01% hydrogen per-oxide. After a final rinse, the cells were mounted under acoverslip.
Preparation of cellular extracts
Caco2 cells were scraped in the following lysis buffer: TRISpH 7,4 50 mmol/L, NaCl 150 mmol/L, Nonidet P 40 0,2%,CHAPS 1%, EDTA 2 mmolM/L dissolved in tetradistilledwater. A mix of protease inhibitors (PMSF 200 µmol/L, Le-upeptin 1 µmol/L, Pepstatin A 1 µmol/L, Calpain inhibitor5 µmol/L) was added just before use. Cytoplasmic extractswere then sonicated and centrifuged at 6000 rpm and theprotein content of supernatants was determined using theBradford assay.
Western blot analysis
Cellular extracts were separated on SDS-PAGE gels with aconcentration of acrylamide specific for the protein studied.Proteins were blotted onto nitrocellulose membranes (BIO-RAD, Bio-Rad Laboratories, Hercules, CA) and probed withthe following antibodies: anti-RB (sc-50), anti -E2F1 sc-251,anti-c-Myc (sc-40), anti –cFLIPS sc-5276, all from SantaCruz biotechnology, anti-cFLIPL, from Upstate Biotech.(Saranac Av., Lake Placid NY) and anti-alpha-tubulin, fromSigma. Antigens were detected with an enhanced chemo-luminescence (ECL) kit from Amersham (Amersham Bio-sciences, Little Chalfont, Buckinghamshire, England) ac-cording to the manufacturer’s instructions.
Densitometry
All Western-blot images were acquired and analyzed throughImaging Fluor S densitometer (Biorad-Hercules). The opti-cal density of each condition was normalized against thesignal of the internal control alpha-tubulin.
Statistical analysis
Quantitative experiments were analysed using the Student’spaired t test.
Results
Antiproliferative effect of embryo proteins on Caco2cells
To assess the inhibitory effect of both ZEPEP and ZEPGA
on cell growth, human Caco2 cells were exposed to variousconcentrations of embryo (from 0.003 to 3 µg/ml), afterwhich cell proliferation was measured by the Srb-c assay andcell count at different times. In ZEPEP-treated samples, cellgrowth was significantly and dose-dependently reduced after24 hrs in all the concentrations tested (Fig. 1(a)). The IC50
was experimentally identified at a concentration of 0.3 µg/ml(ZEP1EP). The embryo fraction obtained before the onset ofgastrulation (ZEP1GA) had no effect on cell proliferation(Fig. 1(b)).
Embryo treatment causes an accumulation of cells in theG2/M phase of the cell cycle
Caco2 cells, treated with two different concentrations ofZEPEP at 0.3 µg/ml (ZEP1EP) and 3 µg/ml (ZEP2EP), showeda marked increase in the G2/M fraction (Figs. 2(a) and (b)),with a corresponding reduction in both the G1 and S frac-tions. The G2/M fraction increased in both the ZEP1EP- andZEP2EP-treated Caco2 cells (up to 60% after 72 hrs). Thiseffect was more evident for ZEP2EP and shows a moderatebut significant time-dependent progression (35% of cells inG2/M at 72 hrs versus 27% at 24 hrs). No effects on the cell-cycle distribution were recorded in ZEPGA-treated samples.
Apoptotic effects of ZEPEP proteins on Caco2 cells
Data obtained with both the DNA-fragmentation-test andcytofluorimetric assays demonstrated a significant apoptoticeffect (five and seven-fold increase for ZEP1EP and ZEP2EP
respectively) in ZEPEP-treated cancer cells, reaching valuessimilar to those obtained with Camptothecin (Figs. 3(a) and(b)). Differences in the apoptotic rate between ZEP1EP andZEP2EP were significantly correlated in a dose-dependentmanner. The apoptotic index (i.e. the ratio between apoptoticand viable cells) was significantly higher in Camptothecin-and ZEPEP-treated cells than in controls, while there wasno significant difference in the apoptotic index betweenCamptothecin- and ZEPEP-treated cells (data not shown).Exposure of Caco2 cells to ZEPGA did not reveal any signif-icant apoptotic effect when compared with control values.
Springer

Apoptosis (2006) 11:1617–1628 1621
Fig. 1 ZEPEP-dependentinhibition of Caco-2 cancergrowth. (a) Caco-2 cells wereexposed for 24 hrs to ZEPEP
(embryo cell extracts at 50% ofthe epiboly stage) and (b) toembryo extracts from thegastrula stage (ZEPGA) atconcentrations ranging from0.03 µg/ml to 3 µg/ml. Results,expressed as percentage ofcontrols, are the mean ± SD ofeight independent cell-countexperiments performed by aparticle count and size analyzer(Beckman Coulter). ∗p <
0.001;∗∗p < 0.01 by Student’s ttest
Apoptosis-related effectors
Programmed cell death may be mediated by activation ofdeath receptors (death receptor pathway), resulting in anup-regulation of the caspase-8 activity, or by the releaseof cytochrome c from mitochondria (mitochondrial path-way), involving the p53 protein and members of the Bcl-2 family of proteins. Activation of effector caspases, suchas caspase-3, is believed to be a highly specific key eventduring apoptosis, resulting in the cleavage of the vast ma-jority of polypeptides that undergo proteolysis in apoptoticcells. A significant role in apoptotic pathways is shared bythe regulation of c-Myc expression, E2F1 and pRb (hyper-phosphorylated and un-phosphorylated) status, FLIP inhibi-tion and the CD95/FAS/Apo-1 signalling complexes (hence-forth called CD95).
Active caspase-8 levels, which were determined by bothcolorimetric assay (Fig. 4(a)) and immunocytochemistry(Figs. 4(c)–(e)), were significantly higher 24 hrs aftertreatment with ZEPEP, though not with ZEPGA. No sig-nificant differences were observed between ZEP1EP andZEP2EP.
A significant activation of caspase-3, induced by bothZEP1EP and ZEP2EP 24 hrs after treatment, was revealed by
both colorimetric assay (Fig. 4(b)) and immunocytochem-ical analysis (Figs. 4(f)–(h)). No significant differences incaspase-3 activity were observed in the ZEPGA-treated cells.
No detectable levels of p53 (data not shown) wererecorded in either control or treated colon cancer cells, asexpected for the Caco2 cell line, in which the p-53 gene isdeleted [15].
The CD95 signalling complex is a 45-kDa cell sur-face protein that belongs to the tumour necrosis factor(TNF)/nerve growth factor (NGF) receptor family. Followingstimulation by its cognate ligand (FasL), CD95 oligomerizesand recruits several death–associated molecules and caspase-8 into a multimolecular signalling platform (DISC, death-inducing signalling complex). Further activation of caspase-8 initiates the apoptotic signalling cascade. CD95 expressionwas unaffected by ZEPEP or ZEPGA treatment, there being nosignificant modifications in any of the conditions tested (datanot shown).
FLIP
The anti-apoptotic protein c-FLIP inhibits death receptor-induced apoptosis, presumably by blocking the interactionbetween CD95 and procaspase-8, which in turn reduces
Springer
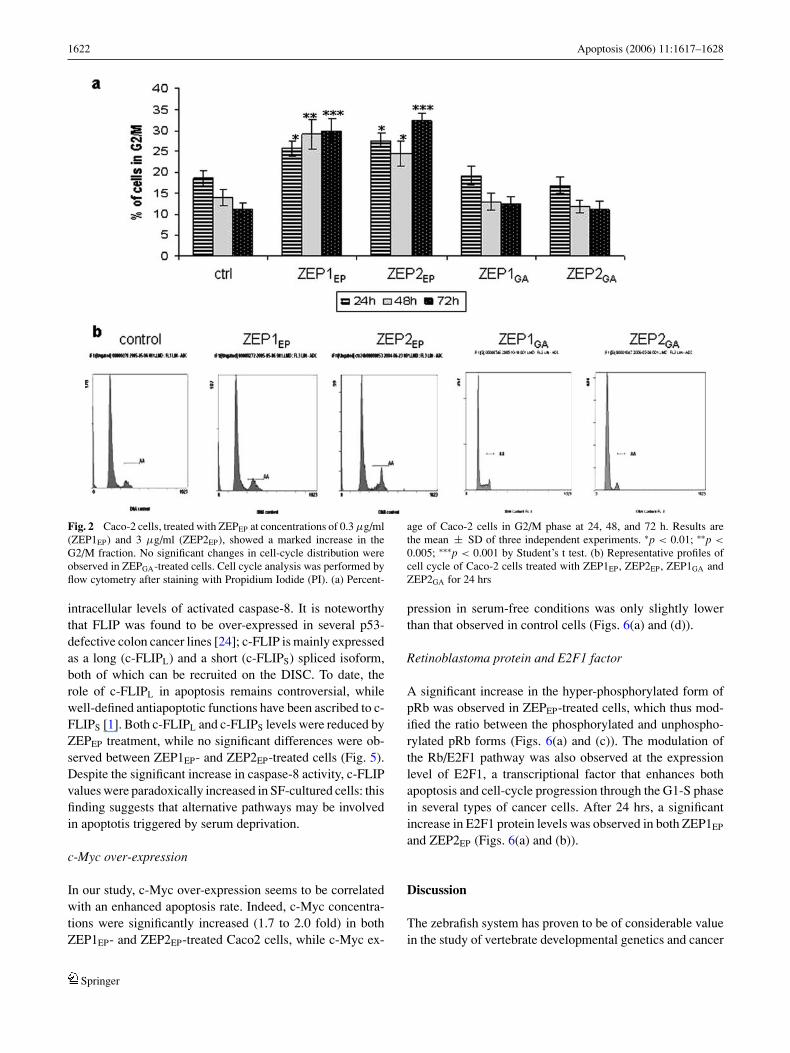
1622 Apoptosis (2006) 11:1617–1628
Fig. 2 Caco-2 cells, treated with ZEPEP at concentrations of 0.3 µg/ml(ZEP1EP) and 3 µg/ml (ZEP2EP), showed a marked increase in theG2/M fraction. No significant changes in cell-cycle distribution wereobserved in ZEPGA-treated cells. Cell cycle analysis was performed byflow cytometry after staining with Propidium Iodide (PI). (a) Percent-
age of Caco-2 cells in G2/M phase at 24, 48, and 72 h. Results arethe mean ± SD of three independent experiments. ∗p < 0.01; ∗∗p <
0.005; ∗∗∗p < 0.001 by Student’s t test. (b) Representative profiles ofcell cycle of Caco-2 cells treated with ZEP1EP, ZEP2EP, ZEP1GA andZEP2GA for 24 hrs
intracellular levels of activated caspase-8. It is noteworthythat FLIP was found to be over-expressed in several p53-defective colon cancer lines [24]; c-FLIP is mainly expressedas a long (c-FLIPL) and a short (c-FLIPS) spliced isoform,both of which can be recruited on the DISC. To date, therole of c-FLIPL in apoptosis remains controversial, whilewell-defined antiapoptotic functions have been ascribed to c-FLIPS [1]. Both c-FLIPL and c-FLIPS levels were reduced byZEPEP treatment, while no significant differences were ob-served between ZEP1EP- and ZEP2EP-treated cells (Fig. 5).Despite the significant increase in caspase-8 activity, c-FLIPvalues were paradoxically increased in SF-cultured cells: thisfinding suggests that alternative pathways may be involvedin apoptotis triggered by serum deprivation.
c-Myc over-expression
In our study, c-Myc over-expression seems to be correlatedwith an enhanced apoptosis rate. Indeed, c-Myc concentra-tions were significantly increased (1.7 to 2.0 fold) in bothZEP1EP- and ZEP2EP-treated Caco2 cells, while c-Myc ex-
pression in serum-free conditions was only slightly lowerthan that observed in control cells (Figs. 6(a) and (d)).
Retinoblastoma protein and E2F1 factor
A significant increase in the hyper-phosphorylated form ofpRb was observed in ZEPEP-treated cells, which thus mod-ified the ratio between the phosphorylated and unphospho-rylated pRb forms (Figs. 6(a) and (c)). The modulation ofthe Rb/E2F1 pathway was also observed at the expressionlevel of E2F1, a transcriptional factor that enhances bothapoptosis and cell-cycle progression through the G1-S phasein several types of cancer cells. After 24 hrs, a significantincrease in E2F1 protein levels was observed in both ZEP1EP
and ZEP2EP (Figs. 6(a) and (b)).
Discussion
The zebrafish system has proven to be of considerable valuein the study of vertebrate developmental genetics and cancer
Springer

Apoptosis (2006) 11:1617–1628 1623
Fig. 3 ZEPEP induces apoptosis in Caco-2 cells. Caco-2 cells weretreated with ZEPEP and ZEPGA at concentrations of 0.3 µg/ml (ZEP1EP,ZEP1GA) and 3 µg/ml (ZEP2EP, ZEP2GA) for 24 hrs and with Camp-tothecin (10 µg/ml) as positive control. (a) Quantification of nucleoso-mal DNA fragmentation was performed by the Cell Death DetectionELISA plus Kit (Roche). The rate of apoptosis is demonstrated by theenrichment of nucleosomes in the cytoplasm, shown on the y axis;an enrichment factor of 1 is equivalent to background apoptosis. Re-
sults are the mean ± SD of six independent experiments performedin triplicate. ∗p < 0.05; ∗∗p < 0.01 by Student’s t test. (b-c) Apop-tosis determination in Caco-2 cells by Annexin V-FITC and 7-AADstaining flow cytometry. (b) Percentage of apoptotic cells. Results arethe mean ± SD of three independent experiments. ∗∗∗p < 0.001 byStudent’s t test. (c) Representative profiles of Annexin V-FITC stainingflow cytometry
toxicology [26]. More recently, some preliminary reportshave highlighted the relevance of stem cell differentiatingproteins extracted from zebrafish embryos in inducing cancerinhibition and apoptosis [14].
In this study we investigate whether protein factors ob-tained from a specific differentiating stage (50% epiboly) ofzebrafish embryos may inhibit colon cancer growth. Our re-sults support the hypothesis that ZEPEP suppresses Caco2growth and induces apoptosis, mainly via the pRb/E2F1apoptotic pathway: loss of or reduced pRb availability, due tohyperphosporylation, leads to the deregulation of E2F1 ac-tivity and, in the absence of other genetic changes, to deathin many cell lineages [17, 27, 28] Inactivation, loss or hyper-phosphorylation of Rb leads specifically to E2F1-dependentapoptosis, which requires p73 [29].
In other colon cancer cell lines, several apoptotic factorshave induced cell arrest in the G1 phase, mainly through theinduction of p53 and the cell-cycle inhibitor p21 [30]. It is
well known that p53 controls cell-cycle arrest at the G1-Stransition in response to DNA damage, involving both p53[31] and the p53 downstream effector protein p21 [32] as keyfactors in mediating negative regulation of cell proliferation.Nevertheless, as previously described [15], the p53 gene inCaco2 cells is deleted and, in our experiments, no accumula-tion of the corresponding protein was observed. The absenceof p53 may explain why apoptosis in Caco2 cells cannotbe triggered by blocking cell proliferation in the G1 phase.This means that ZEPEP increases the apoptosis rate in Caco2cells, presumably through the activation of p53-independentmechanisms that lead to an unexpected, specific accumula-tion of growing cells in the G2/M phase, as observed formesalazine, a non-steroidal anti-inflammatory drug [33], orafter over-expression of E2F1 in colon cancer p53-defectivecells [34]. Thus, although wild-type p53 may be requiredfor G1 arrest, E2F1-associated G2-M arrest and apoptosisappear to be able to proceed regardless of the status of p53
Springer

1624 Apoptosis (2006) 11:1617–1628
Fig. 4 ZEPEP induces activation of caspase-8 and caspase-3 in Caco-2cells. Caco-2 cells were treated with ZEPEP or ZEPGA at concentrationsof 0.3 µg/ml (ZEP1EP, ZEP1GA) and 3 µg/ml (ZEP2EP, ZEP2GA) for24 hrs and with serum-free medium as positive apoptotic control. (a)Activity of caspase-8, determined by a colorimetric assay kit (Chemi-con), was measured by optical density at 405 nm and presented as thefold increase when compared with an uninduced, negative control. Re-sults are the mean ± SD of three independent experiments performedin triplicate. ∗p <0.001 by Student’s t test. (b) Activity of caspase-3, determined by a colorimetric assay kit (Chemicon), was measured
by optical density at 405 nm and presented as the fold increase whencompared with an uninduced, negative control. Results are the mean± SD of three independent experiments performed in triplicate. ∗p< 0.001 by Student’s t test. (c-e) Caspase-8 immunocytochemical lo-cation in Caco-2 control cells (c) and in cells treated with ZEP1EP
(d) and ZEP2EP (e). The data shown are representative of three inde-pendent experiments. (f-h) Caspase-3 immunocytochemical locationin Caco-2 control cells (f) and in cells treated with ZEP1EP (g) andZEP2EP(h). The data shown are representative of three independentexperiments
in these cell lines. Indeed, previous studies have shown thatp53-deficient colon cancer cell lines undergo apoptosis andarrest in the G2/M phase of the cell cycle when exposed tochemotherapeutic drugs or DNA-damaging agents [35].
E2F1 stimulates both proliferation and apoptosis.Whether E2F1 contributes to proliferation or apoptosis maydepend on E2F1 levels in the cell [36]. Increased levelsof phosphorylated pRb lead to an increased release (10-to 17-fold) of E2F1 in colon cancer cells and increasedapoptotic activity, while over-expression of unphosphory-lated pRb in colorectal cancer cells may provide a home-ostatic mechanism that protects them from growth inhibi-tion and apoptosis by counterbalancing death signals dueto excessive E2F1 activity [2]. It is known that increasedE2F1 release in colon cancers is linked to the activation ofapoptosis, via both the p53-dependent and p53-independent
pathways. In the p53-independent pathways, apoptosis maybe mediated by the p53 homologue p73, which leads tocaspase-3 activation, a key-factor in the “executive phase”of apoptosis. As previously stated, this specific E2F1-relatedapoptotic pathway requires acetylation for E2F1 recruitmenton the P1p73 promoter and for its transcriptional activation[37]. Furthermore, it has been demonstrated that a paradox-ical increase in unpRb levels or forced E2F1 expression,even though associated with the G1-S transition, eventuallyleads to G2/M cell-cycle arrest [38]. Furthermore, E2F1 mayalso activate caspase-8-dependent apoptosis by directly up-regulating c-Myc expression in colon cancer cells [39]. Theover-expression of c-Myc, documented by our results, mayenhance the pRb/E2F1 apoptotic pathway through an alterna-tive biochemical mechanism i.e. the inhibition of c-FLIP andthe subsequent activation of caspase-8-dependent apoptosis.
Springer

Apoptosis (2006) 11:1617–1628 1625
Fig. 5 ZEPEP reduces proteinlevels of c-FLIPL and c-FLIPS
in Caco-2 cells. (a)Representative Western blotshowing the reduction in levelsof c-FLIPL and c-FLIPS inCaco-2 cells, after 72 hrs oftreatment with ZEPEP atconcentrations of 0.3 µg/ml(ZEP1EP) and 3 µg/ml(ZEP2EP), compared withuntreated control cells. Alphatubulin was used as a loadingcontrol. (b) Densitometricquantification of protein levelsof c-FLIPL, obtained by opticaldensity (O.D.) of protein bandsnormalized with O.D. of alphatubulin bands. Results are themean ± SD of threeindependent experiments. ∗p <
0.05 by Student’s t test. (c)Densitometric quantification ofprotein levels of c-FLIPS,obtained by O.D. of proteinbands normalized with O.D. ofalpha tubulin bands. Results arethe mean ± S.D. of threeindependent experiments. ∗p <
0.05 by Student’s t test
The gene encoding the caspase activation inhibitor (c-FLIP)appears to be a direct target of c-Myc-mediated transcrip-tional repression: over-expression of c-Myc decreases FLIPlevels both in vitro and in vivo and enhances TRAIL-inducedcaspase-8 cleavage, sensitizing cells to death receptor stim-uli [40], even if in the absence of any major increase inCD95 complexes; as previously shown, the inducible synthe-sis of CD95 is not a prerequisite for drug-induced caspase-8activation and apoptosis [41]. Moreover, previous studieshave suggested that the ability of c-Myc to induce apop-tosis is E2F1-dependent and is markedly reduced in cellslacking E2F1 [42]; indeed, E2F1 induces a specific decreasein c-FLIPS mRNA, thereby facilitating caspase-8 activation[19], restoring tumour cell sensitivity to death-receptor stim-uli. Nevertheless, as c-Myc exerts other apoptotic effects,which are both p53-dependent and p53-independent [43],the caspase-8 activation demonstrated by our results is un-likely to be related exclusively to c-FLIPS inhibition or pre-liminary caspase-3 activation, as suggested by Wieder andco-workers [44].
Alternative pathways are probably triggered, which isprobably what occurs in the case of serum-free culturedCaco2 cells, in which the significant increase in caspase-8plays a major role in inducing apoptosis, even if in the pres-ence of a paradoxical increase in c-FLIPS. This phenomenonmay be independent of both the activation of caspase-3, asdocumented for other colon cancer cell lines [45], and over-expression of c-Myc.
It is now well known that a number of oncoproteins in-duce apoptosis when over-expressed in cells. The examplescharacterized best are the adenovirus protein E1A and thetranscription factor c-Myc [46]. Since these oncoproteinspromote cell proliferation, their proapoptotic activity has of-ten been interpreted as arising from some kind of abortive orfailed attempt to go through the cell cycle, thereby inducinga mitotic or S phase catastrophe [47]. Our results support thishypothesis: the G2/M arrest observed in ZEPEP-treated cellspresumably leads to mitotic arrest, followed by subsequentapoptosis driven mainly by an E2F1-dependent pathway thatultimately up-regulates the expression of caspase-3.
Springer

1626 Apoptosis (2006) 11:1617–1628
Fig. 6 ZEPEP induces an increase in protein levels of pRb, E2F1 andc-Myc in Caco-2 cells. (a) Representative Western blot showing the in-crease in the levels of pRb, E2F1 and c-Myc in Caco-2 cells, after 72 hrsof treatment with ZEPEP at concentrations of 0.3 µg/ml (ZEP1EP) and3 µg/ml (ZEP2EP), compared with untreated control cells. Alpha tubulinwas used as a loading control. (b) ppRb (phosphorylated pRb)/unpRb(unphosphorylated pRb) ratio obtained by the O.D. ratio of the ppRbbands and unpRb bands, both normalized with the O.D. of alpha tubulinbands. Results are the mean ± SD of three independent experiments.
∗p < 0.01; ∗∗p < 0.005 by Student’s t test. (c) Densitometric quantifi-cation of protein levels of E2F1, obtained by the O.D. of protein bandsnormalized with the O.D. of alpha tubulin bands. Results are the mean± SD of three independent experiments. ∗p < 0.01 by Student’s t test.(d) Densitometric quantification of protein levels of c-Myc, obtainedby the O.D. of protein bands normalized with the O.D. of alpha tubulinbands. Results are the mean ± S.D. of three independent experiments.∗p < 0.01; ∗∗∗p < 0.001 by Student’s t test
We find that the biological activity of ZEPEP is sharedby a semi-purified protein fraction whose molecular weightranges from 20 to 60 kDa. Proteins were rigorously obtainedat 50% of the epiboly stage (onset of gastrulation). Extractsobtained during the late gastrula period fail to show anygrowth-inhibitory or pro-apoptotic actions. This is notsurprising if we consider that zebrafish embryo cells atthis stage produce several signal transducers and activatorsof transcription that mediate biological actions such ascell proliferation and survival in response to cytokinesand growth factors in a variety of biological processes,such as gastrulation, organogenesis, wound healing andcancer progression [48]. ZEPEP are ineffective in normalhuman cells, and are probably tissue- and cancer-specific, asreported for mouse embryo extracts [49]; moreover, differentbiochemical pathways may be activated in cancer arising
from different tissues, even though the pRb/E2F1 complexappears to be one of the main targets [50]. In our study, wedemonstrated that ZEPEP modify the ratio between phospho-rylated and unphosphorylated pRb, there being a significantincrease in phosphorylated pRb which would presumablylead to an increased release of E2F1 accompanied byc-Myc over-expression. As a consequence, c-FLIPS activitydecreases and caspase-8 increases, while E2F1 results, viathe p73 apoptotic pathway, in straightforward activation ofcaspase-3. These combined effects, thus, presumably leadto growth arrest in the G2/M phase and apoptosis. So, evenin the absence of p53, alternative apoptotic pathways aresignificantly activated in Caco2 cells by ZEPEP.
As it is unlikely that apoptotic effectors (i.e. caspases)[51] or hortologue tumor-suppressor gene products [52]can be extracted from zebrafish embryos at this stage of
Springer

Apoptosis (2006) 11:1617–1628 1627
development, we suggest that ZEPEP be considered an inte-grated molecular network belonging to what may be referredto as an “apoptotic and developmental field”, even thoughany attempt to elucidate this network is, for the time be-ing, merely speculative. Proteomic studies on bi-dimensionalelectrophoresis, chromatographic analysis and mass-spectrometric studies designed to shed light on the structureof ZEPEP are currently being conducted in our laboratories.
Several studies have suggested that the capacity of malig-nant cells to induce apoptosis or differentiation is a propertyshared by a number of embryonic factors. Indeed, injec-tion of leukaemia cells into the placenta of a 10-day-oldmouse foetus induced cancer-growth inhibition, followed byhaematopoietic maturation and the appearance of normalwhite blood cells carrying the leukaemia cell marker [53].Melanoma [54] and neuroblastoma [55] cells are similarlyregulated if placed in “embryonal developmental fields”,where normal differentiation of these cell types occurs. Morerecently, embryonic proteins extracted from the pregnantuterus [13] or from Zebrafish embryos have been shown toexert significant apoptotic and antiproliferative effects onseveral cancer lines, though not when the extract is takenfrom a mainly replicative (gastrula) stage [14]. Thus, sometumour cell types appear to be regulated or committed toapoptosis by their respective embryonic fields, which are spe-cific to given types of tumour. Under normal circumstances,during development both cell proliferation and apoptosis areongoing processes that occur as the cells of the embryo in-teract to form the differentiating organs. May it possible toapply the process that controls the development of normalcells to tumour cells? Although further research is requiredto answer this question, there is no doubt that an understand-ing of the cellular signals that control stem cell proliferationand differentiation may ultimately lead to novel integratedtherapeutic strategies in oncology [56].
Conclusion
In conclusion, our study shows that zebrafish embryoproteins significantly inhibit Caco2 growth and induceprogrammed cell death by activation of p53-independentmechanisms, acting mainly through the pRb/E2F1 apoptotic-pathway.
Acknowledgements This study was partly supported by a grant ob-tained from NGC-Spa, Milano (Italy). We thank Lewis Baker for re-viewing the English in the manuscript
References
1. Ellis RE, Yuan JY, Horvitz HR (1991) Mechanisms and functionsof cell death. Annu Rev Cell Biol 7:663–698
2. Einhorn L (1982) Are there factors preventing cancer developmentduring embryonic life? Oncodev Biol Med 4:219–229
3. Yu C-L, Tsai M-H (2001) Fetal fetuin selectively induces apoptosisin cancer cell lines and shows anti-cancer activity in tumor animalmodels. Cancer Lett 166:173–184
4. Lakshmi MS, Shebert GV (1974) Embryonic and Tumor CellsInteractions, Karger Basel, p 380–399
5. Brent RL (1980) Radiation teratogenesis. Teratology 21:281–2986. Sell S (2004) Stem cell origin of cancer and differentiation therapy.
Crit Rev Oncol/Haemato l51:1–287. Martin G (1980) Teratocarcinomas and mammalian embryogene-
sis. Science 209:768–7768. Warrell Jr RP, Frankel SR, Miller Jr WH, Scheinberg DA, Itri LM,
Hittelman WN (1991) Differentiation therapy of acute promye-locytic leukemia with tretinoid (all-trans-retinoic acid). N Engl JMed 324:1385–1393
9. Pierce GB (1983) The cancer cell and its control by the embryo.Am J Pathol 113:117–124
10. Michaeli J, Rifkind RA, Marks PA (1993) Differentiating agentsfor transformed cells. In: Pinedo HM, Longo DL, Chabner BA(eds) Cancer Chemotheraphy and Biological Response ModifiersAnnual 14, Elsevier, New York, p 330–352
11. Livraghi T, Meloni F, Frosi A, Lazzaroni S, Bizzarri M, Frati L,Biava PM (2005) Treatment with stem cell differentiation stage fac-tors of intermediate-advanced hepatocellular carcinoma: an openrandomized clinical trial. Oncol Res 15:399–408
12. Bizzarri M, Facco R, Ielapi T. Frati L (2002) The embryonic andmaternal regulatory factor as a palliative therapy for advanced solidtumours. J Tumour Marker Oncol 17(3):31–36
13. Biava PM, Fiorito A, Negro C, Mariani M (1988) Effects of treat-ment with embryonic and uterine tissue homogenates on Lewislung carcinoma development. 41:265–270
14. Biava PM, Bonsignorio D, Hoxha M (2001) Cell proliferationcurves of different human tumor lines after in vitro treatmentwith Zebrafish embryonic extracts. J Tumor Marker Oncol 16:195–201
15. Djelloul S, Forgue-Lafitte M-E, Hermelin B (1997) Enterocytedifferentiation is compatible with SV40 large T expression andloss of p53 function in human colonic Caco2 cells. FEBS Lett406:234–242
16. Yamamoto H, Soh J-W, Monden T (1999) Paradoxical increase inRetinoblastoma Protein in colorectal carcinomas may protect cellsfrom apoptosis. Clin Cancer Res 5:1805–1815
17. Ezhevsky SE, Nagahara H, Vocero-Akbani AM, Gius DR, WeiMC, Dowdy SF (1997) Hypo-phosphorilation of the retinoblas-toma protein (pRb) by cyclin D:Cdk4/6 complexes results in activepRb. Proc Natl Acad Sci USA 94:10699–10704
18. Putzer BM, Stiewe T, Parssanedjad Reega S, Esche H (2000) E1Ais sufficient by itself to induce apoptosis independent of p53 andother adenoviral gene products. Cell Death Differ 7:177–188
19. Ginsberg D (2002) E2F1 pathways to apoptosis. FEBS Lett529:122–125
20. Salon C, Eymin B, Micheau O, Chaperot L, Plumas J, Bram-billa C, Brambilla E, Gazzeri S. (2005) E2F1 induces apoptosisand sensitizes human lung adenocarcinoma cells to death-receptor-mediated apoptosis through specific downregulation of c-FLIPshort.Cell Death Differ 3:1–13
21. Biava PM, Carluccio A (1997) Activation of anti-oncogene p53produced by embryonic extracts in vitro tumor cells. J TumorMarker Oncol 12:9–15
22. Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, VisticaD, Warren JT, Bokesch H, Kenney S, Boyd MR (1990) New col-orimetric cytotoxicity assay for anticancer-drug screening. J NatlCancer Inst 82:1107–12
23. Aragane Y, Kulms D, Metze D, Wilkes G, Poppelmann B, LugerTA, Schwarz T (1998) Ultraviolet light induces apoptosis via di-rect activation of CD95 (FAS/APO-1) independently of its ligand
Springer

1628 Apoptosis (2006) 11:1617–1628
CD95L. J Cell Biol 140:171–18224. Zhou XD, Yu JP (2005) Chen HX. Expression of FLICE-inhibitory
protein an dits association with p53 mutation in colon cancer. WorldJ Gastroenterol 28(11):2482–2485
25. Krueger A, Schmitz I, Baumann S, Rammer PH, Kirchoff S (2001)Cellular FLICE-inhibitory protein splice variants inhibit differentsteps of caspases-8 activation at the CD95 death-inducing sig-nalling complex. J Biol Chem 276:20633–20640
26. Stern HM, Zon LI (2003) Cancer genetics and drug discovery inthe zebrafish. Nat Rev Cancer 3:1–7
27. Qin X-Q, Livingston D, Kaelin WG Jr., Adams PD (1994) Dereg-ulated transcription factor E2F1 expression leads to S-phase entryand p53 mediated apoptosis. Proc Natl Acad Sci USA 91:10918–10922
28. Jacks T, Fazeli A, Schimdt EM, Bronson RT, Goodell MA, Weim-berg RA (1992) Effects of an Rb mutation in the mouse. Nature359:295–300
29. Alexander K, Yang H-S, Hinds PW (2003) PRb inactivation insenescent cells leads to an E2F-dependent apoptosis requiring p73.Mol Cancer Res 1:716–728
30. Goldberg Y, Nassif I, Pittas A (1996) The antiproliferative ef-fect of sundilac and sundilac disulfide on Ht-29 colon cancercells:alterations in tumor suppressor and cell cycle-regulatory pro-teins. Oncogene 12:893–901
31. Schuler M, Green DR (2001) Mechanisms of p53-dependent apop-tosis. 29(6):684–688
32. Dotto GP (2000) p21WAF/Cipi :more than a break in the cell cycle?Biochem. Biophys Acta 1471:43–56
33. Reinacher-Schick A, Schoeneck A, Graeven U, Schwarte-WadhoffI, Schmiegel W (2003) Mesazaline causes a mitotic arrest and in-duces caspase-dependent apoptosis in colon carcinoma cells. Car-cinog 24(3):443–451
34. Bates S, Phillips AC, Clark PA (1998) p14ARF links the tumoursuppressors RB and p53. Nature 395:124–125
35. Arita D, Kambe M, Ishioka C, Kanamaru R (1997) Induction ofp53-independent apoptosis associated with G2M arrest followingDANN damage in human colon cancer lines. Jpn J Cancer Res88:39–43
36. Stevens C, La Thangue NB (2003) E2F and cell cycle control:adouble-edged sword. Arch Biochem Bioph 412:157–169
37. Pediconi N, Ianari A, Costanzo A (2003) Differential regulation ofE2F1 apoptotic target genes in response to DNA damage. Nat CellBiol 5(6):552–558
38. Vorburger SA, Pater A, Yoshida K (2003) The mithocondrialapoptosis-inducing factor plays a role in E2F1 induced apopto-sis in human colon cancer cells. Ann Surg Oncol 10:314–322
39. Elliot MJ, Dong YB, Yang H, McMasters KM (1999) E2F1 up-regulates c-Myc and p14 (ARF) and induces apoptosis in coloncancer cells. Clin Cancer Res 5:3590–3597
40. Ricci MS, Jin Z, Yu D, Thomas-Tikhonenko A, Dicker DT, El-Deiry WS (2004) Direct repression of FLIP expression by c-Myc is a major determinant of TRAIL sensitivity. Mol Cell Biol24(19):8541–8555
41. Wesselborg S, Engels IH, Rossmann E, Los M, Schulze-OshoffK (1999) Anticancer drugs induce caspase-8/FLICE activationand apoptosis in the absence of CD95 receptor/ligand interaction.Blood 93(9):3053–3063
42. Leone G, Sears R, Huang E (2001) Myc requires distinct E2Factivities to induce S phase and apoptosis. Mol Cell 8:105–113
43. Sakamuro D, Eviner V, Elliot KJ, Showe L, Prendergast GC(1995) C-Myc induces apoptosis in epithelial cells by both p53-dependent and p53-independent mechanisms. Oncogene 11:2411–2418
44. Wieder T, Essmann F, Prokop A (2001) Activation of caspase-8 indrug-induced apoptosis of B-lymphoid is independent of CD95/Fasreceptor-ligand interaction and occurs downstream of caspase-3.Blood 97(5):1378–1387
45. Diaz GD, Li Q, Dashwood RH (2003) Caspase 8 and apoptosisinducing factor mediate a cytochrome c-indipendent pathway ofapoptosis in human colon cancer cells induced by dietary phyto-chemical chlorophyllin. Cancer Res 63:1254–1261
46. Nilsson JA, Cleveland JL (2003) Myc pathways provoking cellsuicide and cancer. Oncogene 22:9007–9021
47. Evan GI, Brown L, Whyte M, Harrington E (1995) Apoptosis andthe cell cycle. Curr Biol 7:825–834
48. Yamashita S, Miyagi C, Fukada T, Zagara N, Cho YS, Hirano T(2004) Zinc transporter LM controls epithelial-mesenchimal tran-sition in zebrafish gastrula organizer. Nature 429:298–302
49. Wells RS, Miotto KA (1986) Widespread inhibition of neuroblas-toma cells in the 13- to 17-day-old mouse embryo. Cancer Res46:1659–1662
50. Biava PM, Bonsignorio D, Hoxha M, Ielapi T, Frati L, Bizzarri M(2002) Post-translational modifications of the Retinoblastoma Pro-tein (pRb) induced by in vitro administration of Zebrafish embry-onic extracts on human kidney adenocarcinoma cell line. J TumorMarker Oncol 17(3):19–24
51. Yabu T, Kishi S, Okazaki T, Yamashita M (2001) Characterizationof zebrafish caspase-3 and induction of apoptosis through ceramidegeneration in fish fathead minnow tailbud cells and zebrafish em-bryo. Biochem J 360:39–47
52. Bertrand S, Pinte S, Stankovic-Valentin N (2004) Identificationand developmental expression of the zebrafish orthologue of thetumor-suppressor gene HIC1. Biochem Biophys Acta 1678:57–66
53. Gootwine E, Webb CG, Sachs L (1982) Partecipation of myeloidleukemia cells injected into embryos in haematopoietic differenti-ation in adult mice. Nature 299:63–65
54. Gerschenson M, Graves K, Carson SD, Wells RS, Pierce GB (1986)Regulation of melanoma by the embryonic skin. Proc Natl AcadSci USA 83:7303–7310
55. Podesta AN, Mullins J, Pierce GB, Sell RS (1984) The neurula statemouse embryos in control of neuroblastomas. Proc Natl Acad SciUSA 81:7608–7611
56. Al-Hajj M, Becker MW, Wicha M, Weissman, Clarke MF (2004)Therapeutic implications of cancer stem cells. Curr Op Gen Dev14:43–47
Springer




















