
Fatty Acids, IL6, and TNFα Polymorphisms: An Example of Nutrigenetics in Crohn's Disease

Virus-Plus-Susceptibility GeneInteraction Determines Crohn’s DiseaseGene Atg16L1 Phenotypes in IntestineKen Cadwell,1 Khushbu K. Patel,1 Nicole S. Maloney,1 Ta-Chiang Liu,1 Aylwin C.Y. Ng,3,4 Chad E. Storer,5
Richard D. Head,5 Ramnik Xavier,3,4 Thaddeus S. Stappenbeck,1,* and Herbert W. Virgin1,2,6,*1Department of Pathology and Immunology2Department of Molecular Microbiology
Washington University School of Medicine, St. Louis, MO 63110, USA3Center for Computational and Integrative Biology and Gastrointestinal Unit, Massachusetts General Hospital, Harvard Medical School,
Boston, MA 02114, USA4Program in Medical and Population Genetics, Broad Institute of MIT and Harvard, Cambridge, MA 02142, USA5Inflammation and Immunology Research Unit, Pfizer Global Research and Development, St. Louis, MO 63017, USA6Midwest Regional Center of Excellence for Biodefense and Emerging Infectious Diseases Research, St. Louis, MO 63110, USA
*Correspondence: [email protected] (T.S.S.), [email protected] (H.W.V.)
DOI 10.1016/j.cell.2010.05.009
SUMMARY
It is unclear why disease occurs in only a smallproportion of persons carrying common risk allelesof disease susceptibility genes. Here we demon-strate that an interaction between a specific virusinfection and a mutation in the Crohn’s disease sus-ceptibility gene Atg16L1 induces intestinal patholo-gies in mice. This virus-plus-susceptibility geneinteraction generated abnormalities in granule pack-aging and unique patterns of gene expression inPaneth cells. Further, the response to injury inducedby the toxic substance dextran sodium sulfate wasfundamentally altered to include pathologies resem-bling aspects of Crohn’s disease. These pathologiestriggered by virus-plus-susceptibility gene interac-tion were dependent on TNFa and IFNg and wereprevented by treatment with broad spectrum antibi-otics. Thus, we provide a specific example of howa virus-plus-susceptibility gene interaction can, incombination with additional environmental factorsand commensal bacteria, determine the phenotypeof hosts carrying common risk alleles for inflamma-tory disease.
INTRODUCTION
Common genetic polymorphisms predispose to complex dis-
eases such as type I diabetes, multiple sclerosis, Crohn’s
disease, and ulcerative colitis (The Wellcome Trust Case Control
Consortium, 2007; Altshuler et al., 2008). It is not clear why some
individuals with a given polymorphism acquire disease whereas
others remain unaffected. The concept that environmental
factors including infections trigger disease in individuals with
certain genetic backgrounds is broadly recognized. In animal
models, autoimmune disease can be influenced by viral infec-
tions. For example, glomerulonephritis is exacerbated by lym-
phocytic choriomeningitis virus and polyoma virus infections in
certain inbred backgrounds (Tonietti et al., 1970), and lympho-
cytic choriomeningitis virus infection inhibits development of dia-
betes in NOD mice or BB rats (Dyrberg et al., 1988; Oldstone,
1988). The genes responsible for these differences in outcome
have not been defined, but these studies indicate that virus infec-
tion can alter disease in specific genetic backgrounds.
Crohn’s disease is a common type of inflammatory bowel
disease involving mucosal ulceration and inflammation occur-
ring in the distal small intestine (ileum) and variable discontin-
uous regions of the colon. A distinguishing feature of inflamma-
tion in Crohn’s disease is involvement of the entire thickness of
the bowel wall. This transmural inflammation leads to atrophy
of ileal villi, fibrosis, hypertrophy of smooth muscle and auto-
nomic nerve cells in the outer layer of the bowel wall, and an
inflammatory response including lymphoid aggregates and
granulomas (Day et al., 2003). Polymorphisms in over 30 loci
have been associated with increased risk of Crohn’s disease
(Barrett et al., 2008). Yet these genetic components individually,
or in combination (Weersma et al., 2008), confer limited risk.
Environmental factors such as exposure to pathogens are
potential cofactors for disease development, but the etiology
of Crohn’s disease remains a controversial topic (Packey and
Sartor, 2009).
One Crohn’s disease susceptibility allele is in the autophagy
gene ATG16L1 (The Wellcome Trust Case Control Consortium,
2007; Rioux et al., 2007; Hampe et al., 2007). The ATG16L1
disease variant is frequent (�50% in European-derived popula-
tions) and confers less than a 2-fold increase in susceptibility.
We previously established two mouse lines in which Atg16L1
expression is disrupted by gene trap mutagenesis (Cadwell
et al., 2008a). Mutant mice displayed hypomorphic (HM)
Atg16L1 protein expression and reduced autophagy (Cadwell
Cell 141, 1135–1145, June 25, 2010 ª2010 Elsevier Inc. 1135

et al., 2008a; Ju et al., 2009). Conventionally raised Atg16L1HM
mice display striking abnormalities in Paneth cells, epithelial cells
at the base of ileal crypts that are important in mucosal immunity
(Porter et al., 2002; Ouellette, 2006; Vaishnava et al., 2008).
In addition to aberrant packaging and exocytosis of antimicrobial
granules, Paneth cells from Atg16L1HM mice display a surprising
gain-of-function transcriptional profile in which transcripts asso-
ciated with lipid metabolism, proinflammatory cytokines, and
other pathways were enriched. Importantly, we observed similar
Paneth cell abnormalities in Crohn’s disease patients homozy-
gous for the risk allele of ATG16L1 but not in control patients
(Cadwell et al., 2008a). Therefore, these aspects of intestinal
pathology link Crohn’s disease patients carrying the suscepti-
bility allele of ATG16L1 to mice that are hypomorphic for
Atg16L1.
Here, we report a striking genetic interaction between Atg16L1
mutation and a specific strain of an enteric virus, murine norovi-
rus (MNV). This interaction determines multiple pathologic
abnormalities in the intestine including several similar to those
observed in Crohn’s disease patients. Noroviruses are positive-
sense encapsidated RNA viruses responsible for the majority of
epidemic nonbacterial gastroenteritis in humans (Mead et al.,
1999). Multiple strains of MNV are prevalent in mouse facilities
around the world (Hsu et al., 2005; Pritchett-Corning et al.,
2009; Goto et al., 2009), and some MNV strains persist for
months after initial infection (Thackray et al., 2007). We demon-
strate that Paneth cell abnormalities in Atg16L1HM mice are
triggered by infection with an MNV strain that establishes persis-
tent infection (persistent strain). This virus-plus-susceptibility
gene interaction alters the transcriptional signature of Paneth
cells and the nature of the inflammatory response in mice
treated with the toxic substance dextran sodium sulfate (DSS).
The mechanism for this pathologic response driven by the
virus-plus-susceptibility gene interaction involved the cytokines
TNFa and IFNg as well as commensal bacteria. Our results
provide evidence for a multi-hit model of inflammatory dis-
ease defined in a highly specific way by a viral infection only in
the presence of a mutation in a disease susceptibility gene.
This observation provides a basis for understanding how a
common allele can be linked to an infrequent severe disease,
and why mice carrying mutations in human disease susceptibil-
ity genes do not always spontaneously reproduce human
pathology.
RESULTS
Viral Infection Triggers Paneth Cell Abnormalitiesin Atg16L1HM MiceAtg16L1HM mice raised in a conventional barrier facility were
rederived into an enhanced barrier facility (Experimental Proce-
dures) by embryo transfer. Surprisingly, in contrast to observa-
tions made in mice from a conventional barrier (Cadwell et al.,
2008a), Paneth cells in rederived Atg16L1HM mice were indistin-
guishable morphologically from those in littermate wild-type
control mice (herein referred to as WT) (Figure 1A) and failed to
display aberrant packaging of the granule protein lysozyme
(Figure 1B). All subsequent experiments were performed in
mice raised in the enhanced barrier facility.
1136 Cell 141, 1135–1145, June 25, 2010 ª2010 Elsevier Inc.
These results suggest that Paneth cell abnormalities require
an exogenous factor present in the conventional barrier facility.
We previously identified the first murine norovirus (MNV) in
mice from this facility (Karst et al., 2003). We therefore consid-
ered the possibility that MNV triggers Paneth cell abnormalities
in Atg16L1HM mice. To test this, mice were orally inoculated
with an MNV strain that establishes persistent infection (MNV
CR6) (Thackray et al., 2007). Seven days postinoculation,
Atg16L1HM but not WT mice displayed Paneth cell abnormalities
including aberrant granule numbers, size, and distribution (Fig-
ure 1A). UV-inactivated virus did not induce these abnormalities
(Figure S1A, available online) indicating that productive virus
infection was required.
MNV CR6 infection for 7 days also induced abnormal lyso-
zyme distribution and abnormal ultrastructural morphology in
Paneth cells from Atg16L1HM mice (Figure 1B and Figures S1B
and S1C). We used a previously established scale to blindly
identify cells with increasingly abnormal lysozyme staining (Fig-
ure 1C) (Cadwell et al., 2008a). Only Atg16L1HM mice that were
infected with MNV CR6 displayed Paneth cells with the two
most severe abnormalities (Figure 1D). Analysis of Paneth cells
by transmission electron microscopy also revealed a substantial
proportion of cells that were depleted of secretory granules in
MNV CR6-infected Atg16L1HM mice (Figures S1B and S1C).
Moreover, many of the Paneth cells from these mice contained
distended rough endoplasmic reticulum (Figures S1B and
S1C). Other cytoplasmic organelles including mitochondria
were not obviously altered in virally-infected Atg16L1HM mice.
Thus, Paneth cell morphological and granule packaging abnor-
malities were induced by the combination of viral infection and
mutation in the Crohn’s disease susceptibility gene Atg16L1.
Hereinafter we will refer to this as a virus-plus-susceptibility
gene interaction.
Properties of MNV Associated with Paneth CellAbnormalitiesSince autophagy is important in innate immunity (Virgin and
Levine, 2009), a mutation in Atg16L1 might lead to enhanced
viral replication and cytopathicity in Paneth cells. When com-
paring WT and Atg16L1HM mice, we did not observe significant
differences in viral shedding in the stool or viral titers in organs
including the distal ileum where Paneth cell abnormalities were
observed (Figure S2A). Consistent with a previous report in
which MNV was detected in the lamina propria (Mumphrey
et al., 2007), we did not detect MNV in Paneth cells by immuno-
histochemistry, and viral RNA was not detected in Paneth cell
RNA procured by laser capture microdissection (data not shown,
Experimental Procedures). Thus, direct infection of Paneth cells
is not responsible for triggering the abnormalities described
above.
To determine if Paneth cell abnormalities develop after infec-
tion with any virus, we investigated MNV CW3 that does not
persist in immunocompetent mice despite sharing 95% amino
acid sequence identity across the genome with MNV CR6
(Mumphrey et al., 2007; Thackray et al., 2007). As in WT mice,
shedding of MNV CW3 in Atg16L1HM mice is initially high but
reduced or undetectable at later time points (Figure S2B). MNV
CW3 failed to trigger aberrant Paneth cell morphology 7 days
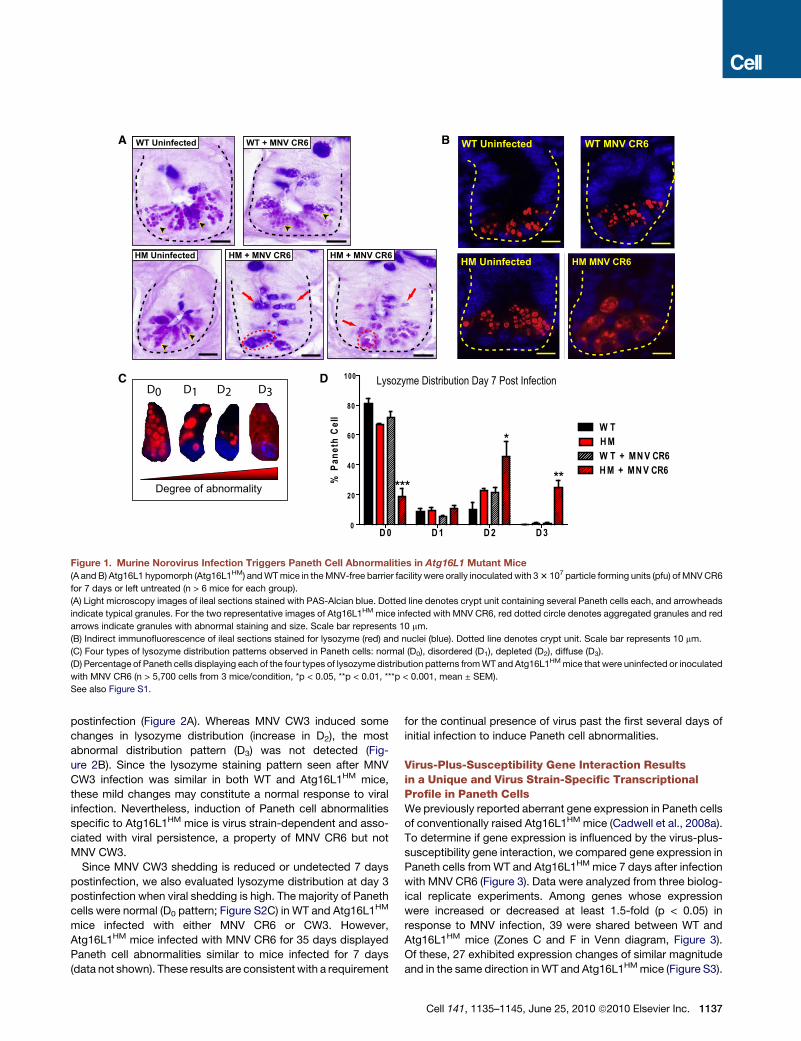
A WT Uninfected BWT Uninfected
HM Uninfected
WT MNV CR6
HM MNV CR6
WT + MNV CR6
HM Uninfected HM + MNV CR6 HM + MNV CR6
CD0 D1 D2 D3
D
H M + MN V CR6
W T
W T + MN V CR6H M
D 0 D 1 D 2 D 30
20
40
60
80
100
% P
anet
h C
ell
***
**
*
Degree of abnormality
Lysozyme Distribution Day 7 Post Infection
Figure 1. Murine Norovirus Infection Triggers Paneth Cell Abnormalities in Atg16L1 Mutant Mice
(A and B) Atg16L1 hypomorph (Atg16L1HM) and WT mice in the MNV-free barrier facility were orally inoculated with 3 3 107 particle forming units (pfu) of MNV CR6
for 7 days or left untreated (n > 6 mice for each group).
(A) Light microscopy images of ileal sections stained with PAS-Alcian blue. Dotted line denotes crypt unit containing several Paneth cells each, and arrowheads
indicate typical granules. For the two representative images of Atg16L1HM mice infected with MNV CR6, red dotted circle denotes aggregated granules and red
arrows indicate granules with abnormal staining and size. Scale bar represents 10 mm.
(B) Indirect immunofluorescence of ileal sections stained for lysozyme (red) and nuclei (blue). Dotted line denotes crypt unit. Scale bar represents 10 mm.
(C) Four types of lysozyme distribution patterns observed in Paneth cells: normal (D0), disordered (D1), depleted (D2), diffuse (D3).
(D) Percentage of Paneth cells displaying each of the four types of lysozyme distribution patterns from WT and Atg16L1HM mice that were uninfected or inoculated
with MNV CR6 (n > 5,700 cells from 3 mice/condition, *p < 0.05, **p < 0.01, ***p < 0.001, mean ± SEM).
See also Figure S1.
postinfection (Figure 2A). Whereas MNV CW3 induced some
changes in lysozyme distribution (increase in D2), the most
abnormal distribution pattern (D3) was not detected (Fig-
ure 2B). Since the lysozyme staining pattern seen after MNV
CW3 infection was similar in both WT and Atg16L1HM mice,
these mild changes may constitute a normal response to viral
infection. Nevertheless, induction of Paneth cell abnormalities
specific to Atg16L1HM mice is virus strain-dependent and asso-
ciated with viral persistence, a property of MNV CR6 but not
MNV CW3.
Since MNV CW3 shedding is reduced or undetected 7 days
postinfection, we also evaluated lysozyme distribution at day 3
postinfection when viral shedding is high. The majority of Paneth
cells were normal (D0 pattern; Figure S2C) in WT and Atg16L1HM
mice infected with either MNV CR6 or CW3. However,
Atg16L1HM mice infected with MNV CR6 for 35 days displayed
Paneth cell abnormalities similar to mice infected for 7 days
(data not shown). These results are consistent with a requirement
for the continual presence of virus past the first several days of
initial infection to induce Paneth cell abnormalities.
Virus-Plus-Susceptibility Gene Interaction Resultsin a Unique and Virus Strain-Specific TranscriptionalProfile in Paneth CellsWe previously reported aberrant gene expression in Paneth cells
of conventionally raised Atg16L1HM mice (Cadwell et al., 2008a).
To determine if gene expression is influenced by the virus-plus-
susceptibility gene interaction, we compared gene expression in
Paneth cells from WT and Atg16L1HM mice 7 days after infection
with MNV CR6 (Figure 3). Data were analyzed from three biolog-
ical replicate experiments. Among genes whose expression
were increased or decreased at least 1.5-fold (p < 0.05) in
response to MNV infection, 39 were shared between WT and
Atg16L1HM mice (Zones C and F in Venn diagram, Figure 3).
Of these, 27 exhibited expression changes of similar magnitude
and in the same direction in WT and Atg16L1HM mice (Figure S3).
Cell 141, 1135–1145, June 25, 2010 ª2010 Elsevier Inc. 1137
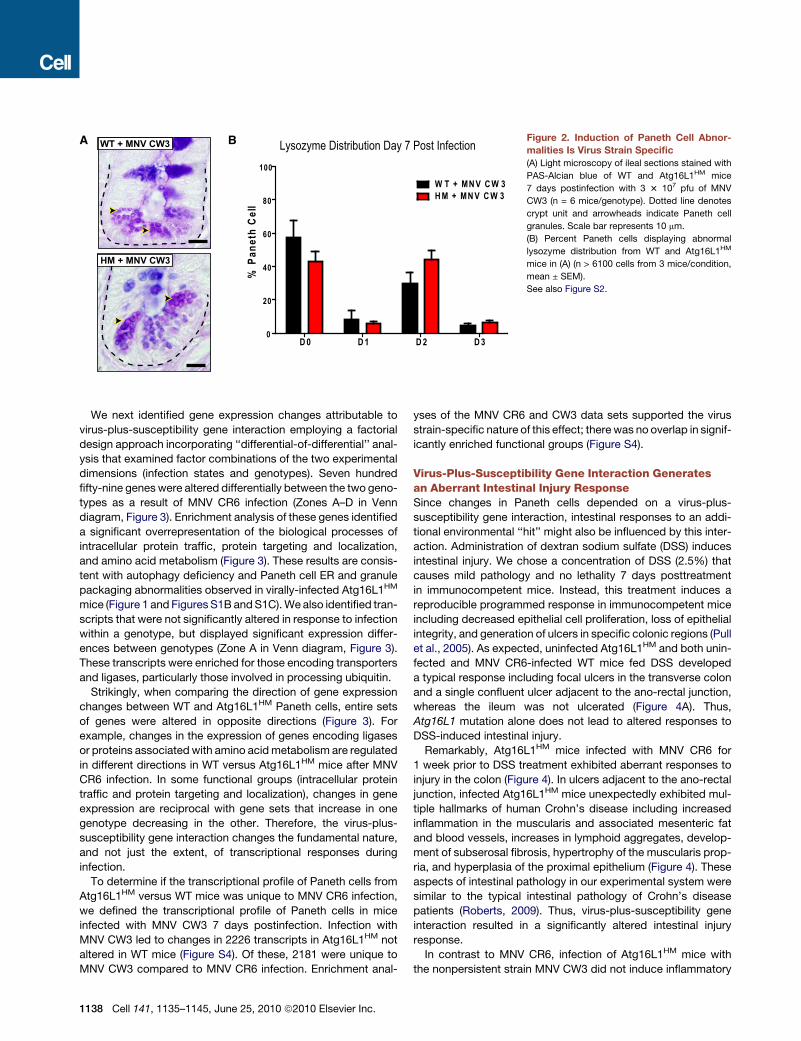
D 0 D 1 D 2 D 30
20
40
60
80
100
W T + MN V C W 3H M + MN V C W 3
% P
anet
h C
ell
WT + MNV CW3
HM + MNV CW3
Lysozyme Distribution Day 7 Post InfectionA B Figure 2. Induction of Paneth Cell Abnor-
malities Is Virus Strain Specific
(A) Light microscopy of ileal sections stained with
PAS-Alcian blue of WT and Atg16L1HM mice
7 days postinfection with 3 3 107 pfu of MNV
CW3 (n = 6 mice/genotype). Dotted line denotes
crypt unit and arrowheads indicate Paneth cell
granules. Scale bar represents 10 mm.
(B) Percent Paneth cells displaying abnormal
lysozyme distribution from WT and Atg16L1HM
mice in (A) (n > 6100 cells from 3 mice/condition,
mean ± SEM).
See also Figure S2.
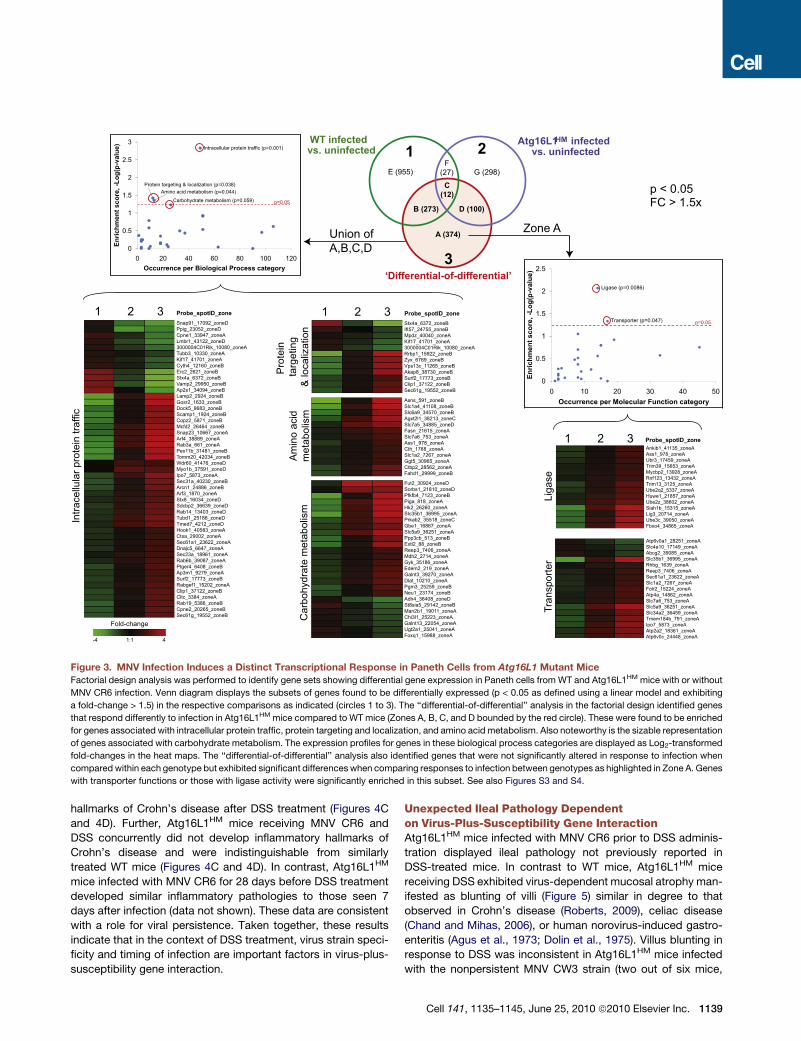
We next identified gene expression changes attributable to
virus-plus-susceptibility gene interaction employing a factorial
design approach incorporating ‘‘differential-of-differential’’ anal-
ysis that examined factor combinations of the two experimental
dimensions (infection states and genotypes). Seven hundred
fifty-nine genes were altered differentially between the two geno-
types as a result of MNV CR6 infection (Zones A–D in Venn
diagram, Figure 3). Enrichment analysis of these genes identified
a significant overrepresentation of the biological processes of
intracellular protein traffic, protein targeting and localization,
and amino acid metabolism (Figure 3). These results are consis-
tent with autophagy deficiency and Paneth cell ER and granule
packaging abnormalities observed in virally-infected Atg16L1HM
mice (Figure 1 and Figures S1B and S1C). We also identified tran-
scripts that were not significantly altered in response to infection
within a genotype, but displayed significant expression differ-
ences between genotypes (Zone A in Venn diagram, Figure 3).
These transcripts were enriched for those encoding transporters
and ligases, particularly those involved in processing ubiquitin.
Strikingly, when comparing the direction of gene expression
changes between WT and Atg16L1HM Paneth cells, entire sets
of genes were altered in opposite directions (Figure 3). For
example, changes in the expression of genes encoding ligases
or proteins associated with amino acid metabolism are regulated
in different directions in WT versus Atg16L1HM mice after MNV
CR6 infection. In some functional groups (intracellular protein
traffic and protein targeting and localization), changes in gene
expression are reciprocal with gene sets that increase in one
genotype decreasing in the other. Therefore, the virus-plus-
susceptibility gene interaction changes the fundamental nature,
and not just the extent, of transcriptional responses during
infection.
To determine if the transcriptional profile of Paneth cells from
Atg16L1HM versus WT mice was unique to MNV CR6 infection,
we defined the transcriptional profile of Paneth cells in mice
infected with MNV CW3 7 days postinfection. Infection with
MNV CW3 led to changes in 2226 transcripts in Atg16L1HM not
altered in WT mice (Figure S4). Of these, 2181 were unique to
MNV CW3 compared to MNV CR6 infection. Enrichment anal-
1138 Cell 141, 1135–1145, June 25, 2010 ª2010 Elsevier Inc.
yses of the MNV CR6 and CW3 data sets supported the virus
strain-specific nature of this effect; there was no overlap in signif-
icantly enriched functional groups (Figure S4).
Virus-Plus-Susceptibility Gene Interaction Generatesan Aberrant Intestinal Injury ResponseSince changes in Paneth cells depended on a virus-plus-
susceptibility gene interaction, intestinal responses to an addi-
tional environmental ‘‘hit’’ might also be influenced by this inter-
action. Administration of dextran sodium sulfate (DSS) induces
intestinal injury. We chose a concentration of DSS (2.5%) that
causes mild pathology and no lethality 7 days posttreatment
in immunocompetent mice. Instead, this treatment induces a
reproducible programmed response in immunocompetent mice
including decreased epithelial cell proliferation, loss of epithelial
integrity, and generation of ulcers in specific colonic regions (Pull
et al., 2005). As expected, uninfected Atg16L1HM and both unin-
fected and MNV CR6-infected WT mice fed DSS developed
a typical response including focal ulcers in the transverse colon
and a single confluent ulcer adjacent to the ano-rectal junction,
whereas the ileum was not ulcerated (Figure 4A). Thus,
Atg16L1 mutation alone does not lead to altered responses to
DSS-induced intestinal injury.
Remarkably, Atg16L1HM mice infected with MNV CR6 for
1 week prior to DSS treatment exhibited aberrant responses to
injury in the colon (Figure 4). In ulcers adjacent to the ano-rectal
junction, infected Atg16L1HM mice unexpectedly exhibited mul-
tiple hallmarks of human Crohn’s disease including increased
inflammation in the muscularis and associated mesenteric fat
and blood vessels, increases in lymphoid aggregates, develop-
ment of subserosal fibrosis, hypertrophy of the muscularis prop-
ria, and hyperplasia of the proximal epithelium (Figure 4). These
aspects of intestinal pathology in our experimental system were
similar to the typical intestinal pathology of Crohn’s disease
patients (Roberts, 2009). Thus, virus-plus-susceptibility gene
interaction resulted in a significantly altered intestinal injury
response.
In contrast to MNV CR6, infection of Atg16L1HM mice with
the nonpersistent strain MNV CW3 did not induce inflammatory
Intra
cellu
lar p
rote
in tr
affic
1 2 3Snap91_17092_zoneDPpig_23052_zoneDCpne1_33947_zoneALmbr1_43122_zoneD3000004C01Rik_10080_zoneATubb3_10330_zoneAKif17_41701_zoneACyth4_12160_zoneBErc2_2621_zoneBStx4a_6372_zoneBVamp2_29950_zoneBAp2s1_34094_zoneBLamp2_2924_zoneBGosr2_1633_zoneBDock5_9683_zoneBScamp1_1924_zoneBCopz2_5871_zoneBMcfd2_26464_zoneBSnap23_10667_zoneAArf4_38889_zoneARab3a_661_zoneAPex11b_31481_zoneBTomm20_42034_zoneBWdr60_41476_zoneDMyo1b_37591_zoneDIpo7_5873_zoneASec31a_40230_zoneBArcn1_24886_zoneBArf3_1870_zoneAStx8_16034_zoneDSdcbp2_36639_zoneDRab14_13403_zoneDTubd1_25186_zoneDTmed7_4212_zoneDHook1_40583_zoneACtsa_29002_zoneASec61a1_23622_zoneADnajc5_6647_zoneASec23a_18961_zoneARab6b_39087_zoneAPtger4_6408_zoneBAp3m1_9279_zoneASurf2_17773_zoneBRabgef1_15202_zoneAClip1_37122_zoneBCltc_3384_zoneARab19_5386_zoneBCpne2_20265_zoneBSec61g_19552_zoneB
Probe_spotID_zone
WT infected
vs. uninfected
Atg16L1HM infected
vs. uninfected
‘Differential-of-differential’
Union of A,B,C,D
E (955) G (298)
A (374)
B (273) D (100)
C
(12)
F(27)
1 2
3
p < 0.05FC > 1.5x
1
1.5
2
2.5
nt sco
re, -L
og
(p
-valu
e)
Ligase (p=0.0086)
Transporter (p=0.047)
0
0.5
0 10 20 30 40 50
En
rich
men
Occurrence per Molecular Function category
p=0.05
Zone A1
1.5
2
2.5
3
nt s
co
re
, -L
og
(p
-v
alu
e)
Intracellular protein traffic (p=0.001)
Protein targeting & localization (p=0.038)Amino acid metabolism (p=0.044)
Carbohydrate metabolism (p=0.059)
0
0.5
0 20 40 60 80 100 120
En
ric
hm
en
Occurrence per Biological Process category
p=0.05
Liga
se
1 2 3Ankib1_41135_zoneAAss1_978_zoneAUbr3_17459_zoneATrim39_15653_zoneAMycbp2_13928_zoneARnf123_13432_zoneATrim13_3125_zoneAUbe2q2_5337_zoneAHuwe1_21857_zoneAUbe2z_38602_zoneASiah1b_15315_zoneALig3_20714_zoneAUbe3c_39050_zoneAFbxo4_34865_zoneA
Tran
spor
ter
Atp6v0a1_28251_zoneASlc4a10_17149_zoneAAbcg2_39085_zoneASlc35b1_36995_zoneARhbg_1639_zoneAReep3_7406_zoneASec61a1_23622_zoneASlc1a2_7267_zoneAFolr2_15224_zoneAAtp4a_14862_zoneASlc7a6_753_zoneASlc5a9_36251_zoneASlc34a2_36459_zoneATmem184b_791_zoneAIpo7_5873_zoneAAtp2a2_18361_zoneAAtp6v0c_24448_zoneA
Pro
tein
targ
etin
g&
loca
lizat
ion
Am
ino
acid
met
abol
ism
Car
bohy
drat
e m
etab
olis
m
Asns_591_zoneBSlc1a4_41108_zoneBSlc6a9_34570_zoneBAgxt2l1_38213_zoneCSlc7a5_34885_zoneDFasn_21615_zoneASlc7a6_753_zoneAAss1_978_zoneACth_1768_zoneASlc1a2_7267_zoneAGgt5_30965_zoneACtbp2_28562_zoneAFahd1_29999_zoneB
Fut2_30924_zoneDSorbs1_21810_zoneDPfkfb4_7123_zoneBPiga_818_zoneAHk2_26260_zoneASlc35b1_36995_zoneAPrkab2_35518_zoneCGbe1_16867_zoneASlc5a9_36251_zoneAPpp3cb_513_zoneBExtl2_88_zoneBReep3_7406_zoneAMdh2_2714_zoneAGyk_35186_zoneAEdem2_219_zoneAGalnt3_39270_zoneADlat_10210_zoneAPgm3_25259_zoneBNeu1_23174_zoneBAdh4_36408_zoneDSt8sia5_29142_zoneBMan2b1_19011_zoneAChi3l1_25223_zoneAGalnt13_22054_zoneAUgt2a1_25041_zoneAFoxq1_15988_zoneA
Stx4a_6372_zoneBIft57_24755_zoneBMpdz_40040_zoneAKif17_41701_zoneA3000004C01Rik_10080_zoneARrbp1_15922_zoneBZyx_6769_zoneBVps13c_11265_zoneBAkap8_38730_zoneBSurf2_17773_zoneBClip1_37122_zoneBSec61g_19552_zoneB
-4 1:1 4
Fold-change
1 2 3 Probe_spotID_zone
Probe_spotID_zone
Figure 3. MNV Infection Induces a Distinct Transcriptional Response in Paneth Cells from Atg16L1 Mutant Mice
Factorial design analysis was performed to identify gene sets showing differential gene expression in Paneth cells from WT and Atg16L1HM mice with or without
MNV CR6 infection. Venn diagram displays the subsets of genes found to be differentially expressed (p < 0.05 as defined using a linear model and exhibiting
a fold-change > 1.5) in the respective comparisons as indicated (circles 1 to 3). The ‘‘differential-of-differential’’ analysis in the factorial design identified genes
that respond differently to infection in Atg16L1HM mice compared to WT mice (Zones A, B, C, and D bounded by the red circle). These were found to be enriched
for genes associated with intracellular protein traffic, protein targeting and localization, and amino acid metabolism. Also noteworthy is the sizable representation
of genes associated with carbohydrate metabolism. The expression profiles for genes in these biological process categories are displayed as Log2-transformed
fold-changes in the heat maps. The ‘‘differential-of-differential’’ analysis also identified genes that were not significantly altered in response to infection when
compared within each genotype but exhibited significant differences when comparing responses to infection between genotypes as highlighted in Zone A. Genes
with transporter functions or those with ligase activity were significantly enriched in this subset. See also Figures S3 and S4.
hallmarks of Crohn’s disease after DSS treatment (Figures 4C
and 4D). Further, Atg16L1HM mice receiving MNV CR6 and
DSS concurrently did not develop inflammatory hallmarks of
Crohn’s disease and were indistinguishable from similarly
treated WT mice (Figures 4C and 4D). In contrast, Atg16L1HM
mice infected with MNV CR6 for 28 days before DSS treatment
developed similar inflammatory pathologies to those seen 7
days after infection (data not shown). These data are consistent
with a role for viral persistence. Taken together, these results
indicate that in the context of DSS treatment, virus strain speci-
ficity and timing of infection are important factors in virus-plus-
susceptibility gene interaction.
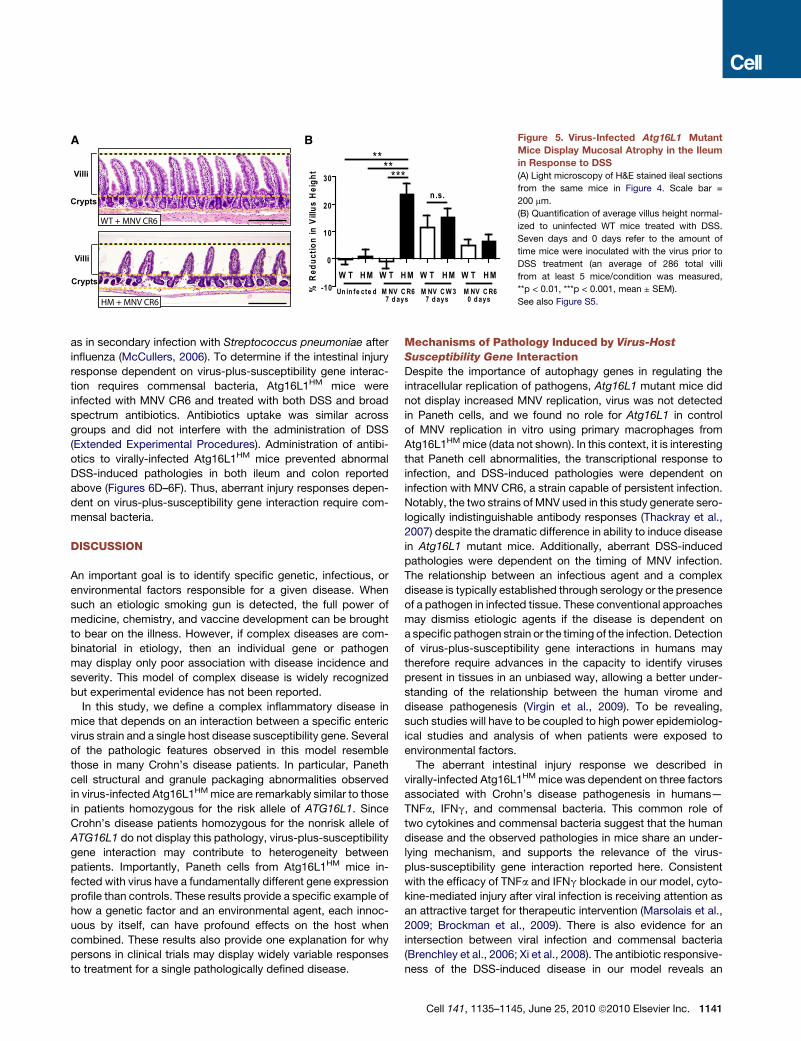
Unexpected Ileal Pathology Dependenton Virus-Plus-Susceptibility Gene InteractionAtg16L1HM mice infected with MNV CR6 prior to DSS adminis-
tration displayed ileal pathology not previously reported in
DSS-treated mice. In contrast to WT mice, Atg16L1HM mice
receiving DSS exhibited virus-dependent mucosal atrophy man-
ifested as blunting of villi (Figure 5) similar in degree to that
observed in Crohn’s disease (Roberts, 2009), celiac disease
(Chand and Mihas, 2006), or human norovirus-induced gastro-
enteritis (Agus et al., 1973; Dolin et al., 1975). Villus blunting in
response to DSS was inconsistent in Atg16L1HM mice infected
with the nonpersistent MNV CW3 strain (two out of six mice,
Cell 141, 1135–1145, June 25, 2010 ª2010 Elsevier Inc. 1139

A
B
HM + MNV CR6
WT Uninfected
WT+ MNV CR6
HM Uninfected
C
D
-20
0
20
40
60
80
100
****
*
W T H M W T H M W T H M W T H M
Un in fe cte d M NV C R67 d ays
M NV C W 37 d ays
M NV C R60 d ays
% In
crea
se in
mu
scle
wid
th
0
1
2
3
W T H M W T H M W T H M W T H M
Un in fe cte d M NV C R67 d ays
M NV C W 37 d ays
M NV C R60 d ays
******
***
Lym
ph
oid
ag
gre
gat
es
N.D.
M N V C R 6 for 7 days D S S for 7 days
M N V CW 3 for 7 days D S S for 7 days
M N V C R 6 for 7 days
D S S for 7 daysS am e D ay
D S S for 7 days
In flam m atory H a llm arks o f C D
D S S Treatm ent P rotoco l W T H M
+M N V C R 6 for 7 days D S S for 7 days
M N V CW 3 for 7 days D S S for 7 days
M N V C R 6 for 7 days
D S S for 7 daysS am e D ay
D S S for 7 daysD S S for 7 days
In flam m atory H a llm arks o f C D
D S S Treatm ent P rotoco l W TW T H M
+
H M
+
Figure 4. Atg16L1 Mutant Mice Display
a Virus-Dependent Aberrant Response to
DSS in the Colon
(A) WT and Atg16L1HM mice were orally inoculated
with MNV CR6 for 7 days or uninfected, and
subsequently given 2.5% DSS for an additional
7 days at which point intestines were harvested.
Light microscopy images of H&E stained sections
of ulcerated regions located immediately adjacent
to the ano-rectal junction are shown. Yellow
double-headed arrows indicate the muscularis
propria thickness. In the MNV CR6-infected
Atg16L1HM sample, lymphoid aggregates are
indicated by yellow dashed circles, and the black
dashed region contains submucosal fibrosis and
inflammation. Scale bar represents 500 mm.
(B) Table summarizing the outcome of DSS treat-
ment. All intestines were harvested at the end of
DSS treatment for analysis. + and � refer to the
presence or absence of inflammatory hallmarks
of Crohn’s disease respectively (n R 6 mice/
condition).
(C) Quantification of the muscularis propria thick-
ness in the region adjacent to the ano-rectal junc-
tion from DSS-treated mice. Seven days and
0 days refer to the amount of time mice were inoc-
ulated with the virus prior to DSS treatment.
The increase in muscle thickness was normalized
to the average of uninfected WT mice treated with
DSS (*p < 0.05, ***p < 0.001, mean ± SEM).
(D) Number of lymphoid aggregates in the region
adjacent to the ano-rectal junction from DSS-
treated mice (***p < 0.001, mean ± SEM). N.D.
refers to not detected.
See also Figure S5.
Figure 5B). MNV CR6 infection for 1 week prior to DSS adminis-
tration was required for induction of villus blunting (Figure 5B).
One explanation for aberrant DSS-induced pathologies
observed in virus-infected Atg16L1HM mice would be uncon-
trolled viral replication. However, there were no significant differ-
ences in viral replication in the ileum and colon where aberrant
pathology is observed prior to or after DSS treatment (Figures
S2A and S5). Less MNV CR6 was detected in the mesenteric
lymph nodes from Atg16L1HM compared to WT mice receiving
DSS, indicating that virus-plus-susceptibility gene effects were
not mediated by increased viral replication.
TNFa, IFNg, and Commensal Bacteria MediateIntestinal Injury Responses Dependenton Virus-Plus-Susceptibility Gene InteractionTo identify mediators of DSS-induced pathology dependent on
virus-plus-susceptibility gene interaction, we examined the role
of cytokines. TNFa is a major mediator of inflammation in Crohn’s
disease; administration of blocking antibodies that interfere with
1140 Cell 141, 1135–1145, June 25, 2010 ª2010 Elsevier Inc.
ly
IF
c
(S
D
o
m
to
(F
v
b
D
w
il
g
d
d
TNFa signaling is a powerful therapeutic
approach (Targan et al., 1997). Further-
more, excess TNFa induces villus blunt-
ing as observed in Figure 5 (Piguet et al.,
1999). Upon in vitro stimulation, intestinal
mphocytes from Crohn’s disease patients secrete increased
Ng (interferon-g) (Fuss et al., 1996; Fais et al., 1991), which
an function in concert with TNFa in inflammatory processes
uk et al., 2001; Lake et al., 1994), and IFNg may have a role in
SS-induced intestinal injury (Brem-Exner et al., 2008).
MNV CR6-infected, DSS-treated Atg16L1HM mice given TNFa
r IFNg blocking antibodies displayed dramatically reduced
uscular hypertrophy, fewer lymphoid aggregates adjacent
the ano-rectal junction, and decreased ileal villus blunting
igures 6A–6C). Importantly, only the pathologies specific to
irally-infected Atg16L1HM mice were altered as blocking anti-
odies had no effect on the superficial ulceration typical of
SS treatment present in all conditions. Thus, TNFa and IFNg
ere each necessary for the DSS-induced pathologies in both
eum and colon that were dependent on virus-plus-susceptibility
ene interaction.
Commensal bacteria are important in inflammatory bowel
isease (Sartor, 1997; Kang et al., 2008), and there is prece-
ence for a role of bacteria in disease triggered by viral infection
WT + MNV CR6
Villi
-10
0
10
20
30 *****
**
W T H M W T H M W T H M W T H M
Un in fe cte d M NV C R67 d ays
M NV C W 37 d ays
M NV C R60 d ays
n.s.
% R
edu
ctio
n in
Vill
us
Hei
gh
tHM + MNV CR6
Crypts
Villi
Crypts
A B Figure 5. Virus-Infected Atg16L1 Mutant
Mice Display Mucosal Atrophy in the Ileum
in Response to DSS
(A) Light microscopy of H&E stained ileal sections
from the same mice in Figure 4. Scale bar =
200 mm.
(B) Quantification of average villus height normal-
ized to uninfected WT mice treated with DSS.
Seven days and 0 days refer to the amount of
time mice were inoculated with the virus prior to
DSS treatment (an average of 286 total villi
from at least 5 mice/condition was measured,
**p < 0.01, ***p < 0.001, mean ± SEM).
See also Figure S5.
as in secondary infection with Streptococcus pneumoniae after
influenza (McCullers, 2006). To determine if the intestinal injury
response dependent on virus-plus-susceptibility gene interac-
tion requires commensal bacteria, Atg16L1HM mice were
infected with MNV CR6 and treated with both DSS and broad
spectrum antibiotics. Antibiotics uptake was similar across
groups and did not interfere with the administration of DSS
(Extended Experimental Procedures). Administration of antibi-
otics to virally-infected Atg16L1HM mice prevented abnormal
DSS-induced pathologies in both ileum and colon reported
above (Figures 6D–6F). Thus, aberrant injury responses depen-
dent on virus-plus-susceptibility gene interaction require com-
mensal bacteria.
DISCUSSION
An important goal is to identify specific genetic, infectious, or
environmental factors responsible for a given disease. When
such an etiologic smoking gun is detected, the full power of
medicine, chemistry, and vaccine development can be brought
to bear on the illness. However, if complex diseases are com-
binatorial in etiology, then an individual gene or pathogen
may display only poor association with disease incidence and
severity. This model of complex disease is widely recognized
but experimental evidence has not been reported.
In this study, we define a complex inflammatory disease in
mice that depends on an interaction between a specific enteric
virus strain and a single host disease susceptibility gene. Several
of the pathologic features observed in this model resemble
those in many Crohn’s disease patients. In particular, Paneth
cell structural and granule packaging abnormalities observed
in virus-infected Atg16L1HM mice are remarkably similar to those
in patients homozygous for the risk allele of ATG16L1. Since
Crohn’s disease patients homozygous for the nonrisk allele of
ATG16L1 do not display this pathology, virus-plus-susceptibility
gene interaction may contribute to heterogeneity between
patients. Importantly, Paneth cells from Atg16L1HM mice in-
fected with virus have a fundamentally different gene expression
profile than controls. These results provide a specific example of
how a genetic factor and an environmental agent, each innoc-
uous by itself, can have profound effects on the host when
combined. These results also provide one explanation for why
persons in clinical trials may display widely variable responses
to treatment for a single pathologically defined disease.
Mechanisms of Pathology Induced by Virus-Host
Susceptibility Gene InteractionDespite the importance of autophagy genes in regulating the
intracellular replication of pathogens, Atg16L1 mutant mice did
not display increased MNV replication, virus was not detected
in Paneth cells, and we found no role for Atg16L1 in control
of MNV replication in vitro using primary macrophages from
Atg16L1HM mice (data not shown). In this context, it is interesting
that Paneth cell abnormalities, the transcriptional response to
infection, and DSS-induced pathologies were dependent on
infection with MNV CR6, a strain capable of persistent infection.
Notably, the two strains of MNV used in this study generate sero-
logically indistinguishable antibody responses (Thackray et al.,
2007) despite the dramatic difference in ability to induce disease
in Atg16L1 mutant mice. Additionally, aberrant DSS-induced
pathologies were dependent on the timing of MNV infection.
The relationship between an infectious agent and a complex
disease is typically established through serology or the presence
of a pathogen in infected tissue. These conventional approaches
may dismiss etiologic agents if the disease is dependent on
a specific pathogen strain or the timing of the infection. Detection
of virus-plus-susceptibility gene interactions in humans may
therefore require advances in the capacity to identify viruses
present in tissues in an unbiased way, allowing a better under-
standing of the relationship between the human virome and
disease pathogenesis (Virgin et al., 2009). To be revealing,
such studies will have to be coupled to high power epidemiolog-
ical studies and analysis of when patients were exposed to
environmental factors.
The aberrant intestinal injury response we described in
virally-infected Atg16L1HM mice was dependent on three factors
associated with Crohn’s disease pathogenesis in humans—
TNFa, IFNg, and commensal bacteria. This common role of
two cytokines and commensal bacteria suggest that the human
disease and the observed pathologies in mice share an under-
lying mechanism, and supports the relevance of the virus-
plus-susceptibility gene interaction reported here. Consistent
with the efficacy of TNFa and IFNg blockade in our model, cyto-
kine-mediated injury after viral infection is receiving attention as
an attractive target for therapeutic intervention (Marsolais et al.,
2009; Brockman et al., 2009). There is also evidence for an
intersection between viral infection and commensal bacteria
(Brenchley et al., 2006; Xi et al., 2008). The antibiotic responsive-
ness of the DSS-induced disease in our model reveals an
Cell 141, 1135–1145, June 25, 2010 ª2010 Elsevier Inc. 1141

A
B
D
-20
0
20
40
60
80
100 *
W T H M W T H M
+Antibiotics-Antibiotics
% In
crea
se in
mu
scle
wid
th
0
50
100*
*
WT H M WT H M WT H M
Is o typ e An ti-TN Fα An ti- IF Nγ
% In
crea
se in
mu
scle
wid
th
E
C F
-10
0
10
20
30*
***
W T H M W T H M W T H M
Isotype Anti-T N Fα Anti-IFN γ% R
edu
ctio
n in
Vill
us
Hei
gh
t
-10
0
10
20
30*
W T H M W T H M
-Antibiotics + Antibiotics% R
edu
ctio
n in
Vill
us
Hei
gh
t
0
1
2
3
4
WT H M WT H M WT H M
Is o typ e An ti-TN Fα An ti- IF N γ
******
Lym
ph
oid
ag
gre
gat
es
0
1
2
3
W T H M W T H M
+ Antibiotics-Antibiotics
***
Lym
ph
oid
ag
gre
gat
es
Figure 6. TNFa and IFNg Inhibition or Anti-
biotics Treatment Ameliorates DSS-
Induced Disease in Virus-Infected Atg16L1
Mutant Mice
(A) Isotype control or blocking antibodies against
TNFa and IFNg were administered to WT and
Atg16L1HM mice infected with MNV CR6 for
7 days followed by additional 7 days of DSS treat-
ment at which point intestines were harvested.
The muscularis propria thickness in the region
adjacent to the ano-rectal junction was quantified
and normalized to the average of WT mice
receiving isotype control antibodies (n R 6 mice/
condition, *p < 0.05, mean ± SEM).
(B) Number of lymphoid aggregates in the region
adjacent to the ano-rectal junction from mice in
(A) (*p < 0.001, mean ± SEM).
(C) Quantification of average villus height from
mice in (A) normalized to the average of WT mice
receiving isotype control antibodies (an average
of 263 total villi from at least 5 mice/condition
was measured, *p < 0.05, ***p < 0.001, mean ±
SEM).
(D) WT and Atg16L1HM mice were orally inoculated
with MNV CR6 for 7 days and then given 2.5%
DSS and broad spectrum antibiotics concurrently
for another 7 days at which point intestines were
harvested. The muscularis propria thickness was
normalized to the average of uninfected WT mice
treated with DSS from Figure 4D (n = 6 mice/
condition, *p < 0.05, mean ± SEM).
(E) Number of lymphoid aggregates from mice in
(D) (***p < 0.001, mean ± SEM).
(F) Quantification of average villus height from
mice in (D) normalized to the average of uninfected
WT mice treated with DSS from Figure 5B (an
average of 320 total villi from at least 5 mice/condi-
tion was measured, *p < 0.05, mean ± SEM).
See also Figure S5.
important bacterial component to the virus-plus-susceptibility
gene interaction that may be exploited therapeutically.
The gene expression and morphological changes in Paneth
cells we report are consistent with an important role of
Atg16L1 in the secretory pathway and the response of epithelial
cells to infection. These functions of Atg16L1 may be related to
the membrane trafficking component of autophagy. Consistent
with a cell autonomous role of Atg16L1 in Paneth cells, intestinal
epithelium-specific deletion of two other autophagy genes also
leads to Paneth cell granule defects (Cadwell et al., 2008a;
Cadwell et al., 2008b). Since Atg16L1 and its binding partner
Atg5 can limit cytokine production (Saitoh et al., 2008; Jounai
et al., 2007; Tal et al., 2009), viral infection may trigger an altered
cytokine response in Atg16L1 mutant mice. Although the
changes we see are in the epithelium, the complex inflammatory
phenotype reported here may also depend on Atg16L1 function
in other cell types. For example, the necessity of autophagy
genes for T-cell function (Pua et al., 2007; Nedjic et al., 2008)
may be relevant given the role of T cells in colitis after adoptive
1142 Cell 141, 1135–1145, June 25, 2010 ª2010 Elsevier Inc.
transfer into Rag2-deficient mice (Barnes and Powrie, 2009);
the role of MNV has not been explored in that model.
It is tempting to speculate that Paneth cell abnormalities
contribute to aberrant DSS-induced pathologies. Local pro-
duction of inflammatory molecules by Paneth cells may drive
villus blunting in the ileum. Moreover, virally-infected Atg16L1HM
mice can have alterations in commensal bacteria since mice
with Paneth cells that have lost a-defensin activation develop
intestinal dysbiosis including the emergence of segmented fila-
mentous bacteria (Salzman et al., 2010) that stimulate the devel-
opment of Th17 lymphocytes (Ivanov et al., 2009). Paneth cells
are not present in the mouse colon where viral infection alters
injury responses in Atg16L1 mutant mice. However, the effects
of Paneth cells need not be restricted to the ileum since active
peptides from their granules can be detected in the distal colonic
lumen (Mastroianni and Ouellette, 2009). Since it is unclear how
diverse pathologies are related to one another in Crohn’s disease
patients, it will be important to address the relationship between
Paneth cells, the microbiome, and the virome in the context

of the aberrant injury response dependent on the virus-plus-
susceptibility gene interaction in our model.
Disease Penetrance in Mouse Modelsof Mucosal ImmunityBased on our observations in Atg16L1 mutant mice, we specu-
late that other traits attributed to specific mutations are also
dependent on viral infections. The presence of an infectious
agent can lead to different experimental outcomes between
laboratories. For example, experimental allergic encephalomy-
elitis correlates with mouse facility-specific pathogen exposure
(Goverman et al., 1993). Experimental discrepancies between
facilities are particularly germane for models of intestinal disease
since MNV is an enteric virus commonly detected in specific
pathogen free facilities (Hsu et al., 2005; Pritchett-Corning
et al., 2009; Goto et al., 2009).
The effect of viral infection can be remarkably specific. In our
model, the MNV CR6 strain altered experimental outcomes only
in Atg16L1HM mice. MNV CR6 infection does not alter immune
responses in C57/BL6 mice challenged with Friend leukemia
virus, influenza, vaccinia, or MCMV (Ammann et al., 2009; Hens-
ley et al., 2009; Doom et al., 2009). In contrast, MNV infection of
Mdra1�/�mice enhances immune responses to Helicobacter bilis
infection (Lencioni et al., 2008). Thus, the effect that a particular
pathogen has on the host cannot be generalized. Although we
focus on a single virus and gene, multiple interactions may exist
between different susceptibility genes and an array of commen-
sals and pathogens. Much remains to be learned about the inter-
actions between susceptibility genes and specific pathogens to
understand what may be a general ‘‘microbe plus susceptibility
gene’’ contribution to a range of complex diseases.
Understanding the Contributions of Geneticand Environmental Factors in Complex DiseasesEpidemiological studies have not identified a specific infectious
cause for inflammatory bowel disease. Reported associations
between disease risk and infectious gastroenteritis are correla-
tive (Porter et al., 2008; Garcia Rodriguez et al., 2006; Gradel
et al., 2009), but are of interest given our data since human
noroviruses related to MNV cause human gastroenteritis (Mead
et al., 1999). Since many bacteria and viruses cause gastroenter-
itis, the infectious trigger of a complex disease need not be
a single specific agent. Rather several agents that affect similar
immunologic pathways can be involved. Interestingly, the
Crohn’s disease associated gene NOD2 may recognize viral
RNA in addition to bacterial peptidoglycan (Sabbah et al.,
2009; Shapira et al., 2009), raising the possibility that a viral infec-
tion can interact with multiple susceptibility genes.
Although Atg16L1 mutant mice do not display all pathology
found in Crohn’s disease patients, we have reproduced several
disease hallmarks in a mouse with a mutation in a single suscep-
tibility gene. It is commonly assumed that the failure to recreate
the entirety of Crohn’s disease in various mouse models is due
to inherent differences between humans and mice. However, in
the combinatorial view of complex disease supported by our
results, reproducing full disease may require combinations of
specific alleles of multiple genes with certain environmental
agents. It is worth noting that not all Crohn’s disease patients
exhibit identical symptoms or pathologies, and the nature of
Crohn’s disease varies over time even within one individual.
In addition, therapeutic interventions that improve conditions for
some do not always alleviate disease in others. Therefore, com-
plex diseases may represent a combinatorial confluence of path-
ologic responses, each with overlapping but nonidentical genetic
and environmental causes and therefore therapeutic responses.
This paradigm has profound implications for how we view
the relationship between genetic heterogeneity and phenotype
in humans. We propose that studies examining associations
between disease susceptibility and genetic variation should
consider the history and current status of viral infections in the
individuals. Similarly, studies examining the correlation between
viral infections and disease would benefit from sorting individ-
uals based on genetic background. If we can improve our knowl-
edge in this area, the concept of personalized medicines may
become closer to clinical application.
EXPERIMENTAL PROCEDURES
Mice
Atg16L1HM mice were described (Cadwell et al., 2008a). The Atg16L1HM line
housed in a conventional barrier facility was rederived by embryo transfer
into the enhanced facility with the following specialized features: all cages,
bedding, chow, and water are autoclaved prior to use; access to breeding
mice in this facility is restricted to specially trained personnel; and sentinel
mice are routinely screened for common mouse pathogens including MNV.
Mice were generated by mating heterozygotes for the gene trap mutagenized
Atg16L1 locus. Progeny homozygous for the mutation and wild-type litter-
mates aged 7–15 weeks were used in experiments.
Viruses
Generation of concentrated stocks was described (Chachu et al., 2008).
Twenty-five microliters of concentrated stocks (3 3 107 pfu) of MNV CR6
and CW3 was orally inoculated into mice. For virus quantification, total RNA
was harvested per one stool pellet, 1 cm of intestine, or total mesenteric lymph
nodes. qRT-PCR for MNV genome copy numbers was as described (Thackray
et al., 2007). For detection of virus in Paneth cells, �300 crypts from three
mice/genotype were procured by laser capture microdissection (Extended
Experimental Procedures) 7 days postinfection with MNV CR6. As a positive
control, MNV was detected in whole gut RNA from similarly prepared tissue.
Statistical Analysis
Lysozyme distribution, muscle thickness, lymphoid aggregates, and villus
height were analyzed by two-tailed unpaired t tests. Viral genome copies
were analyzed with the nonparametric Mann-Whitney test. Analyses except
for microarray data used GraphPad Prism (version 5.00).
ACCESSION NUMBERS
Microarray files have been deposited in Array Express with accession codes
E-TABM-957 and E-TABM-958.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Extended Experimental Procedures and
five figures and can be found with this article online at doi:10.1016/j.cell.
2010.05.009.
ACKNOWLEDGMENTS
This research was supported by grant U54 AI057160 Project 5 and the Broad
Foundation (K.C., N.M., and H.W.V.), R01 AI084887 (T.S.S. and H.W.V.), the
Lallage Feazel Wall Fellowship DRG-1972-08 from the Damon Runyon Cancer
Cell 141, 1135–1145, June 25, 2010 ª2010 Elsevier Inc. 1143

Research Foundation (K.C.), training grant NIH T32-AI007172 (N.M.), the Pew
Foundation (K.K.P. and T.S.S.), Washington University Digestive Diseases
Research Core Center DK52574, Pfizer biomedical agreement with Washing-
ton University, fellowship award from the Crohn’s and Colitis Foundation of
America (A.C.Y.N), and grants DK83756, DK086502, and DK043351 (R.J.X.).
Washington University holds U.S. patents 7,041,444 B2, 7,264,923, and US
7,455,972 related to growth and detection of MNV. Washington University
and H.W.V. receive income based on licenses for MNV technology.
Received: December 9, 2009
Revised: February 26, 2010
Accepted: April 19, 2010
Published: June 24, 2010
REFERENCES
Agus, S.G., Dolin, R., Wyatt, R.G., Tousimis, A.J., and Northrup, R.S. (1973).
Acute infectious nonbacterial gastroenteritis: intestinal histopathology. Histo-
logic and enzymatic alterations during illness produced by the Norwalk agent
in man. Ann. Intern. Med. 79, 18–25.
Altshuler, D., Daly, M.J., and Lander, E.S. (2008). Genetic mapping in human
disease 4. Science 322, 881–888.
Ammann, C.G., Messer, R.J., Varvel, K., Debuysscher, B.L., Lacasse, R.A.,
Pinto, A.K., and Hasenkrug, K.J. (2009). Effects from acute and chronic murine
norovirus infections on immune responses and recovery from Friend retrovirus
infection. J Virol. 83, 13037–13041.
Barnes, M.J., and Powrie, F. (2009). Regulatory T cells reinforce intestinal
homeostasis. Immunity 31, 401–411.
Barrett, J.C., Hansoul, S., Nicolae, D.L., Cho, J.H., Duerr, R.H., Rioux, J.D.,
Brant, S.R., Silverberg, M.S., Taylor, K.D., Barmada, M.M., et al. (2008).
Genome-wide association defines more than 30 distinct susceptibility loci
for Crohn’s disease. Nat. Genet. 40, 955–962.
Brem-Exner, B.G., Sattler, C., Hutchinson, J.A., Koehl, G.E., Kronenberg, K.,
Farkas, S., Inoue, S., Blank, C., Knechtle, S.J., Schlitt, H.J., et al. (2008).
Macrophages driven to a novel state of activation have anti-inflammatory
properties in mice. J. Immunol. 180, 335–349.
Brenchley, J.M., Price, D.A., Schacker, T.W., Asher, T.E., Silvestri, G., Rao, S.,
Kazzaz, Z., Bornstein, E., Lambotte, O., Altmann, D., et al. (2006). Microbial
translocation is a cause of systemic immune activation in chronic HIV infection.
Nat. Med. 12, 1365–1371.
Brockman, M.A., Kwon, D.S., Tighe, D.P., Pavlik, D.F., Rosato, P.C., Sela, J.,
Porichis, F., Le, G.S., Waring, M.T., Moss, K., et al. (2009). IL-10 is up-regu-
lated in multiple cell types during viremic HIV infection and reversibly inhibits
virus-specific T cells. Blood 114, 346–356.
Cadwell, K., Liu, J.Y., Brown, S.L., Miyoshi, H., Loh, J., Lennerz, J.K., Kishi, C.,
Kc, W., Carrero, J.A., Hunt, S., et al. (2008a). A key role for autophagy and the
autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature
456, 259–263.
Cadwell, K., Patel, K.K., Komatsu, M., Virgin, H.W., and Stappenbeck, T.S.
(2008b). A common role for Atg16L1, Atg5, and Atg7 in small intestinal Paneth
cells and Crohn’s disease. Autophagy 5, 250–252.
Chachu, K.A., LoBue, A.D., Strong, D.W., Baric, R.S., and Virgin, H.W. (2008).
Immune mechanisms responsible for vaccination against and clearance of
mucosal and lymphatic norovirus infection. PLoS Pathog. 4, e1000236.
Chand, N., and Mihas, A.A. (2006). Celiac disease: current concepts in diag-
nosis and treatment. J. Clin. Gastroenterol. 40, 3–14.
Day, D.W., Morson, B.C., and Williams, G.T. (2003). Morson and Dawson’s
Gastrointestinal Pathology (Malden, MA: Blackwell Publishing).
Dolin, R., Levy, A.G., Wyatt, R.G., Thornhill, T.S., and Gardner, J.D. (1975).
Viral gastroenteritis induced by the Hawaii agent. Jejunal histopathology and
serologic response. Am. J. Med. 59, 761–768.
Doom, C.M., Turula, H.M., and Hill, A.B. (2009). Investigation of the impact of
the common animal facility contaminant murine norovirus on experimental
murine cytomegalovirus infection. Virology 392, 153–161.
1144 Cell 141, 1135–1145, June 25, 2010 ª2010 Elsevier Inc.
Dyrberg, T., Schwimmbeck, P.L., and Oldstone, M.B. (1988). Inhibition of
diabetes in BB rats by virus infection. J. Clin. Invest. 81, 928–931.
Fais, S., Capobianchi, M.R., Pallone, F., Di, M.P., Boirivant, M., Dianzani, F.,
and Torsoli, A. (1991). Spontaneous release of interferon gamma by intestinal
lamina propria lymphocytes in Crohn’s disease. Kinetics of in vitro response to
interferon gamma inducers. Gut 32, 403–407.
Fuss, I.J., Neurath, M., Boirivant, M., Klein, J.S., de La, M.C., Strong, S.A.,
Fiocchi, C., and Strober, W. (1996). Disparate CD4+ lamina propria (LP)
lymphokine secretion profiles in inflammatory bowel disease. Crohn’s disease
LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis
LP cells manifest increased secretion of IL-5. J. Immunol. 157, 1261–1270.
Garcia Rodriguez, L.A., Ruigomez, A., and Panes, J. (2006). Acute gastroen-
teritis is followed by an increased risk of inflammatory bowel disease. Gastro-
enterology 130, 1588–1594.
Goto, K., Hayashimoto, N., Yasuda, M., Ishida, T., Kameda, S., Takakura, A.,
and Itoh, T. (2009). Molecular detection of murine norovirus from experimen-
tally and spontaneously infected mice. Exp. Anim. 58, 135–140.
Goverman, J., Woods, A., Larson, L., Weiner, L.P., Hood, L., and Zaller, D.M.
(1993). Transgenic mice that express a myelin basic protein-specific T cell
receptor develop spontaneous autoimmunity. Cell 72, 551–560.
Gradel, K.O., Nielsen, H.L., Schonheyder, H.C., Ejlertsen, T., Kristensen, B.,
and Nielsen, H. (2009). Increased short- and long-term risk of inflammatory
bowel disease after salmonella or campylobacter gastroenteritis. Gastroenter-
ology 137, 495–501.
Hampe, J., Franke, A., Rosenstiel, P., Till, A., Teuber, M., Huse, K., Albrecht,
M., Mayr, G., De La Vega, F.M., Briggs, J., et al. (2007). A genome-wide asso-
ciation scan of nonsynonymous SNPs identifies a susceptibility variant for
Crohn disease in ATG16L1. Nat. Genet. 39, 207–211.
Hensley, S.E., Pinto, A.K., Hickman, H.D., Kastenmayer, R.J., Bennink, J.R.,
Virgin, H.W., and Yewdell, J.W. (2009). Murine norovirus infection has no
significant effect on adaptive immunity to vaccinia virus or influenza A virus.
J. Virol. 83, 7357–7360.
Hsu, C.C., Wobus, C.E., Steffen, E.K., Riley, L.K., and Livingston, R.S. (2005).
Development of a microsphere-based serologic multiplexed fluorescent
immunoassay and a reverse transcriptase PCR assay to detect murine norovi-
rus 1 infection in mice. Clin. Diagn. Lab. Immunol. 12, 1145–1151.
Ivanov, I.I., Atarashi, K., Manel, N., Brodie, E.L., Shima, T., Karaoz, U., Wei, D.,
Goldfarb, K.C., Santee, C.A., Lynch, S.V., et al. (2009). Induction of intestinal
Th17 cells by segmented filamentous bacteria. Cell 139, 485–498.
Jounai, N., Takeshita, F., Kobiyama, K., Sawano, A., Miyawaki, A., Xin, K.Q.,
Ishii, K.J., Kawai, T., Akira, S., Suzuki, K., and Okuda, K. (2007). The Atg5
Atg12 conjugate associates with innate antiviral immune responses. Proc.
Natl. Acad. Sci. USA 104, 14050–14055.
Ju, J.S., Miller, S.E., Jackson, E., Cadwell, K., Piwnica-Worms, D., and Weihl,
C.C. (2009). Quantitation of selective autophagic protein aggregate degrada-
tion in vitro and in vivo using luciferase reporters. Autophagy 5, 511–519.
Kang, S.S., Bloom, S.M., Norian, L.A., Geske, M.J., Flavell, R.A., Stappen-
beck, T.S., and Allen, P.M. (2008). An antibiotic-responsive mouse model of
fulminant ulcerative colitis. PLoS Med. 5, e41.
Karst, S.M., Wobus, C.E., Lay, M., Davidson, J., and Virgin, H.W. (2003).STAT1-
dependent innate immunity to a Norwalk-like virus. Science 299, 1575–1578.
Lake, F.R., Noble, P.W., Henson, P.M., and Riches, D.W. (1994). Functional
switching of macrophage responses to tumor necrosis factor-alpha (TNF
alpha) by interferons. Implications for the pleiotropic activities of TNF alpha.
J. Clin. Invest. 93, 1661–1669.
Lencioni, K.C., Seamons, A., Treuting, P.M., Maggio-Price, L., and Brabb, T.
(2008). Murine norovirus: an intercurrent variable in a mouse model of
bacteria-induced inflammatory bowel disease. Comp. Med. 58, 522–533.
Marsolais, D., Hahm, B., Walsh, K.B., Edelmann, K.H., McGavern, D., Hatta,
Y., Kawaoka, Y., Rosen, H., and Oldstone, M.B. (2009). A critical role for the
sphingosine analog AAL-R in dampening the cytokine response during influ-
enza virus infection. Proc. Natl. Acad. Sci. USA 106, 1560–1565.

Mastroianni, J.R., and Ouellette, A.J. (2009). Alpha-defensins in enteric innate
immunity: functional Paneth cell alpha-defensins in mouse colonic lumen.
J. Biol. Chem. 284, 27848–27856.
McCullers, J.A. (2006). Insights into the interaction between influenza virus and
pneumococcus. Clin. Microbiol. Rev. 19, 571–582.
Mead, P.S., Slutsker, L., Dietz, V., McCaig, L.F., Bresee, J.S., Shapiro, C.,
Griffin, P.M., and Tauxe, R.V. (1999). Food-related illness and death in the
United States. Emerg. Infect. Dis. 5, 607–625.
Mi, H., Guo, N., Kejariwal, A., and Thomas, P.D. (2007). PANTHER version 6:
protein sequence and function evolution data with expanded representation
of biological pathways. Nucleic Acids Res. 35, D247–D252.
Mumphrey, S.M., Changotra, H., Moore, T.N., Heimann-Nichols, E.R., Wobus,
C.E., Reilly, M.J., Moghadamfalahi, M., Shukla, D., and Karst, S.M. (2007).
Murine norovirus 1 infection is associated with histopathological changes in
immunocompetent hosts, but clinical disease is prevented by STAT1-depen-
dent interferon responses. J. Virol. 81, 3251–3263.
Nedjic, J., Aichinger, M., Emmerich, J., Mizushima, N., and Klein, L. (2008).
Autophagy in thymic epithelium shapes the T-cell repertoire and is essential
for tolerance. Nature 455, 396–400.
Oldstone, M.B. (1988). Prevention of type I diabetes in nonobese diabetic mice
by virus infection. Science 239, 500–502.
Ouellette, A.J. (2006). Paneth cell alpha-defensin synthesis and function.
Curr. Top. Microbiol. Immunol. 306, 1–25.
Packey, C.D., and Sartor, R.B. (2009). Commensal bacteria, traditional and
opportunistic pathogens, dysbiosis and bacterial killing in inflammatory bowel
diseases. Curr. Opin. Infect. Dis. 22, 292–301.
Piguet, P.F., Vesin, C., Donati, Y., and Barazzone, C. (1999). TNF-induced
enterocyte apoptosis and detachment in mice: induction of caspases and
prevention by a caspase inhibitor, ZVAD-fmk. Lab. Invest. 79, 495–500.
Porter, C.K., Tribble, D.R., Aliaga, P.A., Halvorson, H.A., and Riddle, M.S.
(2008). Infectious gastroenteritis and risk of developing inflammatory bowel
disease. Gastroenterology 135, 781–786.
Porter, E.M., Bevins, C.L., Ghosh, D., and Ganz, T. (2002). The multifaceted
Paneth cell. Cell. Mol. Life Sci. 59, 156–170.
Pritchett-Corning, K.R., Cosentino, J., and Clifford, C.B. (2009). Contemporary
prevalence of infectious agents in laboratory mice and rats. Lab. Anim. 43,
165–173.
Pua, H.H., Dzhagalov, I., Chuck, M., Mizushima, N., and He, Y.W. (2007). A crit-
ical role for the autophagy gene Atg5 in T cell survival and proliferation. J. Exp.
Med. 204, 25–31.
Pull, S.L., Doherty, J.M., Mills, J.C., Gordon, J.I., and Stappenbeck, T.S.
(2005). Activated macrophages are an adaptive element of the colonic epithe-
lial progenitor niche necessary for regenerative responses to injury. Proc. Natl.
Acad. Sci. USA 102, 99–104.
Rioux, J.D., Xavier, R.J., Taylor, K.D., Silverberg, M.S., Goyette, P., Huett, A.,
Green, T., Kuballa, P., Barmada, M.M., Datta, L.W., et al. (2007). Genome-wide
association study identifies new susceptibility loci for Crohn disease and impli-
cates autophagy in disease pathogenesis. Nat. Genet. 39, 596–604.
Roberts, M.E. (2009). Inflammatory disorders of the small intestine. In Surgical
Pathology of the GI Tract, Liver, Biliary Tract and Pancreas, R.D. Odze and J.R.
Goldblum, eds. (Philadelphia, PA: Saunders Elsevier).
Sabbah, A., Chang, T.H., Harnack, R., Frohlich, V., Tominaga, K., Dube, P.H.,
Xiang, Y., and Bose, S. (2009). Activation of innate immune antiviral responses
by Nod2. Nat. Immunol. 10, 1073–1080.
Saitoh, T., Fujita, N., Jang, M.H., Uematsu, S., Yang, B.G., Satoh, T., Omori, H.,
Noda, T., Yamamoto, N., Komatsu, M., et al. (2008). Loss of the autophagy
protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature
456, 264–268.
Salzman, N.H., Hung, K., Haribhai, D., Chu, H., Karlsson-Sjoberg, J., Amir, E.,
Teggatz, P., Barman, M., Hayward, M., Eastwood, D., et al. (2010). Enteric
defensins are essential regulators of intestinal microbial ecology. Nat. Immu-
nol. 11, 76–83.
Sartor, R.B. (1997). The influence of normal microbial flora on the development
of chronic mucosal inflammation. Res. Immunol. 148, 567–576.
Scholtens, D., Miron, A., Merchant, F.M., Miller, A., Miron, P.L., Iglehart, J.D.,
and Gentleman, R. (2004). Analyzing factorial designed microarray experi-
ments. J. Multivariate Anal. 90, 19–43.
Schreiber, R.D., Hicks, L.J., Celada, A., Buchmeier, N.A., and Gray, P.W.
(1985). Monoclonal antibodies to murine gamma interferon which differentially
modulate macrophage activation and antiviral activity. J. Immunol. 134,
1609–1618.
Shapira, S.D., Gat-Viks, I., Shum, B.O., Dricot, A., de Grace, M.M., Wu, L.,
Gupta, P.B., Hao, T., Silver, S.J., Root, D.E., et al. (2009). A physical and regu-
latory map of host-influenza interactions reveals pathways in H1N1 infection.
Cell 139, 1255–1267.
Sheehan, K.C., Ruddle, N.H., and Schreiber, R.D. (1989). Generation and char-
acterization of hamster monoclonal antibodies that neutralize murine tumor
necrosis factors. J. Immunol. 142, 3884–3893.
Smyth, G.K. (2005). Limma: linear models for microarray data. In Bioinfor-
matics and Computational Biology Solutions Using R and Bioconductor, R.
Gentleman, V. Carey, S. Dudoit, R. Irizarry, and W. Huber, eds. (New York:
Springer).
Stappenbeck, T.S., Mills, J.C., and Gordon, J.I. (2003). Molecular features of
adult mouse small intestinal epithelial progenitors. Proc. Natl. Acad. Sci.
USA 100, 1004–1009.
Suk, K., Kim, S., Kim, Y.H., Kim, K.A., Chang, I., Yagita, H., Shong, M., and Lee,
M.S. (2001). IFN-gamma/TNF-alpha synergism as the final effector in autoim-
mune diabetes: a key role for STAT1/IFN regulatory factor-1 pathway in
pancreatic beta cell death. J. Immunol. 166, 4481–4489.
Tal, M.C., Sasai, M., Lee, H.K., Yordy, B., Shadel, G.S., and Iwasaki, A. (2009).
Absence of autophagy results in reactive oxygen species-dependent amplifi-
cation of RLR signaling. Proc. Natl. Acad. Sci. USA 106, 2770–2775.
Targan, S.R., Hanauer, S.B., Van Deventer, S.J., Mayer, L., Present, D.H.,
Braakman, T., DeWoody, K.L., Schaible, T.F., and Rutgeerts, P.J. (1997).
A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis
factor alpha for Crohn’s disease. Crohn’s Disease cA2 Study Group.
N. Engl. J. Med. 337, 1029–1035.
Thackray, L.B., Wobus, C.E., Chachu, K.A., Liu, B., Alegre, E.R., Henderson,
K.S., Kelley, S.T., and Virgin, H.W. (2007). Murine noroviruses comprising
a single genogroup exhibit biological diversity despite limited sequence diver-
gence. J. Virol. 81, 10460–10473.
The Wellcome Trust Case Control Consortium. (2007). Genome-wide associ-
ation study of 14,000 cases of seven common diseases and 3,000 shared
controls. Nature 447, 661–678.
Tonietti, G., Oldstone, M.B., and Dixon, F.J. (1970). The effect of induced
chronic viral infections on the immunologic diseases of New Zealand mice.
J. Exp. Med. 132, 89–109.
Vaishnava, S., Behrendt, C.L., Ismail, A.S., Eckmann, L., and Hooper, L.V.
(2008). Paneth cells directly sense gut commensals and maintain homeostasis
at the intestinal host-microbial interface. Proc. Natl. Acad. Sci. USA 105,
20858–20863.
Virgin, H.W., and Levine, B. (2009). Autophagy genes in immunity. Nat. Immu-
nol. 10, 461–470.
Virgin, H.W., Wherry, E.J., and Ahmed, R. (2009). Redefining chronic viral
infection. Cell 138, 30–50.
Weersma, R.K., Zhernakova, A., Nolte, I.M., Lefebvre, C., Rioux, J.D., Mulder,
F., van Dullemen, H.M., Kleibeuker, J.H., Wijmenga, C., and Dijkstra, G. (2008).
ATG16L1 and IL23R are associated with inflammatory bowel diseases but not
with celiac disease in the Netherlands. Am. J. Gastroenterol. 103, 621–627.
Xi, Z., Ramirez, J.L., and Dimopoulos, G. (2008). The Aedes aegypti toll
pathway controls dengue virus infection. PLoS Pathog. 4, e1000098.
Yang, Y.H., Dudoit, S., Luu, P., Lin, D.M., Peng, V., Ngai, J., and Speed, T.P.
(2002). Normalization for cDNA microarray data: a robust composite method
addressing single and multiple slide systematic variation. Nucleic Acids Res.
30, e15.
Cell 141, 1135–1145, June 25, 2010 ª2010 Elsevier Inc. 1145

Supplemental Information
EXTENDED EXPERIMENTAL PROCEDURES
Administration of DSSAqueous solution of DSS (TDB Consultancy, Uppsala, Sweden) was filtered using a 0.22-mm cellulose acetate filter prior to admin-
istration through drinking water. For antibiotics experiments, 1 g/L ampicillin (Columbus Serum Company, Columbus, OH), 500 mg/L
vancomycin (Sigma, St. Louis, MO), 1 g/L neomycin sulfate (Sigma, St. Louis, MO), and 1 g/L metronidazole (Sigma, St. Louis, MO)
were dissolved in drinking water along with 2.5% DSS, then filtered using a 0.22-mm cellulose acetate filter. 20 mg/ml sugar-sweet-
ened grape Kool-Aid Mix (Kraft Foods) was included in the water to encourage consumption. All mice receiving antibiotics displayed
a similar degree of focal ulceration in the descending colon compared to mice receiving DSS alone, indicating that the presence of
antibiotics did not interfere with the administration of DSS through water and that bacteria are not essential for this aspect of DSS-
induced intestinal injury. Additionally, both WT and Atg16L1HM mice displayed epithelial hypo-proliferation in non-ulcerated regions
of the rectum that is similar to findings in DSS-treated germ-free mice (Pull et al., 2005), thus confirming successful antibiotic uptake.
For TNFa neutralization, mice received 1 mg of either hamster anti-TNFa (TN3–19.12) or hamster anti-PIP isotype control antibodies 1
day prior to DSS treatment, then received a second injection of 0.5 mg 5 days later (4 days into DSS treatment). For IFNg neutrali-
zation, mice received one injection of 0.2 mg of hamster anti-IFNg (H22) 1 day prior to DSS treatment. Blocking antibodies were kindly
provided by Dr. Robert Schreiber (Sheehan et al., 1989; Schreiber et al., 1985).
UV Inactivation of MNV CR6UV-inactivated virus was generated by placing 600 ml of 1.2x109 pfu/ml MNV CR6 in a 6 well plate and subjected to 1000 mJ of UV in
a UV Stratalinker 2400 (Stratagene). The inactivation was confirmed by qRT-PCR for the presence of viral genome in stool specimens
7 days post infection.
MicroscopyThe distal ileum and colon were removed, cut open along the length, and pinned on black wax. The ileum was fixed in 10% formalin
and the colon was fixed in Bouin’s fixative overnight at 4�C. 2 cm strips of intestinal tissues were embedded in agar to enrich for well-
oriented crypt-villus units. To determine villus blunting, villus height was quantified in ileal sections that were cut perpendicular to the
villus-crypt axis as defined by the presence of a visible crypt lumen from the orifice to the base of the crypt, thereby minimizing tissue
orientation-based artifacts of villus height measurements (Stappenbeck et al., 2003). Images were taken on an Olympus BX51 micro-
scope. Immunohistochemistry for lysozyme was previously described (Cadwell et al., 2008). Sections were viewed with a Zeiss Ax-
iovert 200 inverted fluorescence microscope and quantified on an Olympus AX70 epi-fluorescence microscope. Electron micros-
copy of ileal crypts was previously described (Cadwell et al., 2008).
Laser Capture MicrodissectionThe distal ileum was removed, cut open along the length, pinned on black wax, and fixed in methacarn. The tissue was blocked and
embedded as described above and 0.6 mm serial sections were cut and deparaffinized. The crypt-base epithelial cells enriched for
Paneth cells were captured and RNA was isolated by previously described techniques (Stappenbeck et al., 2003; Pull et al., 2005). 25
ng of total RNA from each sample was amplified using the Transplex Complete Whole Transcriptome Amplification Kit (Sigma, St
Louis MO), labeled using a modified protocol with the ULS aRNA Fluorescent Cy5 labeling kit (Kreatech Diagnostics, The
Netherlands); and purified using DNA Clean and Concentrate spin columns according to manufacturer’s protocol (Zymo Research
Corporation, Orange CA).
MicroarraycDNA was hybridized to 4x44K Mouse Whole Genome Microarrays (Agilent) for 18 hr following the manufacturer’s protocol. Micro-
arrays were washed in 0.01x SSC/0.005% Triton X-102 at 40�C, dried with HEPA filtered compressed nitrogen and scanned on an
Agilent Technologies DNA Microarray Scanner at 5 mm resolution. Data were extracted from the scanned image using Agilent Tech-
nologies Feature Extraction Software, background subtracted, and normalized by the Lowess method (Yang et al., 2002). Microarray
files have been submitted to Array Express, accession numbers E-TABM-957 (WT + MNV CR6 and Atg16L1HM + MNV CR6) and E-
TABM-958 (WT uninfected, Atg16L1HM uninfected, WT + MNV CW3, and Atg16L1HM + MNV CW3). A factorial design approach
(Scholtens et. al., 2004) was implemented in the R programming language, in which a linear model (Smyth, 2005) was fitted with
a coefficient for each of the 4 factor combinations and then simultaneously extracted as contrasts for each of the three comparisons
(summarized in the Venn diagram of Figure 3) including the interaction (‘differential-of-differential’) term. Sets of differentially ex-
pressed genes (p < 0.05 and fold-change > 1.5) for each comparison were examined for enrichment in terms of biological process
or molecular function categories compared to their representation in the whole genome using the Panther classification system (Mi
et al., 2007). Enrichment p-values were computed using the hypergeometric distribution and implemented in the R language.
Cell 141, 1135–1145, June 25, 2010 ª2010 Elsevier Inc. S1

SUPPLEMENTAL REFERENCES
Cadwell, K., Liu, J.Y., Brown, S.L., Miyoshi, H., Loh, J., Lennerz, J.K., Kishi, C., Kc, W., Carrero, J.A., Hunt, S., et al. (2008). A key role for autophagy and the
autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 456, 259–263.
Mi, H., Guo, N., Kejariwal, A., and Thomas, P.D. (2007). PANTHER version 6: protein sequence and function evolution data with expanded representation of bio-
logical pathways. Nucleic Acids Res. 35 (Database issue), D247–D252.
Pull, S.L., Doherty, J.M., Mills, J.C., Gordon, J.I., and Stappenbeck, T.S. (2005). Activated macrophages are an adaptive element of the colonic epithelial progen-
itor niche necessary for regenerative responses to injury. Proc. Natl. Acad. Sci. USA 102, 99–104.
Scholtens, D., Miron, A., Merchant, F.M., Miller, A., Miron, P.L., Iglehart, J.D., and Gentleman, R. (2004). Analyzing Factorial Designed Microarray Experiments. J.
Multivariate Anal. 90, 19–43.
Schreiber, R.D., Hicks, L.J., Celada, A., Buchmeier, N.A., and Gray, P.W. (1985). Monoclonal antibodies to murine gamma-interferon which differentially modulate
macrophage activation and antiviral activity. J. Immunol. 134, 1609–1618.
Sheehan, K.C., Ruddle, N.H., and Schreiber, R.D. (1989). Generation and characterization of hamster monoclonal antibodies that neutralize murine tumor
necrosis factors. J. Immunol. 142, 3884–3893.
Smyth, G.K. (2005). Limma: linear models for microarray data. In Bioinformatics and Computational Biology Solutions using R and Bioconductor, R. Gentleman,
V. Carey, S. Dudoit, R. Irizarry, and W. Huber, eds. (New York: Springer).
Stappenbeck, T.S., Mills, J.C., and Gordon, J.I. (2003). Molecular features of adult mouse small intestinal epithelial progenitors. Proc. Natl. Acad. Sci. USA 100,
1004–1009.
Yang, Y.H., Dudoit, S., Luu, P., Lin, D.M., Peng, V., Ngai, J., and Speed, T.P. (2002). Normalization for cDNA microarray data: a robust composite method ad-
dressing single and multiple slide systematic variation. Nucleic Acids Res. 30, e15.
S2 Cell 141, 1135–1145, June 25, 2010 ª2010 Elsevier Inc.
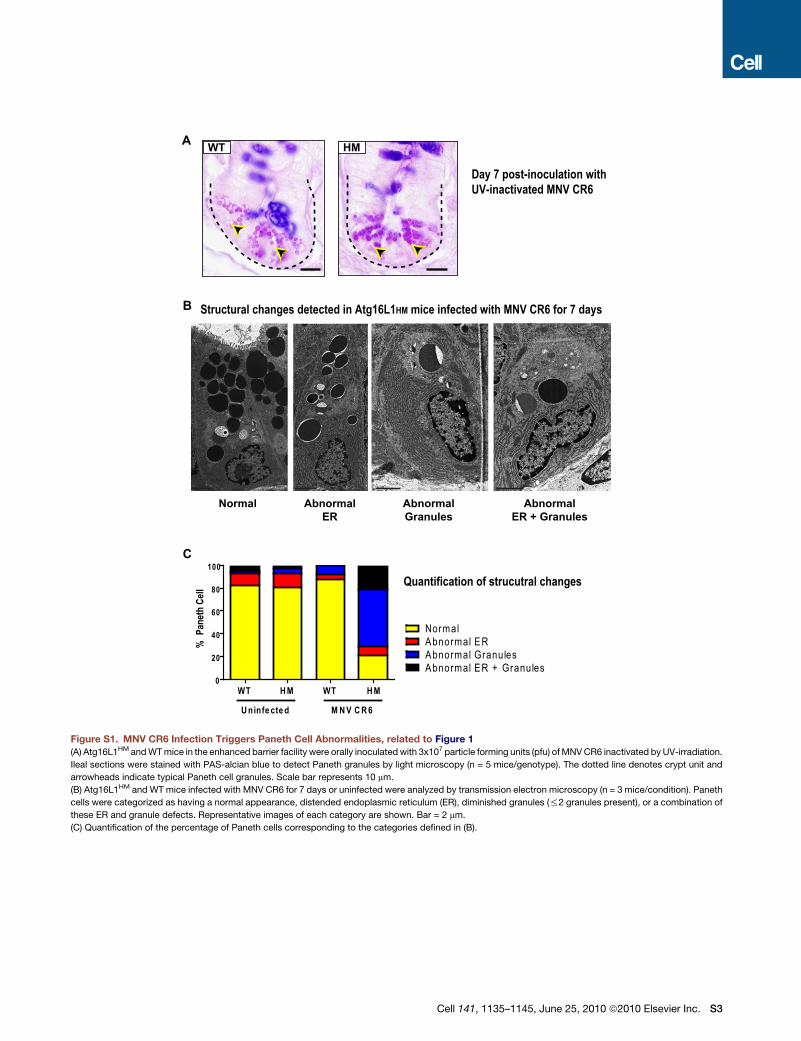
A
B
Normal Abnormal
Granules
Abnormal
ER
Abnormal
ER + Granules
0
20
40
60
80
100
NormalAbnormal ERAbnormal GranulesAbnormal ER + Granules
WT H M WT H M
U ninfe cte d M N V C R 6
% P
anet
h C
ell
Day 7 post-inoculation withUV-inactivated MNV CR6
WT HM
Structural changes detected in Atg16L1HM mice infected with MNV CR6 for 7 days
C
Quantification of strucutral changes
Figure S1. MNV CR6 Infection Triggers Paneth Cell Abnormalities, related to Figure 1
(A) Atg16L1HM and WT mice in the enhanced barrier facility were orally inoculated with 3x107 particle forming units (pfu) of MNV CR6 inactivated by UV-irradiation.
Ileal sections were stained with PAS-alcian blue to detect Paneth granules by light microscopy (n = 5 mice/genotype). The dotted line denotes crypt unit and
arrowheads indicate typical Paneth cell granules. Scale bar represents 10 mm.
(B) Atg16L1HM and WT mice infected with MNV CR6 for 7 days or uninfected were analyzed by transmission electron microscopy (n = 3 mice/condition). Paneth
cells were categorized as having a normal appearance, distended endoplasmic reticulum (ER), diminished granules (%2 granules present), or a combination of
these ER and granule defects. Representative images of each category are shown. Bar = 2 mm.
(C) Quantification of the percentage of Paneth cells corresponding to the categories defined in (B).
Cell 141, 1135–1145, June 25, 2010 ª2010 Elsevier Inc. S3
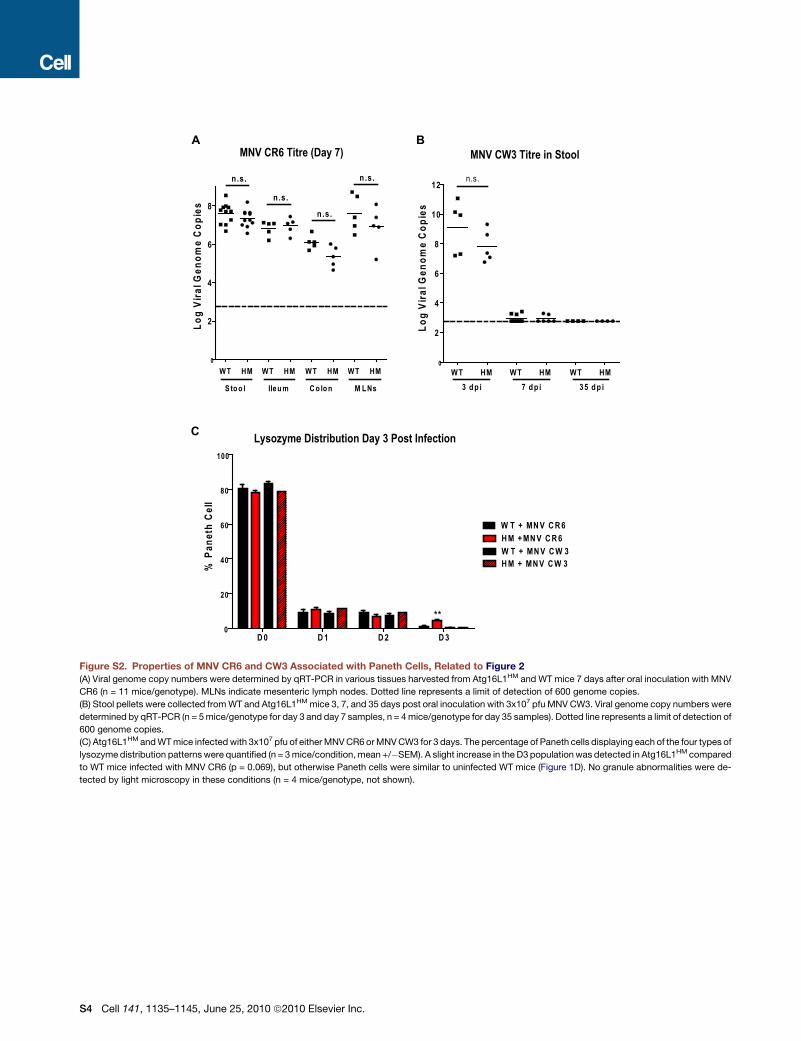
A B
0
2
4
6
8
WT HM WT HM WT HM WT HM
n.s.
n .s.
n .s.
n .s.
S to o l Ileu m C o lo n M L Ns
Lo
g V
iral
Gen
om
e C
op
ies
0
2
4
6
8
10
12n.s .
WT HM WT HM WT HM
3 d p i 7 d p i 35 d p i
Lo
g V
iral
Gen
om
e C
op
ies
C
D 0 D 1 D 2 D 30
20
40
60
80
100
W T + MN V C R 6
H M + MN V C R 6
W T + MN V C W 3H M + MN V C W 3%
Pan
eth
Cel
l
**
Lysozyme Distribution Day 3 Post Infection
MNV CR6 Titre (Day 7) MNV CW3 Titre in Stool
Figure S2. Properties of MNV CR6 and CW3 Associated with Paneth Cells, Related to Figure 2
(A) Viral genome copy numbers were determined by qRT-PCR in various tissues harvested from Atg16L1HM and WT mice 7 days after oral inoculation with MNV
CR6 (n = 11 mice/genotype). MLNs indicate mesenteric lymph nodes. Dotted line represents a limit of detection of 600 genome copies.
(B) Stool pellets were collected from WT and Atg16L1HM mice 3, 7, and 35 days post oral inoculation with 3x107 pfu MNV CW3. Viral genome copy numbers were
determined by qRT-PCR (n = 5 mice/genotype for day 3 and day 7 samples, n = 4 mice/genotype for day 35 samples). Dotted line represents a limit of detection of
600 genome copies.
(C) Atg16L1HM and WT mice infected with 3x107 pfu of either MNV CR6 or MNV CW3 for 3 days. The percentage of Paneth cells displaying each of the four types of
lysozyme distribution patterns were quantified (n = 3 mice/condition, mean +/�SEM). A slight increase in the D3 population was detected in Atg16L1HM compared
to WT mice infected with MNV CR6 (p = 0.069), but otherwise Paneth cells were similar to uninfected WT mice (Figure 1D). No granule abnormalities were de-
tected by light microscopy in these conditions (n = 4 mice/genotype, not shown).
S4 Cell 141, 1135–1145, June 25, 2010 ª2010 Elsevier Inc.

4932438A13Rik_29881_zoneFAbca13_17292_zoneFOas1a_17392_zoneFOasl2_39261_zoneFD5Wsu178e_19585_zoneFMgll_17073_zoneFPrr8_14020_zoneFRmi1_36673_zoneFCamta1_40861_zoneFEct2_9211_zoneFCcpg1_40046_zoneFAcsl1_21776_zoneFFam3c_6701_zoneFPrps2_6911_zoneFFnip2_14653_zoneFKlhl22_13959_zoneFNcoa4_35500_zoneFDnm1l_28024_zoneFHspbap1_22892_zoneFPkn2_25979_zoneF1110038D17Rik_29024_zoneFRnf139_365_zoneFDok7_36970_zoneFShisa2_6111_zoneF5730601F06Rik_26834_zoneFPapola_23141_zoneFSipa1l3_7324_zoneF
1 2 3
-4 1:1 4
Fold-change
WT infected vs. uninfected
Atg16L1HM infected vs. uninfected
‘Differential-of-differential’(Atg16L1HM infected vs. uninfected)
vs. (WT infected vs. uninfected)
Figure S3. Genes that Display Similar Changes in Expression in Paneth Cells from WT and Atg16L1 Mutant Mice in Response to MNV CR6,
Related to Figure 3Heatmap of genes represented in Zone F of the Venn diagram from Figure 3. Changes in gene expression within each comparison are represented as Log2-trans-
formed fold-changes (p < 0.05, fold-change > 1.5).
Cell 141, 1135–1145, June 25, 2010 ª2010 Elsevier Inc. S5
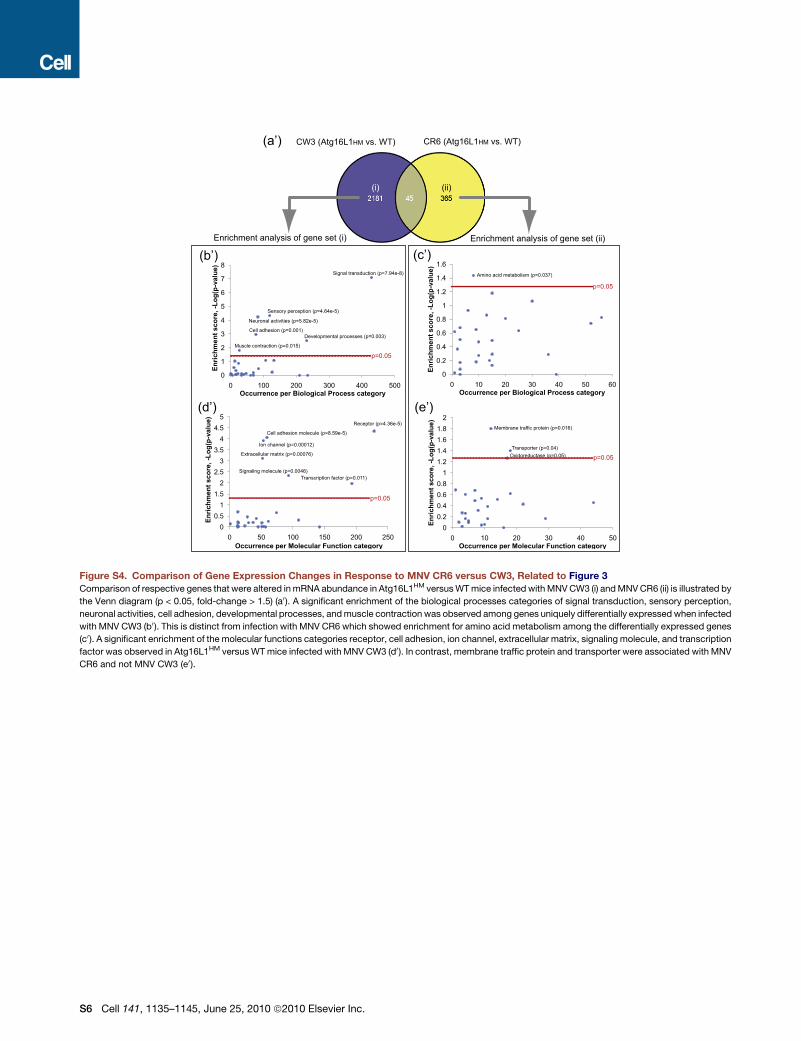
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
0 10 20 30 40 50 60
En
rich
men
t sco
re
,-L
og
(p
-va
lu
e)
Occurrence per Biological Process category
0
1
2
3
4
5
6
7
8
0 100 200 300 400 500
En
ric
hm
en
t s
co
re
,-L
og
(p
-valu
e)
Occurrence per Biological Process category
00.20.40.60.8
11.21.41.61.8
2
0 10 20 30 40 50
En
ric
hm
en
t s
co
re
,-L
og
(p
-va
lu
e)
Occurrence per Molecular Function category
00.5
11.5
22.5
33.5
44.5
5
0 50 100 150 200 250
En
ric
hm
en
t s
co
re
,-L
og
(p
-va
lu
e)
Occurrence per Molecular Function category
CW3 (Atg16L1HM vs. WT) CR6 (Atg16L1HM vs. WT)
Enrichment analysis of gene set (i) Enrichment analysis of gene set (ii)
Receptor (p=4.36e-5)
Cell adhesion molecule (p=8.59e-5)
Ion channel (p=0.00012)
Extracellular matrix (p=0.00076)
Signaling molecule (p=0.0046)Transcription factor (p=0.011)
Signal transduction (p=7.94e-8)
Sensory perception (p=4.64e-5)
Neuronal activities (p=5.82e-5)
Cell adhesion (p=0.001)
Muscle contraction (p=0.015)
Developmental processes (p=0.003)
Amino acid metabolism (p=0.037)
Membrane traffic protein (p=0.016)
Transporter (p=0.04)Oxidoreductase (p=0.05)
(i) (ii)
(a’)
p=0.05
p=0.05
p=0.05
p=0.05
(b’) (c’)
(d’) (e’)
Figure S4. Comparison of Gene Expression Changes in Response to MNV CR6 versus CW3, Related to Figure 3
Comparison of respective genes that were altered in mRNA abundance in Atg16L1HM versus WT mice infected with MNV CW3 (i) and MNV CR6 (ii) is illustrated by
the Venn diagram (p < 0.05, fold-change > 1.5) (a0). A significant enrichment of the biological processes categories of signal transduction, sensory perception,
neuronal activities, cell adhesion, developmental processes, and muscle contraction was observed among genes uniquely differentially expressed when infected
with MNV CW3 (b0 ). This is distinct from infection with MNV CR6 which showed enrichment for amino acid metabolism among the differentially expressed genes
(c0). A significant enrichment of the molecular functions categories receptor, cell adhesion, ion channel, extracellular matrix, signaling molecule, and transcription
factor was observed in Atg16L1HM versus WT mice infected with MNV CW3 (d0). In contrast, membrane traffic protein and transporter were associated with MNV
CR6 and not MNV CW3 (e0).
S6 Cell 141, 1135–1145, June 25, 2010 ª2010 Elsevier Inc.
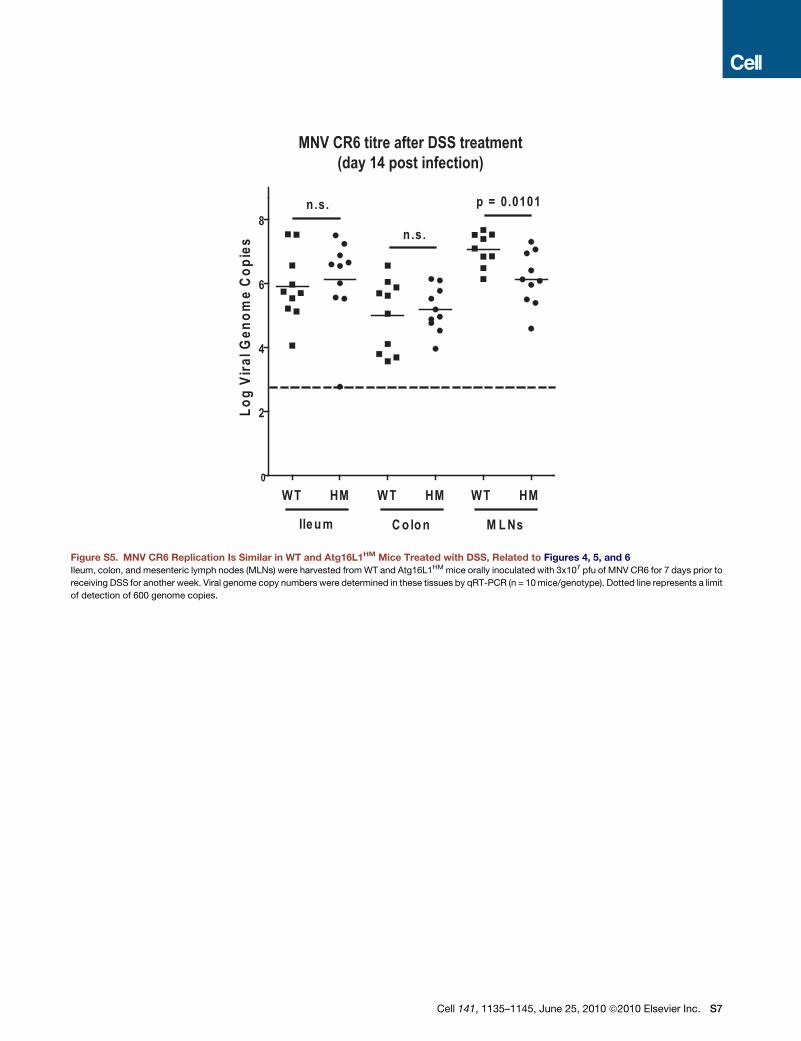
0
2
4
6
8
WT HM WT HM
n.s.
n .s.
Ileu m C o lo n
p = 0.0101
WT HM
M L Ns
Log
Vira
l Gen
ome
Cop
ies
MNV CR6 titre after DSS treatment(day 14 post infection)
Figure S5. MNV CR6 Replication Is Similar in WT and Atg16L1HM Mice Treated with DSS, Related to Figures 4, 5, and 6Ileum, colon, and mesenteric lymph nodes (MLNs) were harvested from WT and Atg16L1HM mice orally inoculated with 3x107 pfu of MNV CR6 for 7 days prior to
receiving DSS for another week. Viral genome copy numbers were determined in these tissues by qRT-PCR (n = 10 mice/genotype). Dotted line represents a limit
of detection of 600 genome copies.
Cell 141, 1135–1145, June 25, 2010 ª2010 Elsevier Inc. S7
Copyright © 2022 FDOKUMEN




















