
Hydrochemical tracers in the middle Rio Grande Basin, USA: 1. Conceptualization of groundwater flow
Upload
independentCategory
view
3download
0
ARTICLE IN PRESS
Applied Radiation and Isotopes 66 (2008) 1531– 1542
Contents lists available at ScienceDirect
Applied Radiation and Isotopes
0969-80
doi:10.1
� Corr
E-m
journal homepage: www.elsevier.com/locate/apradiso
Uranium series disequilibria in ground waters from a fractured bedrockaquifer (Morungaba Granitoids—Southern Brazil): Implications to thehydrochemical behavior of dissolved U and Ra
Erika Reyes, Leila S. Marques �
Departamento de Geofısica, Instituto de Astronomia, Geofısica e Ciencias Atmosfericas, Universidade de Sao Paulo, Rua do Matao, 1226, CEP 05508-090 Sao Paulo, SP, Brazil
a r t i c l e i n f o
Keywords:
Uranium
Radium
Ground waters
Alpha spectrometry
Gamma-ray spectrometry
Fractured aquifers
43/$ - see front matter & 2008 Elsevier Ltd. A
016/j.apradiso.2008.04.003
esponding author. Tel.: +551130914755; fax:
ail address: [email protected] (L.S. Marques).
a b s t r a c t
Activity concentrations of dissolved 234U, 238U, 226Ra and 228Ra were determined in ground waters from
two deep wells drilled in Morungaba Granitoids (Southern Brazil). Sampling was done monthly for little
longer than 1 year. Significant disequilibrium between 238U, 234U and 226Ra were observed in all
samples. The variation of 238U and 234U activity concentrations and 234U/238U activity ratios is related to
seasonal changes. Although the distance between the two wells is short (about 900 m), systematic
differences of activity concentrations of U isotopes, as well as of 234U/238U, 226Ra/234U and 228Ra/226Ra
activity ratios were noticed, indicating distinct host rock–water interactions. Slightly acidic ground
water percolation through heterogeneous host rock, associated with different recharge processes, may
explain uranium and radium isotope behavior.
& 2008 Elsevier Ltd. All rights reserved.
1. Introduction
The radioisotopes 238U, 235U and 232Th are the sources of threenatural decay chains, which comprise several radioisotopespresenting distinct chemical and physical properties, and arelargely used in geochemical, geochronological and environmentalstudies (Ivanovich and Harmon, 1992). In undisturbed systems, forsufficiently long time (�1 Ma), the decay products of each chainachieve secular radioactive equilibrium, in which the activity ofthe parent is equal to those of all its daughters. Conversely to rocksystems, in which secular radioactive equilibrium is common,surface and ground waters are characterized by significantdisequilibria.
Concerning ground water systems, 238U and 234U are of greatimportance. Both isotopes belong to the same radioactive decaychain and disequilibrium between them is due mainly towater–host rock interaction, leading to preferential mobilizationof 234U, caused by the recoil of its parent. Therefore, thedetermination of 238U and 234U has been extensively used tostudy characteristics of host rocks and host rock–water interac-tions, including the determination of rock dissolution rates andother related physical and chemical processes (e.g. Osmond andCowart, 1992; Luo et al., 2000; Lee et al., 2001; Bourdon et al.,2003; Porcelli and Swarzenski, 2003; Robinson et al., 2004). The
ll rights reserved.
+551130915034.
behavior of both radioisotopes also allows investigating aquiferrecharge dynamics, such as meteoric contribution and mixing ofground waters from different sources, and to estimate residencetimes (e.g. Dabous and Osmond, 2000; Puigdomenech, 2001;Henderson et al., 2006; Bonotto and Jimenez-Rueda, 2007).Additionally, uranium isotopes are often employed to investigateheavy metal mobilization in hydrological systems, whose knowl-edge is essential in order to define metal (radioactive or not)waste disposal areas.
In hydrological systems, radium also originates from interac-tion between waters and geological materials (rocks and soils)containing this element and/or its radioactive parents. In addition,significant amounts of this element in waters come from theexploitation of radioactive minerals containing U and Th, whichare usually employed in nuclear energy generation, in thephosphate industry, as well as from the addition of phosphatefertilizers to agricultural terrains.
In nature, Ra is found as several radioisotopes with verydistinct half-lives, varying from seconds to thousands of years,and in the investigation of aquifer systems, activity concentrationsof 226Ra, 228Ra and 224Ra are the most used, since they provideconsiderable information about the dynamics of this element, aswell as that of its progenies, on different time scales (Dickson,1990). Although 228Ra/226Ra activity ratios may vary between 0.1and 2 in ground waters (Osmond and Cowart, 1992), 228Ra isusually the most abundant radium isotope dissolved in groundwaters, because its parent 232Th is naturally enriched incomparison to 238U, which only after five decays gives rise to

ARTICLE IN PRESS
E. Reyes, L.S. Marques / Applied Radiation and Isotopes 66 (2008) 1531–15421532
226Ra. Activity ratios of 228Ra/226Ra may provide importantconstraints on Ra lixiviation, ground water percolation in uraniumenriched zones, or on areas where Ra was accumulated duringearlier mobilization processes, as well as on the identification ofmixing between ground waters originated in different systems(Rihs and Condomines, 2002 and references therein).
This paper describes in detail the analytical methods used fordetermination of dissolved U and Ra concentrations, along with234U, 238U, 226Ra and 228Ra activity concentrations, present indrinking ground waters drawn from two deep wells drilled in afractured granitic body located in southern Brazil (MorungabaGranitoids). Alpha spectrometry was used for measurements ofactivity concentrations of uranium radioisotopes, whereas for thedetermination of radium isotopes the high-resolution gamma-rayspectrometry method was employed. Results obtained during 1year of sampling (the first in December, 2002, thereafter monthly,from March, 2003 to February, 2004), are also reported, allowinginvestigation of the geochemical behavior of those isotopes,specially the dynamic processes related to seasonal variationsand water–host rock interaction. Other physico-chemical para-meters, such as temperature and pH, were also measured, alongwith 234U/238U, 228Ra/226Ra and 234U/226Ra activity ratios.
2. Geological background
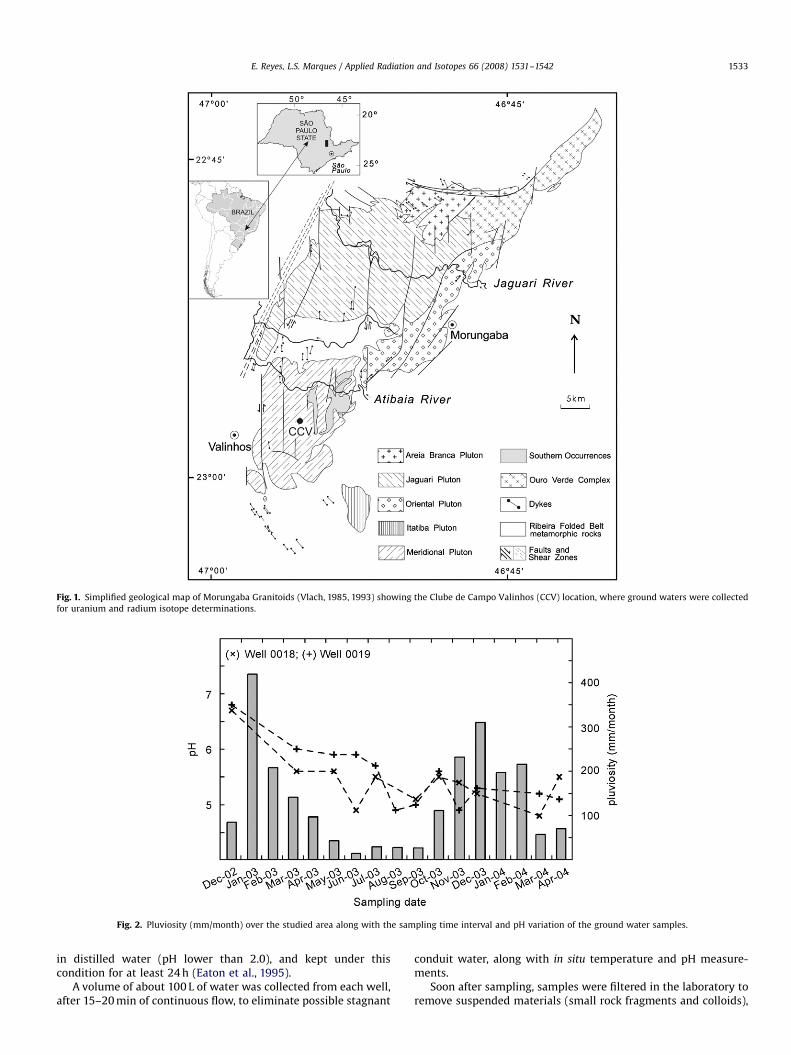
The analyzed ground waters were collected from two deepwells drilled in Morungaba Granitoids, which outcrop in EasternSao Paulo State, located 100 km from Sao Paulo City (Fig. 1), andclose to the border of Parana Sedimentary Basin (Southern Brazil).The granitoids encompass an area of about 330 km2, presenting aroughly rectangular shape of about 15 by 40 km and NNE trending(Vlach, 1985, 1993).
Morungaba Granitoids outcrop as large rounded blocks(boulders) of different sizes, forming a steep relief with heightsvarying from 950 to 1150 m (Vlach, 1993). These intrusive igneousrocks, along with many other granitoids found in the east of SaoPaulo State, were generated during the final stages of thermal-tectonic events related to the agglutination of Gondwana (Pan-African Cycle), during Neoproterozoic to Cambrian times, and areclassified as late- to post-orogenic rocks (620–570 Ma).
Detailed studies carried out by Vlach (1985, 1993) onMorungaba Granitoids showed important differences regardingtheir mode of occurrence, petrography, geochemistry, and age,allowing dividing them in large stratigraphic units (Fig. 1). Jaguari
and Meridional plutons are the largest units, corresponding toabout 37% and 25% of the exposed rocks, respectively (Fig. 1). Areia
Branca (7%), Oriental (13%), and Itatiba (o3%) plutons, as well asOuro Verde Complex (10%) also belong to Morungaba magmatism.Leucomonzogranites and hybrid facies, encompassing about 4% ofthe exposed rocks, outcrop on the domain of Meridional andOriental plutons.
Radiometric age determinations, using K–Ar and Rb–Srmethods, indicated that emplacement-crystallization of magmas,which gave rise to Morungaba Granitoids started at 620–600 Maago. The data also show that the units which outcrop in theSouthern area are younger, with ages of 590710 Ma (Vlach, 1985,1993).
The investigated ground waters were drawn from two deepwells drilled in rocks of Meridional Pluton (Fig. 1), at Clube deCampo Valinhos. The water is used by a house condominium forgeneral supply and domestic use, and as drinking water. Thispluton belongs to the Southern units and its rocks are mainlybiotite monzogranites and biotite quartz monzonites, which aresubordinately accompanied by syenogranites and diorites. Mainminerals of those rocks are potassic and calcic-sodic feldspars,
quartz and biotite. In more mafic lithologies, in addition to biotite,are found, as accessory mafic minerals, allanite, titanite, magne-tite, apatite and zircon. Muscovite, magnetite, ilmenite, titanite,monazite, garnet and fluorite are present as accessories in themore felsic rocks. Meridional Pluton rocks are pink, except formicrogranite dykes, which vary from gray to pink in color, andhave equigranular to inequigranular textures, including porphyri-tic types (Vlach, 1985, 1993; Batista et al., 1986).
Investigations about the distribution of natural radioactiveelements U, Th and K in Morungaba Granitoids (Vlach, 1998)showed that calc-alkaline varieties, which outcrop in the Northernarea, have the highest radioactivity levels, presenting uraniumconcentrations from 1 to 9mg/g, and thorium contents varyingfrom 6 to 40mg/g, whereas K2O concentrations vary between 4.7and 6.5 wt%. Conversely, rocks from Meridional Pluton arecharacterized by relatively low contents of radioactive elements,having uranium concentrations lower than 6 mg/g, whereasthorium and K2O contents show a variation from 4 to 14mg/gand 4.2 to 5.5 wt%, respectively.
3. Deep well characteristics and climate variations
The ground water samples were drawn from two deep wells,registered as well number 0018 and 0019 in the files of theDepartment of Water and Electrical Energy of Sao Paulo State(DAEE). The location coordinates of well 0018 are 22157.1370S and46155.5960W, whereas those of well 0019 are 22157.5020S and46155.2210W, implying a relatively short distance (about 900 m)between them.
Well 0018 is 72 m deep, cutting 9 m of colluvial and residualsoil, 9 m of weathered granitic rock and 54 m of fresh grayish-pinkgranite, and its water outflow is about 5 m3/h. Well 0019 has thesame depth, cutting 2 m earth materials, 3 m of alluvial andcolluvial soil, 15 m of weathered granite and 52 m of fresh pinkishgranite, and its water outflow is 12 m3/h. Both wells arecontinuously pumped to supply a house condominium.
The climate of the region is classified as subtropical, withannual average temperatures of 22.8 1C (maximum) and 13.6 1C(minimum). The rainy and dry seasons are very well defined. Theformer corresponds to the period from October to March, whereasthe latter begins in April and ends in September.
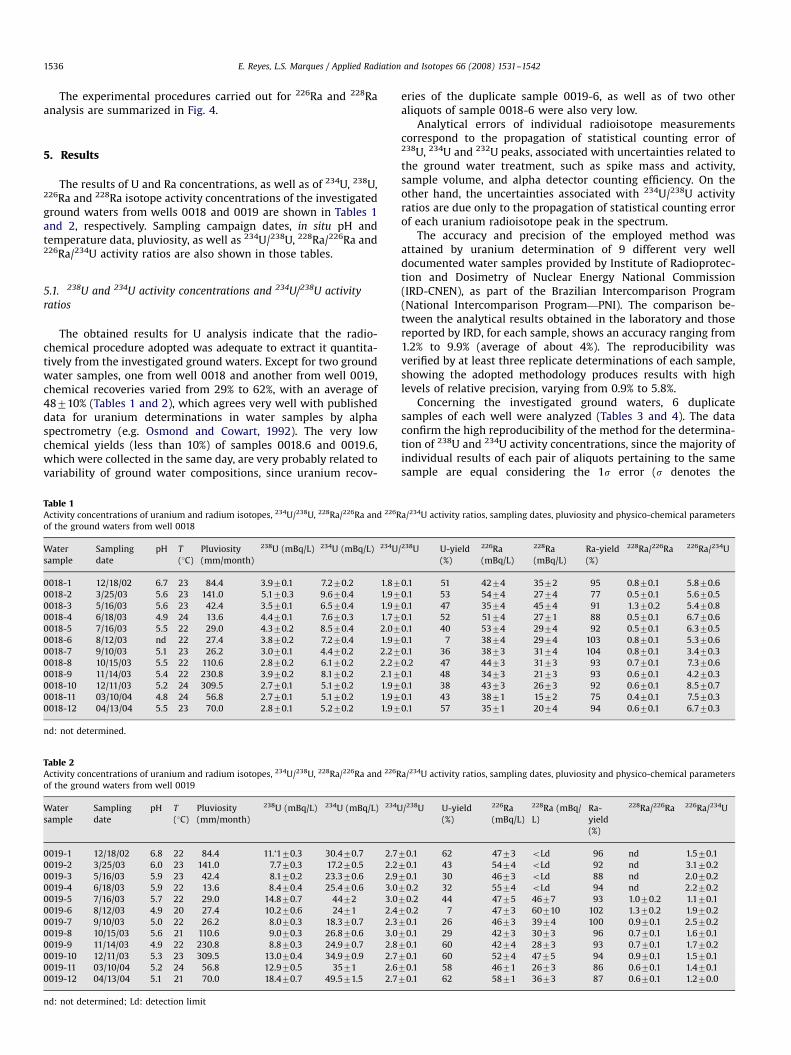
During the period of ground water sampling (from December2002 to April 2004) average pluviosity was 134 mm/month, with amaximum of 419 mm/month and a minimum of about 13.6 mm/month. Monthly precipitation in the area, during this period(Fig. 2; Instituto Agronomico de Campinas), shows a progressivedecrease from January to June, when it registered the lowestvalue. It is also observed that June, July, August and Septemberwere characterized by very low pluviosity levels (about 20 mm/month); thereafter precipitation increases progressively.
4. Sampling and analytical techniques
Two analytical methods were used to determine uranium andradium in the investigated ground waters. Alpha spectrometrywas used in order to measure activity concentrations of 234U and238U isotopes, as well to determine 234U/238U activity ratios,whereas high-resolution gamma-ray spectrometry was employedto ascertain activity concentrations of 226Ra and 228Ra isotopes.
Those analytical techniques were applied to the analysis of 12ground water samples, collected from each well, during a period alittle longer than 1 year. The samples were stored in 50 L capacitypolyethylene containers, which were previously washed withdistilled water. Then the containers were filled with HNO3 diluted
ARTICLE IN PRESS
Fig. 1. Simplified geological map of Morungaba Granitoids (Vlach, 1985, 1993) showing the Clube de Campo Valinhos (CCV) location, where ground waters were collected
for uranium and radium isotope determinations.
Fig. 2. Pluviosity (mm/month) over the studied area along with the sampling time interval and pH variation of the ground water samples.
E. Reyes, L.S. Marques / Applied Radiation and Isotopes 66 (2008) 1531–1542 1533
in distilled water (pH lower than 2.0), and kept under thiscondition for at least 24 h (Eaton et al., 1995).
A volume of about 100 L of water was collected from each well,after 15–20 min of continuous flow, to eliminate possible stagnant
conduit water, along with in situ temperature and pH measure-ments.
Soon after sampling, samples were filtered in the laboratory toremove suspended materials (small rock fragments and colloids),

ARTICLE IN PRESS
E. Reyes, L.S. Marques / Applied Radiation and Isotopes 66 (2008) 1531–15421534
since this investigation is focused on the determination ofdissolved uranium and radium in the ground waters.
The first filtration was carried out by using an AP20 glass fiberfilter, with 47 mm diameter and 45 mm porosity, to remove organicand detrital material. In order to eliminate most of the suspendedsolids, a second filtration was done employing an HA celluloseester membrane, with 0.45mm porosity and 47 mm diameter. It isconsidered by convention that all the dissolved fraction corre-sponds to material which is not retained by a filter with pores of0.45mm (Benes, 1990), including most colloids.
After filtration, the samples were acidified with HNO3 to pHabout 2, to ensure that both uranium and radium remained insolution and to reduce biological growth. Under those conditionswater samples may be preserved for several months (Smithson,1990). Subsequently, the samples were stored in the cleanedvessels, for about 15 days, which is the time interval required toconcentrate the initial volume of 30–1 L by evaporation.
4.1. Uranium determination by alpha spectrometry
Spectrometric measurements of alpha emitting radioisotopesin geological matrices, such as 238U and 234U, require very pure,uniform, compact and thin sources. Therefore, a meticulousradiochemical processing is necessary in order to separateuranium from all other interfering elements (radioactive or not)present in the sample, since alpha particles have very shortpenetration, being easily absorbed by impurities present in thesample. Special attention is essential to remove iron, whichthickens the targets, hindering the escape of alpha particles(e.g. Kressin, 1977).
Before radiochemical processing, 30 L of each sample wasevaporated to 1 L volume, which was subsequently divided in twoaliquots of 500 mL, to analyze each sample in duplicate. There-after, for determination of 238U and 234U activity concentrationsan analytical method based on Hallstadius (1984), Vera Tome andMartin Sanches (1991), Camargo (1994), Santos et al. (2002), andSantos and Marques (2007) was employed.
The radiochemical processing comprises the following steps:addition of 232U tracer and FeCl3 (carrier), uranium pre-concen-tration, uranium separation from interfering elements, uraniumpurification by ion exchange chromatography, and alpha sourcepreparation by electrodeposition on stainless steel disks.
About 0.4 g of 232U tracer, with specific activity of0.31370.005 Bq/g, and 6 mL of a solution of Fe carrier (about25 mg of Fe3+ for sample) were added with vigorous stirring toeach aliquot, in order to attain chemical equilibrium. After that,the aliquot was heated at about 60 1C and uranium was co-precipitated with Fe(OH)3 by the addition of concentratedammonium hydroxide until pH reaches 7–8. After precipitationthe sample was kept at rest for about 12 h in order to ensure aquantitative precipitation and its complete decantation. Subse-quently, the solution was filtered through a fast flow ratequantitative filter paper, with 7.5mm porosity and 185 mmdiameter. The precipitate retained on the filter was first washedwith 300 mL distilled water (pH ¼ 7) and then dissolved with40 mL of 9 M HCl.
That solution was passed through an ion exchange column,10 cm long and 15 mm internal diameter, packed with approxi-mately 10 cm3 of AG 1-X8 anionic resin, 100–200 mesh, pre-conditioned with 40 mL 9 M HCl, in order to separate uraniumfrom iron, thorium and other elements present in the sample. Theflow rate was adjusted to 8 drops per minute. Under thoseconditions U and Fe are quantitatively retained by the resin(Anderson and Fleer, 1982). Thereafter, the column was washedtwice with 10 mL of 9 M HCl. In order to elute iron from the resin,
it was washed with 40 mL of 8 M HNO3 and the eluate discarded.Thereafter, uranium was eluted by percolation of 100 mL of 0.1 MHCl.
Once purified, uranium was electrodeposited on a polishedstainless steel planchet, with 25 mm diameter and a thickness of0.5 mm, according to the procedure proposed by Hallstadius(1984). The solution containing uranium was evaporated untildryness. In case the solution turned yellowish during evaporation,the volume of 8 M HNO3 used was not enough to eliminatecompletely the iron and the ion exchange chromatography stepmust be repeated. When almost dry, the residue was recovered in1 mL of 0.3 M Na2SO4. This solution was dried, the residue treatedwith 0.3 mL of concentrated H2SO4 and, then, 5 mL of distilledwater were added to it, along with 2 drops of thymol blue. Thissolution was heated and its pH was adjusted to 2.1–2.4 by theaddition of some drops of concentrated NH4OH. The solution wasthen transferred to a lucite electrodeposition cell (Sturchio et al.,2001, and references therein). The electrolysis was accomplishedduring 1 h, under current density of 1.2 A/cm2. One minute beforethe end of electrolysis, 1 mL of concentrated NH4OH was added, inorder to assure uranium deposit attachment on the stainless steelcathode disk (Vasconcellos et al., 1987).
The obtained radioactive target was submitted to alphaparticle counting in order to determine 238U, 234U, and 232Uactivity concentrations, as well 234U/238U activity ratio. Thecounting system is composed of a silicon surface barrier detector,with nominal active area of 450 mm2, operating under pressure ofapproximately 10�2 mbar, in order to avoid absorption of alphaparticles by air. The detection system, calibrated to operate in theenergy range from 3 to 7 MeV, is controlled by a microcomputer,which simulates a 4096 multichannel analyzer. Ten independentmeasurements of calibrated alpha sources (241Am and 233U)indicated the counting system has efficiency of 3371% andresolution of 2571 keV (indicated errors correspond to onestandard deviation).
Each sample was submitted to counting for a period varyingfrom 2 to 6 days, depending on the activity of the source. In orderto ensure high precision (about 3%) of activity concentration data,each measurement was carried out until the peak of the lowestactivity radioisotope (in general 238U) accumulated at least 1000counts. Fig. 3 summarizes the experimental procedures carriedout for uranium isotope analysis.
4.2. Radium determination by high-resolution gamma-ray
spectrometry
Gamma-ray spectrometry has numerous applications in theanalysis of geological and environmental samples. It is a highlysensitive technique that allows identifying and quantifyingsimultaneously several gamma-ray emitters present in lowconcentrations in different matrices (Ivanovich and Murray,1992). In comparison to other nuclear techniques it has a greatadvantage, since gamma radiation has a very high penetration,making it possible to apply it in non-destructive analyses or withminimal chemical processing of samples.
High-resolution gamma-ray spectrometry was used for deter-mining activity concentrations of 226Ra, which belongs to the 238Useries decay, as well as of 228Ra, which is a radioisotope from the232Th radioactive chain. Since those Ra isotopes are present in verylow concentrations in the investigated ground waters, it wasnecessary to carry out chemical processing in order to concentrateRa in a relatively small volume aliquot (500 mL), as required forgamma-ray counting.
The radiochemical methodology used for the analysis ofradium isotopes was based on Oliveira (1998, 2001). At the

ARTICLE IN PRESS
Ground water sampling
Sample filtering and acidification with HNO3 at pH ~ 2
Evaporation from 30 L to volume of 1 L
Addition of 232U tracer and FeCl3 (carrier)
Addition of concentrated NH4OH until pH reaches 7-8
Sample kept in rest for about 12 h
Filtering and precipitate dilution with 40 mL of 9M HCl
Solution percolated in anionic AG 1-X8 resin pre-conditionedwith 40 mL of 9M HCl
Iron elution from the resin with 40 mL of 8M HNO3
Uranium elution with 100 mL of 0.1M HCl
Uranium electrodeposition with Na2SO4 andconcentrated H2SO4 (pH = 2.1 - 2.4)
Alpha particle counting for about 4 days
Fig. 3. Radiochemical procedure for uranium analysis in the ground water
samples.
Ground water sampling
Sample filtering and acidification with HNO3 at pH ~ 2
Addition of 133Ba tracer and 50 mL of a barium carrier solution
Solution transferring to a polyethylene flask (500 mL)and sealing with adhesive tape
Addition of 250 mL of concentrate HCl
Addition of 250 mL of concentrated H2SO4under vigorous stirring
Filtering and precipitate dilution with 0.25 M EDTA dissodicsalt and 100 mL of concentrated NH4OH
Gamma counting for 48 hours after at least 45 days
Sample kept in rest for about 12 h
Fig. 4. Radiochemical procedure for radium analysis in the ground water samples.
E. Reyes, L.S. Marques / Applied Radiation and Isotopes 66 (2008) 1531–1542 1535
beginning of the analytical work, radium was directly separatedfrom about 25 L of water. However, because of low concentrationsof 228Ra it was necessary to increase this volume to about 45 L, inorder to determine its activity concentrations. Then, the groundwater samples were divided in two polyethylene containers with30 L capacity each. Initially, about 0.3 g of 133Ba tracer, withspecific activity of 2.05570.014 Bq/g, along with 50 mL of a 20 mgBa2+/mL solution of barium diluted in 16 M HNO3 were added toeach aliquot, which was, then, homogenized with vigorousstirring and kept at rest for at least 1 h. A volume of 250 mL ofconcentrated HCl was then added to the solution under agitation.In order to precipitate Ba and Ra, 250 mL of concentrated H2SO4
were added to the solution, under vigorous stirring. Sufficientvolumes of Ba carrier, HCl and H2SO4 were added to the solutionto ensure complete Ba(Ra)SO4 precipitation.
After keeping the supernatant and the precipitate at rest forabout 15 h, the former was discarded using a pipette connected toa vacuum pump, until the content of each container reached about2 L. Then, the content of each container was passed throughquantitative (slow flow rate) 2.0 mm porosity paper filter. Subse-quently, the precipitate collected on filter paper was dissolved in500 mL of 0.25 M EDTA dissodic salt (C10H14N2Na2O8 �2H2O), intowhich 100 mL of concentrated NH4OH were added. The solution,then, was heated to the boiling point, in order to ensure thecomplete dissolution of the precipitate. The solution was thenevaporated to reduce its volume to 500 mL, and kept at rest toachieve room temperature. After that, the solution containing Rawas transferred to a 500 mL capacity polyethylene flask, whichwas tightly sealed with adhesive tape.
Gamma counting was carried out after at least 45 days restperiod, necessary for the 238U series to reestablish secularradioactive equilibrium from 226Ra to 210Pb, since 222Rn gas hasa half-life of 3.38 days. Gamma radiation measurements weredone by using a high-resolution gamma-ray spectrometer,composed of a hyperpure Ge detector, with 252 cm3 volume and
operating at liquid nitrogen temperature. The detection systemhas a relative efficiency of 70% and nominal resolution of 2.0 keVfor 60Co peak of 1332.49 keV.
Each sample was submitted to counting for 48 h. In order tocalculate 226Ra and 228Ra activity concentrations, two in-housestandards of 226Ra and 228Ra, with activity concentrations of19.570.7 and 5.170.7 Bq/g, respectively, were prepared from theprimary standards provided by Institute of Radioprotection andDosimetry (Instituto de Radioprotec- ao e Dosimetria-IRD) ofNational Nuclear Energy Commission (Comissao Nacional deEnergia Nuclear-CNEN). Both radioisotopes were diluted in500 mL of EDTA.
A blank of 500 mL of 0.25 M EDTA solution was also used toaccount for background counting. Samples, in-house standardsand the blank were all conditioned in plastic bottles of the sametype, to assure identical counting geometry.
Although activity concentrations of 226Ra could be determinedby its 186 keV peak, since the radiochemical procedure eliminatedthe interference from 235U (185.7 keV), the measured activitieswere often below the detection limit Ld (Currie, 1968), caused bythe low probability (�4%) of gamma-ray emission. Therefore, inorder to determine 226Ra, it was necessary to use gamma raysemitted by its daughters 214Bi and 214Pb, since these threeradioisotopes are in secular radioactive equilibrium. Because ofthe relatively low emission probability of 1120 and 1764 keVgamma rays from 214Bi, in most cases their measured activitieswere below Ld. Gamma rays of 352 keV emitted by 214Pb were notalso used because they suffer interference from the 133Ba(356 keV) tracer.
Due to the difficulties noted, the activity concentrations of226Ra were, in general, determined by the arithmetic means of theresults obtained by the 609 keV gamma peak from 214Bi and the295 keV peak from 214Pb.
Gamma-ray spectrometry only allows the determination ofactivity concentrations of 228Ra indirectly, through the measure-ment of its daughters 228Ac (911.2 keV), 212Pb (238.6 keV) and 208Tl(583.2 keV), since secular radioactive equilibrium is attained. Dueto the low concentrations of 228Ra in the analyzed samples,reproducible results of its activity concentrations were obtainedthrough 228Ac (in most cases the measured 212Pb and 208Tlactivities were below the detection limit Ld).
ARTICLE IN PRESS
E. Reyes, L.S. Marques / Applied Radiation and Isotopes 66 (2008) 1531–15421536
The experimental procedures carried out for 226Ra and 228Raanalysis are summarized in Fig. 4.
5. Results
The results of U and Ra concentrations, as well as of 234U, 238U,226Ra and 228Ra isotope activity concentrations of the investigatedground waters from wells 0018 and 0019 are shown in Tables 1and 2, respectively. Sampling campaign dates, in situ pH andtemperature data, pluviosity, as well as 234U/238U, 228Ra/226Ra and226Ra/234U activity ratios are also shown in those tables.
5.1. 238U and 234U activity concentrations and 234U/238U activity
ratios
The obtained results for U analysis indicate that the radio-chemical procedure adopted was adequate to extract it quantita-tively from the investigated ground waters. Except for two groundwater samples, one from well 0018 and another from well 0019,chemical recoveries varied from 29% to 62%, with an average of48710% (Tables 1 and 2), which agrees very well with publisheddata for uranium determinations in water samples by alphaspectrometry (e.g. Osmond and Cowart, 1992). The very lowchemical yields (less than 10%) of samples 0018.6 and 0019.6,which were collected in the same day, are very probably related tovariability of ground water compositions, since uranium recov-
Table 1Activity concentrations of uranium and radium isotopes, 234U/238U, 228Ra/226Ra and 226R
of the ground waters from well 0018
Water
sample
Sampling
date
pH T
(1C)
Pluviosity
(mm/month)
238U (mBq/L) 234U (mBq/L) 234U
0018-1 12/18/02 6.7 23 84.4 3.970.1 7.270.2 1.870018-2 3/25/03 5.6 23 141.0 5.170.3 9.670.4 1.970018-3 5/16/03 5.6 23 42.4 3.570.1 6.570.4 1.970018-4 6/18/03 4.9 24 13.6 4.470.1 7.670.3 1.770018-5 7/16/03 5.5 22 29.0 4.370.2 8.570.4 2.070018-6 8/12/03 nd 22 27.4 3.870.2 7.270.4 1.970018-7 9/10/03 5.1 23 26.2 3.070.1 4.470.2 2.270018-8 10/15/03 5.5 22 110.6 2.870.2 6.170.2 2.270018-9 11/14/03 5.4 22 230.8 3.970.2 8.170.2 2.170018-10 12/11/03 5.2 24 309.5 2.770.1 5.170.2 1.970018-11 03/10/04 4.8 24 56.8 2.770.1 5.170.2 1.970018-12 04/13/04 5.5 23 70.0 2.870.1 5.270.2 1.97
nd: not determined.
Table 2Activity concentrations of uranium and radium isotopes, 234U/238U, 228Ra/226Ra and 226R
of the ground waters from well 0019
Water
sample
Sampling
date
pH T
(1C)
Pluviosity
(mm/month)
238U (mBq/L) 234U (mBq/L) 234U
0019-1 12/18/02 6.8 22 84.4 11.‘170.3 30.470.7 2.770019-2 3/25/03 6.0 23 141.0 7.770.3 17.270.5 2.270019-3 5/16/03 5.9 23 42.4 8.170.2 23.370.6 2.970019-4 6/18/03 5.9 22 13.6 8.470.4 25.470.6 3.070019-5 7/16/03 5.7 22 29.0 14.870.7 4472 3.070019-6 8/12/03 4.9 20 27.4 10.270.6 2471 2.470019-7 9/10/03 5.0 22 26.2 8.070.3 18.370.7 2.370019-8 10/15/03 5.6 21 110.6 9.070.3 26.870.6 3.070019-9 11/14/03 4.9 22 230.8 8.870.3 24.970.7 2.870019-10 12/11/03 5.3 23 309.5 13.070.4 34.970.9 2.770019-11 03/10/04 5.2 24 56.8 12.970.5 3571 2.670019-12 04/13/04 5.1 21 70.0 18.470.7 49.571.5 2.77
nd: not determined; Ld: detection limit
eries of the duplicate sample 0019-6, as well as of two otheraliquots of sample 0018-6 were also very low.
Analytical errors of individual radioisotope measurementscorrespond to the propagation of statistical counting error of238U, 234U and 232U peaks, associated with uncertainties related tothe ground water treatment, such as spike mass and activity,sample volume, and alpha detector counting efficiency. On theother hand, the uncertainties associated with 234U/238U activityratios are due only to the propagation of statistical counting errorof each uranium radioisotope peak in the spectrum.
The accuracy and precision of the employed method wasattained by uranium determination of 9 different very welldocumented water samples provided by Institute of Radioprotec-tion and Dosimetry of Nuclear Energy National Commission(IRD-CNEN), as part of the Brazilian Intercomparison Program(National Intercomparison Program—PNI). The comparison be-tween the analytical results obtained in the laboratory and thosereported by IRD, for each sample, shows an accuracy ranging from1.2% to 9.9% (average of about 4%). The reproducibility wasverified by at least three replicate determinations of each sample,showing the adopted methodology produces results with highlevels of relative precision, varying from 0.9% to 5.8%.
Concerning the investigated ground waters, 6 duplicatesamples of each well were analyzed (Tables 3 and 4). The dataconfirm the high reproducibility of the method for the determina-tion of 238U and 234U activity concentrations, since the majority ofindividual results of each pair of aliquots pertaining to the samesample are equal considering the 1s error (s denotes the
a/234U activity ratios, sampling dates, pluviosity and physico-chemical parameters
/238U U-yield
(%)
226Ra
(mBq/L)
228Ra
(mBq/L)
Ra-yield
(%)
228Ra/226Ra 226Ra/234U
0.1 51 4274 3572 95 0.870.1 5.870.6
0.1 53 5474 2774 77 0.570.1 5.670.5
0.1 47 3574 4574 91 1.370.2 5.470.8
0.1 52 5174 2771 88 0.570.1 6.770.6
0.1 40 5374 2974 92 0.570.1 6.370.5
0.1 7 3874 2974 103 0.870.1 5.370.6
0.1 36 3873 3174 104 0.870.1 3.470.3
0.2 47 4473 3173 93 0.770.1 7.370.6
0.1 48 3473 2173 93 0.670.1 4.270.3
0.1 38 4373 2673 92 0.670.1 8.570.7
0.1 43 3871 1572 75 0.470.1 7.570.3
0.1 57 3571 2074 94 0.670.1 6.770.3
a/234U activity ratios, sampling dates, pluviosity and physico-chemical parameters
/238U U-yield
(%)
226Ra
(mBq/L)
228Ra (mBq/
L)
Ra-
yield
(%)
228Ra/226Ra 226Ra/234U
0.1 62 4773 oLd 96 nd 1.570.1
0.1 43 5474 oLd 92 nd 3.170.2
0.1 30 4673 oLd 88 nd 2.070.2
0.2 32 5574 oLd 94 nd 2.270.2
0.2 44 4775 4677 93 1.070.2 1.170.1
0.2 7 4773 60710 102 1.370.2 1.970.2
0.1 26 4673 3974 100 0.970.1 2.570.2
0.1 29 4273 3073 96 0.770.1 1.670.1
0.1 60 4274 2873 93 0.770.1 1.770.2
0.1 60 5274 4775 94 0.970.1 1.570.1
0.1 58 4671 2673 86 0.670.1 1.470.1
0.1 62 5871 3673 87 0.670.1 1.270.0
ARTICLE IN PRESS
Table 3Comparison of uranium activity concentrations, 234U/238U activity ratios and
chemical yields obtained in duplicate analyses of ground waters from well 0018
Water sample 238U (mBq/L) 234U (mBq/L) 234U/238U U-Yield (%)
0018-1 3.970.1 7.270.2 1.870.1 51
4.170.3 7.770.4 1.970.1 47
0018-3 3.570.1 6.570.4 1.970.1 47
3.270.1 6.170.3 1.970.1 49
0018-5 4.370.2 8.570.4 2.070.1 40
4.770.2 8.670.4 1.870.1 47
0018-7 3.070.1 4.47 0.2 1.570.2 36
7.070.2 9.370.3 1,470.2 68
0018-9 3.970.2 8.170.2 2.170.1 48
3.570.1 7.570.2 2,170.1 46
0018-11 2.770.1 5.170.2 1.970.1 43
3.070.2 5.470.2 1.870.1 57
Table 4Comparison of uranium activity concentrations, 234U/238U activity ratios and
chemical yields obtained in duplicate analyses of ground waters from well 0019
Water sample 238U (mBq/L) 234U (mBq/L) 234U/238U U-yield (%)
0019-1 11.170.3 30.470.7 2.770.1 62
9.770.3 27.170.5 2.870.1 60
0019-3 8.170.2 23.370.6 2.970.1 30
8.570.2 24.170.6 2,870.2 33
0019-5 14.870.7 4472 3.070.2 44
12.270.6 3871 3,170.2 41
0019-7 8.070.3 26.870.6 3.070.1 29
8.270.3 26.170.6 3.270.1 34
0019-9 8.870.3 24.970.7 2.870.1 60
8.770.4 24.270.9 2.870.1 57
0019-11 12.970.5 3371 2.670.1 58
13.670.7 3571 2.670.1 62
Table 5Activity concentrations of 226Ra and 228Ra of replicate analyses of two samples
from ground waters from well 0019
Water Sample 226Ra (mBq/L) 228Ra (mBq/L)
0019-7 45.5 39.0
43.1 36.6
46.2 37.3
44.4 30.6
42.2 36.7
44.2 40.0
Average 4471 3674
Relative precision (%) 3 10
0019-9 42.4 28.4
48.6 26.2
45.1 30.6
46.1 24.6
41.9 27.5
49.8 24.6
Average 4673 2772
Relative precision (%) 7 9
E. Reyes, L.S. Marques / Applied Radiation and Isotopes 66 (2008) 1531–1542 1537
analytical error of each result, as described above). For samples0018.3, 0018.9 and 0019.4 the results of each pair of aliquots areequal at the 2s level, whereas for 0018.7 and 0019.1 the activityconcentrations can be considered equal only at the 3s level. For allduplicates, 234U/238U activity ratios are equal at the 1s level.
The results show that ground waters from each well havesignificant and systematic differences in 238U and 234U activityconcentrations, as well as in 234U/238U activity ratios (Tables 1 and2). Samples from well 0018 are characterized by relatively lowactivity concentrations of 238U and 234U, which vary from 2.770.1to 5.170.3 mBq/L (average ¼ 4.070.8 mBq/L) and from 5.170.2 to11.270.4 mBq/L (average ¼ 772 mBq/L), respectively. Groundwaters from well 0019 have 238U activity concentrations spanningfrom 7.770.3 to 18.470.7 mBq/L (average ¼ 1173 mBq/L) and234U activity concentrations varying between 17.270.5 and5072 mBq/L (average ¼ 29710 mBq/L).
In all analyzed samples uranium isotopes are out of secularradioactive equilibrium, exhibiting significant enrichment of 234Urelative to 238U. However, 234U/238U ratios of ground waters fromwell 0019, whose average is 2.770.3 (range: 2.270.1–3.070.2),
are in general larger and more variable than those of well 0018,whose average is 2.070.2 (range: 1.770.1–2.270.2).
5.2. 226Ra and 228Ra activity concentrations and 226Ra/228Ra activity
ratios
The adopted analytical procedure proved very efficient in theprocess of separation and isolation of Ra from the investigatedsamples. The chemical recoveries, which were assumed to be thesame as those obtained for the 133Ba spike, varied from 75% to104% (average of 9377%). Although a series of replicate analyseswere not carried out for the determination of the precision of themethod, the reproducibility of gamma-ray measurements wasevaluated by counting two samples (0019.7 and 0019.9) for sixtimes each, whose results indicated a relative precision of 10% for228Ra, and in the range from 3% to 7% for 226Ra (Table 5). Theseresults were considered as an estimate of the relative precision ofthe data. It is also important to remark that the first four samplesfrom well 0019 (0019.1, 0019.2, 0019.3 and 0019.4) presented228Ra activity concentrations lower than Ld (detection limit) of themethod. As previously explained, for the analysis of the remainingsamples this problem was solved by processing larger volumes ofwater (about 45 L).
Regarding the radium analysis, the investigated ground watersshowed similar activity concentrations, although samples fromwell 0018 presented the smallest values for both 226Ra and 228Raisotopes. Activity concentrations of 226Ra in ground waterstaken from this well varied from 3473 to 5474 mBq/L(average ¼ 4277 mBq/L), and of 228Ra from 1572 to 4574 mBq/L(average ¼ 2878 mBq/L). For ground waters from well 0019 226Raand 228Ra activity concentrations varied from 4273 to5871 mBq/L (average ¼ 4975 mBq/L), and from 2673 to60710 mBq/L (average ¼ 39712 mBq/L), respectively.
Taking into account analytical uncertainties, 226Ra activityconcentrations obtained in the present work agree with thosedetermined by radon emanometry (Lucas and Ribeiro, 2006) onthe same ground water samples. According to these authors, theaverages of 226Ra activity concentrations are 4577 and5279 mBq/L for ground waters from wells 0018 and 0019,respectively.
Results of all analyzed samples show that 226Ra activityconcentrations are usually larger than those of 228Ra, resultingin 228Ra/226Ra activity ratios lower than unity. For ground watersextracted from well 0018, 228Ra/226Ra ratios varied from 0.470.1to 0.870.2 (average ¼ 0.670.1; except for sample 0018-3),
ARTICLE IN PRESS
E. Reyes, L.S. Marques / Applied Radiation and Isotopes 66 (2008) 1531–15421538
whereas those from well 0019 presented 228Ra/226Ra in the rangebetween 0.670.1 and 1.070.2 (average ¼ 0.870.2; except forsample 0019-6). Although, there is some overlapping of228Ra/226Ra activity ratios of ground waters extracted from eachwell, slightly higher values are observed in those from well 0019(Tables 1 and 2).
6. Discussion
Uranium activities determined in all analyzed ground watersamples are within the range reported in the literature (Bonottoand Andrews, 2000; Gafvert et al., 2002; Benedetti et al., 2003;Calsteren and Thomas, 2006; Gomez et al., 2006). The results for238U and 234U activity concentrations show, for both ground waterwells, a significant variation during the sampling period. Althoughsome scatter is noticed, the largest concentrations were observed
Fig. 5. Variation of 234U and 238U activity concentrations (mBq/L) along with 234U/238U
Solid line: activity ratios; dashed line: activity concentrations.
Fig. 6. Variation of 234U and 238U activity concentrations (mBq/L), along with 234U/238U
Solid line: activity ratios; dashed line: activity concentrations.
during the dry season, which starts on May and finishes onSeptember (Figs. 5 and 6).
It is noteworthy to observe that the pH of the analyzed groundwaters is in general moderately acidic (Tables 1 and 2), favoringweathering processes and uranium lixiviation. During the sam-pling period the ground water pH varied from 4.8 to 6.7(average ¼ 5.470.5) for well 0018, whereas the range of well0019 was from 4.9 to 6.8 (average ¼ 5.570.6). Although there wasa slightly irregular pH variation, ground waters from both wellsexhibited similar behavior, with minimal values during the dryseason (Fig. 2).
The data also show an important disequilibrium between bothuranium isotopes in the investigated ground waters. The sig-nificant 234U enrichment is probably due to its instability incrystalline lattices after recoil following alpha emission from 238U.In this process, the chemical bond is weakened and the 234Uoxidation state changes from tetravalent to a more soluble
activity ratios of ground waters from well 0018 during the sampling time interval.
activity ratios, of ground waters from well 0019 during the sampling time interval.
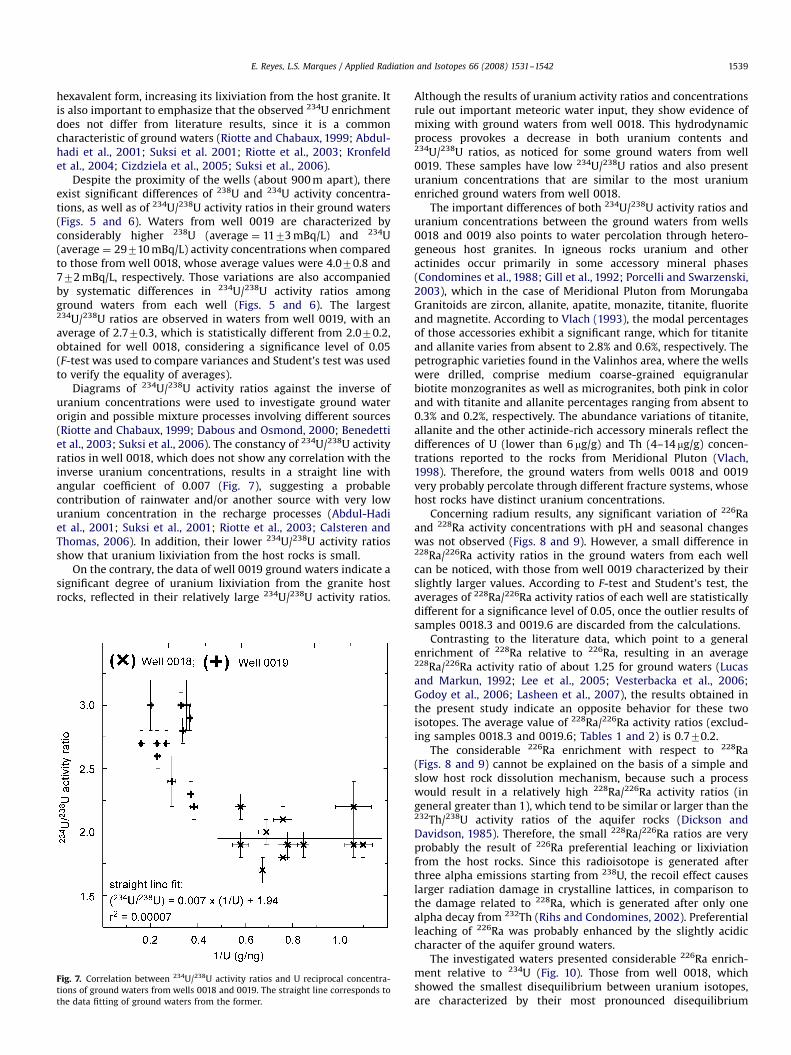
ARTICLE IN PRESS
E. Reyes, L.S. Marques / Applied Radiation and Isotopes 66 (2008) 1531–1542 1539
hexavalent form, increasing its lixiviation from the host granite. Itis also important to emphasize that the observed 234U enrichmentdoes not differ from literature results, since it is a commoncharacteristic of ground waters (Riotte and Chabaux, 1999; Abdul-hadi et al., 2001; Suksi et al. 2001; Riotte et al., 2003; Kronfeldet al., 2004; Cizdziela et al., 2005; Suksi et al., 2006).
Despite the proximity of the wells (about 900 m apart), thereexist significant differences of 238U and 234U activity concentra-tions, as well as of 234U/238U activity ratios in their ground waters(Figs. 5 and 6). Waters from well 0019 are characterized byconsiderably higher 238U (average ¼ 1173 mBq/L) and 234U(average ¼ 29710 mBq/L) activity concentrations when comparedto those from well 0018, whose average values were 4.070.8 and772 mBq/L, respectively. Those variations are also accompaniedby systematic differences in 234U/238U activity ratios amongground waters from each well (Figs. 5 and 6). The largest234U/238U ratios are observed in waters from well 0019, with anaverage of 2.770.3, which is statistically different from 2.070.2,obtained for well 0018, considering a significance level of 0.05(F-test was used to compare variances and Student’s test was usedto verify the equality of averages).
Diagrams of 234U/238U activity ratios against the inverse ofuranium concentrations were used to investigate ground waterorigin and possible mixture processes involving different sources(Riotte and Chabaux, 1999; Dabous and Osmond, 2000; Benedettiet al., 2003; Suksi et al., 2006). The constancy of 234U/238U activityratios in well 0018, which does not show any correlation with theinverse uranium concentrations, results in a straight line withangular coefficient of 0.007 (Fig. 7), suggesting a probablecontribution of rainwater and/or another source with very lowuranium concentration in the recharge processes (Abdul-Hadiet al., 2001; Suksi et al., 2001; Riotte et al., 2003; Calsteren andThomas, 2006). In addition, their lower 234U/238U activity ratiosshow that uranium lixiviation from the host rocks is small.
On the contrary, the data of well 0019 ground waters indicate asignificant degree of uranium lixiviation from the granite hostrocks, reflected in their relatively large 234U/238U activity ratios.
Fig. 7. Correlation between 234U/238U activity ratios and U reciprocal concentra-
tions of ground waters from wells 0018 and 0019. The straight line corresponds to
the data fitting of ground waters from the former.
Although the results of uranium activity ratios and concentrationsrule out important meteoric water input, they show evidence ofmixing with ground waters from well 0018. This hydrodynamicprocess provokes a decrease in both uranium contents and234U/238U ratios, as noticed for some ground waters from well0019. These samples have low 234U/238U ratios and also presenturanium concentrations that are similar to the most uraniumenriched ground waters from well 0018.
The important differences of both 234U/238U activity ratios anduranium concentrations between the ground waters from wells0018 and 0019 also points to water percolation through hetero-geneous host granites. In igneous rocks uranium and otheractinides occur primarily in some accessory mineral phases(Condomines et al., 1988; Gill et al., 1992; Porcelli and Swarzenski,2003), which in the case of Meridional Pluton from MorungabaGranitoids are zircon, allanite, apatite, monazite, titanite, fluoriteand magnetite. According to Vlach (1993), the modal percentagesof those accessories exhibit a significant range, which for titaniteand allanite varies from absent to 2.8% and 0.6%, respectively. Thepetrographic varieties found in the Valinhos area, where the wellswere drilled, comprise medium coarse-grained equigranularbiotite monzogranites as well as microgranites, both pink in colorand with titanite and allanite percentages ranging from absent to0.3% and 0.2%, respectively. The abundance variations of titanite,allanite and the other actinide-rich accessory minerals reflect thedifferences of U (lower than 6 mg/g) and Th (4–14 mg/g) concen-trations reported to the rocks from Meridional Pluton (Vlach,1998). Therefore, the ground waters from wells 0018 and 0019very probably percolate through different fracture systems, whosehost rocks have distinct uranium concentrations.
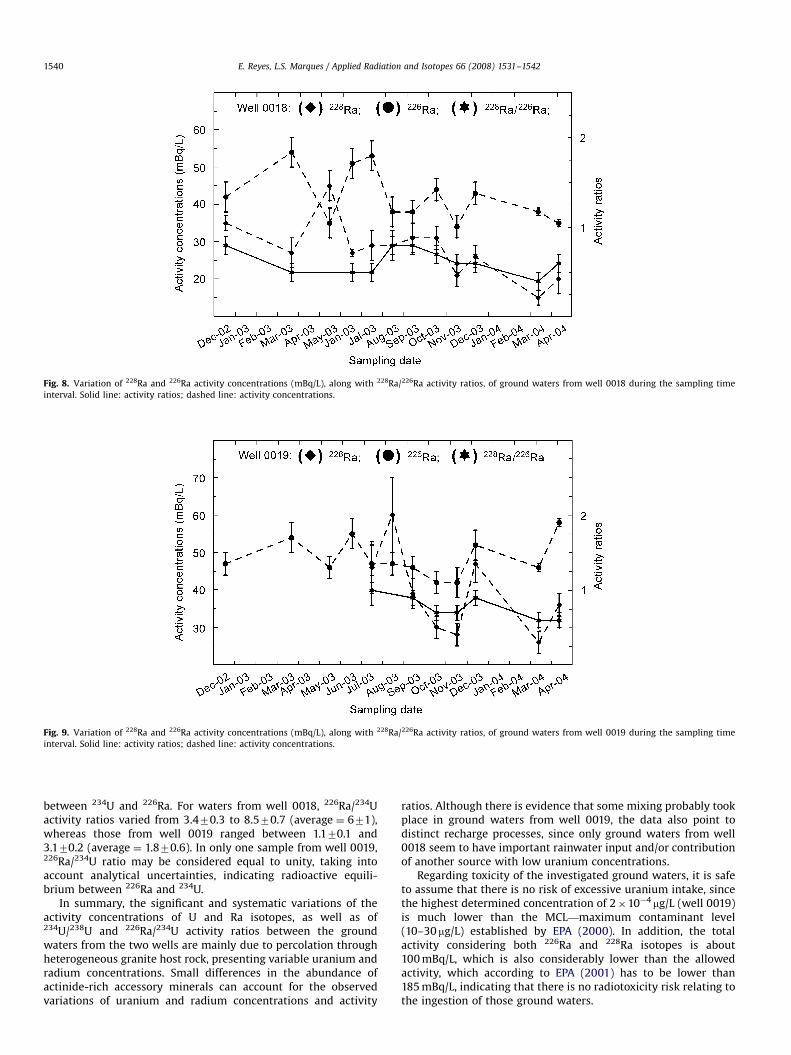
Concerning radium results, any significant variation of 226Raand 228Ra activity concentrations with pH and seasonal changeswas not observed (Figs. 8 and 9). However, a small difference in228Ra/226Ra activity ratios in the ground waters from each wellcan be noticed, with those from well 0019 characterized by theirslightly larger values. According to F-test and Student’s test, theaverages of 228Ra/226Ra activity ratios of each well are statisticallydifferent for a significance level of 0.05, once the outlier results ofsamples 0018.3 and 0019.6 are discarded from the calculations.
Contrasting to the literature data, which point to a generalenrichment of 228Ra relative to 226Ra, resulting in an average228Ra/226Ra activity ratio of about 1.25 for ground waters (Lucasand Markun, 1992; Lee et al., 2005; Vesterbacka et al., 2006;Godoy et al., 2006; Lasheen et al., 2007), the results obtained inthe present study indicate an opposite behavior for these twoisotopes. The average value of 228Ra/226Ra activity ratios (exclud-ing samples 0018.3 and 0019.6; Tables 1 and 2) is 0.770.2.
The considerable 226Ra enrichment with respect to 228Ra(Figs. 8 and 9) cannot be explained on the basis of a simple andslow host rock dissolution mechanism, because such a processwould result in a relatively high 228Ra/226Ra activity ratios (ingeneral greater than 1), which tend to be similar or larger than the232Th/238U activity ratios of the aquifer rocks (Dickson andDavidson, 1985). Therefore, the small 228Ra/226Ra ratios are veryprobably the result of 226Ra preferential leaching or lixiviationfrom the host rocks. Since this radioisotope is generated afterthree alpha emissions starting from 238U, the recoil effect causeslarger radiation damage in crystalline lattices, in comparison tothe damage related to 228Ra, which is generated after only onealpha decay from 232Th (Rihs and Condomines, 2002). Preferentialleaching of 226Ra was probably enhanced by the slightly acidiccharacter of the aquifer ground waters.
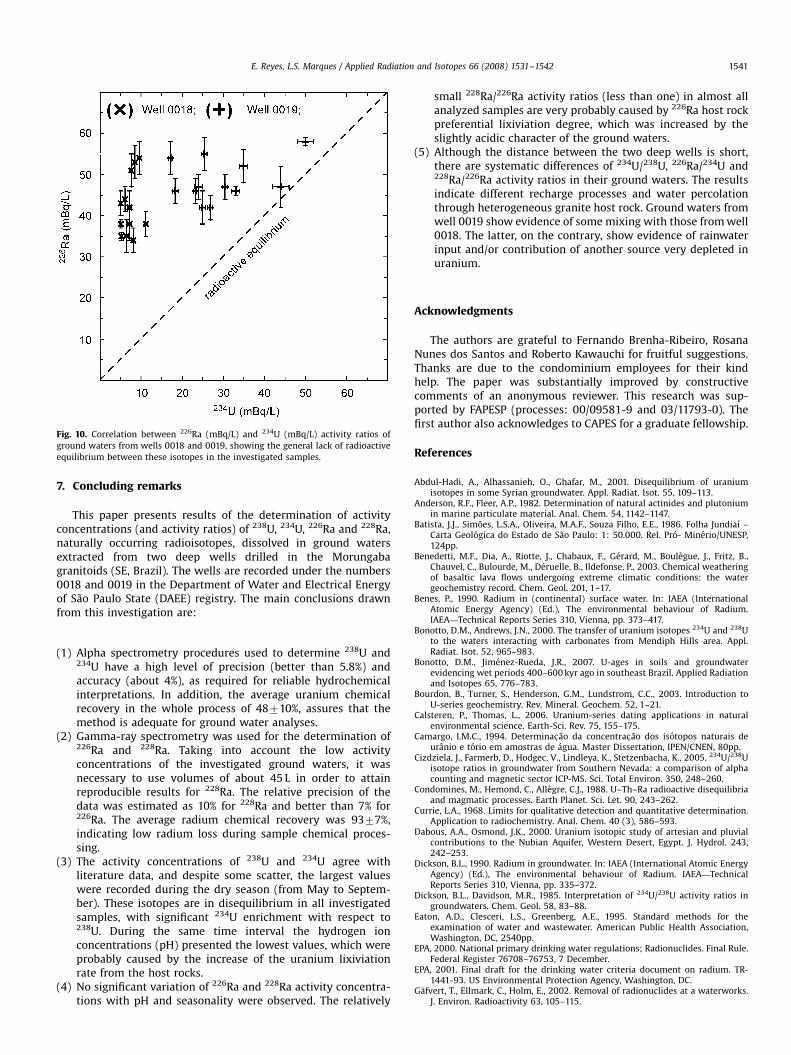
The investigated waters presented considerable 226Ra enrich-ment relative to 234U (Fig. 10). Those from well 0018, whichshowed the smallest disequilibrium between uranium isotopes,are characterized by their most pronounced disequilibrium
ARTICLE IN PRESS
Fig. 8. Variation of 228Ra and 226Ra activity concentrations (mBq/L), along with 228Ra/226Ra activity ratios, of ground waters from well 0018 during the sampling time
interval. Solid line: activity ratios; dashed line: activity concentrations.
Fig. 9. Variation of 228Ra and 226Ra activity concentrations (mBq/L), along with 228Ra/226Ra activity ratios, of ground waters from well 0019 during the sampling time
interval. Solid line: activity ratios; dashed line: activity concentrations.
E. Reyes, L.S. Marques / Applied Radiation and Isotopes 66 (2008) 1531–15421540
between 234U and 226Ra. For waters from well 0018, 226Ra/234Uactivity ratios varied from 3.470.3 to 8.570.7 (average ¼ 671),whereas those from well 0019 ranged between 1.170.1 and3.170.2 (average ¼ 1.870.6). In only one sample from well 0019,226Ra/234U ratio may be considered equal to unity, taking intoaccount analytical uncertainties, indicating radioactive equili-brium between 226Ra and 234U.
In summary, the significant and systematic variations of theactivity concentrations of U and Ra isotopes, as well as of234U/238U and 226Ra/234U activity ratios between the groundwaters from the two wells are mainly due to percolation throughheterogeneous granite host rock, presenting variable uranium andradium concentrations. Small differences in the abundance ofactinide-rich accessory minerals can account for the observedvariations of uranium and radium concentrations and activity
ratios. Although there is evidence that some mixing probably tookplace in ground waters from well 0019, the data also point todistinct recharge processes, since only ground waters from well0018 seem to have important rainwater input and/or contributionof another source with low uranium concentrations.
Regarding toxicity of the investigated ground waters, it is safeto assume that there is no risk of excessive uranium intake, sincethe highest determined concentration of 2�10�4 mg/L (well 0019)is much lower than the MCL—maximum contaminant level(10–30 mg/L) established by EPA (2000). In addition, the totalactivity considering both 226Ra and 228Ra isotopes is about100 mBq/L, which is also considerably lower than the allowedactivity, which according to EPA (2001) has to be lower than185 mBq/L, indicating that there is no radiotoxicity risk relating tothe ingestion of those ground waters.
ARTICLE IN PRESS
Fig. 10. Correlation between 226Ra (mBq/L) and 234U (mBq/L) activity ratios of
ground waters from wells 0018 and 0019, showing the general lack of radioactive
equilibrium between these isotopes in the investigated samples.
E. Reyes, L.S. Marques / Applied Radiation and Isotopes 66 (2008) 1531–1542 1541
7. Concluding remarks
This paper presents results of the determination of activityconcentrations (and activity ratios) of 238U, 234U, 226Ra and 228Ra,naturally occurring radioisotopes, dissolved in ground watersextracted from two deep wells drilled in the Morungabagranitoids (SE, Brazil). The wells are recorded under the numbers0018 and 0019 in the Department of Water and Electrical Energyof Sao Paulo State (DAEE) registry. The main conclusions drawnfrom this investigation are:
(1)
Alpha spectrometry procedures used to determine 238U and234U have a high level of precision (better than 5.8%) andaccuracy (about 4%), as required for reliable hydrochemicalinterpretations. In addition, the average uranium chemicalrecovery in the whole process of 48710%, assures that themethod is adequate for ground water analyses.(2)
Gamma-ray spectrometry was used for the determination of226Ra and 228Ra. Taking into account the low activityconcentrations of the investigated ground waters, it wasnecessary to use volumes of about 45 L in order to attainreproducible results for 228Ra. The relative precision of thedata was estimated as 10% for 228Ra and better than 7% for226Ra. The average radium chemical recovery was 9377%,indicating low radium loss during sample chemical proces-sing.(3)
The activity concentrations of 238U and 234U agree withliterature data, and despite some scatter, the largest valueswere recorded during the dry season (from May to Septem-ber). These isotopes are in disequilibrium in all investigatedsamples, with significant 234U enrichment with respect to238U. During the same time interval the hydrogen ionconcentrations (pH) presented the lowest values, which wereprobably caused by the increase of the uranium lixiviationrate from the host rocks.(4)
No significant variation of 226Ra and 228Ra activity concentra-tions with pH and seasonality were observed. The relativelysmall 228Ra/226Ra activity ratios (less than one) in almost allanalyzed samples are very probably caused by 226Ra host rockpreferential lixiviation degree, which was increased by theslightly acidic character of the ground waters.
(5)
Although the distance between the two deep wells is short,there are systematic differences of 234U/238U, 226Ra/234U and228Ra/226Ra activity ratios in their ground waters. The resultsindicate different recharge processes and water percolationthrough heterogeneous granite host rock. Ground waters fromwell 0019 show evidence of some mixing with those from well0018. The latter, on the contrary, show evidence of rainwaterinput and/or contribution of another source very depleted inuranium.Acknowledgments
The authors are grateful to Fernando Brenha-Ribeiro, RosanaNunes dos Santos and Roberto Kawauchi for fruitful suggestions.Thanks are due to the condominium employees for their kindhelp. The paper was substantially improved by constructivecomments of an anonymous reviewer. This research was sup-ported by FAPESP (processes: 00/09581-9 and 03/11793-0). Thefirst author also acknowledges to CAPES for a graduate fellowship.
References
Abdul-Hadi, A., Alhassanieh, O., Ghafar, M., 2001. Disequilibrium of uraniumisotopes in some Syrian groundwater. Appl. Radiat. Isot. 55, 109–113.
Anderson, R.F., Fleer, A.P., 1982. Determination of natural actinides and plutoniumin marine particulate material. Anal. Chem. 54, 1142–1147.
Batista, J.J., Simoes, L.S.A., Oliveira, M.A.F., Souza Filho, E.E., 1986. Folha Jundiaı –Carta Geologica do Estado de Sao Paulo: 1: 50.000. Rel. Pro- Minerio/UNESP,124pp.
Benedetti, M.F., Dia, A., Riotte, J., Chabaux, F., Gerard, M., Boulegue, J., Fritz, B.,Chauvel, C., Bulourde, M., Deruelle, B., Ildefonse, P., 2003. Chemical weatheringof basaltic lava flows undergoing extreme climatic conditions: the watergeochemistry record. Chem. Geol. 201, 1–17.
Benes, P., 1990. Radium in (continental) surface water. In: IAEA (InternationalAtomic Energy Agency) (Ed.), The environmental behaviour of Radium.IAEA—Technical Reports Series 310, Vienna, pp. 373–417.
Bonotto, D.M., Andrews, J.N., 2000. The transfer of uranium isotopes 234U and 238Uto the waters interacting with carbonates from Mendiph Hills area. Appl.Radiat. Isot. 52, 965–983.
Bonotto, D.M., Jimenez-Rueda, J.R., 2007. U-ages in soils and groundwaterevidencing wet periods 400–600 kyr ago in southeast Brazil. Applied Radiationand Isotopes 65, 776–783.
Bourdon, B., Turner, S., Henderson, G.M., Lundstrom, C.C., 2003. Introduction toU-series geochemistry. Rev. Mineral. Geochem. 52, 1–21.
Calsteren, P., Thomas, L., 2006. Uranium-series dating applications in naturalenvironmental science. Earth-Sci. Rev. 75, 155–175.
Camargo, I.M.C., 1994. Determinac- ao da concentrac- ao dos isotopos naturais deuranio e torio em amostras de agua. Master Dissertation, IPEN/CNEN, 80pp.
Cizdziela, J., Farmerb, D., Hodgec, V., Lindleya, K., Stetzenbacha, K., 2005. 234U/238Uisotope ratios in groundwater from Southern Nevada: a comparison of alphacounting and magnetic sector ICP-MS. Sci. Total Environ. 350, 248–260.
Condomines, M., Hemond, C., Allegre, C.J., 1988. U–Th–Ra radioactive disequilibriaand magmatic processes. Earth Planet. Sci. Let. 90, 243–262.
Currie, L.A., 1968. Limits for qualitative detection and quantitative determination.Application to radiochemistry. Anal. Chem. 40 (3), 586–593.
Dabous, A.A., Osmond, J.K., 2000. Uranium isotopic study of artesian and pluvialcontributions to the Nubian Aquifer, Western Desert, Egypt. J. Hydrol. 243,242–253.
Dickson, B.L., 1990. Radium in groundwater. In: IAEA (International Atomic EnergyAgency) (Ed.), The environmental behaviour of Radium. IAEA—TechnicalReports Series 310, Vienna, pp. 335–372.
Dickson, B.L., Davidson, M.R., 1985. Interpretation of 234U/238U activity ratios ingroundwaters. Chem. Geol. 58, 83–88.
Eaton, A.D., Clesceri, L.S., Greenberg, A.E., 1995. Standard methods for theexamination of water and wastewater. American Public Health Association,Washington, DC, 2540pp.
EPA, 2000. National primary drinking water regulations; Radionuclides. Final Rule.Federal Register 76708–76753, 7 December.
EPA, 2001. Final draft for the drinking water criteria document on radium. TR-1441-93. US Environmental Protection Agency, Washington, DC.
Gafvert, T., Ellmark, C., Holm, E., 2002. Removal of radionuclides at a waterworks.J. Environ. Radioactivity 63, 105–115.

ARTICLE IN PRESS
E. Reyes, L.S. Marques / Applied Radiation and Isotopes 66 (2008) 1531–15421542
Gill, J.B., Pyle, D., Williams, R.W., 1992. Igneous rocks. In: Ivanovich, M., Harmon,R.S. (Eds.), Uranium Series Disequilibrium: Applications to Earth, Marine, andEnvironmental Science, second ed. Claredon Press, Oxford, p. 910pp.
Godoy, J.M., Carvalho, Z.L., de Fernandes, F.C., da Danelon, O.M., Godoy, M.L.D.P.,Ferreira, A.C.M., Roldao, L.A., 2006. 228Ra and 226Ra in coastal seawater samplesfrom the Ubatuba region—Brazilian Southeastern coastal region. J. Braz. Chem.Soc. 17 (4), 730–736.
Gomez, A., Garralon, A., Buil, B., Turrero, M.J., Sanchez, A., Cruz, B., 2006. Modelingof geochemical processes related to uranium mobilization in the groundwaterof a uranium mine. Sci. Total Environ. 366, 295–309.
Hallstadius, L., 1984. A method for the eletrodepostion of actinides. Nucl. Instrum.Methods 223, 266–267.
Henderson, G.M., Hall, B.L., Smith, A., Robinson, L.F., 2006. Control on (234U/238U) inlake water: a study in the dry valleys of Antarctica. Chem. Geol. 226, 298–308.
Ivanovich, M., Murray, A., 1992. Spectroscopic methods. In: Ivanovich, M., Harmon,R. (Eds.), Uranium Series Disequilibrium; Applications to Earth, Marine, andEnvironmental Science, second ed. Claredon Press, Oxford, p. 910pp.
Kressin, I.K., 1977. Eletrodeposition of plutonium and americium for highresolution alpha spectrometry. Anal. Chem. 49 (6), 842–846.
Kronfeld, J., Godfrey-Smith, D.I., Johannessen, D., Zentilli, M., 2004. Uranium seriesisotopes in the Avon Valley, Nova Scotia. J. Environ. Radioactivity 73, 335–352.
Lasheen, Y.F., Seliman, A.F., Abdel-Rassoul, A.A., 2007. Simultaneous measurementof 226Ra and 228Ra in natural water by liquid cintillation counting. J. Environ.Radioactivity 95, 86–97.
Lee, M.H., Choi, G.S., Cho, Y.H., Lee, C.W., Shin, H.S., 2001. Concentrations andactivity ratios of uranium isotopes in the groundwater of the Okchun Belt inKorea. J. Environ. Radioactivity 57, 105–116.
Lee, J.S., Kim, K.H., Moon, D.S., 2005. Radium isotopes in the Ulsan Bay. J. Environ.Radioactivity 82, 129–141.
Lucas, H.F., Markun, F., 1992. The determination of 226Ra and 228Ra in water andsolids by the least squares gamma spectrometric method. J. Environ. Radio-activity 15, 1–18.
Lucas, F.O., Ribeiro, F.B., 2006. Radium content in ground water from a graniticbatholith of the metamorphic basement, eastern Sao Paulo State, Brazil. Appl.Radiat. Isot. 64, 735–749.
Luo, S., Teh-lung, K., Roback, R., Murrell, M., Mcling, T.L., 2000. In-situ radionuclidetransport and preferential groundwater flows at INEEL (Idaho): decay-seriesdisequilibrium studies. Geochim. Cosmochim. Acta 64 (5), 867–881.
de Oliveira, J., 1998. Determinac- ao dos nıveis de radioatividade natural presentenas aguas utilizadas para abastecimento publico no estado de Sao Paulo. Ph.D.Thesis, IPEN/CNEN, 292pp.
de Oliveira, J., Mazzilli, B.P., de Sampa, M.H.O., Bambalas, E., 2001. Naturalradionuclides in drinking water supplies of Sao Paulo State, Brazil andconsequent population doses. J. Environ. Radioactivity 53, 99–109.
Osmond, J.K., Cowart, J.B., 1992. Ground water. In: Ivanovich, M., Harmon, R.S.(Eds.), Uranium Series Disequilibrium—Applications to Environmental Pro-blems, second ed. Claredon Press, Oxford, p. 910pp.
Porcelli, D., Swarzenski, P.W., 2003. The behaviour of U and Th series nuclides ingroundwater. Rev. Mineral. Geochem. 52, 317–362.
Puigdomenech, I., 2001. Hydrochemical stability of groundwaters surrounding aspent nuclear fuel repository in a 100000 year perspective. SKB TechnicalReport TR-01-28, 83pp.
Rihs, S., Condomines, M., 2002. An improved method for Ra isotope (226Ra, 228Ra,224Ra) measurements by gamma spectrometry in natural waters: applicationto CO2-rich thermal from the French Massif Central. Chem. Geol. 182,409–421.
Riotte, J., Chabaux, F., 1999. (234U/238U) activity ratios in freshwaters as tracers ofhydrological processes: the Strengbach watershed (Vosges, France). Geochim.Cosmochim. Acta 63, 1263–1275.
Riotte, J., Chabaux, F., Benedetti, M., Dia, A., Gerard, M., Boulegue, J., Etame, J., 2003.Uranium colloidal transport and origin of the 234U–238U fractionation insurface waters: new insights from Mount Cameroon. Chem. Geol. 202,365–381.
Robinson, L.F., Henderson, G.M., Hall, L., Matthews, I., 2004. Climatic control ofriverine and seawater uranium-isotope ratios. Science 305, 851–854.
Santos, R.N., Marques, L.S., 2007. Investigation of 238U–230Th–226Ra and232Th–228Ra–228Th radioactive disequilibria in volcanic rocks from Trindadeand Martin Vaz Islands (Brazil; Southern Atlantic Ocean). J. Volcanol.Geotherm. Res. 161, 215–233.
Santos, R.N., Marques, L.S., Brenha-Ribeiro, F., 2002. Determination of uraniumconcentrations and activity ratios in silicates by alpha spectrometry: applica-tion to the volcanic rocks from the Trindade and Martin Vaz Islands (Brazil).Appl. Radiat. Isot. 56, 741–750.
Smithson, G., 1990. Sampling and selection of analytical methods forradium. In: IAEA (International Atomic Energy Agency) (Ed.), The environ-mental behaviour of Radium. IAEA—Technical Reports Series 310, Vienna,pp. 257–271.
Sturchio, N.C., Banner, J.L., Binz, C.M., Heraty, L.B., Musgrove, M., 2001. Radiumgeochemistry of ground waters in Paleozoic carbonate aquifers, midcontinent,USA. Appl. Geochem. 16, 109–122.
Suksi, J., Rasilainen, K., Casanova, J., Ruskeeniemi, T., Blomqvist, T., Smellie, J.A.T.,2001. U-series disequilibria in a groundwater flow route as an indicator ofuranium migration processes. J. Contam. Hydrol. 47, 187–196.
Suksi, J., Rasilainen, K., Pitkanen, P., 2006. Variations in 234U/238U activity ratios ingroundwater—a key to flow system characterization? Phys. Chem. Earth 31,556–571.
Vasconcellos, M.B.A., Armelin, M.J.A., Figueiredo, A.M.G., Mazzilli, B.P., Saiki, M.,1987. A comparative study of some nuclear methods for 235U/238U isotopicratio determination. J. Radioanal. Nucl. Chem. 113, 357–370.
Vera Tome, F., Martin Sanches, A., 1991. Optimizing the parameters affecting theyield and energy resolution in the electrodeposition of uranium. Appl. Radiat.Isot. 42, 135–140.
Vesterbacka, P., Turtiainen, T., Heinavaara, S., Arvela, H., 2006. Activity concentra-tions of 226Ra and 228Ra in drilled well water in Finland. Radiat. Prot. Dosim.121 (4), 406–412.
Vlach, S.R.F., 1985. Geologia, petrologia e geocronologia das regioes meridional eoriental do Complexo de Morungaba, SP. Master Dissertation, IGc-University ofSao Paulo, 253pp.
Vlach, S.R.F., 1993. Geologia e petrologia dos granitoides de Morungaba, SP. Ph.D.Thesis, IGc-University of Sao Paulo, 414pp.
Vlach, S.R.F., 1998. Teores de U, Th e K dos granitoides de Morungaba, SP, e implicac-oes para a produc- ao de calor radiogenico e para radiac- ao natural. In: XLCongresso Brasileiro de Geologia, 395. Belo Horizonte, MG, Brazil.
Copyright © 2022 FDOKUMEN




















