
Synthesis and Vibrational Circular Dichroism Analysis ... - MDPI
Upload
independentCategory
view
2download
0
JOURNAL OF CHEMICAL PHYSICS VOLUME 115, NUMBER 9 1 SEPTEMBER 2001
Ultrafast dichroism spectroscopy of anthracene in solution.I. Inertial versus diffusive rotation in benzyl alcohol
Yunhan Zhang, Mikhail I. Sluch, Mark M. Somoza, and Mark A. Berga)
Department of Chemistry and Biochemistry, University of South Carolina, Columbia, South Carolina 29208
~Received 5 January 2001; accepted 11 June 2001!
Dichroism experiments with 150 fs time resolution on anthracene in benzyl alcohol are presented asa function of viscosity from 14.4 cP~274 K! to 2.7 cP~329 K!. These measurements test aqualitative prediction of the viscoelastic picture of liquid dynamics, specifically that earlier‘‘inertial’’ dynamics have a viscosity independent rate, whereas later ‘‘diffusive’’ dynamics have arate directly proportional to viscosity. This paper focuses on two components of the dichroism decaythat are assigned to rotational motion. A third component is assigned to electronic-state solvationand is analyzed in a companion paper@J. Chem. Phys.115, 4231~2001!#. The longest componentis due to rotational diffusion and is very well described by a hydrodynamic model with slipboundary conditions. A fast decay component in the subpicosecond region is found and shown tohave a viscosity-independent rate. It is assigned to inertial rotation by comparison to the computersimulations of Jaset al. @J. Chem. Phys.107, 8800~1997!#. Inertial rotation extends out to at least1 ps, longer than the range commonly assumed for inertial dynamics. Over much of this range, theinertial rotation is not free-rotor-like, but is strongly modified by interaction with the solvent. Theinertial rotation also accounts for the ‘‘missing’’ anisotropy found when the rotational diffusion fitsare extrapolated to zero time. ©2001 American Institute of Physics.@DOI: 10.1063/1.1389295#
o-
ymer
a
thociois-icpe
engmec
tiay-mo
reb
othf
-l:’’
ion
oa
on
’’l-lid,
ith
ndca-can
vis-ll-
hortiondentnrvessves
8u
I. INTRODUCTION
One of the major developments in the recent studymolecular dynamics in liquids is the recognition of the importance of a region of rapid ‘‘inertial’’ dynamics at vershort times. These dynamics are distinct from long-ti‘‘diffusive’’ dynamics, whose properties are much bettestablished.1 In recent theoretical work, we have developedviscoelastic~VE! interpretation of these two regimes.2–6 In-ertial dynamics occur during times short compared toshear-relaxation time of the solvent; diffusive dynamicscur on times similar to or longer than the shear-relaxattime. This picture leads to an experimental criterion for dtinguishing inertial from diffusive dynamics; inertial dynamics have a viscosity independent rate, diffusive dynamhave a rate inversely proportional to the viscosity. This paand its companions@papers II~Ref. 7! and III ~Ref. 8!# areaimed at testing this idea quantitatively. Dynamics are msured over both picosecond and subpicosecond time raand as a function of solvent viscosity by transient dichroisIndividual components of the dynamics are readily classifias diffusive or inertial based on their viscosity dependenInertial dynamics are seen over a 1 ps orlonger time range,a much longer time than is usually associated with inerdynamics. Moreover, it is shown explicitly that inertial dnamics cannot be equated to gaslike, single-moleculetions. Over much of the inertial time range, interactionsthe solute with the solvent are important.
The concept of a very early time regime in liquids chaacterized by inertial dynamics has existed for a long timHowever, it came to renewed prominence following the o
a!Author to whom correspondence should be addressed. Telephone:777-1514; Fax: 803-777-1456; electronic mail: [email protected]
4210021-9606/2001/115(9)/4212/11/$18.00
Downloaded 01 Sep 2003 to 139.78.100.86. Redistribution subject to A
f
e
e-n-
sr
a-es.de.
l
o-f
-.-
servation of very strong responses in the 100 fs region, bin computer simulations9 and in the then emerging area ofemtosecond spectroscopy.10–13 Several interrelated, but distinct, ideas have become associated with the term ‘‘inertia~1! the fact that the leading term in the decay of correlatfunctions does not involve intermolecular forces,14,15 i.e., in-ertial motion is ‘‘free’’ or ‘‘ballistic’’ 16 motion; ~2! the qua-dratic behavior of correlation functions in the limit of zertime;14,15 ~3! the extension of this quadratic behavior intoGaussian approximation for the inertial portion of correlatifunctions;9 ~4! a ‘‘rigid-cage’’ approximation in which eachsolvent molecule moves under the forces present att50, i.e.,inertial motion is a superposition of ‘‘single-moleculemotions;17 ~5! a viscoelastic approximation in which the soute interacts with a solvent that responds like an elastic sobut cannot undergo structural rearrangement;2–6 ~6! the ob-servation of a clearly distinguishable decay component wa time constant near 100 fs.9–13 It is not always clear whichof these ideas is defining, which are rigorous corollaries awhich are approximations subject to experimental verifition. In fact, many of these ideas are not equivalent andbe valid over different time ranges.
In a recent series of papers, we have developed acoelastic picture of liquid dynamic that leads to a wedefined and testable definition of inertial dynamics.2–6 In theVE picture, the solvent behaves as an elastic solid at stimes and as a viscous liquid at long times. The transitbetween these time ranges is governed by a time-depenshear modulusG(t) with a characteristic shear relaxatiotime tsh and initial valueG`. At times shorter than the shearelaxation time, the solvent can support propagating wa~acoustic phonons!. In a physical picture, inertial dynamicare defined as the interaction of a solute with these wa
03-
2 © 2001 American Institute of Physics
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

tac-iutia.,umn
tin
-s-neth
–eg
erlidc
le
eiiffue-ca
thdoioorad
esatit
he
thctatest
seics
tioa
abroo
en aityhetest-ge
ec-hro-nald.-
e toen-
ex-pers.om-
er, a-n.n
tialight
ns,nts,a-tua-le-
r-les,lventIR
ce-
e-nt
ionionthe
ab-le,
dis-e’slace-on-ncelute
4213J. Chem. Phys., Vol. 115, No. 9, 1 September 2001 Inertial versus diffusive rotation. I
during the time before shear relaxation becomes impor@idea~5! above#. In a mathematical treatment of the VE piture, the solvent is approximated as a continuous medgoverned by a viscoelastic equation of motion. The inerdynamics arise from the ‘‘inertial’’ term in this equation, i.ethe term contains the mass and acceleration of each volof the solvent. This term allows coherent transport amemory of momentum through the solvent, i.e., propagawaves.
This VE definition of inertial dynamics is distinctly different from ideas~1!–~3! above. The isolated molecule, galike picture of dynamics contained in these ideas is confito a shorter time period, which we prefer to distinguish as‘‘ballistic’’ regime.2 The VE theory is related to the rigidcage model@~4! above# in that both models include solventsolute interactions during the inertial dynamics. Howevthe VE theory goes one step further than the rigid-camodel by allowing solvent molecules to affect each othmotion, at least to the extent that they do in an elastic soConsequently, correlated, phononlike solvent motions ocin the VE model, but not in the rigid-cage model.
In experiments, a simple distinction based on time scahas usually been used to define inertial dynamics@~6! above#.Decays of 100 fs or shorter are generally accepted as binertial; times of 1 ps or greater are accepted as being dsive. This definition suffers from subjectivity in the intermdiate time range. Although the observation of a short detime is sometimes equated with ideas~1!–~3! above, thisconnection is difficult to make rigorous. The shortness of1/e decay time or a bifurcation of the relaxation dynamicsnot in themselves imply that the zero-time limiting behavof the correlation function has been observed. Furthermthe extension of the zero-time limiting behavior intoGaussian correlation function to describe the entire fastcay component is a completely empirical approximation.9
In contrast, the VE definition of inertial dynamics makan objective and verifiable experimental prediction—the rof inertial dynamics is independent of the solvent viscosh. Viscosity is a direct result of shear relaxation within tliquid
h5G`tsh. ~1!
If a time dependent viscosity is defined, it is zero duringtime before shear relaxation occurs, and it cannot be a fain solute dynamics.~The viscosity is zero because there hbeen no energy dissipation. The processes that contribuviscosity at long times only cause a nondissipative, elaresponse at short times.!
In general, diffusive dynamics are defined by contraWhatever is longer than inertial is diffusive. The VE modsharpens this definition as well. It defines diffusive dynamas those governed by the relaxation of the shear modulu@orthe similar relaxation of the bulk modulusK(t)#. Diffusivedynamics occur on the time scale of the shear relaxaitself or on longer times when multiple shear relaxations leto the observed phenomenon. The experimentally testcharacteristic of diffusive dynamics is that their rate is pportional to the shear relaxation time and, therefore, proptional to the solvent viscosity.
Downloaded 01 Sep 2003 to 139.78.100.86. Redistribution subject to A
nt
ml
edg
de
r,es.
ur
s
ng-
y
e
re,
e-
ey
eorsto
ic
t.ls
ndle-r-
Although these ideas are appealing, there has not beclear experimental demonstration of the differing viscosdependence of inertial and diffusive dynamics within tsame system. The purpose of this series of papers is tothe validity of the VE-based definitions of inertial and diffusive by monitoring dynamics over the 150 fs to 290 ps ranas a function of solvent viscosity. Transient dichroism sptroscopy is used as the probe of dynamics. Transient dicism is usually associated with measurement of rotatiodiffusion.1,18–20 We will find decay components associatewith both rotational diffusion and with inertial rotationMoreover, we will show that under the correct circumstances, dichroism also shows a decay component duelectronic-state solvation. Our initial experiments are in bzyl alcohol, a solvent of moderate viscosity~3–15 cP!, whereinertial and diffusive times are well separated. For clearposition, these experiments are discussed in three paThis paper focuses on the rotational components. The cpanion papers~papers II and III! focus on the theory andanalysis of the solvation component. A forthcoming pap~paper IV! will present dichroism measurement in toluenelow viscosity ~0.3–1 cP! solvent where the distinction between inertial and diffusive dynamics begins to break dow
The current observation of inertial rotation in itself is aadvance over existing studies of inertial dynamics. Inereffects have been most widely observed in depolarized lscattering21,22 and optical Kerr-effect experiments.10,23Theseexperiments measure a combination of density fluctuatiosingle-molecule rotation and, in the case of neat solvecollective rotation.24–27 Because the current dichroism mesurements are resonant with a dilute solute, density fluctions and collective motions do not interfere with the singmolecule rotation effects.
More fundamentally, light scattering and optical Kereffect experiments are largely focused on small molecui.e., solutes that are the same size or smaller than the somolecules. The same is true of most studies of rotation byor Raman line shape analysis.22 An exception is the detailedstudy by Pereriaet al. of the inertial rotation of aniline usingfluorescence depolarization.28 Although aniline is relativelylarge, its nearly circular profile causes little solvent displament during its rotation. In small molecule~or low solvent-displacement! systems, inertial effects become prominent bcause the coupling of the solute rotation to solvedisplacement is weak. In the limit where the solute rotatrequires very little solvent displacement, the solute’s motis nearly free, and the solvent acts as a perturbation ongaslike rotation. This perspective is evident in many estlished theoretical treatments of inertial rotation, for exampGordon’s well-known extended diffusion model.29
In the current experiments, the solute, anthracene, istinctly larger than the solvent, benzyl alcohol. Anthracenaspherical shape also guarantees that large solvent dispments are needed for overall rotation. In this case, a ctinuum model of the solvent is a more reasonable referesystem than a gas-phase model. Inertial rotation of the so
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

ota
luigwntamh
eeE
ha
tat-
ecbe
thp
one
Wsint te
dyelsribth
dd
ancaut
tho
cuontc-
thE
ald.
tan-
21tion. Ay
ong.asrobeingn alorza-ol-ntoamolli-er.as
l fil-tedfil-ndamst of
pli-amre-the
a-
de-reastore-
tingalgethe
ionlse-d byonlse
gedcann-ef-se
line
4214 J. Chem. Phys., Vol. 115, No. 9, 1 September 2001 Zhang et al.
arises from the inherent properties of the early dynamicsthe solvent, not because the solute is weakly coupled tosolvent. The current experiments are the first to make qutitative measurements of this important case.
In all these experiments, anthracene is used as the soIn terms of rotational properties, anthracene is rigid, has hsymmetry, and a very compact geometry. Consequently,find that the diffusive rotation is near single exponential avery well described by hydrodynamic models. The free rotion of anthracene can be calculated easily and will be copared to the early rotational behavior that we observe. Tcomparison provides a direct test of the distinction betwballistic and inertial dynamics that is suggested by Vtheory.
Anthracene’s rotation on the picosecond time scalebeen studied by depolarized light scattering,30 fluorescencedepolarization,30,31 and UV/IR double resonance.32 This pa-per extends the time range of these studies into the femsecond region. Anthracene’s rotation has also been simulin computer experiments,31 and the results of these simulations will be compared to our early time results. Femtosond dichroism studies of substituted anthracenes havereported by Hartmanet al.,33 but they did not include theunsubstituted anthracene studied here.
The experimental methods are described in Sec. II ofpaper. The data show three well-separated decay comnents. The slowest of these times is assigned to rotatidiffusion and is discussed in Sec. III. A hydrodynamic modprovides a very good explanation of this component.calibrate the amplitude of the dichroism measurements ufluorescence depolarization measurements and find thaamplitude of the rotational anisotropy does not extrapolat0.4 at zero time as predicted theoretically.
The intermediate decay time is assigned to solvationnamics. It is discussed in detail in paper III. Paper II devops the spectroscopic basis for seeing solvation dynamicdichroism measurements. In this paper, we only deschow this component is subtracted from the data to leaverotational components.
The third and fastest decay component is discusseSec. IV. It has a decay constant of about 0.5 ps that is inpendent of viscosity. By the VE definition, it comes frominertial process, despite the long decay time. This dematches the decay time of inertial rotation seen in compsimulations of anthracene rotation.31 The size of this decaycomponent also accounts for the missing amplitude intime-zero anisotropy. For these reasons, the fast decay cponent is assigned to inertial rotation.
In Sec. V, the free-rotor behavior of anthracene is callated and compared to the experimental inertial rotatiFree-rotor~i.e., ballistic! behavior only occurs for the firs100 fs of the total 1 ps range of inertial motion, and it acounts for only the first half of the total inertial decay amplitude. All these results are in good agreement withqualitative picture of liquid dynamics contained in the Vmodel.
The role of inertial rotation in reducing experimentvalues of the initial anisotropyr 0 is discussed in Sec. VI, anthe conclusions of the paper are summarized in Sec. VII
Downloaded 01 Sep 2003 to 139.78.100.86. Redistribution subject to A
fhen-
te.he
d--
isn
s
o-ed
-en
iso-allegheto
--inee
ine-
yer
em-
-.
-
e
II. EXPERIMENTAL METHODS
The dichroism measurements were performed in a sdard polarization-spectroscopy setup.20 The light pulses weregenerated by a mode-locked Ti:sapphire laser with LAKLprisms. The pulses had a temporal width of 60 fs, a repetirate of 78 MHz, and a typical average power of 850 mW1 mm LBO crystal produced 25 mW of light at 384 nm bsecond harmonic generation. This wavelength is on the lwavelength edge of the anthracene absorption spectrum
The 384 nm beam was split into two portions: 90% wused as the pump beam and 10% as the probe. The pbeam passed through a Glan–Taylor polarizer before befocused into the sample. The pump beam traveled dowvariable optical delay line and through another Glan–Taypolarizer oriented at 45° with respect to the probe polarition. The pump beam was reflected to travel nearly clinearly with the probe, and both beams were focused ithe sample jet by a 75 mm focal length lens. The pump bewas blocked after the sample. The probe beam was cmated and directed to an analyzing Glan–Taylor polarizThe extinction ratio for the probe beam without sample wbetter than 131027 and increased to 531026 with thesample jet in place.
The transmitted probe beam passed through a spatiater to eliminate scattered pump light and then was detecby a photomultiplier tube. Bandpass and neutral densityters before the photomultiplier blocked background light aadjusted the signal intensity. Both the pump and probe bewere modulated by a mechanical chopper, and the outputhe photomultiplier tube was processed by a lock-in amfier. The chopping frequency was 1 kHz for the probe beand 832 Hz for the pump. Detection was at the sum fquency. The signal was recorded for each position ofdelay line and stored in a computer.
To avoid the inherent complexity of analyzing a qudratic signal~as observed in a homodyne experiment! and toincrease the experimental sensitivity, optical heterodynetection was used.20 By rotating the analyzing polarizeslightly from the position of maximum extinction toward thpump polarization, a positive in-phase local oscillator wproduced. The local oscillator amplitude was adjusted30–40 times the homodyne signal intensity. Scans werepeated with a negative local oscillator generated by rotathe analyzer in the opposite direction. The residuhomodyne-dichroism, birefringence, and polarizer-leakasignals were eliminated by taking the difference betweenpositive and negative scans.
A large signal peak occurs in the pulse-overlap regand is about ten times the size of the signal in the puseparated region. The pulse autocorrelation was measureelectronic Kerr birefringence in silica at the sample positiand gave a 73 fs FWHM Gaussian, indicating that the puoverlap effects should be complete by;100 fs. However, thedetails of the signal in the 100–150 fs time range chanday-to-day, suggesting that the tail of the coherence peakleak into this region if the pulse properties are not well cotrolled. In this paper, we avoid the region of coherencefects entirely and do not report data below 150 fs. After theexperiments were complete, improvements in the delay
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp
n
it
iiowadd
atw
t frnthb
re
eO
a
omlardf
-
e
(-
wa
lcielotstangfi
s-anon
is
a-ta-
fu-
rin-
ebe
ta-
l ats
and
thevan
, it
4215J. Chem. Phys., Vol. 115, No. 9, 1 September 2001 Inertial versus diffusive rotation. I
and in pulse-chirp control stabilized the signal in this regiowithout changing results at times.150 fs. Paper IV exam-ines the coherence peak in more detail and shows thatwell distinguished from the real decays reported here.34
The sample consisted of a solution of anthracenebenzyl alcohol in a flowing jet. The anthracene concentratwas 5 mM, and the absorption at the pump wavelength30%. At this concentration and below, the decays were inpendent of concentration, although artifacts were observehigher concentrations.
The sample had to be placed in a flowing jet to eliminthermal effects and artifacts from photo-by-products. At lotemperatures, the jet system was fully enclosed, excepsmall holes around the laser beams. A small flow of dnitrogen into the circulating system excluded room air aprevented condensation and absorption of water intosample. The temperature of the sample was measuredthermistor placed on the jet nozzle.
The viscosity of benzyl alcohol was taken from literatusources for temperatures above 300 K.35–38Below 300 K, wemade measurements with a standard Ubbelohde viscomused according to the manufacturer’s specifications.measurements were interpolated with the following fit:
lnS h
cPD521.61471368
~T/K!2188.31~274 K,T,301 K!.
~2!
Fluorescence depolarization measurements were musing time-resolved single photon counting.39,40 The staticsample was excited by low intensity pulses at 375 nm frsecond harmonic generation of a different, but simiTi:sapphire laser. The instrument response function haFWHM of 50 ps. Fluorescence decays were measuredpolarizations parallelI i(t) and perpendicularI'(t) to theexcitation polarization. The sum signalI S(t)5I i(t)12I'(t) was fit by iterative reconvolution with the instrument response function to the response functionRS(t)5Ae2t/tfl1(12A)e2t/t2. This fit gives the excited statlifetime of tfl52.67 ns with an amplitudeA50.992 in asingle exponential. The secondary component is small2A50.008), fast (t25236 ps) and unassigned, but improved the fits to the sum signal. This response functionfixed in a reconvolution fit of the difference signalI d
5I i(t)2I'(t) to the response functionRd(t)5r (t)RS(t).
III. DIFFUSIVE ROTATION
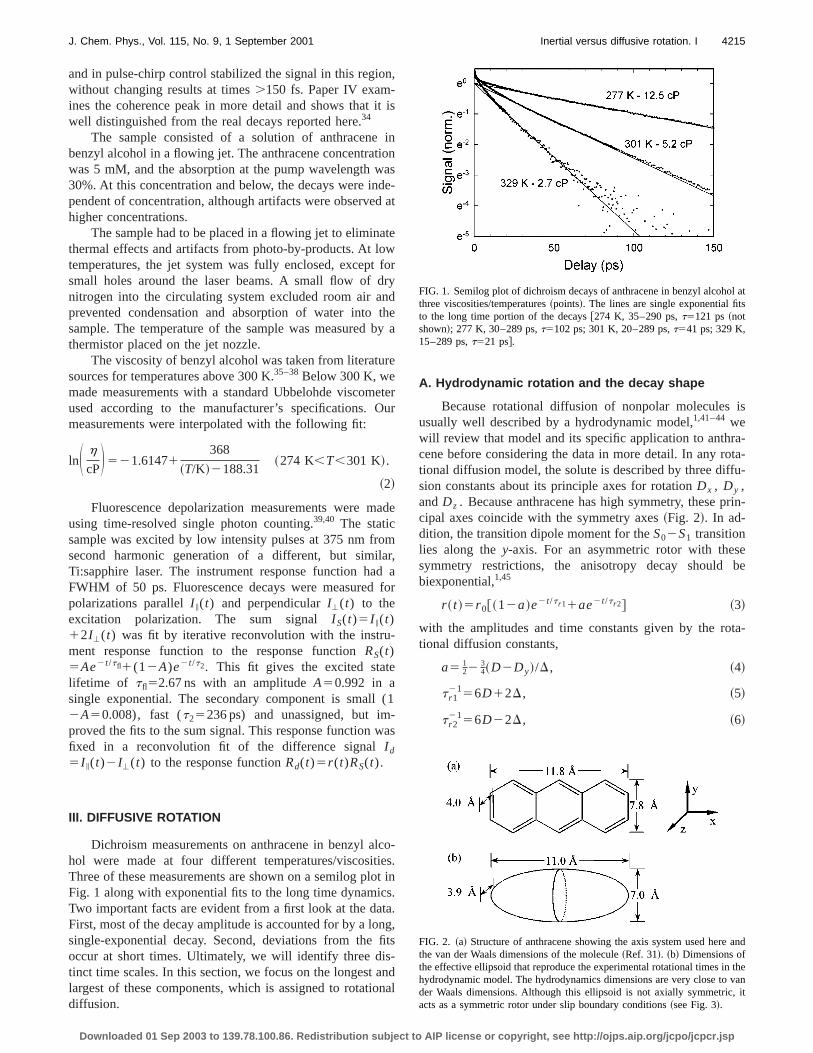
Dichroism measurements on anthracene in benzyl ahol were made at four different temperatures/viscositThree of these measurements are shown on a semilog pFig. 1 along with exponential fits to the long time dynamicTwo important facts are evident from a first look at the daFirst, most of the decay amplitude is accounted for by a losingle-exponential decay. Second, deviations from theoccur at short times. Ultimately, we will identify three ditinct time scales. In this section, we focus on the longestlargest of these components, which is assigned to rotatidiffusion.
Downloaded 01 Sep 2003 to 139.78.100.86. Redistribution subject to A
,
is
nns
e-at
e
oryde
y a
terur
de
,a
or
1
s
o-s.in
..,
ts
dal
A. Hydrodynamic rotation and the decay shape
Because rotational diffusion of nonpolar moleculesusually well described by a hydrodynamic model,1,41–44 wewill review that model and its specific application to anthrcene before considering the data in more detail. In any rotional diffusion model, the solute is described by three difsion constants about its principle axes for rotationDx , Dy ,andDz . Because anthracene has high symmetry, these pcipal axes coincide with the symmetry axes~Fig. 2!. In ad-dition, the transition dipole moment for theS02S1 transitionlies along they-axis. For an asymmetric rotor with thessymmetry restrictions, the anisotropy decay shouldbiexponential,1,45
r ~ t !5r 0@~12a!e2t/tr11ae2t/tr2# ~3!
with the amplitudes and time constants given by the rotional diffusion constants,
a5 122 3
4~D2Dy!/D, ~4!
t r12156D12D, ~5!
t r22156D22D, ~6!
FIG. 1. Semilog plot of dichroism decays of anthracene in benzyl alcohothree viscosities/temperatures~points!. The lines are single exponential fitto the long time portion of the decays@274 K, 35–290 ps,t5121 ps~notshown!; 277 K, 30–289 ps,t5102 ps; 301 K, 20–289 ps,t541 ps; 329 K,15–289 ps,t521 ps#.
FIG. 2. ~a! Structure of anthracene showing the axis system used herethe van der Waals dimensions of the molecule~Ref. 31!. ~b! Dimensions ofthe effective ellipsoid that reproduce the experimental rotational times inhydrodynamic model. The hydrodynamics dimensions are very close toder Waals dimensions. Although this ellipsoid is not axially symmetricacts as a symmetric rotor under slip boundary conditions~see Fig. 3!.
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

th
tein
ec
idalsin
ob
f
-
e
ire-
a-
ny
e
netrex
haotmti
h--,n
allD,tricons
them-nottwo
e aeli-urves
-
e aa-
hro-
ene
x-
y
-
etric
4216 J. Chem. Phys., Vol. 115, No. 9, 1 September 2001 Zhang et al.
D5~Dx1Dy1Dz!/3, ~7!
D25Dx21Dy
21Dz22DxDy2DyDz2DxDz . ~8!
The initial anisotropyr 0 has a theoretical value ofr 050.4,although experimental measurements often fall short ofvalue.
In the hydrodynamic model, the solute is approximaas an ellipsoid rotating in a viscous continuum. The resultdiffusion constants are
Di5kT
l iVhh. ~9!
The unitless shape factorl i depends on the axial ratios of thellipsoid and on the boundary conditions. For a companonpolar solute, slip boundary conditions are expectedapply.41–44Ideally, the hydrodynamic volume of the ellipsoVh would be rigorously calculated from the van der Wasize of the solute. In practice, there is some flexibilitymatching the true van der Waals shape of the molecule tellipsoid, soVh becomes a fitting parameter constrained tonear the van der Waals volume of the solute.
Within the rotational diffusion model, the amplitude othe second exponential of Eq.~3! will vanish if the diffusionconstants obeyDx5Dz , i.e., in the case of a symmetric rotor. In this limit, the anisotropy decay simplifies to
r ~ t !5r 0e2t/tr1, ~10!
t r151
6D'
5l'Vh
6kTh, ~11!
whereD'5Dx5Dz . In the hydrodynamic model, this castranslates to a condition on the shape factors,lx5lz . If thesolute ellipsoid is described byx21y2/a21z2/b251, thiscase obviously holds for the pointa5b, i.e., for a symmetricellipsoid. However, this condition also occurs for an entline of other points in thea–b plane. In other words, asymmetric ellipsoids can be symmetric rotors.
A portion of the symmetric rotor line relevant to anthrcene is shown in Fig. 3. The line corresponding toDx5Dz
was found by interpolating the calculations of Youngren aAcrivos46 for slip boundary conditions as corrected bSension and Hochstrasser.47 The point corresponding to thratios of the van der Waals axes31 is shown on Fig. 3 as anopen square. Given the expanded scales in Fig. 3, the vaWaals shape predicts that anthracene is a nearly symmrotor. The values ofa andb that correspond well to computesimulations of anthracene rotation in cyclohexane liesactly on the symmetric rotor line~Fig. 3, open circle!.31
These estimates of a nearly symmetric rotor show tthe observation of a single exponential decay due to rtional diffusion is very reasonable. The current dichroisexperiments are not the only ones to find single exponendecays for anthracene. Lettenbergeret al. found single expo-nential r (t)’s for anthracene by an entirely different tecnique, IR/UV double resonance.32 Our fluorescence depolarization experiments~Sec. III B! also find this result. Thusanthracene is a nearly symmetric rotor due to an accidedegeneracy ofDx andDz .
Downloaded 01 Sep 2003 to 139.78.100.86. Redistribution subject to A
is
dg
t,to
ane
d
derric
-
ta-
al
tal
In principle, there is a second and longer decay timet r2
due to deviations from a perfectly symmetric rotor@Eq.~10!#,
t r251
4Dy12D'
53
112~l' /ly!t r1 . ~12!
For a nearly symmetric rotor, this component has a smamplitude that vanishes in the symmetric limit. In Sec. IIIwe will find that a good model for anthracene is a symmerotor with dimensions close to the van der Waals dimensi~a50.35 andb50.64, filled circle in Fig. 3!. These param-eters gively54.43 andl'51.12, ort r252.0 t r1 . Thus ifthe second decay were observed, it would be longer thanprimary decay time. It cannot explain the faster decay coponents seen in Fig. 1. Also, note that the second decay isobserved because its amplitude is small, not because thedecay times are difficult to distinguish.
B. Fluorescence depolarization and the initialanisotropy
Our fluorescence depolarization measurements havlimited instrument response time of 50 ps and are only rable for measuring rotational diffusion at the high end of oviscosity range. However, fluorescence depolarization githe absolute orientation functionr (t) directly, whereas di-chroism givesDAr(t), the product of the orientation function and the absorption change. The magnitude ofDA is notknown, so the dichroism measurements do not providmeasurement ofr 0 . We use fluorescence depolarization mesurements to calibrate the absolute magnitude of the dicism measurements.
The fluorescence depolarization curves from anthracin benzyl alcohol at 274 K~14.4 cP! are shown in Fig. 4. Thecurves are fit with an orientation function that is single eponential, consistent with a symmetric rotor@Eq. ~10!#. The
FIG. 3. Certain ratios of the ellipsoid dimensions@see Fig. 2~b!# give asymmetric rotor (Dx5Dz) in the hydrodynamic model with slip boundarconditions. The curve shows some of these values on a plot ofa, the ratio ofthe y- to x-semiaxes, againstb, the ratio ofz- to x-semiaxes. The curve isinterpolated from the calculation in Refs. 46 and 47.~h!, van der Waalsdimensions@Fig. 2~a!#. ~s!, dimensions from matching computer simulations ~Ref. 31!. ~j!, our hydrodynamic model@Fig. 2~b!#. Note that thescales are expanded, and all these points correspond to nearly symmrotors.
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp
ten
ialthad
s-tu
inil
en
tioll
--see
a
cera
m-riza-
terndthe
ive
icstedthepyre-as--
40.ent
enweonre-
ata
theeeThefm
scale
4217J. Chem. Phys., Vol. 115, No. 9, 1 September 2001 Inertial versus diffusive rotation. I
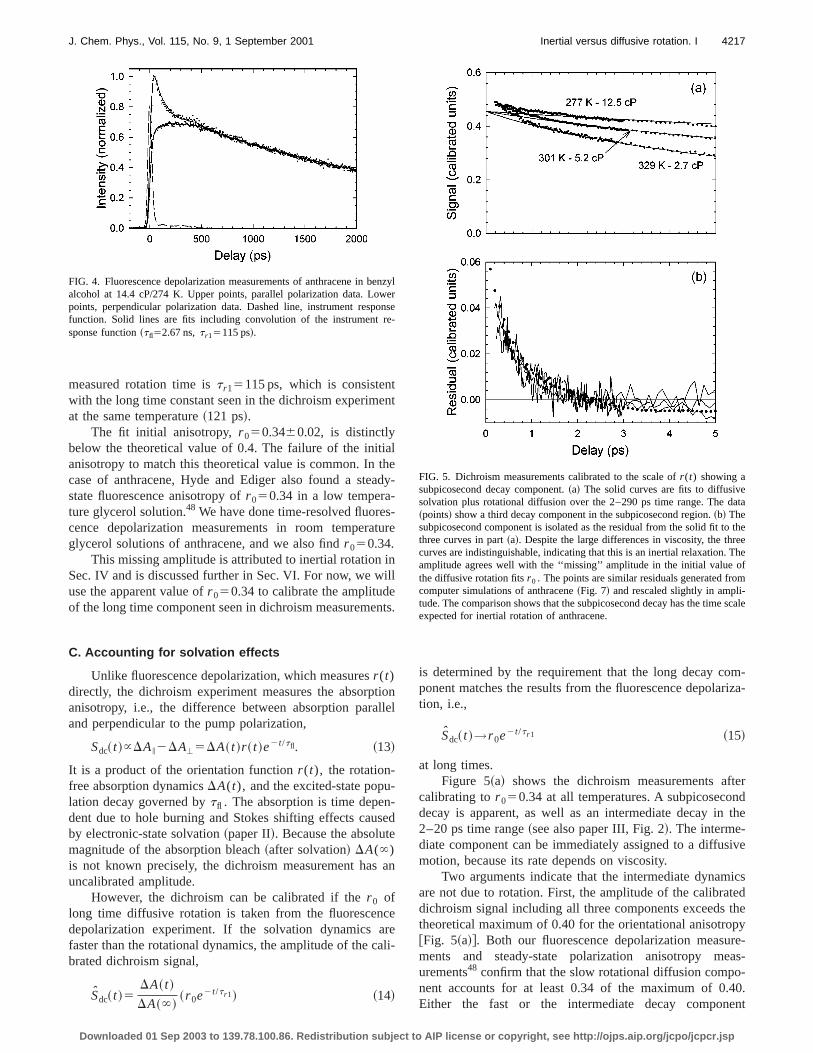
measured rotation time ist r15115 ps, which is consistenwith the long time constant seen in the dichroism experimat the same temperature~121 ps!.
The fit initial anisotropy,r 050.3460.02, is distinctlybelow the theoretical value of 0.4. The failure of the initanisotropy to match this theoretical value is common. Incase of anthracene, Hyde and Ediger also found a stestate fluorescence anisotropy ofr 050.34 in a low tempera-ture glycerol solution.48 We have done time-resolved fluorecence depolarization measurements in room temperaglycerol solutions of anthracene, and we also findr 050.34.
This missing amplitude is attributed to inertial rotationSec. IV and is discussed further in Sec. VI. For now, we wuse the apparent value ofr 050.34 to calibrate the amplitudof the long time component seen in dichroism measureme
C. Accounting for solvation effects
Unlike fluorescence depolarization, which measuresr (t)directly, the dichroism experiment measures the absorpanisotropy, i.e., the difference between absorption paraand perpendicular to the pump polarization,
Sdc~ t !}DAi2DA'5DA~ t !r ~ t !e2t/tfl. ~13!
It is a product of the orientation functionr (t), the rotation-free absorption dynamicsDA(t), and the excited-state population decay governed bytfl . The absorption is time dependent due to hole burning and Stokes shifting effects cauby electronic-state solvation~paper II!. Because the absolutmagnitude of the absorption bleach~after solvation! DA(`)is not known precisely, the dichroism measurement hasuncalibrated amplitude.
However, the dichroism can be calibrated if ther 0 oflong time diffusive rotation is taken from the fluorescendepolarization experiment. If the solvation dynamics afaster than the rotational dynamics, the amplitude of the cbrated dichroism signal,
Sdc~ t !5DA~ t !
DA~`!~r 0e2t/tr1! ~14!
FIG. 4. Fluorescence depolarization measurements of anthracene in balcohol at 14.4 cP/274 K. Upper points, parallel polarization data. Lopoints, perpendicular polarization data. Dashed line, instrument respfunction. Solid lines are fits including convolution of the instrumentsponse function~tfl52.67 ns,t r15115 ps!.
Downloaded 01 Sep 2003 to 139.78.100.86. Redistribution subject to A
t
ey-
re
l
ts.
nel
d
n
eli-
is determined by the requirement that the long decay coponent matches the results from the fluorescence depolation, i.e.,
Sdc~ t !→r 0e2t/tr1 ~15!
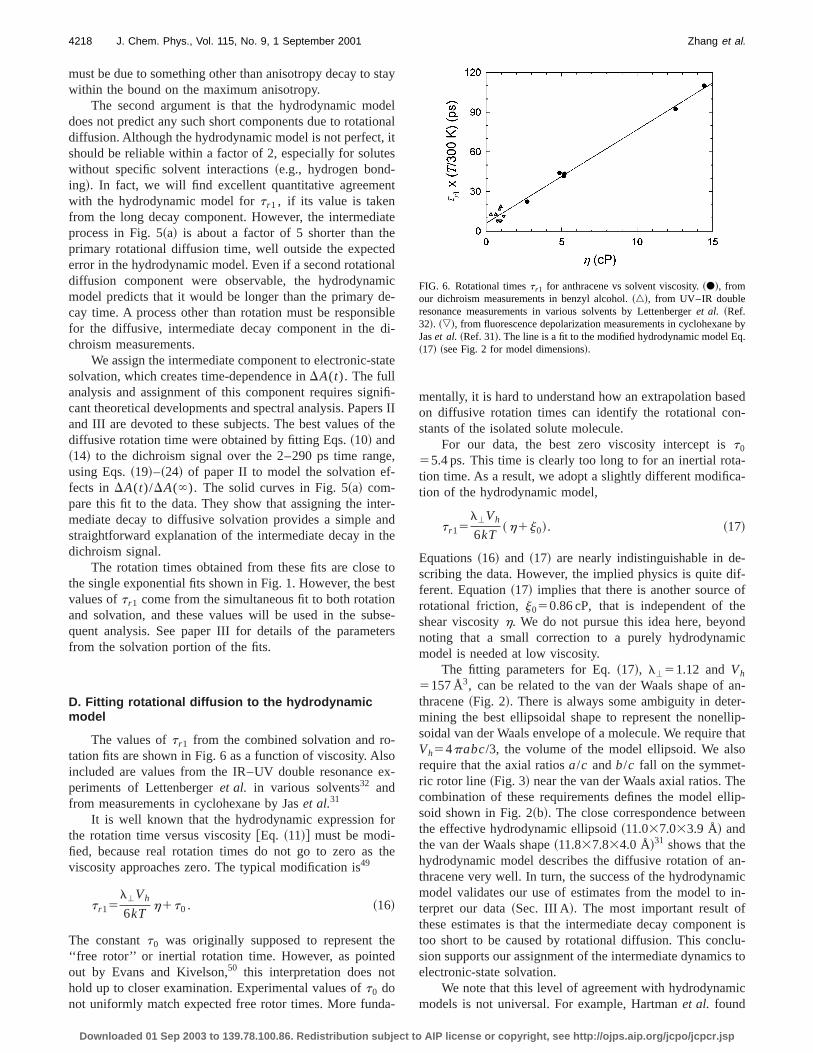
at long times.Figure 5~a! shows the dichroism measurements af
calibrating tor 050.34 at all temperatures. A subpicosecodecay is apparent, as well as an intermediate decay in2–20 ps time range~see also paper III, Fig. 2!. The interme-diate component can be immediately assigned to a diffusmotion, because its rate depends on viscosity.
Two arguments indicate that the intermediate dynamare not due to rotation. First, the amplitude of the calibradichroism signal including all three components exceedstheoretical maximum of 0.40 for the orientational anisotro@Fig. 5~a!#. Both our fluorescence depolarization measuments and steady-state polarization anisotropy meurements48 confirm that the slow rotational diffusion component accounts for at least 0.34 of the maximum of 0.Either the fast or the intermediate decay compon
zylrse
FIG. 5. Dichroism measurements calibrated to the scale ofr (t) showing asubpicosecond decay component.~a! The solid curves are fits to diffusivesolvation plus rotational diffusion over the 2–290 ps time range. The d~points! show a third decay component in the subpicosecond region.~b! Thesubpicosecond component is isolated as the residual from the solid fit tothree curves in part~a!. Despite the large differences in viscosity, the thrcurves are indistinguishable, indicating that this is an inertial relaxation.amplitude agrees well with the ‘‘missing’’ amplitude in the initial value othe diffusive rotation fitsr 0 . The points are similar residuals generated frocomputer simulations of anthracene~Fig. 7! and rescaled slightly in ampli-tude. The comparison shows that the subpicosecond decay has the timeexpected for inertial rotation of anthracene.
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp
st
don, ite-nt
iaeedna
eibdi
ta
nierf t
ge-
tenth
tens
te
-soex
or
th
hedt
a
sedn-
a-a-
-if-
ofendic
an-r-llip-thato
ellip-en
n-micin-
fnt islu-s to
ic
e byq.
4218 J. Chem. Phys., Vol. 115, No. 9, 1 September 2001 Zhang et al.
must be due to something other than anisotropy decay towithin the bound on the maximum anisotropy.
The second argument is that the hydrodynamic modoes not predict any such short components due to rotatidiffusion. Although the hydrodynamic model is not perfectshould be reliable within a factor of 2, especially for solutwithout specific solvent interactions~e.g., hydrogen bonding!. In fact, we will find excellent quantitative agreemewith the hydrodynamic model fort r1 , if its value is takenfrom the long decay component. However, the intermedprocess in Fig. 5~a! is about a factor of 5 shorter than thprimary rotational diffusion time, well outside the expecterror in the hydrodynamic model. Even if a second rotatiodiffusion component were observable, the hydrodynammodel predicts that it would be longer than the primary dcay time. A process other than rotation must be responsfor the diffusive, intermediate decay component in thechroism measurements.
We assign the intermediate component to electronic-ssolvation, which creates time-dependence inDA(t). The fullanalysis and assignment of this component requires sigcant theoretical developments and spectral analysis. Papand III are devoted to these subjects. The best values odiffusive rotation time were obtained by fitting Eqs.~10! and~14! to the dichroism signal over the 2–290 ps time ranusing Eqs.~19!–~24! of paper II to model the solvation effects in DA(t)/DA(`). The solid curves in Fig. 5~a! com-pare this fit to the data. They show that assigning the inmediate decay to diffusive solvation provides a simple astraightforward explanation of the intermediate decay indichroism signal.
The rotation times obtained from these fits are closethe single exponential fits shown in Fig. 1. However, the bvalues oft r1 come from the simultaneous fit to both rotatioand solvation, and these values will be used in the subquent analysis. See paper III for details of the paramefrom the solvation portion of the fits.
D. Fitting rotational diffusion to the hydrodynamicmodel
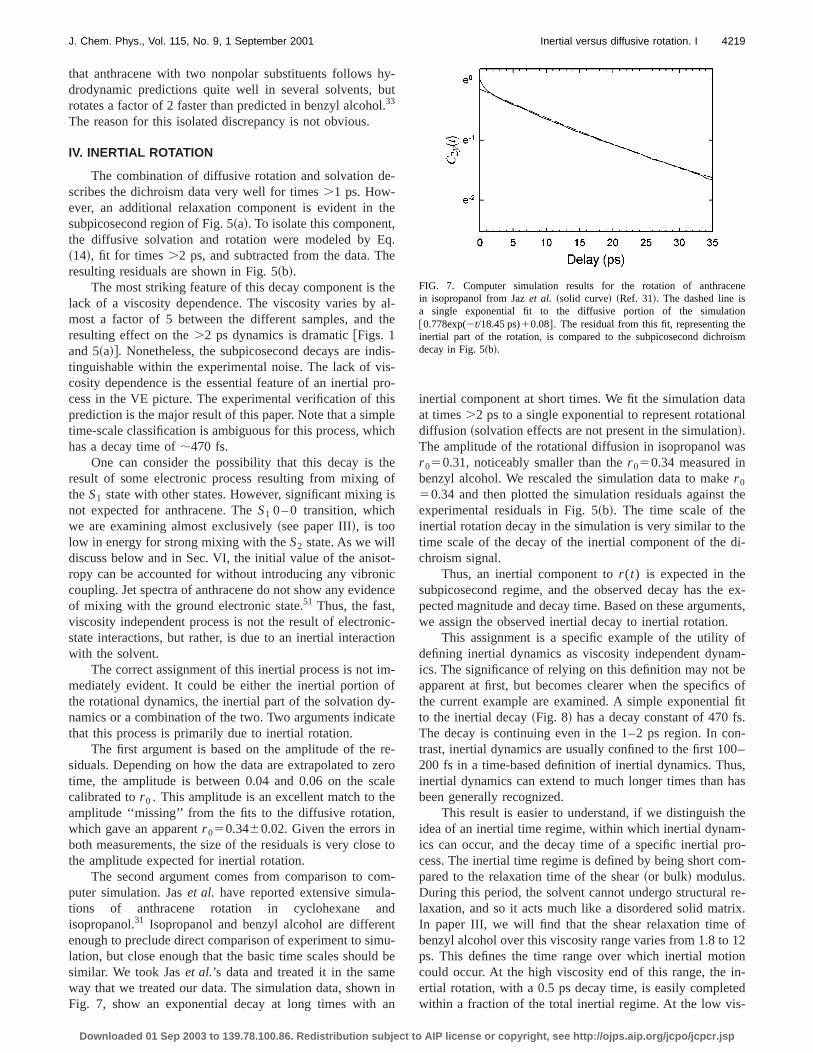
The values oft r1 from the combined solvation and rotation fits are shown in Fig. 6 as a function of viscosity. Alincluded are values from the IR–UV double resonanceperiments of Lettenbergeret al. in various solvents32 andfrom measurements in cyclohexane by Jaset al.31
It is well known that the hydrodynamic expression fthe rotation time versus viscosity@Eq. ~11!# must be modi-fied, because real rotation times do not go to zero asviscosity approaches zero. The typical modification is49
t r15l'Vh
6kTh1t0 . ~16!
The constantt0 was originally supposed to represent t‘‘free rotor’’ or inertial rotation time. However, as pointeout by Evans and Kivelson,50 this interpretation does nohold up to closer examination. Experimental values oft0 donot uniformly match expected free rotor times. More fund
Downloaded 01 Sep 2003 to 139.78.100.86. Redistribution subject to A
ay
elal
s
te
lic-le-
te
fi-s IIhe
,
r-de
ost
e-rs
-
e
-
mentally, it is hard to understand how an extrapolation baon diffusive rotation times can identify the rotational costants of the isolated solute molecule.
For our data, the best zero viscosity intercept ist0
55.4 ps. This time is clearly too long to for an inertial rottion time. As a result, we adopt a slightly different modifiction of the hydrodynamic model,
t r15l'Vh
6kT~h1j0!. ~17!
Equations~16! and ~17! are nearly indistinguishable in describing the data. However, the implied physics is quite dferent. Equation~17! implies that there is another sourcerotational friction, j050.86 cP, that is independent of thshear viscosityh. We do not pursue this idea here, beyonoting that a small correction to a purely hydrodynammodel is needed at low viscosity.
The fitting parameters for Eq.~17!, l'51.12 andVh
5157 Å3, can be related to the van der Waals shape ofthracene~Fig. 2!. There is always some ambiguity in detemining the best ellipsoidal shape to represent the nonesoidal van der Waals envelope of a molecule. We requireVh54pabc/3, the volume of the model ellipsoid. We alsrequire that the axial ratiosa/c andb/c fall on the symmet-ric rotor line ~Fig. 3! near the van der Waals axial ratios. Thcombination of these requirements defines the model esoid shown in Fig. 2~b!. The close correspondence betwethe effective hydrodynamic ellipsoid~11.037.033.9 Å! andthe van der Waals shape~11.837.834.0 Å!31 shows that thehydrodynamic model describes the diffusive rotation of athracene very well. In turn, the success of the hydrodynamodel validates our use of estimates from the model toterpret our data~Sec. III A!. The most important result othese estimates is that the intermediate decay componetoo short to be caused by rotational diffusion. This concsion supports our assignment of the intermediate dynamicelectronic-state solvation.
We note that this level of agreement with hydrodynammodels is not universal. For example, Hartmanet al. found
FIG. 6. Rotational timest r1 for anthracene vs solvent viscosity.~d!, fromour dichroism measurements in benzyl alcohol.~n!, from UV–IR doubleresonance measurements in various solvents by Lettenbergeret al. ~Ref.32!. ~,!, from fluorescence depolarization measurements in cyclohexanJaset al. ~Ref. 31!. The line is a fit to the modified hydrodynamic model E~17! ~see Fig. 2 for model dimensions!.
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp
hyul.
e-
th,q
he
thath
disprhiplic
thois
oticn
nitio
imoy
at
rezecae,
e
om-ndnt
udena
atanalnas
the
edi-
ex-ents,
ofm-
bes ofl fits.on-0–s,
has
the-ro-m-
re-rix.f
12onin-ted-
ne
neism
4219J. Chem. Phys., Vol. 115, No. 9, 1 September 2001 Inertial versus diffusive rotation. I
that anthracene with two nonpolar substituents followsdrodynamic predictions quite well in several solvents, brotates a factor of 2 faster than predicted in benzyl alcoho33
The reason for this isolated discrepancy is not obvious.
IV. INERTIAL ROTATION
The combination of diffusive rotation and solvation dscribes the dichroism data very well for times.1 ps. How-ever, an additional relaxation component is evident insubpicosecond region of Fig. 5~a!. To isolate this componentthe diffusive solvation and rotation were modeled by E~14!, fit for times .2 ps, and subtracted from the data. Tresulting residuals are shown in Fig. 5~b!.
The most striking feature of this decay component islack of a viscosity dependence. The viscosity varies bymost a factor of 5 between the different samples, andresulting effect on the.2 ps dynamics is dramatic@Figs. 1and 5~a!#. Nonetheless, the subpicosecond decays are intinguishable within the experimental noise. The lack of vcosity dependence is the essential feature of an inertialcess in the VE picture. The experimental verification of tprediction is the major result of this paper. Note that a simtime-scale classification is ambiguous for this process, whhas a decay time of;470 fs.
One can consider the possibility that this decay isresult of some electronic process resulting from mixingtheS1 state with other states. However, significant mixingnot expected for anthracene. TheS1 0 – 0 transition, whichwe are examining almost exclusively~see paper III!, is toolow in energy for strong mixing with theS2 state. As we willdiscuss below and in Sec. VI, the initial value of the anisropy can be accounted for without introducing any vibroncoupling. Jet spectra of anthracene do not show any evideof mixing with the ground electronic state.51 Thus, the fast,viscosity independent process is not the result of electrostate interactions, but rather, is due to an inertial interacwith the solvent.
The correct assignment of this inertial process is notmediately evident. It could be either the inertial portionthe rotational dynamics, the inertial part of the solvation dnamics or a combination of the two. Two arguments indicthat this process is primarily due to inertial rotation.
The first argument is based on the amplitude of thesiduals. Depending on how the data are extrapolated totime, the amplitude is between 0.04 and 0.06 on the scalibrated tor 0 . This amplitude is an excellent match to thamplitude ‘‘missing’’ from the fits to the diffusive rotationwhich gave an apparentr 050.3460.02. Given the errors inboth measurements, the size of the residuals is very closthe amplitude expected for inertial rotation.
The second argument comes from comparison to cputer simulation. Jaset al. have reported extensive simulations of anthracene rotation in cyclohexane aisopropanol.31 Isopropanol and benzyl alcohol are differeenough to preclude direct comparison of experiment to simlation, but close enough that the basic time scales shoulsimilar. We took Jaset al.’s data and treated it in the samway that we treated our data. The simulation data, showFig. 7, show an exponential decay at long times with
Downloaded 01 Sep 2003 to 139.78.100.86. Redistribution subject to A
-t
e
.
el-e
is--o-seh
ef
-
ce
c-n
-f-e
-role
to
-
-be
inn
inertial component at short times. We fit the simulation dat times.2 ps to a single exponential to represent rotatiodiffusion ~solvation effects are not present in the simulatio!.The amplitude of the rotational diffusion in isopropanol wr 050.31, noticeably smaller than ther 050.34 measured inbenzyl alcohol. We rescaled the simulation data to maker 0
50.34 and then plotted the simulation residuals againstexperimental residuals in Fig. 5~b!. The time scale of theinertial rotation decay in the simulation is very similar to thtime scale of the decay of the inertial component of thechroism signal.
Thus, an inertial component tor (t) is expected in thesubpicosecond regime, and the observed decay has thepected magnitude and decay time. Based on these argumwe assign the observed inertial decay to inertial rotation.
This assignment is a specific example of the utilitydefining inertial dynamics as viscosity independent dynaics. The significance of relying on this definition may notapparent at first, but becomes clearer when the specificthe current example are examined. A simple exponentiato the inertial decay~Fig. 8! has a decay constant of 470 fThe decay is continuing even in the 1–2 ps region. In ctrast, inertial dynamics are usually confined to the first 10200 fs in a time-based definition of inertial dynamics. Thuinertial dynamics can extend to much longer times thanbeen generally recognized.
This result is easier to understand, if we distinguishidea of an inertial time regime, within which inertial dynamics can occur, and the decay time of a specific inertial pcess. The inertial time regime is defined by being short copared to the relaxation time of the shear~or bulk! modulus.During this period, the solvent cannot undergo structurallaxation, and so it acts much like a disordered solid matIn paper III, we will find that the shear relaxation time obenzyl alcohol over this viscosity range varies from 1.8 tops. This defines the time range over which inertial moticould occur. At the high viscosity end of this range, theertial rotation, with a 0.5 ps decay time, is easily complewithin a fraction of the total inertial regime. At the low vis
FIG. 7. Computer simulation results for the rotation of anthracein isopropanol from Jazet al. ~solid curve! ~Ref. 31!. The dashed line isa single exponential fit to the diffusive portion of the [email protected](2t/18.45 ps)10.08#. The residual from this fit, representing thinertial part of the rotation, is compared to the subpicosecond dichrodecay in Fig. 5~b!.
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

bl
mhe
m,
tfowreheatbetio-bic
r-tueinlao
syoana
toonns
inior.letheticaseand
ofde-
lsoffotot-ta
--n.opy
ion00e
tatee
ng
be
leat
ayll’’tonde-inge-ys iset-
-ted,theas
sor
ecfr
en
4220 J. Chem. Phys., Vol. 115, No. 9, 1 September 2001 Zhang et al.
cosity end, the inertial rotation covers most of time availafor inertial dynamics.
Note that the inertial time varies with the process exained. For example, inertial solvation is not seen with tcurrent time resolution of 150 fs. It is estimated to havetime constant of 70 fs~see paper III!, about seven timesfaster than the inertial rotation. Despite the difference in tiscales, both processes are faster than shear relaxationtherefore, both are inertial.
Extrapolating these results to lower viscosity leadssome interesting predictions. The range of viscositiescommon liquids extends another factor of 10 below the loest viscosity examined here. The duration of the inertialgime also shrinks proportionately. For inertial solvation, treduced inertial range has few consequences; the relaxwill still be essentially complete before shear relaxationcomes important. However, shear relaxation and solvawill begin to compete with the inertial rotation in low viscosity liquids. The consequences of this competition willexamined in more detail in experiments in toluene, whwill be presented in paper IV.
V. BALLISTIC ROTATION
Another result of the viscosity-based definition of inetial dynamics is that inertial dynamics are not equivalentthe quasifree dynamics expected at very early times. Wethe term ‘‘ballistic’’ to refer specifically to these quasifredynamics. Ballistic dynamics are still an important andevitable feature of short time dynamics. However, the retionship between the inertial and ballistic dynamics is npredetermined. It can vary from process to process andtem to system, and it is subject to experimental testing. Nthat we have quantitative data on the inertial rotation ofthracene in benzyl alcohol, we are in a position to evaluhow much of the inertial dynamics are also ballistic.
One approach to determining the ballistic contributiondynamics is to rely on the known properties of correlatifunctions in the limit of zero time. In practice, this mea
FIG. 8. The inertial decay of the dichroism data~solid lines! is representedby dividing out the slower diffusive [email protected]., the fits in Fig. 5~a!#. Thedata shown here are the same as in Fig. 5. An exponential fit to this d~–1–! gives a time constant of 0.47 ps. The data are compared to therotor behavior of anthracene~dashed curve!. The ‘‘ballistic’’ free rotationcorrectly describes the first 200–400 fs, but does not encompass theregion of inertial dynamics.
Downloaded 01 Sep 2003 to 139.78.100.86. Redistribution subject to A
e
-
a
eand
or--
ion-n
eh
ose
--ts-
w-
te
determining the initial curvature of the experimental datasolution and using it to predict the free-molecule behavThis is a difficult task even with ideal data and is impossibwith the pulse overlap effects present in our data. We takedifferent approach of independently calculating the ballisrotation of anthracene and comparing it to the solution-phdata. This procedure is possible because of the simplerigid geometry of anthracene.
Such a comparison is shown for the inertial rotationanthracene in Fig. 8. The inertial rotational response isfined by decomposing the total dichroism signal by
Sdc~ t !5DA~ t !
DA~`!~r i~ t !1r 0e2t/tr1!. ~18!
@This definition differs slightly from the empirical residuashown in Fig. 5~b!.# Figure 8 shows the inertial responsethe system,r i(t)1r 0 , i.e., the rotational anisotropy free odiffusive decay. Note that the diffusion-free signal is nr i(t) itself. In the absence of diffusive processes, the anisropy decays tor 0 , not to zero. Figure 8 shows the actual dacorrected using the best-fit values oft r1 and DA(t) foundabove.
The ballistic curve~dashed in Fig. 8! is calculated fromthe moments of inertial of anthracene~I x51.84310244, I y
54.97310245, I z52.34310244kg m2! using anthracene dimensions from Cruickshank,52 and the early-time approximation give by St. Pierre and Steele for gas-phase rotatio53
This approximation is a Gaussian decay to zero anisotrand is good for the first;80% of the gas-phase decay.
The ballistic approximation is a reasonable extrapolatof the inertial rotation data in the 0–200 fs region. After 4fs, the inertial rotation and ballistic approximation divergrapidly. In the gas phase, the molecule continues to rofreely after this time, whereas in the liquid, the initially frerotation is halted by collision with the cage of surroundisolvent molecules.
In the post-200 fs region, the inertial dynamics candescribed with a 470 fs exponential decay~Fig. 8!. The ex-trapolation of this decay to,200 fs results in a reasonabamplitude at zero time, but an unrealistic nonzero slopet50.
In the postballistic region, the inertial anisotropy decis determined by collisions of the anthracene with the ‘‘waof the solvent cavity. Significant questions remain on howdescribe these dynamics. If that wall were smooth, rigid auniform from solute molecule to solute molecule, the rbound from this wall would be elastic and coherent, resultin oscillations of the anisotropy. If the entire solvent is rgarded as a rigid cage, the fact that the anisotropy decadue to ‘‘dephasing’’ of these oscillations, either due to herogeneity of the cage structure~inhomogeneous dephasing!or due to the corrugation of the wall~homogeneous dephasing!. In a VE picture, these processes would be neglecand the decay would result from energy exchange withsolvent due to the absorption and emission of ‘‘phonons’’the anthracene hit the elastic solvent.
The stochastic cage model54–56 and the instantaneounormal mode theory57,58 are other possible approaches f
ayee
tire
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

ate
stiaanneann
aa
stia
deratlint
obtt
i-nteAe
u-isatath-
erw
oalarst
si
u-ary
ryes.
ceer
abethe
tofini-tey-
isin
alethislyis
ive
arebe
volveIVerona-n
ocu-na-
tynitewlypro-
ics
ortt ay-ite
ouldoryx-
-
onrkdere-
4221J. Chem. Phys., Vol. 115, No. 9, 1 September 2001 Inertial versus diffusive rotation. I
analyzing these data. However, both of these latterproaches require significant input information from compusimulations, so these analyses are not pursued here.
In summary, the ballistic regime lasts for;200 fs,whereas the inertial regime lasts for at least 1 ps. Ballimotion accounts for approximately half of the total inertdecay amplitude. This result is specific to this processthis system. For example, consider the rotation of aniliwhich is much more weakly coupled to the solvent thanthracene is. Pereiraet al. have shown that aniline rotation iisopentane is mostly inertial~viscosity independent! and bal-listic ~near free-rotor times!.28 In another example, Pererand Berkowitz have shown in computer simulations that blistic motion accounts for;70% of the inertial decay of amodel polar solvation process.59 The current system providea counterpoint to those examples by showing that inerdynamics need not be dominated by ballistic effects.
VI. INITIAL VALUE OF THE ANISOTROPY
Another point that Fig. 8 illustrates is that the amplitu‘‘missing’’ in fits to diffusive rotation is nicely accounted foby the inertial component of rotation. It is well known ththe theoretical value ofr 0 is 0.40. However, experimentameasurements ofr 0 , either by steady-state fluorescenceglassy media or by extrapolating diffusive time-decayszero time, often fall slightly short of this value.60 A com-monly cited reason for a small value ofr 0 is absorption to adifferent electronic state than the emitting state. This prlem can be avoided by using an excitation wavelength thalonger than the origin of theS2 state, as we do in the currenexperiments.61 The value ofr 0 also can be reduced if exctation or emission occur on vibronic lines with significavibronic coupling.62 Again, this problem is not present if thexcitation and detection are done on the 0–0 transition.paper III shows in more detail, these conditions are wsatisfied in the current experiment.
Even when these problems are avoided, careful measments ofr 0 frequently give values slightly below the theoretical maximum.60 In the current system, we show that thdeficit is due to an early component of inertial rotation thcannot be accounted for by extrapolating the diffusive rotional decay to zero time. This mechanism for reducingobservedr 0 is quite evident in computer simulation of rotation, but does not seem to be widely recognized in expmental studies. The results presented here provide adocumented experimental example of this effect.
VII. CONCLUSIONS
In previous theoretical work, we suggested a divisionliquid dynamics into three time regimes: ballistic, inertiand diffusive.2 More importantly, we suggested testable chacteristics of each of these regions. These characterihave been experimentally verified in this study.
The diffusive region is characterized by dynamics whorate is proportional to the solvent viscosity. This behaviorquite familiar in many processes, including rotational diffsion. The solute studied here, anthracene, is an unusugood example of this behavior. Its diffusive rotation is ve
Downloaded 01 Sep 2003 to 139.78.100.86. Redistribution subject to A
p-r
icld,-
l-
l
o
-is
sll
re-
t-e
i-ell
f
-ics
es
lly
well described by a hydrodynamic model with slip boundaconditions and dimensions close to van der Waals valuThe ability to model the diffusive dynamics with confidenis a major aid in extracting the solvation dynamics in papIII and the inertial rotation in this paper.
In the inertial time region, the experiments providequantitative demonstration that inertial dynamics canidentified based on their viscosity independence. At first,viscosity independence of inertial dynamics does not seembe a controversial assertion. However, using this as a detion of inertial, we find inertial rotation that is not compleuntil after 1 ps. Most previous identifications of inertial dnamics have been confined to the first 100–200 fs.
The observation of relatively long inertial dynamicseasily understood from the perspective of VE theory. Withthis theory, inertial dynamics can occur on any time scshorter than the shear relaxation time of the solvent, andtime becomes arbitrarily long if the viscosity is sufficienthigh. In the current solvent, benzyl alcohol, the viscositymoderately high, and the separation of inertial and diffusdynamics is clean.
Moreover, the solvation and rotation times scalesalso well separated. As a result, solvation and rotation canregarded as independent processes, even though both ininterrelated reorganizations of the local solvent. In paperof this series, we will look at toluene, a solvent with a lowand more typical viscosity. In that system, inertial rotatiand diffusive solvation dynamics overlap in time. The sepration of inertial and diffusive and of solvation and rotatiobecomes less sharp in this case. The simpler situation dmented here will provide a valuable reference point in alyzing the toluene data.
The definition of inertial dynamics in terms of viscosidependence also means inertial dynamics have an indeficonnection with free-molecule or ballistic dynamics. Homuch of an inertial process is also ballistic is an entiresystem dependent question. The results presented herevide an example where the majority of the inertial dynamare outside the ballistic regime.
These findings support the VE approach to treating shtime dynamics in liquids. However, the tests are only aqualitative level; a quantitative VE theory of rotational dnamics does not yet exist. Such a theory would be qudesirable, and the data presented here indicate that it whave a good chance of succeeding. If a successful VE theof rotation is developed, it could be combined with the eisting VE theories of nonpolar solvation2,4–6,63,64and vibra-tional dephasing3,65,66to provide a unified description of molecular dynamics in solution.
ACKNOWLEDGMENTS
We thank Professor Kuczera for providing his simulatiresults on anthracene rotation in numerical form. This wowas supported by the National Science Foundation unGrant No. CHE-9809719 and by the Office of Naval Rsearch through the University Research Initiative~Grant No.N00014-97-1-0806!.
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

.,
n-
ys
hy
m
n
,
he
he
em
W.
y,
ch-
.
m.
les
4222 J. Chem. Phys., Vol. 115, No. 9, 1 September 2001 Zhang et al.
1G. R. Fleming,Chemical Applications of Ultrafast Spectroscopy~OxfordUniversity Press, Oxford, 1986!.
2M. Berg, J. Phys. Chem. A102, 17 ~1998!.3M. A. Berg and H. W. Hubble, Chem. Phys.233, 257 ~1998!.4M. A. Berg, J. Chem. Phys.110, 8577~1999!.5J. Ma, J. T. Fourkas, D. A. Vanden Bout, and M. Berg, inSupercooledLiquids: Advances and Novel Applications, edited by J. T. Fourkas, DKivelson, U. Mohanty, and K. A. Nelson~American Chemical SocietyWashington, D.C., 1997!, Vol. 676, p. 199.
6M. Berg, Chem. Phys. Lett.228, 317 ~1994!.7Y. Zhang and M. A. Berg, J. Chem. Phys.115, 4223 ~2001!, followingpaper.
8Y. Zhang and M. A. Berg, J. Chem. Phys.115, 4231~2001!, this issue.9E. A. Carter and J. T. Hynes, J. Chem. Phys.94, 5961~1991!.
10D. McMorrow, W. T. Lotshaw, and G. A. Kenney-Wallace, IEEE J. Quatum Electron.24, 443 ~1988!.
11S. J. Rosenthal, X. L. Xie, M. Du, and G. R. Fleming, J. Chem. Phys.95,4715 ~1991!.
12R. Jimenez, G. R. Fleming, P. V. Kumar, and M. Maroncelli, Nature~Lon-don! 369, 471 ~1994!.
13M. L. Horng, J. A. Gardecki, A. Papazyan, and M. Maroncelli, J. PhChem.99, 17311~1995!.
14B. J. Berne and G. D. Harp, Adv. Chem. Phys.17, 63 ~1970!.15J. P. Boon and S. Yip,Molecular Hydrodynamics~Dover, New York,
1980!.16A. M. Walsh and R. F. Loring, Chem. Phys. Lett.186, 77 ~1991!.17M. Maroncelli, J. Chem. Phys.94, 2084~1991!.18C. V. Shank, E. P. Ippen, O. Teschke, and K. B. Eisenthal, J. Chem. P
67, 5547~1978!.19D. Waldeck, A. J. Cross, D. B. McDonald, and G. R. Fleming, J. Che
Phys.74, 3381~1981!.20D. S. Alvari, R. S. Harman, and D. H. Waldeck, J. Chem. Phys.92, 4055
~1990!.21R. Pecora,Dynamic Light Scattering: Applications of Photon Correlatio
Spectroscopy~Plenum, New York, 1985!.22W. A. Steele, Adv. Chem. Phys.34, 1 ~1976!.23D. McMorrow, M. Thantu, J. S. Melinger, D. K. Kim, and W. T. Lotshaw
J. Phys. Chem.100, 10389~1996!.24L. C. Geiger and B. M. Ladanyi, J. Chem. Phys.89, 6588~1988!.25H. Stassen and W. A. Steele, J. Chem. Phys.103, 4408~1995!.26B. M. Ladanyi and Y. Q. Liang, J. Chem. Phys.103, 6325~1995!.27R. L. Murry, J. T. Fourkas, W. X. Li, and T. Keyes, Phys. Rev. Lett.83,
3550 ~1999!.28M. A. Pereira, P. E. Share, M. J. Sarisky, and R. M. Hochstrasser, J. C
Phys.94, 2513~1991!.29R. G. Gordon, inAdvances in Magnetic Resonance, edited by J. S. Waugh
~Academic, New York, 1968!, Vol. 3.30T. Dorfmuller, B. Daum, and A. Hanschmidt, J. Chem. Phys.95, 813
~1991!.31G. S. Jas, Y. Wang, S. W. Pauls, C. K. Johnson, and K. Kuczera, J. C
Phys.107, 8800~1997!.32M. Lettenberger, F. Emmerling, N. H. Gottfried, and A. Laubereau, Ch
Phys. Lett.240, 324 ~1995!.
Downloaded 01 Sep 2003 to 139.78.100.86. Redistribution subject to A
.
s.
.
m.
m.
.
33R. S. Hartman, W. M. Konitsky, D. H. Waldeck, Y. J. Chang, and E.Castner, J. Chem. Phys.106, 7920~1997!.
34Y. Zhang and M. A. Berg~in preparation!.35CRC Handbook of Thermophysical and Thermochemical Data, edited by
D. R. Lide and H. V. Kehiaian~CRC Press, Boca Raton, 1994!.36CRC Handbook of Chemistry and Physics, edited by D. R. Lide and H. V.
Kehiaian~CRC Press, Boca Raton, 1998!.37Lange’s Handbook of Chemistry, edited by J. A. Dean~McGraw–Hill,
New York, 1992!.38Lange’s Handbook of Chemistry, edited by J. A. Dean~McGraw–Hill,
New York, 1999!.39D. S. Birch and R. E. Imhof, inTopics in Fluorescence Spectroscop
Volume I: Techniques, edited by J. R. Lakowicz~Plenum, New York,1991!, p. 1.
40E. W. Small, in Topics in Fluorescence Spectroscopy, Volume I: Teniques, edited by J. R. Lakowicz~Plenum, New York, 1991!, p. 97.
41D. Ben-Amotz and T. W. Scott, J. Chem. Phys.87, 3739~1987!.42D. Ben-Amotz and J. M. Drake, J. Chem. Phys.89, 1019~1988!.43M. Roy and S. Doraiswamy, J. Chem. Phys.98, 3213~1993!.44A. M. Williams, Y. Jiang, and D. Ben-Amotz, Chem. Phys.180, 119
~1994!.45T. J. Chuang and K. B. Eisenthal, J. Chem. Phys.57, 5094~1972!.46G. K. Youngren and A. Acrivos, J. Chem. Phys.63, 3846~1975!.47R. J. Sension and R. M. Hochstrasser, J. Chem. Phys.98, 2490~1992!.48P. D. Hyde and M. D. Ediger, J. Chem. Phys.92, 1036~1990!.49G. R. Alms, D. R. Bauer, J. I. Brauman, and R. Pecora, J. Chem. Phys58,
5570 ~1973!.50G. T. Evans and D. Kivelson, J. Chem. Phys.84, 385 ~1986!.51W. R. Lambert, P. M. Felker, J. A. Syage, and A. H. Zewail, J. Che
Phys.81, 2195~1984!.52W. J. Cruickshank, Acta Crystallogr.9, 915 ~1956!.53A. G. St. Pierre and W. A. Steele, Phys. Rev.184, 172 ~1969!.54A. Polimeno, G. J. Moro, and J. H. Freed, J. Chem. Phys.102, 8094
~1995!.55A. Polimeno, G. J. Moro, and J. H. Freed, J. Chem. Phys.104, 1090
~1996!.56G. J. Moro and A. Polimeno, J. Chem. Phys.107, 7884~1997!.57J. Y. Jang and R. M. Stratt, J. Chem. Phys.112, 7524~2000!.58J. Y. Jang and R. M. Stratt, J. Chem. Phys.112, 7538~2000!.59L. Perera and M. L. Berkowitz, J. Chem. Phys.97, 5253~1992!.60J. R. Lakowicz,Principles of Fluorescence Spectroscopy~Plenum, New
York, 1983!.61I. B. Berlman,Handbook of Fluorescence Spectra of Aromatic Molecu
~Academic, New York, 1971!.62A. C. Albrecht, J. Mol. Spectrosc.6, 84 ~1961!.63J. T. Fourkas, A. Benigno, and M. Berg, J. Non-Cryst. Solids172, 234
~1994!.64J. Ma, D. A. Vanden Bout, and M. Berg, J. Chem. Phys.103, 9146~1995!.65H. W. Hubble, T. Lai, and M. A. Berg, J. Chem. Phys.114, 3662~2001!.66K. D. Rector, M. A. Berg, and M. D. Fayer, J. Phys. Chem. B105, 1081
~2001!.
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp
Copyright © 2022 FDOKUMEN







![Photochemical reaction of 9-nitro-substituted anthracene-like molecules 9-methyl-10-nitroanthracene and 12-methyl-7-nitrobenz[a]anthracene](https://static.fdokumen.com/doc/165x107/6336a9004e9c1ac02e082ee9/photochemical-reaction-of-9-nitro-substituted-anthracene-like-molecules-9-methyl-10-nitroanthracene.jpg)












