
Cysteine S-glycosylation, a new post-translational modification found in glycopeptide bacteriocins
Upload
independentCategory
view
1download
0
JMB—MS 514 Cust. Ref. No. CAM 453/94 [SGML]
J. Mol. Biol. (1995) 248, 901–909
Truncated Profibrillin of a Marfan Patient is ofApparent Similar Size as Fibrillin: IntracellularRetention Leads to over- N-glycosylation
Michael Raghunath 1*, Cay M. Kielty 2 and Beat Steinmann 1
We studied profibrillin-1 (proFib) synthesis and microfibril formation in1Division of Metabolismcultured fibroblasts from an individual with severe Marfan syndromeDept. of Paediatricsharboring a premature stop codon (W2756ter) in one FBN1 allele. RotaryUniversity of Zurichshadowing analysis of extracellular matrix produced by these cells revealedSteinwiesstr, 75, CH-8032the presence of only a very few intact microfibrils which showed markedZurich, Switzerlanddisorganisation within the interbeaded domains. Metabolic pulse-chase2School of Biological Sciences studies identified intracellularly a population of truncated proFib molecules
University of Manchester which were secreted more slowly than the normal proFib derived from theOxford Rd, Manchester normal allele. Culture media contained strikingly reduced amounts ofM13 9PT, U.K. wild-type proFib in comparison to fibrillin (Fib). Our findings imply that (1)
the truncated proFib is secreted and disturbs microfibril assembly; (2) themutation is probably close to a putative cleavage site in the proFib C terminusnecessary for the conversion of proFib to Fib; (3) the truncated proFib isover-N-glycosylated due to intracellular retention rather than incompletecleavage of proFib with persistence of N-glycosylated sites; (4) not allpotential N-glycosylation sites in proFib seem to be normally used, since wecould produce over-N-glycosylated proFib in normal cells by brefeldin Amediated intracellular captivation and subsequent appearance of over-glyco-sylated Fib in culture medium upon removal of the compound. It isconceivable that post-translational over-modification might be important formodulating the phenotype of FBN1 mutations in Marfan syndrome.
Keywords: profibrillin 1; glycosylation; extracellular matrix; Marfan*Corresponding author syndrome
Introduction
The large cysteine-rich glycoprotein fibrillin-1(Fib) has been shown to be a major constituent of10–12 nm microfibrils of the extracellular matrix(ECM; Sakai et al., 1986, 1991). These microfibrils arewidely distributed in the human body (Low, 1962;Maddox et al., 1989) and are believed to function asa scaffold for the deposition and formation of elasticfibres (Rosenbloom, 1993). It is now well establishedthat defects in the gene for Fib, FBN1 (Pereira et al.,1993), cause Marfan syndrome (MFS: Dietz et al.,
1991), an autosomal dominant connective tissuedisorder characterized by cardiovascular, ocular andskeletal manifestations (Pyeritz, 1993).
Recently, a nonsense mutation was reported thatalters the codon for a tryptophan residue at position2756 (numbering according to Pereira et al., 1993) intoa premature termination signal (Kainulainen et al.,1992). The predicted truncated Fib molecules,however, could apparently not be immunoprecipi-tated from culture medium of fibroblasts harboringthis mutation and it was concluded that the truncatedmolecules, which were not found in the culturemedium (Kainulainen et al., 1992), were not secretedat all (Ramirez et al., 1993). According to thisassumption, only normal Fib molecules would bepresent in the ECM albeit in reduced amounts. Thiswould be equivalent to a functional null allele, asituation which has been described in anotherheritable generalized connective tissue disorder,osteogenesis imperfecta. This condition, character-ized by brittleness of bones and weakness of othertissues, is caused by mutations in the genes coding
Present address: M. Raghunath, Institute ofPhysiological Chemistry and Pathobiochemistry,Waldeyerstr. 15, D-48149 Munster, Germany.
Abbreviations used: Fib, fibrillin-1; ECM, extracellularmatrix; MFS, Marfan syndrome; proFib, profibrillin-1;EM, electron microscopy; MEM, minimal essentialmedium; FCS, fetal calf serum; BFA, brefeldin A; EGTA,ethylene glycol bis(2-aminoethyl ether)-N,N,N',N'-tetra-acetic acid; MSH, mercaptoethanol; HBSS, Hank’sbalanced salt solution; NP-40, Nonidet P-40.
0022–2836/95/200901–09 $08.00/0 7 1995 Academic Press Limited
JMB—MS 514
Post-translational Modification of Profibrillin 1902
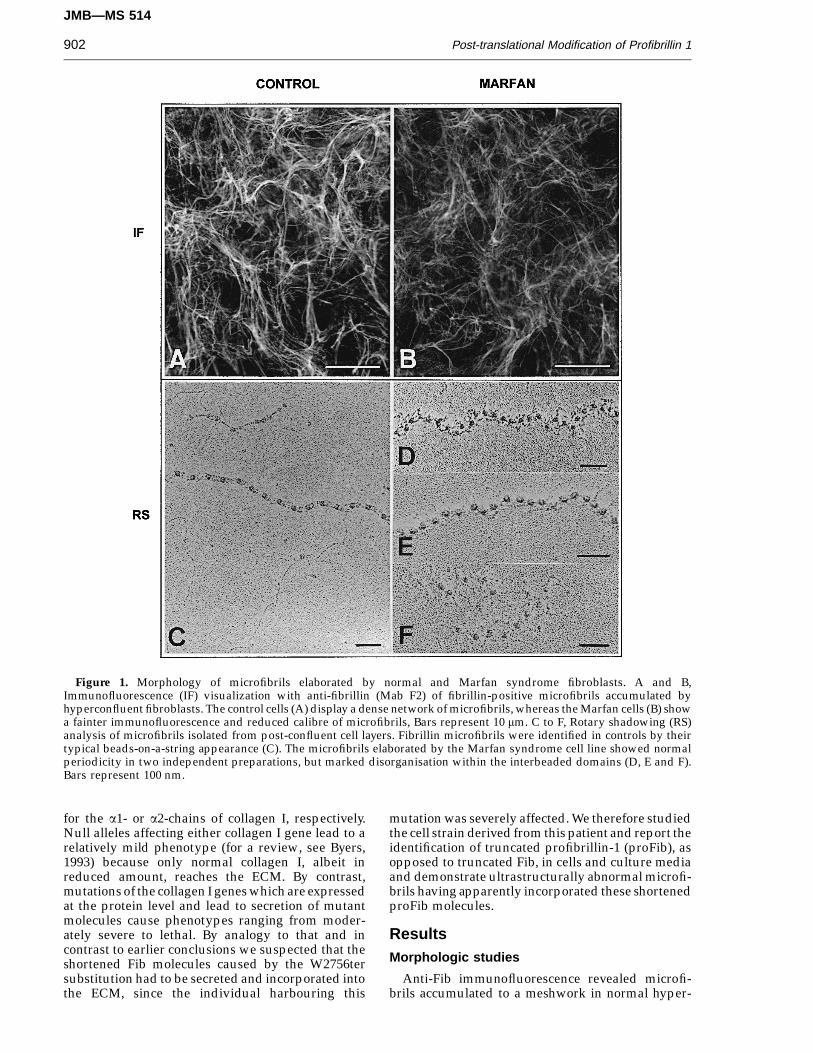
Figure 1. Morphology of microfibrils elaborated by normal and Marfan syndrome fibroblasts. A and B,Immunofluorescence (IF) visualization with anti-fibrillin (Mab F2) of fibrillin-positive microfibrils accumulated byhyperconfluent fibroblasts. The control cells (A) display a dense network of microfibrils, whereas the Marfan cells (B) showa fainter immunofluorescence and reduced calibre of microfibrils, Bars represent 10 mm. C to F, Rotary shadowing (RS)analysis of microfibrils isolated from post-confluent cell layers. Fibrillin microfibrils were identified in controls by theirtypical beads-on-a-string appearance (C). The microfibrils elaborated by the Marfan syndrome cell line showed normalperiodicity in two independent preparations, but marked disorganisation within the interbeaded domains (D, E and F).Bars represent 100 nm.
for the a1- or a2-chains of collagen I, respectively.Null alleles affecting either collagen I gene lead to arelatively mild phenotype (for a review, see Byers,1993) because only normal collagen I, albeit inreduced amount, reaches the ECM. By contrast,mutations of the collagen I genes which are expressedat the protein level and lead to secretion of mutantmolecules cause phenotypes ranging from moder-ately severe to lethal. By analogy to that and incontrast to earlier conclusions we suspected that theshortened Fib molecules caused by the W2756tersubstitution had to be secreted and incorporated intothe ECM, since the individual harbouring this
mutation was severely affected. We therefore studiedthe cell strain derived from this patient and report theidentification of truncated profibrillin-1 (proFib), asopposed to truncated Fib, in cells and culture mediaand demonstrate ultrastructurally abnormal microfi-brils having apparently incorporated these shortenedproFib molecules.
ResultsMorphologic studies
Anti-Fib immunofluorescence revealed microfi-brils accumulated to a meshwork in normal hyper-

JMB—MS 514
Post-translational Modification of Profibrillin 1 903
Figure 2. SDS-PAGE analysis of profibrillin synthesis by Marfan syndrome fibroblasts harboring a W2756ter mutationin one FBN1 allele. Cell cultures of a control (C) and of the patient (P) were pulsed for 30 min with [35S]Met/Cys andthen chased for 0, 1, 2 and 4 h. Only the truncated parts of the 4% layer of the step gel are shown. Cells, the patient’s(P) cells contain both the normal and truncated proFib population (filled arrowhead). The normal population is secretedefficiently and cleared from the cells (open arrowhead), whereas the shortened chains are secreted with delay. ‘‘x’’ indicatesan anonymous intracellular protein as a representative of cell mass and monitor for equal loading of radiolabelled proteinsper lane. Medium, both cell strains have released maximal amounts of proFib/Fib after 4 h chase into the culture medium.The normal cells (C) show a strong Fib and a weaker proFib band, whereas virtually no proFib is detectable in the patient’scells (P) culture medium. In addition, the molecules in the Fib position show a slightly retarded migration (dashes). ECM,no significant differences in the amount of Fib extracted from the extra/pericellular matrix are obvious.
confluent fibroblast cultures. The mutant cellsdeposited microfibrils of roughly comparableorganization, although the intensity of fluorescenceand calibre of individual microfibrils appearedreduced (Figure 1A, B). Examination by rotaryshadowing EM of high-Mr material solubilized bybacterial collagenase from post-confluent cell layersdemonstrated the presence of extensive andabundant microfibrils in control cell layers (Figure1C), which were similar in morphology to thoseisolated previously from tissues (Kielty et al., 1991,1993). Matrix extracts from the mutant cells containedonly a few intact microfibrils with normal periodicitybut marked disorganisation within the interbeadeddomains (Figure 1D to F). In addition, short beadedaggregates were also present. The identity ofmicrofibrillar assemblies in extracts of control andmutant cells was confirmed using immunogoldlocalization of fibrillin epitopes as described(Raghunath et al., 1994; results not shown).
Findings at protein level—metabolic labellingstudies
Cell lysates
Control cells contained the normal intracellularproFib population (approx. 350 kDa), whereas themutant cells showed an additional, faster migratingband representing the product of the mutant
transcript (Figure 2) which was immunoprecipitablewith anti-Fib (Figure 3). This population ofmolecules was intracellularly retained, whereasnormal proFib produced by the mutant cellswas cleared normally with increasing chase time(Figure 2).
Culture media
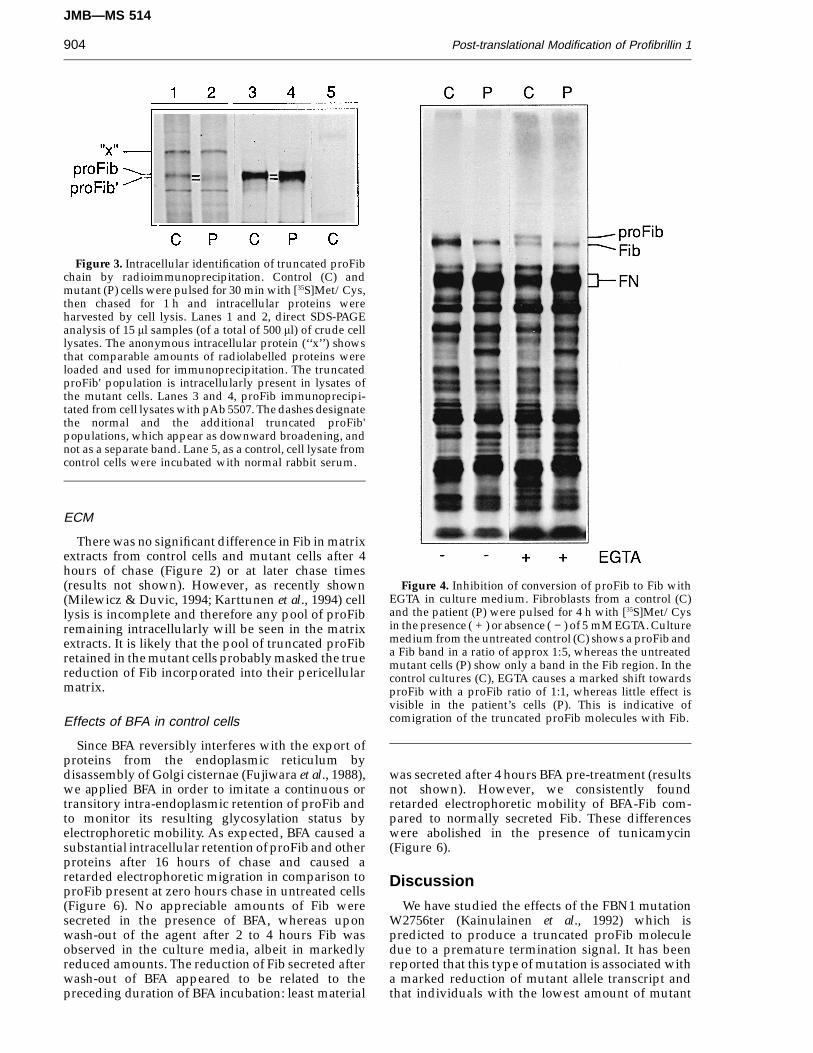
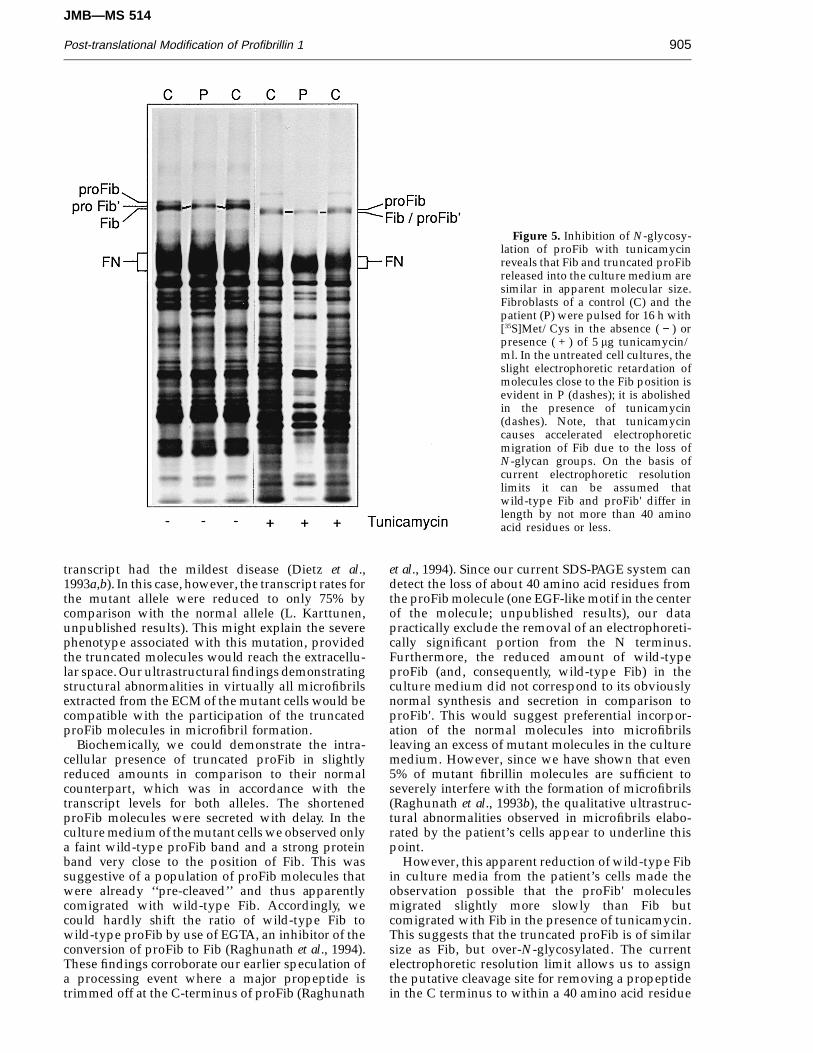
During pulse–chase experiments a remarkablelack of normal proFib was evident in the culturemedium of mutant cells and no truncated Fibpopulation was observed (Figures 2 and 4). In controlcells, the cleavage of proFib to Fib was stronglyreduced in the presence of 5 mM EGTA whichresulted in a significant proFib:Fib ratio shift of 1:5to 1:1 (Figure 4). In contrast, when the mutant cellswere treated with EGTA, normally migrating proFibmolecules were only faintly visible, and theproFib:Fib ratio never exceeded 1:10. In addition, theband apparently representing Fib consistentlyshowed slight electrophoretic retardation (Figures 2and 4). Tunicamycin caused a markedly fasterelectrophoretic migration of proFib and Fib in bothcontrol and mutant cells suggesting an apparentdecrease in Mr of about 30 kDa. In the presence of thisN-glycosylation inhibitor, the apparent Fib popu-lation produced by the mutant cells achieved thesame electrophoretic mobility as Fib produced by thecontrol cells (Figure 5).
JMB—MS 514
Post-translational Modification of Profibrillin 1904
Figure 3. Intracellular identification of truncated proFibchain by radioimmunoprecipitation. Control (C) andmutant (P) cells were pulsed for 30 min with [35S]Met/Cys,then chased for 1 h and intracellular proteins wereharvested by cell lysis. Lanes 1 and 2, direct SDS-PAGEanalysis of 15 ml samples (of a total of 500 ml) of crude celllysates. The anonymous intracellular protein (‘‘x’’) showsthat comparable amounts of radiolabelled proteins wereloaded and used for immunoprecipitation. The truncatedproFib' population is intracellularly present in lysates ofthe mutant cells. Lanes 3 and 4, proFib immunoprecipi-tated from cell lysates with pAb 5507. The dashes designatethe normal and the additional truncated proFib'populations, which appear as downward broadening, andnot as a separate band. Lane 5, as a control, cell lysate fromcontrol cells were incubated with normal rabbit serum.
Figure 4. Inhibition of conversion of proFib to Fib withEGTA in culture medium. Fibroblasts from a control (C)and the patient (P) were pulsed for 4 h with [35S]Met/Cysin the presence ( + ) or absence ( − ) of 5 mM EGTA. Culturemedium from the untreated control (C) shows a proFib anda Fib band in a ratio of approx 1:5, whereas the untreatedmutant cells (P) show only a band in the Fib region. In thecontrol cultures (C), EGTA causes a marked shift towardsproFib with a proFib ratio of 1:1, whereas little effect isvisible in the patient’s cells (P). This is indicative ofcomigration of the truncated proFib molecules with Fib.
ECM
There was no significant difference in Fib in matrixextracts from control cells and mutant cells after 4hours of chase (Figure 2) or at later chase times(results not shown). However, as recently shown(Milewicz & Duvic, 1994; Karttunen et al., 1994) celllysis is incomplete and therefore any pool of proFibremaining intracellularly will be seen in the matrixextracts. It is likely that the pool of truncated proFibretained in the mutant cells probably masked the truereduction of Fib incorporated into their pericellularmatrix.
Effects of BFA in control cells
Since BFA reversibly interferes with the export ofproteins from the endoplasmic reticulum bydisassembly of Golgi cisternae (Fujiwara et al., 1988),we applied BFA in order to imitate a continuous ortransitory intra-endoplasmic retention of proFib andto monitor its resulting glycosylation status byelectrophoretic mobility. As expected, BFA caused asubstantial intracellular retention of proFib and otherproteins after 16 hours of chase and caused aretarded electrophoretic migration in comparison toproFib present at zero hours chase in untreated cells(Figure 6). No appreciable amounts of Fib weresecreted in the presence of BFA, whereas uponwash-out of the agent after 2 to 4 hours Fib wasobserved in the culture media, albeit in markedlyreduced amounts. The reduction of Fib secreted afterwash-out of BFA appeared to be related to thepreceding duration of BFA incubation: least material
was secreted after 4 hours BFA pre-treatment (resultsnot shown). However, we consistently foundretarded electrophoretic mobility of BFA-Fib com-pared to normally secreted Fib. These differenceswere abolished in the presence of tunicamycin(Figure 6).
Discussion
We have studied the effects of the FBN1 mutationW2756ter (Kainulainen et al., 1992) which ispredicted to produce a truncated proFib moleculedue to a premature termination signal. It has beenreported that this type of mutation is associated witha marked reduction of mutant allele transcript andthat individuals with the lowest amount of mutant
JMB—MS 514
Post-translational Modification of Profibrillin 1 905
Figure 5. Inhibition of N-glycosy-lation of proFib with tunicamycinreveals that Fib and truncated proFibreleased into the culture medium aresimilar in apparent molecular size.Fibroblasts of a control (C) and thepatient (P) were pulsed for 16 h with[35S]Met/Cys in the absence ( − ) orpresence ( + ) of 5 mg tunicamycin/ml. In the untreated cell cultures, theslight electrophoretic retardation ofmolecules close to the Fib position isevident in P (dashes); it is abolishedin the presence of tunicamycin(dashes). Note, that tunicamycincauses accelerated electrophoreticmigration of Fib due to the loss ofN-glycan groups. On the basis ofcurrent electrophoretic resolutionlimits it can be assumed thatwild-type Fib and proFib' differ inlength by not more than 40 aminoacid residues or less.
transcript had the mildest disease (Dietz et al.,1993a,b). In this case, however, the transcript rates forthe mutant allele were reduced to only 75% bycomparison with the normal allele (L. Karttunen,unpublished results). This might explain the severephenotype associated with this mutation, providedthe truncated molecules would reach the extracellu-lar space. Our ultrastructural findings demonstratingstructural abnormalities in virtually all microfibrilsextracted from the ECM of the mutant cells would becompatible with the participation of the truncatedproFib molecules in microfibril formation.
Biochemically, we could demonstrate the intra-cellular presence of truncated proFib in slightlyreduced amounts in comparison to their normalcounterpart, which was in accordance with thetranscript levels for both alleles. The shortenedproFib molecules were secreted with delay. In theculture medium of the mutant cells we observed onlya faint wild-type proFib band and a strong proteinband very close to the position of Fib. This wassuggestive of a population of proFib molecules thatwere already ‘‘pre-cleaved’’ and thus apparentlycomigrated with wild-type Fib. Accordingly, wecould hardly shift the ratio of wild-type Fib towild-type proFib by use of EGTA, an inhibitor of theconversion of proFib to Fib (Raghunath et al., 1994).These findings corroborate our earlier speculation ofa processing event where a major propeptide istrimmed off at the C-terminus of proFib (Raghunath
et al., 1994). Since our current SDS-PAGE system candetect the loss of about 40 amino acid residues fromthe proFib molecule (one EGF-like motif in the centerof the molecule; unpublished results), our datapractically exclude the removal of an electrophoreti-cally significant portion from the N terminus.Furthermore, the reduced amount of wild-typeproFib (and, consequently, wild-type Fib) in theculture medium did not correspond to its obviouslynormal synthesis and secretion in comparison toproFib'. This would suggest preferential incorpor-ation of the normal molecules into microfibrilsleaving an excess of mutant molecules in the culturemedium. However, since we have shown that even5% of mutant fibrillin molecules are sufficient toseverely interfere with the formation of microfibrils(Raghunath et al., 1993b), the qualitative ultrastruc-tural abnormalities observed in microfibrils elabo-rated by the patient’s cells appear to underline thispoint.
However, this apparent reduction of wild-type Fibin culture media from the patient’s cells made theobservation possible that the proFib' moleculesmigrated slightly more slowly than Fib butcomigrated with Fib in the presence of tunicamycin.This suggests that the truncated proFib is of similarsize as Fib, but over-N-glycosylated. The currentelectrophoretic resolution limit allows us to assignthe putative cleavage site for removing a propeptidein the C terminus to within a 40 amino acid residue
JMB—MS 514
Post-translational Modification of Profibrillin 1906
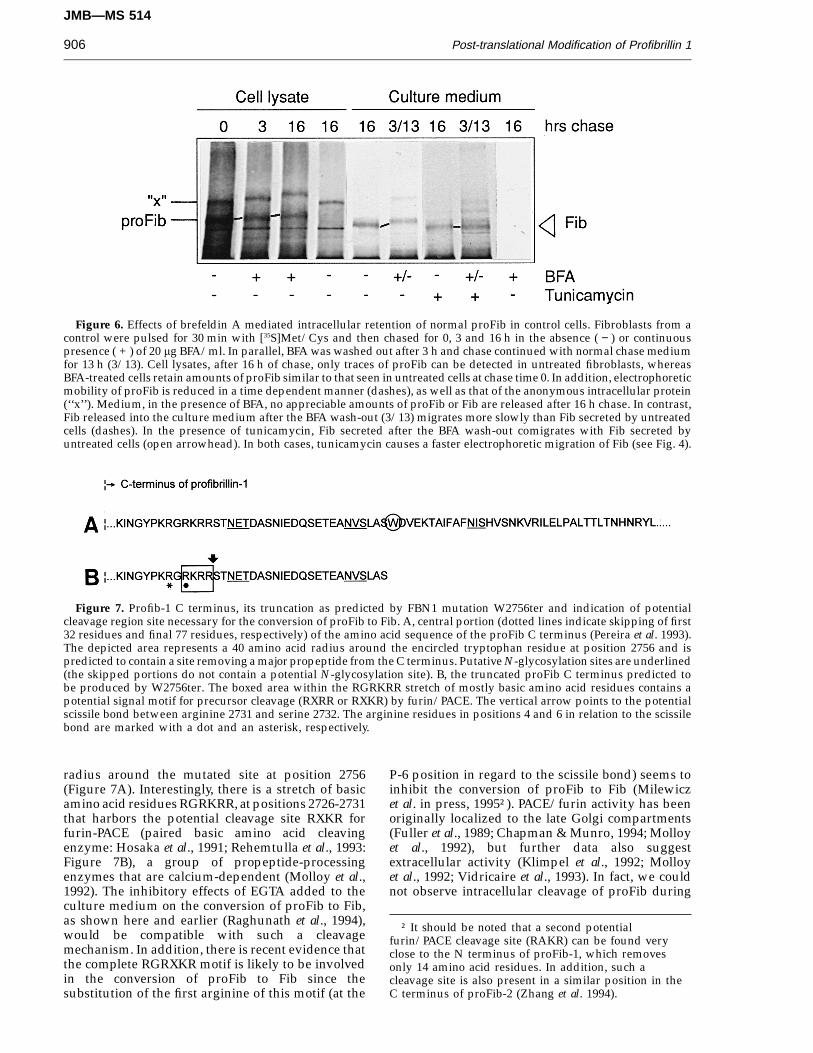
Figure 6. Effects of brefeldin A mediated intracellular retention of normal proFib in control cells. Fibroblasts from acontrol were pulsed for 30 min with [35S]Met/Cys and then chased for 0, 3 and 16 h in the absence ( − ) or continuouspresence ( + ) of 20 mg BFA/ml. In parallel, BFA was washed out after 3 h and chase continued with normal chase mediumfor 13 h (3/13). Cell lysates, after 16 h of chase, only traces of proFib can be detected in untreated fibroblasts, whereasBFA-treated cells retain amounts of proFib similar to that seen in untreated cells at chase time 0. In addition, electrophoreticmobility of proFib is reduced in a time dependent manner (dashes), as well as that of the anonymous intracellular protein(‘‘x’’). Medium, in the presence of BFA, no appreciable amounts of proFib or Fib are released after 16 h chase. In contrast,Fib released into the culture medium after the BFA wash-out (3/13) migrates more slowly than Fib secreted by untreatedcells (dashes). In the presence of tunicamycin, Fib secreted after the BFA wash-out comigrates with Fib secreted byuntreated cells (open arrowhead). In both cases, tunicamycin causes a faster electrophoretic migration of Fib (see Fig. 4).
Figure 7. Profib-1 C terminus, its truncation as predicted by FBN1 mutation W2756ter and indication of potentialcleavage region site necessary for the conversion of proFib to Fib. A, central portion (dotted lines indicate skipping of first32 residues and final 77 residues, respectively) of the amino acid sequence of the proFib C terminus (Pereira et al. 1993).The depicted area represents a 40 amino acid radius around the encircled tryptophan residue at position 2756 and ispredicted to contain a site removing a major propeptide from the C terminus. Putative N-glycosylation sites are underlined(the skipped portions do not contain a potential N-glycosylation site). B, the truncated proFib C terminus predicted tobe produced by W2756ter. The boxed area within the RGRKRR stretch of mostly basic amino acid residues contains apotential signal motif for precursor cleavage (RXRR or RXKR) by furin/PACE. The vertical arrow points to the potentialscissile bond between arginine 2731 and serine 2732. The arginine residues in positions 4 and 6 in relation to the scissilebond are marked with a dot and an asterisk, respectively.
radius around the mutated site at position 2756(Figure 7A). Interestingly, there is a stretch of basicamino acid residues RGRKRR, at positions 2726-2731that harbors the potential cleavage site RXKR forfurin-PACE (paired basic amino acid cleavingenzyme: Hosaka et al., 1991; Rehemtulla et al., 1993:Figure 7B), a group of propeptide-processingenzymes that are calcium-dependent (Molloy et al.,1992). The inhibitory effects of EGTA added to theculture medium on the conversion of proFib to Fib,as shown here and earlier (Raghunath et al., 1994),would be compatible with such a cleavagemechanism. In addition, there is recent evidence thatthe complete RGRXKR motif is likely to be involvedin the conversion of proFib to Fib since thesubstitution of the first arginine of this motif (at the
P-6 position in regard to the scissile bond) seems toinhibit the conversion of proFib to Fib (Milewiczet al. in press, 1995†). PACE/furin activity has beenoriginally localized to the late Golgi compartments(Fuller et al., 1989; Chapman & Munro, 1994; Molloyet al., 1992), but further data also suggestextracellular activity (Klimpel et al., 1992; Molloyet al., 1992; Vidricaire et al., 1993). In fact, we couldnot observe intracellular cleavage of proFib during
† It should be noted that a second potentialfurin/PACE cleavage site (RAKR) can be found veryclose to the N terminus of proFib-1, which removesonly 14 amino acid residues. In addition, such acleavage site is also present in a similar position in theC terminus of proFib-2 (Zhang et al. 1994).

JMB—MS 514
Post-translational Modification of Profibrillin 1 907
BFA treatment, which would allow us to exclude theendoplasmic reticulum, the salvage compartmentand the cis Golgi (Pelham, 1991) as intracellular sitesfor the conversion of proFib to Fib; however, morework is needed to determine where the conversion ofproFib to Fib takes place and to characterize theenzyme involved.
As regards the over-N-glycosylation of thetruncated proFib molecules, we would favor thehypothesis that the mutation, as such, primarilycauses intracellular retention due to conformationalchanges of the C terminus without compromising thecorrect cleavage of the truncated proFib moleculesafter export from the endoplasmic reticulum. In fact,we could over-N-glycosylate normal proFib incontrol cells through transitory captivation in theendoplasmic reticulum by BFA; this is compatiblewith the assumption that not all putative N-glycosy-lation sites in normal proFib are used during itsregular intracellular post-translational processing.Consequently, any event that prolonged the intra-cellular presence of proFib would increase thenumber of N-glycan groups attached to themolecule. It appears noteworthy, that analogousobservations have been made with the glycosylationof hydroxylysyl residues of collagen I which are onlysaturated under conditions of intracellular retentioncaused by structural abnormalities of the collagentriple helix (Steinmann et al., 1984; Byers, 1993). It iscurrently a matter of debate to what extent thisovermodification contributes to the pathogenesis ofosteogenesis imperfecta (Tenni et al., 1993). Sinceintracellular retention and delayed secretion ofmutant proFib has been reported in several instances(Milewicz, 1992; Aoyama et al., 1993; Milewicz &Duvic, 1994) it is conceivable that the resultingover-N-glycosylation of the mutant proFib might bean important modulator of the phenotype of a MFSpatient.
Methods
Cell cultures
We analysed dermal fibroblast cultures, Ma122,harboring a heterozygous point mutation W2756ter inFBN1. This cell strain was established from an adultFinnish patient with severe Marfan syndrome, thephenotype of which has been described earlier (patientE. Y. in Kainulainen et al., 1992). As a control we useddermal fibroblasts derived from a normal adult. These cellstrains were grown under standard conditions in minimalessential medium (MEM: Gibco) supplemented with 10%(v/v) fetal calf serum (FCS: Gibco) and antibiotics.
Immunofluorescence studies
Cells (106/35 mm dish) were grown on glass coverslipsfor 72 hours to hyperconfluency, fixed in absolute methanoland processed for immunofluorescence visualization ofaccumulated microfibrils by Mab anti-fibrillin F2 (agenerous gift from Dr M. Godfrey, Omaha) as described(Raghunath et al., 1993a,b).
Microfibril extraction from cell culture andprocessing for rotary shadowing electronmicroscopy
Cells were maintained at post-confluence for up to 3weeks. For microfibril extractions, cell layers were washedin 0.05 M Tris-HCl (pH 7.4) containing 0.4 M NaCl(Tris-saline), and digested for 2 hours at 20°C with 0.1 mgbacterial collagenase/ml (type 1A: Sigma) in Tris-salinesupplemented with 5 mM CaCl2, 2 mM phenylmethylsul-fonyl fluoride and 5 mM N-ethyl maleimide. Solubleextracts were clarified by centrifugation for 15 minutes at7500 g on a bench microfuge. The extracts werechromatographed directly without concentration undernon-reducing, non-denaturing conditions on a SepharoseCL-2B column equilibrated and eluted at room tempera-ture using Tris-saline (Kielty et al., 1991). High Mr materialpresent in the excluded volume was retained andvisualized for its microfibril content by rotary shadowingelectron microscopy (EM) using a modification of the micasandwich technique (Kielty et al., 1993). Immunogold EMof fibrillin microfibrils was carried out as described (Kielty& Shuttleworth, 1993).
Metabolic labelling of proFib
Cells (100,000) were seeded on day 1 in 15 mm dishes(4-well dishes from Nunclon, 1.8 cm2) in MEM (Gibco)with 10% FCS, 5 mM glutamine and antibiotics. On day 2,the medium was removed, the cells were washed twice anddepleted for 30 minutes in MEM lacking methionine,cysteine and FCS (MEM-/-/-: Northumbria), and finallypulsed with 50 mCi of TranSlabel [35S]Met/Cys in 200 mlMEM-/-/-for 4 hours or 20 mCi for 16 hours. Forpulse–chase experiments, the cells were pulsed on day 3 for30 minutes with 50 mCi TranSlabel in 200 ml MEM-/-/-.The pulse medium was removed, the cells were washedtwice with MEM-/-/- supplemented with 10 mMmethionine and 1 mM cysteine and chased with 200 ml ofthis solution for 0, 1, 2, 4 and 20 hours.
Inhibition of N-linked glycosylation
Mutant and control cells were preincubated for 60minutes with 5 mg/ml tunicamycin in 200 ml MEM-/-/-,after which 20 mCi of TranSlabel were added for a 16 hourcontinuous pulse or 50 mCi for 30 minutes for pulse–chasestudies. Only culture media were processed for SDS-PAGEafter continuous pulse.
Radioimmunoprecipitation of proFib and Fib
Cells (5 × 105) of the mutant and the control strain,respectively, were seeded in 35 mm dishes and grown for48 hours. On day 3 cells were depleted, pulsed for 30minutes with 250 mCi TranSlabel in 300 m MEM-/-/-, andchased for 1, 2 and 3 hours with 500 ml chase medium. Thecells were lysed with 500 ml lysis buffer (see below) for 20minutes on ice. Radioactive profibrillin was immuno-precipitated from cell lysates and culture media asdescribed (Raghunath et al., 1994).
Continuous and transitory intracellular captivationof proFib by brefeldin A during pulse–chase
Control cells were seeded and depleted for 60 minutesas described, in the absence or presence of 20 mg brefeldinA (BFA)/ml (Fluka) in 200 ml MEM-/-/-. The pulse–chase

JMB—MS 514
Post-translational Modification of Profibrillin 1908
was performed as described in the presence of BFA, whichwas also present throughout the washing steps. Chasetimes were 0, 2, 3, 4 and 20 hours. In some instancestunicamycin (5 mg/ml) was added to BFA and non-BFAtreated cells. In order to achieve a final release ofintracellularly retained proteins, BFA was washed out after2, 3 or 4 hours of chase by 3 rinses with chase medium orchase medium containing tunicamycin, and incubationwas continued in 200 ml of normal chase medium, with orwithout tunicamycin, overnight.
Precursor-product relationship of proFib and Fib
Mutant and control fibroblasts were continuouslypulsed for 3 hours with 50 mCi TranSlabel in 200 mlMEM-/-/- in the presence of 5 mM ethylene glycolbis(2-aminoethyl ether)-N,N,N',N'-tetraacetic acid (EGTA)(Raghunath et al., 1994). Only the media were processed forSDS-PAGE.
Processing of samples for SDS-PAGE analysis ofradiolabelled Fib
The medium (200 ml) was aspirated and added to 200 mlof 2 × sample buffer and 40 ml mercaptoethanol (MSH).The wells were washed twice with ice-cold Hank’sbalanced salt solution (HBSS) lacking Ca and Mg, and thecells lysed for 20 minutes on ice in 200 ml of 10 mM Hepes(pH 7.4), containing 2 mM o-phenanthroline-HCl, 1%(w/v), Nonidet P-40 (NP-40), 1 mM PMSF and 1 mMN-ethylmaleimide. Cell lysates were transferred to doublestrength sample buffer/MSH as described. The remainderin the dish which was, by definition, extracellular matrix,was washed twice with HBSS and extracted using 440 ml ornormal strength sample buffer (0.5 M urea and 10% (w/v)MSH) for 10 min at room temperature. Then 200 ml ofdouble strength sample buffer (Raghunath et al., 1993)supplemented with 1 M urea was added to mediumsamples and cell lysates, were immediately boiled togetherwith the ECM extracts for 3 minutes, centrifuged, and 30 mlof each was used for 4%/7% step gel SDS-PAGE andfluorography as described (Raghunath et al., 1993). Fluoro-graphic bands representing vimentin and an anonymousintracellular non-secretory protein migrating aboveproFib (Milewicz et al., 1992) harvested from the celllysates were used as calibrators for densitometricquantification.
Acknowledgements
We are very grateful to Dr Dianna Milewicz for fruitfuldiscussion and for sharing data and the cell line P 139harboring the R2726W mutation with us. We are indebtedto Dr Maurice Godfrey for mAb F2 to Fib and to L. Burgerand Susanne Staubli for excellent photographic and artwork. This work was supported by the Swiss NationalFoundation (grant 32-27884.89/2) and the MedicalResearch Council (UK). Parts of this work were presentedat the 3rd International Symposium on the MarfanSyndrome in Berlin (September 6–9, 1994) with thegenerous support of the Swiss Marfan Foundation.
ReferencesAoyama, T., Tynan, K., Dietz, H. C., Francke, U. &
Furthmayr, H. (1993). Missense mutations impair
intracellular processing of fibrillin and microfibrilassembly in Marfan syndrome. Hum. Mol. Gen. 12,2135–2140.
Byers, P. H. (1993). Osteogenesis imperfects. In ConnectiveTissue and Its Heritable Disorders: Molecular, Genetic andMedical Aspects. (Royce, P. M. & Steinmann, B., eds),pp. 317–350, Wiley-Liss, New York.
Chapman, R. E. & Munro, S. (1994). Retrieval of TGNproteins from the cell surface requires endosomalacidification. EMBO J. 13, 2305–2312.
Dietz, H. C., Cutting, G. R., Pyeritz, R. E., Maslen, C. L.,Sakai, L. Y., Corson, G. M., Puffenberger, P. E.,Hamosh, A., Nanthakumar, E., Curristin, S. M.,Stetten, G., Meyers, D. A. & Francomano, C. A. (1991).Marfan syndrome caused by a recurrent de novomissense mutation in the fibrillin gene. Nature(London), 352, 337–339.
Dietz, H. C., Valle, D., Francomano, C. A., Kendzior, R. J.,Pyeritz, R. E. & Cutting, G. R. (1993a). The skipping ofconstitutive exons in vivo induced by nonsensemutations. Science, 259, 680–682.
Dietz, H. C., McIntosh, I., Sakai, L. Y., Corson, G. M.,Chalberg, S. C., Pyeritz, R. E. & Francomano, C. A.(1993b). Four novel FBN1 mutations: significance formutant transcript level and EGF-like domain calciumbinding in the pathogenesis of Marfan syndrome.Genomics, 17, 468–475.
Fujiwara, T., Oda, K., Yokota, S., Takatsuki, A. & Ikehara,Y. (1988). Brefeldin A causes disassembly of the Golgicomplex and accumulation of secretory proteins in theendoplasmic reticulum. J. Biol. Chem. 263, 18545–18552.
Fuller, R. S., Brake, A. J. & Thorner, J. (1989). Intracellulartargeting and structural conservation of a prohor-mone-processing endoprotease. Science, 246, 482–486.
Hosaka, M., Nagahama, M., Kim, W. S., Watanabe, T.,Hatsuzawa, K., Ikemizu, J., Murakami, K. &Nakayama, K. (1991). Arg-X-Lys/Arg-Arg motif as asignal for precursor cleavage catalyzed by furinwithin the constitutive secretory pathway. J. Biol.Chem. 266, 12127–12130.
Kainulainen, K., Sakai, L. Y., Child, A., Pope, F. M., Puhaka,L., Ryhanen, L., Palotie, A., Kaitila, I. & Peltonen, L.(1992). Two unique mutations in Marfan syndromeresulting in truncated polypeptide chains of fibrillin.Proc. Nat. Acad. Sci., U.S.A. 89, 5917–5921.
Karttunen, L., Raghunath, M., Lonnqvist, L. & Peltonen, L.(1994). Compound heterozygous Marfan patient:point mutations in both fibrilin alleles result in lethalphenotype. Am. J. Hum. Genet. 55, 1083–1091.
Kielty, C. M. & Shuttleworth, C. A. (1993). Synthesis andassembly of fibrillin by fibroblast and smooth musclecells. J. Cell Sci., U.S.A. 89, 10277–10281.
Kielty, C. M., Cummings, C., Whittaker, S. P., Shuttleworth,C. A. & Grant, M. E. (1991). Isolation andultrastructural analysis of microfibrillar structuresfrom bovine elastic tissues. J. Cell Sci. 99, 797–807.
Kielty, C. M., Berry, L., Whittaker, S. P. & Shuttleworth,C. A. (1993). Microfibrillar assemblies of foetal bovineskin. Developmental distribution and relative abun-dance of type VI collagen and fibrillin. Matrix, 13,103–112.
Klimpel, K. R., Molloy, S. S., Thomas, G. & Leppla, S. H.(1992). Anthrax toxin protective antigen is activatedby a cell surface protease with the sequence specificityand catalytic properties of furin. Proc. Nat. Acad. Sci.,U.S.A. 89, 10277–10281.
Low, F. N. (1962). Microfibrils: fine filamentous com-ponents of the tissue space. Anat. Rec. 142, 131–137.

JMB—MS 514
Post-translational Modification of Profibrillin 1 909
Maddox, K. B., Sakai, L. Y., Keene, D. R. & Glanville, R. W.(1989). Connective tissue microfibrils. J. Biol. Chem.264, 21381–21385.
Milewicz, D.M. & Duvic, M. (1994). Severe neonatalMarfan syndrome resulting from a de novo 3-bpinsertion into the fibrillin gene on chromosome 15.Am. J. Hum. Genet. 54, 447–453.
Milewicz, D. M., Pyeritz, R. E., Crawford, E. S. & Byers,P. H. (1992). Marfan syndrome: defective synthesis,secretion, and extracellular matrix formation offibrillin by cultured dermal fibroblasts. J. Clin. Invest.89, 79–86.
Milewicz, D. M., Grossfield, J., Cao, S.-H., Kielty, C.,Covitz, W. & Jewett, T. (1995). A mutation in FBN1disrupts profibrillin processing and results in isolatedskeletal features of the Marjan syndrome. J. Clin.Invest. In the press.
Molloy, S. S., Bresnahan, P. A., Leppla, S. H., Klimpel, K. R.& Thomas, G. (1992). Human furin is a calcium-dependent serine endoprotease that recognizes thesequence Arg-X-X-Arg and efficiently cleaves anthraxtoxin protective antigen. J. Biol. Chem. 267, 16396–16402.
Pelham, H. R. B. (1991). Multiple targets for brefeldin A.Cell, 67, 449–451.
Pereira, L., D’Alessio, M., Ramirez, F., Lynch, J. R., Sykes,B., Pangilinan, T. & Bionadio, J. (1993). Genomicorganization of the sequence coding for fibrillin, thedefective gene product in Marfan syndrome. Hum.Mol. Genet. 2, 961–968.
Pyeritz, R. E. (1993). The Marfan syndrome. In ConnectiveTissue and Its Heritable Disorders: Molecular, Genetic andMedical Aspects (Royce, P. M. & Steinmann, B., eds),pp. 437–468, Wiley-Liss, New York.
Raghunath, M., Superti-Furga, A., Godfrey, M. &Steinmann, B. (1993a). Decreased deposition offibrillin and decorin in neonatal Marfan syndromefibroblasts. Hum. Genet. 90, 511–515.
Raghunath, M., Godfrey, M. & Steinmann, B. (1993b).Mutant fibrillin from fibroblasts from a patient withneonatal Marfan syndrome disturbs the deposition ofmicrofibrils in cocultured normal cells. Am. J. Hum.Genet. 47, 143.
Raghunath, M., Kielty, C. M., Kainulainen, K., Child, A.,
Peltonen, L. & Steinmann, B. (1994). Analyses oftruncated fibrillin caused by a 366 bp deletion in theFBN1 gene resulting in Marfan syndrome. Biochem. J.302, 889–896.
Ramirez, F., Pereira, L., Zhang, H. & Lee, B. (1993). Thefibrillin-Marfan syndrome connection. BioEssays, 9,589–594.
Rehemtulla, A., Barr, P. J., Rhodes, C. J. & Kaufman, R.(1993). PACE4 is a member of the mammalianpropeptidase family that has overlapping but notidentical substrate specificity to PACE. Biochemistry,32, 11586–11590.
Rosenbloom, J. (1993). Elastin. in Connective Tissue and ItsHeritable Disorders: Molecular, Genetic and MedicalAspects (Royce, P. M. & Steinmann, B., eds),pp. 167–188, Wiley-Liss, New York.
Sakai, L. Y., Keene, D. R. & Engvall, E. (1986). Fibrillin, anew 350 kD glycoprotein, is a component ofextracellular microfibrils. J. Cell Biol. 103, 2499–2509.
Sakai, L. Y., Keene, D. R., Glanville, R. W. & Bachinger, H. P.(1991). Purification and partial characterization offibrillin, a cystein-rich structural component ofconnective tissue microfibrils. J. Biol. Chem. 266,14763–14770.
Steinmann, B., Rao, V. H., Vogel, A., Bruckner, P.,Gitzelmann, R. & Byers, P. H. (1984). Cysteine in thetriple-helical domain of one allelic product of the a1(I)gene of type I collagen produces a lethal form ofosteogenesis imperfecta. J. Biol. Chem. 259, 11129–11138.
Tenni, R., Valli, M., Rossi, A. & Cetta, G. (1993). Possible roleof overglycosylation in the type I collagen triplehelical domain in the molecular pathogenesis ofosteogenesis imperfects. Am. J. Med. Genet. 45,252–256.
Vidricaire, G., Denault, J. B. & Leduc, R. (1993).Characterization of a secreted form of human furinendoprotease. Biochem. Biophys. Res. Commun. 195,1011–1018.
Zhang, H., Apfelroth, S. D., Hu, W., Davis, E. D.,Sanguineti, C., Bonadio, J., Mecham, R. P. & Ramirez,F. (1994). Structure and expression of fibrillin-2, anovel microfibrillar component preferentially locatedin elastic matrices. J. Cell Biol. 124, 885–863.
Edited by J. Karn
(Received 3 October 1994; accepted 21 February 1995)
Copyright © 2022 FDOKUMEN




















