
Viral 5′-triphosphate RNA and non-CpG DNA aggravate autoimmunity and lupus nephritis via distinct...
12
Viral 5 0 -triphosphate RNA and non-CpG DNA aggravate autoimmunity and lupus nephritis via distinct TLR-independent immune responses Ramanjaneyulu Allam 1 , Rahul D. Pawar 1 , Onkar P. Kulkarni 1 , Veit Hornung 2 , Gunter Hartmann 2 , Stephan Segerer 1 , Shizuo Akira 3 , Stefan Endres 4 and Hans-Joachim Anders 1 1 Medical Policlinic, University of Munich, Germany 2 Division of Clinical Pharmacology, Department of Medicine, University of Bonn, Bonn, Germany 3 Department of Host Defense, Research Institute for Microbial Diseases, Osaka University, Osaka, Japan 4 Department of Clinical Pharmacology, University of Munich, Munich, Germany Certain viral nucleic acids aggravate autoimmunity through nucleic acid-specific TLR. Viral 5 0 -triphosphate RNA (3P-RNA) and double-stranded non-CpG DNA induce antiviral immunity via TLR-independent pathways but their role in autoimmunity is unknown. Transient exposure of 16-wk-old MRL lpr/lpr mice to 3P-RNA aggravated lupus nephritis by increasing IFN signaling and decreasing CD4 1 CD25 1 T cells. By contrast, transient exposure to non-CpG DNA exacerbate lupus nephritis in association with splenomegaly, lymphoproliferation, hypergammaglobulinaemia and increased B220 1 CD138 1 plasma cells. Both, 3P-RNA and non-CpG DNA increased glomerular complement factor C3c deposits but both nucleic acid formats were less potent in aggravating renal pathology as compared with CpG DNA. 3P-RNA and non-CpG DNA also localized to the glomerular mesangial cells and activated cultured mesangial cells to produce IL-6. We conclude, 3P-RNA or non-CpG DNA both trigger autoimmune disease in MRL lpr/lpr mice by specifi- cally activating adaptive immunity but similarly enhance inflammation on the tissue level. Key words: Autoimmunity . Innate immunity . Lupus . Lupus nephritis . TLR Introduction Viral infection can aggravate disease activity of autoimmune diseases like systemic lupus erythematosus (SLE) but the molecular mechanisms remain elusive. Antiviral immune responses might drive autoimmune tissue injury via type I IFN, Th1 cytokines or CD8 1 T cells as these mediators regulate both conditions [1, 2]. Antiviral immunity is triggered by conserved viral nucleic acid formats via two classes of innate recognition receptors. These are the nucleic acid-specific TLR, i.e. TLR3, TLR7, TLR8 and TLR9, which detect viral RNA (TLR3, TLR7, TLR8) or viral CpG DNA (TLR9) in intracellular endosomes of antigen-presenting cells [3–7]. Signaling via these nucleic acid-specific TLR involves the intracellular adaptor molecules myeloid differentiation primary-response protein (MyD)-88 (TLR7, TLR8, TLR9) or translation initiation region domain- containing adaptor protein-inducing-IFN beta TRIF (TLR3) and subsequent activation of IFN-regulatory factors (IRF) and NF-kB [3]. The second class of viral recognition receptors includes the retinoic-acid-inducible gene (RIG)-I, and melanoma-differentiation-associated gene (MDA)-5 that detect viral RNA in the intracellular cytosol and induce type 1 IFN via Correspondence: Dr. H.-J. Anders e-mail: [email protected] & 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu Eur. J. Immunol. 2008. 38: 3487–3498 DOI 10.1002/eji.200838604 Innate immunity 3487
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Viral 5′-triphosphate RNA and non-CpG DNA aggravate autoimmunity and lupus nephritis via distinct...

Viral 50-triphosphate RNA and non-CpG DNA aggravateautoimmunity and lupus nephritis via distinctTLR-independent immune responses
Ramanjaneyulu Allam1, Rahul D. Pawar1, Onkar P. Kulkarni1,
Veit Hornung2, Gunter Hartmann2, Stephan Segerer1, Shizuo Akira3,
Stefan Endres4 and Hans-Joachim Anders1
1 Medical Policlinic, University of Munich, Germany2 Division of Clinical Pharmacology, Department of Medicine, University of Bonn, Bonn,
Germany3 Department of Host Defense, Research Institute for Microbial Diseases, Osaka University,
Osaka, Japan4 Department of Clinical Pharmacology, University of Munich, Munich, Germany
Certain viral nucleic acids aggravate autoimmunity through nucleic acid-specific TLR.
Viral 50-triphosphate RNA (3P-RNA) and double-stranded non-CpG DNA induce antiviral
immunity via TLR-independent pathways but their role in autoimmunity is unknown.
Transient exposure of 16-wk-old MRLlpr/lpr mice to 3P-RNA aggravated lupus nephritis by
increasing IFN signaling and decreasing CD41CD251 T cells. By contrast, transient
exposure to non-CpG DNA exacerbate lupus nephritis in association with splenomegaly,
lymphoproliferation, hypergammaglobulinaemia and increased B2201CD1381 plasma
cells. Both, 3P-RNA and non-CpG DNA increased glomerular complement factor C3c
deposits but both nucleic acid formats were less potent in aggravating renal pathology as
compared with CpG DNA. 3P-RNA and non-CpG DNA also localized to the glomerular
mesangial cells and activated cultured mesangial cells to produce IL-6. We conclude,
3P-RNA or non-CpG DNA both trigger autoimmune disease in MRLlpr/lpr mice by specifi-
cally activating adaptive immunity but similarly enhance inflammation on the tissue level.
Key words: Autoimmunity . Innate immunity . Lupus . Lupus nephritis . TLR
Introduction
Viral infection can aggravate disease activity of autoimmune
diseases like systemic lupus erythematosus (SLE) but the
molecular mechanisms remain elusive. Antiviral immune
responses might drive autoimmune tissue injury via type I IFN,
Th1 cytokines or CD81 T cells as these mediators regulate both
conditions [1, 2]. Antiviral immunity is triggered by conserved
viral nucleic acid formats via two classes of innate recognition
receptors. These are the nucleic acid-specific TLR, i.e. TLR3,
TLR7, TLR8 and TLR9, which detect viral RNA (TLR3, TLR7,
TLR8) or viral CpG DNA (TLR9) in intracellular endosomes
of antigen-presenting cells [3–7]. Signaling via these nucleic
acid-specific TLR involves the intracellular adaptor molecules
myeloid differentiation primary-response protein (MyD)-88
(TLR7, TLR8, TLR9) or translation initiation region domain-
containing adaptor protein-inducing-IFN beta TRIF (TLR3)
and subsequent activation of IFN-regulatory factors (IRF)
and NF-kB [3]. The second class of viral recognition
receptors includes the retinoic-acid-inducible gene (RIG)-I, and
melanoma-differentiation-associated gene (MDA)-5 that detect
viral RNA in the intracellular cytosol and induce type 1 IFN viaCorrespondence: Dr. H.-J. Anderse-mail: [email protected]
& 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu
Eur. J. Immunol. 2008. 38: 3487–3498 DOI 10.1002/eji.200838604 Innate immunity 3487

the mitochondrial IFN-b promoter stimulator (IPS)-1. These
helicases have been identified to recognize viral dsRNA [8] and
single-stranded 50-triphosphate (3P)-RNA in the cytosol, respec-
tively [9, 10]. In addition, recently identified cytosolic DNA
receptor DAI and yet unknown cytosolic DNA receptors must
exist to mediate the immunostimulatory effects of viral dsDNA
[11–14]. TLR7, TLR8 and TLR9 are only expressed in a narrow
subset of immune cells, while almost all nucleated cells can
activate antiviral responses upon viral infection [15]. In fact,
TLR3 and TLR-independent recognition of viral RNA or dsDNA is
a potent inducer of type I IFN and proinflammatory cytokines in
non-immune cells [12, 16–19].
While the role of cytosolic and TLR-independent nucleic acid
recognition for antiviral immunity becomes increasingly recog-
nized, its potential role in activating disease activity of auto-
immunity and autoimmune tissue injury, similar to what has
been documented for the nucleic acid-specific TLR, remains
unknown [20–22]. We therefore hypothesized that exposure to
viral 3P-RNA or non-CpG DNA would modulate autoimmune
tissue injury in vivo and we questioned whether this would be
mediated via similar or different types of immune responses.
Results
3P-RNA and non-CpG DNA induce IL-6 independent ofMyd88 in vivo
Innate RNA and DNA recognition can involve TLR-dependent or
independent pathways [3]. Triphosphate RNA(3P-RNA) and non-
CpG DNA have both been reported to trigger cytokine release via
TLR-independent pathways in cultured DC [9, 11]. We replicated
the same results from cultured Myd88�/� and TLR9�/� BMDC
(data not shown). To extend these results in vivo we compared
serum IL-6 levels before and 6 h after a single injection of 3P-RNA
or non-CpG DNA to Myd88-deficient and wild-type mice. We
found that Myd88 was not required to induce IL-6 with either of
these two ligands in vivo (Fig. 1A).
Non-CpG DNA induces serum cytokines in nephriticMRLlpr/lpr mice
In order to test the effects of transient exposure to 3P-RNA and
non-CpG DNA in mice with pre-existing autoimmune disease we
repeatedly injected 16-wk-old nephritic MRLlpr/lpr mice with
3P-RNA and non-CpG DNA. Pilot studies with different doses of
3P-RNA and non-CpG DNA revealed that a single dose of either
20mg 3P-RNA or 50mg non-CpG DNA, but not higher doses,
potently increased IL-6 or CCL2 serum levels in 16-wk-old
MRLlpr/lpr mice (Fig. 1B). As this response was not dose-dependent
we assumed the doses tested to be at the plateau-level of the dose
response. Based on these data we injected 16-wk-old MRLlpr/lpr
mice with either 5% glucose, 20mg of 3P-RNA, 50mg of non-CpG
DNA or 40mg CpG DNA for 2 wk. Serum cytokine levels were
determined 3 h after the last injection. Only non-CpG DNA
significantly increased serum TNF and IL-12 levels at 18 wk,
although not as high as compared with CpG DNA (Fig. 2). By
contrast, 3P-RNA and non-CpG DNA had no effect on levels of
IFN-a and IFN-g, but IFN-g was significantly induced by CpG DNA
(Fig. 2). Together, 3P-RNA and non-CpG DNA are less potent than
CpG DNA to increase serum levels of TNF, IFN-a, IFN-g, IL-12, IL-6
and TNF in nephritic MRLlpr/lpr mice.
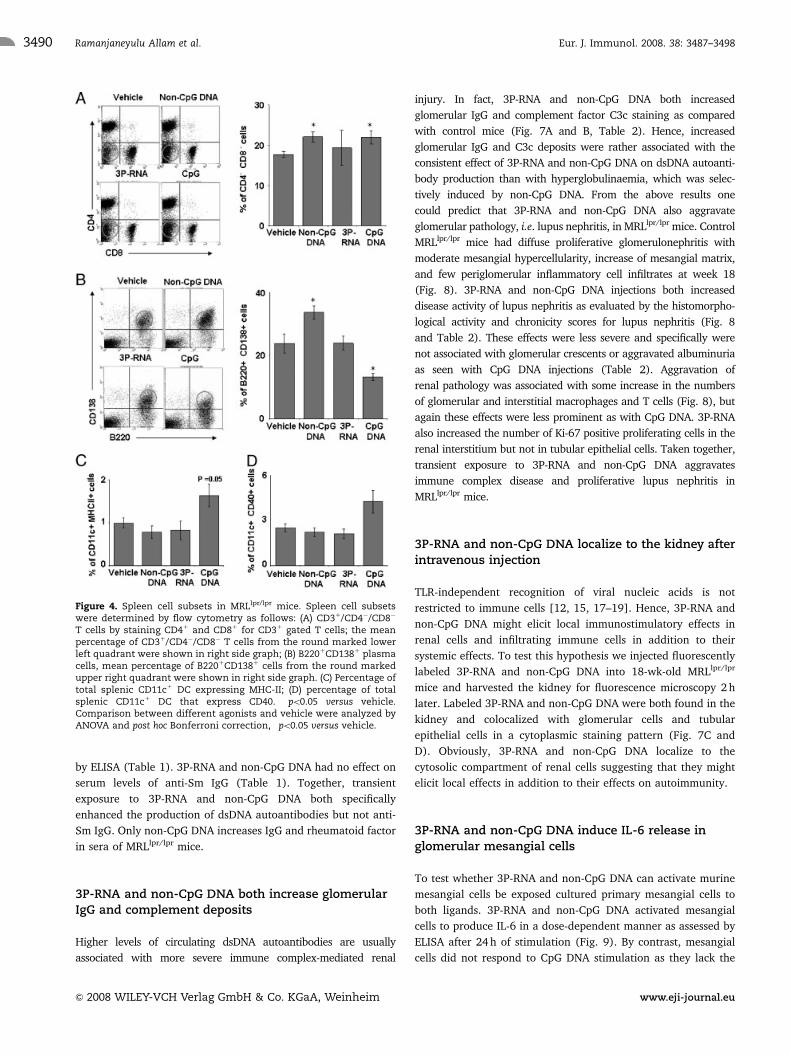
Non-CpG DNA but not 3P-RNA induces lympho-proliferation in MRLlpr/lpr mice
Repeated exposure to CpG DNA can trigger significant lympho-
proliferation [23], hence, we assessed the weight of spleens
and mesenteric lymphnodes at the time of sacrifice in all mice.
Non-CpG DNA but not 3P-RNA increased the weight of spleen
and mesenteric lymphnodes in MRLlpr/lpr mice to the same extent
Figure 1. Serum IL-6 response in Myd88 knockout mice and dosestudies in MRLlpr/lpr mice. (A) C57BL/6 mice with (black bars) or withoutMyD88 (white bars) were injected with a single dose of 3P-RNA, non-CpG DNA or CpG DNA. After 6 h, serum samples were obtained and IL-6levels were determined by ELISA. Values are means7SEM from threemice in each group, each performed in duplicate. Note that non-CpGDNA and 3P-RNA injected Myd88-deficient mice significantly increaseserum IL-6 levels 6 h after injection as compared with time point zerobut no response to CpG DNA. (B) Different concentrations of 3P-RNAand non-CpG DNA were injected intraperitoneally into 12-wk-oldMRLlpr/lpr mice as indicated. Serum IL-6 (B) and CCL2 (C) weremeasured at 6 h after injection by ELISA. Data represent means7SEMfrom four mice in each group. Comparison between different doseswere analyzed by ANOVA and post hoc Bonferroni correction, �po0.05.N.d. 5 not detectable.
Eur. J. Immunol. 2008. 38: 3487–3498Ramanjaneyulu Allam et al.3488
& 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

as CpG DNA (Fig. 3A and B). Non-CpG DNA increased the
percentage of splenic CD4/CD8 double negative T cells and
B220/CD138 double positive plasma cells in MRLlpr/lpr mice
(Fig. 4A and B). Furthermore, we assessed the activation of
CD11c1 DC by flow cytometry for MHC class II and CD40 surface
expression. 3P-RNA and non-CpG DNA did not increase surface
expression of MHC II and CD40 as it was observed with CpG DNA
(Fig. 4C and D). Thus, non-CpG DNA but not 3P-RNA induces
lymphoproliferation in nephritic MRLlpr/lpr mice.
Spleen mRNA expression of inflammatory mediatorsand transcription factors
How do 3P-RNA and non-CpG DNA affect the expression of
inflammatory mediators? 3P-RNA but not non-CpG DNA induced
Mx1, a marker of type I IFN signaling, in spleens of MRLlpr/lpr
mice, while IFN-g and TNF mRNA were not induced (Fig. 5).
Neither 3P-RNA nor non-CpG DNA induced IL-6 and CCL2 mRNA
as seen with CpG DNA (Fig. 5). The mRNA levels of TBX21 and
GATA2, respective markers of Th1 or Th2 responses, were rather
decreased upon 3P-RNA exposure (Fig. 5). 3P-RNA and non-CpG
DNA did not significantly affect the mRNA expression of RIG-I,
MDA5 (and DAI, data not shown) but both nucleic acids reduced
IPS-1 mRNA levels (Fig. 5). 3P-RNA suppressed Foxp3 mRNA,
crucial regulator in the development and function of regulatory
T cells [24]. This was consistent with reduced numbers of CD4/
CD251 T cells in 3P-RNA-treated MRLlpr/lpr mice (Table 1). IL-15
mRNA levels were not altered by any of the three ligands (Fig. 5).
Do 3P-RNA and non-CpG DNA modulate Th17-dependent
autoimmunity [25, 26]? Both, the Th17-related factors Rorc
and IL-23a, were found not to be induced in spleens of MRLlpr/lpr
mice (data not shown). Thus, 3P-RNA induces type I IFN-
dependent MX1 signaling and impairs Foxp3-driven regulatory T
cells in spleens of MRLlpr/lpr mice.
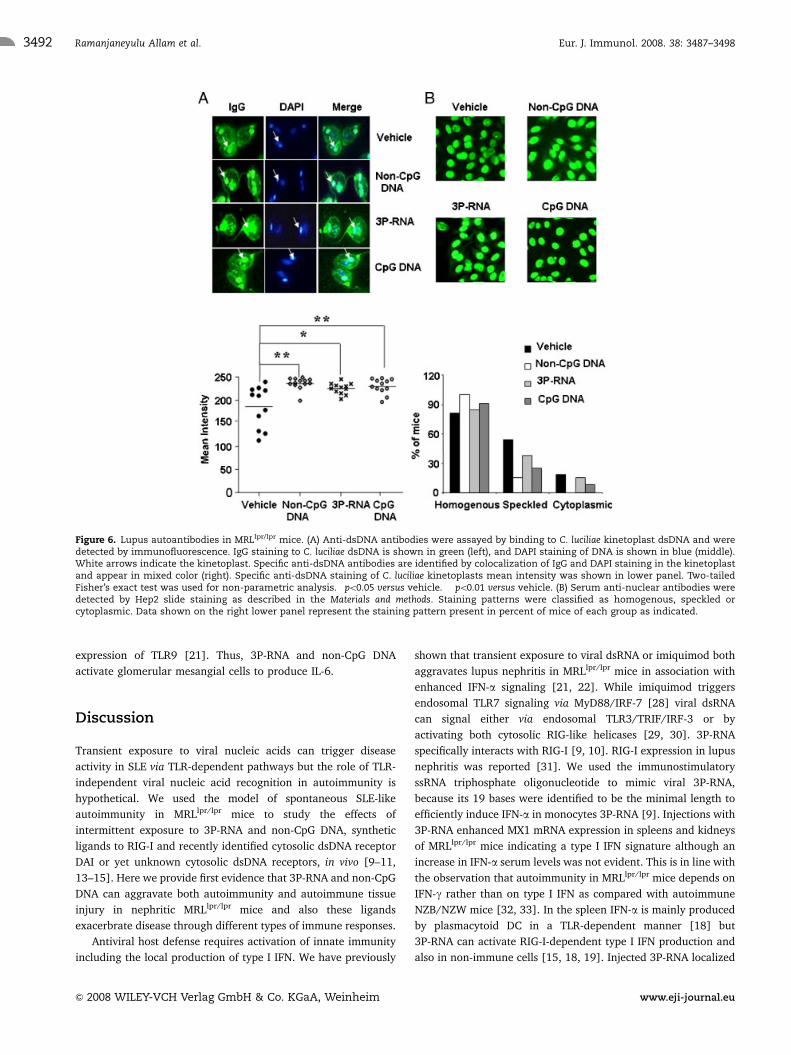
Hypergammaglobulinaemia and DNA autoantibodiesin MRLlpr/lpr mice
TLR signaling contributes to B-cell activation and the production
of selected autoantibodies in lupus [27]. Hence, we meticulously
analyzed the effects of 3P-RNA and non-CpG DNA on serum IgG
levels and a number of common lupus autoantibodies. Consistent
with its effect on spleen plasma cells only non-CpG DNA
significantly increased serum levels of total IgG including all
isotypes and that of rheumatoid factor (Table 1). Critidiae luciliae
kinetoplast dsDNA binding was used to detect and quantify anti-
dsDNA IgG. A digital quantitative analysis of C. luciliae
kinetoplast positivity revealed that both 3P-RNA- and non-CpG
DNA-injected MRLlpr/lpr mice had little but statistically significant
higher levels of anti-dsDNA IgG as compared with vehicle
controls (Fig. 6A). This was associated with increased homo-
genous ANA staining pattern on Hep2 cells (Fig. 6B). Increased
anti-dsDNA IgG antibodies were observed with both ligands also
Figure 2. Serum cytokine levels in MRLlpr/lpr mice. Serum cytokinelevels of 18-wk-old MRLlpr/lpr mice for TNF, IFN-g, IL-6, IL-12, IFN-a weremeasured by ELISA. Data represent means7SEM from 10 to 12 mice ineach group. Comparison between different agonists and vehicle wereanalyzed by ANOVA and post hoc Bonferroni correction, �po0.05 versusvehicle.
Figure 3. Spleen and lymphnodes in MRLlpr/lpr mice. Weights ofspleens (A) and of mesenterial lymphnodes (B) were determined inall mice at the end of the study. Comparison between differentagonists and vehicle were analyzed by ANOVA and post hoc Bonferronicorrection, �po0.05 versus vehicle, ��po0.01 versus vehicle.
Eur. J. Immunol. 2008. 38: 3487–3498 Innate immunity 3489
& 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu
by ELISA (Table 1). 3P-RNA and non-CpG DNA had no effect on
serum levels of anti-Sm IgG (Table 1). Together, transient
exposure to 3P-RNA and non-CpG DNA both specifically
enhanced the production of dsDNA autoantibodies but not anti-
Sm IgG. Only non-CpG DNA increases IgG and rheumatoid factor
in sera of MRLlpr/lpr mice.
3P-RNA and non-CpG DNA both increase glomerularIgG and complement deposits
Higher levels of circulating dsDNA autoantibodies are usually
associated with more severe immune complex-mediated renal
injury. In fact, 3P-RNA and non-CpG DNA both increased
glomerular IgG and complement factor C3c staining as compared
with control mice (Fig. 7A and B, Table 2). Hence, increased
glomerular IgG and C3c deposits were rather associated with the
consistent effect of 3P-RNA and non-CpG DNA on dsDNA autoanti-
body production than with hyperglobulinaemia, which was selec-
tively induced by non-CpG DNA. From the above results one
could predict that 3P-RNA and non-CpG DNA also aggravate
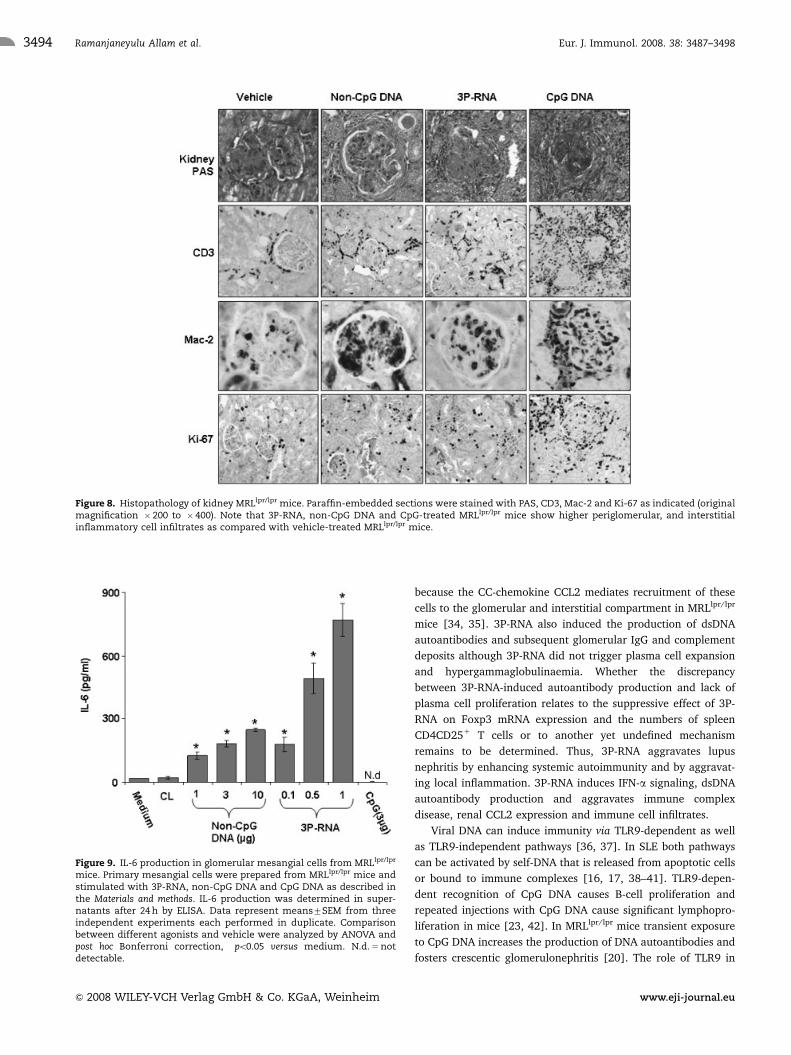
glomerular pathology, i.e. lupus nephritis, in MRLlpr/lpr mice. Control
MRLlpr/lpr mice had diffuse proliferative glomerulonephritis with
moderate mesangial hypercellularity, increase of mesangial matrix,
and few periglomerular inflammatory cell infiltrates at week 18
(Fig. 8). 3P-RNA and non-CpG DNA injections both increased
disease activity of lupus nephritis as evaluated by the histomorpho-
logical activity and chronicity scores for lupus nephritis (Fig. 8
and Table 2). These effects were less severe and specifically were
not associated with glomerular crescents or aggravated albuminuria
as seen with CpG DNA injections (Table 2). Aggravation of
renal pathology was associated with some increase in the numbers
of glomerular and interstitial macrophages and T cells (Fig. 8), but
again these effects were less prominent as with CpG DNA. 3P-RNA
also increased the number of Ki-67 positive proliferating cells in the
renal interstitium but not in tubular epithelial cells. Taken together,
transient exposure to 3P-RNA and non-CpG DNA aggravates
immune complex disease and proliferative lupus nephritis in
MRLlpr/lpr mice.
3P-RNA and non-CpG DNA localize to the kidney afterintravenous injection
TLR-independent recognition of viral nucleic acids is not
restricted to immune cells [12, 15, 17–19]. Hence, 3P-RNA and
non-CpG DNA might elicit local immunostimulatory effects in
renal cells and infiltrating immune cells in addition to their
systemic effects. To test this hypothesis we injected fluorescently
labeled 3P-RNA and non-CpG DNA into 18-wk-old MRLlpr/lpr
mice and harvested the kidney for fluorescence microscopy 2 h
later. Labeled 3P-RNA and non-CpG DNA were both found in the
kidney and colocalized with glomerular cells and tubular
epithelial cells in a cytoplasmic staining pattern (Fig. 7C and
D). Obviously, 3P-RNA and non-CpG DNA localize to the
cytosolic compartment of renal cells suggesting that they might
elicit local effects in addition to their effects on autoimmunity.
3P-RNA and non-CpG DNA induce IL-6 release inglomerular mesangial cells
To test whether 3P-RNA and non-CpG DNA can activate murine
mesangial cells be exposed cultured primary mesangial cells to
both ligands. 3P-RNA and non-CpG DNA activated mesangial
cells to produce IL-6 in a dose-dependent manner as assessed by
ELISA after 24 h of stimulation (Fig. 9). By contrast, mesangial
cells did not respond to CpG DNA stimulation as they lack the
Figure 4. Spleen cell subsets in MRLlpr/lpr mice. Spleen cell subsetswere determined by flow cytometry as follows: (A) CD31/CD4�/CD8�
T cells by staining CD41 and CD81 for CD31 gated T cells; the meanpercentage of CD31/CD4�/CD8� T cells from the round marked lowerleft quadrant were shown in right side graph; (B) B2201CD1381 plasmacells, mean percentage of B2201CD1381 cells from the round markedupper right quadrant were shown in right side graph. (C) Percentage oftotal splenic CD11c1 DC expressing MHC-II; (D) percentage of totalsplenic CD11c1 DC that express CD40. �po0.05 versus vehicle.Comparison between different agonists and vehicle were analyzed byANOVA and post hoc Bonferroni correction, �po0.05 versus vehicle.
Eur. J. Immunol. 2008. 38: 3487–3498Ramanjaneyulu Allam et al.3490
& 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu
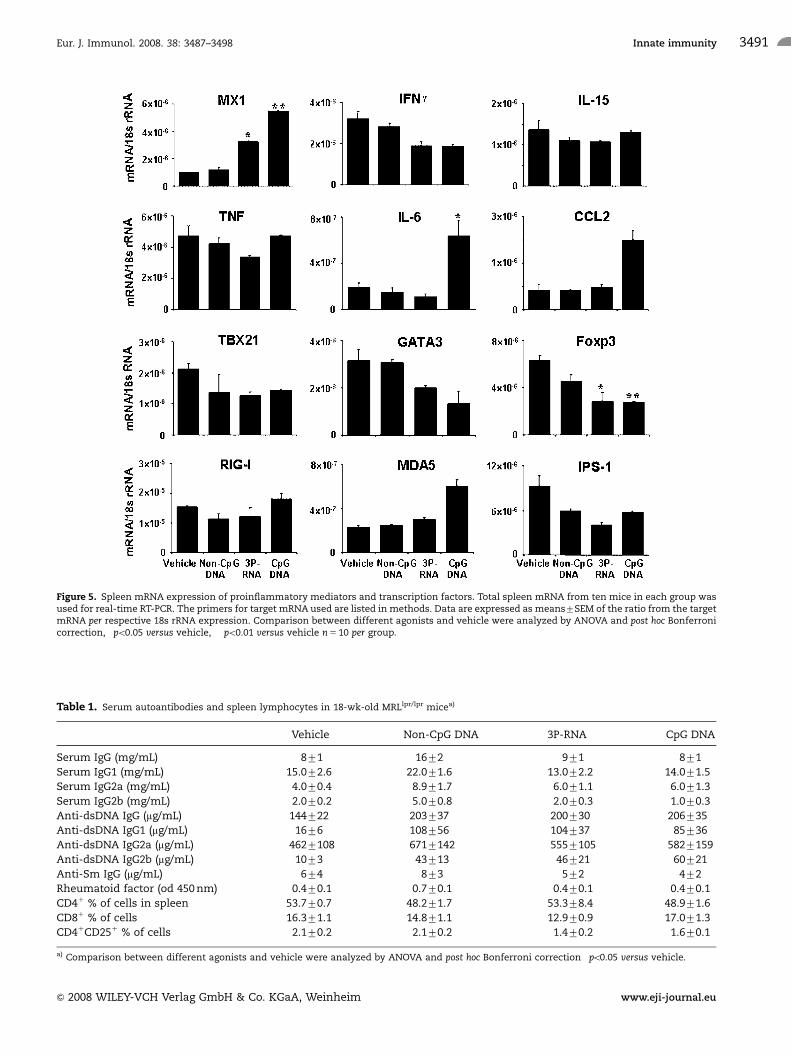
Figure 5. Spleen mRNA expression of proinflammatory mediators and transcription factors. Total spleen mRNA from ten mice in each group wasused for real-time RT-PCR. The primers for target mRNA used are listed in methods. Data are expressed as means7SEM of the ratio from the targetmRNA per respective 18s rRNA expression. Comparison between different agonists and vehicle were analyzed by ANOVA and post hoc Bonferronicorrection, �po0.05 versus vehicle, ��po0.01 versus vehicle n 5 10 per group.
Table 1. Serum autoantibodies and spleen lymphocytes in 18-wk-old MRLlpr/lpr micea)
Vehicle Non-CpG DNA 3P-RNA CpG DNA
Serum IgG (mg/mL) 871 1672�� 971 871
Serum IgG1 (mg/mL) 15.072.6 22.071.6� 13.072.2 14.071.5
Serum IgG2a (mg/mL) 4.070.4 8.971.7� 6.071.1 6.071.3
Serum IgG2b (mg/mL) 2.070.2 5.070.8� 2.070.3 1.070.3
Anti-dsDNA IgG (mg/mL) 144722 203737 200730 206735
Anti-dsDNA IgG1 (mg/mL) 1676 108756 104737� 85736
Anti-dsDNA IgG2a (mg/mL) 4627108 6717142 5557105 5827159
Anti-dsDNA IgG2b (mg/mL) 1073 43713� 46721 60721�
Anti-Sm IgG (mg/mL) 674 873 572 472
Rheumatoid factor (od 450 nm) 0.470.1 0.770.1� 0.470.1 0.470.1
CD41 % of cells in spleen 53.770.7 48.271.7 53.378.4 48.971.6
CD81 % of cells 16.371.1 14.871.1 12.970.9 17.071.3
CD41CD251 % of cells 2.170.2 2.170.2 1.470.2 1.670.1
a) Comparison between different agonists and vehicle were analyzed by ANOVA and post hoc Bonferroni correction �po0.05 versus vehicle.
Eur. J. Immunol. 2008. 38: 3487–3498 Innate immunity 3491
& 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu
expression of TLR9 [21]. Thus, 3P-RNA and non-CpG DNA
activate glomerular mesangial cells to produce IL-6.
Discussion
Transient exposure to viral nucleic acids can trigger disease
activity in SLE via TLR-dependent pathways but the role of TLR-
independent viral nucleic acid recognition in autoimmunity is
hypothetical. We used the model of spontaneous SLE-like
autoimmunity in MRLlpr/lpr mice to study the effects of
intermittent exposure to 3P-RNA and non-CpG DNA, synthetic
ligands to RIG-I and recently identified cytosolic dsDNA receptor
DAI or yet unknown cytosolic dsDNA receptors, in vivo [9–11,
13–15]. Here we provide first evidence that 3P-RNA and non-CpG
DNA can aggravate both autoimmunity and autoimmune tissue
injury in nephritic MRLlpr/lpr mice and also these ligands
exacerbrate disease through different types of immune responses.
Antiviral host defense requires activation of innate immunity
including the local production of type I IFN. We have previously
shown that transient exposure to viral dsRNA or imiquimod both
aggravates lupus nephritis in MRLlpr/lpr mice in association with
enhanced IFN-a signaling [21, 22]. While imiquimod triggers
endosomal TLR7 signaling via MyD88/IRF-7 [28] viral dsRNA
can signal either via endosomal TLR3/TRIF/IRF-3 or by
activating both cytosolic RIG-like helicases [29, 30]. 3P-RNA
specifically interacts with RIG-I [9, 10]. RIG-I expression in lupus
nephritis was reported [31]. We used the immunostimulatory
ssRNA triphosphate oligonucleotide to mimic viral 3P-RNA,
because its 19 bases were identified to be the minimal length to
efficiently induce IFN-a in monocytes 3P-RNA [9]. Injections with
3P-RNA enhanced MX1 mRNA expression in spleens and kidneys
of MRLlpr/lpr mice indicating a type I IFN signature although an
increase in IFN-a serum levels was not evident. This is in line with
the observation that autoimmunity in MRLlpr/lpr mice depends on
IFN-g rather than on type I IFN as compared with autoimmune
NZB/NZW mice [32, 33]. In the spleen IFN-a is mainly produced
by plasmacytoid DC in a TLR-dependent manner [18] but
3P-RNA can activate RIG-I-dependent type I IFN production and
also in non-immune cells [15, 18, 19]. Injected 3P-RNA localized
Figure 6. Lupus autoantibodies in MRLlpr/lpr mice. (A) Anti-dsDNA antibodies were assayed by binding to C. luciliae kinetoplast dsDNA and weredetected by immunofluorescence. IgG staining to C. luciliae dsDNA is shown in green (left), and DAPI staining of DNA is shown in blue (middle).White arrows indicate the kinetoplast. Specific anti-dsDNA antibodies are identified by colocalization of IgG and DAPI staining in the kinetoplastand appear in mixed color (right). Specific anti-dsDNA staining of C. luciliae kinetoplasts mean intensity was shown in lower panel. Two-tailedFisher’s exact test was used for non-parametric analysis. �po0.05 versus vehicle. ��po0.01 versus vehicle. (B) Serum anti-nuclear antibodies weredetected by Hep2 slide staining as described in the Materials and methods. Staining patterns were classified as homogenous, speckled orcytoplasmic. Data shown on the right lower panel represent the staining pattern present in percent of mice of each group as indicated.
Eur. J. Immunol. 2008. 38: 3487–3498Ramanjaneyulu Allam et al.3492
& 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

to glomerular cells and tubular epithelial cells in kidneys of
MRLlpr/lpr mice cytosolic staining pattern and cultured mesangial
cells produced IL-6 upon 3P-RNA and non-CpG DNA stimulation.
Consistently, RIG-I expression was reported to localize to
mesangial cells in human lupus nephritis [32]. Kidneys
of 3P-RNA-injected mice expressed increased levels of CCL2 (data
not shown), which was consistent with the moderate increase in
glomerular and interstitial macrophage and T-cell infiltrates,
Figure 7. Glomerular immune complex deposits and distribution of 3P-RNA and non-CpG DNA in kidneys of MRLlpr/lpr mice. (A) Immunostainingfor total IgG was performed on paraffin-embedded renal sections from of 18-wk-old MRLlpr/lpr mice of all groups. Original magnification � 400.(B) Frozen sections of the same kidneys were used for immunostaining for complement factor C3 with a FITC-labeled antibody (green). Cell nucleiare stained with DAPI (blue). Original magnification � 400. (C) and (D) Rhodamine-labeled 3P-RNA and non-CpG DNA were intravenously injectedinto 16-wk-old MRLlpr/lpr mice. Renal tissue was harvested 2 h later and frozen sections underwent fluorescence microscopy. Rhodamin-labelednucleic acids appear in red. FITC phalloidin staining appears in green and DAPI-stained cell nuclei are blue. Glomeruli are encircled. Originalmagnification �200.
Table 2. Parameters of lupus nephritis in 18-wk-old MRLlpr/lpr micea)
Vehicle Non-CpG DNA 3P-RNA CpG DNA
Proteinuria (mg/mg)
Albumin/creatinine ratio 33711 37714 67739 108755
Glom. deposit scores (0–3)
IgG 1.270.1 1.570.1 1.970.2 2.170.2�
C3c 0.870.1 1.570.1� 1.870.1� 1.870.1�
Histological scores
Activity index 6.371.5 12.472.0� 11.071.7� 15.471.7�
Chronicity index 0.670.5 2.570.9� 1.870.6� 3.670.9�
Cellular response (cells/glom. or hpf)
Glom. Mac21 (cells/glom.) 6.970.9 8.570.8� 10.170.9� 15.371.6�
Glom. CD31 (cells/glom.) 0.770.1 1.470.2� 1.270.2 2.270.2�
Glom. Ki671 (cells/glom.) 2.370.4 2.570.2 2.170.2 4.670.2�
Interst. Mac21 (cells/hpf) 8.071.3 10.371.0 17.473.4 29.374.6�
Interst. CD31 (cells/hpf) 11.671.8 16.071.9 17.173.0 34.674.1�
Interst. Ki671 (cells/hpf) 5.871.4 6.971.2 10.471.6 35.371.5�
Tubular Ki671 (cells/hpf) 13.973.1 12.571.7 10.571.4 40.273.8�
a) �po0.05 versus vehicle.
Eur. J. Immunol. 2008. 38: 3487–3498 Innate immunity 3493
& 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu
because the CC-chemokine CCL2 mediates recruitment of these
cells to the glomerular and interstitial compartment in MRLlpr/lpr
mice [34, 35]. 3P-RNA also induced the production of dsDNA
autoantibodies and subsequent glomerular IgG and complement
deposits although 3P-RNA did not trigger plasma cell expansion
and hypergammaglobulinaemia. Whether the discrepancy
between 3P-RNA-induced autoantibody production and lack of
plasma cell proliferation relates to the suppressive effect of 3P-
RNA on Foxp3 mRNA expression and the numbers of spleen
CD4CD251 T cells or to another yet undefined mechanism
remains to be determined. Thus, 3P-RNA aggravates lupus
nephritis by enhancing systemic autoimmunity and by aggravat-
ing local inflammation. 3P-RNA induces IFN-a signaling, dsDNA
autoantibody production and aggravates immune complex
disease, renal CCL2 expression and immune cell infiltrates.
Viral DNA can induce immunity via TLR9-dependent as well
as TLR9-independent pathways [36, 37]. In SLE both pathways
can be activated by self-DNA that is released from apoptotic cells
or bound to immune complexes [16, 17, 38–41]. TLR9-depen-
dent recognition of CpG DNA causes B-cell proliferation and
repeated injections with CpG DNA cause significant lymphopro-
liferation in mice [23, 42]. In MRLlpr/lpr mice transient exposure
to CpG DNA increases the production of DNA autoantibodies and
fosters crescentic glomerulonephritis [20]. The role of TLR9 in
Figure 8. Histopathology of kidney MRLlpr/lpr mice. Paraffin-embedded sections were stained with PAS, CD3, Mac-2 and Ki-67 as indicated (originalmagnification � 200 to � 400). Note that 3P-RNA, non-CpG DNA and CpG-treated MRLlpr/lpr mice show higher periglomerular, and interstitialinflammatory cell infiltrates as compared with vehicle-treated MRLlpr/lpr mice.
Figure 9. IL-6 production in glomerular mesangial cells from MRLlpr/lpr
mice. Primary mesangial cells were prepared from MRLlpr/lpr mice andstimulated with 3P-RNA, non-CpG DNA and CpG DNA as described inthe Materials and methods. IL-6 production was determined in super-natants after 24 h by ELISA. Data represent means7SEM from threeindependent experiments each performed in duplicate. Comparisonbetween different agonists and vehicle were analyzed by ANOVA andpost hoc Bonferroni correction, �po0.05 versus medium. N.d. 5 notdetectable.
Eur. J. Immunol. 2008. 38: 3487–3498Ramanjaneyulu Allam et al.3494
& 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

SLE is still unclear because lack of TLR9 aggravates SLE in
MRLlpr/lpr mice despite a reduction of dsDNA autoantibodies
[43–46]. Hence, other pathways might be responsible for DNA
autoantibody production. In the present study this 45 bases long,
double-stranded, phosphodiester non-CpG DNA induced plasma
cell expansion, lymphoproliferation and dsDNA autoantibodies in
MRLlpr/lpr mice similar to CpG DNA. Stetson and Medzhitov [11]
have described overlapping but unique gene expression programs
by CpG- and non-CpG DNA in DC, e.g. in terms of IL-12 induc-
tion. Consistent with these data, CpG DNA strongly induced
serum IL-12 levels and reduced spleen GATA3 mRNA expression
in MRLlpr/lpr mice, which was not observed in non-CpG DNA-
injected MRLlpr/lpr mice. IL-12 production has been implicated
with crescent glomerular lesions in murine glomerulonephritis. In
fact, non-CpG DNA did not induce crescentic glomerulonephritis
and did not induce IL-6 and CCL2 mRNA in kidneys as did CpG
DNA. This may explain why non-CpG DNA was less potent in
aggravating lupus nephritis as compared with CpG DNA.
However, the induction of hypergammaglobulinaemia, rheuma-
toid factors and dsDNA autoantibodies was associated with
increased glomerular IgG and C3c deposits and glomerular
pathology. Obviously, Myd88-dependent recognition of CpG DNA
and Myd88-independent recognition of non-CpG DNA share the
potential to trigger B-cell activation in vivo. CpG DNA and dsDNA
differ in terms of shifting T-cell function to the Th1-type, which is
associated with crescent formation in mice.
In summary, the antiviral immune responses triggered by
3P-RNA and non-CpG DNA differently modulate autoimmunity
but both aggravate autoimmune tissue injury such as lupus-like
nephritis in MRLlpr/lpr mice. These data extend the previous
finding that TLR-independent recognition of RNA and DNA
initiate overlapping but still different gene expression patterns
[11]. RNA and DNA both enhance the production of dsDNA
autoantibodies, the glomerular deposition of IgG and subsequent
complement activation in the glomerulus. However, the immu-
nostimulatory effects of 3P-RNA and non-CpG DNA differ in
terms of type I IFN signaling, Foxp3-mediated regulatory T cells
and plasma cell proliferation in MRLlpr/lpr mice. These data
suggest that viral 3P-RNA and dsDNA differently modulate
autoimmunity but still both aggravate autoimmune tissue injury,
e.g. by activating non-immune cells at the tissue level.
Materials and methods
Animals and experimental protocol
Ten-week-old female MRLlpr/lpr mice were obtained from
Harlan Winkelmann (Borchen, Germany) and kept in
filter top cages under a 12 h light and dark cycle. Water and
standard chow (Sniff, Soest, Germany) were available
ad libitum. All experimental procedures have been approved by
the local government authorities. Mice were distributed into
four groups, each group consisting of 10–14 mice. From weeks
16 to 18 of age, groups of 12 MRLlpr/lpr mice received
intraperitoneal injections every other day with either 5%
glucose or 20 mg phosphodiester 3P-RNA (50-PPP-GAAAAGGGGA-
CACACACACACACACACACAC-30) dissolved in transfection
agent in vivo-JetPEITM (Polyplus-transfection, IIIkirch, France)
according to the manufacturer’s recommendations. 3P-RNA,
devoid of TLR7 activation, was prepared as previously described
[9]. A third group of MRLlpr/lpr mice was injected with
50 mg phosphodiester non-CpG dsDNA (sense 50-TACAGATCTAC-
TAGTGATCTATGACTGATC TGTACATGATCTACA-30). Non-CpG
DNA was generated by annealing complementary single
strands of DNA as previously described [11] dissolved in JetPEI.
JetPEI was used to facilitate the uptake of both ligands
into the cellular cytosol in vivo. As a positive control for the
immunostimulatory effects of DNA, a group of mice were
injected with 40mg of the immunostimulatory A-class phosphothio-
ate CpG-ODN 1668, 50-TCG ATG ACG TTC CTG ATG CT-30
(TIB Molbiol, Berlin, Germany) dissolved in saline as described
[20]. Phosphorothioate or phosphodiester backbones of
synthetic CpG oligonucleotides can modulate their binding
affinity to DNA autoantibodies [47]. Neither of the aforementioned
ODN bound to serum IgG from C57BL/6 mice but all of them
bound to serum IgG of MRLlpr/lpr mice. Binding affinity was
strongest for phosphorothioate CpG DNA particularly at lower
concentrations. Binding affinity of phosphodiester non-CpG DNA
was less potent and was not much affected by complexing non-CpG
DNA to JetPEI. At a concentration of 10mg/mL the binding
affinity of all nucleic acid formats tested was almost identical (data
not shown). All mice were sacrificed by cervical dislocation
at the end of week 18 of age. Endotoxin levels of all synthetic
nucleic acids were negligible as tested by the Limulus Assay.
To assess the renal distribution of injected 3P-RNA and dsDNA in
vivo 50-rhodamine-labeled forms of 3P-RNA/JetPEI and non-CpG
DNA/JetPEI complexes were intravenously injected into MRLlpr/lpr
mice at 18 wk of age. Frozen renal sections were collected 2 h later,
stained with FITC-labeled phalloidin (Invivogen, San Diego, CA,
USA, 1:50) and 40,6-diamidino-2-phenylindol (DAPI, Vector
Laboratories, Burlingame, CA, USA) and subjected to immunofluor-
escence imaging. In some experiments single dose studies
were performed in C57BL/6 mice that were obtained from
Charles River, Sulzfeld, Germany, or in MyD88-deficient mice
that had been backcrossed for six generations to the same
background [48].
Evaluation of systemic lupus
The weight ratio of spleen and the bulk of mesenterial lymphnodes
to total body weight were calculated as markers of the
lupus-associated lymphoproliferative syndrome. Kidneys
from all mice were fixed in 10% buffered formalin, processed and
embedded in paraffin. Three micrometer sections for periodic
acid-Schiff stains and immunostaining were prepared following
routine protocols. The severity of the renal lesions was graded
using the indices for activity and chronicity as described
Eur. J. Immunol. 2008. 38: 3487–3498 Innate immunity 3495
& 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

for human lupus nephritis [49]. The following primary antibodies
were used for immunostaining: rat anti-Mac2 (macrophages,
Cederlane, Ontario, Canada, 1:50), anti-mouse CD3 (T cells,
1:100, clone 500A2, Serotec, Oxford, UK), anti-Ki-67 (cell
proliferation, Dianova, Hamburg, Germany, 1:25), anti-mouse IgG
(1:100, M32015, Caltag Laboratories, Burlingame, CA, USA), anti-
mouse C3c (complement, GAM/C3c/FITC, Nordic Immunological
Laboratories, Tilburg, The Netherlands, 1:200). Negative controls
included incubation with a respective isotype antibody. For
quantitative analysis glomerular cells were counted in 15 cortical
glomeruli per section. Glomerular Ig and C3c deposits were scored
from 0 to 3 on 15 cortical glomerular sections. Albuminuria was
determined by ELISA and expressed as urinary albumin/creatinine
ratio as described [22].
In vitro studies with BMDC and mesangial cells
BMDC cells were isolated from C57BL/6 wild-type mice or Myd88-,
TLR9-mutant mice backcrossed to the C57BL/6 background for at
least six generations as previously described [7, 48, 50]. BM isolates
were cultured with Flt3 ligand (Immuno Tools GmbH, Friesoythe,
Germany) for 1 wk as described [51]. For the preparation of
primary mesangial cells capsule and medulla of the kidney were
removed and the renal cortices were diced in cold phosphate
buffered saline, and sequentially passed through a series of stainless
steel sieves (150, 103, 63, 50 and 45mm) and treated with a 1mg/mL
solution of type IV collagenase (Worthington, Lakewood, NY, USA)
for 15min at 371C. Finally, the digested glomeruli were seeded into
(6-well plates with) RPMI 1640 containing 20% FCS, 1% ITS
(insulin, transferrine, selenium; Roche, Mannheim, Germany). After
five passages mesangial cells were positive for smooth muscle actin
and negative for cytokeratin 18. Cells were treated with either
medium or various concentrations of non-CpG DNA or 3P-RNA
complexed with lipofectamine as well as CpG DNA. Supernatants
were harvested after 24h for ELISA.
Cytokine ELISA
Serum and cell culture supernatant cytokine levels were
determined using commercial ELISA kits: IL-6, IL-12p70, IFN-g(all OptEiA, BD, Biosciences, Heidelberg, Germany), IFN-a (PBL
Biomedical Labs, NJ, USA), TNF-a (BioLegend, San Diego, CA,
USA) following the manufacturers protocols.
Flow cytometry
Splenocyte flow cytometry was performed as previously
described [51]. Anti-mouse CD3, CD4, CD8, CD25, B220 and
CD138 (BD) antibodies were used to detect CD31CD4�CD8�
double negative T cells, CD41CD251 regulatory T cells and
B2201CD1381 plasma cell populations in spleens. CD11c has
been stained to identify plasmacytoid and myeloid DC and their
activation was assessed by costaining for CD40 (BD) and MHCII
(eBioscience, San Diego, CA, USA). Respective isotype antibodies
were used to demonstrate specific staining of cell subpopulations.
Autoantibody analysis
Serum IgG isotype concentrations were determined as previously
described [52]. Anti-nuclear antibodies were determined by
incubating serum samples (1:200 dilutions) with Hep-2 slides
(Biosystems S.A, Barcelona, Spain). The fluorescent patterns
were classified as described [50]. Serum of 6–10-wk-old C57BL/6
mice was used as negative control. For anti-dsDNA, 1:100 diluted
serum was applied to fixed C. luciliae slides (Bio-Rad Labora-
tories, Munich, Germany). FITC-labeled goat anti-mouse IgG
(Molecular Probes, Leiden, The Netherlands) was used as a
detection reagent. C. luciliae DNA colocalized with DAPI and the
kinetoplast staining intensity was quantified from digital images
with Adobe Photoshop software (Adobe, San Jose, CA, USA).
Anti-Sm ELISA: NUNC maxisorb ELISA plates were coated with
Smith (Sm) antigen (Immunovision, Springdale, AR, USA)
overnight. An anti-Sm IgG (Y12) antibody (GeneTex, San
Antonia, TX, USA) was used as standard. HRP-conjugated goat
anti-mouse IgG (Rockland, Gilbertsville, PA, USA) was used for
detection as previously described [22]. Rheumatoid factor: ELISA
plates were coated with 10 mg/mL rabbit IgG (Jackson Immunor-
esearch, West Grove, PA, USA) overnight at 41C. Serum samples
were diluted 1:100 times, 10 wk C57BL/6 mouse serum was used
as negative control. HRP-conjugated anti-mouse IgG was used as
secondary antibody. For antibody binding assay ELISA plates
were coated overnight at 41C with CpG DNA, non-CpG DNA or
non-CpG DNA complexed with JetPEI and mouse genomic DNA
diluted to various concentrations in SSC solution. 1:100 diluted
sera from 20-wk-old MRLlpr/lpr or C57BL/6 mice were added for
1 h. After washing HRP-conjugated anti-mouse IgG was used as
secondary antibody. Reading was taken at OD 450 nm.
Real-time quantitative (TaqMan) RT-PCR
Real-time RT was performed on total spleen and renal mRNA as
previously described [22]. 18S rRNA was used as a housekeeper.
Controls consisting of ddH2O were negative for target and
housekeeper genes. Oligonucleotide primer (300 nM) and probes
(100 nM) were from Applied Biosystems (Darmstadt, Germany):
IL-6: ID Mm00446190_m1 FAM 50-AAATGAGAAAA
GAGTTGTGCAATGG-30,
Mx1: ID Mm00487796_m1 FAM 50-TGTACTGCTAAGTC
CAAAATTAAAG-30,
Ccl2: ID Mm00441242_m1 FAM 50-GCTCAGCCA
GATGCAGTTAACGCCC-30,
Ccl5: ID Mm01302428_m1 FAM 50-CCAATCTTGCAG
TCGTGTTTGTCAC-30,
Tbx21: ID Mm00450960_m1 FAM 50-GCAAGGACGGC
GAATGTTCCCATTC-30,
Eur. J. Immunol. 2008. 38: 3487–3498Ramanjaneyulu Allam et al.3496
& 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

Gata3: ID Mm00484683_m1 FAM 50-CCCACCACGGGAGC
CAGGTATGCCG-30,
FoxP3: ID Mm00475156_m1 FAM 50-ACCCAGCCACTC
CAGCTCCCGGCAA-30,
Rorc: ID Mm00441139_m1 FAM 50-CCCACACCTCA
CAAATTGAAGTGAT-30,
Ddx58/RIG-I ID Mm00554529_m1 FAM 50-CCAAACCA
GAGGCCGAGGAAGAGCA-30,
Ifih1/Mda-5 ID Mm00459183_m1 FAM 50-GACACCAGA
GAAAATCCATTTAAAG-30,
Ifn-g: ID Mm00801778_m1 FAM 50-CTATTTTAACT
CAAGTGGCATAGAT-30,
IPS-1 ID Mm00523168_m1FAM 50-AGTGACCAGGATC
GACTGCGGGCTT-30,
TNF ID Mm00443258_m1 FAM 50-GTCCCCAAAGGGATGA
GAAGTTCCC-30,
IL-23 ID Mm00518984_m1 FAM 50-CAAGGACAACAGC-
CAGTTCTGCTTG-30,
DAI Forward primer 50-CAG GGA AGC ACC CCT CTT AT-30,
Reverse primer 50-GAA TGA AGC TCC TGG GTC AG-30,
Core sequence 50-CCC CCA GAA GTG TCA ACC ACC ACT-30.
Acknowledgements: The work was supported by grants from the
Deutsche Forschungsgemeinschaft (AN372/9-1, GRK 1202,
SE888/4-1) and by the Else Kroner-Fresenius Foundation. We
thank Jana Mandelbaum, Ewa Radomska, Stephanie Pfeiffer and
Dan Draganovic for their expert technical assistance. Parts of this
project were prepared as a doctoral thesis at the Faculty of
Medicine, University of Munich, by R. A.
Conflict of interest: The authors declare no financial or
commercial conflict of interest.
References
1 Ronnblom, L. and Alm, G. V., An etiopathogenic role for the type I IFN
system in SLE. Trends Immunol. 2001. 22: 427–431.
2 Singh, R. R., SLE translating lessons from model systems to human
disease. Trends Immunol. 2006. 26: 572–579.
3 Akira, S., Uematsu, S. and Takeuchi, O., Pathogen recognition and innate
immunity. Cell 2006. 124: 783–801.
4 Alexopoulou, L., Holt, A. C., Medzhitov, R. and Flavell, R. A., Recognition
of double-stranded RNA and activation of NF-kappaB by Toll-like receptor
3. Nature 2001. 413: 732–738.
5 Diebold, S. S., Kaisho, T., Hemmi, H., Akira, S. and Reis e Sousa, C., Innate
antiviral responses by means of TLR7-mediated recognition of single-
stranded RNA. Science 2004. 303: 1529–1531.
6 Heil, F., Hemmi, H., Hochrein, H., Ampenberger, F., Kirschning, C., Akira,
S., Lipford, G. et al., Species-specific recognition of single-stranded RNA
via toll-like receptor 7 and 8. Science 2004. 303: 1526–1529.
7 Hemmi, H., Takeuchi, O., Kawai, T., Kaisho, T., Sato, S., Sanjo, H.,
Matsumoto, M. et al., A Toll-like receptor recognizes bacterial DNA. Nature
2000. 408: 740–745.
8 Kato, H., Takeuchi, O., Sato, S., Yoneyama, M., Yamamota, M., Matsui, K.,
Uematsu, S. et al., Differential roles of MDA5 and RIG-I helicases in the
recognition of RNA viruses. Nature 2006. 441: 101–105.
9 Hornung, V., Ellegast, J., Kim, S., Brzozka, K., Jung, A., Kato, H., Poek, H.
et al., 50-Triphosphate RNA is the ligand for RIG-I. Science 2006. 314:
994–997.
10 Pichlmair, A., Schulz, O., Tan, C. P., Naslund, T. I., Liljestrom, P., Weber, F.
and Reis e Sousa, C., RIG-I-mediated antiviral responses to single-
stranded RNA bearing 50-phosphates. Science 2006. 314: 997–1001.
11 Stetson, D. B. and Medzhitov, R., Recognition of cytosolic DNA activates
an IRF3-dependent innate immune response. Immunity 2006. 24: 93–103.
12 Ishii, K. J., Coban, C., Kato, H., Takahashi, K., Torii, Y., Takeshita, F.,
Ludwig, H. et al., A Toll-like receptor-independent antiviral response
induced by double-stranded B-form DNA. Nat. Immunol. 2006. 7: 40–48.
13 Takaoka, A., Wang, Z., Choi, M. K., Yanai, H., Negishi, H., Ban, T., Lu, Y.
et al., DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of
innate immune response. Nature 2007. 448: 501–505.
14 Chi, H. and Flavell, R. A., Immunology: sensing the enemy within. Nature
2007. 448: 423–424.
15 Suzuki, K., Mori, A., Ishii, K. J., Saito, J., Singer, D. S., Klinman, D. M.,
Krause, P. R. et al., Activation of target-tissue immune-recognition
molecules by double-stranded polynucleotides. Proc. Natl. Acad. Sci. USA
1999. 96: 2285–2290.
16 Ishii, K. J. and Akira, S., Innate immune recognition of and regulation by
DNA. Trends Immunol. 2006. 27: 525–532.
17 Okabe, Y., Kawane, K., Akira, S., Taniguchi, T. and Nagata, S., Toll-like
receptor-independent gene induction program activated by mammalian
DNA escaped from apoptotic DNA degradation. J. Exp. Med. 2005. 202:
1333–1339.
18 Cheng, G., Zhong, J., Chung, J. and Chisari, F. V., Double-stranded DNA
and double-stranded RNA induce a common antiviral signaling pathway
in human cells. Proc. Natl. Acad. Sci. USA 2007. 104: 9035–9040.
19 Kato, H., Sato, S., Yoneyama, M., Yamamoto, M., Uematsu, S., Matsui, K.,
Tsujimura, T. et al., Cell type-specific involvement of RIG-I in antiviral
response. Immunity 2005. 23: 19–28.
20 Anders, H. J., Vielhauer, V., Eis, V., Linde, Y., Kretzler, M., Perez de Lema,
G., Strutz, F. et al., Activation of toll-like receptor-9 induces progression of
renal disease in MRL-Fas(lpr) mice. FASEB J. 2004. 18: 534–536.
21 Patole, P. S., Grone, H. J., Segerer, S., Ciubar, R., Belemezova, E.,
Henger, A., Kretzler, M. et al., Viral double-stranded RNA aggravates
lupus nephritis through Toll-like receptor 3 on glomerular mesangial cells
and antigen-presenting cells. J. Am. Soc. Nephrol. 2005. 16: 1326–1338.
22 Pawar, R. D., Patole, P. S., Zecher, D., Segerer, S., Kretzler, M., Schlondorff,
D. and Anders, H. J., Toll-like receptor-7 modulates immune complex
glomerulonephritis. J. Am. Soc. Nephrol. 2006. 17: 141–149.
23 Heikenwalder, M., Polymenidou, M., Junt, T., Sigurdson, C., Wagner, H.,
Akira, S., Zinkernagel, R. et al., Lymphoid follicle destruction and
immunosuppression after repeated CpG oligodeoxynucleotide adminis-
tration. Nat. Med. 2004. 10: 187–192.
24 Zheng, Y. and Rudensky, A. Y., Foxp3 in control of the regulatory T cell
lineage. Nat. Immunol. 2007. 8: 457–462.
25 Iwakura, Y. and Ishigame, H., The IL-23/IL-17 axis in inflammation. J. Clin.
Invest. 2006. 116: 1218–1222.
Eur. J. Immunol. 2008. 38: 3487–3498 Innate immunity 3497
& 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

26 Afzali, B., Lombardi, G., Lechler, R. I. and Lord, G. M., The role of T helper
17 (Th17) and regulatory T cells (Treg) in human organ transplantation
and autoimmune disease. Clin. Exp. Immunol. 2007. 148: 32–46.
27 Marshak-Rothstein, A. and Rifkin, I. R., Immunologically active auto-
antigens: the role of toll-like receptors in the development of chronic
inflammatory disease. Annu. Rev. Immunol. 2007. 25: 419–441.
28 Hemmi, H., Kaisho, T., Takeuchi, O., Sato, S., Sanjo, H., Hoshino, K., Horiuchi,
T. et al., Small anti-viral compounds activate immune cells via the TLR7
MyD88-dependent signaling pathway. Nat. Immunol. 2002. 3: 196–200.
29 Yoneyama, M., Kikuchi, M., Natsukawa, T., Shinobu, N., Imaizumi, T.,
Miyagishi, M., Taira, K. et al., The RNA helicase RIG-I has an essential
function in double-stranded RNA-induced innate antiviral responses.
Nat. Immunol. 2004. 5: 730–737.
30 Yoneyama, M., Kikuchi, M., Matsumoto, K., Imaizumi, T., Miyagishi, M.,
Taira, K., Foy, E. et al., Shared and unique functions of the DExD/H-Box
helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol.
2005. 175: 2851–2858.
31 Suzuki, K., Imaizumi, T., Tsugawa, K., Ito, E. and Tanaka, H., Expression
of retinoic acid-inducible gene-I in lupus nephritis. Nephrol. Dial.
Transplant. 2007. 22: 2407–2409.
32 Balomenos, D., Rumold, R. and Theofilopoulos, A. N., Interferon-gamma
is required for lupus-like disease and lymphoaccumulation in MRL-lpr
mice. J. Clin. Invest. 1998. 101: 364–371.
33 Lu, Q., Shen, N., Li, X. M. and Chen, S. L., Genomic view of IFN-alpha
response in pre-autoimmune NZB/W and MRL/lpr mice. Genes Immun.
2007. 8: 590–603.
34 Tesch, G. H., Maifert, S., Schwarting, A., Rollins, B. J. and Kelley, V. R.,
Monocyte chemoattractant protein 1-dependent leukocytic infiltrates are
responsible for autoimmune disease in MRL-Fas(lpr) mice. J. Exp. Med.
1999. 190: 1813–1824.
35 Kulkarni, O., Pawar, R. D., Purschke, W., Eulberg, D., Selve, N.,
Buchner, K., Ninichuk, V. et al., Spiegelmer inhibition of CCl2/Mcp-1
ameliorates lupus nephritis in MRL-(Fas)lpr mice. J. Am. Soc. Nephrol. 2007.
18: 2350–2358.
36 Hochrein, H., Schlatter, B., O’Keeffe, M., Wagner, C., Schmitz, F.,
Schiemann, M., Bauer, S. et al., Herpes simplex virus type-1 induces
IFN-alpha production via Toll-like receptor 9-dependent and -indepen-
dent pathways. Proc. Natl. Acad. Sci. USA 2004. 101: 11416–11421.
37 Malmgaard, L., Melchjorsen, J., Bowie, A. G., Mogensen, S. C. and
Paludan, S. R., Viral activation of macrophages through TLR-dependent
and -independent pathways. J. Immunol. 2004. 173: 6890–6898.
38 Boule, M. W., Broughton, C., Mackay, F., Akira, S., Marshak-Rothstein, A.
and Rifkin, I. R., Toll-like receptor 9-dependent and -independent
dendritic cell activation by chromatin-immunoglobulin G complexes.
J. Exp. Med. 2004. 199: 1631–1640.
39 Yasuda, K., Yu, P., Kirschning, C. J., Schlatter, B., Schmitz, F., Heit, A.,
Bauer, S. et al., Endosomal translocation of vertebrate DNA activates
dendritic cells via TLR9-dependent and -independent pathways.
J. Immunol. 2005. 174: 6129–6136.
40 Decker, P., Singh-Jasuja, H., Haager, S., Kotter, I. and Rammensee, H. G.,
Nucleosome: the main autoantigen in systemic lupus erythematosus,
induces direct dendritic cell activation via a MyD88-independent path-
way: consequences on inflammation. J. Immunol. 2005. 174: 3326–3334.
41 Leadbetter, E. A., Rifkin, I. R., Hohlbaum, A. M., Rifkin, I. R., Hohlbaum,
A. M., Beaudette, B. C. and Shlomchik, M. J., Chromatin-IgG complexes
activate B cells by dual engagement of IgM and Toll-like receptors. Nature
2002. 416: 603–607.
42 Krieg, A. M., Yi, A. K., Matson, S., Waldschmidt, T. J., Bishop, G. A.,
Teasdale, R., Koretzky, G. A. et al., CpG motifs in bacterial DNA trigger
direct B-cell activation. Nature 1995. 374: 546–549.
43 Yu, P., Wellmann, U., Kunder, S., Quintanilla-Martinez, L., Jennen, L.,
Dear, N., Amann, K. et al., Toll-like receptor 9-independent aggravation of
glomerulonephritis in a novel model of SLE. Int. Immunol. 2006. 18:
1211–1219.
44 Wu, X. and Peng, S. L., Toll-like receptor 9 signaling protects against
murine lupus. Arthritis. Rheum. 2006. 54: 336–342.
45 Lartigue, A., Courville, P., Auquit, I., Francois, A., Arnoult, C., Tron, F.,
Gilbert, D. et al., Role of TLR9 in anti-nucleosome and anti-DNA antibody
production in lpr mutation-induced murine lupus. J. Immunol. 2006. 177:
1349–1354.
46 Christensen, S. R., Kashgarian, M., Alexopoulou, L., Flavell, R. A., Akira,
S. and Shlomchik, M. J., Toll-like receptor 9 controls anti-DNA autoanti-
body production in murine lupus. J. Exp. Med. 2005. 202: 321–331.
47 Pisetsky, D. S. and Reich, C. F., 3rd, The binding of anti-DNA antibodies to
phosphorothioate oligonucleotides in a solid phase immunoassay. Mol.
Immunol. 1998. 35: 1161–1170.
48 Adachi, O., Kawai, T., Takeda, K., Takeda, K., Matsumoto, M., Tsutsui, H.,
Sakagami, M. et al., Targeted disruption of the MyD88 gene results in loss
of IL-1- and IL-18-mediated function. Immunity 1998. 9: 143–150.
49 Austin, H. A., 3rd, Muenz, L. R., Joyce, K. M., Antonovych, T. T. and Balow,
J. E., Diffuse proliferative lupus nephritis: identification of specific
pathologic features affecting renal outcome. Kidney Int. 1984. 25: 689–695.
50 Hoebe, K., Du, X., Georgel, P., Janssen, E., Tabeta, K., Kim, S. O., Goode, J.
et al., Identification of Lps2 as a key transducer of MyD88-independent
TIR signalling. Nature 2003. 424: 743–748.
51 Pawar, R. D., Ramanjaneyulu, A., Kulkarni, O. P., Lech, M., Segerer, S. and
Anders, H. J., Inhibition of Toll-like receptor -7 (TLR-7) or TLR-7 plus TLR-9
attenuates glomerulonephritis and lung injury in experimental lupus.
J. Am. Soc. Nephrol. 2007. 18: 1721–1731.
52 Lech, M., Kulkarni, O. P., Pfeiffer, S., Savarese, E., Krug, A., Garlanda, C.,
Mantovani, A. and Anders, H. J., Tir8/Sigirr prevents murine lupus by
suppressing the immunostimulatory effects of lupus autoantigens. J. Exp.
Med. 2008. 205: 1879–1888.
Abbreviations: IPS: IFN-b promoter stimulator � 3P-RNA: triphosphate
RNA � RIG: retinoic-acid-inducible gene � SLE: systemic lupus
erythematosus
Full correspondence: Dr. H.-J. Anders, Medizinische Poliklinik, Klinikum
der Universitat–Innenstadt, Pettenkoferstr. 8a, 80336 Munich, Germany
Fax: 149-89-218075860
e-mail: [email protected]
Received: 11/6/2008
Revised: 1/8/2008
Accepted: 9/9/2008
Eur. J. Immunol. 2008. 38: 3487–3498Ramanjaneyulu Allam et al.3498
& 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu




















