
IL4 Therapy Prevents the Development of Proteinuria in Active Heymann Nephritis by Inhibition of Tc1...
10
of April 15, 2014. This information is current as Inhibition of Tc1 Cells Proteinuria in Active Heymann Nephritis by IL-4 Therapy Prevents the Development of Hodgkinson and Bruce M. Hall Nicole Carter, Giang Tran, Mark J. Penny, Suzanne J. S. Timothy Spicer, Hong Ha, Rochelle A. Boyd, Xiao Y. He, http://www.jimmunol.org/content/167/7/3725 2001; 167:3725-3733; ; J Immunol References http://www.jimmunol.org/content/167/7/3725.full#ref-list-1 , 18 of which you can access for free at: cites 58 articles This article Subscriptions http://jimmunol.org/subscriptions is online at: The Journal of Immunology Information about subscribing to Permissions http://www.aai.org/ji/copyright.html Submit copyright permission requests at: Email Alerts http://jimmunol.org/cgi/alerts/etoc Receive free email-alerts when new articles cite this article. Sign up at: Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved. Copyright © 2001 by The American Association of 9650 Rockville Pike, Bethesda, MD 20814-3994. The American Association of Immunologists, Inc., is published twice each month by The Journal of Immunology by guest on April 15, 2014 http://www.jimmunol.org/ Downloaded from by guest on April 15, 2014 http://www.jimmunol.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
8 -
download
0
Transcript of IL4 Therapy Prevents the Development of Proteinuria in Active Heymann Nephritis by Inhibition of Tc1...

of April 15, 2014.This information is current as
Inhibition of Tc1 CellsProteinuria in Active Heymann Nephritis by IL-4 Therapy Prevents the Development of
Hodgkinson and Bruce M. HallNicole Carter, Giang Tran, Mark J. Penny, Suzanne J. S. Timothy Spicer, Hong Ha, Rochelle A. Boyd, Xiao Y. He,
http://www.jimmunol.org/content/167/7/37252001; 167:3725-3733; ;J Immunol
Referenceshttp://www.jimmunol.org/content/167/7/3725.full#ref-list-1
, 18 of which you can access for free at: cites 58 articlesThis article
Subscriptionshttp://jimmunol.org/subscriptions
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/ji/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/cgi/alerts/etocReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 2001 by The American Association of9650 Rockville Pike, Bethesda, MD 20814-3994.The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on April 15, 2014
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on April 15, 2014
http://ww
w.jim
munol.org/
Dow
nloaded from

IL-4 Therapy Prevents the Development of Proteinuria inActive Heymann Nephritis by Inhibition of Tc1 Cells1
S. Timothy Spicer,2 Hong Ha, Rochelle A. Boyd, Xiao Y. He, Nicole Carter, Giang Tran,Mark J. Penny, Suzanne J. Hodgkinson, and Bruce M. Hall
The role of IL-4, a key Th2 cytokine, in promoting or inhibiting active Heymann nephritis (HN) was examined. HN is induced byimmunization with Fx1A in CFA, and proteinuria in HN is associated with subepithelial IgG and C3 deposition and infiltrationof CD8� T-cytotoxic 1 (Tc1) cells and macrophages into glomeruli, as well as induction of Abs to Crry. Treatment with rIL-4 fromthe time of Fx1A/CFA immunization stimulated an earlier IgG1 response to Fx1A, induced anti-Crry Abs, and up-regulated IL-4mRNA in lymphoid tissue, but did not alter proteinuria. Treatment with MRCOx-81, an IL-4-blocking mAb, resulted in greaterproteinuria, which suggests endogenous IL-4 regulated the autoimmune response. Delay of rIL-4 treatment until 4 wk post-Fx1A/CFA immunization and just before the onset of proteinuria prevented the development of proteinuria and reduced Tc1 cellinfiltrate in glomeruli. Delayed treatment with IL-4 had no effect on titer or isotype of Abs to Fx1A or on Ig, C3, and C9accumulation in glomeruli. Treatment with rIL-13, a cytokine that alters macrophage function such as rIL-4, but has no directeffect on T or B cell function, reduced glomerular macrophage infiltrate, but did not prevent proteinuria or CD8� T cell infiltrate.Anti-Crry Abs were paradoxically only induced with rIL-4 therapy, not in HN controls with proteinuria. It was concluded thatthe rIL-4 effect was probably by inhibition of Tc1 cells, which normally mediate the glomerular injury that results inproteinuria. The Journal of Immunology, 2001, 167: 3725–3733.
I mmune injury in glomerulonephritis is categorized as eitherTh1 with infiltration of Th1 cells and macrophages (1–4) orTh2 with Ab deposition (5, 6). Active Heymann nephritis
(HN)3 is a rat model of human membranous glomerulonephritisthat has Ab/complement deposition in glomeruli and would thus beexpected to be due to a Th2 response. HN is induced in susceptiblerat strains by immunization with a rat renal tubular Ag (Fx1A) inCFA (7, 8). Anti-Fx1A Ab titers appear 2–4 wk later, and auto-antibody binds to antigenic determinants on Gp330 (Megalin) atthe base of glomerular epithelial cells (GEC) (9). Activation of C3by anti-Fx1A Abs leads to formation of the membrane attack com-plex (C5b-9/MAC), which is thought to mediate sublytic injury toGEC, synthesis of abnormal glomerular basement membrane(GBM) matrix, and proteinuria (10, 11). In passive HN (PHN), C3depletion with cobra venom factor abrogates proteinuria, suggest-ing complement is the principal mediator of glomerular injury (12,13). Ab responses that block the function of the complement reg-ulatory molecule Crry, the rodent analogue of human decay-accel-erating factor and monocyte chemoattractant protein, are also nec-essary for the development of proteinuria in HN (14).
Against an essential role for MAC in HN is the finding that it canbe induced in C6-deficient PVG rats that are unable to assemble C5b-9/MAC (15). Our group also observed that the onset of proteinuria inHN is associated with glomerular infiltration of T-cytotoxic 1 (Tc1)cells and macrophages (16). Furthermore, permanent depletion ofCD8� T cells prevents the onset of proteinuria (17). These findingssuggest that Th1 responses such as Tc1 cytotoxic cells, not MAC,may mediate glomerular injury. It has been proposed that the T-cytotoxicTc1 response only develops 4–6 wk after immunization in response tothe inflammation caused by Ab deposition and C activation in the glo-merulus (17).
IL-4 is the key Th2 cytokine in that it both promotes the devel-opment of Th2 responses and induces IgE production and Ig iso-type switching to IgG1 (18), as well as inhibiting Th1 responses.IL-4 blocks Tc1 cell development by making them noncytolytic(19). IL-4 also inhibits macrophage activation and their productionof cytotoxic molecules, including TNF-� and NO (20–22).
This study examined the effects on HN of both IL-4 therapy andinhibition of endogenous IL-4 with anti-IL-4 mAb. The aim was tosee whether Ab/complement responses or Th1 and cytotoxic Tc1responses were more important. Therapies were given at threephases in the disease: first, at the time of immunization when theinitial anti-Fx1A Ig response is induced and there is glomerulardeposition of Ig with complement activation; second, between 4and 6 wk after Fx1A immunization, at a time just before the onsetof proteinuria, when Tc1 cells and macrophages infiltrate into glo-meruli; and third, from 8–10 wk post-Fx1A immunization, afterproteinuria has developed and all inflammatory mediators are inthe glomerulus. These studies showed that rIL-4 administrationjust before onset of proteinuria prevented proteinuria, inhibitedCD8� T cell and macrophage infiltrate, induced anti-Crry Abs, buthad no effect on glomerular Ig and C3 deposition. Treatment withanti-IL-4 mAb resulted in greater proteinuria. As the antiinflam-matory effect of rIL-4 could be due to inhibition of activation ofeither CD8� T cells or macrophages, we also examined the effectsof treatment with rIL-13. IL-13 has a similar effect as IL-4 on
Department of Medicine, University of New South Wales, Liverpool Hospital, Liv-erpool, New South Wales, Australia
Received for publication March 16, 2001. Accepted for publication July25, 2001.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 Portions of this work have been presented as abstracts (65, 66). S.T.S. and B.M.H.are supported by Australian National Health and Medical Research Council grants.2 Address correspondence and reprint requests to Dr. Timothy Spicer, Department ofRenal Medicine, Liverpool Hospital, Liverpool, Sydney, 2170 Australia. E-mail ad-dress: [email protected] Abbreviations used in this paper: HN, Heymann nephritis; CHO, Chinese hamsterovary; GBM, glomerular basement membrane; GEC, glomerular epithelial cell;iNOS, inducible NO synthase; MAC, membrane attack complex; PHN, passive HN;Tc1, T-cytotoxic 1.
Copyright © 2001 by The American Association of Immunologists 0022-1767/01/$02.00
by guest on April 15, 2014
http://ww
w.jim
munol.org/
Dow
nloaded from

macrophages, but does not inhibit Th1 and Tc1 cells, nor promoteTh2 cytokines and Ig isotype switching (23, 24).
Materials and MethodsExperimental animals and induction of HN
Lewis rats (LEW/Ssn) were bred in the animal facility at Liverpool Hos-pital, and Sprague Dawley rats and BALB/c mice were purchased from theAnimal Breeding and Holding Unit (New South Wales, Australia). Allanimals had water and standard chow available ad libitum. All procedureshave been previously described, including preparation and immunizationwith renal tubular Ag (Fx1A) in CFA (IFA; Sigma, St. Louis, MO) andMycobacterium tuberculosis H37RA (Difco, Detroit, MI) (25), monitoringwith kidney biopsies, 24-h urine protein estimations, and sera for anti-Fx1A Abs (16). The Animal Care and Ethics Committee of University ofNew South Wales approved animal experimental protocols.
Production and administration of rat rIL-4 and rIL-13
Rat rIL-4 was produced as cell culture supernatant from a Chinese hamsterovary (CHO-K1) cell line transfected with rat IL-4 cDNA (a kind gift of D.Mason, Medical Research Council, Cellular Immunology Unit, Oxford,U.K.) (26), as described (27). One unit of rIL-4 was defined as the amountof rIL-4 required to promote 50% of the maximal MHC class II inductionon 5 � 105 B cells (28). A daily dose of 32,000 U rIL-4 was administeredas twice-daily i.p. injections for 10 days. This protocol is based on thepublished use of rat IL-4 in anti-GBM Ab-mediated glomerulonephritis (3)and in prolongation of neonatal heart allograft survival (29). Rat rIL-13was produced as cell culture supernatant from a CHO-K1 cell line trans-fected with rat IL-13 cDNA (30). One unit of rIL-13 was defined as theconcentration inducing half-maximal proliferation of a dependent humanerythroleukemia cell line (TF1), as described (31). A daily dose of 5000 UrIL-13 was administered as a twice-daily i.p. injection for 10 days, basedon the dose of rIL-13 used to prolong allograft survival.4
As a control for administration of cytokines, supernatant from nontrans-fected CHO-K1 cells was concentrated and administered. All preparationsof rIL-4, rIL-13, CHO-K1 supernatant and mAb preparations were assayedfor endotoxin levels and had �0.006 U/ml in a Limulus amebocyte lysateassay (CoA test Gel-LAL; Chromogenix, Molndal, Sweden).
Production and administration of mAbs
Clones used were MRCOx-81, IgG1 anti-rat IL-4 (a kind gift of D. Mason)(26), and isotype control A6 (IgG1) mouse anti-human CD45R0 with noreactivity to rats (32). These mAb were produced as described (27), and i.p.injection of 7 mg/kg was performed in five doses over 10 days. Sera takenfrom rats 10 days after treatment with one dose of MRCOx-81 inhibitedIL-4-induced MHC class II up-regulation on B cells (27).
ELISA assay for anti-Fx1A and Crry Abs
Anti-Fx1A Ab titers (total Ig) were determined by ELISA, as described(33). Fx1A for ELISA was solubilized and purified by fractional salt pre-cipitation, and plates were prepared (25). Triplicate sample ODs were readat 405 nm, corrected for a control sample of known strongly positive se-rum, and expressed as a percentage of a control positive serum OD (i.e.,sample OD/control positive serum OD � 100). The anti-Fx1A Ab titer ofthe control positive serum was 1:250. IgG subclasses were assessed by thesame method using alkaline phosphatase-conjugated mouse mAb to ratIgG1, IgG2a, and IgG2b (BD PharMingen, San Diego, CA). IgE anti-Fx1Atiters were assayed using biotin-conjugated anti-rat IgE Abs and a strepta-vidin/alkaline phosphatase conjugate (BD PharMingen).
Anti-Crry Abs were assessed by ELISA, as described (14). Briefly,plates were coated with purified Crry (a gift of R. Quigg, University ofChicago, Chicago, IL), and titers of anti-Crry Ab (total Ig) in experimentalsera were compared with control sera and anti-rat Crry/p65 mAb (IgG1)(BD PharMingen).
Immunoperoxidase cytochemistry of renal cortex and isolatedwhole glomeruli
An indirect peroxidase-antiperoxidase complex technique was used on re-nal cortical wedge biopsies or isolated glomeruli, as described (16). Glo-meruli were stained with MRCOx-12, which recognizes rat Ig �-L chains(34), C9 polyclonal rabbit anti-rat Ab (35) (a kind gift of S. Piddlesden,
The Austin Research Institute, Heidelberg, Victoria, Australia), and goatanti-rat C3 peroxidase conjugate (Nordik Immunology, Tilberg, The Neth-erlands). The degree of glomerular Ig, C3, and C9 staining was scored as0, 1�, 2�, or 3�.
Cellular infiltrates were characterized with the following mAb: R73 forthe TCR-�� receptor on T cells (36); W3/25 for CD4� T cells and somemacrophages (37); MRCOx-8 for CD8� T cytotoxic and NK cells (38);3.2.3 for NK cells (39); ED-1, for most macrophages, and some dendriticcells (40); and MRCOx-33 for the CD45A isoform found only on B cells(41) (all purchased from PharMingen). Cellular infiltrates were counted,with labels covered, under �40 magnification, and results were ex-pressed as the mean � SD of cells per high power field or glomerulus,as described (17).
Staining of lymph node cell subpopulations
Popliteal lymph node cells were prepared and directly stained with acombination of FITC- and PE-conjugated mouse anti-rat mAb, includ-ing CD4�/CD3� T cells (MRCOx-35�/G4.18�), CD8�/CD3� T cells(MRCOx-8�/G4.18�), and B cells (MRCOx-33�/G4.18�) before theywere analyzed on FACScan (BD Biosciences, San Jose, CA), asdescribed (41).
Semiquantitative RT-PCR
RT-PCR to assess the amount of cytokine mRNA present in isolated glomer-uli, renal cortex, and popliteal lymph nodes was performed, as previouslydescribed (16). Primer oligonucleotide sequences for IL-2, IL-4, IL-5, IL-6,IL-10, IFN-�, granzyme A, granzyme B, perforin, TNF-�, TNF-�, andGAPDH were as described (16). Amplification of the housekeeping geneGAPDH was used as a positive control for intact RNA and efficiency of re-verse transcriptase. Reaction and amplification conditions were optimized foreach primer set. FTS-960 thermal sequencers (Corbett Research, Sydney, NewSouth Wales, Australia) were used to amplify products, which were then an-alyzed by m.w. on 6% polyacrylamide gels, and stained with ethidium bro-mide. Southern transfer and hybridization with digoxigenin 3� end-labeled oli-gonucleotide probes verified specificity of the products. Hybridized probe wasdetected using the digoxigenin luminescent detection kit (Boehringer Mann-heim, Germany). The semiquantitative RT-PCR method modified from Dall-man (42) involved a cycle titration at 5-cycle intervals, with the cycle of firstappearance of PCR products being used to compare levels of mRNA expres-sion. Duplicate amplifications were performed at each cycle. This technique isreproducible (17, 43) and can detect 2- to 5-fold differences in cytokinemRNA. Within experimental groups, similar results were obtained for indi-vidual rats. Negative controls without cDNA and positive controls of cDNAfrom Con A-stimulated rat lymphocytes were included in all experiments. Gelswere photographed with a Kodak DC40 digital camera and saved as digitalimages using Kodak Digital Science software, version 1.6 (Kodak,Rochester, NY).
Experimental protocol
The effect on HN of rIL-4 and MRCOx-81 treatment at the time of, as well asat 4 and 8 wk after immunization with Fx1A was assessed. In another exper-iment, the effect of rIL-13 treatment given 4 wk post-Fx1A immunization wasalso assessed. In all experiments, groups of male Lewis rats were immunizedin the hind footpads with the same batch of Fx1A in CFA, and were comparedwith a group of CFA-treated control animals over a 12-wk period. Serumanti-Fx1A Ab titers were determined at 2, 4, 6, 8, and 10 wk, and renal biopsieswere taken at wk 8 and 12. Animals were sacrificed at 12 wk, and glomeruliwere isolated for RT-PCR and i.p. cytochemistry. All experiments were re-peated two or three times with similar results.
Statistical analysis
Data were analyzed using Statview for Macintosh version 5.0 (AbacusConcepts, Berkeley, CA). Urine protein estimations, anti-Fx1A Ab levels,leukocyte counts, lymph node weight, and lymphocyte FACS analysis foreach group were expressed as mean � SEM. Comparisons between groupswere made by ANOVA, with Bonferroni-Dunn multiple comparisons posthoc test. Semiquantitative RT-PCR results were treated as parametric data,with comparisons between groups made by ANOVA and a Bonferroni-Dunn post hoc test, and expressed graphically as the median PCR cyclenumber at which PCR product was first identified. This is an intentionallyconservative analysis of the data, as differences in expression of PCR prod-uct are exponential. A p value of �0.05 was considered significant.
4 X. Y. He, N. Verma, C. Davidson, C. Robinson, J. Chen, K. Plain, G. T. Tran, S. J.Hodgkinson, and B. M. Hall. IL-13 prolongs allograft survival associated with in-duction of IL-12 p35, iNOS and TNF-�. Submitted for publication.
3726 IL-4 THERAPY IN HN
by guest on April 15, 2014
http://ww
w.jim
munol.org/
Dow
nloaded from
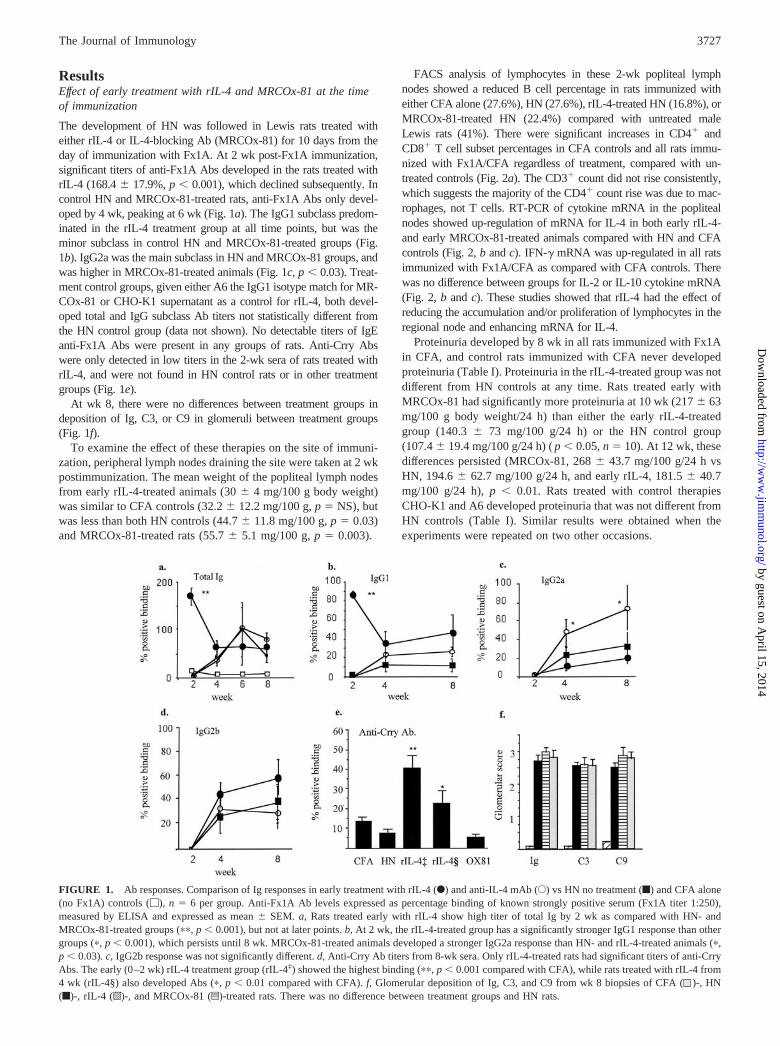
ResultsEffect of early treatment with rIL-4 and MRCOx-81 at the timeof immunization
The development of HN was followed in Lewis rats treated witheither rIL-4 or IL-4-blocking Ab (MRCOx-81) for 10 days from theday of immunization with Fx1A. At 2 wk post-Fx1A immunization,significant titers of anti-Fx1A Abs developed in the rats treated withrIL-4 (168.4 � 17.9%, p � 0.001), which declined subsequently. Incontrol HN and MRCOx-81-treated rats, anti-Fx1A Abs only devel-oped by 4 wk, peaking at 6 wk (Fig. 1a). The IgG1 subclass predom-inated in the rIL-4 treatment group at all time points, but was theminor subclass in control HN and MRCOx-81-treated groups (Fig.1b). IgG2a was the main subclass in HN and MRCOx-81 groups, andwas higher in MRCOx-81-treated animals (Fig. 1c, p � 0.03). Treat-ment control groups, given either A6 the IgG1 isotype match for MR-COx-81 or CHO-K1 supernatant as a control for rIL-4, both devel-oped total and IgG subclass Ab titers not statistically different fromthe HN control group (data not shown). No detectable titers of IgEanti-Fx1A Abs were present in any groups of rats. Anti-Crry Abswere only detected in low titers in the 2-wk sera of rats treated withrIL-4, and were not found in HN control rats or in other treatmentgroups (Fig. 1e).
At wk 8, there were no differences between treatment groups indeposition of Ig, C3, or C9 in glomeruli between treatment groups(Fig. 1f).
To examine the effect of these therapies on the site of immuni-zation, peripheral lymph nodes draining the site were taken at 2 wkpostimmunization. The mean weight of the popliteal lymph nodesfrom early rIL-4-treated animals (30 � 4 mg/100 g body weight)was similar to CFA controls (32.2 � 12.2 mg/100 g, p � NS), butwas less than both HN controls (44.7 � 11.8 mg/100 g, p � 0.03)and MRCOx-81-treated rats (55.7 � 5.1 mg/100 g, p � 0.003).
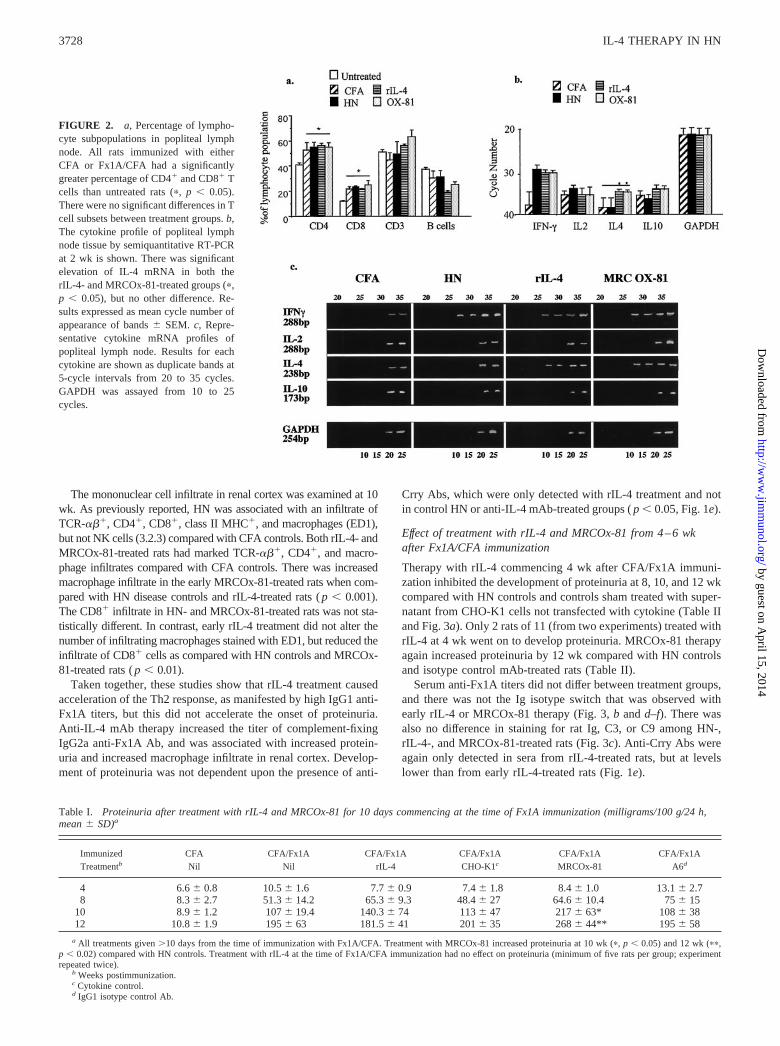
FACS analysis of lymphocytes in these 2-wk popliteal lymphnodes showed a reduced B cell percentage in rats immunized witheither CFA alone (27.6%), HN (27.6%), rIL-4-treated HN (16.8%), orMRCOx-81-treated HN (22.4%) compared with untreated maleLewis rats (41%). There were significant increases in CD4� andCD8� T cell subset percentages in CFA controls and all rats immu-nized with Fx1A/CFA regardless of treatment, compared with un-treated controls (Fig. 2a). The CD3� count did not rise consistently,which suggests the majority of the CD4� count rise was due to mac-rophages, not T cells. RT-PCR of cytokine mRNA in the poplitealnodes showed up-regulation of mRNA for IL-4 in both early rIL-4-and early MRCOx-81-treated animals compared with HN and CFAcontrols (Fig. 2, b and c). IFN-� mRNA was up-regulated in all ratsimmunized with Fx1A/CFA as compared with CFA controls. Therewas no difference between groups for IL-2 or IL-10 cytokine mRNA(Fig. 2, b and c). These studies showed that rIL-4 had the effect ofreducing the accumulation and/or proliferation of lymphocytes in theregional node and enhancing mRNA for IL-4.
Proteinuria developed by 8 wk in all rats immunized with Fx1Ain CFA, and control rats immunized with CFA never developedproteinuria (Table I). Proteinuria in the rIL-4-treated group was notdifferent from HN controls at any time. Rats treated early withMRCOx-81 had significantly more proteinuria at 10 wk (217 � 63mg/100 g body weight/24 h) than either the early rIL-4-treatedgroup (140.3 � 73 mg/100 g/24 h) or the HN control group(107.4 � 19.4 mg/100 g/24 h) ( p � 0.05, n � 10). At 12 wk, thesedifferences persisted (MRCOx-81, 268 � 43.7 mg/100 g/24 h vsHN, 194.6 � 62.7 mg/100 g/24 h, and early rIL-4, 181.5 � 40.7mg/100 g/24 h), p � 0.01. Rats treated with control therapiesCHO-K1 and A6 developed proteinuria that was not different fromHN controls (Table I). Similar results were obtained when theexperiments were repeated on two other occasions.
FIGURE 1. Ab responses. Comparison of Ig responses in early treatment with rIL-4 (F) and anti-IL-4 mAb (E) vs HN no treatment (f) and CFA alone(no Fx1A) controls (�), n � 6 per group. Anti-Fx1A Ab levels expressed as percentage binding of known strongly positive serum (Fx1A titer 1:250),measured by ELISA and expressed as mean � SEM. a, Rats treated early with rIL-4 show high titer of total Ig by 2 wk as compared with HN- andMRCOx-81-treated groups (��, p � 0.001), but not at later points. b, At 2 wk, the rIL-4-treated group has a significantly stronger IgG1 response than othergroups (�, p � 0.001), which persists until 8 wk. MRCOx-81-treated animals developed a stronger IgG2a response than HN- and rIL-4-treated animals (�,p � 0.03). c, IgG2b response was not significantly different. d, Anti-Crry Ab titers from 8-wk sera. Only rIL-4-treated rats had significant titers of anti-CrryAbs. The early (0–2 wk) rIL-4 treatment group (rIL-4‡) showed the highest binding (��, p � 0.001 compared with CFA), while rats treated with rIL-4 from4 wk (rIL-4§) also developed Abs (�, p � 0.01 compared with CFA). f, Glomerular deposition of Ig, C3, and C9 from wk 8 biopsies of CFA ( )-, HN(f)-, rIL-4 (o)-, and MRCOx-81 (z)-treated rats. There was no difference between treatment groups and HN rats.
3727The Journal of Immunology
by guest on April 15, 2014
http://ww
w.jim
munol.org/
Dow
nloaded from
The mononuclear cell infiltrate in renal cortex was examined at 10wk. As previously reported, HN was associated with an infiltrate ofTCR-���, CD4�, CD8�, class II MHC�, and macrophages (ED1),but not NK cells (3.2.3) compared with CFA controls. Both rIL-4- andMRCOx-81-treated rats had marked TCR-���, CD4�, and macro-phage infiltrates compared with CFA controls. There was increasedmacrophage infiltrate in the early MRCOx-81-treated rats when com-pared with HN disease controls and rIL-4-treated rats ( p � 0.001).The CD8� infiltrate in HN- and MRCOx-81-treated rats was not sta-tistically different. In contrast, early rIL-4 treatment did not alter thenumber of infiltrating macrophages stained with ED1, but reduced theinfiltrate of CD8� cells as compared with HN controls and MRCOx-81-treated rats ( p � 0.01).
Taken together, these studies show that rIL-4 treatment causedacceleration of the Th2 response, as manifested by high IgG1 anti-Fx1A titers, but this did not accelerate the onset of proteinuria.Anti-IL-4 mAb therapy increased the titer of complement-fixingIgG2a anti-Fx1A Ab, and was associated with increased protein-uria and increased macrophage infiltrate in renal cortex. Develop-ment of proteinuria was not dependent upon the presence of anti-
Crry Abs, which were only detected with rIL-4 treatment and notin control HN or anti-IL-4 mAb-treated groups ( p � 0.05, Fig. 1e).
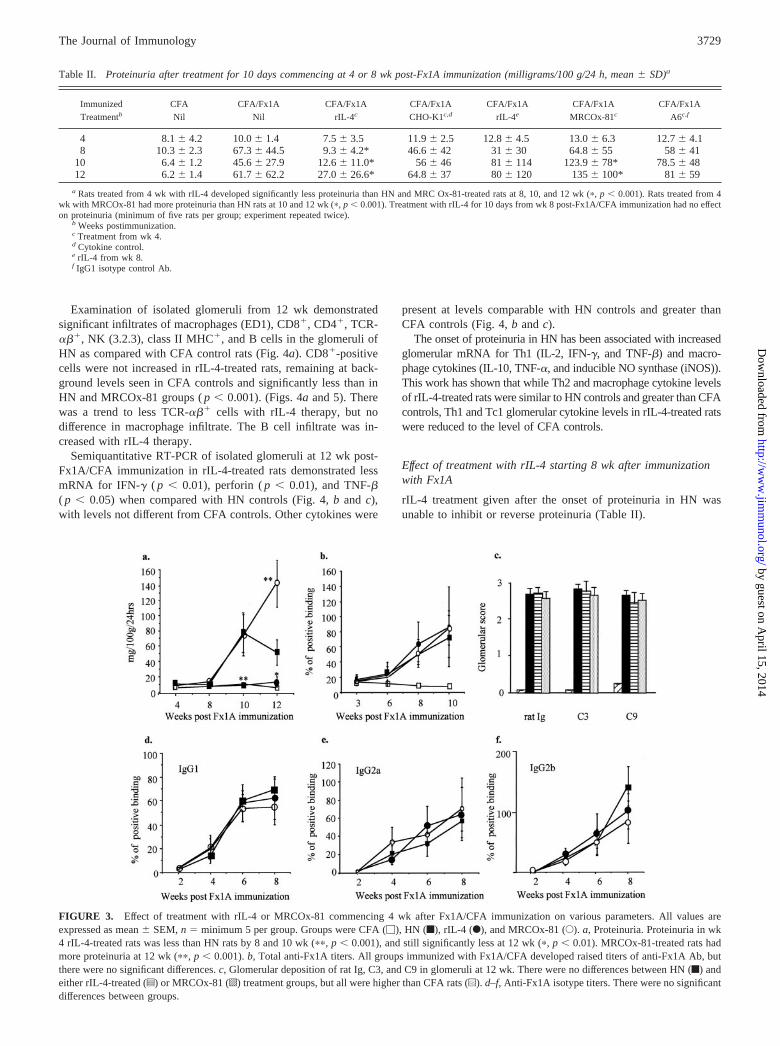
Effect of treatment with rIL-4 and MRCOx-81 from 4–6 wkafter Fx1A/CFA immunization
Therapy with rIL-4 commencing 4 wk after CFA/Fx1A immuni-zation inhibited the development of proteinuria at 8, 10, and 12 wkcompared with HN controls and controls sham treated with super-natant from CHO-K1 cells not transfected with cytokine (Table IIand Fig. 3a). Only 2 rats of 11 (from two experiments) treated withrIL-4 at 4 wk went on to develop proteinuria. MRCOx-81 therapyagain increased proteinuria by 12 wk compared with HN controlsand isotype control mAb-treated rats (Table II).
Serum anti-Fx1A titers did not differ between treatment groups,and there was not the Ig isotype switch that was observed withearly rIL-4 or MRCOx-81 therapy (Fig. 3, b and d–f). There wasalso no difference in staining for rat Ig, C3, or C9 among HN-,rIL-4-, and MRCOx-81-treated rats (Fig. 3c). Anti-Crry Abs wereagain only detected in sera from rIL-4-treated rats, but at levelslower than from early rIL-4-treated rats (Fig. 1e).
FIGURE 2. a, Percentage of lympho-cyte subpopulations in popliteal lymphnode. All rats immunized with eitherCFA or Fx1A/CFA had a significantlygreater percentage of CD4� and CD8� Tcells than untreated rats (�, p � 0.05).There were no significant differences in Tcell subsets between treatment groups. b,The cytokine profile of popliteal lymphnode tissue by semiquantitative RT-PCRat 2 wk is shown. There was significantelevation of IL-4 mRNA in both therIL-4- and MRCOx-81-treated groups (�,p � 0.05), but no other difference. Re-sults expressed as mean cycle number ofappearance of bands � SEM. c, Repre-sentative cytokine mRNA profiles ofpopliteal lymph node. Results for eachcytokine are shown as duplicate bands at5-cycle intervals from 20 to 35 cycles.GAPDH was assayed from 10 to 25cycles.
Table I. Proteinuria after treatment with rIL-4 and MRCOx-81 for 10 days commencing at the time of Fx1A immunization (milligrams/100 g/24 h,mean � SD)a
Immunized CFA CFA/Fx1A CFA/Fx1A CFA/Fx1A CFA/Fx1A CFA/Fx1ATreatmentb Nil Nil rIL-4 CHO-K1c MRCOx-81 A6d
4 6.6 � 0.8 10.5 � 1.6 7.7 � 0.9 7.4 � 1.8 8.4 � 1.0 13.1 � 2.78 8.3 � 2.7 51.3 � 14.2 65.3 � 9.3 48.4 � 27 64.6 � 10.4 75 � 15
10 8.9 � 1.2 107 � 19.4 140.3 � 74 113 � 47 217 � 63* 108 � 3812 10.8 � 1.9 195 � 63 181.5 � 41 201 � 35 268 � 44** 195 � 58
a All treatments given �10 days from the time of immunization with Fx1A/CFA. Treatment with MRCOx-81 increased proteinuria at 10 wk (�, p � 0.05) and 12 wk (��,p � 0.02) compared with HN controls. Treatment with rIL-4 at the time of Fx1A/CFA immunization had no effect on proteinuria (minimum of five rats per group; experimentrepeated twice).
b Weeks postimmunization.c Cytokine control.d IgG1 isotype control Ab.
3728 IL-4 THERAPY IN HN
by guest on April 15, 2014
http://ww
w.jim
munol.org/
Dow
nloaded from
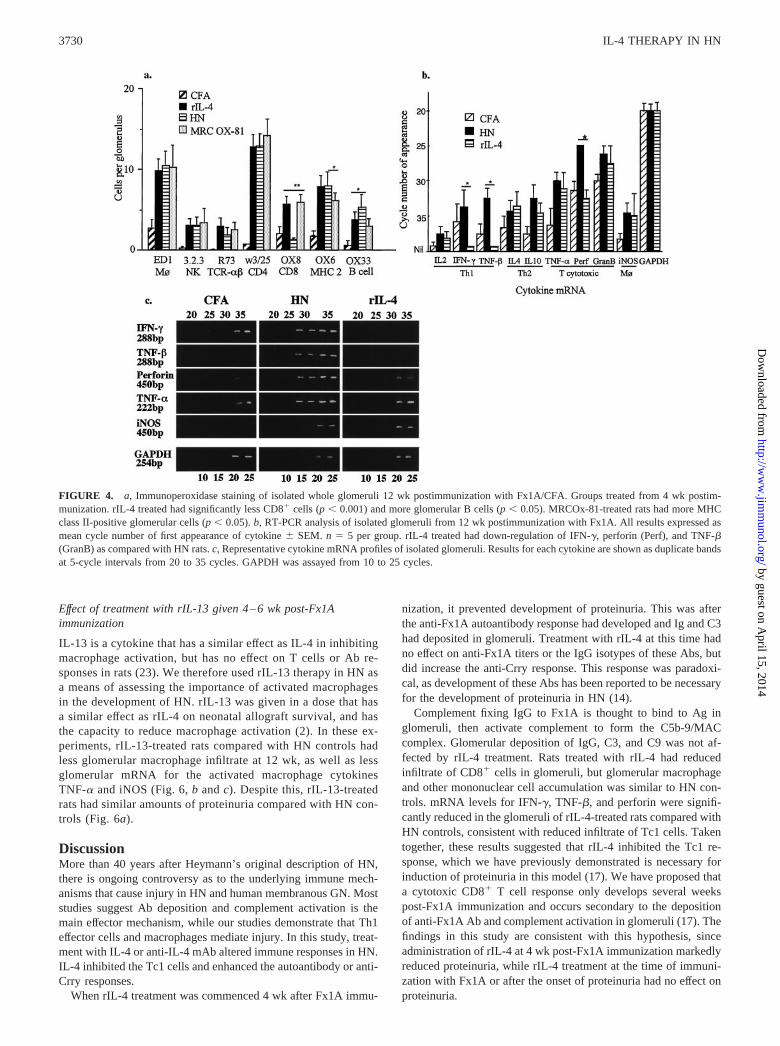
Examination of isolated glomeruli from 12 wk demonstratedsignificant infiltrates of macrophages (ED1), CD8�, CD4�, TCR-���, NK (3.2.3), class II MHC�, and B cells in the glomeruli ofHN as compared with CFA control rats (Fig. 4a). CD8�-positivecells were not increased in rIL-4-treated rats, remaining at back-ground levels seen in CFA controls and significantly less than inHN and MRCOx-81 groups ( p � 0.001). (Figs. 4a and 5). Therewas a trend to less TCR-��� cells with rIL-4 therapy, but nodifference in macrophage infiltrate. The B cell infiltrate was in-creased with rIL-4 therapy.
Semiquantitative RT-PCR of isolated glomeruli at 12 wk post-Fx1A/CFA immunization in rIL-4-treated rats demonstrated lessmRNA for IFN-� ( p � 0.01), perforin ( p � 0.01), and TNF-�( p � 0.05) when compared with HN controls (Fig. 4, b and c),with levels not different from CFA controls. Other cytokines were
present at levels comparable with HN controls and greater thanCFA controls (Fig. 4, b and c).
The onset of proteinuria in HN has been associated with increasedglomerular mRNA for Th1 (IL-2, IFN-�, and TNF-�) and macro-phage cytokines (IL-10, TNF-�, and inducible NO synthase (iNOS)).This work has shown that while Th2 and macrophage cytokine levelsof rIL-4-treated rats were similar to HN controls and greater than CFAcontrols, Th1 and Tc1 glomerular cytokine levels in rIL-4-treated ratswere reduced to the level of CFA controls.
Effect of treatment with rIL-4 starting 8 wk after immunizationwith Fx1A
rIL-4 treatment given after the onset of proteinuria in HN wasunable to inhibit or reverse proteinuria (Table II).
FIGURE 3. Effect of treatment with rIL-4 or MRCOx-81 commencing 4 wk after Fx1A/CFA immunization on various parameters. All values areexpressed as mean � SEM, n � minimum 5 per group. Groups were CFA (�), HN (f), rIL-4 (F), and MRCOx-81 (E). a, Proteinuria. Proteinuria in wk4 rIL-4-treated rats was less than HN rats by 8 and 10 wk (��, p � 0.001), and still significantly less at 12 wk (�, p � 0.01). MRCOx-81-treated rats hadmore proteinuria at 12 wk (��, p � 0.001). b, Total anti-Fx1A titers. All groups immunized with Fx1A/CFA developed raised titers of anti-Fx1A Ab, butthere were no significant differences. c, Glomerular deposition of rat Ig, C3, and C9 in glomeruli at 12 wk. There were no differences between HN (f) andeither rIL-4-treated (z) or MRCOx-81 (o) treatment groups, but all were higher than CFA rats (u). d–f, Anti-Fx1A isotype titers. There were no significantdifferences between groups.
Table II. Proteinuria after treatment for 10 days commencing at 4 or 8 wk post-Fx1A immunization (milligrams/100 g/24 h, mean � SD)a
Immunized CFA CFA/Fx1A CFA/Fx1A CFA/Fx1A CFA/Fx1A CFA/Fx1A CFA/Fx1ATreatmentb Nil Nil rIL-4c CHO-K1c,d rIL-4e MRCOx-81c A6c,f
4 8.1 � 4.2 10.0 � 1.4 7.5 � 3.5 11.9 � 2.5 12.8 � 4.5 13.0 � 6.3 12.7 � 4.18 10.3 � 2.3 67.3 � 44.5 9.3 � 4.2* 46.6 � 42 31 � 30 64.8 � 55 58 � 41
10 6.4 � 1.2 45.6 � 27.9 12.6 � 11.0* 56 � 46 81 � 114 123.9 � 78* 78.5 � 4812 6.2 � 1.4 61.7 � 62.2 27.0 � 26.6* 64.8 � 37 80 � 120 135 � 100* 81 � 59
a Rats treated from 4 wk with rIL-4 developed significantly less proteinuria than HN and MRC Ox-81-treated rats at 8, 10, and 12 wk (�, p � 0.001). Rats treated from 4wk with MRCOx-81 had more proteinuria than HN rats at 10 and 12 wk (�, p � 0.001). Treatment with rIL-4 for 10 days from wk 8 post-Fx1A/CFA immunization had no effecton proteinuria (minimum of five rats per group; experiment repeated twice).
b Weeks postimmunization.c Treatment from wk 4.d Cytokine control.e rIL-4 from wk 8.f IgG1 isotype control Ab.
3729The Journal of Immunology
by guest on April 15, 2014
http://ww
w.jim
munol.org/
Dow
nloaded from
Effect of treatment with rIL-13 given 4–6 wk post-Fx1Aimmunization
IL-13 is a cytokine that has a similar effect as IL-4 in inhibitingmacrophage activation, but has no effect on T cells or Ab re-sponses in rats (23). We therefore used rIL-13 therapy in HN asa means of assessing the importance of activated macrophagesin the development of HN. rIL-13 was given in a dose that hasa similar effect as rIL-4 on neonatal allograft survival, and hasthe capacity to reduce macrophage activation (2). In these ex-periments, rIL-13-treated rats compared with HN controls hadless glomerular macrophage infiltrate at 12 wk, as well as lessglomerular mRNA for the activated macrophage cytokinesTNF-� and iNOS (Fig. 6, b and c). Despite this, rIL-13-treatedrats had similar amounts of proteinuria compared with HN con-trols (Fig. 6a).
DiscussionMore than 40 years after Heymann’s original description of HN,there is ongoing controversy as to the underlying immune mech-anisms that cause injury in HN and human membranous GN. Moststudies suggest Ab deposition and complement activation is themain effector mechanism, while our studies demonstrate that Th1effector cells and macrophages mediate injury. In this study, treat-ment with IL-4 or anti-IL-4 mAb altered immune responses in HN.IL-4 inhibited the Tc1 cells and enhanced the autoantibody or anti-Crry responses.
When rIL-4 treatment was commenced 4 wk after Fx1A immu-
nization, it prevented development of proteinuria. This was afterthe anti-Fx1A autoantibody response had developed and Ig and C3had deposited in glomeruli. Treatment with rIL-4 at this time hadno effect on anti-Fx1A titers or the IgG isotypes of these Abs, butdid increase the anti-Crry response. This response was paradoxi-cal, as development of these Abs has been reported to be necessaryfor the development of proteinuria in HN (14).
Complement fixing IgG to Fx1A is thought to bind to Ag inglomeruli, then activate complement to form the C5b-9/MACcomplex. Glomerular deposition of IgG, C3, and C9 was not af-fected by rIL-4 treatment. Rats treated with rIL-4 had reducedinfiltrate of CD8� cells in glomeruli, but glomerular macrophageand other mononuclear cell accumulation was similar to HN con-trols. mRNA levels for IFN-�, TNF-�, and perforin were signifi-cantly reduced in the glomeruli of rIL-4-treated rats compared withHN controls, consistent with reduced infiltrate of Tc1 cells. Takentogether, these results suggested that rIL-4 inhibited the Tc1 re-sponse, which we have previously demonstrated is necessary forinduction of proteinuria in this model (17). We have proposed thata cytotoxic CD8� T cell response only develops several weekspost-Fx1A immunization and occurs secondary to the depositionof anti-Fx1A Ab and complement activation in glomeruli (17). Thefindings in this study are consistent with this hypothesis, sinceadministration of rIL-4 at 4 wk post-Fx1A immunization markedlyreduced proteinuria, while rIL-4 treatment at the time of immuni-zation with Fx1A or after the onset of proteinuria had no effect onproteinuria.
FIGURE 4. a, Immunoperoxidase staining of isolated whole glomeruli 12 wk postimmunization with Fx1A/CFA. Groups treated from 4 wk postim-munization. rIL-4 treated had significantly less CD8� cells (p � 0.001) and more glomerular B cells (p � 0.05). MRCOx-81-treated rats had more MHCclass II-positive glomerular cells (p � 0.05). b, RT-PCR analysis of isolated glomeruli from 12 wk postimmunization with Fx1A. All results expressed asmean cycle number of first appearance of cytokine � SEM. n � 5 per group. rIL-4 treated had down-regulation of IFN-�, perforin (Perf), and TNF-�(GranB) as compared with HN rats. c, Representative cytokine mRNA profiles of isolated glomeruli. Results for each cytokine are shown as duplicate bandsat 5-cycle intervals from 20 to 35 cycles. GAPDH was assayed from 10 to 25 cycles.
3730 IL-4 THERAPY IN HN
by guest on April 15, 2014
http://ww
w.jim
munol.org/
Dow
nloaded from
Therapy with rIL-4 at the time of Fx1A immunization causedan accelerated autoantibody response with isotype switching to en-hance IgG1 and reduce complement-fixing IgG2a, as well as in-creased IL-4 mRNA expression in the draining lymph nodes. Thesefindings demonstrated rIL-4 induced Th2 cells. Even so, these ratsdeveloped proteinuria. The failure of early rIL-4 treatment to stop
proteinuria also supports our proposal that a delayed CD8� T cellresponse is the mediator of injury in HN, as the short-acting rIL-4would not be available at the later time when cytotoxic T cells areactivated.
Anti-IL-4 mAb (MRCOx-81) therapy when given at the time ofimmunization or 4–6 wk after immunization with Fx1A resulted
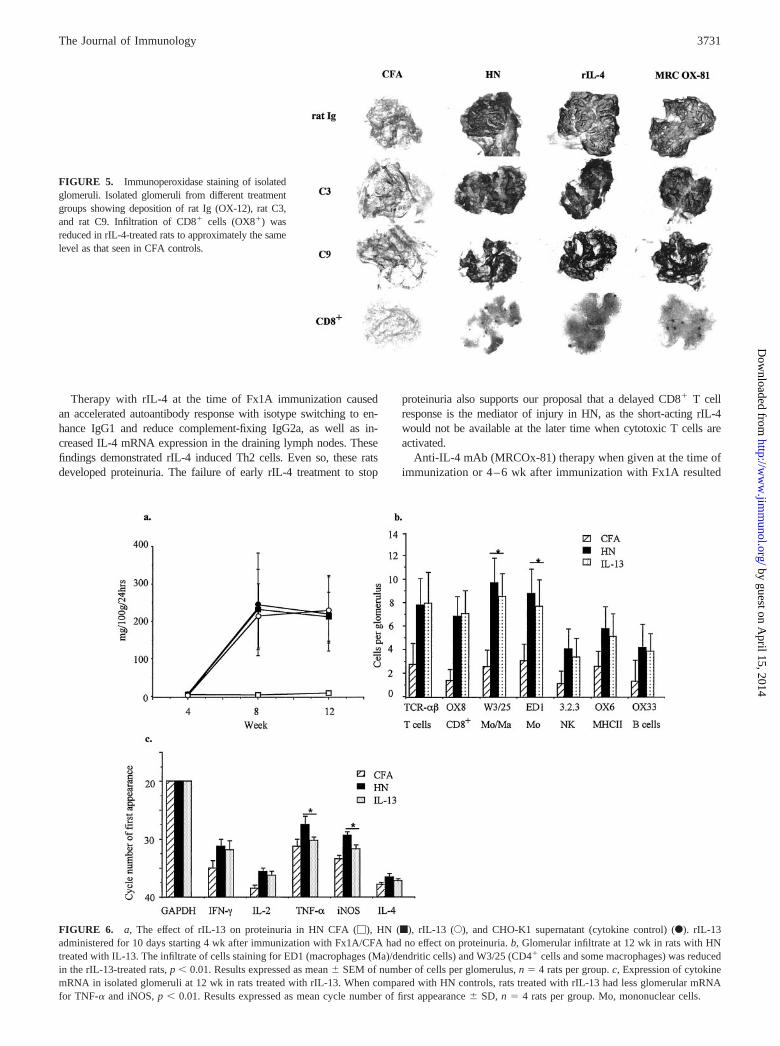
FIGURE 5. Immunoperoxidase staining of isolatedglomeruli. Isolated glomeruli from different treatmentgroups showing deposition of rat Ig (OX-12), rat C3,and rat C9. Infiltration of CD8� cells (OX8�) wasreduced in rIL-4-treated rats to approximately the samelevel as that seen in CFA controls.
FIGURE 6. a, The effect of rIL-13 on proteinuria in HN CFA (�), HN (f), rIL-13 (E), and CHO-K1 supernatant (cytokine control) (F). rIL-13administered for 10 days starting 4 wk after immunization with Fx1A/CFA had no effect on proteinuria. b, Glomerular infiltrate at 12 wk in rats with HNtreated with IL-13. The infiltrate of cells staining for ED1 (macrophages (Ma)/dendritic cells) and W3/25 (CD4� cells and some macrophages) was reducedin the rIL-13-treated rats, p � 0.01. Results expressed as mean � SEM of number of cells per glomerulus, n � 4 rats per group. c, Expression of cytokinemRNA in isolated glomeruli at 12 wk in rats treated with rIL-13. When compared with HN controls, rats treated with rIL-13 had less glomerular mRNAfor TNF-� and iNOS, p � 0.01. Results expressed as mean cycle number of first appearance � SD, n � 4 rats per group. Mo, mononuclear cells.
3731The Journal of Immunology
by guest on April 15, 2014
http://ww
w.jim
munol.org/
Dow
nloaded from

in greater proteinuria than untreated HN controls. Early therapywith MRCOx-81 caused increased IgG2a and reduced IgG1 anti-Fx1A titers consistent with it blocking IL-4 and allowing Th1 cellsto facilitate isotype switching to complement-fixing Ig. MRCOx-81-treated rats had increased macrophage infiltrate into renal cor-tex, which may have been due to an enhanced Th1 response or thegreater proteinuria. The higher IL-4 mRNA in popliteal lymphnode is consistent with MRCOx-81 blocking released IL-4, but notproduction of IL-4 at a cellular level. Similar increased IL-4mRNA has been observed in MRCOx-81-treated transplant recip-ients (29). The loss of negative feedback from IL-4 may enhanceits production by Th2 cells, resulting in higher mRNA for IL-4.The worsening of proteinuria seen with blocking IL-4 is consistentwith IL-4 having a natural role in regulation of severity of disease.
Therapy with rIL-13 reduced both glomerular macrophage ac-cumulation and expression of mRNA for activated macrophagecytokines TNF-� and iNOS in glomeruli, but rIL-13 had no effecton proteinuria in HN or on Tc1 cell accumulation in glomeruli.This suggests that the glomerular macrophages seen in HN are notthe primary cause of proteinuria, and supports the finding thatrIL-4 therapy reduced proteinuria by inhibition of Tc1 cell, but notmacrophage infiltrate into glomeruli. This finding is in distinctionto the disease-suppressive effect of IL-13 therapy in experimentalautoimmune encephalomyelitis, a disease in which macrophageinjury is important and Tc1 cells play little or no role (23).
Taken together, the results of this study support the role of cel-lular immune mechanisms in the glomerular injury of HN. Earlywork by Heymann and others showed that splenic or lymph nodecells from proteinuric rats could transfer nephrosis to naive rats,suggesting the importance of cellular immune mechanisms in thepathogenesis of this model (44, 45). However, subsequent study ofactive and PHN has focused on the effects of IgG deposition andcomplement activation, concluding that sublytic MAC injury ofthe GEC leads to the typical GBM thickening and proteinuria ofHN (11). The presence of subepithelial IgG and C3 in the GBM isone of the hallmarks of HN and human membranous nephropathy,and the importance of the early complement components in PHNis well established. Rats depleted of complement with cobravenom factor do not develop proteinuria in PHN (13, 46, 47), andBaker et al. (48) showed that depletion of C6 with a mAb pre-vented proteinuria in PHN. However, the pathogenic role of theMAC complex has been questioned with the induction of bothactive HN (15) and PHN (49, 50) in a C6-deficient PVG rat strain.Furthermore, a Th1/Tc1 lymphocyte and macrophage infiltrate intothe glomeruli of HN rats parallels the course of proteinuria (16),and rats depleted of CD8� T cells do not develop proteinuria (17).
Since it has been assumed that the pathogenic role of comple-ment is entirely due to MAC, little attention has been paid to otherpotential immunostimulatory or chemotactic effects of early com-plement components deposited in the GBM. The finding that earlycomplement components are required for development of PHN isstill consistent with our proposal that secondary cellular immunemechanisms are operative in HN, as cellular immune injury isstimulated at sites of immune complex formation or deposition.C3b and C4b are important in facilitating Ag presentation andactivation of T cell responses (51–53).
Recent support for complement-mediated injury in HN camefrom the finding that proteinuria in HN required development ofAbs to Crry that blocked the function of this complement-deacti-vating molecule and allowed complement-mediated injury to pro-ceed (14). It is important to note that Crry (the rodent equivalentof decay-accelerating factor and monocyte chemoattractant pro-tein) inhibits complement activation at the C3 level, while anothermolecule, CD59, is required for inhibition of the MAC. In the
present study, anti-Crry Abs were only detected in rats treated withrIL-4. They were not detected in HN controls or our anti-IL-4 mAb(MRCOx-81)-treated groups, all of which developed massive pro-teinuria. An alternative explanation for the anti-Crry relationshipto proteinuria is that less pure Ag preparations elicit autoantibodiesto many glomerular Ags other than to gp330, including Crry. It isthese Abs that may be required to trigger the secondary CD8� Tcell response. CD8� T cell-mediated injury has been found to bethe principal mechanism of injury in experimental interstitial ne-phritis (54–56). Furthermore, antitubular basement membrane-specific CD8� T cell clones with low IFN-� and perforin expres-sion do not induce interstitial nephritis (57). Perforin has beenshown to effect sublytic cellular injury in nucleated cells (58) anal-ogous to the effects of MAC, and we propose that sublytic injuryof the GEC is conducted by nephritogenic Tc1 clones.
IL-4 is the prototypic Th2 cytokine (18) that has multiple effectson activated T lymphocytes, including directing the developmentof Th2 cells and blocking the development of Th1 cells (59, 60).IL-4 has been used successfully as an antiinflammatory agent inTh1-type animal models of autoimmune disease, such as rodentmodels of crescentic (61) and anti-GBM glomerulonephritis (3),experimental autoimmune encephalomyelitis (62), arthritis (63),and diabetes (64). It is generally thought that the antiinflammatoryeffect of IL-4 in these models is due to induction of a Th2 phe-notype combined with direct inhibition of the Th1 response. IL-4is known to cause specific phenotypic as well as functional inhi-bition of CD8� Th1 cells (Tc1 cells): CD8� cells activated in thepresence of IL-4 stop expressing CD8, IFN-�, and perforin (19),and are thus rendered noncytotoxic. The reduction in glomerularCD8� T cell infiltration, but not macrophage infiltration seen withrIL-4 therapy suggests that the rIL-4 effect in HN is mediatedthrough specific inhibition of nephritogenic Tc1 lymphocytes.
AcknowledgmentsWe thank Moheb Boutros for expert work as the director of the animalfacility at Liverpool Hospital.
References1. Santiago, M. L., L. Fossati, C. Jacquet, W. Muller, S. Izui, and L. Reininger.
1997. Interleukin-4 protects against a genetically linked lupus-like autoimmunesyndrome. J. Exp. Med. 185:65.
2. Coelho, S. N., S. Saleem, B. T. Konieczny, K. R. Parekh, F. K. Baddoura, andF. G. Lakkis. 1997. Immunologic determinants of susceptibility to experimentalglomerulonephritis; role of cellular immunity. Kidney Int. 51:646.
3. Tam, F. W., J. Smith, A. M. Karkar, C. D. Pusey, and A. J. Rees. 1997. Inter-leukin-4 ameliorates experimental glomerulonephritis and up-regulates glomer-ular gene expression of IL-1 decoy receptor. Kidney Int. 52:1224.
4. Tipping, P. G., A. R. Kitching, X. R. Huang, D. A. Mutch, and S. R. Holdsworth.1997. Immune modulation with interleukin-4 and interleukin-10 prevents cres-cent formation and glomerular injury in experimental glomerulonephritis. Eur.J. Immunol. 27:530.
5. Saoudi, A., M. Castedo, D. Nochy, C. Mandet, R. Pasquier, P. Druet, and L. Pelletier.1995. Self-reactive anti-class II T helper type 2 cell lines derived from gold salt-injected rats trigger B cell polyclonal activation and transfer autoimmunity in CD8-depleted normal syngeneic recipients. Eur. J. Immunol. 25:1972.
6. Van der Meide, P. H., R. J. Groenestein, M. C. de Labie, J. Aten, andJ. J. Weening. 1995. Susceptibility to mercuric chloride-induced glomerulone-phritis is age-dependent: study of the role of IFN-�. Cell. Immunol. 162:131.
7. Heymann, W., D. B. Hackel, S. Harwood, S. G. Wilson, and J. L. P. Hunter.1959. Production of nephrotic syndrome in rats by Freund’s adjuvants and ratkidney suspensions. Proc. Soc. Exp. Biol. Med. 100:660.
8. Heymann, W., D. B. Hackel, S. Harwood, S. G. Wilson, and J. L. Hunter. 2000.Production of nephrotic syndrome in rats by Freund’s adjuvants and rat kidneysuspensions. 1951. J. Am. Soc. Nephrol. 11:183.
9. Kerjaschki, D., and M. G. Farquhar. 1983. Immunocytochemical localization ofthe Heymann nephritis antigen (GP330) in glomerular epithelial cells of normalLewis rats. J. Exp. Med. 157:667.
10. Quigg, R. J., A. V. Cybulsky, J. B. Jacobs, and D. J. Salant. 1988. Anti-Fx1Aproduces complement-dependent cytotoxicity of glomerular epithelial cells. Kid-ney Int. 34:43.
11. Couser, W. G. 1998. Pathogenesis of glomerular damage in glomerulonephritis.Nephrol. Dial. Transplant. 13:10.
3732 IL-4 THERAPY IN HN
by guest on April 15, 2014
http://ww
w.jim
munol.org/
Dow
nloaded from

12. Salant, D. J., S. Belok, M. P. Madaio, and W. G. Couser. 1980. A new role forcomplement in experimental membranous nephropathy in rats. J. Clin. Invest.66:1339.
13. Gabbai, F. B., C. A. Mundy, C. B. Wilson, and R. C. Blantz. 1988. An evaluationof the role of complement depletion in experimental membranous nephropathy inthe rat. Lab. Invest. 58:539.
14. Schiller, B., C. He, D. J. Salant, A. Lim, J. J. Alexander, and R. J. Quigg. 1998.Inhibition of complement regulation is key to the pathogenesis of active Hey-mann nephritis. J. Exp. Med. 188:1353.
15. Leenaerts, P. L., B. M. Hall, D. B. J. Van, M. R. Daha, and Y. F. Vanrenterghem.1995. Active Heymann nephritis in complement component C6 deficient rats.Kidney Int. 47:1604.
16. Penny, M. J., R. A. Boyd, and B. M. Hall. 1997. Role of T cells in the mediationof Heymann nephritis. ii. Identification of Th1 and cytotoxic cells in glomeruli.Kidney Int. 51:1059.
17. Penny, M. J., R. A. Boyd, and B. M. Hall. 1998. Permanent CD8� T cell de-pletion prevents proteinuria in active Heymann nephritis. J. Exp. Med. 188:1775.
18. Paul, W. E. 1991. Interleukin-4: a prototypic immunoregulatory lymphokine.Blood 77:1859.
19. Erard, F., M. T. Wild, J. A. Garcia-Sanz, and G. Le Gros. 1993. Switch of CD8T cells to noncytolytic CD8�CD4� cells that make TH2 cytokines and help Bcells. Science 260:1802.
20. Haas, M., B. H. Spargo, and S. Coventry. 1995. Increasing incidence of focal-segmental glomerulosclerosis among adult nephropathies: a 20-year renal biopsystudy. Am. J. Kidney Dis. 26:740.
21. Hart, P. H., G. F. Vitti, D. R. Burgess, G. A. Whitty, D. S. Piccoli, andJ. A. Hamilton. 1989. Potential antiinflammatory effects of interleukin 4: sup-pression of human monocyte tumor necrosis factor �, interleukin 1, and prosta-glandin E2. Proc. Natl. Acad. Sci. USA 86:3803.
22. Jungi, T. W., M. Brcic, H. Sager, D. A. Dobbelaere, A. Furger, and I. Roditi.1997. Antagonistic effects of IL-4 and interferon-� (IFN-�) on inducible nitricoxide synthase expression in bovine macrophages exposed to Gram-positive bac-teria. Clin. Exp. Immunol. 109:431.
23. Cash, E., A. Minty, P. Ferrara, D. Caput, D. Fradelizi, and O. Rott. 1994. Mac-rophage-inactivating IL-13 suppresses experimental autoimmune encephalomy-elitis in rats. J. Immunol. 153:4258.
24. Chomarat, P., and J. Banchereau. 1998. Interleukin-4 and interleukin-13: theirsimilarities and discrepancies. Int. Rev. Immunol. 17:1.
25. Edgington, T. S., R. J. Glassock, and F. J. Dixon. 1968. Autologous immunecomplex nephritis induced with renal tubular antigen. I. Identification and isola-tion of the pathogenetic antigen. J. Exp. Med. 127:555.
26. McKnight, A. J., and B. J. Classon. 1992. Biochemical and immunological prop-erties of rat recombinant interleukin-2 and interleukin-4. Immunology 75:286.
27. Hall, B. M., L. Fava, J. Chen, K. M. Plain, R. A. Boyd, S. T. Spicer, andM. F. Berger. 1998. Anti-CD4 monoclonal antibody-induced tolerance to MHC-incompatible cardiac allografts maintained by CD4� suppressor T cells that arenot dependent upon IL-4. J. Immunol. 161:5147.
28. Ramirez, F., D. J. Fowell, M. Puklavec, S. Simmonds, and D. Mason. 1996.Glucocorticoids promote a TH2 cytokine response by CD4� T cells in vitro.J. Immunol. 156:2406.
29. He, X. Y., J. Chen, N. Verma, K. Plain, G. Tran, and B. M. Hall. 1998. Treatmentwith interleukin-4 prolongs allogeneic neonatal heart graft survival by inducingT helper 2 responses. Transplantation 65:1145.
30. He, X. Y., N. Verma, K. Plain, and B. M. Hall. 1999. Interleukin 13 cloning fromDA rats. Transplant. Proc. 31:1572.
31. Lakkis, F. G., E. N. Cruet, G. M. Nassar, K. F. Badr, and D. W. Pascual. 1997.Expression of recombinant rat interleukin-13 (IL-13) and generation of a neu-tralizing rat IL-13 antiserum. Biochim. Biophys. Acta 235:529.
32. Aversa, G., J. A. Waugh, and B. M. Hall. 1994. A monoclonal antibody (A6)recognizing a unique epitope restricted to CD45RO and RB isoforms of theleukocyte common antigen family identifies functional T cell subsets. Cell. Im-munol. 158:314.
33. Cheng, I. K., S. E. Dorsch, and B. M. Hall. 1984. Lymphocyte subsets in Hey-mann nephritis. Lab. Invest. 51:286.
34. Hunt, S. V., and M. H. Fowler. 1981. A repopulation assay for B and T lym-phocyte stem cells employing radiation chimeras. Cell Tissue Kinet. 14:445.
35. Linington, C., H. Lassmann, B. P. Morgan, and D. A. Compston. 1989. Immu-nohistochemical localization of terminal complement component C9 in experi-mental allergic encephalomyelitis. Acta Neuropathol. 79:78.
36. Hunig, T., H. J. Wallny, J. K. Hartley, A. Lawetzky, and G. Tiefenthaler. 1989.A monoclonal antibody to a constant determinant of the rat T cell antigen receptorthat induces T cell activation: differential reactivity with subsets of immature andmature T lymphocytes. J. Exp. Med. 169:73.
37. Williams, A. F., G. Galfre, and C. Milstein. 1977. Analysis of cell surfaces byxenogenic myeloma-hybrid antibodies: differentiation antigens of rat lympho-cytes. Cell 12:663.
38. Brideau, R. J., P. B. Carter, W. R. McMaster, D. W. Mason, and A. F. Williams.1980. Two subsets of rat T lymphocytes defined with monoclonal antibodies.Eur. J. Immunol. 10:609.
39. Dijkstra, C. D., E. A. Dopp, P. Joling, and G. Kraal. 1985. The heterogeneity ofmononuclear phagocytes in lymphoid organs: distinct macrophage subpopula-tions in the rat recognized by monoclonal antibodies ED1, ED2 and ED3. Im-munology 54:589.
40. Woollett, G. R., A. N. Barclay, and M. Pulkavec. 1985. Molecular and antigenicheterogeneity of the rat leukocyte common antigen from thymocytes and T andB lymphocytes. Eur. J. Immunol. 15:168.
41. Hall, B. M., I. DeSaxe, and S. E. Dorsch. 1983. The cellular basis of allograftrejection in vivo. III. Restoration of first-set rejection of heart grafts by T helpercells in irradiated rats. Transplantation 36:700.
42. Dallman, M. J., and A. C. G. Porter. 1991. Semi-quantitative PCR for the analysisof gene expression. In PCR: A Practical Approach. M. J. McPherson, P. Quirke,and G. R. Taylor, eds. Oxford University Press, Oxford, p. 215.
43. Plain, K. M., J. Chen, S. Merten, X. Y. He, and B. M. Hall. 1999. Induction ofspecific tolerance to allografts in rats by therapy with non-mitogenic, non-de-pleting anti-CD3 monoclonal antibody: association with TH2 cytokines not an-ergy. Transplantation 67:605.
44. Heymann, W., J. L. P. Hunter, D. B. Hackel, and F. Cuppage. 1962. Transfer ofexperimental autoimmune nephrosis in rats. Proc. Soc. Exp. Biol. Med. 111:568.
45. Hess, E. V., C. T. Ashworth, and M. Ziff. 1962. Transfer of an autoimmunenephrosis in the rat by means of lymph node cells. J. Exp. Med. 115:421.
46. Lianos, E. A., and B. Noble. 1989. Glomerular leukotriene synthesis in Heymannnephritis. Kidney Int. 36:998.
47. Hara, M., S. R. Batsford, M. J. Mihatsch, S. D. Bitter, and A. Vogt. 1991. Com-plement and monocytes are essential for provoking glomerular injury in passiveHeymann nephritis in rats: terminal complement components are not the solemediators of proteinuria. Lab. Invest. 65:168.
48. Baker, P. J., R. F. Ochi, M. Schulze, R. J. Johnson, C. Campbell, andW. G. Couser. 1989. Depletion of C6 prevents development of proteinuria inexperimental membranous nephropathy in rats. Am. J. Pathol. 135:185.
49. Minto, A. W. M., C. D. Andry, and D. J. Salant. 1996. C5b-9-independent glo-merular basement membrane expansion in experimental membranous nephropa-thy. In American Society of Nephrology 29th Annual Scientific Meeting, J. Am.Soc. Nephrol. Vol. 7. New Orleans, p. 1740A.
50. Spicer, S. T., M. J. Penny, M. Killinsworth, K. Paizis, D. Power, and B. M. Hall.1999. Induction of PHN in the C6-deficient PVG rat: glomerular injury and pro-teinuria in PHN is independent of the membrane attack complex. In AmericanSociety of Nephrology 32nd Annual Scientific Meeting, J. Am. Soc. Nephrol. Vol.10, Miami Beach, p. 538A.
51. Arvieux, J., H. Yssel, and M. G. Colomb. 1988. Antigen-bound C3b and C4benhance antigen-presenting cell function in activation of human T-cell clones.Immunology 65:229.
52. Jacquier-Sarlin, M. R., F. M. Gabert, M. B. Villiers, and M. G. Colomb. 1995.Modulation of antigen processing and presentation by covalently linked comple-ment C3b fragment. Immunology 84:164.
53. Miletic, V. D., and M. M. Frank. 1995. Complement-immunoglobulin interac-tions. Curr. Opin. Immunol. 7:41.
54. Neilson, E. G., E. McCafferty, R. Mann, L. Michaud, and M. Clayman. 1985.Murine interstitial nephritis. III. The selection of phenotypic (Lyt and L3T4) andidiotypic (RE-Id) T cell preferences by genes in Igh-1 and H-2K characterizes thecell-mediated potential for disease expression: susceptible mice provide a uniqueeffector T cell repertoire in response to tubular antigen. J. Immunol. 134:2375.
55. Kelly, C. J., M. D. Clayman, and E. G. Neilson. 1986. Immunoregulation inexperimental interstitial nephritis: immunization with renal tubular antigen inincomplete Freund’s adjuvant induces major histocompatibility complex-re-stricted, OX8� suppressor T cells which are antigen-specific and inhibit the ex-pression of disease. J. Immunol. 136:903.
56. Meyers, C. M., and C. J. Kelly. 1991. Effector mechanisms in organ-specificautoimmunity. I. Characterization of a CD8� T cell line that mediates murineinterstitial nephritis. J. Clin. Invest. 88:408.
57. Meyers, C. M., and C. J. Kelly. 1994. Immunoregulation and TGF-�1: suppres-sion of a nephritogenic murine T cell clone. Kidney Int. 46:1295.
58. Jones, J., M. B. Hallett, and B. P. Morgan. 1990. Reversible cell damage by T-cellperforins: calcium influx and propidium iodide uptake into K562 cells in theabsence of lysis. Biochem. J. 267:303.
59. Swain, S. L., A. D. Weinberg, M. English, and G. Huston. 1990. IL-4 directs thedevelopment of Th2-like helper effectors. J. Immunol. 145:3796.
60. Swain, S. L. 1993. IL4 dictates T-cell differentiation. Res. Immunol. 144:616.61. Kitching, A. R., P. G. Tipping, X. R. Huang, D. A. Mutch, and S. R. Holdsworth.
1997. Interleukin-4 and interleukin-10 attenuate established crescentic glomeru-lonephritis in mice. Kidney Int. 52:52.
62. Racke, M. K., A. Bonomo, D. E. Scott, B. Cannella, A. Levine, C. S. Raine,E. M. Shevach, and M. Rocken. 1994. Cytokine-induced immune deviation as atherapy for inflammatory autoimmune disease. J. Exp. Med. 180:1961.
63. Van Roon, J. A., J. L. van Roy, F. H. Gmelig-Meyling, F. P. Lafeber, andJ. W. Bijlsma. 1996. Prevention and reversal of cartilage degradation in rheu-matoid arthritis by interleukin-10 and interleukin-4. Arthritis Rheum. 39:829.
64. Tominaga, Y., M. Nagata, H. Yasuda, N. Okamoto, K. Arisawa, H. Moriyama,M. Miki, K. Yokono, and M. Kasuga. 1998. Administration of IL-4 preventsautoimmune diabetes but enhances pancreatic insulitis in NOD mice. Clin. Im-munol. Immunopathol. 86:209.
65. Spicer, S. T., R. Boyd, C. He, M. Penny, and B. M. Hall. 2000. Th2 immunedeviation prevents active Heymann nephritis: the role of Th1 cytotoxic cells andmacrophages: The 36th Australian and New Zealand Society of Nephrology An-nual Meeting. Melbourne, Australia, March 2000.
66. Spicer, S. T., R. Boyd, C. He, M. Penny, and B. M. Hall. 1999. Interleukin-4therapy prevents active Heymann nephritis, The 32nd American Society of Ne-phrology Annual Meeting. Miami Beach, FL, J. Am. Soc. Nephrol., Vol 10,p521A.
3733The Journal of Immunology
by guest on April 15, 2014
http://ww
w.jim
munol.org/
Dow
nloaded from




















