
Transgenes Targeted to Growth Hormone Cells - UCL Discovery
261
Transgenes Targeted to Growth Hormone Cells Lindsay McGuinness 2002 Thesis submitted in accordance with the requirements of the UNIVERSITY OF LONDON for the degree of DOCTOR OF PHILOSOPHY Department of Molecular Neuroendocrinology National Institute for Medical Research The Ridgeway Mill Hill London NW7 lAA
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of Transgenes Targeted to Growth Hormone Cells - UCL Discovery

Transgenes Targeted to Growth Hormone Cells
Lindsay McGuinness
2002
Thesis submitted in accordance with the requirements of the
UNIVERSITY OF LONDON
for the degree of
DOCTOR OF PHILOSOPHY
Department of Molecular Neuroendocrinology
National Institute for Medical Research
The Ridgeway
Mill Hill
London NW7 lAA

ProQuest Number: U643219
All rights reserved
INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted.
In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed,
a note will indicate the deletion.
uest.
ProQuest U643219
Published by ProQuest LLC(2016). Copyright of the Dissertation is held by the Author.
All rights reserved.This work is protected against unauthorized copying under Title 17, United States Code.
Microform Edition © ProQuest LLC.
ProQuest LLC 789 East Eisenhower Parkway
P.O. Box 1346 Ann Arbor, Ml 48106-1346

A b s t r a c t
The relevance of targeting eGFP to GH vesicles is to study a particular dominant-negative GH
mutation, which causes dwarfism in children. Green fluorescent proteins (GFPs) are useful in
vivo reporter molecules, which can be used to identify and isolate living cells in which they are
expressed. I used an expression cassette in which the signal peptide and the first 22 residues of
hGH protein were linked in frame with an enhanced GFP (eGFP) sequence. Pituitary GC cells
were transfected with a construct linking the GH signal peptide to eGFP and stable fluorescent
lines were established. Confocal microscopy confirmed a granular localisation of eGFP. This
eGFP reporter cassette was then placed in a cosmid construct containing the locus control region
for human GH, and used to generate transgenic mice. Anterior pituitaries from GH-eGFP mice
showed numerous clusters of strongly fluorescent somatotrophs; EM showed co-localisation of
GFP with granules in pituitary somatotrophs. GH content was lower in pituitaries of transgenic
animals compared to wild type litter mates, but this did not appear to impair their growth. GH
cells could be purified by FACS.
The dominant-negative mutation is an intronic splice site G-^A transition causing exon 3 to be
skipped, producing a smaller inactive form of GH (del32-71). The mechanism by which the
mutated allele product prevents the release of GH from the normal allele is unknown. Pituitary
GC cells were transfected with a hGH gene containing the wild-type hGH or dominant-negative
mutation (hGH-IVS3). Stable cell lines were established and hGH-IVS3 transcribed the exon 3
skip mRNA. When co-transfected with GH-eGFP, TIRF (total internal reflection) and confocal
microscopy showed a reduction of GFP granules in hGH-IVS3 mutant GC cells and a disruption
in cell morphology. RIA of media for rGH from hGH-IVS3 was significantly reduced compared
to wild type GC cells. hGH-IVS3 was then placed in the LCR cosmid and expressed in mouse
pituitary somatotrophs, and induced profound pituitary GH deficiency and autosomal dominant
dwarfism, reduced in weight and length. As expected GH deficiency was associated with an
increase in GHRH expression in the arcuate nuclei in these mice. In adult mice from the most
severely affected line, the anterior pituitary glands were profoundly hypoplastic with few
recognizable somatotrophs, and their morphology showed a similar granular disruption seen in
cell lines with evident macrophage invasion. These transgenic models provide us with the means
to study the development and the progression of the pathology of human dominant-negative
dwarfism in a rodent model in vivo.
- I -

Transgenes Targeted to Growth Hormone Cells
Contents
Abstract IList o f figures VIIAbbreviations XAcknowledgements XIII
Introduction1.1 Aims o f this thesis 11.2 The hypothalamo-pituitary axis 3
1.2.1 The pituitary gland 31.2.2 Hypothalamo-pituitary interaction 31.2.3 Hypothalamus architecture 51.2.4 Anterior Pituitary Cell Differentiation 5
1.3 Growth hormone 11.3.1 Growth hormone 71.3.2 Transcriptional regulation of GH gene expression and
somatotroph development 91.4 Growth hormone releasing hormone (GHRH): the gene, its expression
and the receptor 111.4.1 GHRH gene 111.4.2 GHRH expression 121.4.3 The GHRH receptor 13
1.5 Somatostatin 151.6 Growth hormone releasing peptides (GHRP) and artificial growth
hormone secretogogues (GHS) 171.7 Physiological mechanisms o f GH release 18
1.7.1 Regulation of pulsatile GH secretion 181.7.2 Secretion of GH is sexually dimorphic 191.7.3 GH feedback mechanisms 19
1.8 The human growth hormone gene cluster 231.8.1 Physical linkage of the hGH gene cluster with CD79b and SCN4a 25
1.9 Locus Control Regions (LCRs) 261.9.1 hGH gene is controlled by a multi-component locus control
region (LCR) 281.10 Growth Hormone Receptor (GH-R), Growth Hormone Binding
Proteins (GHBP) and Insulin-like Growth Factors (IGF) 291.10.1 The tertiary structure of human growth hormone (hGH) 291.10.2 The Growth hormone receptor and GH signal transduction 321.10.3 Growth hormone binding protein 3 51.10.4 GH’s action in growth: Insulin-like Growth Factors (IGF’s) 38
1.11 GH-N gene product variants and isoforms 3 91.11.1 22KDahGH 411.11.2 20KDahGH 41
- I I -

1.11.3 17.5KDahGHorhGH-IVS3 421.12 Protein hormone storage in secretory granules and the mechanisms
fo r storage and sorting 421.12.1 Sorting of soluble proteins 431.12.2 Functioning of the Golgi complex and formation of
secretory granules 461.12.3 Hormone aggregates in neuroendocrine cells 48
1.13 Isolated growth hormone deficiency (IGHD) and short stature 531.13.1 Isolated Growth Hormone Deficiency Type 1A 531.13.2 Isolated Growth Hormone Deficiency Type IB 541.13.3 Biodefective Growth Hormone and GH-R’s 56
i) Mutant GH with an antagonistic effect 56ii) Laron Syndrome - mutations in the GH-R 57
1.13.4 Isolated Growth Hormone Deficiency Type II 577.14 Rodent models o f mutations in the hypothalamo-pituitary GH axis 581.15 Green Fluorescent Protein (GFP) as an endogenous cell marker 61
1.15.1 Properties of wild-type and mutant GFPs 621.15.2 GFP-tagged proteins and their subcelllular localisation 63
Chapter 2 Materials and Methods2.1 Preparation o f DNA 64
2.1.1 Bacterial cultures 642.1.2 Preparation o f plasmid and cosmidDNA 642.1.3 Preparation o f genomic DNA from animal tissue 642.1.4 Purification o f DNA 65
2.2 Subcloning of DNA fragments 652.2.1 Restriction digest 652.2.2 ‘Blunt-ending ’ o f DNA fragments 652.2.3 Vector dephosphorylation 652.2.4 Insertion o f linkers 652.2.5 Gel electrophoresis 662.2.6 Purification o f DNA fragments from agarose gels 662.2.7 Ligation o f DNA fragments 662.2.8 Transformation o f competent cells 662.2.9 Packaging o f cosmid DNA into bacteriophage 67
2.3 DNA sequencing 672.4 Polymerase Chain Reaction (PGR) 672.5 Detection of DNA sequences by Southern blotting 67
2.5.1 Southern blotting 682.5.2 Radioactive random prime labelling 682.5.3 Hybridisation o f Southern blots 68
2.6 Determination of protein content 682.7 Generation o f transgenic mice 69
2.7.1 Purification o f large DNA fragments fo r micro-injection 692.7.2 Superovulation, micro-injection and embryo transfers 69
2.8 Reverse transcriptase polymerase chain reaction (RT-PCR) 692.9 RNAse Protection Assay (RPA) 702.10 Northern Blotting 702.11 Radioimmunoassay (RIA) 71
- I I I -

2.11.1 Preparation o f pituitaries 712.11.2 Pituitary mGH, mPRL, mLH, mTSH, eGFP RIA 71
2.12 In Situ Hybridisation (ISH) 732.12.1 Sectioning o f tissue fo r in situ analysis 732.12.2 In vitro transcription 742.12.3 Riboprobe hybridisation 742.12.4 Image analysis 74
2.13 Immunocytochemi stry 752.13.1 Preparation o f GH-GFP Pituitaries fo r ICC 75
2.14 Preparation of in vitro systems 762.14.1 Culture o f rat GC cells 762.14.2 GC cell transfections 762.14.3 Dispersion o f mouse pituitary cells 772.14.4 Primary culture o f mouse pituitary cells 77
2.15 Fluorescence Activated Cell Sorting (FACS) 782.15.1 GH- GFP GC cell FA CS analys is 782.15.2 FACS sorting o f GH-GFP dispersed pituitaries 78
2.16 In vitro GH release studies 782.17 Electron microscopy 792.18 Total Internal Reflection (TIRF) Microscopy 79
2.18.1 The Evanescent Wave 802.18.2 The advantages o f TIRF 812.18.3 Exocytosis in action 822.18.4 The TIRF Microscope 822.18.5 Image acquisition 842.18.6 Image Processing 842.18.7 Calibration 84
2.19 Data Analysis 85
Chapter 3 In vitro studies in a GH cell line stably transfected with eGFP3.1 Introduction 863.2 Construction of hOH-eOFP plasmids for transfection of GC cells 883.3 Expression of p8GH-eGFP and p48GH-eGFP in rat GC cells 903.4 Growth rates of GH-eGFP cells v WT GC cells 943.5 Fluorescent activated cell sorting 943.6 Total Internal Refraction Microscopy (TIRF) 983.7 Discussion 102
Chapter 4 Targeting fluorescent reporters to pituitary somatotrophs in transgenic mice
4.1 Introduction 1064.2 hGH-GFP cosmid construct for generating transgenic animals 1074.3 Generation and identification of hGH-GFP transgenic mice 1084.4 Expression of the hGH-GFP transgene 1104.5 EM of GH-GFP pituitaries immuno-stained with GFP, GH, PRL 1124.6 Physiological Studies of hGH-GFP transgenic mice 118
4.6.1 Stimulation o f hGH-eGFP transgenic pituitaries with HGRF 118
- I V -

4.6.2 Pituitary hormone content and eGFP 1184.6.3 mGH mRNA levels in transgenic HGH-eGFP pituitary 1214.6.4 GHRH and somatostatin expression in hGH-eGFP hypothalamus 124
4.7 Fluorescent Activated Cell Sorting (FACS) of somatotrophs fromhGH-eGFP transgenic pituitaries 124
4.8 TIRP Microscopy of primary cultures of GH-GFP pituitaries 1284.9 Patterns of spontaneous [Ca^^]i transients in hGH-eGFP cells 1284.10 Discussion 133
Chapter 5 In Vitro studies of stably transfected rat GC cells expressing humangrowth hormone (hGH) dominant-negative mutation (IVS3 +1 G—>A)
5.1 Introduction 1395.2 Construction of hGH-WT and hGH-IVS3 constructs for transfection
in rat GC cells 1405.3 Characterisation of phGH-IVS3(+l G-^A) and phGH-WT in rat
GC cells 1415.4 Growth rates of phGH-WT and pGH-IVS3 GC cells 1445.5 GH content in hGH-IVS3 compared to hGH-WT transfected cells 1445.6 EM of hGH-IVS3 GC cells 147
5.6.1 Morphology o f GH-IVS3 transfected GC cells 1475.6.2 Immuno-gold labelling o f GH in hGH- IVS3 transfected cells 151
5.7 Co-transfection studies using confocal and TIRF microscopy 1515.8 Discussion 156
Chapter 6 Transgenic mice expressing a dominant-negative human growth hormone mutation
6.1 Introduction 1646.2 Targeting the hGH-IVS3 mutation to pituitary somatotrophs in mice 1656.3 Generation and identification of transgenic mice 1686.4 Physiological studies of GH-IVS3 transgenic mice 168
6.4.1 Growth parameters ofhGH-IVS3 transgenic v WT littermates 1686.4.2 Pituitary hormone (GH; PRL; TSH; LH) content 1726.4.3 GHRH and somatostatin mRNA in the hypothalamus 178
6.5 EM studies of pituitary sections from GH-IVS3 transgenic mice 1816.5.1 Ultra structural morphology o f hGH-IVS3 somatotrophs 1816.5.2 EM immuno-labelling o f mGH in pituitary somatotrophs
from h GH-I VS3 trans gen ic mice 1816.6 Discussion 186
Chapter 7 Final Discussion7.1 Transgenes targeted to growth hormone cells 1957.2 The hGH-eGFP transgenic mouse 1957.3 IGHD-II - a dominant-negative growth hormone 1977.4 hGH-IVS3 transgenic mice 1997.5 The role of zinc in GH dimérisation 200
- V -

AppendixI Engineering an Mlul linker into cos.GH 206II List o f Primers 209III NCBI GH sequence alignment 210
Bibliography 211
- V I -

List of Figures
Chapter 1Figure 1 Figure 1 Figure 1 Figure 1 Figure 1 Figure 1 Figure 1 Figure 1 Figure 1 Figure 1 Figure 1 Figure 1 Figure 1 Figure 1 Figure 1
12345678910 11 12131415
Figure 1.16
Table 1.1 Table 1.2
Hypothalamic-pituitary axis 4Hypothalamus architecture 6Development of anterior pituitary cell lineages 8GHRH signalling pathway 14Regulation of pituitary GH secretion 21Genomic organisation of the seven genes in the hGH gene cluster 24 The hGH gene cluster and LCR 27The tertiary structure of hGH and proposed zinc binding sites 30Back-bone structure of the hGH«(hGHbp)2 complex 34Sequential dimérisation model 36Schematic representation of the GHR and GHBP 37hGH-N mRNA Splicing 40The endoplasmic reticulum and protein folding 45Two models of transport through the Golgi complex 47Schematic of 3-D electron microscopy of the Golgi apparatus of a rat lactotroph 49Multiple stages of the exocytotic pathway 52
Isolated Growth Hormone Deficiency 55Rodent models with mutations in the GH axis 60
Chapter 2Figure 2.1 Figure 2.2
Typical mGH RIA Standard Curve A Schematic of the TIRF set-up
7283
Chapter 3Figure 3.1 hGH-eGFP constructs 89Figure 3.2 Expression of eGFP in GC cell lines 91Figure 3.3 Confocal Microscopy imaging of p48hGH-eGFP GC cells 92Figure 3.4 Confocal microscopy scanning of p48hGH-eGFP GC cells 93Figure 3.5 Growth rates and rGH content of p48hGH-eGFP + WT GC cells 95Figure 3.6 FACS analysis of hGH-eGFP cell lines 96Figure 3.7 FACS sorting of hGH-eGFP cell lines 97Figure 3.8 TIRF microscopy of GH granule motion in p48hGH-eGFP
GC cells 100Figure 3.9 Analysis of single granule trajectories in p48hGH-eGFP GC cells 101Figure 3.10 Expression of eGFP in GHRH neurones from rGH-eGFP
transgenic mice 104
Chapter 4Figure 4.1 Insertion of Mlu\ linkered hGH-eGFP fragment into cosGH.M 109
- V I I -

Figure 4.2 Analysis of eGFP expression in transgenic hGH-eGFP mice 111Figure 4.3 eGFP expression in pituitary GH cells from transgenic mice 113Figure 4.4 eGFP localisation in pituitary GH cells in transgenic mice 114Figure 4.5 Immunoelectron microscopy of eGFP in hGH-eGFP transgenic
pituitary cells 115Figure 4.6 Double immunoelectron microscopy of eGFP and mGH
co-localised in GH granules 116Figure 4.7 Double immunoelectron microscopy of eGFP and mPrl
co-localised in granules in mammosomatotrophs 117Figure 4.8 eGFP is secreted from GH cells in hGH-eGFP transgenic mice 119Figure 4.9 Pituitary hormone content in hGH-eGFP transgenic mice 120Figure 4.10 Growth curve of hGH-eGFP vs WT transgenic mice 122Figure 4.11 RNAse protection analysis of pituitary extracts from WT and
hGH-eGFP transgenic mice 123Figure 4.12 In situ hybridisation of mGHRH mRNA levels in hGH-eGFP
transgenic and WT mice 125Figure 4.13 In situ hybridisation of mSRIF mRNA levels in hGH-eGFP
transgenic and WT mice 126Figure 4.14 FACS purification of somatotrophs from hGH-eGFP transgenic
pituitaries 127Figure 4.15 TIRF microscopy of GH granule motion in hGH-eGFP
somatotrophs in primary culture 130Figure 4.16 TIRF microscopy of single granule trajectories in somatotrophs
of hGH-eGFP transgenic pituitaries 131Figure 4.17 Patterns of spontaneous [Ca^^Ji transients in hGH-eGFP pituitary
cells 132Figure 4.18 Expression of eGFP in d3 transgenic pituitaries 138
Chapter 5Figure 5.1 hGH-IVS3(+lG—>A) and hGH WT constructs for transfection
into rat GC cells 142Figure 5.2 Expression of phGH-WT and phGH-IVS3 in rat GC cells 143Figure 5.3 Growth rates o f hGH-WT v hGH-IVS3 GC cells 145Figure 5.4 RIA of GH production in hGH-WT vs hGH-IVS3 transfected
GC cells 146Figure 5.5 EM of rat GC cells transfected with hGH-WT and hGh-IVS3 148-9Figure 5.6 E.M of hGH-IVS3 transfected GC cells showing distribution
of large lipid vesicles 150Figure 5.7 EM immuno-labelling o f rGH in hGH-IVS3 transfected GC cells 152Figure 5.8 Confocal microscopy o f hGH-IVS3 GC cells cotransfected with
hGH-eGFP 154Figure 5.9 TIRF microscopy of hGH-WT vs hGH-IVS3 GC cells,
co-transfected with eGFP 155
Chapter 6Figure 6.1 Figure 6.2
hGH-IVS3 cosmid construct for microinjection Characterisation of IVS3+1G>A Mutation in GH cosmid
166167
-VIII

Figure 6.3 Genotyping and identification of hOH-IVS3 transgenic mice 169Figure 6.4 hOH-IVS3 transgenic mouse vs wild type littermate 170Figure 6.5 Growth Curves for HGH-IVS3 transgenic mice 171Figure 6.6 Body and tibia length in hGH-IVS3 transgenic males
V non-transgenic littermates 173Figure 6.7 hGH-IVS3 vs wild type pituitary gland 174Figure 6.8 RIA of pituitary hormone content (GH, Prl, LH and TSH) 175Figure 6.9 RIA of pituitary GH content in hGH-IVS3 (F23) mice 177Figure 6.10 In situ hybridisation of mouse GHRH mRNA levels in hGH-IVS3
transgenic and WT mice 179Figure 6.11 In situ hybridisation of mouse SRIF mRNA levels in hGH-IVS3
transgenic and WT mice 180Figure 6.12 EM of somatotroph from hGH-IVS3 transgenic pituitary 182Figure 6.13 Dominant-negative hGH-IVS3 phenotype affects other
cell types in the anterior pituitary 183Figure 6.14 Intermediate lobe/anterior pituitary boundary contains perivascular
macrophages 184Figure 6.15 EM immuno-labelling o f mGH in pituitary somatotrophs from
hGH-IVS3 transgenic mice 185Figure 6.16 NCBI Sequence Viewer - hGH v mGH and rGH 192
- I X -

ABBREVIATIONS
A alanineAa amino acidAC adenyl cyclaseACTH adrenocorticotrophic hormoneAla alanineAP anterior pituitaryARC arcuate nucleusArg arginineAY? arginine vasopressinBAC bacterial artificial chromosomebp base pairBSA bovine serum albuminC cysteinecAMP cyclic adenosine 3’,5’-monophosphatecDNA complementary DNACRE(B) cAMP response element (binding protein)CRH corticotrophin releasing hormonecpm counts per minuteCys cysteineDAB diaminobenzidine tetrahydrochlorideDAG diacetyl glyceroldHzO distilled waterD-MEM Dulbecco’s Modified Eagles MediumDNA deoxyribonucleic acidDNAse deoxyribonucleaseDM dorsomedial nucleusdl post-natal day 1el embryonic day 1E glutamateEDTA ethylenediaminetetracetic acideGFP enhanced green fluorescent proteinER endoplasmic reticulumERE oestrogen response elementF phenylalanineFACS fluorescent activated cell sortingFITC Fluorescein-isothionateFM family memberFSH follicle stimulating hormoneG glycineGABA y-aminobutyric acidGH growth hormoneGPCR G-protein coupled receptorbGH bovine growth hormonehGH human growth hormonemGH mouse growth hormone
- X -

rGH rat growth hormoneGHRH growth hormone releasing hormoneGHRH-R growth hormone releasing hormone receptorGHRP growth hormone releasing peptideGHS growth hormone secretagogueGHS-R growth hormone secretagogue receptorGRF growth hormone releasing factorHESS Hank’s balanced salt solutionH histidineHis histidineHS hypersensitive siteIGHD isolated growth hormone deficiencyi.c.v intracerebroventricularIGF-1 insulin growth factor-1i.p intraperitonealIP3 inositol-tris-phosphatelU international unitsi.v intravenousIVS intervening sequenceJAK Janus KinaseK lysineKDa kilodaltonKb kilobaseKO knock outL leucineLCR locus control regionLH luteinising hormoneLHN lateral hypothalamic nucleusmRNA messenger RNAME median eminenceMSG monosodium glutamateMSH melanocyte stimulating hormoneMX metallothioneinNGS normal goat serumNMS normal monkey serumNPY neuropeptide YNRS normal rabbit serumn.s. not significantNSS normal sheep serumNSG neurosecretory granuleNT non transgenicGRF open reading frameOT oxytocinP prolinePBS phosphate buffered salinePGR polymerase chain reactionPEG polyethylene glycolPEN periventricular nucleusPeVN periventricular nucleus
- X I -

Phe phenylalaninePit-1 pituitary specific transcription factorPKA proteinase KPLC phosophlipase CPOMC pro-opiomelanocortinPP posterior pituitaryPrl prolactinPVN paraventricular nucleusProp-1 Prophet of Pit-1R arginineRIA radioimmunoassayRNA ribonucleic acidRPA RNAse protection AssayRT-PCR reverse-transcriptaseSDR spontaneous dwarf ratsem standard error of the meanSCN suprachiasmatic nucleusSON supraoptic nucleusSRIF somatostatin release inhibiting factors s somatostatinSSTR/SST somatostatin receptorSTAT signal transducer and activator of transcriptionT transgenicTBS tris buffered salineTON trans golgi networkTOR transgenic growth retardedTIRF Total Internal Reflection MicroscopyTris Tris (hydromethyl)aminoethaneTRE thyroid response elementTSH thyroid stimulating hormoneV voltsV valineVMN ventromedial nucleusWT wild-typeZI zona inserta
XI I -

Acknowledgements
I am greatly indebted and very grateful to my PhD supervisor, Prof. Iain Robinson, for
his continuous support, enthusiasm and confidence in me over the past 4 years. I would
also like to thank Dr. Babis Magoulas for his supervision especially in the first couple of
years, without whom molecular biology would still be a myth! My friends in Molecular
Neuroendocrinology at the NIMR, particularly Abdul, Randip and Béné, made my four
years fun and go far too quickly. I would like to take this opportunity to thank Dr. Simon
Luckman, for my time at The Babraham Institute and persuading me that a PhD is not
such a bad idea!
I am grateful to Dr. Kathleen Mathers for her teaching, time and patience during
micro injection sessions. Thanks to Dr. Helen Christian in Oxford, who became my
mentor in EM and pituitary morphology and to Lynn and Sarah who cut all my sections.
Warm thanks to Danielle Carmignac for all her help and advice with RIA and science in
general. Many thanks to Dr. Pam Houston who helped and guided me with in situ even
when 8 months pregnant! I would like to thank Patrice Mollard kindly for his time and
patience with electrophysiology and for making me feel very welcome in Montpellier. I
would also like to mention and thank staff in Dunkin Red, who cared for my transgenic
mice; Chris Atkins in the FACS lab; Jean-Baptiste Manneville for TIRF microscopy and
physics lessons; Nancy Cooke for the donation o f the hGH cosmid and John Phillips III
for donation of the hOH-IVS3 gene.
Many thanks to all my fantastic Edinburgh Uni friends and friends from home who have
pulled me through this time, especially the writing up.
And, lastly, my very special love and thanks to my family at home in Bridge of Allan for
always, always being there for me.
-XIII

X IV -

[jJiJJlL'JLL____________________________________________ I m r o d i i c l i o n
Chapter 1
Introduction
1.1 Aims of this thesis
The aims of the work described in this thesis are (i) to target growth hormone cells with a
fluorescent reporter to facilitate the analysis of living somatotrophs and (ii) to express in
these cells a product of a dominant-negative growth hormone gene mutation, in order to
elucidate the mechanisms o f growth hormone suppression. The dominant-negative
growth hormone mutation I chose to study is manifested by a single base pair mutation in
the donor splice site o f intron 3 (IV S3+1G ^A ), resulting in the mis-splicing o f exon 3
and a truncated GH protein. To investigate the pathophysiology o f this, and other GH
mutations in the growth hormone axis, I chose an approach that combines transgenesis
with physiology.
Firstly, I wished to engineer transgenic mice targeting the fluorescent marker, green
fluorescent protein (eGFP), to GH granules in pituitary somatotrophs to identify and
isolate these cells in vivo. I aimed to target eGFP to somatotrophs in two systems; firstly
by transfection into a rodent GH cell line (rat GC cells) in vitro, and secondly by highly
directed transgenesis to mouse pituitary somatotrophs in vivo. Ideally, the components
that were to make up the constructs targeting eGFP to these cells in vivo should first be
tested in other simpler systems.
Two constructs containing eGFP were to be engineered for eGFP expression studies in
hGH-eGFP stably transfected GC cells. The first of which contained only the first 8
residues of the hGH signal peptide; the second construct was engineered to contain the
intact signal peptide and a further 22 N-terminal residues of hGH to target the expression
of eGFP specifically to GH granules. The transfection of eGFP into rat GC cells would be
performed with the aim of targeting of eGFP to GH granules in a GH cell and to assess
the effects of expressing such a human GH construct (p48GH-eGFP) in a rodent cell line
in vitro.
1 -

k Inlrocliuiion
To generate hGH-eGFP mice, we aimed to combine highly directed transgenesis to
somatotrophs (using the locus control region for hGH [Jones et al., 1995]) with the
granule-targeting eGFP fusion reporter constructs. The advantage of such an approach is
that transgenic hGH-eGFP mice would be useful tools, as one of the problems when
working with pituitary cells is identifying these cells in vivo. Transgenic hGH-eGFP mice
expressing eGFP in GH cells would overcome this problem and would greatly facilitate
the study of GH cell physiology (particularly with reference to the dominant-negative GH
mutation, which is thought to disrupt the secretory process) by visualising intracellular
distribution and secretory processes directly in pre-identified somatotrophs.
I wished to test whether inserting a human GH-IVS3 mutated gene into a rodent GH cell
line would cause dominant suppression of endogenous rat GH from GC cells - necessary
for the production of a transgenic rodent model carrying a human GH mutation. A cell
system would also allow further elucidation of the dominant suppression of wild type GH
in vitro. In parallel, I wished to generate transgenic mice bearing the hGH-IVS3
mutation (which results in dwarfism in human patients) using the same cosmid construct
that directs transgenes to pituitary somatotrophs. The dominant suppression o f
endogenous wild type GH by hGH-IVS3 would be studied in isolation in a somatotroph
cell line. A transgenic mouse bearing the hGH-IVS3 gene could provide a useful model
in which to study the mechanisms of the disease in vivo in the presence of other paracrine
effects in situ and subsequent physiological effects in the whole animal.
In my introduction, I provide a brief overview of the general physiology o f growth
hormone. I also introduce and describe the regulatory sequences controlling the
expression of human growth hormone as I utilised the locus control region (LCR) of
human growth hormone to target transgenes specifically to pituitary somatotrophs in
transgenic mice. Since one of the objectives o f my thesis is to investigate the mechanism
o f a dominant-negative GH mutation, I also introduce and discuss in more detail, the
structure o f hGH and the role o f zinc in GH hetero-dimerisation in the aggregation of GH
to form condensed GH secretory vesicles.
- 2 -

( ' lu i i 'l c r /_____________________________________ __________________________________________________ I n i r o d u c l i o n
1.2 The hypothalamo-pituitary axis
1.2.1 The pituitary gland
The pituitary gland is functionally and anatomically divided into three regions, the two
major regions being the anterior lobe (adenohypophysis) and the posterior lobe
(neurohypophysis). The former originates from ectodermal cells and is formed from an
invagination of the developing pharyngeal tissue, whereas the posterior pituitary is
derived from neuroectodermal tissue of the diencephalon (Everitt et al., 1986; Ikeda et
a l, 1988). A third region (pars intermedia) has a species specific role and is of more
importance in lower vertebrates (Lemer et al., 1981)
1.2.2 Hypothalamo-pituitary interaction
The core o f the neuroendocrine system is represented by the hypothalamic-pituitary
complex. The hypothalamus acts through efferent neural pathways to autonomic nuclei
in the brainstem and spinal cord, and through an intimate neuroendocrine relationship
with the pituitary gland. Two different mechanisms o f hypothalamo-pituitary
communications are used. The hypothalamo-neurophysial system comprises of large
magnocellular neurones which project to the posterior lobe of the pituitary gland (figure
1.1). The cell bodies of these neurosecretory cells produce two hormones, oxytocin and
arginine vasopressin, which are transported down the axons to the posterior pituitary by
carrier proteins called neurophysins and stored in neurosecretory granules in the axon
terminals. In response to nerve impulses the hormones are then released from the nerve
endings to enter capillary vessels in the posterior pituitary and then into the peripheral
circulation (Leng et al., 1999, for review). The anterior pituitary, most relevant to this
thesis, contains at least five different endocrine cell types, with their hormone production
under tight influence of hypothalamic regulating hormones or factors. The hypothalamic
stimulatory or inhibitory hormones are produced in the cell bodies o f parvocellular
neurones located in several different hypothalamic nuclei, from which they project their
axons to the median eminence (ME) depicted in figure 1.1. The neurones release their
product from the terminals in the median eminence into the hypophyseal blood system,
which delivers them to anterior pituitary cells. There, they interact with specific
receptors in the endocrine cells to stimulate or inhibit hormone release into the blood in
the pituitary sinusoids, before entry into the peripheral circulation.
3 -

Chapter I Iiitrochiciioti
Hypothalamus
MedianEminence
Magnocellular Neurones
Posterior PituitaryHypophyseal Portal Vessel
Posterior Hypophyseal Veins
Anterior Pituitary
Target Cell
Figure 1.1 Hypothalamo-pituitary Axis
Hypothalamic neurones (yellow) projecting to the median eminence release their
neuropeptides or amines into the hypophyseal blood system, which transports them to
their site of action in the anterior pituitary, where they stimulate or inhibit hormone
release by acting on specific receptors in pituitary endocrine cells. Magnocellular
neurones (blue) project directly to the posterior pituitary and release their neurosecretory
product into capillary veins.
- 4 -

___________________________________ _ ______________ In /roJ in -lion
1.2.3 Hypothalamus architecture
The hypothalamus contains two major subdivisions, a medial, nuclear component and a
lateral component containing the medial forebrain bundle. The medial zone is subdivided
into three regions: anterior, tuberal and mamillary. Relevant nuclei o f the medial zone
for this thesis (GH axis) are the periventricular (PeN) nucleus in the anterior region, and
the arcuate (ARC), ventromedial (VMN) and dorsomedial (DM) nuclei of the tuberal
region (figure 1.2). Hypothalamic function is diverse - it regulates food intake, water
balance, energy metabolism, growth, reproduction, circadian rhythm and stress responses
to name but a few. In keeping with its array of functions, hypothalamic nuclei contain
neurones expressing numerous neuropeptides, transmitters and receptors. Hypothalamic
factors regulating the anterior pituitary include corticotrophin-, thyrotrophin-,
gonadotrophin-, growth hormone releasing hormone, somatostatin and dopamine.
Together they influence systems as diverse as steroid metabolism, thyroid function,
reproduction, lactation and growth. In addition to expressing several o f the GH
regulating hormones and receptors, the hypothalamic arcuate nucleus (ARC) is also
involved in the hypothalamic control o f feeding. Numerous peptides expressed in the
arcuate nucleus are implemented in metabolic control, most o f these under the control of
the anti-obesity factor leptin (Ahima et al., 2000). Neuropeptide Y (NPY) is one of
these, but in addition to its function in energy metabolism, it may play a role in the
feedback mechanism of GH secretion (Kamegai et al., 1994). Importantly, hypothalamic
neurones not only project directly to the median eminence to release their neuropeptide
from the nerve terminals, but also to other neuronal targets in and beyond the
hypothalamus, e.g. brainstem.
1.2.4 Anterior Pituitary Cell Differentiation
Pituitary ontogeny gives rise to five different endocrine cell types as defined by the
hormones these cells produce and secrete. These are:
Somatotrophs - growth hormone (GH)
Lactotrophs - prolactin (PRL)
Corticotrophs - adrenocortico troph ic horm one (A CTH ) and melanocyte
stimulating hormone (MSH)
5 -

( 'fia p ter I Introcliiclion
Anterior Zone
PVN
SCNSON
Tubero-infundlbular Zone
mDM
ARCM E
Figure 1.2 Hypothalamic Architecture
The brain sections above are typical coronal sections through the anterior and tubero-
infundibular region of the medial hypothalamus. Nuclei mentioned in this thesis are
highlighted. Abbreviations are: supraoptic (SON), suprachiasmatic (SCN),
periventricular (PEN), paraventricular (PVN), arcuate (ARC), ventromedial (VMN) and
dorsomedial (DM), median eminence (ME).
- 6 -

_____ I m r o d u c t in n
Thyrotrophs - thyroid stimulating hormone (TSH)
Gonadotrophs - luteinising hormone (LH) and follicle stimulating hormone (FSH).
These cells originate from an invagination o f the neural ectoderm beneath the
diencephalon called Rathke’s Pouch (Everitt et al., 1986; Ikeda et a l , 1988). Both the
combined pattern of expression of different transcription factors (mainly homeotic genes)
and proliferative induction of signals from cells of the floor o f the diencephalon (gives
rise to the hypothalamus) account for organogenesis of the anterior pituitary, detailed in
figure 1.3 (reviewed in el Amaoui & Dubois (1993) and Asa & Ezzat (1999). Several
studies have indicated that PRL expressing cells are derived from GH expressing cells
(Hoeffler et a l , 1985; Behringer et al., 1988). These cells are derived from the Pit-1
lineage, which is discussed in more detail in 1.3.2.
1.3 Growth hormone
1.3.1 Growth hormone
GH was one of the first anterior pituitary hormones to be discovered. The association of
pituitary tumours composed of acidophilic cells with gigantism was first noted in the late
19 century and can be credited to Pierre Marie, 1886. The first experimental links
between the pituitary gland and body growth were established at the turn of the century
from work on dogs by Aschner and Cushing. Subsequently, Evans and Long (1921)
demonstrated that bovine anterior pituitary extract administered intraperitoneally to
normal rats produced an increase in growth. In 1941 Silberberg showed that injected
extract also accelerated the closure o f growth plates in young mice (Silberberg et al.,
1941). In 1945, the growth promoting factor was isolated in a purified form from bovine
pituitaries (Li et al., 1945). The protein proved to be a hormone, and was observed to
induce somatic growth in hypophysectomised rats as well as in normal rats, with little or
no effect on the gonads or thyroid gland. It was termed somatotropin or more commonly
growth hormone. Following this, (Li & Papkoff, 1956) isolated human growth hormone
(hGH) and its amino acid sequence was determined thirteen years later (Li et al., 1969).
Now, more than 50 years after its isolation, GH is widely used to treat growth disorders,
but it is still not fully understood how GH exerts its effects on longitudinal growth.

( Im ptcr I hiiroduclioii
NEURAL ECTODERM
RATHKE’SPOUCH
CUTE
H esx l
H esx l
T/EBP
ro s tr a l
Bm -4 dependent pro lifera tion
ANTERIORPITUITARY
P-OTX dependent pathw ay
Prop-1dependent
pathw ay
Bm -4dependen t
p ro lifera tion
P-OTX ACTH
© LHFSH
GH
PRL
P-TSHcaudom ed ia !
Figure 1.3 Development of Anterior Pituitary lineages
Transcription factors implicated in the development o f Rathke’s pouch and subsequently o f the individual pituitary cell types are shown above. The proposed schema involves early determination o f pituitary development from the oral ectoderm by a number o f factors, including P tx-\, Lhx-\, Lhx-A, P-LIM, Rpx and Prop-\. Molecular determinants o f corticotrophs, the first to occur, remain speculative but likely involve CUTE (corticotroph upstream transcription binding element). This is followed by P it-\ expression that designates a somatotroph stem cell which in the absence o f other transcription factors, retains somatotroph morphology and function. Expression o f the oestrogen receptor (ER) enhances prolactin gene expression in mammosomatotrophs; a putative GH repressor is implicated in silencing GH transcription to allow the emergence o f mature lactotrophs. Thyroid embryonic factor (TEF) is required for cells in the P it-\ lineage to develop into thyrotrophs. The third pathway o f cytodifferentiation is dictated by SF-1 (steroidogenic factor-1) which in conjunction with ER, determines gonadotroph differentiation and gonadotropin gene transcription. Reproduced from Asa & Ezzat, (1999).

( 'hi!l'il. /____________________________________________________________ /nir(j(/nc>i()ii
hGH is comprised of a chain of 191 amino acids, cross-linked by two disulphide bridges.
The rat growth hormone (rGH) is only 190 amino acids long and exhibits 66% homology
with its human counterpart (Seeburg et a l, 1977a,b). Mouse GH also consists o f 190
amino acids and is highly homologous to rat GH (95%) (Linzer & Talamantes, 1985).
The tertiary structure of hGH and its interaction with the GH receptor is discussed later in
more detail. While the rat possesses a single GH gene, the human GH gene is comprised
o f a 5 gene cluster, spanning 48kb (Barsh et al., 1983; Chen et al., 1989).
Phylogenetically lower animals tend to respond to GH from higher animals, but not vice
versa. In rats, human GH binds to both GH and PRL receptors (lactogenic receptors),
whereas bovine GH binds to GH-receptors exclusively (somatogenic receptors) (Ranke et
a l, 1976). Growth hormone is the major endocrine regulator o f post-natal grov^h in
mammals. Human growth hormone deficiency causes growth failure in young animals
and metabolic alterations in later life. Familial isolated growth hormone deficiency
(IGHD) is a heterogeneous disease that result from perturbations in different steps in the
expression of the GH-N gene (Cogan et al., 1994) and is described in more detail in 1.13.
1.3.2 Transcriptional regulation o f GH gene expression and somatotroph development
Research into the molecular mechanisms underlying somatotroph differentiation has
demonstrated cw-acting elements essential for cell specific expression of the GH gene
and a cell specific transcription factor that binds these elements was isolated (Bodner et
al., 1988; Ingraham et a l, 1988). Pit-1 (also called GHF-1) is the most important
transcriptional activation factor that contributes to the specific expression of GH in the
pituitary. It contains 291 amino acids, and has a transcriptionally active domain, a POU
specific DNA-binding domain and a homeo DNA-binding domain. The DNA binding
domain resemble those of oncogenes Oct and Une, hence the designation o f POU (Pit-
Onc-Unc) (Parks et a l, 1995). The Snell {dw/dw) and Jackson {dw^) allelic murine dwarf
models established that mutations in this POU homeodomain gene, Pit-1/GHF-1, result
in dwarfism (Li et a l, 1990). These mice are deficient in GH, PRL and TSH and have no
detectable somatotrophs, lactotrophs or thyrotrophs, the three cell types in which Pit-1
expression is observed in the normal pituitary (Simmons et a l, 1990). Pit-1 is later
required for the continued expression of the Pit-1 gene itself and the proliferation and
survival of these three cell types (Castrillo et a l, 1991). A mouse genetic defect, referred
- 9

( 'humer ! Inlrudmiiu!]
to as the Ames dwarf {dj), is located on chromosome 11 (Somson et a l, 1996) and results
in a hypoplastic anterior pituitary phenotypically similar to that of the Snell and Jackson
dwarfs. It was shown that with a mutation of the Prop-1 gene {Prophet o f Pit-1), a
pituitary specific paired-like homeodomain factor, Pit-1 transcription failed to activate,
thus leading to GH, PRL and TSH deficiency, although TSH was detectable (Somson et
al., 1996). Mutations in many of the genes have been proven to give rise to hypopituitary
phenotypes in man {Pit-\, P rop-l, H esX l, Lhx3). Besides Pit-1, the growth hormone
releasing hormone receptor (GHRH-R) is also required for normal somatotroph
development. However, the GHRH-R is needed at a later point in the development of the
pituitary cell lineage than Pit-\; GHRH-R is only necessary for somatotroph and not for
lactotroph and thyrotroph development. Little mice {lit/lit), which have a GHRH-R
mutation (Godfrey et a l, 1993; Lin et al., 1993) and their human equivalents (Wajnrajch
et al., 1996) only lack somatotrophs and GH (Lin et al., 1992).
Although GH gene transcription is dependent on the presence of the homeodomain
protein Pit-1, additional factors must be required to restrict GH to the somatotroph
lineage (Lira et al., 1993).
Simplified schema of transcriptional regulation of the GH gene in rats
[TAT;
Sp-1 Zn-16Pit-1TRE Pit-1
-184bp -85bp
A conserved element between the two Pit-1 binding sites referred to as Zn-16 has been
identified and it has been demonstrated that mutations within this site strongly represses
GH reporter gene expression in transgenic animals (Lipkin et al., 1993). Zn-16 is a
member of the Cys/His Zinc finger superfamily; its binding domain comprises three zinc
fingers separated by unusually long linker sequences that would be expected to interrupt
specific DNA site recognition. Zn-16 synergises with Pit-1 to activate the GH promoter
in heterologous cells. Other studies in rats have defined elements by which cAMP-
dependent pathways and SP-1, in co-ordination with Pit-1, regulate GH gene activity
(Shepard et a l, 1994). Further stimulation of this gene by the thyroid hormone receptor
- 1 0 -

_______________________________________ ____________________________________________________ / n iracJucf ioii
has also been characterised (Tansey et a l, 1993). It is thought to bind to the retinoic acid
receptor as a heterodimer to interact with the retinoid X receptor on their respective
binding sites.
Previous studies have suggested that PRL expressing cells are derived from GH
expressing cells (Hoeffler et aL, 1985). In agreement with these observations, Behringer
et al., (1988) found that expression of a GH-diptheria toxin chimeric gene in transgenic
mice led to ablation of GH expressing cells as well as most o f the PRL expressing cells.
According to this model, a stem cell synthesising neither GH nor PRL gives rise to cells
that synthesise GH. These cells give rise to mature somatotrophs synthesising only GH,
which cannot undergo further differentiation, and to mammosomatotrophs expressing
both GH and PRL. The latter cell type gives rise to mature lactotrophs that produce PRL;
the original stem cell rarely differentiates into PRL only cells (Behringer et al., 1988).
There is plasticity of gene regulation in the adult pituitary, dependent on influencing
hormones e.g. chronic oestogen treatment shifts somatotrophs to mammosomatotrophs or
lactotrophs. However, the relationships between these cell types is not really defined,
and some other observations suggest that trans-differentiation could occur. This will be
discussed in more detail in results chapters.
1.4 Growth hormone releasing hormone (GHRH): the gene, its expression and
the receptor
1.4.1 GHRH gene
The first evidence for hypothalamic control of GH secretion came from in vivo and in
vitro experiments by Reichlin in 1960 demonstrating that lesions in the ventromedial
hypothalamus led to impaired linear growth in rats. GHRH was isolated 20 years later
from pancreatic tumours causing acromegaly (Guillemin et al., 1982; Rivier et al., 1982)
and later from human hypothalamus (Ling et al., 1984). Human GHRH is a single gene
and is a 44 residue peptide, but various shorter forms exist (Rivier et al., 1982; Miki et
al., 1994). Rat GHRH-43 (Speiss et a l, 1983) and mouse GHRH-42 (Suhr et ah, 1989)
are structurally different to human GHRH, nevertheless, all GHRH peptides can release
GH across species. The active GH stimulating GHRH is derived by multiple post-
- 1 1 -

__________________________________________________________ I n l r o d u c l i o n
translational processing steps (Nillni et a l, 1999). The signal leader sequence (amino
acids (aa) 1-30) and a 30 aa C-terminal peptide are cleaved from the 104aa GHRH
precursor to form the active GHRH molecule (31-73aa) (Mayo et a l , 1985a). In humans
the formation of the active GHRH is followed by amidation o f the terminal glycine,
giving the major GHRH form produced by the hypothalamus (Frohman et al., 1989).
1.4.2 GHRH expression
In 1983, Jacobowitz et al., were the first to demonstrate GHRH-like immunoreactivity in
the rat brain following treatment with Colchicine, a mitosis inhibitory factor which
arrests axonal transport and causes accumulation of products synthesised in the cell body.
GHRH cell bodies were principally located in the arcuate nucleus of the hypothalamus
(Jacobowitz et al., 1983) with greatest numbers in the ventrolateral regions or in the
ventromedial nucleus (VMN) (Sawchenko et al., 1985). These cell bodies project their
terminals to the median eminence (ME) releasing peptides into the hypophyseal portal
blood. Hypothalamic GHRH mRNA was shown to be sexually dimorphic, with males
expressing greater levels than females (Argente et al., 1991).
Pituitary GH gene expression, GH synthesis and secretion (Barinaga et al., 1985a,b;
Bilezikjian & Vale, 1984; Fukata et al., 1985; Tanner et al., 1990) are increased by
GHRH. Furthermore, GHRH induces the expression of the proto-oncogene c-fos and
stimulates the proliferation o f pituitary somatotroph cells (Billestrup et al., 1987;
Billestrup et al., 1986). These data suggest a strong trophic effect of GHRH on the
p ituitary. Transgenic mice over-expressing human GHRH under ubiquitous
metallothionein (MT) promoter control (Hammer et al., 1985) show enhanced growth,
high serum GH levels, pituitary hyperplasia and adenoma (Mayo et al., 1988), thereby
reflecting GHRH control of pituitary GH. Ectopic GHRH over-expressing tumours in
acromegalic patients cause a very similar phenotype which diminishes after removal of
the tumour (Thomer et a l, 1982).
An important factor in GHRH mRNA control is feedback regulation through GH,
mediated directly by GH-R expressed in the hypothalamus. GH deficiency is associated
with increased GHRH mRNA levels (Chomczynski et al., 1988), while GH treatment
- 1 2 -

CjjtH'lyj:J_____________________________________ ________________ _____________________________hiirodiii 'lion
decreases GHRH levels (de Gennaro Colonna et al., 1988). Regulation based on GH
status has been observed in genetically manipulated GH deficiency and excess syndromes
(for review see Phelps and Hurley, 1999). In accordance with their low GH levels, Ames
and Snell dwarf mice demonstrate increased levels of GHRH expression (Phelps et a l,
1993), while transgenic mice expressing GH under the ubiquitous MT promoter or the
phosphoenolpyruvate carboxykinase (PEPCK) promoter (Steger et al., 1991) show
markedly reduced GHRH mRNA levels (Phelps & Hurley, 1999).
1.4.3 The GHRH receptor
Three groups achieved the molecular cloning o f anterior pituitary GHRH-receptors
(GHRH-R) in human, rat and mouse (Gaylinn et al., 1993; Lin et al., 1992; Mayo, 1992).
The GHRH-R is a 423 aa seven trans-membrane G protein-coupled receptor in all three
species and its expression was localised to GH cells of the anterior pituitary (Lin et al.,
1992). Expression levels o f GHRH-R mRNA are up-regulated by GHRH itself (Miller
and Mayo, 1997) and by Pit-1 (Lin et al., 1992). The GHRH-R is absent in dwarf Snell
and Ames mice which are impaired in Pit-1 expression, suggesting that GHRH-R
expression might be Pit-1 dependent (Lin et al., 1992). Early studies show that GH
release in response to GHRH was associated with stimulation o f cAMP production
(Bilezikjian and Vale, 1984) and that extracellular Ca^^ was required for this cAMP
accumulation, suggesting that Ca^^ might act independently o f cAMP (Brazeau et al.,
1982b). Transfection of COS or human kidney 293 cells with the GHRH-R demonstrated
that GHRH stimulated the accumulation of cAMP as well as transcription o f cAMP-
responsive reporter gene (Gaylinn et al., 1993; Mayo et al., 1992), consistent with the
predicted coupling o f the GHRH-R to a Gs protein. Although no complete cAMP
responsive elements have been identified in the GH gene promoter (Bertherat et al.,
1995), the importance o f cAMP response element binding protein (CREB) was
demonstrated by pituitary hypoplasia and dwarfism in transgenic mice over expressing an
inactive CREB (Struthers et a l , 1991). GHRH signalling in pituitary somatotrophs is
illustrated in figure 1.4.
13

Cliupler I IniroJuclion
GHRH
GH
osGH AC
AAA cAMPGH
GHHRH-R
CREB-PGHA A A
Pit-1 Pit-1
AAA
Figure 1.4 GHRH signalling pathway
GHRH interaction with its receptor leads rapidly to GH secretion, possibly by a G-
protein mediated interaction with ion channels or through ion channel phosphorylation,
and to stimulation of intracellular cAMP through stimulation of adenylate cyclase (AC)
by the G§ protein (Mayo et al., 1996). Elevated cAMP results in the phosphorylation
and activation of CREB by protein kinase A (PKA) (Gonzalez and Montminy, 1989)
and then enhanced Pit-1 transcription (Ingraham et at., 1988; McCormick et al., 1991).
Pit-1 activates transcription of the GH gene to elevate GH mRNA and protein and also
activates GHRH-R transcription (Lin et al., 1992). DNA; III ' mRNA:
(Modified from Mayo et al., 1996)
AA
- 14 -

( 'h(ij>h‘r I _____________________________________________________________________________________I m n n l u c tion
The importance of GHRH-R function for pituitary somatotroph development is illustrated
by the dwarfed little mouse. A point mutation in the GHRH-R gene is responsible for it’s
severe GH deficiency (Godfrey et a l, 1993; Lin et a l, 1993). Its pituitary somatotroph
(Wilson et al., 1988) and GH levels (Cheng et al., 1983) are markedly reduced, but
detectable. The few somatotrophs left are unresponsive to GHRH, but do release GH in
response to cAMP (Clark & Robinson, 1985; Jansson et al., 1986a). Recently, Gaylinn
and co-workers reported that the little mutation did not dramatically affect the expression
level, glycosylation, or cellular localisation of the receptor protein, but that it blocked
specific GHRH binding and therefore inhibited signalling (Gaylinn et al., 1999). In
humans, mutation o f the GHRH-R causes a similar GH phenotype (Baumann &
Maheshwari, 1997; Wajnrajch etal., 1996).
1.5 Somatostatin
Somatostatin (SS) or somatotropin release inhibitory factor (SRIF) was identified much
earlier than GHRH (Brazeau et al., 1973). SS exists in two major forms consisting of 14
or 28 amino acids (aa), derived by endopeptidase cleavage of a 116aa precursor, pre-pro
SS (Van Italie & Femstrom, 1983). Both SS14 and SS28 inhibit spontaneous and GHRH
induced GH secretion in vivo and in vitro (Brazeau et al., 1981). In contrast to GHRH,
the sequence of SS has remained highly conserved between species.
SS producing neurones are widely present throughout the central and peripheral nervous
system, the gut, the endocrine pancreas and in smaller numbers in the thyroid, adrenals,
submandibular glands, kidney, prostate and placenta (Patel, 1990; Reichlin, 1983).
Within the hypothalamus, the majority of SS neurones lie in the PEN (Alpert et al., 1976)
but these are also seen in the ARC and VMN (Finley et al., 1978). 78% of SS PEN
neurones project to the external layer of the ME (Kawano & Daikoku, 1988); other
neurones interconnect with several hypothalamic nuclei, including the ARC and VMN.
The SS axons and perikarya in the ARC and VMN, two areas where GHRH neurones
originate, indicate the existence o f anatomical and functional peptide interactions.
Furthermore, several somatostatinergic axon varicosities cluster around the GHRH
synthesising cells, suggesting synaptic associations (Liposits et a l , 1988). Co
localisation studies finally showed that SS receptor 1 (sstrl) and sstr2 were co-localised
1 5 -

( Jhr>icr J _ _ h u r o d u d ton
in part with ARC neurones expressing GHRH mRNA (M cCarthy et al., 1992;
Tannenbaum et a l , 1998). These data provide strong anatomical support for a direct
hypothalamic interaction o f SS with GHRH to modulate GH secretion. Zheng and co
workers supported this hypothesis with sstr2 knock-out (KO) mice (Zheng et a l, 1997).
Pre-treatment with GH prevents the activation of c-fos by GH secretagogues (GHS) in
ARC neurones (GHS’s activate neurones expressing c-fos in the ARC) in wild-type but
not sstr2 KG mice, the authors suggested that GH induced SS release from PEN
neurones, suppressed the activity o f ARC neurones. This interpretation is further
supported by electrophysiological experiments, where stimulation o f PEN neurones
resulted in inhibition of ARC neurones (Dickson et al., 1994). Willoughby and co
workers demonstrated that only very few SS neurones were closely approached by
GHRH immuno-reactive fibres in the PEN, providing possible evidence o f scant
innervation o f SS neurones by GHRH cells (Willoughby et al., 1989).
SS’s lack of trophic inhibitory effect on the pituitary was shown by the absence of an
effect on basal levels o f GH synthesis, gene transcription, mRNA or somatotroph
proliferation in rat or bovine pituitary cell cultures (Baringa et al., 1985a,b; Bilestrup et
al., 1986; Fukata et al., 1985; Simard et al., 1986; Tanner et al., 1990). Conversely, SS
could partially attenuate the GHRH-induced increases in somatotroph proliferation and c-
fo s response in the rat (Billestrup et a l, 1986). At the pituitary level, SS was shown to
inhibit spontaneous GH release and GHRH induced GH secretion (Brazeau et ah, 1982a,
Fukata er a/., 1985).
As reported for GHRH, there is a sex-related difference in hypothalamic SS expression,
with males expressing higher SS mRNA and protein levels than females (Argente et a l,
1991; Nurhidayat et a l, 1999). In contrast to GHRH, SS mRNA is down-regulated in
GH deficiency and up-regulated in GH excess. Contrasting intrahypothalamic GHRH
and SRIH expression has been demonstrated in two dwarf rat models with primary
{dw/dw, Charlton et a l , 1988) or secondary (TGR, Flavell et a l , 1996) GH deficiency
with a subcutaneous implant of rat GC cells, which form encapsulated GH secreting
tumours that maintain high rat GH levels for several weeks (Pellegrini et a l, 1997). In
both strains, GC cell tumours stimulated growth. In dw /dw rats, GC cell implants
increased SRIF expression in the PEN, but not in the ARC nucleus and their high GHRH
- 16 -

L 'IiUJ2l}:':l_______________________________________________________________________________________________ I n n in J i ic lK m
expression in ARC was decreased by GC cells. In contrast, GC cell implants in TGR rats
had little effect on the already high SRIF expression in PEN or low GHRH expression in
ARC, although they did reduce SRIF expression the ARC nucleus.
The actions of SS are mediated by a family of G-protein coupled receptors (sstrl-5; for
review see Patel, 1997). All five genes are expressed by the majority o f rat pituitary cell
sub-types, with high levels of sstr4 and 5 in rat somatotrophs and sstr2 in thyrotrophs
(O’Carroll & Kremples, 1995). CNS sstr expression is wide spread, with a particular
pattern for each receptor sub-type (Bruno et al., 1993). The functional role of the
individual sstr sub-types is however unknown. In situ hybridisation showed that all sstr
are expressed within the hypothalamus, sstrl and 2 being particularly abundant (Beaudet
et al., 1995; Breder et al., 1992). sstrl, 2 and 3 have been located in the ARC (Senaris et
al., 1994) and mRNA regulation by GH has been shown for sstrl, implicating its
potential involvement in feedback regulation o f GH secretion (Guo et al., 1996).
Interestingly, sstrl expression was reported to co-localise with SS expressing neurones;
the sstrl might thus act as an auto-receptor (Lanneau et a l , 2000). As described
previously, sstr2 KO mice implicated this sstr in PEN-ARC communication (Zheng et a l,
1997).
Most sstr are coupled to a G^i -protein; ligand binding results in a decrease in adenylate
cyclase, cAMP and PKA levels (for review see Patel et al., 1995). Therefore activation
of sstr causes activation of K^ channels, hyperpolarisation of the membrane and reduction
of intracellular Ca^^ (Sims et al., 1991). This effect is enhanced by the blocking o f Ca "
channels (Ikeda & Schofield, 1989). These opposite effects to GHRH thus cause
inhibition of GH release, even in the presence of GHRH or GH secretagogues.
1.6 Growth hormone releasing peptides (CHRP) and artificial growth hormone
secretagogues (GHS)
Apart from GHRH there is another class of peptides that can release GH with their own
endogenous signalling cascade. In 1990, Bowers and colleagues showed that a synthetic
hexapeptide, called GHRP (growth hormone releasing peptide-6), specifically released
GH and was orally active (Bowers et al., 1980; Bowers et al., 1984). Many different
- 1 7 -

( P sinh 'l^L ___________________________________________________________________________________lu lrotliictioii
synthetic GHRP’s now exist and potent non-peptide secretagogues have been developed
which demonstrate high bioavailability and sustained increase in GH and IGF-1 levels
after a single oral dose (MK-0677 - Patchett et al., 1995). GHRP activity is associated
with increased c-fos immunoreactivity and electrical activity in GHRH containing
neurones in the arcuate nucleus (Dickson et a l, 1995; Dickson & Luckman, 1997).
20 years after the discovery of GHRP’s, an endogenous receptor (GHRP-R/GHS-R) was
cloned (Howard, 1996). The GHS-R belongs to the family of 7 transmembrane looped,
G-protein coupled receptors and is located mainly in the arcuate and ventromedial
nucleus of the hypothalamus (Bennett et a i, 1997), but also in the pituitary (Howard et
al., 1996; Pong et al., 1996). The discovery of the GHS-R suggested the existence of an
endogenous ligand involved in GHS physiology. Finally, an endogenous ligand for the
GHS-R was found and termed ghrelin (Kojima et al., 1999). Ghrelin was a surprising
discovery, as it was shown to be a 27 or 28 amino acid peptide with a bulky octanoylated
SerineS, the first peptide ever to show this kind of post-translational modification,
essential for its GH releasing activity. Also surprising was its pattern of expression, with
very high levels of expression in the stomach and low levels of detectable peptide in the
hypothalamus. GHS are not as specific as first thought: apart from GH release, PRL,
ACTH and adrenal steroid release is stimulated, although in a much smaller magnitude
(Thomas et al., 1997). Several lines of investigation indicate that this effect is exerted
indirectly via the hypothalamus, as hypothalamo-pituitary stalk lesions abolished the
ACTH response to GHS and no ACTH was released in vitro from isolated pituitaries
(Cheng et al., 1993; Schleim et al., 1996). The physiological role o f ghrelin in GH
physiology remains unclear - it may have more to do with metabolic regulation.
1.7 Physiological mechanisms of GH release
1.7.1 Regulation o f pulsatile GH secretion
GH secretion from the pituitary is pulsatile in all species studied so far, including sheep
(Davis et al., 1977), rats (Tannenbaum et al., 1976), mice (Macleod et al., 1991),
hamsters (Borer et al., 1982), guinea pigs (Gabrielsson et al., 1990) and man (Finkelstein
et al., 1972). In man, GH bursts are maximal during slow wave sleep, but secretion is
1 8 -

C 'lhii'li'r / ____ Inirtjclndion
also episodic during the day. This pulsatility is important for regulating growth
(Hindmarsh et al., 1987) and metabolism (Hindmarsh et al., 1997) and requires the co
ordinated interaction of the hypothalamic peptides GHRH and SS. Most mechanistic
studies have been carried out in the rat, since GH secretory rhythm in the rat is extremely
regular and episodic. In the male rat, large GH pulses appear every 3-3.5 hours and
troughs between pulses are very low, often below detection level (Tannenbaum et al.,
1976).
The low levels in pulses are thought to be due to somatostatin release, since passive
immunisation against SRIF results in elevation of baseline levels, with intact intervening
pulses (Terry et al., 1981). Immunisation against GHRH abolishes GH surges and
inhibits growth (Wehrenberg et al., 1984), thus GHRH seems responsible for GH pulses.
However, ability o f somatotrophs to respond to GHRH depends on underlying SRIF
tonus (Clark et al., 1985a); during trough levels (high SRIF tonus), there is a
refractoriness for GHRH. This suggests a cyclic SRIF release, with GH pulses occuring
when SRIF tonus is low. Withdrawal of SRIF results in a rebound GH release (Clark et
al., 1988a). This could partly be due to a build up of GH stores, but is also mediated by
GHRH, since immunisation against GHRH abolishes the rebound release (Clark et al.,
1988a). SRIF is thought to mediate an inhibitory tone on GHRH secretion; immunisation
against SRIF results in increased GHRH levels in the portal circulation, supporting this
concept (Plotsky et al., 1985). It has been shown that the most effective way to promote
growth in GH deficient dwarf dw/dw rats (Charlton et al., 1988) is by infusion of brief,
large GH pulses (Gevers et al., 1996).
1.7.2 Secretion o f GH is sexually dimorphic
Body growth, GH secretory pattern, hepatic GH receptor (GHR), and plasma GH binding
protein (GHBP) levels are all sexually dimorphic in the rat. As previously mentioned,
male rats show regular GH secretory pulses every 3-3.5 hours, with very low trough GH
levels between the secretory bursts (Tannenbaum & Martin, 1976; Edén, 1979), whereas
females show a more continuous GH secretion with frequent, irregular pulses
superimposed on high baseline levels, which rarely fall to undetectable levels (Clark et
al., 1987; Edén, 1979). Both GHRH and SS are necessary for generating the GH pulse
pattern, since studies with anti-serum and antagonists for SS and GHRH have shown that
- 1 9 -

( 'iu ir içr !__________________________________________________________________ In lro d u c lio n
abolishing either one interrupts episodic GH secretion (Baumbach, W.R., Fairhall, K.M.,
Carmignac, D.F. & Robinson, I.C.A.F., unpublished data; Wehrenberg et al., 1982).
Trains of GH pulses will entrain the GH pulse generator in conscious male rats (Carlsson
& Jansson, 1990), whilst continuous exposure to GH will suppress GH secretion (Abe et
al., 1983; Clark et al., 1988b). These models exclude other inputs, i.e. ghrelin and the
GH feedback mechanism, which suggest that the system is in reality, far more complex.
The relationship between peak amplitude, peak frequency, base-line levels of the GH
pattern is far from clear. Most studies have concentrated on the peak height of the GH
pulse and pulse frequency as the principle factors governing the growth response.
However, trough levels appear to be as important, shown in studies by Gevers and co
workers. Continuous infusion of GH, which promotes growth to a lesser extent than
pulsatile exposure o f GH, leads to increased levels of GH receptor and GH-binding
protein levels (Gevers et al., 1996). One explanation which has been put forward is
increased GHBP levels in-between GH pulses could serve as a reservoir to prolong the
time of low-level GH exposure (Baumann et al., 1987). As in rats, GH base-line levels
are higher in women than in men (Winer et al., 1990; Chapman et al., 1994) although the
magnitude is less apparent. Short-term comparisons of continuous vs. pulsatile GH
treatment in man have revealed only minor differences in metabolic parameters
(Jorgensen et a l, 1993; Laursen et al., 1995). However, longer treatment in GH deficient
children shows not only growth, but induction of GHBP after continuous but not pulsatile
GH treatment (Tauber et al., 1993). The exact mechanism that determines the
spontaneous pulse frequency o f GH (and other horm ones) rem ains elusive.
Electrophysiological studies in the hypothalamus might give novel insights into the
regulation o f pulsatile hormone secretion. Dickson et al., have shown that co-ordinated
firing in a population of synchronised hypothalamic ARC cells elicit a pulse of GH
release (Dickson et al., 1993). A similar system has been described for magnocellular
oxytocin neurones (for review see, Leng et al., 1999).
1.7.3 GHfeedback mechan isms
As previously mentioned, a key regulator of GH secretion is GH itself. GH feedback
mechanisms, involved in the control of GH secretion are summarised schematically in
figure 1.5 and described in brief below.
2 0 -

( h a p ie r I IniroJiicliofi
HYPOTHALAMUSGHS and Ghrelin
NPYARC PeVNGHS-R
GHRH + ? SRIF
GH-RGH-R
SRIF-R
GHRH /o SRIF
^ GHRl F-R
ACAC
GHS
PKA PKAGH Production
Somatotroph Proliferation
cAMP cAMPGhrelin
GUT /GHANTERIOR PITUITARY
GH Release
HEART FUNCTION FAT MASSBONE GROWTH MUSCLE MASS
+
Figure 1.5 Regulation of pituitary GH releaseGHRH is produced in neurones in the arcuate nucleus (ARC) and SRIF in the periventricular nucleus (PeVN) o f the hypothalamus. GHRH stimulates the production and release of GH from the somatotroplis in the anterior pituitary, via the GHRH-R and the signal transduction pathway of adenyl cyclase (AC), protein kinase A (PKÂ) and cyclic AMP (cAMP). SRIF binds to the SRlF-R and uses tire same signal transduction pathway as described for GHRH and inhibits GH release, but has flo effect on GH production and somatotroph proliferation. Neuropeptides and neurotransmitters influence the production o f GHRH and SRIF. Tire GHS-R is located on neurons in the ARC nucleus and in the anterior pituitary, where ghrelin and GHS potentially affect GH secretion via these sites. SRIF inhibits GHRH production via the SRIF receptor on the ARC nucleus, forming a negative feedback loop for GH secretion. GH release also results in negative feedback via GH-R. probably invoviing altered production of NPY and galanin there. Not showm is negative feedback regulation by IGF-I. GHS = growth hormone secretagogue; GHS-R = growth hormone secretagogue receptor; GHRH = growth hormone releasing hormone; GHRH-R = growth hormone releasing hormone-receptor; SRIF = somatostatin; SRIF-R = somatostatin-receptor; GH = growth hormone; NPY = neuropeptide Y; AC = adenyl cyclase; PKA = protein kinase A; cAMP = cyclic adenosine-mono-phosphate
-21 -

( [hiJ!>lLr_ L_____________________________ _______ _________________________________I iu ro i l i i r tK in
The feedback mechanisms of GH have been described as (1) a short loop, involving the
control of hypothalamic neuropeptides by GH, and (2) a long loop, involving IGF-1. GH
secretion and probably SRIF and GHRH are under feedback control of GH itself. GH’s
actions are mediated by the GH receptor (GH-R). In the adult CNS, the main sites of
expression are the hypothalamus and hippocampus. The hypothalamic GH-R expression
was found to be in PEN SS neurones and a small percentage o f GHRH neurones in the
ARC (Burton et al., 1995; Minami et al., 1992) but predominantly in the ARC NPY
neurones (Kamegai et al., 1994). In females, the GH response to GHRH is not
diminished during a GH infusion and the negative feedback of GH on its own secretion is
due to an inhibition of GHRH release; in males, GH response is suppressed, as GH
feedback causes SRIF to increase (Clark et al., 1988b; Pellegrini et a l, 1997). However,
GH also down-regulates GHRH-R in the pituitary (Horikawa et a l , 1996). This negative
feedback is shown very clearly in transgenic rats over-expressing GH in the
hypothalamus, which are dwarfed as a consequence (Flavell et a l , 1996). Furthermore,
SRIF and GHRH autoregulate their own secretion: GHRH inhibits its own secretion and
stimulates SRIF, while SRIF inhibits its own secretion.
GH-R’s found in the arcuate nucleus, on NPY neurons (Kamegai et a l, 1996) and in PEN
of the hypothalamus are regulated by GH itself and are mediators of the GH-feedback of
GH secretion (Bennett et a l , 1995). In an in vitro hypothalamus explant system, NPY
decreased GHRH release and increased somatostatin, consistent with NPY’s role as a GH
feedback mediator (Korbonits et a l, 1999). It is interesting to note that NPY expression
is GH sensitive (Chan et a l, 1996), NPY expression being reduced in GH deficiency
(Bennett et a l, 1997). GH in turn is sensitive to NPY, with i.c.v. NPY injections causing
reduced plasma and pituitary GH (Rettori et a l , 1990), but i.v. injections of NPY
increasing GH release (Catzeflis et a l, 1993). In summary current reports suggest that in
addition to a direct feedback of GH onto SS and GHRH neurones, negative GH feedback
is mediated by NPY. However, the GH effects caused by NPY might be primarily in the
regulation of food intake and utilisation rather than growth regulation (Bennett et al,
1999). Hypothalamic GH-R expression itself is also regulated by GH status, being
decreased in GH deficiency and increased in GH treatment (Bennett et a l , 1995).
Physiologically the short-loop GH mechanism becomes apparent in chronic cannulation
- 2 2 -

U j j j I Js 'Jl L_____________________________ _______ ______ ___ ____ ___ ___ _______I n i r o d i i c l i n i ';
experiments in rats, where GH inhibits its own secretion, blocking GH pulsatilty (Abe et
al., 1983; Chihara etal., 1981; Clark e tal., 1988b; Willoughby etal., 1980).
The long-loop feedback mechanism is operated by IGF-I, at least via the pituitary (in
ewes) (Fletcher et a i, 1995) but may also be via the hypothalamus since IGF-receptors
are located in both hypothalamus (Bondy et al., 1992) and pituitary (Goodyer et al.,
1984). IGF-I acts directly on the pituitary to inhibit GH gene expression and GHRH
induced GH release (Yamashita and Melmed, 1986; Ceda et al., 1987). In the
hypothalamus IGF-I has been reported to stimulate somatostatin synthesis and release
(Sato & Frohman, 1993). By all these mechanisms, IGF-I inhibits GH production and
reduces GH exocytosis. Other hormones can affect GH secretion, either by affecting the
GHRH/SRIF system or by affecting GH-transcription through binding to the promoter.
In the rat GH promoter a specific thyroid hormone response element (TRE) is present,
although this is not true for human GH. The retinoic acid receptor (RAR) can also bind
to the TRE and also has its own binding site in the Pit-1 promoter. A glucocorticoid
responsive element is also present in the hGH promoter. Activin inhibits GH
transcription by a decrease in Pit-\ binding to the GH promoter (Bertherat et al., 1995).
Thus, GH production is affected by many endocrine regulators.
1.8 The human growth hormone gene cluster
The human growth hormone (GH) locus contains 5 structurally related genes spanning 47
kb (Barsh et a l, 1983; Chen et a l, 1989) on chromosome 17q22-q24 (George et a l ,
1981), see figure 1.6. The 5 GH genes are all organised in the same transcriptional
orientation and are each composed of 5 exons. These genes display more than 90%
nucleotide sequence homology in their coding and immediate flanking regions (Seeburg,
1982). Despite linkage and homology, the GH genes are expressed in two mutually
exclusive tissue-specific patterns: hGH-N solely in the som atotrophs and
mammosomatotrophs of the anterior pituitary and the hCS-L, hCS-A, hCS-V and hCS-B
specifically in the syncytiotrophoblastic layer of the placenta (MacLeod et a l , 1992; for
review see Cooke & Liebhaber, 1995).
- 2 3

hurndliC'-i}:
SCN4a Gene I 7 q 2 3 .1-25.3
3 2 .5kb
III 1 1 IIII II I I
CD79b Gene Human Growth Horm one Gene ClusterI7q23 !7 q 2 2 -2 43 .5kb 47kb
-► M-P- ^ ►hGH-N CS-L CS-A liGH-V CS-B
t
5.2 9.7kb 6kb 13kb 13kb 6kb
n-54
Skeletal m uscle
-13.2 0 +7.6 +22.2 +36.9 +45.1
I------------------------------------------------------------------------1
B-Lym phocyte Placental Syncytiotrophoblast
Pituitary somatotroph
Figure 1.6 Genomic organisation of the seven genes in the 100-kb SCN4A,
CD79b, hGH cluster locus on human chromosome 17q23.
The top of the diagram contains the names of each of the genes, their independently
determined chromosomal localisation, and the sizes of each of the transcription units,
SCN4A, CD79b, hGH-N. On the next level is the map of the region showing the 7
contiguous genes. The exons are represented as vertical rectangles; 24 exons in SCN4A,
6 exons in CD79b and 5 exons for each gene in the hGH cluster. The distances between
the genes (two headed arrows) represent the distances from the polyadenylation site of
the upstream gene to the transcriptional start site of the downstream gene. The positions,
orientations and tissues of expression for each promoter are noted at the bottom of the
figure (taken from Bennani-Baiti et a l, 1998b). *
- 2 4 -

. _______ In lr o d i i i ■lion
The hGH-N encodes two alternatively spliced mRNAs (DeNoto et a l , 1981) that are
translated into 22- and 20-kDa GH proteins (Lewis et al., 1980). The placental genes
hCS-A and hGH-V are alternatively spliced and predicted to encode 22- and 26-kDa
proteins, while hCS-B encodes a single 22kDa protein product (MacLeod et ah, 1992).
The expression o f an hCS-L protein has not yet been documented, although this gene
produces several alternatively spliced mRNA transcripts in placenta (Misra-Press et al.,
1993). The GH genes are abundantly transcribed; hGH-N mRNA composes 3% of the
pituitary mRNA, while hCS-A and hCS-B constitute 3.5% of placental mRNA.
1.8.1 Physical linkage o f the hGH gene cluster with CD79b and SCN4a
The SCN4a locus was also mapped to chromosome 17 [17q23.1-q25.3] (George et al.,
1993) 21.5kb up stream of the hGH-N gene (Bennani-Baiti et al., 1995). The SCN4a
gene contains 24 exons that span 32.5kb and encodes the 260kDa a-subunit of the human
skeletal muscle voltage gated sodium channel. The hGH locus control region (LCR)
which is explained in more detail in 1.9, contains the major transcriptional control
elements of the pituitary-specific hGH-N gene and spans -14.5 to -31.5 kb 5’ to the hGH
cap site. Surprisingly, two o f the DNAse hypersensitive sites, HS III and IV are
embedded within the muscle specific SCN4a gene also seen in figure 1.6 and 1.7.
An extensive body of data suggests that hGH is also slightly expressed in lymphocytes
and may be involved in important autocrine circuits involved in the immune response.
GH mRNA has been detected in the human thymus, spleen an and lymph nodes (Wu et
al., 1996); GH receptors are expressed on the surface of lymphocytes (Badolato et al.,
1994) and GH has been shown to mediate a GH-receptor-generation o f IGF-1 in
lymphocyte lines (Geffner et a l , 1993). Wood and co-workers mapped the human
CD79b to chromosome 17q23 (Wood et al., 1993). The complete genomic sequence of
the human CD79b was determined by Hashimoto et al. (1994) and was found to contain 6
exons spanning 3.5 kb. During sequence analysis of the LCR between hGH-N gene and
its proximal hypersensitive site (HS I) (Bennani-Baiti et al., 1998a,b) revealed a perfect
match to the B-lymphocyte specific gene CD79b (Igp/B29). The CD79b start site is
located 8.2kb downstream of the SCN4a gene, and its polyadenylation signal lies 9.7kb
- 2 5 -

C J m r i c r / __________________ _ Jniro(-!uc;;on
upstream of the hGH-N transcription start site. HSI and HSII of the hGH LCR are also
located 5’ to the CD79b gene, which can be seen in figure 1.6 and 1.7.
1.9 Locus Control Regions (LCRs)
The genome is largely packaged into inaccessible chromatin conformation. When
eukaryotic genes become transcriptionally active, the structure of the chromatin in their
vicinity changes to accommodate trans-acting factors and the passage o f RNA
polymerase. (Felsenfeld, 1996). Many promoter and enhancer sequences, capable of
driving tissue-specific, high-level expression in eell-transfeetion systems are subject to
site-of-integration or position effects after integration into the genome of transgenic mice.
These effects reflect the influence of neighbouring chromatin on the integrated transgene
which results in failure o f the transgene to be reproducibly and appropriately expressed
(Palmiter & Brinster, 1986).
Genetic evidence demonstrates that cw-aeting sequences remote from a gene promoter
are critical mediators o f tissue-specific changes in chromatin structure that presage the
activation of promoter-proximal elements. In particular, certain limited deletions far
upstream of the (3-globin gene cluster completely inactivate expression o f all genes in the
cluster (Grosveld et al., 1987; reviewed by Dillon & Grosveld, 1993). The chromatin
domain encompassing this inactivated (3-globin gene cluster is DNAse I resistant and late
replicating. Conversely, the normal |3-globin gene cluster in erythroid cells replicates
early in the cell cycle and resides in an open, DNase I-sensitive chromatin domain
(DNAse I hypersensitive-sites) (Forrester et ah, 1990). This suggested that P-globin
expression is mediated by upstream regulatory elements that function to establish an open
chromatin domain essential for subsequent transcriptional activation. Due to their crucial
gene activation function in the p-globin gene cluster, such dominant regulatory elements
have been termed locus control regions (LCR’s) (Grosveld et al., 1987; Forrester et al.,
1990). LCR’s are powerful regulatory elements that are characterised phenotypically by
three major properties. They confer tissue-specific and physiological levels of expression
on linked genes, activate the transcription of transgenes in a position-independent, copy-
number dependent manner, and determine DNA replication timing and the origin utilised
(Grosveld et al., 1987). Thus tissue-specific control of basal transcription might reside in
- 2 6 -

In lnxl la lK il:
hGH LCR I------------------------------------ 1
Pituitar)' V III II II I uPlacenta V IV III
IIISkeletal muscle
lOOkb
CD 79b
Growth Horm one Gene Cluster
i -li-Lym phocyte Pituitary
somatotroph
hGH-N .CS-L CS-A hGH-V CS-B,
Placentalsyncytiotrophoblasts
-40 -30 -20 -10
Figure 1.7 The human growth hormone gene cluster and locus control region.
The growth hormone gene (hGH-N) is shown as a black box. The growth hormone
variant (hGH-V) and the three chorionic somatomammotropin (hCS) genes are shown as
dark grey boxes. The unrelated but closely linked SCN4a and CD79b genes are shown as
white boxes (Bennani-Baiti et a l, 1995, 1998). An arrow above each gene shows its
transcriptional orientation. The postition of the hypersensitive sites (HS I-V) in pituitary
and/or placental chromatin are marked by the downward pointing arrows.
- 27 -

; _ _ _ l iu r o iiiiciion
the promoter, whereas tissue-specific enhancement of gene expression is a property of the
LCR. The identification of locus control regions that were able to overcome position
effects was thus a major discovery. For my purposes, the availability of such sequences
would be powerful tools to establish reliable, physiologically regulated transgene
expression.
1.9.1 hGH gene is controlled by a multi-component locus control region (LCR)
Cooke and Liebhaber have studied the human growth hormone gene cluster intensively,
identifying 4 hypersensitive sites (HS I, II, III and V) located 5 ’ to the hGH gene cluster
that form selectively in pituitary chromatin and together function as an LCR after germ-
line transformation into the mouse genome. As previously mentioned, the hGH-N gene
is the most 5 ’ member of a cluster of 5 structurally related genes sharing greater than
95% sequence identity (Seeburg, 1982). The hGH-N is expressed exclusively in
somatotrophs and mammosomatotrophs o f the anterior pituitary, whereas the other 4
genes -hCS-L, hCS-A, hCS-V and hCS-B- are expressed in the syncytiotrophoblastic
layer o f the placenta (MacLeod et al., 1992). This element must also allow for
appropriate silencing of CD79b in pituitary cells, and GH in lymphocytes.
Cw-acting regulatory elements adjacent to both hGH-N and hCS, defined in cell
transfection assays are unable to consistently direct appropriate expression in vivo.
Transcription of the transfected hGH-N in rat pituitary GH3 cells and GC cells is
dependent on promoter proximal elements including a single site for Zn-16 (Lipkin et al.,
1993) and 2 sites for Pit-1 (Ingraham et al., 1988). When tested for expression in
transgenic mice, hGH-N directed by its proximal promoter, is either not expressed or
only poorly expressed, in transgenic pituitaries (Palmiter and Brinster, 1986). Likewise,
an hCS-transgene including all defined control elements, is shown to be expressed poorly
in the mouse placenta (Jones et a l, 1995). The sporadic expression of hGH-N and hCS
transgenes suggested that additional regulatory elements required for reproducible, high-
level expression were lacking.
The DNAse mapping studies carried out in Cooke and Liebhaber’s lab revealed two
DNAse I hypersensitive sites at co-ordinates -32.5 and -27.5 which were common to
- 2 8 -

( _ _ _ _ I n i m d u c l i o i i
placenta and pituitary and a third placenta-specific HS at co-ordinate -30.5. Two
additional HS specific to the pituitary at Kb -14.6 and -15.4 were also detected. A
summary of these sites can be seen in figure 1.7. Functional analyses of these regulatory
elements in transgenic mice demonstrated that inclusion o f 40Kb o f contiguous 5’-
flanking regions resulted in reproducible, copy-number dependent, and pituitary-specific
expression of hGH-N at levels that were comparable to those of endogenous mOH (Jones
et al., 1995).
The gene proximal set of HS (HS I & II [-22.5hOH]) contained potent enhancer activity
in the pituitary although there was evidence for loss of tissue-specificity which was not
present with the addition of HS III and V [-40hGH]. Within the set o f HS I & II, multiple
binding sites for the pituitary homeodomain transcription factor Pit-1 have been
identified (Shewchuk et al., 1999). Using transgenic studies, these sites were shown to
be required for high level, position-independent, and somatotroph-specific expression of
a linked hGH-N transgene. However, the Pit-1 sites alone are insufficient for gene
activation in vivo and appear to play a unique chromatin-mediated developmental role in
the hGH-N LCR (Shewchuk et a l , 1999). The ability of the HS III and V region to
overcome chromatin position effects suggests it may function to establish site-of-
integration independence.
1.10 The tertiary structure of Growth Hormone and its Receptor (GH-R), Growth
Hormone Binding Proteins (GHBP) and Insulin-like Growth Factors (IGF)
1.10.1 The tertiary structure o f human growth hormone (hGH)
Human growth hormone is a member of a family of homologous hormones that includes
the placental lactogens, prolactins, and other genetic and species variants of GH. Among
these, hGH alone exhibits broad species specificity and binds monomerically to either the
clones somatogenic liver (Leung et al., 1987) or prolactin receptor (Boutin et al., 1988).
The gene for human GH has been expressed in a secreted from in E.Coli, and the three
dimensional folding pattern for hGH has been resolved (Cunningham et al., 1990a) based
upon the x-ray structure of porcine GH (Abdel-Meguid et al., 1987).
2 9 -

NH
H2
Z n t
Figure 1.8 Tertiary structure of human growth hormone and zinc binding sites
Location and structure of zinc binding site. Folding model for hGH based upon a 2.8Â
structure for porcine GH that shows the putative location of the zinc ligands (solid black
circles) and binding regions for the extracellular domains of the hPRL (dashed eclipse)
and hGH (large solid circle) receptors. S-S represent disulphide bridges.
(Cunningham etal., 1991).
- 3 0 -

__________ I n l r o d u c l i o n
The crystal structure o f hGH and the extracellular domain o f its receptor have been
characterised by Ultsch and de Vos, (1992). The major structural feature of human
growth hormone molecule is a four helical bundle, with unusual connectivity (figure 1.8);
the helices run up-up-down-down, in contrast to the more usual up-down-up-down case.
The NH2- and COOH- terminal helices (helices 1 and 4) are longer than the other two (26
and 30 residues compared to 21 and 23 residues), and helix 2 is kinked at Pro^^. A long
crossover connection, consisting of residues 35 to 71, links helix 1 to helix 2, and a
similar connection (residues 129 to 154) is found between helices 3 and 4. The first
connection is disulphide-bonded to helix 4 through Cys^^ and Cys'^^. In contrast, helix 2
is linked to helix 3 by a much shorter segment (residues 93 to 105).
In addition to the four helices in the core, three much shorter segments o f helix are found
in the connecting loops: one each at the beginning and end o f the connection between
helices 1 and 2 (residues 38 to 47 and 64 to 70, respectively) and one in the shorter
connection between helices 2 and 3 (residues 94 to 100). The NH 2-terminal eight
residues extend away from the remainder of the molecule whereas the COOH-terminus is
linked to helix 4 with a disulphide bond between Cys^^^ and Cys**^. The core of the four
helix bundle is made up of mostly hydrophobic residues and is tightly packed, therefore
mutations in such buried positions are generally destabilising (Cunningham et al.,
1989a,b).
Furthermore, scanning mutational analysis performed by Cunningham and colleagues
identified three residues in hGH (His’*, His^’ and Glu’ '’) which form a plausible site for
the binding o f Zn^^. These three residues are clustered when mapped upon a model of
hGH (Cunningham et al., 1989b; Cunningham et al., 1990a) which can also be seen in
figure 1.8. Cunningham and colleagues performed size exclusion chromatography and
sedimentation equilibrium studies, demonstrating that the zinc ion (Zn^^) forms a 1:1
complex with hGH. Binding of one Zn^^ ion promotes the binding of another Zn " ion
(positive cooperativity) and induced the dimérisation o f human growth hormone
(Cunningham et a l, 1991a). This subject is important to this thesis and is discussed in
more detail in 1.12 and chapters 5 and 6.
- 31 -

[ iLUl li ' t l 'LJ- _______________________________________________________________________ I n H i J i l n c h o i i
1.10.2 The Growth hormone receptor and GH signal transduction
The rabbit and human growth hormone receptor were isolated from liver and its sequence
deduced in 1987 (Leung et a l , 1987); its genomic organisation was resolved in 1989
(Godowski et a l , 1989). The GH-R is a 620aa protein with an extracellular domain
(246aa), a single transmembrane domain (24aa) and an intracellular domain (350aa)
without a tyrosine kinase domain. It is a member of the haematopoietic or cytokine/GH-
R/PRL-R superfamily (Bazan, 1989), that also includes the receptors for erythropoietin,
interleukins 2-7, IL-9, granulocyte-macrophage colony stimulating factor, granulocyte
colony stimulating factor, myeloproliferative leukaemia virus, ciliary neutrotropic factor,
leukocyte inhibitory factor, prolactin and interferon-alpha and -gamma. All family
members have a pair of cysteines (Fuh et al., 1990) in the N-terminal region of the
extracellular part, and all receptors except the GH-R, have a WSXWS motif (Kelly et al.,
1993). The cysteine pairs are involved in ligand binding (Bass et al., 1991). Although
the WSXWS box does not touch the ligand, mutations in this box decrease ligand binding
in cases o f IL-2, erythropoietin and prolactin (Wells et al., 1993). In GH-R/BP the
WSXWS is replaced by YGEFS (Wells et al., 1993). A conserved proline rich region
(box 1) is present in the GH-R and is required for association with the protein kinase
Janus kinase 2 (JAK2) (Tanner et al., 1995).
hGH-R is encoded by an 87 Kb spanning region on the long arm of chromosome 5.
Exons 2 through 7 encode for the extracellular domain, exon 8 for the transmembrane
domain and exons 9 and 10 for the cytoplasmic region (Barton et al., 1989). Exon 1 is
not translated (5’ untranslated region). Several exon 1 variants exist (Leung et a l, 1987)
of which some might be tissue-specific (Baumbach et al., 1995; Southard et a l , 1995)
and which may be regulated differentially (Gabrielsson et a l, 1995). A few years before
the cloning of GH-R, Herington and colleagues found that GH was bound to a binding
protein in plasma (Herington et a l , 1986). With the cloning of GH-R, it became clear
that GHBP is the soluble extracellular part of the GH-R (Leung et a l , 1987). The
existence o f a soluble receptor is not unique to GH-R; other members of the haemopoietic
receptor family have long membrane-spanning and short soluble forms, while for some
members, even short membrane anchored forms exist (Carlsson et a l , 1991).
- 3 2 -

( ’hcipU'i- I________ _ _ _ In ir( j ( / i (c f ion
To fully understand GH-GH-R interactions, we have been enormously helped by the
knowledge of the 3-dimensional structure of the complex (seen in figure 1.9) and the sites
important for binding. With the production of recombinant GH and GHBP (Fuh et al.,
1990), the group at Genentech had the tools to resolve the GH-GHBP structure (Ultsch &
de Vos et a l, 1992). Initial GH/GH-R interaction studies by Cunningham and co-workers
using a strategy termed homolog-scanning mutagenesis (systematic replacement of
segments of hGH with sequences derived from non-binding GH homologs) defined a
binding patch on a structural model of hGH that included the NH 2-terminal portion of
helix 1, a loop between residues 54 and 74 and the COOH-terminal portion of helix 4.
These disruptive mutations were found to not only remove favourable interactions but
introduce unfavourable ones. This was the case with the N12R mutation in the hPRL
homolog, which not only changed the hydrogen bonding amide function of Asn*^, but the
Arg substitution introduced a bulkier side chain that was positively charged (Cunningham
et al., 1989b). This analysis provided a general outline of the receptor binding site, but
did not identify the specific residues involved in receptor binding.
Alanine scanning mutagenesis (replacement of residues encompassed in the binding
patch with alanine), was used to identify specific side chains in hGH that strongly
modulate binding to the hGH receptor. The results o f the alanine scan were consistent
with those from the homolog scan in showing that the middle and COOH-terminal
segments are more important in binding than the NH^-terminal segment. The most
disruptive alanine substitutions form a patch confined to three and a half turns of helix 4,
which appear to be facing in the same direction in the model o f hGH. Alanine scanning
also identified at least one side chain in hGH (E l74A) that disrupted binding to the hGH
receptor. Interestingly, this glutamate is thought to play an important role in zinc
dimérisation o f hGH (Cunningham et al., 1989a).
There is only one binding domain in the receptor/BP, consisting of-lOOaa, including the
four cysteines (Bass et al., 1991). Im m unoprécipitation and gel filtration
chromatography together with fluorescent “homoquenching” experiments suggested that
one molecule o f GH binds to 2 GHBP’s. This homodimerisation o f two receptors after
GH binding suggests receptor dimérisation is required for signal transduction.
Crystallography o f the hGH-hGHBP complex confirmed a GH-GHBP2 complex.
- 3 3 -

I
-/ J
Figure 1.9 Back-bone structure of the hGH»(hGHbp)2 complex
The hormone is shown as yellow cylinders representing the helices connected by red
tubes. The ^-strands of the binding proteins are shown in brown, the loops are green
(hGHbp I) and blue (hOHbp 11). The viewing direction is approximately down the four
helix bundle of hGH. In this orientation, the COOH-termini of the extracellular domains
and therefore the cell membrane, are at the bottom (de Vos et a l, 1992).
- 3 4 -

. . _ ____________________________
Binding o f GH to GH-R/BP occurs sequentially: the first GHBP is bound to site 1 of GH,
after which a second GHBP binds to site 2 (Cunningham et aL, 1991b). This is
represented in figure 1.10. Human GH can also bind the prolactin receptor (in a 1:1
stoichiometry) (Somers et al., 1994) but with lower affinity and requiring Zn^^
(Cunningham et a i , 1990a; Cunningham et aL, 1991b). Prolactin cannot bind to the GH-
R (Cunningham et aL, 1990b).
Such a sequential dimérisation model predicts self-antagonism of high dose GH and this
was indeed proven in GH-R containing cells, showing a typical bell-shaped dose-
response curve: at high concentrations, all receptor molecules are occupied by site 1 of
the GH molecules and there are no receptors available for dimérisation (see figure 1.10)
(Fuh et aL, 1992; Silva et aL, 1993). Human GH mutants with a non-functional site 2
(Takahashi et aL, 1996a,b) which renders dimérisation impossible but occupy receptors
due to site 1 binding, are potent antagonists of GH in GH-R expressing cell assays (Fuh
et aL, 1992) and are being used clinically. Thus receptor dimérisation is essential for
signal transduction. This is not the only case in humans (Duquesnoy et aL, 1994), but
also in rats, at least in adipocytes (Silva et aL, 1993; Hondo et aL, 1994).
Upon GH-R homodimerisation, the cytoplasmic tyrosine kinase JAK2 becomes
phosphorylated and is recruited to the receptor which in turn is phosphorylated. Tyrosine
phosphorylation o f STAT’s (Signal Transducers and Activators of Transcription) leads to
STAT activation of DNA binding activity and initiation of transcription o f immediate
early genes like c-fos, c-jun and the serine protease inhibitor S p il .l (Argetsinger et aL,
1993; Gronowski et aL, 1994; Horseman et aL, 1994; Gouilleux et aL, 1995). These in
turn can bind and activate promoters of numerous genes. We now know that also other
signal transduction pathways are activated upon GH-R dimérisation: the MAP-kinase
pathway is activated, both via JAK2 - small G-proteins (Grb, Sos, Ras, Raf) and via
diacylglycerol - protein kinase C; furthermore IRS-1 is phosphorylated and Ca^^-influx is
increased after GH-R dimérisation (Postel-Vinay et aL, 1996).
1.10.3 Growth hormone binding protein
Human plasma contains two GH binding proteins: a low affinity binding protein, not
- 3 5 -

h u r o J i u ' l i o n
GHGHBP
GH-R
I
Figure 1.10 Sequential dimérisation model
In the circulation some GH is bound to GHBP. On finding a GH-R, GH binds via site 1
to the receptor: GH then binds via site 2 to a second receptor, so that homodimerisation
o f both receptors occurs, which is the start for signal transduction. In the case of GH
mutants with disrupted site 2, or theoretically high GH levels, receptor dimérisation will
not occur. Redrawn from Fuh & Cunningham, 1992
- 3 6 -

( 'lliinli/r I InirocitidK'ti:
related to the GH-R (Baumann et aL, 1990b) and the high affinity, principal GHBP,
which is the soluble extracellular part of the GH-R (Leung et a l , 1987). The low affinity
binding protein has been suggested to be a transformed a 2-macroglobulin (Kratzsh et aL,
1995) which may bind preferentially the 20kDa GH variant, which is described in more
detail in 1.11. The high affinity GHBP is a 50-60 kDa glycoprotein. The glycosylation
accounts for about half of the molecular mass, but is not required for binding to GH (Fuh
et aL, 1990). It binds 22kDa GH with a higher affinity than 20kDa (Hansen et aL, 1993).
22kDa GH binds with the same affinity to GHBP as to the GH-R. The Kd is 1-3 xlO'^M
for 22kDa GH and its capacity is approximately 20ng hGH/ml plasma (Baumann et aL,
1996; Amit et aL, 1990). Plasma levels of GHBP are low (nanomolar levels) so
circulating GH might mostly be bound in a 1:1 complex, whereas at the cell surface
(relatively) high concentrations of GH-R exist, making 2:1 complex-formation possible,
required for cell signalling (Baumann et aL, 1994).
In rodents, GHBP has in the position of the transmembrane domain of the GH-R, a 17aa
hydrophilic tail (figure 1.11) (Baumbach et aL, 1989), encoded by an alternative exon 8A
(Zhou et aL, 1994) and GHBP arises from alternative splicing (Linzer & Talamantes,
1985; Baumbach et aL, 1989) in rodents.
Figure 1.11 Schematic representation of the GHR and GHBP
extracell t.m intracell
"tail"rodent GHBP
GHR
human GHBP
GHR consists o f an intracellular, lipophilic transmembrane (t.m) and extracellular part. Rodent GHBP
arise through alternative splicing o f the GHR gene and consists o f the same extracellular part as GHR and
an additional hydrophilic “tail” o f 17aa. Human GHBP arises at least partly, from proteolytic cleavage and
only consists o f the extracellular part
- 3 7 -

( ' ' A r V i v - / ________ _ ______________ _____________________________ ___________ ______ ___________________________________________________________ I n i r i j J u L-lioii
In rabbit and human, GHBP does not have such a hydrophilic tail and only one form of
mRNA has been found suggesting that alternative splicing is not the mechanism of
GHBP production in humans. GHBP seems to arise mostly from proteolytic cleavage of
the receptor in human (Sotiropoulos et aL, 1993).
The function of the GHBP is still not clear. It could play a role to transport GH to target
tissues (figure 1.10). GHBP could also function as a reservoir o f GH: it delays the
clearance of GH, probably because the GHBP complex is too big for renal clearance and
GHBP keeps GH longer in the circulation (Baumann et aL, 1987; Fairhall et aL, 1992).
Probably based on this increased half life o f GH, in vivo experiments indeed showed that
co-treatment with GHBP of GH treated rats enhanced body weight gain and tibial growth
(Clark et aL, 1991, Clark et aL, 1996). GHBP could also be involved in the local
regulation of GH sensitivity of peripheral tissues, since locally GHBP can compete with
GH-R for binding to GH (Lim et aL, 1990; Mannor et aL, 1991; Wang et aL, 1993;
Dattani et aL, 1994); in this way, GHBP inhibits GH’s action. GHBP could also dampen
the peaks and increase the troughs of the GH-profile and thereby modify the pattern of
GH exposure (Veldhuis et aL, 1993). GHBP infusions have shown to be able to alter
GH-pattern dependent hepatic cytochrome gene expression (Wells et aL, 1994).
Furthermore, there is evidence that GHBP is transported to the nucleus where it could
function as a transcription factor (Lobie et aL, 1990; Lobie et aL, 1991; Lobie et aL,
1994).
1.10.4 G H ’s action in growth: Insulin-like Growth Factors (IGF’s)
One of the main effects of GH is the hepatic production and secretion of insulin-like
growth factors (IGF-I and IGF-II) mediated by hepatic GH-R. The IGF’s have a similar
structure as insulin and therefore members o f the insulin family of proteins and can also
have insulin like effects. Traditionally, IGF’s were believed to have primarily an
endocrine role to stimulate longitudinal growth, but it is now believed that at least IGF-I
affects many cells in auto- and paracrine fashions.
Whether GH exerts its growth promoting action directly on the growth plate or via
hepatic IGF-I production is still controversial. The somatomedin hypothesis of Salmon
- 3 8 -

( hunwr I ______ _ _ ______ __________ ______
and Daughday proposes that longitudinal growth is mediated through the circulating
insulin-like growth factors (Salmon et aL, 1997). Isaksson and colleagues on the other
hand favoured Green’s dual effector theory o f GH’s action (Green et aL, 1985)
postulating a direct effect of GH on differentiation of stem cells, inducing local IGF-I
production that in turn stimulates proliferation of these cells (Isaksson et aL, 1987). The
importance of both IGF’s for prenatal growth is proven by KO mice models in which
IGF-I and -I I and /or IGF-I-R are mutated and all show severe growth retardation. In
postnatal growth stimulation IGF-I seems not as efficient as GH (Skottner et aL, 1987).
Treatment with IGF-II has no effect (Schoenle et aL, 1985) and similarly in Snell dwarf
mice over-expressing IGF-II does not increase growth (Van Buul-Offers et aL, 1995)
indicating that systemic IGF-II has only a minor role, if at all in postnatal growth.
1.11 GH-N gene product variants and isoforms
Human growth hormone consists of multiple molecular variants. Numerous factors
contribute to GH multiplicity, including multiple genes, multiple post-transcriptional
pathways, protein-protein interaction (aggregation or polymerisation), binding to at least
two binding proteins in blood and fragment generation. The interest in polypeptide
hormone heterogeneity was stimulated in the early 1970’s. Several factors appeared to
contribute to this: first, the discovery of pro-insulin (Steiner & Oyer, 1967) and the
finding o f “big insulin” in plasma (Roth et al., 1968) suggested that molecular
heterogeneity is a physiological phenomenon. These observations were extended to other
peptide hormones, with the result that virtually all were found to exist in more than one
molecular form (Yalow, 1974). In the case of GH, the “little”, “big”, “big-big” (or “pre
big”) GH forms in the pituitary corresponded to similar molecular size variants in plasma
(Goodman et aL, 1972; Gordon et aL, 1973). Second, interest in GH heterogeneity was
stimulated by the observation that some of the acidic GH forms in pituitary extracts might
have several fold higher biological activity than pure 22KDa GH (Singh et aL, 1974),
suggesting that they may be of physiological significance. Third, the concept that certain
activities o f GH (e.g. growth promoting and diabetogenic activity) could be dissociated
from each other received support from the observations that the two activities could be
physically separated, and that diabetogenic activity could be generated by proteolytic
digestion o f GH (Lewis et aL, 1974). More recently, the issue of GH heterogeneity has
- 3 9 -

* -'u/b'''I - - . -----.............. - - __ _______ ___________
received further refinement through the detailed study o f the GH gene locus, the
production o f biosynthetic recombinant GH, the discovery of placental GH and the
discovery of GH binding proteins.
As previously mentioned, the human genome contains 2 GH genes, GH-N and GH-V,
clustered on chromosome 17 together with the highly homologous placental lactogen
(hPrl) genes (Chen et aL, 1989). The work described in this thesis has focused on the
GH-N gene and the mature mRNAs that are produced from distinct splicing sites within
the GH-N transcript, shown in figure 1.12.
Figure 1.12 GH-N mRNA Splicing
Transcribe, cap, polyadenylate
nForm lariats
B\
Splice
t ^-26
A I
2 l 3
L /u 3’
AAAAA.
-26
-26
sp
sp
- 1 1
-1 1 32 46
191
191
22K, 191 aa, hGH
20K, 176aa, hGH
Transcripts o f the hGH-N gene are spliced into two different mature mRNAs coding for 22kDa and 20kDa GH respectively. The 22K splicing site is preferentially used and both are produced in the pituitary. 20K differs from 22K by an internal deletion o f a 15 aa sequence corresponding residues 32-46. sp = signal peptide.
- 4 0 -

' .. , _ _ ____________ . Jniruchicnon
1.11.1 22KDahGH
Pre-GH/pre-somatotrophin is produced from exons 1-5 as a 217 amino acid protein. The
first 26 amino acids serve as a signal peptide and are enzymatically cleaved to produce
the mature 191 amino acid, 22kDa GH molecule. This 191 amino acid, 22kDa GH (22K)
is the most abundant product of the hGH-N gene transcript (70-75%). The monomeric
22KDa GH is made from the “regular” splice site at the transition between intron B and
exon 3. It contains four cysteines that form two disulphide bridges (Cys^^-Cys^^^ and1QO 1QÛ
Cys -Cys ), with two corresponding loops, one large and one small (Li & Dixon,
1971). This overall structure is highly conserved among mammalian GHs. The tertiary
structure has been determined for human GH based on a 2.8Â structure of porcine GH
(de Vos et a l, 1992). Crystal structure of hGH has been defined at 2Â resolution (Ultsch
et a l, 1994) and it corresponds to a twisted bundle of four a-helices in anti-parallel
arrangement (figure 1.8 and 1.9), connected by flexible extended segments that are
involved in receptor and antibody binding (Abdel-Meguid et a l , 1987).
1.11.2 20KDahGH
A secondary product of the GH-N gene is the 20,000-dalton variant (20K) [seen in fig
1.12], which arises from alternative pre-mRNA splicing at a site within exon 3 (Chapman
et a l, 1981; DeNoto et a l, 1981). It is a single chain 176 amino acid protein, similar to
22K except for an internal deletion of amino acids 32-46, with a molecular weight of
20,269 (Lewis et a l , 1980). It is the second most abundant GH in the pituitary (5-10%).
20K has a propensity to dimerise and forms both homodimers and heterodimers with 22K
(Lewis et a l, 1980; Chapman et a l , 1981) although it has substantially reduced affinity
for GH receptors from various species when compared to 22K (Smal et a l , 1985; Closset
et a l, 1983). Human plasma does contain a binding protein (low affinity hGHBP) that
binds specifically to 20K, but there is no known relationship between this binding protein
and a tissue specific receptor (Baumann & Shaw, 1990a). Despite the low affinity of
20K for GH receptors, its growth promoting activity, IGF-generating, and lactogenic
activity in rats in vivo is similar to 22K (Kostyo et a l , 1987; Cameron et a l , 1988),
attributed to its slower metabolic clearance and longer biological persistence in vivo
(Baumann et a l , 1985). The 20kDa form is more diabetogenic than the 22kDa form
(Culler er a/., 1988).
- 4 1 -

( j 'iiC 'L iT y ______ l / i l r o c h ic l io n
1.11.3 17.5KDahGHorhGH-IVS3
The other potential alternative splicing product is the 17.5kDa, 151 amino acid hGH form
that lacks residues 32 to 71, predicted from an mRNA that completely lacks exon 3
(Lecomte et al., 1987). This is thought to arise in a specific form of GHD, although it is
presently not known if the protein exists. Patients suffering from IGHD-II respond well
to GH therapy, without the development of GH antibodies, suggesting the presence of
some GH protein (Cogan et a l , 1994; Cogan et aL, 1995; Phillips III & Cogan, 1994;
Hayashi et al., 1999a; Binder et aL, 2001). This exon 3 skip is more commonly known as
the hGH-IVS3 dominant-negative mutation and has been reported to cause severe
isolated growth hormone deficiency (IGHD-type II). This is the subject of the work
described in this thesis and is therefore described in more detail in 1.13.
1.12 Protein hormone storage in secretory granules and the mechanisms for
storage and sorting
Protein secretion is a basic function of all animal cells. In the constitutive secretory
pathway, which is common to all cells, proteins are continuously secreted without
intracellular storage. In the regulated secretory pathway, which is present in addition to
the constitutive secretory pathway, in exocrine cells, endocrine cells, mast cells and
neurones, a subset of secretory proteins is sorted from the constitutive secretory proteins
to highly specialised storage organelles called secretory granules (Tooze & Huttner,
1990). Cells retain the secretory vesicles until stimulated, when they release their
contents through exocytosis, so that large amounts o f hormones may be rapidly available
when needed. Concentration of hormones in granules is extensive, GH is 200 times more
concentrated in the dense cores of secretory granules than in the lumen of the
endoplasmic reticulum (Dannies, 1999). Morphological studies have revealed several
steps in the formation of secretory granules, summarised simply as follows: Proteins in
this pathway are synthesised on polysomes attached to the endoplasmic reticulum and
pass through its membranes into its lumen. Vesicular or tubular structures transport
proteins from there to the cw-Golgi region, and the proteins pass through the stacks of the
Golgi complex to the trans-Go\g\ side. The formation o f a dense-core aggregates
concentrates specific proteins in the /rara-Golgi network (TGN), which are enveloped by
- 4 2

. . . „ ________________ J n i r o < l u d i o n
membrane and bud off from the TGN which yields a dense-core immature secretory
granule (ISO). Using specific marker molecules present in the TGN in a cell-free system,
Tooze & Huttner demonstrated that constitutive and regulated secretory proteins are
already sorted upon exit from the TGN. The formation of ISGs from the TGN occurs
rapidly and these too, are subject to maturation thus are a vesicular intermediate en route
to mature secretory granules.
1.12.1 Sorting o f soluble proteins
How cells secrete products, is a major unanswered question in cell biology. Models have
been proposed based on the possibility of a sorting signal to direct or retain proteins in
secretory granules and on the possibility that aggregation o f hormones serves both a
concentration and a sorting function. It is not even clear whether there is any sorting
process, other than aggregation. The best characterised system o f sorting soluble proteins
is that of lysosomal hydrolases, which are separated from other proteins after transport
through the Golgi complex. This is thought to be the mechanism of sorting and transport
for all secretory proteins (Dannies, 1999) including growth hormone and is summarised
in simplified form here.
Proteins in the secretory pathway proceed from the cw-Golgi to the trans-Go\g\ side of
this complex and in the outermost layer called the trans-GoXgi network (TGN), proteins
are sorted and carried to different locations in the cell (Keller & Simons, 1997).
Lysosomal hydolases have mannose-6 -phosphate goups attached to their carbohydrate
moieties, which are recognised by a transmembrane protein, the mannose-6 -phosphate
receptor. Lysosomal hydrolases bind to this transmembrane protein in the lumen of the
Golgi cistemae, and a protein called adapter protein 1 (AP-1) binds to this protein on the
cytosolic side (Schmid, 1997; Dannies, 1999). Clathrin, a component o f a caged
structure around certain small membrane vesicles, binds to AP-1, and the membrane in
this area begins to invaginate (Schmid, 1997). The invagination buds off into a separate
vesicle through an active process that involves dynamin (Jones et aL, 1998) and the
vesicle carries the enclosed lysosomal hydrolases to endosomes. These two processes,
sorting and transport are linked, because the mannose-6 -phospate receptor that binds the
vesicular cargo of lysosmal enzymes on the lumen side also binds the proteins necessary
- 4 3

_________ ______________________ _______________________________hiiroclHdion
to form the vesicle on the cytosolic side. AP-1 links clathrin to transmembrane proteins
and the mannose-6 -phosphate receptor links specific soluble proteins to part o f the
machinery necessary for forming vesicles.
Clathrin-coated vesicles are only involved in certain phases o f transport, and additional
vesicle classes have been characterised. Vesicles with completely different coats
designated COPII (coat protein complex II) transport proteins from the ER to the region
where the cis-Golgi layer forms, and vesicles with coats designated COPI transport
vesicles back from the Golgi complex to the ER as well as through the Golgi cistemae
(Salama & Schekman, 1995; Lewis & Pelham 1996; Scales et aL, 1997).
In neuroendocrine cells, clathrin-coated patches occur on parts o f the TGN where dense
cores of secretory granules form and on parts of the membrane of immature secretory
granules (ISG) (Orci et aL, 1987c; Steiner et aL, 1987). ISG contain lysosomal enzymes
that are removed as granules mature (Kuliawat & Arvan, 1996) and clathrin-coated
vesicles may remove lysosomal enzymes from ISGs as well as from the TGN. Sorting of
these enzymes from immature secretory granules uses the same system involving the
mannose-6 -phosphate receptor as else where in the TGN (Kuliawat & Arvan, 1996).
It has been shown that sorting linked to transport also occurs earlier in the endoplasmic
reticulum (Pelham, 1995). Soluble chaperone proteins in the ER that assist in protein
folding are retained there or are returned to that location if they do leave (Gething &
Sambrook, 1992). Retention usually occurs through a KDEL sequence on the retained
proteins, and the initial view was that soluble proteins are passively carried from the ER
unless they are actively retained by this sequence (seen in figure 1.13). A more recent
idea is that sorting not only occurs by retention, but also that proteins not meant to stay in
the ER may be selected and actively transported from there. It has been known for
sometime, that growth hormone is proteolytically cleaved from its signal peptide and
must be folded correctly to leave the ER and proceed further along the secretory pathway
(Blobel, 1975). Mutations within secreted proteins resulting in disruption o f folding
would consequently lead to their retention in the ER, activate an unfolded protein
response and removal for degradation. There is now an alternate interpretation: that a
sorting signal necessary for transport out o f the ER may have been affected.
- 4 4 -

Membr
Lumen
1Protein — >
2 I Chaperones Correct folding ^and assembly ^
~ ^ A Mutations^ or stress
\ 8 .
Mlsfolding and misassembly
Dissociation of chaperons from proteins
6Accumulation of proteins bound to chaperons
Synthesis of new
chaperons
Vesiculartransport
Degradation
Golgi a(#aratus
Figure 1.13 The Endoplasmic Reticlulum (ER) and protein folding
During synthesis, secretory and membrane proteins are co-translationally translocated into the lumen of the
ER through an aqueous gated channel (1). They bind to molecular chaperones (2) and begin the folding
process, which is facilitated by chaperones and by folding an processing enzymes (3). After completion o f
folding and other post-translational modifications (5), the proteins (7) dissociate from the chaperones (6)
and are transported to the Golgi apparatus. When folding or assembly o f proteins is not completed
successfully (4) i.e. if the protein structure is altered by a mutation or stress, the mis-folded proteins, bound
to the chaperones, are retained in the ER (8). This retention also signals the synthesis o f new chaperones
(12) and in many cases, helps the cell to survive the stress by ensuring prompt disposal o f abnormal
proteins. Misfolded proteins are transported out of the ER to the cytoplasm, by way o f protein-conducting
channels (9 and 10); they are then degraded in the cytoplasm (11) X indicates blocking o f the process o f
vesicluar transport (reproduced from Kuznetsov & Nigam, 1998).
- 4 5 -

C ü l i j j A 'L À . . ____ . .. . . ______________________________ J i i l r o d i i c l i o n
Prosequences of secretory proteins have been shown to facilitate transport from the ER
(Mains et aL, 1995). Such facilitation could be caused because the sequence increases
the rate of correct folding, allows the formation of oligomers, or reacts with adaptor
proteins to increase active transport from the ER. Evidence that some sorting may occur
earlier comes from the work of Ladinsky et aL, (1994), who found that individual regions
of the TGN with no tubular connections to each other, produced vesicles of only one
type, either clathrin-coated or not. In vesicular transport, different coats imply different
cargos.
1.12.2 Functioning o f the Golgi complex and formation o f secretory granules
Two models for transport through the Golgi-complex have been described. The first is
the vesicular transport model, in which proteins in the secretory pathway are carried
forward from one layer o f the Golgi complex to the next by COPI coated vesicles
(Rothman, 1994; Orci et aL, 1997a). The second is a directed maturation model, in
which the Golgi stacks are made of progressively maturing, discontinuous compartments
with no forward transport by vesicles. These models can be seen in figure 1.14. The
directed maturation model for protein traffic through the Golgi-complex may have
implications for sorting. It has become clear that secretory granules within a single
endocrine cell are not always homogeneous. Somatomammotrophs have three different
types o f granules that contain either prolactin, growth hormone or secretogranins (Reaves
& Dannies, 1991). Separation occurs in the trans-Go\g\ and has been attributed to
preferential self-aggregation or different susceptibility of aggregation o f hormones to
factors such as the change in pH that occurs in moving from the ER to the TGN
(Hashimoto et aL, 1987). If this second directed maturation model is correct, it is easier
to envisage mechanisms by which separate sorting mechanisms for transport from the ER
might result in the kind of segregation in the trans-go\g\ region in somatotrophs. In
addition, if the first model of vesicular traffic forward through the Golgi-complex occurs,
then aggregation of proteins that will be concentrated in secretory granules must be
restricted to a size that will fit the small COPI-coated vesicles, until the last layer is
reached. In the directed maturation model, no such restriction applies and formation of
large aggregates may begin earlier.
- 46 -

COPI?COPI
CGN
VTC
COPII
B
COPI _
f CO PIIOqO Oq O
Figure 1.14 Two models o f transport through the Golgi complex
A V esicular transport m odel. COPII ves ic les transport proteins from the endoplasm ic reticulum
(ER ) to vesicular-tubular clusters (V T C ) and the c/^-G olgi network (C G N ). COPI v es ic les
retrieve proteins and membrane back from each layer o f the G olgi com plex, cis, m edial and trans
and the trans-Go\g \ network (TG N) to the ER. Transport o f secretory proteins through the stack
o f G olgi cistem ae are mediated by small vesic les at each step. Each compartment is distinct.
B Directed maturation m odel. A ssem b ly o f COPII vesic les form vesicular-tubular clusters that
m erge to ultimately form the first layer, the cw -G olgi cistem ae. Secretory proteins do not exit the
first layer; instead, COPI vesic les, carry processing enzym es to it from more mature layers. As
new layers form behind, the layer m oves up through the stack until it is consum ed by sorting in
the TGN (reference: Banykh & Balch, 1997)
- 4 7 -

(. 'hd/Vcr !________ __________________________________ ______________ _______ ___________________ l i ili-OiJitd i o n
Rambourg and co-workers examined PRL-producing cells o f lactating rats by 3-D
electron microscopy and found PRL aggregates in the lumen of the trans-Golgi cistemae;
the lumps of PRL were found in the plane of the layer of trans-Go\gi complex, scattered
throughout, and not in a sequestered region projecting above the layer (Rambourg et a i,
1992). The morphology is consistent with this transAayQT beginning to peel away from
the Golgi stack as small vesicles form from it, some clathrin-coated and some not,
removing membrane that is not directly around the aggregated PRL. As these vesicles
are removed, the remaining membrane of the layer, no longer an uninterrupted surface
takes on the tubular-vesicular appearance of the trans-Golgi network. What has been
termed ISGs are in this view, those aggregates in which removal of membrane by clathrin
coated and other vesicles is not complete (figure 1.15). What has been interpreted as the
ISGs that have fused to each other may have been two or more protein aggregates that
were drawn near each other as the trans-Golgi layer is consumed, although this does not
rule out the fusion of separate vesicles also. Further removal o f membrane is thought to
lead to mature secretory granules. The only endocrine cell type to be examined by 3-D
EM is the lactotroph, it would be interesting to discover whether the morphology of
somatotrophs is similar.
1.12.3 Hormone aggregates in neuroendocrine cells
If everything buds off, leaving membrane containing aggregated protein inside, there is
no need for a separate budding mechanism for secretory granules from the TGN. Also, if
aggregation occurs before vesicle budding occurs, the potential for aggregation in itself is
an excellent sorting mechanism. If the directed maturation model of Golgi transport is
correct, the aggregate need not form all at once in the trans-Golgi layer, but may begin to
form as the protein enters the Golgi complex, developing in size until it reaches the large
size detected in the ^ra«5 -Golgi lumen. It is also possible that aggregation is not just a
sorting mechanism, but in addition, an important factor driving formation of secretory
granules.
Whether the aggregates get packaged into secretory granules or not depends on whether
the cell can form granules, for example, transfected cells with secretory granules will
package expressed proteins into a regulated pathway if that is their normal fate, even if
the cells do not normally make the proteins. Kelly and co-workers demonstrated this in
- 4 8 -

j Early w Progranule
Mature v Granule
Polynodular Tubular Progranules
PolymorphousGranule
Figure 1.15 Schematic of 3-D electron microscopy of the Golgi apparatus of a rat
lactotroph
PRL is made in the endoplasm ic reticulum (ER) and transported to the G olgi com plex.
(W ) W ell, a gap between the c/5-eIements (CE) o f the G olgi apparatus. (S) Saccule, also
called a G olgi-cisternae. PRL aggregates are detectd in the trans-Qo\g\ cistem ae, showTi
in 2- and 3-D . What appear in 2-D to be fused immature granules are neighbouring
aggregates o f PRL in the lumen. The trans-Go\g\ layer progressively vesiculates through
v esic le s (V ) budding off, until the mature secretory’ granules remain. (Reproduced from
Dannies , 1999).
- 4 9 -

i ______ ______ _ _________ _________ ______ Inirocliii-non
AtT20 cells, a mouse pituitary cell line that makes ACTH. When these cells are
transfected with cDNA for proinsulin, GH or trypsinogen, which are normally secreted in
a regulated pathway, the cells duly package these non-native proteins into a regulated
pathway (Burgess & Kelly, 1987). It could be hypothesised that cells with secretory
granules, but not other cell types, have a mechanism for forming granules at the trans-
Golgi cistemae; that as the granules form, they enclose what is present in the trans-Golgi
cistemae; with no sorting signal required. The trans-Golgi is more acidic than the
endoplasmic reticulum [it decreases to ~pH6 ] and it is thought that acidification enhances
aggregation (Orci et aL, 1986; Seksek et aL, 1995; Colomer et aL, 1996). Kelly’s finding
that sorting into secretory granules is not restrieted to proteins that secretory cells
normally make, may reflect the similarity of the /ra«5 -Oolgi environment, so that eertain
proteins will always tend to aggregate in the trans-Golgi cistemae.
Environmental conditions in part of the secretory pathway in which aggregates form will
also influence the process of aggregation. Calcium concentrations in the endoplasmic
reticulum are high in all cells, since intracellular stores of Ca^^ for signalling are kept
there (Berridge & Irvine, 1989; Sambrook, 1990). The effects o f calcium ions on the
export o f proteins from the ER can be explained by a model based on the following
assumptions: lowering the ealcium in the lumen causes vésiculation of the ER membrane
and consequent packaging of secretory proteins (Booth & Koch, 1989). Second, the
concentration of calcium ions in the ER fluctuates in a cyclical manner as waves of ions
flow from the luminal reservoirs either into the cytoplasm or into another membrane
bound compartment and are then retumed to the ER (Berridge& Galione, 1988). Insulin
containing granules have been isolated and have very high cation concentrations: in
addition to 42mM insulin, they eontain 23mM Zn^^, 120mM Ca^^ and 42mM Mg^^
(Hutton et aL, 1983).
As previously mentioned in 1.10.1 with reference to GH dimérisation, Cunningham and
colleagues performed size exclusion chromatography and sedimentation equilibrium
studies, demonstrating that zinc ion (Zn^^) forms a 1:1 complex with hGH. Binding of
one Zn^^ ion promotes the binding of another Zn^^ ion (positive cooperativity) and
induced the dimérisation o f human growth hormone. Therefore, formation of GH
aggregates is dependent upon the presence of zinc. Aggregates of protein hormones, to
- 5 0

liilrodiictton
make a concentrated, insoluble masses must be able to dissociate reversibly and relatively
rapidly after exocytosis. It is not clear how GH is freed from its condensed form in order
to be exocytosed into the circulation. However, recently Han et aL, (1999) have
described GFP-tagged peptides inside secretory vesicles in rat proANF. They
demonstrated that Ca^^ influx into the cell’s cytoplasm rapidly alkalinises the contents of
the peptidergic secretory vesicle increasing the intensity of GFP fluorescence. This rapid
Ca^^-induced deprotonation caused by the rise in pH inside vesicles may facilitate
solubilisation o f peptides and their release. This mechanism could be applied to the
exocytosis of GH, although its role in the release of this hormone is yet to be confirmed.
It is important to note that aggregates alone are not sufficient to form secretory granules.
There is another whole literature on exocytotic mechanisms, beyond the scope of this
thesis, however, I would like to mention there are transmembrane proteins which are
necessary for regulated exocytosis, such as synaptotagmin and synaptobrevin. The
exocytotic pathway and stage specific roles of proteins are depicted in figure 1.16.
Synaptotagmin and synaptobrevin (VAMP) are found in both secretory granule
membranes and the membranes of small vesicles found in neuroendocrine cells, termed
synaptic-like microvesicles (Thomas-Reetz & Camilli, 1994). A class of transmembrane
proteins that resemble protein tyrosine phosphatase receptors ICA512 (or IA2) and
phogrin, have a location primarily on secretory granules and not in synaptic-like
microvesicles (Solimena et aL, 1996; Lan et aL, 1994; Wasmeier & Hutton, 1996).
Increasing the number of secretory granules in a cell has been found to induce
stabilisation o f 1CA512 (Lee et aL, 1998). Phogrin and 1CA512 must have a way of
recognising dense core granules, as they preferentially localise in granule membranes.
One mechanism would be for phogrin and 1CA512 to recognise some aspect of the cargo,
or to recognise the adaptor molecules that recognise the cargo in their aggregated state. If
the aggregate has properties o f a colloid, it may have specific structural attributes that
couls serve as properties to be recognised (Lekkerkerker & Stroobants, 1998; Grier,
1998). Alternatively, recognition of protein cargo may be due to differences in
membrane lipid composition. If the membrane lipid composition changes around
aggregates because they induce membrane curvature or change other properties, then
certain transmembrane proteins may preferentially localise in this region, as described for
sphingo-cholestrol domains for some apical proteins (Simons & Ikonen, 1997).
- 5 1 -

AnchoredDocked Primed Triggered
Recruitment pool
Fused
Mg-ATPMg-ATP \ (1)
° » A c t i n • S c i n d e r i n
^ S y n a p t o t a g m i n ^ S y n t a x i n
^ V A M P / • S N A P - 2 5s y n a p t o b r e v i n a S y n a p t o p h y s i n
• S y n a p s i n ^ M y o s i n II
Figure 1.16 Multiple stages of the exocytotic pathway
Stages indicated are (1) ATP dependent recruitment, (2) docking, (3) ATP-dependent priming
(reversible) and (4) Ca^"^-triggered fusion. In this sim plified figure, recruitm ent (1) in volves
proteins that disassem ble the actin cytoslelton, regulate vesicle-actin association (synapsin) and
translocate v es ic les relative to actin (m yosin II). SNA R E proteins (V A M P/synaptobrevin ,
S N A P -25 , syntaxin) are shown to form com p lexes during or fo llow in g dock ing (2) with a
preceding dissociation o f V A M P/synaptobrevin-synaptophysin com plexes. SN A R E com p lexes
in the docked state are shown to contain SNA P proteins, w hich act during A TP-dependent
priming (3) to d isassem ble com plexes and promote conformational changes in SN A R E proteins.
ATP-dependent priming (3) occurs before a Ca^^-triggered step that leads to fusion (4) w hich is
shown to in volve synaptotagm in interactions with syntaxin and SN A P -25 (reproduced from
Martin, 1997)
- 5 2 -

A hu i 'o d u c l io n
A specific protein, Carboxy peptidase E (CPE) has been identified in the secretory
pathway which has the ability to bind soluble secretory granule proteins. It has been
reported that in CPE ^ mice in which a mutation in this protein keeps it inactive in the
ER, POMC, proinsulin and GH are not sorted into regulated secretory pathways (Cool et
aL, 1997; Normant & Loh, 1998; Shen & Loh, 1999). The N-terminal end of POMC
binds to CPE, and this sequence in POMC is necessary for proper sorting (Cool et aL,
1997). However, there are contradictory lines of evidence for the role o f CPE from other
labs showing that although mutant CPE ^ mice do not process proinsulin to insulin well,
the unprocessed proinsulin is concentrated into secretory granules (Varlamov et aL, 1997;
Irminger et aL, 1997).
1.13 Isolated growth hormone deficiency (IGHD) and short stature
O f the many causes of GHD in children, 11% are due to craniospinal irradiation, 22% are
due to organic causes (i.e. septo-optic dysplasia, pituitary tumours, CNS insults) or are
associated with multiple deficiencies (panhypopituitary dwarfism), and 65% are
idiopathic with an isolated deficiency (Carel et aL, 1997). Most cases o f IGHD are the
only known case in the family {de novo mutation), estimates of the proportion of cases
with an affected parent, sibling or child range from 3 to 30% in different studies
suggesting that a significant proportion of GHD cases have a genetic basis. Although
failure in the GH pathway can be caused by a variety o f defects, they share the common
clinical findings in children of short stature, delayed growth velocity and delayed skeletal
maturation. Familial IGHD is associated with at least six different Mendelian disorders.
These include four autosomal recessive disorders (IGHD lA and IB, biodefective GH,
and GHRH-R defects). In addition there is an autosomal dominant (IGHD-II) and an X-
linked disorder. I am interested in the mutations involved with the GH gene, particularly
IGHD-II, which are summarised in the next few pages.
1.13.1 Isolated Growth Hormone Deficiency Type lA
The most severe form of IGHD, called IGHD-1A has an autosomal recessive mode of
inheritance. Affected neonates occasionally have birth lengths that are shorter than
expected for their birth weights and hypoglycaemia in infancy. By six months in age.
- 5 3 -

( i m i '1er / _ _ _ I n i r o d i i i 'Ho n
severe dwarfism develops in all affected infants. Type lA IGHD is caused by a deletion
o f the GH (GH-N) gene. The initial IGHD-1A cases were homozygous for GH-N gene
deletions and anti-GH antibodies developed in all after treatment with exogenous GH
preparations. However, additional patients have been described who had good responses
to GH replacement (Laron et aL, 1985; Matsuda et aL, 1987). In addition to gene
deletions, frameshift and nonsense mutations have also been found to cause the IGHD-
lA phenotype, which are documented in table 1 .1 .
1.13.2 Isolated Growth Hormone Deficiency Type IB
A second form of IGHD, called IGHD-IB, also has autosomal recessive mode of
inheritance. These cases differ clinically from IGHD-1A by the presence of low but
detectable levels of GH and a continued growth response as a result o f immunologic
tolerance to treatment with exogenous GH. Type lA has been clinically diagnosed in
some patients with IGHD-IB because their GH gene defects result in a mutant protein
that cannot be effectively measured by RIA. The presence o f this mutant protein may
explain their good response to GH therapy (Igarashi et aL, 1993). One example o f IGHD
type IB is the G—>C transversion of the first base o f intron IV of GH-N (Cogan et aL,
1994) which activates a cryptic splice site 73 bases upstream. This causes splicing out of
half of exon IV (aal03-126) and a frameshift in exon V (aal21-191). Previously reported
studies on bovine GH have demonstrated the importance of exons IV and V for proper
targeting o f the GH protein to secretory granules (McAndrew et aL, 1991; Chen et aL,
1991). Importantly, deletion of either of these exons was found to target GH peptide to
the cytoplasm despite proper cleavage of the signal peptide in the endoplasmic reticulum.
These studies suggest a possible mechanism for the IGHD 1 phenotype is that in
heterozygotes the resulting GH protein is not transported to the secretory granules but is
targeted to the cytoplasm, where it cannot interfere with the transport and secretion of the
normal GH allele products. Representative mutations of IGHD-IB are also found in table
1. 1.
54

( in ij 'i^r i h u ro d i ic t io n
Table 1.1 Isolated Growth Hormone Deficiency
IGHDType
LOCATIONNUCLEOTIDECHANGE EFFECT OF M UTATION REFERENCES
lA - Deletion 7.6kb deletion o f GH geneVnencak-Jones et a/., (1990)
lA - Deletion 7.0kb deletion o f GH geneVnencak-Jones et a/., (1990) 1
lA - Deletion 6.7kb deletion o f GH geneVnencak-Jones et a/., (1990)
lA Exon III 5536 del. CFrameshift after I7‘ aa o f signal peptide
Duquesnoy et aL, (1990)
lA Exon III 5543 G ^ AStop codon after 19" aa o f signal peptide
Cogan et al., (1993)
IB Intron IV 6242 G ^ CDonor splice site mutation; frameshift
Cogan et al., (1994); Abdul- L atif(I995)
IB Intron IV 6242 G->T Donor splice site mutation; frameshift
Miller-Davis et al., (1993)
IB Exon III 5938-39 del. AGFrameshift after 55‘*’ aa o f mature GH
Igarashi et al., (1993)
II Intron III 5955 G ^ ADonor splice site mutation; exon III skip
Cogan et al., (1995)
II Intron III 5955 G->CDonor splice site mutation; exon III skip
Binder et al., (1995)
II Intron III5960 T->C; G-^A
Donor splice site mutation; exon III skip
Cogan et a l , (1994); Missarelli et al., (1997)
II Intron III 5982-99 del Splicing mutation; exon III skipCogan et a l , (1995)
II Intron III 5982 G ^ A Splicing mutation; exon III skipCogan et al., (1996)
II Intron V 6664 G—>A Amino acid change (Argl83His)Wajnrach et al., (2000)
II Exon IV 6129 C ^ T Amino acid change (ProI89 Leu);Duquesnoy et al., (1998)
II Exon IV 6I9I G ^ T Amino acid change (Vail lOPhe)Binder et al., (2001)
5 5 -

L h i ip h J ' À _ _____________ _______________________ _ _ , _____________________________ In t iudar f i ' / i i
1.13.3 Biodefective Growth Hormone and GH-Rs
i) Mutant GH with an antagonistic effect
In 1978, Kowarski and co-workers studied two unrelated boys with growth retardation,
delayed bone age, high serum levels of immunoreactive GH and low serum lGF-1.
However, treatment with exogenous GH increased both serum lGF-1 and somatic linear
growth. The precise molecular basis of this disorder was unknown. More recently,
Takahashi and co-workers reported two children with short stature and mutant GH caused
by a single missense mutation in the GH-1 gene. The first child had a base pair
substitution which replaced arginine by cysteine at residue 77 o f the GH-N gene
(Takahashi et al., 1996a,b) which is located in the second a-helix o f the GH molecule
behind binding site 1 to the GHR (Cunningham et a l, 1991b). The substituted cysteine
probably forms new disulphide bonds, changes the charge o f the GH molecule and
contributes to a conformational change of the mutant GH. The affinity of the mutant GH
for GHBP was found to be six times higher than that o f WT GH. As previously
mentioned, the binding of GH to its receptor is thought to proceed sequentially in the
different portion of the GH molecule, first in site 1 and then in site 2 (Cunningham et al.,
1991b). Dimérisation of the GH receptor, induced by ligand binding and sequential
protein phosphorylation in tyrosine residues is necessary for GH-induced signal
transduction. Mutant GH failed to effect tyrosine phosphorylation when bound to the GH
receptor and further inhibited the binding of wild type GH, acting as an antagonist with a
dominant-negative effect. Interestingly, the patient’s father is phenotypically normal,
despite carrying the same genetic abnormality. The mechanism by which the expression
of the mutant GH gene is prevented in him is unclear.
A second child also carried a heterozygous single-base substitution (A—>G) in exon 4 of
the GH-N gene, resulting in the substitution of glycine for aspartic acid at codon 112
(D112G) (Takahashi et al., 1997). The locus of mutation D112G is at the central portion
of helix 3 within binding site 2 of the GH molecule in binding with GHR/GHBP. The
expressed recombinant mutant GH formed 1:1 instead o f 1:2 GH-GHBP complexes
normally produced by wild-type GH, thus preventing dimérisation o f the GHR and
- 5 6 -

( j \ i l 2 h : iL L ________ ________ __ „ . . .. i n i n j J n rlii / i i
subsequent sequential tyrosine phosphorylation of GHR. The mutant GH proved to be
bio-inactive, resulting in GHD and growth retardation.
ii) Laron Syndrome - mutations in the growth hormone receptor (GHR)
The prototype of GH insensitivities is widely known as Laron-type short stature (Laron et
aL, 1966). Laron syndrome is an autosomal recessive disorder caused by resistance to
the action of GH because of proven defects in the GHR gene (Rosenbloom, 1992).
Studies o f GHR genes from patients with Laron syndrome have identified a number of
exon deletions and base substitutions. Berg et aL, (1993) reported ten different GHR
mutations, one of which was a recurrent mutation involving a CpG dinucleotide hot-spot
[CpG dinucleotides are reported to have increased mutation rates due to spontaneous
deamination of a methylated cytosine on the sense or antisense strand (Cooper &
Youssoufian, (1988); Shen et aL, (1992)]. Patients with Laron syndrome have short
stature and delayed bone age and at the clinical level are indistinguishable from those
with GHD. However, biochemically they have low levels of IG F-1, despite normal or
increased levels o f GH. Importantly, exogenous GH does not induce an IGF-1 response
or restore normal growth in patients with Laron syndrome because o f their GHR
dysfunction. The growth hormone binding protein is the extracellular domain of the GH
receptor (Cunningham et aL, 1991b) and serves as a GH reservoir in vivo (Herrington et
aL, 1991; Baumann et aL, 1988). Although plasma levels of the GHBP are usually low
in patients with Laron syndrome. Woods et aL, (1996) reported a homozygous point
mutation in the intracellular domain of the GHR that caused Laron syndrome with
elevated GHBP levels. Their hypothesis was that the mutant GHR would not be
anchored in the cell membrane, but measurable in serum as GHBP, thus explaining the
phenotype of severe GH resistance combined with elevated GHBP.
1.13.4 Isolated Growth Hormone Deficiency Type II
Type II IGHD differs from all forms of IGHD I in that it has an autosomal dominant
mode of inheritance because of dominant-negative mutations in the GH gene. Autosomal
disorders have one obvious difference from recessive disorders. Autosomal recessive
disorders, like many enzyme deficiencies, occur when the mutant protein acts as a
monomer, so that 50% of the protein inactivated by the single mutant gene does not act
- 5 7 -

c . / _____ _____ __________________ _________________________________ I n l r o i l u d i n n
with the other 50% produced by the unaffected allele. By contrast, when proteins form
polymers, one mutant monomer can disrupt the whole polymer, so that only one mutant
allele causes disease. Patients in whom IGHD II is diagnosed are all heterozygous for the
mutation, have a single affected parent, vary in clinical severity between kindreds, and
respond well to GH treatment (Cogan et aL, 1994; Cogan et aL, 1995; Phillips III &
Cogan, 1994; Hayashi et aL, 1999a; Binder et aL, 2001). Almost all o f the GH gene
defects reported to date in IGHD II cases are mutations in intron 3, either in conserved
nucleotides in the 5’ consensus splice site or in a conserved heterogeneous nuclear
ribonuclear protein (hnRNP) binding site. These alter splicing of GH mRNA and cause
deletion of exon 3. It is unclear as to why this mutated, truncated protein might act as a
dominant-negative. Other GH-N missense mutations have also been implicated with
IGHD-II and have recently been reported are P89L (Duquesnoy et aL, 1998), R183H
(Wajnrajch et aL, 2000) and V llO F (Binder et aL, 2001). Again, the mechanism by
which these mutant proteins suppress the production o f wild type GH is unclear,
however, in both splice site and missense mutations, the mutant GH gene product must
somehow prevent normal intracellular protein transport.
1.14 Rodent models of mutations in the hypothalamo-pituitary GH axis
Advances in our understanding of the basic physiological mechanisms have emerged
from studies in rodents bearing spontaneous mutations which result in an altered pituitary
GH production and growth phenotype. Since the ultimate cause o f dwarfism is GH
deficiency, they can invariably be stimulated to grow by direct replacement therapy,
either with exogenous GH (Charlton et aL, 1988) or by a transgene expressing GH
(Hammer et aL, 1984). The most obvious example is the dwarf rat (dr/dr) which carries
a mutation in the GH gene resulting in a truncated, inactive product (Takeuchi et aL,
1990). Our lab has also characterised the spontaneous GH-deficient dwarf {dw/dw) rat
(Charlton et aL, 1988) whose recessive mutation remains unidentified. A summary of
rodent models of GH disease can be seen in table 1.2.
Recent advances in transgenesis have made it possible to create new models of altered
growth rate. Numerous lines of transgenic mice have been generated which express GH
- 5 8 -

( / ' i 7' ' . _ _ l iU r o d i n ' l i r i n
under the eontrol of heterologous promoters, usually resulting in increased growth given
a significant level o f peripheral GH expression (Palmiter et aL, 1982; Morello et aL,
1986; Echini et aL, 1991). There have been a few interesting exceptions to this
phenotype, in which dominant dwarfism results from hGH expression in the CNS
(Hollingshead et aL, 1989; Banjeree et aL, 1994). This arises from unregulated hGH
transgene expression exerting an inappropriate negative feedback via hypothalamic GH
receptors to suppress GHRH activity, which in turn reduces mouse GH production. The
transgenic growth retarded rat (TGR) generated in our lab exploited this GH negative
feedback mechanism more specifically, by targeting expression o f hGH to the GHRH
neurones of the hypothalamus using the rGHRH promoter (Flavell et aL, 1996). The
phenotypic dwarfism which has resulted in these transgenic animals have provided
models for assessing not only similar human deficits, but have provided evidence that
these pituitary hormones have trophic, as well as dynamic feedback effects on the
hypothalamic neurones that regulate GH secretion.
One of the main aims of my thesis is to generate a transgenic mouse, modelling a human
dominant negative mutation which has already been reported to cause IGHD-II and
severe short stature in children to see if the mice were dwarf. Although studies in
children with IGHD-II exist, they can only present the data from screening studies and
hence location o f specific mutations in the GH-N gene, their occurrence and biochemical
and clinical data (Cogan et aL, 1993; Binder et aL, 1995; Missarelli et aL, 1997). Studies
in endocrine cell lines expressing the human dominant-negative GH mutation have
shown that a 17.5kDa mRNA is transcribed (Cogan et aL, 1995). After I began my
thesis, Dannies and eo-workers expressed hGH-IVS3 mutations in GH4C] cell lines and
have showed that the truncated (exon 3 skip) GH product from the mutant gene does not
accumulate in, nor is secreted by neuroendocrine cells. It suppresses production of WT
rGH (Lee et aL, 2000). Although studies in cell lines can shed some light on the
mechanisms o f this GH-IVS3 mutation, they cannot portray a true picture of the
mechanism of the disease in vivo and subsequent physiological effects in the anterior
pituitary. Chapters 5 and 6 begin to explain the mechanisms o f dominant-negative
suppression of GH in both rat GC cells, and transgenic hGH-IVS3 mice.
- 5 9

( 'hiin'iicr In tro d u c tio n
Table 1.2 Rodent models with mutations in the GH axis
Model Defect Phenotype Reference
Snell Mouse{dw /dw )
Point mutation in Pit-1
Hypoplastic pituitary lacking somatotrophs, thyrotrophs and lactotrophs. Sterile.
Snell, (1929); Li e t a l , (1990)
Jackson Mouse{dw'^/dv/)
Gross rearrangement o fP i t -1
Hypoplastic pituitary lacking somatotrophs, thyrotrophs and lactotrophs. Sterile.
Li e ta /. (1990)
Ames Mouse Point mutation in Prop-1
Hypoplastic pituitary lacking somatotrophs, thyrotrophs and lactotrophs. Sterile.
Andersen e t al. (1995) Gage e t al. (1995) Somson e t al. (1996)
Little Mouse {lit/lit)
Point mutation in GRF-R
H ypoplastic pituitary with severely reduced somatotroph number. N o cAMP or GH secretion in response to GRF. Mild PRL deficiency. Mildly reduced fertility (males taking longer to sire young).
Lin e t al. (1993)
1 Spontaneous Dwarf rat (SDR)
Point mutation in GH Pituitary normal weight. Increased population o f nonhormone producing cells. R eduction in lactotroph number.
Takeuchi e t al., 1990 Nogami e t al., 1993
Hypothyroid rat {rdw)
Secretory defect o f the thyroid
Reduced GH and PRL protein and mRNA. Many proteins expressed at different level in thyroid. Sterile.
Koto e t a/. (1988) Oh-Ishi et a/. (1998)
Dwarf rat {dw /dw )
Unknown Reduced somatotroph number. Reduced GH content per somatotroph. Reduced cAMP response to GRF, although secretory response relatively intact. PRL and lactotroph number normal or increased.
Charlton et al. (1988) D ow n s and Frohman, (1991)Carmignac, (1990)
1 Transgenic growth retarded rat {tgr)
hG H tr a n sg e n e expressed in GHRH neurones resulting in GH deficiency via intrahypothalamic feedback suppression o f GHRH.
Decreased GHRH neurones and expression , increased SRIH expression.Increase in TIDA (DA-only) neurones, but no increase in cell number.
Flavell et al. (1996) Thomas et al. (1999)
G H -R knock out (Laron Dwarf)
Targeted disruption o f GHR BP gene - panhypopituitary dwarfism
Severe growth retardation, proportional dwarfism, low circulating IGF-1 le v e ls , elevated GH. Hyperprolactinaemia. D im in ished respon ses to GnRH and LH. Reduced fertility in males.
Zhou et al. (1997) Chandrashekar et al. (1999)
- 6 0 -

( ’i n ip i c r I _______________________________________________ h i l r o i l t i c i io n
1.15 Green Fluorescent Protein (OFF) as an endogenous cell marker
The anterior pituitary gland consists of families of hormone-producing cells that can be
differentiated by their hormone expression, receptor population, secretory product, or the
common expression of certain transcription factors. Many neuroendocrine laboratories
who want to study the regulation of individual pituitary cell types have used tumour cell
lines as models for pituitary cell function (Asa & Ezzat, 2000). Or, if they wanted to use
primary cell, they have either separated and purified them (reviewed in Childs et a/.,
1992; Van Bael et a l , 1996) or applied techniques to identify the cells post-hoc
(Guerineau et al., 1998; Bonnefont et al., 2000), which are laborious and have a low
yield. It has also been recognised that removal of a group o f cells from the pituitary
network has deleterious effects on their function, although many basic secretory and
signal transduction functions remain intact, pituitary cells behave differently in vitro
when they are allowed to “re-aggregate” (Vankelecom & Denef, 1997). Ideally, the cell
should be identified in situ, without altering key regulatory processes or expression of the
genes in question. Such a marker would essentially work in the background, allowing the
pituitary cells to be identified in the living state.
Green fluorescent protein (GFP) was discovered by Shimomura et al., as a companion
protein to aequorin, the chemiluminescent protein from Aequorea Victoria jellyfish
(Shimomura et a l , 1962). The fluorescence is generated by sequential activation o f two
photoproteins, aequorin and green fluorescent protein (GFP) (Prasher, 1995). Upon
calcium binding, aequorin emits blue light, which in turn excites GFP to fluoresce green.
This GFP has a unique property in that it forms a chromophore o f three amino acids
within its primary structure and, in contrast to other bioluminescent molecules, operates
independently of co-factors (Prasher et a l, 1995). Although the properties o f the proteins
have been known for many years (Cubitt et a l , 1995), it was not until the cloning o f the
cDNA of GFP in 1992 (Prasher et al., 1992) and its subsequent heterologous expression
in E.Coli and C. Elegans (Chalfie et a l, 1994) that researchers from many fields became
aware o f the potential of this molecule. Enhanced green fluorescent protein (eGFP) has
been widely used by cell biologists all over the world and is exploited as a marker gene
of expression and protein targeting in intact cells and organisms (for review, Tsien 1998).
Its numerous applications have included: using GFP as a reporter for gene expression
61

________________________________________________________________ _____________ ___________ I n i r o d u c U n n
(Chalfie et al., 1994), as a marker to study cell lineage during development (Zemika-
Goetz et a l, 1995) as a tag to localise proteins in living cells (Kaether & Gerdes, 1995),
or for their isolation and analysis using fluorescence activated cell sorting techniques
(Manjunath et al., 1999 & Kawakami et al., 1999). Since GFP fusion is often unaffected
by fusion to other sequences, intracellular distribution and secretion events can also be
visualised in real-time by tagging GFP with sequences that target it to different sub-
cellular compartments.
1.15.1 Properties o f wild-type and mutant GFPs
GFP, as deduced from its cDNA is a 238 amino acid protein with a molecular weight of
about 27-30kDa on SDS-page (Chalfie, 1995). Its chromophore is formed by cyclisation
and oxidation of the three amino acids Ser65, Tyr66 and Gly67 (Prasher et a l , 1992).
This post-translational modification occurs within 2 to 4 hours after synthesis and is
probably autocatalytic (Heim et a l , 1994). The wild-type GFP has two absorption
maxima, a major peak at 395nm and a minor one at 475nm (Cubitt et a l , 1995).
Excitation of either o f the two wavelengths results in emission of green light at 508nm.
These fluorescent properties have been changed by genetic engineering leading to several
mutants (for review Tsien, 1998). These mutants are of special interest for cell biology.
The Ser65—>Thr (S65T) mutant of Heim et a l, (1994) termed enhanced green fluorescent
protein (eGFP) has three advantages relative to GFP. First, a single excitation peak of
490nm adapts this mutant to common fluorescein isothionate (FITC) filter sets; second,
excitation yields a six fold stronger fluorescence which is more resistant to photo-
bleaching and third, this mutant matures fourfold faster than GFP, thus shortening the lag
time between synthesis and photo-activity. Mutations o f GFP have been further
improvised to emit blue fluorescence (BFP), cyan fluorscence (CFP) or yellow
fluorescence (YFP) when excited at particular wavelengths. Moreover, not only can an
engineered GFP emit light o f a different colour i.e. blue fluorescent protein (BFP), but
the light of BFP can also excite GFP thus allowing fluorescence energy transfer (FRET)
as has been elegantly demonstrated in vitro with the two mutants as donor and acceptor
indicating protein-protein interaction (Heim & Tsien, 1996).
- 62 -

______ ____________________________________________________________________________ In trochic lion
1.15.2 GFP-taggedproteins and the subcellular localisation
Expression of GFP on its own, results in a diffuse distribution throughout the cytoplasm,
in many cases including the nucleus (Ogawa et al., 1995; Olson et al., 1995; Magoulas et
al., 2000). Pioneering work by Wang and Hazelrigg has demonstrated that GFP can be
used as a fluorescent tag for the N- or C-termini o f proteins (Wang & Hazelrigg, 1994).
Although the chromphore is formed by just three amino acids, truncation o f a very few
amino acids at either end of the full length GFP is not tolerated if the protein is to
maintain its fluorescence. Despite the large size of such a GFP tag (238aa), it has shown
in numerous cases that the tagged proteins were functional and were targeted properly
(Tsien, 1998).
For the purpose of my studies, I wished to target eGFP, not just to GH cells, but to the
secretory vesicles o f GH cells in two systems. I fused the sequence encoding GFP in
frame with the sequence encoding human growth hormone and expressed the resulting
GH-GFP fusion in rat GC cell lines, which is described in chapter 3 o f this thesis.
Chapter 4 goes on to describe the expression of GH-GFP in growth hormone granules in
pituitary somatotrophs in transgenic mice. It was of critical importance to include the
entire GH LCR and sufficient signal peptide sequence to target GFP not only to the
correct cell type, but also direct its entry into the endoplasmic reticulum and subsequent
secretory pathway, without interfering with the physiology of the animal. We hoped the
resulting hGH-eGFP transgene, when translated in transfected cells or pituitary
somatotrophs, would identify the GH cell population and its granules, in situ. Thence,
this model would provide an invaluable tool in order to improve our understanding of
how pituitary growth hormone is sorted and processed, and how these processes may be
regulated. To begin to understand the dominant-negative suppression o f GH, it is
necessary understand what causes the malformation o f heterodimers or aggregates of
growth hormone, which results in a dominant-negative effect and thus, toxic
accumulation o f dysfunctional heterodimers in the regulated secretory pathway and
consequently somatotroph cell death. Chapters 5 and 6 investigate the expression of a
human dominant-negative growth hormone mutation using both the in vitro and in vivo
systems I have described above. A transgenic model expressing hGH-IVS3 would
provide us with the means to study the development and the progression of the pathology
of human dominant-negative dwarfism in a rodent model in vivo.
63

i '. lnipicr 2_____ M d lg - iiils a n J .Vfclhuds
Chapter 2
Materials and Methods
2.1 Preparation of DNA
All kits were used according to manufacturer’s instructions, unless otherwise stated.
2.1.1 Bacterial cultures
Bacterial cultures were grown in standard LBroth (Sambrook et al., 1989) with shaking
(250rpm) at 37°C. E. Coli strains were plated onto LBroth agar plates (LBroth with 15g/l
bacto-agar). Medium was supplemented with appropriate antibiotic (Sigma, UK) and
used at the following concentrations: ampicillin - lOOpg/ml, tetracyclin - 50pg/ml,
kanamycin - 50|Lig/ml. Bacterial stocks were stored at -70°C in 50% glycerol.
2.1.2 Preparations o f plasmid and cosmid DNA
Small scale plasmid DNA preparations (2-3ml saturated bacterial culture) were prepared
using the Qiaprep Spin Mini-prep Kit (Qiagen) and DNA from large volumes of saturated
bacterial cultures (300ml) was extracted with the Plasmid Maxi Kit (Qiagen). Cosmid
DNA was specially prepared using Large Construct Kit (Qiagen) to avoid shearing of
DNA.
2.1.3 Preparation o f genomic DNA from animal tissue
Mouse tail biopsies were taken from 18-21 day old animals and placed in ‘tail buffer’
(50mM Tris-HCl pH 8.0, lOOmM EDTA, lOOmM NaCl, 1%SDS). Post-mortem ear and
organ samples were also collected for genotyping. Genomic DNA was extracted using
the method described by Hogan et a l, (1986). Tissues were homogenised and incubated
with proteinase K (20-40pg/ sample. Sigma) at 55°C for 4 hours, followed by RNAse A
(10-20pg/ sample. Sigma) incubation at 37°C for 30 minutes. Digested samples were
then phenol-chloroform extracted, the DNA precipitated with 0.6 volumes of isopropanol
and DNA pellets resuspended in distilled water.
64

_______________ _____________ ___________________________ M üh^iil^jndA klhiih
2.1.4 Purification o f DNA
Aqueous solutions o f DNA were purified by adding an equal volum e o f
phenol:choloroform:isoamyl alcohol (25:24:1) (Life Technologies), vortexing and
centrifuging at 12000rpm for 5 minutes in a bench-top microfuge at room temperature.
The aqueous supernatant was precipitated by adding 3M sodium acetate (pH 5.2) to a
final concentration of 300mM and two volumes o f absolute ethanol. Samples were
incubated at -20°C for 1 hour and the pellet resuspended in distilled water.
2.2 Subcloning of DNA fragments
2.2.1 Restriction digest
Plasmid and cosmid DNA was incubated with restriction enzymes following the
manufacturers instructions (Roche Molecular Biochemicals, UK or New England
Biolabs) for up to 2 hours. Genomic DNA was digested overnight.
2.2.2 ‘Blunt-ending ’ o f DNA fragments by filling the 5 ’ overhang
After restriction digest, DNA was purified by phenol extraction and ethanol precipitation
and resuspended in water. Digestion with a restriction enzyme which leaves a 5’
overhang and single stranded ends are filled using the Klenow fragment of E. Coli DNA
polymerase I (New England Biolabs). Klenow reactions are performed in Ix Klenow
buffer (New England Biolabs) supplemented with dNTP’s (Amersham Pharmacia
Biotech, UK) to a final concentration of 0.25mM and 10 units o f Klenow fragment at
37°C for 30 minutes, DNA was repurified.
2.2.3 Vector dephosphorylation
In order to prevent self-ligation after digestion with a restriction enzyme, the 5’
phosphate groups were removed. DNA was incubated with 5 units o f shrimp alkaline
phosphatase in Ix alkaline phospatase buffer (New England Biolabs) at 37°C for 1 hour
and repurified.
2.2.4 Insertion o f I inkers
Plasmid DNA was digested, blunted and dephosphorylated to prevent self-ligation. A
phosphorylated linker was ligated to the plasmid in Ix ligase buffer (New England
- 65 -

{ 'h j j j 'h r .J ______ S k i t c r i n h a n d M e th o d s
Biolabs) with 400 units of T4 DNA ligase (New England Biolabs) at 16°C overnight.
The reaction was inactivated at 65 °C for 15 minutes, then repurified. The plasmid was
then linearised with a restriction enzyme appropriate to the linker to confirm insertion.
2.2.5 Gel electrophoresis o f DNA fragments
DNA fragments were separated by size in gels o f varying percentages o f agarose in Ix
TAB buffer (0.04M Tris-acetate, ImM EDTA) supplemented with 0.5ng/ml ethidium
bromide. DNA bands were recorded on an ultraviolet transluminator and fragment sizes
estimated by DNA markers (1Kb Plus DNA ladder, Life Technologies).
2.2.6 Purification o f DNA fragments from agarose gels
DNA fragments were excised from agarose gels and purified using the QIAEX II Gel
Extraction Kit (Qiagen).
2.2.7 Ligation o f DNA fragments
DNA fragments were mixed at varying molar ratios (insert:vector, 4:1) in Ix ligase buffer
(New England Biolabs) and 400 units T4 DNA ligase (New England Biolabs). The
reaction was incubated overnight at 16°C and inactivated at 65°C for 15 minutes
preceding repurification.
2.2.8 Transformation o f competent cells
Transformation o f plasmid DNA into competent cells {E.Coli X L-1-Blue, Stratagene)
was performed using the heatshock method, according to manufacturers instructions.
Briefly, l-5p,l ligated DNA were added to 20pl competent cells and incubated on ice for
2 minutes. The mix was transferred to 37°C waterbath for 1 minute then immediately
replaced on ice for a further 5 minutes. 250|l i 1 of SOC media (2% tryptone, 0.5% yeast
extract, lOmM NaCl, 2.5mM KCl, lOmM MgCE, lOmM MgS0 4 , 20 mM glucose) was
added and the reaction incubated at 37°C for 1 hour, before plating onto selective
medium plates.
66 -

UMPhllL-l_______ M airru ils an d M ethods
2.2.9 Packaging o f cosmid DNA into bacteriophage
Packaging o f cosmid constructs into bacteriophage particles was performed using
Gigapack III XL packaging extracts (Stratagene). E.Coli strain VCS257 was infected
following the manufacturer’s instructions.
2.3 DNA Sequencing
Double stranded DNA was sequenced on both strands on an ABI 377 automated
sequencer using the ABI Prism BigDye™ Terminators Sequencing Ready Reaction kit
(Applied Biosystems-Perkin Elmer, UK).
2.4 Polymerase Chain Reaction (PGR)
Amplification of DNA fragments was performed by the polymerase chain reaction
(PGR). Differing amounts of DNA template were added to the IxPCR reaction mix (Ix
PGR reaction buffer containing 1.5mM MgCL, [Perkin Elmer], 0.25mM dNTP’s,
[Amersham Pharmacia Biotech], two specific primers (50ng/reaction, [Oswel DNA
Services, Southhampton, UK]) and 1.25 units of Taq Polymerase (Perkin Elmer). The
reactions were incubated in a Biometra Trio-Thermoblock 48 PGR Machine (Whatman
International, Maidstone, UK) using a typical programme as follows:
Initial dénaturation: 94°C for 5 minutes
Denaturing: 94°C for 1 minute
Annealing: 60°C for 30 seconds
Extension: 72°C for 1 minute 30 seconds
Final Extension: 72°C for 5 minutes
30 cycles
Annealing temperatures and times were varied according to primers and templates used.
All PGR products were analysed on agarose gels.
2.5 Detection of DNA sequences by Southern blotting
2.5.1 Southern blotting
- 6 7

Clnii'icr _ _ \Liicr!(i!s and M clluxh
DNA (10 }xg o f genomic DNA or 0.5|xg o f plasmid/cosmid DNA) was digested with
appropriate restriction enzymes and separated by gel electrophoresis in 0.6% agarose
gels. The DNA was transferred onto Hybond-N^ nylon membranes (Amersham
Pharmacia Biotech) in 0.4M NaOH by a capillary transfer method (Southern, 1975;
Sambrook et al, 1989).
2.5.2 Radioactive random prime labelling
Single stranded, gel purified DNA fragments (~25ng) were radiolabelled with
[a^^P]dCTP and [a^^P]dATP with the random primer labelling kit Prime-a-Gene
(Promega, Southampton, UK). Radiolabelled probes were purified by gel filtration
through Sephadex G-50 columns (Amersham Pharmacia Biotech).
2.5.3 Hybridisation o f Southern blots
Hybridisation solutions:
20xSSC 3M NaCl, 0.3M NaCitrate, pH 7.0
Denhardt’s solution 2% BSA, 2% ficoll 400, 2% Polyvinyl Pyrollidine
20xSSPE 3M NaCl, 0.2M NaH2P0 4 , 25mM EDTA, pH 7.4
The membranes from Southern blotting were pre-hybridised for 4 hours at 65°C in pre
hybridisation mix (5x SSPE, 5x Denhardt’s solution, 0.5% SDS, 250pg/ml heterologous
salmon sperm DNA. The pre-hybridisation mix was then replaced with fresh mix,
supplemented with the single stranded radioactive probe and incubated at 65°C
overnight. Blots were washed in 3x SSC, 0.1%SDS, followed by a 0.3x SSC, 0.1% SDS
wash at 65°C for 1 hour and exposed to Kodak BioMax film at -70°C.
2.6 Determination of Protein content
Protein content in tissue homogenates was measured with the BCA Protein Assay
Reagent (Pierce, Rockford, IE). The OD 562 o f samples and bovine serum albumin
standards (Sigma, UK) was measured in a Beckman DU-64 spectrophotometer.
- 6 8 -

( 'h u 2 l^ L l____________________________________________________________________________ A liilm A iL i. (If u! \ l a h o ds
2.7 Generation of transgenic mice
2.7.1 Purification o f large DNA fragments fo r microinjection
lOOng cosmid DNA was digested with Not\, to release the insert, phenol/chloroform
extracted, ethanol precipitated and resuspended in 200pl TE (lOmM Tris.HCl, ImM
EDTA, pH 8.0). A 5-25% NaCl gradient was prepared using a Hoefer SG 30 gradient
former and the DNA was layered on top o f the gradient. The gradient was spun at
37000rpm for 5.5 hours and harvested in 0.5ml aliquots, which were examined by
electrophoresis. The aliquots containing the desired fragment were pooled, ethanol
precipitated and resuspended in microinjection buffer (lOmM Tris.HCl pH 7.5, ImM
EDTA, pH 8.0).
2.7.2 Superovulation, microinjection and embryo transfers.
Prepubertal (3-4 week old), superovulated, fertilised female mice ([CBA/ca x Bl/lOJFl)
were received from the NIMR SPF Unit. The female mice were culled and their oviducts
transferred into M2 medium (Hogan et a l, 1986). The oocytes were collected from the
dissected oviducts and placed in fresh M2 medium supplemented with 0.5mg/ml
hyaluronidase (Sigma, [to remove cumulus cells from the surface o f the oocytes]). The
oocytes were then washed twice in M2 and transferred into M l6 media (Hogan et a l,
1986) in an incubator at 37°C supplemented with 5% CO2 . With the help o f Dr. K.
Mathers (Biological and Procedural Services, NIMR) DNA (~10ng/pl) was injected into
the pronucleus of fertilised one-cell mouse oocytes using standard methods (Hogan et a l,
1986). Viable oocytes were incubated at 37°C until transfer into the oviducts of pseudo
pregnant mice (supplied by NIMR SPF Unit, ([CBA/ca x Bl/lOJFl). Surgery was
performed under 10% Avertin anaesthetic (0.1 ml/1 Og bodyweight, i.p; 100% solution:
Ig/ml tribromoethanol in amylalcohol [Sigma Aldrich]), with approximately 15 oocytes
transferred into each infundibulum. Tail biopsies from resulting pups were analysed by
PGR and Southern blot.
2.8 Reverse transcriptase polymerase chain reaction (RT-PCR)
RNA was prepared from homogenised mouse tissues and lysed GC cells using SNAP
total RNA Isolation Kit (Invitrogen BY, The Netherlands). 500ng of total RNA were
transcribed with 200 units o f M-LMV reverse transcriptase (Roche M olecular
- 6 9 -

( 'lnii>lcr J ____________________ Mdicridls and M a h a J s
Biochemicals, UK) supplemented with Ipg random primers (Invitrogen), 0.3mM dNTP’s
(Amersham Pharmacia Biotech), 40 units RNAse Inhibitor (Promega) and 5mM DTT.
The mixture was incubated at 37°C for 2 hours and the desired cDNA cloned into pCR-II
vector (TOPO Cloning Kit, Invitrogen BV) and amplified by PGR.
2.9 RNAse Protection Assay (RPA)
Tissue RNA was isolated using a Kinematica Polytron PT 3000 homogeniser and
extracted in TRIzol (Gibco BRL, Paisley, Scotland). Plasmids containing polymerase
promoters were linearised with the relevant restriction enzyme and re-purified using
QIAEX II gel extraction kit (Qiagen). Anti-sense and sense radiolabelled riboprobes
incorporating [a^^S]-UTP were generated using the SP6/T7 transcription kit (Roche
Molecular Biochemicals). Actin was used as an internal RNA control (RPA III, Ambion,
Texas, USA). After transcription probes were treated with RNAse free DNAse I (Roche
Molecular Biochemicals) for 15 minutes at 37°C and separated on a 5% denaturing,
polyacrylam ide sequagel (National diagnostics, Hessle, Hull, UK) and run for
approxim ately 1.5 hours. Transcript bands were visualised by exposure to
autoradiographic film (BioMax MR, Kodak) followed by excision and elution of the
transcript.
RNAse protection assays were performed using the RPA III kit (Ambion). Briefly,
IxlO^cpm labelled probe and lOpg sample RNA were incubated at 42°C over night.
After hybridisation, the mixture was treated with RNAse and protected fragments were
separated on a 5% acrylamide gel. Gels were dried under vacuum at 90°C, then exposed
to a phospor screen (Molecular Dynamics) and digitally analysed using the ImageQuant
(Molecular Dynamics) program and a STORM-860 scanner. The relative quantities of
protected sample RNA was normalised by comparison to p-actin.
2.10 Northern Blotting
Total RNA from pituitaries was isolated using the TRIzol reagent (Life Technologies) as
described in section 2.9. Northern analysis on GH-GFP transgenic mouse pituitaries was
kindly performed by Dr. C. Magoulas (NIMR). Briefly, RNA was electrophoresed in a
1.2% agarose gel containing 8% formaldehyde, blotted onto Hybond-N^ membrane
70

________________________________________________________ M ülcrials an d M el/iods
(Amersham Pharmacia Biotech) and hybridised at 45°C in 5x SSC (20x: 3M NaCl, 0.3M
NaCit, pH 7.0), 5x Denhardt’s solution (2% BSA, 2% ficoll 400, 2% Polyvinyl
Pyrollidine), 50mM phosphate buffer (50mM NaH 2P 0 4 , lOOmM NaCl, 0.6mM
Thimerosal, pH 6.5), 0.1% SDS, salmon sperm DNA (250mg/ml and 50% formamide.
Membranes were washed with O.lx SSC and 0.1% SDS at 65°C. A VOObp Xmal-Notl
fragment of pEGFP-N3 vector was radiolabelled by random priming (Prime-a-Gene,
Promega, Inc.) and used as a hybridsation probe for eGFP sequences.
2.11 Radioimmunoassay (RIA)
Radioimmunoassay solutions:
PBS 50mM NaH2P0 4 , lOOmM NaCl, 0.6mM Thimerosal, pH 7.4
Tris Buffer lOOmM Tris.HCl (pH 8.4), 0.6mM Thimerosal
18% PEG 2ml/l 10% Triton-X, 1.5g y-globulins, 330ml/ Tris buffer, 667ml/l
27% polyethylene glycol
RIA Buffer PBS, 0.3% BSA
2.11.1 Preparation o f pituitaries
Pituitaries were dissected from transgenic and wild type mice and stored in 1.5ml
microcentrifuge tubes at -70°C until further use. Homogenisation was performed after
thawing in a 1ml glass homogeniser in 1ml of PBS.
2.11.2 Pituitary mGH, mPRL, mLH, mTSH, eGFP RIA
The following radioimmunoassays were performed with the help and advice of Danielle
Carmignac (NIMR). Danielle Carmignac kindly labelled the mPRL tracer. I labelled all
other tracers and performed most of the RIA’s. mGH, mPRL, mLH, mTSH reagents
were kindly provided by the National Institute of Health for Diabetes and Digestive and
Kidney Disease, Bethdesa, MD, USA and pituitary extracts were assayed by RIA as
previously described for the rat (Carmignac & Robinson, 1990). NIHDDK antibody
concentrations and standards are detailed in table 2.2. A typical standard curve for mGH
is shown in figure 2.1. For eGFP a new RIA, developed by D. Carmignac was used as
follows: Recombinant eGFP (Clontech), 5pg, was radiolabelled with Nal^^^ using the
lodogen Method as previously described (Robinson 1980) and purified by Sephadex G75
- 7 1 -

( 'haptcr 2 A fate rials and Methods
chromatography. For assay, lOOpl of iodinated eGFP (5-7000cpm) were mixed with
lOOpl of tissue extract or standards (O.l-lOng) of recombinant eGFP and lOOpl
polyclonal antibody against GFP (Molecular Probes Inc.) at a dilution of 1:500,000 for 16
hours at room temperature. Bound and free fractions were seperated by addition of 2
volumes of 18% polyethylene glycol followed after 30 minutes by centrifugation.
Radioactivity in the pellets was determined by gamma counting. The assay sensitivity
was lOpg eGFP.
Figure 2.1 Typical mGH RIA Standard Curve
oCÛ
100 1
0.008 0.031 0.125 0,5 2.0
Standards (ng)
50% Bo = 0.130937ng ABL = 6.71%B o = 45.42%
Each sample was measured in triplicate, and values between Bo 30-80% on the standard curve were read.
These readings would then be multiplied according to dilution factor. (ABL - antibody blank, Bq - Bound
antibody)
- 7 2 -

( 'hunier M iü cr in is a iu l M e lh o d s
Table 2.2 NIHDDK antibodies and standards used for RIA
ASSAY ANTIBODYFINAL
ANTIBODYCONG.
STANDARD STANDARDRANGE
mGHmonkey
a-rat GH NIDDK
1:1,500,000mGH
NIDDKlOng-IOpg
mPRLrabbit
a-m ouse PRL NIDDK
1:400,000mPRL
NIDDK 5ng-20pg
mTSHguinea-pig
a-m ouse TSH NIDDK
1:150,000Rat TSH NIDDK
lOng-IOpg
mLHrabbit
a-rat LH NIDDK
1:750,000mLH
NIDDK I0ng-20pg
2.12 In Situ Hybridisation
2.12.1 Sectioning o f tissue fo r in situ analysis
Mouse tissue was mounted onto chucks in Cryo-M-Bed embedding medium (Bright
Instrument Company, Ltd., Huntingdon, UK) ready for positioning in a Bright Model
OTF cryostat with the chamber held at 16°C. In each case, tissue was cut until a uniform
section representing the majority of the cell types in that tissue was achieved. 12pm
sections were mounted onto cold gelatin and chrome alum-coated slides, placed onto a
warm hotplate for a few minutes to fix the section onto the slide and then stored at -70°C.
In order to cut brain sections, the samples were thawed slightly at room temperature and
the anterior part of each brain in front o f the hypothalamus was removed. An incision
was also made through the brain stem to provide a flat surface for mounting. Coronal
brain sections (12pM) were collected throughout the brain, selecting various nuclei (PeN,
SON, PVN, ARC, VMN, DM) which were recognised by specific land marks i.e. third
ventricle and optic chiasm, for further analysis.
73

(JlnPhrJ'__________________________________________________________
2.12.2 In vitro transcription
Plasmids containing polymerase promoters were linearized with a restriction enzyme and
re-purified. Anti-sense and sense radiolabelled riboprobes, incorporating [a^^S]-UTP
were generated using a SP6/T7 transcription kit (Roche Diagnostics). After transcription
probes were treated with RNAse free DNAse I kit (Roche Diagnostics) for 15 minutes at
37°C and purified on a Sephadex G-50 Nick Column (Amersham Pharmacia Biotech)
and eluted in lOmM Tris (pH 7.5), 0.1% SDS. 6-8 elutions were taken and Ipl of each
counted in 5ml scintillation fluid in a Beckman LS 5000CE counter.
2.12.3 Riboprobe hybridisation
In situ hybridisations were performed by a previously published method (Bennett et a l ,
1995). Briefly, sections were fixed in 4% paraformaldehyde (Sigma), acetylated (8.75%
triethanolamine. Sigma) dehydrated through graded alcohol solutions and delipidated in
chloroform. Sections were hybridised overnight at 45-50°C, under Nescofilm (Bando
Chemical Industries Ltd., Kobe, Japan) in buffer containing 1x10^ cpm probe/slide in
hybridisation buffer (50% formamide, 0.025M Tris pH 7.5, O.OOIM EDTA ph 8.0, 0.4M
NaCl, Ix D enhardfs solution [0.02% Ficoll, 0.02% polyvinyl pyrrolidone and 0.2%
BSA], 10% dextran sulphate [MW 500,000], lOg sheared single stranded homologous
DNA/ml and 5pg yeast tRNA/ml). Following hybridisation, sections were washed three
times in 2x SSC at room temperature, twice in 2x SSC/50% formamide at 45°C for 15
minutes each, twice in 2x SSC at room temperature then dipped in distilled water then
100% ethanol and air dried.
2.12.4 Image analys is
Slides were exposed to autoradiographic film (BioMax MR, Kodak, NY, USA) for one or
two days to determine the strength of the signal. Most films were exposed for 7 days
before quantifying integrated densities, using the Image Program (W. Rasband, NIH,
Bethdesa, MD). The values represent integrated optical density expressed in arbitrary
units. For each transcript, comparisons were made with the same batch of labelled
riboprobe on sections from all animals exposed concurrently.
- 7 4 -

(' 'hap ic r 2_ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ Mulct i(jis a n d M e!h o d s
2.13 Immunocytochemistry (ICC)
Double labelling immunocytochemistry was performed using primary anti-mouse
antibodies kindly supplied by NIHDDK. Concentrations of primary and secondary
antibodies are detailed in table 2.4.
Mouse pituitaries were dissected and fixed in 4% paraformaldehyde for 12 hours and
washed in 100% ethanol for 2-3 hours. Paraffin embedding, tissue sectioning and
mounting onto microscope slides was carried out by the NIMR histology service. Tissue
sections (6pM) were de-waxed in histoclear (National Diagnostics, Georgia, USA), taken
through 100%, 70% and 30% ethanol for 20 seconds each and then washed in distilled
water. Endogenous peroxidase activity was blocked in 1% H2O2 in 40% methanol at
room temperature for 30 minutes. Sections were then washed for 5 minutes in distilled
water and incubated in 0.05% trypsin (Sigma, UK) in 0.2% CaCh (pH 7.4) for 15
minutes, which reduces cross linking of antibody. Sections were washed with distilled
water for 5 minutes and incubated in 0.5% Triton-X for 15 minutes to permeabilise the
cell membrane. Sections were washed in distilled water for 5 minutes, then transferred to
Tris buffer saline (TBS) for 5 minutes. Non-specific binding was blocked by incubation
in 2% normal serum (matched to that of the animal in which the secondary antibody was
raised) in blocking buffer (NEN Life Sciences) for 1 hour. The sections were washed in
TBS, then incubated in the primary antibody at 4°C (diluted in normal serum/5 % BSA)
overnight. For control sections the first antibody was omitted. The sections were washed
twice in TBS and incubated in a biotinylated secondary antibody (diluted in normal
serum/5 % BSA) for 30 minutes at room temperature. After washing, sections were
incubated with TRITC/FITC-avidin for 30 minutes at room temperature. Finally,
sections were washed in TBS and mounted in DPX (BDH).
2. IS. 1 Preparation o f GH-GFP Pituitaries fo r ICC
Due to endogenous green fluorescent protein in pituitaries of transgenic GH-GFP mice, it
was necessary to develop a modified immunocytochemical procedure without harsh
reagents which were found to denature GFP. Therefore, mouse pituitaries were fixed in
4% paraformaldehyde for 12 hours, washed in acetone for 2 hours and embedded in
paraffin wax. Tissue sections (6pM) were de-waxed in histoclear (National Diagnostics,
Georgia, USA), taken through 100%, 70% and 30% acetone for 20 seconds each and then
washed in distilled water. After incubation in a blocking solution (20% normal serum.
- 7 5

( -?__________________________________________________ MiHcrld/s (Hid Mcl l imls
5% BSA in Tris.HCl saline buffer) for 30 minutes at room temperature, they were
exposed to the specific first antibody overnight at 4°C. Sections were washed in
Tris.HCl buffer, then incubated with a biotinylated secondary antibody for 30 minutes at
room temperature. After washing, sections were incubated with TRITC-avidin (1:1000,
Sigma UK) for 30 minutes at room temperature. When co-staining GH-GFP pituitaries
with GH, for co-localisation studies DAPI (Ipg/ml, Molecular Probes Inc.) was added for
2 minutes to stain cell nuclei. Finally, sections were mounted in TBS and sealed with
nail varnish. Sections were analysed on both light microscope and confocal microscopes.
2.14 Preparation of In Vitro Systems
2.14.1 Culture o f rat GC cells
All culture work was carried out in a Biomat Class II microbiological safety cabinet
(Medical Air Technology Ltd.). Cells were cultured using a method derived from
Kineman & Frawley (1994), using reagents obtained from Sigma, unless otherwise
stated. Cells were incubated at 37°C in a 5% CO2 incubator in complete medium
(Dulbecco’s Modified Eagles Medium, 15% horse serum, 2.5% foetal calf serum [PAA,
Weiner Strasse, Austria], 2mM L-Glutamine and Ix antibiotic-antimycotic solution
[penicillin/streptomycin/amphotericin]). Medium was changed every 2-3 days and cells
harvested by centrifugation o f the cell suspension at 130 x g for 10 minutes. Cells were
frozen at -70°C before long-term storage in liquid nitrogen.
2.14.2 GC Cell Transfection
GC cells (200,000/60mm culture dish) were transfected with plasmid DNA (pCDNA3.1,
[Invitrogen BV] containing either hGH-GFP, hGH, hGH-IVS3) using Lipofectamine
(Life Technologies). Briefly, l-2pg plasmid DNA were diluted in lOOpl Opti-MEM
(Life Technologies). This was added to 14pl Lipofectamine and lOOpl Opti-MEM and
incubated for 45 minutes to form DNA-lipid complexes. This solution was diluted in
0.8ml Opti-MEM and added to 50-80% confluent cells and incubated for approximately
16 hours. Opti-MEM medium was then replaced with complete medium for 48 hours.
Stably transfected cells were selected for neomycin resistance by addition of G-418
(250pg/ml, Life Technologies) or zeocin resistance by the addition o f zeocin (200pg/ml,
- 76 -

CJiü12_i_çL 2_____________________________________________________________________ Mi II crin is an d M dhncls
Invitrogen BV). Newly transfected cells were incubated with appropriate antibiotic for at
least 21 days to establish stable cell lines.
2.14.3 Dispersion o f mouse pituitary cells
The dispersion of pituitary cells is fundamentally similar for cell culture, FACS sorting
and FACS analysis. All reagents were obtained from Sigma, UK.
Whole pituitaries (5-10) were removed from mice and placed in a petri dish of Hank’s
Balanced Salt solution (HBSS). If the cells were destined for culture or sterile sort
followed by culture, the following procedures were carried out in a BioMAT Class II
microbiological safety cabinet (Medical Air Technology. Ltd, Manchester, UK). If no
cell culture was involved, the method was performed on the bench.
The cells were enzymatically dispersed using a method modified from Weiner et al.
(1983). The pituitaries were gently minced into small pieces and dispersed in HBSS
containing enzymes (0.1 mg/ml collagenase type lA, 0.25% trypsin) For cell culture, and
sorting, the pituitaries were pooled in 10-20ml and maintained at 37°C, 5% CO2 . After
30 minutes, DNAsel was added to the dipersion to a final concentration of 0.05mg/ml.
The cells were mechanically sheared (using a sterile syringe with a sterile filling tube) at
intervals o f about 10 minutes until no visible cell clumps remained (approximately 1.5
hours). The cell suspension was passed through a 40pM cell strainer (Falcon, Becton
Dickinson) and centrifuged at 130g for 10 minutes. The resulting pellet was processed
for culture, FACS sorting, or FACS analysis.
2.14.4 Primary culture o f mouse pituitary cells
Cells were cultured using a method modified from McNicoll et al. (1990) and Lieberman
et al. (1982), all reagents were obtained from Sigma, UK, unless otherwise stated. The
dispersed cell suspension was centrifuged at 130g for 10 minutes, washed in 10ml of
HBSS and re-centrifuged. The HBSS was then aspirated and the cells resuspended in
complete medium (Dulbecco’s Modified Eagles Medium, 15% horse serum, 2.5% foetal
calf serum [PAA, Weiner Strasse, Austria], 2mM L-Glutamine and Ix antibiotic-
antimycotic solution [penicillin/streptomycin/amphotericin]) and plated in 6 well plates
(Falcon, Beckton Dickinson) with poly-L-lysine coated coverslips at a concentration of
approximately 500,000-1x10^ cells per well.
- 7 7 -

_ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ ^ [ c r ù ils a n d M e lh o d s
2.15 Fluorescence Activated Cell Sorting (FACS)
2.15.1 GH-GFP GC Cell FACS analysis
Dispersed transfected GC-GFP cells were harvested by centrifugation from a confluent
flask and resuspended in 1ml FACS buffer (lOg/1 NaCl, 0.25g/l KCl, 1.37g/l Na2HP0 4 ,
0.25g/l KH2PO4 , lg/1 BSA pH 7.3) and passed through a 40pM cell strainer. The cells
were layered onto 4% BSA in FACS buffer and centrifuged for 5 minutes at lOOg to
remove cell debris. Cells were gently resuspended in 0.5ml of FACS buffer and analysed
on a FACS Star Plus Machine (Becton-Dickinson, CA) with WinMDI software, using the
FITC-cbannel to gate for eGFP fluorescence and the population o f cells was
electronically gated to exclude cell debris. Aliquots of the starting cell suspension, and
pools o f cells sorted by eGFP fluorescence intensity were collected and assayed for
mouse GH (mGH) content.
2.15.2 FACS sorting o f GH-GFP dispersed pituitaries
Dispersed anterior pituitary cells were prepared from 10 mice, as described in section
2.14.3. The pellet of dispersed pituitary cells was resuspended in full medium (not FACS
buffer) prior to sorting (the media was filtered because small particles in serum registered
in the FACS machine). An aliquot of unsorted cells was removed to serve as a control.
The remainder of the cells were sorted for fluorescence on a FACS Vantage (Becton-
Dickinson, Mountain View, CA).Positive and negative cell populations were collected
into FACS tubes (Falcon, Becton-Dickinson) containing 250pl Of complete medium.
2.16 In vitro GH Release Studies
These studies were performed by Danielle Carmignac (NIMR) on GH-GFP transgenic
mouse pituitaries provided by me. In brief, freshly dissected pituitary glands were placed
in 2ml Dulbecco’s Modified Eagle’s Medium (Sigma) without L-Glutamine, rinsed
several times and then incubated for 2 hours at 37°C with the medium changed every 30
minutes. Following this washout period, the pituitaries were incubated in 0.5 ml aliquots
of medium, exposed to Ipg/ml bGRF 1-29 NH2 (Bacbem Inc) and after a further 90
minute recovery period, to 5pg/ml bGRF 1-29 NH 2 . The medium was collected and
assayed for GH and eGFP contents by RIA (see section 2.11).
7 8 -

( _ 7 ? ( , y > / i 7 - ? ___________ _ \ ! i i i c r i ( i i \ ( j i h l VI c i I u h / s
2.17 Electron Microscopy
Electron microscopy was performed by me in the Department of Anatomy, University of
Oxford, with the help and advice of Dr. H. Christian. I am grateful to Prof. John Morris,
for allowing me access to his facilities.
After initial fixation (2.5% glutaraldehyde in phosphate buffer for 2 hours, then 0.25%
glutaraldehyde overnight) pituitary segements were post-fixed in osmium tetroxide
(l% w /v in O.IM phosphate buffer) stained with uranyl acetate (2% w/v in distilled
water), dehydrated through increasing concentrations of ethanol (70%, 80%, 90%, 100%)
and embedded in LR Gold (London Resin Company, Reading, UK) for immuno-staining,
or Spurr resin for cell morphology. EM sections were kindly cut by Sarah Rodgers,
University o f Oxford). Ultrathin sections (50-80nm) were incubated at room temperature
with a primary antibody for 2 hours, then followed by a specific secondary antibody
linked to gold or protein A linked to gold (British Biocell, Cardiff UK) for 1 hour at room
temperature. All antisera were diluted in O.IM phospahte buffer containing 0.1% egg
albumin. For control sections, the primary antibody was replaced by an unrelated
monoclonal antibody. After immunolabelling in LR Gold and Spurr resin, sections were
lightly counter-stained with lead citrate and uranyl acetate and examined with a
transmission electron microscope (JEM-1010, JEOL, Peabody MA).
2.18 Total Internal Reflection Fluorescence (TIRF) Microscopy
All TIRE microscopy was done entirely as collaboration with Jean-Baptiste Manneville
(Department o f Physical Biochemistry, NIMR). Evanescent wave microscopy, also
termed total internal reflection fluorescence microscopy (TIRF) has shed new light on
important cellular processes taking place near the plasma membrane. The specialised
technique of TIRF specifically illuminates fluorophores in the thin optical plane on the
coverslip and provides vertical resolution and signal to noise (S/N) ratio that is
unmatched by any other light microscopy technique. It is particularly well suited for
real-time motion analysis o f growth hormone vesicles in somatotrophs from the GH-
eGFP mice that we have developed within our lab.
- 7 9 -

{Ju,pu Malcriül.' and Mclhads
2.18.1 The E vanescent Wave
The principle, based on S n e ll’s law , is straightforward: i f light travelling in a high
refractive index medium (ni i.e. glass, n=1.5) strikes a lower refractive index medium, (u2
i.e. water, n=1.33) beyond a certain critical angle, 0c, the light w ill undergo TIR. This
critical angle depends on the relative refractive indexes o f the two media.
refracted
TIR(reflected)incident
n\ sin0i = ni s in 02
Cells are grown on glass coverslips, which have a high refractive index, and a laser beam
is optically coupled into the coverslip by a prism. W hen the angle o f incidence is below
the critical angle ( 0 i < 0 c ) , light w aves are refracted into the solution, and are sinusoidal
with a characteristic period. As the angle approaches 0c the period increases and the
refractive beam becom es more parallel to the surface. At the critical angle the period is
infinite, w ith the w avefronts o f the refracted light normal to the surface. O w ing to
interference o f the incident and reflected light a standing w ave is generated in the
optically rarer medium (02), termed the evanescent wave.
The probability o f a fluorophore within the evanescent w ave being excited decreases
exponentially as a function o f distance away from the interface, but the intensity o f the
fluorophore v iew ed varies in a more com plicated fashion, because the fluorescence
lifetim e o f a fluorophore and the angular pattern o f the em itted radiation are affected by
the proximity o f the surface (Axelrod et al., 1992). For my purposes, the net effect is that
on ly fluorophores very near the coverslip (w ithin lOOnm), corresponding to the
“footprint” o f the cell are excited; for this reason, one o f the first applications o f the
- 8 0 -

_ _ _ _ ...____ ______ ________ :IÀ ihiud^AiihLMcilhJih.
technique was to monitor cell-substrate contacts (Thompson et al., 1997, Bummeister et
a l, 1998).
2.18.2 A dvantages o f TIRF
Compared with the more familiar confocal system, TIRF illuminates a vertical slice of
5~100nm as opposed to a slice of ~500-800nm for 1- and 2- photon confocal system
respectively. This thin optical sectioning means that the signal to noise ratio is much
better than with confocal images, and photobleaching is minimised. Confocal
microscopes can generate deep three-dimensional (3-D) images of cells but photo-bleach
areas o f interest. However, when TIRF is used as a complementary approach with other
microscopy techniques, such as bright-field, epifluorescence and confocal microscopy,
good spatial-temporal resolution can be achieved e.g., membrane trafficking and fusion.
Many biochemical, genetic and EM experiments have indicated that regulated and
constitutive vesicles traffic to and fuse with the plasma membrane. The technological
breakthrough of patch clamping opened the door for detecting changes in surface area
(capacitance) or release of oxidative material with high temporal resolution (Neher,
1998), but only the fusion event is monitored and prior vesicle trafficking, tethering and
docking are not detected. In studies of regulated exocytosis in PC 12 cells and chromaffin
cells, specificity is difficult as all fusion events are detected and most studies have been
limited to regulated exocytosis (Henkel & Aimers 1996; Parsons et a l , 1995).
Conversely, EM has exceptional spatial resolution, but only gives “snapshots” of the
process. TIRF offers an encouraging compromise in that both spatial and temporal
resolution can be attained. The somatotrophs from the anterior pituitary o f our GH-eGFP
mouse is a good model to use as somatotrophs are well characterised, and are packed fiill
of large GH granules endogenously labelled with eGFP.
However, this technique has its disadvantages - only the first lOOnm of the cell, attached
to the coverslip can be viewed. This limitation means that only a small area of the cell
surface can be monitored which will not give a true overall representation of exocytotic
events.
- 8 1

L M lUcvkjIs (i>hl M cihods
2.18.3 Exocytos is in A ction
When studying the mechanism of secretion using TIRF microscopy, vesicles, whether
granules, constitutive vesicles or synaptic vesicles, can be followed from where they first
dimly appear in the evanescence field (~100-400nm) until they fuse with the plasma
membrane as bright flashes. In calibrated systems, fluorescence changes can be
extrapolated to changes in axial position. This means that, although the cells are only
imaged in two-dimensions, 3D information on the position o f vesicles can be obtained.
The tracking o f vesicles has led to some interesting observations. Granules and synaptic
vesicles were observed to approach the plasma membrane at an angle, presumably guided
there by the cytoskeleton (Steyer & Aimers 1999; Steyer et al., 1997). The lateral
diffusion of granules and synaptic vesicles decreased approximately five fold when the
granules ‘docked’ near the plasma membrane, suggesting restrictive diffusion, perhaps
due to the cortical actin meshwork (Steyer & Aimers 1999; Steyer et al., 1997; Oheim &
Stuhmer 2000). The priming time or time taken to go from a morphologically docked to
fused state varied considerably (0.25s for synaptic vesicles Zenisek et al., 2000), -40s for
constitutive vesicles (Toomre et al., 2000). Many constitutive vesicles were found to
‘dock’ for several minutes, and a significant fraction (>25%) of docked vesicles did not
fuse and detached from the membrane (Zenisek et al., 2000; Oheim & Stuhmer 2000;
Toomre et al., 2000). A dual-colour study performed by Tsuboi and co-workers indicates
that granules can kiss the membrane and secrete soluble cargo, yet retain membrane
proteins associated with the granule, favouring the “kiss and run” model of vesicle
recycling (Neher, 1993; Aimers & Tse, 1990; Tsuboi et al., 2000).
The questions that may be answered using the GH-eGFP model are numerous. To date,
no other study has targeted eGFP to somatotrophs in anterior pituitary cells. The
mechanism of synthesis and secretion of GH granules in somatotrophs in vivo can now be
visualised in real-time using non-invasive techniques to identify these GH granules in
somatotrophs.
2.18.4 The TIRF Microscope
The experimental set up can be seen in figure 2.2. A beam of frequency doubled Argon
ion laser (488nm, 25mW, Model IMAlOl, Melles Griot, CA, USA) was expanded by a 5
8 2 -

( 'hd/'h'r r \ i i ucrictls an d Mcîhods
ion laser ^
Computer
VideoI-CCD
Band pass filter
Excitation filterDichroic
filterBeamexpander
H g lamp
Objective(xlOO)
Attenuator
\CT Lens 3
hot water circulation
5% CO2 chamber
/E3
Figure 2.2 A Schematic of the TIRF set-up.
The TIRF microscope has been adapted to reproduce cell culture conditions (37°C, 5% CO2) to allow
visualisation o f live cells for extended periods o f time. (M=mirror [45°]).
X beam expander (LBX 001, Melles Groit, CA, USA), passed through an attenuator
(925B, Newport, CA, USA), and focused by a 400mm focal length lens (KPX115,
Newport, CA, USA). The beam then entered the upright microscope (Axiotech^^'°, Zeiss,
Germany) through a truncated triangular flint glass prism (ni = 1.46, UQG ltd., England),
striking a glass slide at an angle of 65-67° (critical angle: sin 0c = n2/ni = 0.887 ; 0c =
62.46°). The gap between the prism and the slide was filled with very low fluorescence
immersion oil. Fluorescence light was collected by a water immersion objective (lOOx,
1.0 NA, Achroplan, Zeiss, Germany). The emitted light then passed through a dichroic
filter (505DRLP01, Omega Optical, VT, USA) and a band pass filter (530DF30, Omega
Optical, VT, USA).
- 83 -

Llli-JJ’lL r 2_____________________________________________________ .\Jii!cri(i/s cuh! S 'iahods
Epi-fluorescence illumination was stimulated by directing the focused beam of a mercury
lamp through a 485±20nm excitation filter vertically through the prism toward the
objective. Brightfleld illumination was also simulated by directing the focused beam
from a white lamp vertically through the prism towards the objective. The emitted light
from both brightfleld and darkfield images were passed through the dichroic filter and
collected with a standard CCD camera (XC-75CE, Sony, Japan). The experimental
temperature was controlled by means of hot water circulation (RTE211, Neslab, UK) and
a controlled heating jacket (NIMR Electronics/Engineering Dept, UK) fitted to both the
prism and the objective, providing a temperature of 37°C.
2.18.5 Image acquisition
Detection of fluorescence was by intensified CCD camera (Remote Head Darkstar, S25
Intensifier, Photonics Science, UK). Video images were digitised at video frame rate by a
frame grabber (IC-PCI 4Mb (AMVS), Imaging Technology, MA, USA) and
simultaneously recorded on SYHS (BR-S800SE, JVC, Japan).
The frame grabber digitises the analogue signal at video standard 25 frames/sec, creating
a sequence of images stored in the computer memory. At the end of each experiment, the
images were then stored to the computer hard drive and subsequently on CD as files in
the .tif format. Control of this process, and subsequent analysis was by the image
processing package Optimas version 6.5 (Optimas Corp. WA, USA).
2.18.3 Image Process ing
Image processing was carried out using Optimas vs. 6.5. using a variety o f analysis
functions available as ALI functions (Analytical Language for Images), the macro control
language used by Optimas. Generally, measurements were made on lines or areas defined
as Regions of Interest (ROI), providing real or integer values. Analysis was performed on
raw images, with greyscale values (0-255) of a range that was defined in the camera and
software settings (gain and offset) at the time of image acquisition.
2.18.4 Calibration
All images were spatially calibrated using a 1mm graticule (Graticules Ltd, UK) using
the ‘Calibrate Spatial‘ function available in Optimas, collecting reference images for both
- 8 4 -

(■ 'Inij' icr J________________________________________________ Mnlcridls a n d M d h o d s
cameras at each magnification used. With a video monitor, the aspect ratio determines the
shape of the image displayed on the screen. A typical computer monitor is four units
wide by three units tall, yielding an aspect ratio of 4:3 or 1.33. Using a reference image of
the graticule spacing, distances were calibrated in both the x and y directions, normalising
for the aspect ratio o f the displayed images. The calibration was checked by summing
together experimental and reference images and drawing a line of known length based on
the reference graticule.
2.19 Data Analysis
Unless otherwise stated, data are shown as Mean ± the standard error o f the mean (SEM).
Differences between groups were analysed by Students t test, if the standard deviations of
the two populations being compared were not statistically different. Where the standard
deviations were different, the non-parametric M ann-W itney U test was used.
Significance levels are shown as p< 0.05 = p<0.01 = p<0.001=
- 8 5 -

( 'h a i 'h ‘1' ^ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ J n V!’rn ii: il ( il I r c l i h i u v / < f / > / r n-iinsjcclc i/ with r G V 'Y'*
Chapter 3
In vitro studies in a GH cell line stably transfected with eGFP
3.1 Introduction
A variety of clonal strains of pituitary tumour cells (MtT/W5) were established from the
Wistar-Furth rat in 1965 (Tashjian et aL, 1968). The GC cell is a cloned variant of the
original GH] cell producing both GH and Prl. The GC clone has a greater basal rate of
GH synthesis than that of GH] cells, but synthesises little detectable Prl (Bancroft, 1973).
The GC cell has specific responses to SRIF and thyroid hormone, but no response to
GHRH (Tashjian et aL, 1970). These cells can also stimulate body weight gain and linear
growth after injection into normal or hypophysectomised Wistar-Furth rats (Tashjian et
a l, 1968) or transgenic growth retarded rats (TGR’s) (Pellegrini et aL, 1997) in which
they form encapsulated tumours.
Studies o f living populations of pituitary GH cells would be greatly facilitated by the
ability to visualise secretory processes directly in cells. As previously mentioned in
chapter 1, enhanced green fluorescent protein (eGFP) has been used widely in cell
biology in order to visualise and study cellular processes in real time (Tsien, 1998). One
way to achieve this is to use GFP as a fusion partner with GH sequences which when
expressed from a cell-specific promoter would allow identification and isolation of
growth hormone producing cells. Rat GC cells were an ideal system to test the
expression of a human growth hormone/eGFP fusion cassette in a rodent GH cell line
before making a transgenic mouse model.
In this chapter, two constructs containing eGFP are discussed. The first contains only the
first 8 residues of the hGH signal peptide, the second construct contains the intact signal
peptide and a further 22 N-terminal residues of hGH. The choice o f the length of the
additional N-terminal peptide was guided by several considerations. With both
constructs, the first intron o f the hGH gene was included, since this contains enhancer
sequences that could be important for efficient transgene expression (Brinster et aL,
1988) and for appropriate transcription in our transgene construct. It was probably not
- 8 6 -

L'/Z'V'/i'/' J '_ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ In viiro in , ; 07/ cc// //;/,/ \lcihlv li\i)]\fci-tc(l with cG h !^
necessary for GC cells in vitro, but we felt it necessary for transgenic mice, more fully
discussed in chapter 4. This intron occurs after the first three residues of the signal
peptide. We aimed to make the shortest peptide possible, but avoiding altering the
nucleotide sequence context close to the splice acceptor site, but with a convenient
cloning site nearby. We chose an extra 15bp 3' including a further 5 residues o f the
signal peptide from exon 2 before fusing with eGFP. The accuracy and efficiency of
splicing is often affected by the nucleotide sequence in the immediate vicinity o f the
splice junction (e.g. the length and/or sequence composition of exons, Reed & Maniatis,
1986; Matsuo et aL, 1991), perhaps as a result o f secondary structure considerations
(Krawczak, M., Reiss, J. & Cooper, D.N., 1992). We felt it was unlikely that this short
N-terminal peptide extension would provide sufficient signal peptide recognition
sequence to alter the cytoplasmic fate of eGFP.
The construct aimed at targeting eGFP to secretory vesicles (p48GH-GFP) included not
only the entire hGH signal peptide, but also the first 22 residues o f the N-terminal
sequence of hGH. This was chosen as it also included the two N-terminal histidine
residues o f hGH (’^His and ^’His). These contribute significant binding activity to
hGH and may be involved in GH dimérisation and subsequent packaging. In previous
hormone secretion studies with GH (Cunningham et aL, 1991a) Prl (Dannies et aL, 1999)
and insulin (Dodson et aL, 1995) zinc has proven to be an important factor in
oligomerisation and packaging of protein hormones for secretion. As these His sites were
only 21 residues downstream of the hGH signal peptide, we thought it unlikely that the
additional residues would affect the expression o f GFP, but only act to enhance any
potential co-packaging of the GH-GFP fusion protein in mouse GH vesicles. However,
they would not be sufficient to confer any GH-bioactivity on the fusion product. There is
another zinc binding site in human growth hormone (^^"^Glu), but this could not be
included in the transgene as it would include too much GH sequence.
C. Magoulas (NIMR) engineered these two eGFP constructs largely before my arrival in
the lab, although I was involved with the final stages o f cloning into the cosmid
construct. GFP requires relatively strong promoters to drive sufficient expression for
detection, especially in mammalian cells, so for the purpose o f expressing GFP in GC
cells I used the constitutive promoter from cytomegalovirus (CMV). These cells were
8 7 -

c hh s tiiiîics in il ( i l l ccU line slah lv Iruns fcc lcd with cGF!^
studied using light and confocal microscopy and analysed using fluorescence activated
cell sorting (FACS) techniques.
3.2 Construction of HGH-eGFP plasmids for transfection of GC cells
As described above, two different lengths of the 5’ coding sequence of the human GH
gene (Jones et aL, 1995) were fused in frame with an enhanced variant o f GFP (eGFP,
Clontech Inc.). The longer version of the hGH-eGFP fusion construct (p48GH-eGFP),
contains a genomic sequence encoding the first 48 amino acids of the hGH gene product
(signal peptide and N-terminal 22 residues o f hGH) fused in frame via a 15mer
oligonucleotide linker to the coding sequence of eGFP. Briefly, an Xmal-Notl fragment
(750bp) of the pEGFP CMV expression plasmid (pEGFP-N3; Clontech Inc.) was blunt-
ended by Klenow and ligated into the Pvull sites o f an hGH genomic clone (Flavell et al.,
1996) containing 5’-and 3 ’ untranslated hGH sequences flanked by an M lul linker (the
choice o f an Mlul adapter was guided by the subsequent transgenic strategy, described
later in chapter 4). This Mlul fragment was then cloned into a version of the pEGFP-N3
expression fragment (pN3/M), modified by insertion o f a Mlul cloning site in place of its
Xmal-Notl fragment (see figure 3.1).
A shorter version of the hGH-eGFP fusion construct (p8GH-eGFP) was derived from
p48GH-GFP and contained genomic sequence encoding only the first 8 amino acids of
the hGH signal peptide 1 inhered in frame with eGFP as described above. This was
engineered by a PCR strategy based on p48GH-eGFP as a template. The forward primer
(primer F 1) was 5’ vector sequence that introduced multiple cloning sites upstream of
the amplified hGH sequence. The reverse primer (Primer R 2) was designed to recognise
the hGH coding sequence at codon 8, flanked by a BamHl cloning site. The PCR product
of this reaction was then inserted in place o f the EcoRl-BamHl fragment of the p48hGH-
eGFP plasmid construct (figure 3.1).

( liapter 3: In vitro siudies in a GH veil Hue siahly Iransfecied w ith eCil'P
Xm cilA TG
N ot!
eGFP
1.M ill!
I_________ A TGP vull
I A TG
CM V48aa
eGFP
1 .3 k B
IP vull M in i
± = 'h G H poly A tail
2 .
P F l
E c o R l M lu l P vullA TG
hGH
48aaBamHl site
, P R l
I
eGFP
8aa
P vu ll M ildI
■HGH poly A tail
CM VA,TG 1 A TG 1
hGH 1 1 eGFP g
hG H poly A tail
3.1 hGH-eGFP Constructs
T w o plasm id constructs w ere engineered and cloned in the m am m aliam expression vector pEGFP
-N 3 containing a C M V promoter driving 5' and 3' sequences o f the hGH gene. Construct 1
contains eG FP linkered in frame to the intact hGH signal peptide (26aa) and a further 22aa o f
hGH (p48hG H -eG FP). The X m aV N oil eGFP fragm ent w as blunt-ended and cloned into P v u ll
sites o f an hGH gen om ic clone, flanked by an M lu l linker. The M lu l fragm ent w as cloned into
pE G FP-N 3, m odified by insertion o f a M lu l cloning site in p lace o f its X m aV N oil fragment.
Construct 2, the shorter version (p8G H -G FP) w as derived from p48G H -G FP and contains
gen om ic sequence encoding only the first 8aa o f the hGH signal peptide linkered in fram e w ith
eG FP. This w as engineered by PCR m utagenesis. The PCR product w as then inserted in p lace o f
the E coR I/B am H I fragm ent o f the p48G H -G FP plasm id construct. H atched bars are vector
sequences. For sim plicity, the intron interrupts the GH signal peptide after residue 3, and has
been om itted from this cartoon. Primers FI and R1 listed in appendix.
- 89 -

In \ni:l in\ in i ell I n w . ^ V i / V r / ranslcclccl wi ih cCih'I’
3.3 Expression of p8GH-eGFP and p48GH-eGFP in rat GC cells
Plasmids containing eGFP fused to two different lengths o f the amino terminus o f hGH
(figure 3.1) were transfected into the GH producing GC cell lines, and several stable lines
were established by incorporation of neomycin into the media for 4 weeks for selection.
Expression of eGFP in these cells was examined by confocal and electron microscopy.
Both constructs produced brightly fluorescent cells, but with a markedly different
distribution o f fluorescence as seen in figure 3.2a and 3.2c.
The construct containing only 8 amino acids of the GH signal peptide fused to eGFP
showed a relatively uniform distribution of fluorescence throughout the cells and it was
observed that some cells fluoresced more brightly than others. I analysed three
independent transfections of this construct, all o f which had cytoplasmic fluorescence,
bright but varying in intensity.
The construct with the intact signal peptide and the first 22 residues o f hGH gave a
completely punctate distribution of fluorescence (fig 3.2c), consistent with vesicular
targeting o f this fusion product. This can be seen at higher magnification in confocal
microscopy scanning through a single GC cell in figure 3.3. To check the distribution of
eGFP throughout the cell, figure 3.4 shows images from scanning confocal microcopy.
Our aim in engineering the p48hGH-eGFP construct was to target eGFP to GH secretory
granules. By comparing the expression of GFP from this p48hGH-eGFP with the control
p8hGH-eGFP construct we have shown that the sequence chosen was sufficient to target
GFP to vesicles in rat GC cells. The following studies in this chapter use the longer
version of the eGFP construct (p48GH-eGFP).
9 0 -

Figure 3.2 Expression of eGFP in GC cell lines
GH producing ce lls w ere transfected w ith C M V prom oter p lasm ids contain ing the
reporter constructs show n in figure 3.1 and stable cell lines were generated. L iving cells
in culture w ere exam ined by confocal m icroscopy. Left panels (A, C) show endogenous
eG FP fluorescence. R ight panels (B , D ), phase contrast im age. The construct with only
8 aa o f the GH signal peptide (A) show ed an intense, even ly distributed, fluorescence
signal. The construct with the entire signal peptide and part o f the am ino term inus o f
hGH fused to eGFP (C) gives a punctate distribution o f fluorescence. Scale bar = 10pm.
- 91 -

( 'lhi/?lL’r3: _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ In viiro .s/iulic.s in a ( iH cell line siiih/y inutsfcvtcd m ///? U P
Figure 3.3 Confocal Microscopy imaging of a p48hGH-cGFP GC cell
Confocal microscopy imaging of p48hGH-eGFP cells shows the typical granular
distribution of eGFP (magnification = xlOO)
- 9 2 -

Figure 3.4 Confocal microscopy scanning of GC cells expressing eGFP targeted to granules (p48hGH-eGFP)
The confocal scan runs from top to bottom (1-6) of a single ph48hGH-eGFP GC cell. The nucleus is easily detected in 2, 3 and 4.
GFP distribution is granular. Scale bar = 10pm.

_ _ _ _ _ _ _ _ ___ _ _ _ _ _ _ _ _ _ _ _ _ _ _ 1j1 studic.', ill (I a n ca'I l ine sidhi]- v / with c G I ' ! ’
3.4 Growth rates and GH-GFP protein content of GH-eGFP cells v WT GC cells
From confocal and light microscopy observations, GC cells transfected with p48GH-
eOFF did not appear to look abnormal compared to wild-type GC cells. However, in
order to check that the expression of GFP had no deleterious effects on cell growth, I
performed simple counting studies. Over the course of 14 days, I maintained two culture
flasks of GC wild type cells, and p48GH-eGFP transfected cells. Both flasks were
inoculated with the same number of cells in 25 ml of complete media and counted every
two days by extracting duplicate aliquots of 1ml of cell suspension which were counted
with a haemocytometer. The results (figure 3.5) confirmed that the GC cells stably
transfected with p48GH-eGFP showed the same growth rates as wild-type GC cells (GC
wild-type 7.85x10^ GC-GFP 7.87x10'').
I performed RIA of GC cell homogenates to ascertain if GFP disrupted rGH production
in hGH-eGFP transfected cells. I counted and collected an equal number o f WT and
hGH-eGFP transfected cells (2x10^), spun the cell suspension, re-suspended in 1ml of PB
and homogenised each before RIA. The rat growth hormone content in hGH-eGFP
transfected cells was similar to that in GC untransfected cells (hGH-eGFP:
117±3|ag/2xl0^cells; GC: 125±5ng/2xl0^cells). Note however, the amount o f eGFP
detected by RIA was approximately 1000 fold less than that of GH (170±9ng/2xl0^cells).
3.5 Fluorescence activated cell sorting
The endogenous eGFP fluorescence could be used to analyse and enrich populations of
GC cells by fluorescence activated cell sorting (FACS). A typical FACS analysis of
control and transfected p48hGH-eGFP GC cells is displayed in figure 3.6. The
untransfected GC cells show little auto-fluorescence (0.1% of total cell population). I
took this to define an arbitrary cut-offline for GFP fluorescence. FACS analysis o f hGH-
GFP transfected GC cell lines (clone 1.2 and 3.2) demonstrate a population of strongly
fluorescent cells, but this did not distribute as a single population, suggesting that some
low expressing GFP cells may also be present in the cut off zone.
- 9 4 -

( Im p ie r 3: /n vitro sti/c/ies in a (}H ce// Une siahly iniusfec/ed w ith eCrI'P
esC
01E3z
8
6
4
2
02 4 6 8 10 12 14
- O — GC W ild-type
^ ...... G Cp48G H -eG FP
Days
B
%
CSXo
200
150 -
100 -
200 ,
100 * -
GC WT hGH-eGFP hGH-eGFP
3.5 Growth rates and rGH content of GH-eGFP cells v WT GC cells
A. Growth rates o f GC wild-type cells and GC-GFP cells were compared by performing simple counting
studies. Throughout 14 days, two culture flasks of GC wild type cells, and p48GH-eGFP transfected cells
were maintained Both flasks were innoculated with approximately the same number o f cells in 25 ml of
complete media counted every two days by extracting duplicate aliquots o f 1ml o f cell suspension and
counting with a haemocytometer. The black line represents the GC wild-type cells and the green line GC-
GFP (p48GH-GFP) transfected cells.
B. RIA o f rGH in WT and transfected cell lines shows no significant difference in amount o f endogenous
rGH with the expression o f GH-GFP construct. RIA o f eGFP content in hGH-eGFP transfected GC cells
(green bar) reveals a 1000 fold reduction in protein content compared to GH.
-9 5 -

( iKipter 3:
A)
SB
g
In v in o s l iu i ies in a ( jH c e l l line sicihlv iransfecicU v i i h e ijI- P
GC controls
GFP channel 0.1%
B)GCGFP #1.2
C)
GFP channel 29.3%
GCGFP #3.2
GFP channel 49.5%
0 10 100 1000 10000
Fluorescence intensity
3,6 FACS analysis of p48hGH-eGFP GC cell lines
(A) FACS analysis o f untransfected GC cells. Fluorescence intensity is plotted along the x-axis and cell
number is plotted along the y-axis. The broken line to the right o f the cell population represents an
arbitrary cut o ff linefor auto-fluorescence (amount o f fluorescence 0.1%).
(B) GC-GFP 1.2 and (C) GC-GFP 3.2 are typical FACS analyses o f transfected hGH-eGFP cells,
illustrating a broad population o f intensely fluorescent cells (B=29.3%; C=49.5%).
- 9 6 -

( In/p ie r 3: fil viiro studies in a ( , / / cell line siah/v iransfecteci with eGFP
10» 10 ' 102 103 10° 1 0 ’ 102 103 1 0 *
1 0 ‘ R17 R18
R19R8
1 0 ' -
10» 1 0 ' 1 0 * 10' 102 1 0 ° 1 0 *
1 0 *R18R17
R20R19 R25R8
10 ' -
10 *1 0 '
247
1 8 5 -
123
10° 1 0 ’ 1 0 *
Figure 3.7 FACS sorting o f hGH-eGFP GC cells lines
This figure shows scatter plots (left panels) and distribution (right panels) o f fluorescent intensities in GH-
eGFP GC cells subjected to FACS sorting and analysis. Top panels. Gates were set arbitrarily to define
GFP-ve (R19=12%) and GFP+ve (R20=88%) populations. Within these quadrants, subpopulations were
collected o f extremely low (R8=4.7%) and highly (R25=38%) fluorescent cells, cultured separately for 3
days and then re-analysed by FACS. Middle panels show cells from the R25 culture. The vast majority
o f these continue to be GFP+ve (R20= 98.1%) and highly fluorescent (62.5% within the R25 gate). Lower
panels show cells from the R8 culture. Very few GFP+ve cells emerge during culture (R20=1.4%;
R25=0.7%).
- 9 7 -

( lu ip le r 3: hi vitro studies m a (iH cell line stably transfecicd n ifh eGhP
In order to investigate the differing intensities of fluorescence within a single population
of transfected GC cells, 1 repeated a FACS experiment but collected the cells, under
sterile conditions, and divided them into populations of “weakly” fluorescing cells and
“strongly” fluorescing cells. This was achieved by setting electronic gates at either end
of the fluorescence scale to collect exclusively the “very bright” cells or the “very dim”
cells. These sorted populations were then re-cultured for 3 days before FACS analysis
(figure 3.7). 1 found that the cells enriched for intense fluorescence contained almost
100% fluorescing cells (98.96% GFP+ve, 1.04% GFP-ve). The cells that were collected
and re-cultured from the opposite end of the scale, containing little or no fluorescence,
recovered a small population of cells (3.36% GFP+ve) which were brightly fluorescing.
3.6 Total Internal Reflection Fluorescence Microscopy (TIRE)
Total internal reflection fluorescence microscopy (TIRF) is particularly well suited for
real-time motion analysis of secretory vesicles in GC cells transfected with p48hGH-
eGFP. As previously described, vesicles in these transfected GC cells contain
endogenous GFP. In collaboration with J-B Manneville (Physical Biochemistry, NIMR)
we have been able to analyse the GH granules in these cells.
The TIRF set up is described in chapter 2 (2.17). The cells imaged were stuck firmly to
the surface of the slide, only the first hundred nanometres were illuminated, as
represented in the diagram below:
GH-GFP vesicles
cell cytoplasm
cell plasma membrane
/(2 )= /oexp (-z /ô )
with Ô ~ 100-500nm
- 9 8 -

( 'InipU' r .■>_________________________ __________________ In vif!u> sfitd i cs in ii ( J L L i n i l Um vA i t J y J l i ‘h i d HvV// c i U 'P
The TIRF images of GH-GFP transfected GC cells show GFP fluorescence in vesicles,
near the plasma membrane of the cells attached to the slide. Some spots fluoresced
brightly, others more dimly. Five hundred frames could be taken without significant
photo-bleaching, allowing time-resolved studies at the level o f single granules. Because
the intensity o f the evanescent wave rapidly declines with distance from the glass
substrate, vesicles closest to the plasmalemma are expected to be the brightest, while
those more than 300nm away are too dim to be resolved. Real-time imaging o f these
GH-GFP granules showed that they moved and could be tracked. Some granules slowly
appeared and faded from view, while they moved into and out of the evanescent wave;
some granules remained stationary. There were also a few granules that could be tracked
successfully, becoming brighter, before stopping and disappearing abruptly. Perhaps
these granules were docking and undergoing exocytosis and the GFP, thus released,
would diffuse away.
Figure 3.8 shows TIRF images of GH-GFP GC cells, which have been captured and
analysed. The raw fluorescent TIRF image (fig 3.8a) was filtered to enhance its contrast
(fig 3.8b). Each frame can be superimposed onto the previous one to visualise the path of
vesicles, which can be seen for 10 frames in figure 3.8c. In order to visualise long-range
movement in GC cells, 500 frames were filtered and superimposed (fig 3.8e) and
skeletonised (fig 3.8f) using custom written macros in imaging analysis software (J.B.
Manneville, NIMR).
Analysis o f single granule trajectories determines whether the motion is random or
directed. This is achieved by computer assisted analysis routines based on the imaging
software Optimas and the ATI (Analytical Language for Images) to quantify in real-time
the two- and three-dimensional motion of the vesicle. Figure 3.9 displays two types of
vesicle movement in GC cells, showing long-range directed motion in 3.9a and short-
range diffusive motion in 3.9b. X and y plots display parallel, horizontal motion, while z
plots vertical motion. The long-range directed motion of vesicles signifies vesicular
transport on the cytoskeleton. Most o f the granules observed displayed short-range
diffusive motion and were most likely bound to the plasma membrane or docked.
- 9 9 -

Figure 3.8 TIRF microscopy of GH granule motion in p48hGH-eGFP GC cells
The raw fluorescent TIRF image of GH-eGFP cells (A) is FFT (Fast Fourier Transform)
filtered (B) to enhance its contrast. Each frame can be superimposed onto the previous
one to visualise the path of vesicles (C shows path of vesicles in 10 frames). Long-range
movement can be visualised by filtering and superimposing 500 frames (E ) and
skeletonising (F). 1 frame = 40ms; scale bar = 5p.m.
- 1 0 0 -

^ long-range % ^V . k motion " #
f ® V * v■ . ^ > • *
\ stow diffusion4 # '
N -0 05
■0 15
-0 2-0 5 0 5
t i m e ( s e c )
-0 5 0 0 51 1
L o n g - r a n g e , d i r e c t e d m o t i o n
B 1
0 20 50 15
'-0 05
- 0.5 20
t i m e ( s e c )
1- 0.5 0 0.51 1
X (nm) S h o r t - r a n g e , d i f f u s i v e m o t i o n
Figure 3.9 Analysis of single granule trajectories in ph48hGH-eGFP GC cells
Analysis o f single granule trajectories in TIRF microscopy determines whether their motion is random or directed. Two types o f vesicle movement in GC cells
are displayed above, showing long-range directed motion in A and short-range diffusive motion in B. and y ploys display parallel, horizontal motion, while z
plots vertical motion. The long-range directed motion of vesicles (shown by the arrow in the top xy plot) signifies vesicular transport on the cytoskeleton. Most
o f the granules observed displayed slow, short-range diffusive motion and were most likely tethered or docked to the plasma membrane.

L _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ . . . . . . . / / ' viirii in (I ( HI ( ' ( . ' / / / / / ; , .\!iih!]- l i i u n l c c icci with. c d l T
3.7 Discussion
GFP has been used widely in cell biology in order to visualise and study cellular
processes in real time (Tsien, 1998). Most studies have used transfection to express GFP,
fused to a variety o f different proteins in cell lines, resulting in a fusion protein that
maintains normal functions and localisations of the host protein but is also fluorescent.
The transfection of eGFP into rat GC cells was performed to achieve targeting of eGFP to
GH granules in a GH cell and to assess the viability of the human GH construct (p48GH-
eGFP) in a rodent cell line in vitro, before making a transgenic mouse.
When expressed alone or with minimal N-terminal peptide extensions, eGFP pervades
throughout the cytoplasm. However, targeting signals may be fused to GFP that direct
localisation o f a fluorescent fusion product to specific sub-cellular structures or
molecules e.g. progesterone receptor (Lim et a l, 1999). In particular, GFP variants
targeted to secretory vesicles have been used to follow the genesis, trafficking and
regulated release from these organelles in endocrine cell lines (Steyer et a l , 1999;
Kaether et a l , 1997; Lang et a l , 2000). The hGH signal peptide (Martial et a l, 1979) is
sufficient to enable heterologous reporter sequences to be processed through the secretory
pathway in cell cultures (Pecceu et a l, 1991; Blam et a l , 1988). For the purpose of my
studies, eGFP was fused with the signal peptide and an additional portion of the N-
terminus o f hGH (p48hGH-eGFP), which directed the fluorescent product to GH
secretory vesicles in rat GC cells.
Since we wished to avoid the possibility that the fusion protein would retain hGH
bioactivity, we chose not to fuse GFP onto the end of the intact hGH to make a full-
length fusion protein. Although less of a concern in cell lines, we intended to use the
same construct in animals, where it would be a concern. Expression of intact human
growth hormone in pituitaries of transgenic mice would cause local feedback at pituitary
and hypothalamic level and disrupt normal endogenous GH production. One example of
local feedback disruption is the Transgenic Growth Retarded (TGR) rat, previously
characterised in our lab (Flavell et a l, 1996) in which the hGH gene was targeted to GRF
neurones in the hypothalamus, inducing dominant-dwarfism by feedback inhibition of
GRF.
- 1 0 2 -

LJ’i 'I ’R 'L . ! - _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ l'i ill II ( / / / Ci'// !inc s ta h lv In in s /c d c d MiJ/i c G F P
Although we included the N-terminal zinc binding sites of GH, my data does not show
whether these residues were in fact necessary for granule packaging of eGFP or merely
fortuitous. The aim of this was not to identify the sequences necessary, merely to find a
construct that did achieve this. A further series of constructs, knocking out each o f the
three relevant histidines in hGH (’^His, ^’His and ^ " His) that are thought to be involved
in packaging o f GH dimers, would be required to address this issue and this is discussed
more thoroughly in chapter 6.
It could be that the N-terminal GH sequences in the eGFP fusion product partially
interacted with rat GH sequences which facilitated co-packaging in GC cells. However,
this cannot be the only explanation since the same construct also gave granular staining
when expressed in other secretory cell types (PCI2 cells, unpublished results) and also in
hypothalamic GHRH neurones (McGuinness et aL, 1999) that do not express endogenous
GH (see figure 3.10). I cannot assume from co-localisation studies that GFP is
‘aggregated’ in GH granules. The p48hGH-eGFP fusion protein contains the first 48
amino acids of GH. After the signal peptide is cleaved, prior to sorting and packaging
into GH granules in the TGN, only 22 residues o f hGH from the hGH-eGFP fusion
protein remain. It is unlikely that the hGH-eGFP fusion protein dimerises with
endogenous rGH, supported by the reduced amount of eGFP compared to rGH in RIA of
GC cells. In my view, eGFP is co-packaged with rGH because it contains the signal
peptide directing protein sorting, and aggregates to some degree with GH; during zinc
mediated GH dimérisation, the hGH-eGFP fusion protein may bind to a few GH
molecules. However, the large differences in amounts should suggest that the hGH-eGFP
cannot fully participate in the aggregation/condensation that achieves a much higher
concentration of itself.
I found the GFP fluorescence to be highly variable in all o f the stably transfected hGH-
eGFP cell lines, which may indicate non-clonal transfection. However, the heterogeneity
of reporter expression has been described in several studies (Frawley et aL, 1994; White
et aL, 1995; Takasuka et aL, 1998) and may demonstrate variability in gene expression,
implying that some cells are transcriptionally silent at given times.
- 103 -

N o t \
A)
Mil lI
M/»
rGHRH LCR I cGFP I
hGH SP
B)
Figure 3.10 Expression of eGFP in GHRH neurones from rGHRH-eGFF
transgenic mice
(A) The cosmid construct engineered to target eGFP to vesicles in GHRH neurones in
transgenic mice. The 1.3kb p48hGH-eGFP fusion fragment was inserted, using Mlul
linkers, into the first exon of the rGHRH gene.
(B) Expression of eGFP in GHRH neurones appeared granular (1) and was transported
along axon fibres (2) to the ME (3). Scale bar = 10pm.
- 104 -

( c r i e r _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ In vitra \ l iicHcs in </ ( III ccH Une s lu h iv t r u n s f c r l a l with cGf-'F
The transfection o f p48GH-GFP into rat GC cells confirmed that a human GH-eGFP
fusion protein can target rat growth hormone secretory granules in vitro. I have shown
that hGH-GFP growth hormone cells can be purified and enriched using FACS.
Endocrine cells release hormones by exocytosis o f secretory vesicles or granules. To
become available for exocytosis, granules must move from the cytosol to the plasma
membrane and dock there. It is of interest to us to observe single granules near the
plasma membrane in live GH cells, which we have accomplished using TIRF
microscopy. In hGH-eGFP transfected GC cells, we have been able to track GH granules
beneath the plasma membrane in three-dimensions to analyse their motion. Previously
this has been done in chromaffin cells (Steyer & Aimers, 1999) and PC 12 cells (Lang et
aL, 1997). When hGH-eGFP cells were analysed on our TIRF set up, we observed two
different types o f motion. Short-range diffusive motion, where granules wander
randomly around a resting position and long-range directed motion, where a granule
would move along a track, becoming increasingly bright, before disappearing abruptly.
Short-range motion could be modelled quantitatively by assuming granules to be attached
or tethered to the plasma membrane prior to docking and fusion. Electron microscopy
studies in chromaffin cells have shown chromaffin granules tethered to the cytoskeleton,
and a filamentous actin mesh work beneath the plasmalemma of chromaffin cells (Nakata
& Hirokawa, 1992). The long-range directed motion of granules which I have observed
in these cells is likely to be vesicular transport on the cytoskeleton, immediately prior to
exocytosis, which might explain the sudden disappearance of GFP. However, this could
also be explained by a granule moving out of the evanescent field and hence out o f view.
Successful targeting o f GFP to GH secretory granules was a significant, preliminary
result for my subsequent studies in transgenic mice in vivo. If transfection of the hGH-
eGFP fusion gene failed to target GFP to rat GH granules due to species specificity, a
double transfection of hGH-GFP in hGH-GC cells and double transgenic hGH animals
would have been necessary. These studies in cell lines encouraged me to proceed to
target GFP to GH granules in mouse somatotrophs in our transgenic hGH-eGFP mouse
for in vivo studies.
105 -

( 'Jjyii]Lf.r_î_i___________________/'(//(jr///;:.! l ' Iu o r cs cc iu rc pc ir lc r s lo n i 'n i l i i i 'v s(>;ii(il()li-()/)/j.s in t r u nsi^cn ii ' i n i a '
Chapter 4
Targeting fluorescent reporters to pituitary somatotrophs in transgenic
mice
4.1 Introduction
Somatotrophs constitute the major endocrine cell type in the anterior pituitary gland, in
which all the processes o f hormone synthesis, storage, stimulus/secretion coupling and
release mechanisms may be studied. In vivo, GH release is usually highly pulsatile
involving large amplitude bursts of secretion and this probably requires the co-ordinated
activation o f many GH cells (Achermann, 1999; Robinson, 2000). One of the main
problems of studying the cellular physiology of pituitary somatotrophs is the difficulty of
identifying the cells in vivo. One of the aims of my project was to develop a model in
which living populations of primary pituitary GH cells could be studied, which could be
facilitated by the ability to visualise secretory processes directly in identified cells in
transgenic animals. The aim of this section was to combine highly directed transgenesis
to somatotrophs (using the locus control region [LCR] for hGH) with the granule
targeting-reporter eGFP fusion constructs, shown in the previous chapter.
As previously described in the introduction (1.6 and 1.7), when designing a construct for
introduction into a transgenic organism the elements o f transcriptional control must be
present to achieve appropriate transgene expression. The cosmid targeting transgenes to
pituitary GH cells had already been proved previously to contain the complete hGH LCR
(Bennani-Baiti et aL, 1998; Jones et aL, 1995; Su et aL, 2000). The inclusion of 40kb of
contiguous 5’-flanking regions resulted in reproducible, copy-number dependent, and
pituitary-specific expression o f hGH-N at levels that were comparable to those of
endogenous mGH (Jones et aL, 1995).
In order to reproduce the GH-GFP in vitro system (discussed in chapter 3) in a transgenic
model, the hGH-GFP fusion cassette must be under the control of an endogenous GH
promoter. Thus, a transgenic mouse expressing eGFP, driven by the human GH LCR
- 106 -

( ' h a p l c r __________________________ 1'( . ' /; /?L' f l i io r c ^ c c n ! I 'c p o r tc r s lo p i l i i i l d iT S(ii)n/l(>lr(>p,hs in I r a n s i s e n i c m i c e
would specifically target GFP to granules in mouse pituitary somatotrophs. The ability to
visualise pituitary cells would then enable me to study the physiological properties and
processes o f pre-identified secretory cells. Others have used similar approaches to target
cells in other endocrine systems (GnRH, Spergel et aL, 1999; TSH, Dromta et aL, 1998).
My studies are the first to describe the generation and characterisation of transgenic mice
that express the enhanced variant o f GFP specifically in pituitary GH cells, and
moreover, in their regulated secretory pathway.
4.2 hGH-GFP cosmid contruct for generating transgenic animals
A 40kb (K2B) cosmid (Jones et aL, 1995), containing the LCR for the human GH gene
was a generous gift from Professor Nancy Cooke (Pennsylvania University). Dr C.
Magoulas (NIMR) modified the cosmid construct inserting a unique site (Mlul) 326bp
upstream of the hGH ATG, which would allow easy insertion o f any Mlu\ linkered
sequence. The Robinson lab had previously developed Mlul linked strategies for similar
transgenes, including an hGH genomic fragment expressed in GRF neurones in
transgenic rats (Flavell et aL, 1996). I have shown that the hGH signal peptide and 22
residues of hGH was sufficient to enable eGFP to be packaged into GH secretory vesicles
in GC cell lines. We aimed to insert this expression cassette into an hGH LCR cosmid
transgene to target GFP specifically to GH vesicles in the mouse somatotroph, which
might allow us to isolate and identify somatotrophs to follow GH/GFP transport within
the secretory process, all under appropriate physiological control, attained by the LCR.
Therefore, construct p48hGH-GFP (described in chapter 3) which targets GFP to GH
granules in rat GC cells, was cloned into the cosmid construct containing the hGH LCR
to make transgenic mice.
The cloning strategy that describes the engineering o f the cosmid construct K2B,
containing the Mlul linker is written in Appendix I, as it was done by Dr. Magoulas
before my arrival at NIMR. However, I was involved in the final stages o f the cosmid
construction and further study. Although complex, it had the design advantage that all
subsequent engineering for future transgenes could be performed as a one-step cloning
method, linked by a common Mlul fragment, and the engineering introduced only a
minimal (2bp) alteration in the new 5’ sequence o f the hGH promoter.
- 1 07 -

( 'Jjj 11 >lcr 4 _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ I'crr^.id in'j: fli itjrcs c u v ij' i lor lcn i ijU-^Wi ilcirv sa iv i i lo lron lis in i ra n sa c n ic m ice
In brief, the insert contained in the K2B cosmid was reversed in orientation (B2K) and a
unique Mlul site was introduced upstream of the coding region of the hGH gene by PCR
site-directed mutagenesis to alter the sequence at -326bp (upstream from hGH ATG start
site) from 5’-CCACGT-3’ to 5’-ACGCGT-3’. The hGH gene sequences of the cosmid
(cosGH.M) could then be excised as a single Mlul fragment (see Appendix I and figure
4.1) and replaced with the M lul linkered hGH-eGFP sequence to give coshGH-eGFP.
The final cosmid thus contained a ~40kB insert containing the LCR, 5 ’ and 3 ’
untranslated sequences for the hGH gene driving expression o f the hGH-eGFP fusion
protein. Figure 4.1 outlines the final stages of the cloning process and the fragment
prepared for micro injection.
4.3 Generation and identification of hGH-GFP transgenic mice
The 40kB hGH-GFP Notl fragment was purified by ultracentrifugation in a 5-20% salt
gradient and brought to a concentration of lOng/pl. With help from Kathleen Mathers
(Biological Services, NIMR) I injected the construct into fertilised oocytes of [(Cba/Ca x
C57B1/10) FI] mice followed by oviductal transfer into pseudopregnant recipients.
Resulting pups were assayed for the presence of the hGH-GFP transgene by PCR and
Southern analysis (figure 4.2a). Due to the different size of the intron I in the mouse and
human GH genes, PCR with primers hGH 5’UTR (F) and hGH exon 2 (R) (see table 2.1)
resulted in two products; with the endogenous mGH product 50bp smaller than the hGH
transgene product (350bp). The reliability of this PCR genotyping method was tested
several times by confirming the genotype of mice by Southern blot; the PCR method
proved to be extremely reliable.
In a total of 29 pups surviving to term from pro-nuclear injection and oviductal transfer, 3
pups were transgenic. All three founder mice were set up to breed with wild-type
[(Cba/Ca x C57B1/10) FI] mice, and 50% transgene transmission was obtained in two of
these lines (1 and 28). The third mouse (17), produced only two transgenic pups in 4
litters, suggesting the initial founder was chimeric or the insertion was deleterious.
Although the third founder did not transmit the transgene efficiently, each o f the three
lines expressed the eGFP phenotype in the pituitary.
- 1 08 -

( ' lu ip le r -f: T a r g c i in g f lu o r e .s c c i i l n 'p o r f c r s l o p i t u i l a r x s o m a to t r o p h s in t r a n s ^ e i l i e m ic e
Mlul Pvull
48aa
Pvull MlulA T G 1 A TG
1 1
a ) hGH eGFP _________ M 1
b)
EcoRl NotlI I
40
c )
EcoRl Notl
40
BamHl
30
BamHl
30
M lul
BamHl SnaBl SnaBlEcoRl I EcoRl I EcoRl I EcoRl
20 10
BamHl EcoRl I N otl I M lul
0
1 kB
M lul
BamHl EcoRl I
SnaBl SnaBl EcoRl I EcoRl I EcoRl
20
BamHl EcoRl I N otl I Mlul
I L - J ^0
1 kB
Figure 4.1 Insertion of Mlul linkered GH-eGFP fragment into cosGH.M
The 1.3kB p48hGH-GFP fusion construct (a) was inserted into the cosmid construct B2K
with Mlul linkers (b). The final transgene cosmid (c) is under the transcriptional control
of the 40kB hGH locus control region. Shaded bars indicate hGH genomic sequences;
hatched bars indicate vector sequence; green bar indicates GFP cDNA. Note that the
fragments are not drawn to scale.
- 1 0 9 -

( Tar'^’c / i n i ’ Ih .u > r c \c c n i r c p o r u rs io j i i l i t i l i i r v s()i}nru>lr<>j>h\ in l i 'a i i s ^ ^ c n ic m i c e
Both the positive lines (1 and 28) termed hGH-eGFP, were maintained and studied
initially to compare transgene expression of GFP and growth rates. Both lines 1 and 28
displayed a comparable, uniform phenotype, therefore line 1 was chosen for intensive
breeding and further study. For simplicity, this is referred to as hGH-eGFP. Sperm from
both lines were cryopreserved.
4.4 Expression of the hCH-GFF transgene
Transgene expression was studied by several methods: analysing eGFP pituitary RNA by
Northern blot, in situ hybridisation, eGFP pituitary protein content and release by RIA,
fluorescence microscopy and electron microscopy.
Northern analysis of pituitary RNA with an eGFP probe showed a single transcript of the
expected size in the transgenic but not wild-type mice (figure 4.2b). A RIA specifically
designed to detect eGFP protein in transgenic pituitaries showed that eGFP immuno-
reactive protein was readily detectable in extracts of pituitary glands from transgenic but
not wild-type animals (figure 4.2c). In order to confirm that the expression of eGFP was
confined to the pituitary, various tissues from hGH-eGFP were cut into 12pM sections on
a cryostat and used in an in situ hybridisation assay with an eGFP anti-sense and sense
riboprobe. A strong hybridisation signal was seen in the anterior pituitary, no eGFP
expression was observed in the brain areas or any other tissue sections examined such as
kidney, stomach, heart in accordance with Jones et al., 1995. Because a B-cell receptor
sub-unit gene (CD79b) was recently discovered to be present within this hGH LCR
(Bennani-Baiti et al., 1998) and thus present in our transgene, we specifically examined
lymphocytes from the spleen and the thymus in GH-eGFP mice. RT-PCR of RNA
extracts from spleen and thymus, and further amplification using primers hGH 5’UTR
and eGFP (R) (appendix) showed hGH-eGFP expression in the pituitary and no
detectable expression in the spleen or thymus.
- 1 1 0 -

A.b p
3 8 2 ^
290 ►
c.U)c>%L_(Ü
' 3
*Q-
0)a
c0)
4—'coÜ
Û.L i.O
70
60
50
40
30
20
10
0
M (+)B.
(-) (+)
28 S
18 S
(-) (+)
Figure 4.2 Analysis of eGFP expression in transgenic hGH-eGFP transgenic mice
(A) Mice carrying a hGH-eGFP transgene could be identified by PCR analysis o f tail DNA. Primers were
chosen to span the first intron o f the GH gene and amplified a 382bp product from the transgene as well as
a smaller 290bp product from the endogenous mGH gene. (-) WT mice; (+) transgenic mice. (B) Northern
blot analysis o f RNA from WT (-) and transgenic (+) mice showed a strong band hybrididing with a probe
corresponding to the eGFP coding region in transgenic progeny only. (C) GFP content was assayed by
RIA in pituitary extracts from WT (-) and transgenic {+) mice.
- I l l -

(. 7/( ip l c i' 4 • ________________________ 7 ( // ii I'.i r c ' /u ir ic f : !o piiii ih irx- ‘<()!in/U)lr (>i>h.s in / / - , ; / 7 \ L f c /7 / c in ic c
Whole pituitaries of hGH-eGFP transgenic mice were sectioned on a cryostat (25pM),
and examined immediately by fluorescent microscopy. As anticipated, a major
population o f the anterior pituitary cells (-40% ) was strongly fluorescent for eGFP,
whereas there was no expression in the intermediate lobe or in the posterior pituitary
(figure 4.3a). As was observed in GC cells transfected with the same fusion construct,
individual pituitary GH cells from hGH-eGFP transgenic mice showed a punctate
distribution o f eGFP fluorescence when examined by scanning confocal microscopy
(figure 4.3b). The cells showing eGFP fluorescence were compared with those
expressing GH, as identified by immunocytochemistry (in collaboration with D.
Carmignac, NIMR). Figure 4.4 shows three-colour confocal microscopy of a 15pm
section from a GH-eGFP mouse anterior pituitary (fig 4.4a) stained with an antibody to
GH (see table 2.2) and visualised with TRITC (fig 4.4b) and also stained with DAPI (fig
4.4c) in order to visualise all cell nuclei. Approximately half o f these cells showed
endogenous eGFP fluorescence, and virtually all of these co-localised with GH immuno-
reactivity (fig 4.4d). The cells showing eGFP fluorescence (fig 4.4e) were also
compared with those expressing PRL. eGFP does not co-localise with cells which stain
for PRL, visualised with TRITC (fig 4.4f).
4.5 EM of hGH-eGFP pituitaries immuno-stained with GFP, GH, PRL
To investigate the punctate localisation of eGFP, transgenic hGH-eGFP pituitaries were
fixed in 2.5% gluteraldehyde and processed for eGFP immunogold electron microscopy.
The ultra-structural morphology of somatotrophs from hGH-eGFP transgenic mice was
indistinguishable from that in non-transgenic animals and showed numerous dense cored
GH secretory vesicles. These secretory granules showed specific immuno-gold labelling
with an anti-GFP antibody (figure 4.5a) and no specific labelling o f any other structure
was apparent. Double immuno-gold labelling with an anti-eGFP antibody and anti-mGH
antibody showed co-localisation o f eGFP and GH in these vesicles (figure 4.6). Sections
from hGH-eGFP transgenic animals were also labelled with a-m PRL and a-GFP. It
would be expected that in transgenic hGH-eGFP pituitaries, mammosomatotrophs would
also contain eGFP, either concurrently or perhaps residually after differentiation into
lactotrophs.
- 1 1 2 -

Figure 4.3 eGFP expression in pituitary GH cells from transgenic mice
(A) Strong fluorescence is observed in many cells of the anterior pituitary (AP) of hGH-
eGFP transgenic mice. Note the absence of eGFP from the posterior pituitary (PP).
(B) Confocal scanning image through a single eGFP positive GH cell showing a highly
granular distribution of eGFP. Scale bar = 10 m.
- 1 1 3 -

Figure 4.4 eGFP localisation in pituitary GH cells from transgenic mice
(A) Confocal microscopy of endogenous eGFP fluorescence in a section of anterior
pituitary from a hGH-eGFP transgenic mouse. (B) The same section after immuno-
staining for mGH followed by a second antibody tagged with TRITC. (C) The same
section stained with DAPI to visualise all cell nuclei and this image superimposed with
that in (A). (D) An overlay of images in (A) and (B) to show co-localisation of eGFP
with GH. (E) Endogenous eGFP fluorescence from a different section shows Prl
(TRITC) does not co-Iocalise with eGFP expressing cells (F). Scale bar = 10pm.
- 1 14 -

( 'lu i p l ç r -i ï a i ‘ t’l in ^ f h i ( ) i \ ‘s c c iU r c p o r f c r s lo p i n n i a r v s o n u i f o l r o p h s lu /n m s 'f 'c n ic rn icc
t y - A ' f . . y
Figure 4.5 Immunoelectron microscopy of cGFP in hGH-cGFP transgenic
mouse pituitary cells
Ultrathin pituitary sections from hGH-eGFP transgenic mice were processed for
immunogold electron microscopy. Numerous dense cored secretoiy/ vesicles could be
seen in somatotrophs. Immunogold labelling, performed using a primary antibody
against eGFP, showed the hGH-eGFP fusion product clearly localised to these
secretory vesicles. No specific labelling of any other structure was obser\ ed and no
labelling was seen in sections from WT mice. Magnification = x l2,000; n=nucleus.
- 115 -

7'iir'^clins: / h i o i c n i r r p o r h ’rs to n i ti i iinrv s o n hila ir/j/'lis m iri.insMmic m ic e
m L
■■■ . 1 ' 'm m
Figure 4.6 Double-immunoelectron microscopy of eGFP and mGH shows co
localisation in somatotroph granules in transgenic hGH-eGFP
pituitaries
Double immunogold labelling was performed using antibodies against eGFP (rabbit
a-eOFF linked to proteinA gold; lOnm) and mGH (goat a-mGH linked to a-goat
gold; 15nm). Both eGFP and mGH are co-localised in granules in somatotrophs from
transgenic pituitaries. Magnification = x25,000; n=nueleus).
-116-

\ ' ■ ( / < : In ; ‘:,.;ihir\ saJihJloirpjihs in Iran'^acim: inu c
.VC" "
#
■ *
•V V *
Figure 4.7 Double immunoelectron microscopy of eGFP and mPRL co
localised in granules in mammosomatotrophs
Double Immunogold labelling was performed using antibodies against eGFP (rabbit
a-eOFP linked to protein A gold; lOnm) and mPRL (goat a-mPRL linked to a-goat
gold; 15nm). Both eGFP and mPRL are co-localised in granules in a few cells
morphologically resembling lactotrophs in pituitaries from transgenic animals. These
cells are likely to be mammosommatotrophs (magnification = x25,000)
- 1 1 7 -

( A 'c /y / r " V . _ _ _ _ / i f ' L ' l v / , v rciu>i-!cr.'- !(> j ’i!nili.ir\' in lr(i;i',<^c!u'c m i c e
I found that eGFP co-localised with PRL in vesicles in mammosomatotrophs (figure 4.7).
The number of cells that co-localised for eGFP and PRL, compared to those labelled for
PRL only, were 5 in 100. I could not triple label these cells with mGH, but from
morphological studies performed by Christian and colleagues, (Christian et al., 1999) it
is clear that these cells are mammosomatotrophs; cells which label for GH and GFP,
morphologically resemble lactotrophs, which can be detected by the smaller, in
consistently shaped granules.
4.6 Physiological Studies of hGH-GFP transgenic mice
4.6.1 Stimulation o f hGH-eGFP transgenic pituitaries with hGRF
I have already verified that eGFP is targeted to GH vesicles within somatotrophs in
transgenic pituitaries. Therefore, if the hGH-eGFP protein product is packaged in the
secretory vesicle, it should be released in response to specific GH secretagogues. To test
this, pituitary glands were isolated from hGH-eGFP transgenic mice, which were
incubated in vitro, before and after challenge with 1 and 5pg/ml hGRF-(l-29)NH2 (a
synthetic fragment of human GRF-(1-44)NH2; Grossman et a l , 1984). The release of
GH and eGFP into the incubate was measured by specific RIA for these proteins (a
specific RIA for GFP was developed by D. Carmignac in the lab) and the results are
shown in figure 4.8. Both eGFP and GH were released in a highly parallel, dose-
dependent manner, in response to this secretagogue in hGH-eGFP transgenic mouse. The
amount of GH measured in transgenic and wild-type animals was similar.
4.6.2 Pituitary hormone content and eGFP
To determine if expression of eGFP in transgenic animals compromised anterior pituitary
function, pituitary GH, PRL, TSH and LH were measured and compared in transgenic
hGH-eGFP and WT littermates. Measurements of pituitary mGH content in transgenic
and wild type mice showed a significant reduction in GH stores in both male and female
transgenic animals compared to wild-type littermates (fig 4.9a). GH levels were tested in
both lines of hGH-eGFP mice at an age of 10 weeks. Levels of PRL were also reduced
- 118 -

( 'honil.'!' y : _ _ _ _ _ _ _ _ J 'l imdnii: llni>rc^ccm i\iy<>nc/\s !c i>liiiih.ir\- S(>iviiloin</’fi\ in tra ns'^cnic nii<:c
2 -1
IXo
1 -
GRF 5pg **
GRF Ijig
**
30 60 90 120 150 180 210 240
Time (min)
- 1
OhUh%
- 0
Figure 4.8 eGFP is secreted from GH cells in hGH-eGFP transgenic mice
Pituitary glands w ere rem oved from groups o f normal (n=6) and hG H -eG FP (=4)
transgenic m ice, incubated in vitro in a succession o f 30 minute incubations, after which
the media were collected and replaced by fresh media. After 90 mins and again after 210
m ins, hGRF 1-29 N H 2 (GRF) Ip g or 5pg/m l w as added to the m edia. The m edia
concentrations o f W T m ouse (grey shaded bars), transgenic m ouse GH (open bars) and
eGFP (closed bars) were measured by RIA. Data show n are mean ±SEM * p<0.05;
**p<0.01 vs sample immediately prior to stimulation.
- 1 1 9 -

( h a p tc r 4 : T a r ^ e t in ^ J J u o r e s c e tU r e p o r t e r s t o p i t u i t a i y s o m a to t r o p h s in f r a n s ^ c i i ic m ic e
80
I 60
coXO
GH PRL
&3 20•t;0.
n=12
6
I .uCeiCL
■3 2
0n=6
8 0 0 -
&S 600
1oX 4 0 0
H
■| 200 CL
n=6
TSH 800
E^ 600
Ï400
X-J
'S 200 0%
LH
n=6WT T
c f
WT
9
4.9 Pituitary hormone content in hGH-eGFP transgenic mice
Pituitary hormone contents were assayed in pituitary homogenates from adult male and
female littermates (age: 10 wks). GH, PRL, TSH and LH were measured. Data are Mean
± SEM, *P<0.05; **P<0.01; ***P<0.001.
- 1 2 0 -

I ' lu !j'ih'v 4 ___________________________ ' l a r i ; c i lii<j i J i in rc sc i i i i fcn(jr[u-< [ o jh l j i i 1 1 ir\' sm iK i lD l i i ip h s in i n v i s ^ i c n i c m ice
(fig 4.9b), slightly in males, more so in female transgenic mice. TSH and LH levels were
not changed in transgenic animals (fig 4.9c,d).
Although pituitary mGH measurements were significantly reduced in transgenic animals
compared to wild-type littermates, it did not affect their growth rates and the animals
appeared phenotypically normal. A typical growth curve can be seen in figure 4.10. I
weighed the animals weekly, for 50 weeks, from both transgenic lines (the growth trend
was the same for both lines; n=12).
The developm ent o f the eGFP RIA in our lab (D.Carmignac, NIMR) enabled
quantification of eGFP produced in the pituitary o f transgenic hGH-eGFP mice. The
eGFP RIA is shown in figure 4.2c (n=6) and the resulting 62ng/pituitary eGFP is
approximately 500-1000 fold less than that of mGH. It is apparent that the presence of
the eGFP transgene alters the steady state levels of endogenous mGH stores the pituitary.
4.6.3 mGH mRNA levels in transgenic hGH-eGFP pituitary
The growth rate of transgenic hGH-eGFP animals is normal, despite significantly reduced
levels o f pituitary GH stores. There are many possible reasons for this. We reasoned that
GH mRNA ought to be higher and mGH production enhanced. To test this idea, RNAse
protection analysis (RPA) using a mouse GH probe was performed on pituitary RNA
extracts to quantify mGH mRNA. RNA was extracted from 12 transgenic and 12 wild-
type animals. Two pituitaries were pooled per sample. The RNA content was
normalised by analysing with p-actin mRNA expression in the same samples and assay.
Quantification, using P-actin as the loading control revealed mouse growth hormone
mRNA levels were 1.5x higher in transgenic mouse pituitary extracts. Figure 4.11a
shows a representative sample of the RPA and analysis (figure 4.11b). Analysis of all
protected fragments showed that GH mRNA was up-regulated in hGH-eGFP transgenic
mice (WT: 10.67 ± 1.50 normalised arbitrary units; hGH-eGFP: 16.28 ± 1.83 normalised
arbitrary units, n=6, Mann-Whitney, p<0.05).
1 2 1

/ t ir ^ç iu_ni Üi(!jrc s i c iU , h> n iu i iu irv ^(.'nuitolri^nhs in i in \ \
( f 9
OX
50
4 0 -
§ 30*S
^ 20oCÛ
10
10 20 30 40 50 10 20 30 40 50
(n=12) Age (weeks) Age (weeks)
Figure 4.10 Growth curve of HGH-eGFP transgenic vs WT mice
The body w eights o f 12 transgenic hGH-eGFP (open bars) and 12 W T (closed bars) m ice
were measured over 50 w eeks in both male and fem ale m ice. N o difference in w eight
between transgenic and w ild-type animals was found.
- 122 -

A)
pCRII-mGH V/AX m n \ __ L_
mGH cDNA (750bn)
h460bp Sp6
B)
WT hGH-eGFP
C)
co-aoH,c
Cu
20
15
10
5
0hGH-eGFPWT
4.11 RNAse Protection analysis of pituitary extracts from WT and hGH-eGFP
mice
lOpg pituitary RNA extract were hybridised with a mGH probe (A) and digested with R N A se (B)
The m iddle panel show s the protected mGH fragment from WT and hGH-eGFP transgenic m ice.
mGH expression is up-regulated in hGH-eGFP transgenic m ice 1.5 tim es as seen in (C) ( n -6 ,
p<0.05 M ann-W hitney non-parametric).
- 123 -

( ' l iJ iUcf -I:__________________ ______/ ( / / fJi'.orc.H d V i\'n(>r!crs lo p i!!iihir\- in l i \ i n s ' j c n ic m i c e
4.6.4 GHRH and somatostatin expression in hGH-eGFP hypothalamus
The increase in GH mRNA in the pituitary was consistent with an increased drive to
synthesise GH. I therefore performed in situ hybridisation with a mouse GHRH
riboprobe on hypothalamic ARC and PVN sections o f hGH-eGFP and WT littermates. I
found that mGHRH mRNA expression levels were upregulated in transgenic mice, as
shown in figure 4.12. A similar argument would suggest that there might be a
concomitant fall in SRIF expression if this is a physiological mechanism to maintain
normal GH output in eGFP transgenic mice. In situ hybridisation o f SRIF mRNA in the
PeN of the hypothalamus revealed no significant change, although SRIF appeared to be
slightly reduced in eGFP transgenic mice (figure 4.13).
4.7 Fluorescence Activated Cell Sorting (FACS) of somatotrophs from hGH-
eGFP transgenic pituitaries
The endogenous GFP fluorescence could be used to analyse and enrich populations of
GH cells from transgenic pituitary isolates by fluorescence activated cell sorting (FACS).
Pituitaries from a group of 10 hGH-eGFP mice were harvested and isolated. The cells
were mechanically and enzymatically dissociated and subjected to FACS. Strongly
fluorescent eGFP containing cells could readily be separated, counted and collected
(figure 4.14a, fraction II). Each pool o f cells were collected and counted then
homogenised in order to quantify GH content. If hGH-eGFP is co-packaged with mGH
vesicles, sorting for GFP fluorescence would potentially purify the somatotroph
population. Measurement of GH by RIA showed the strongly fluorescent population
(Fraction II) to be markedly enriched in GH content compared to the unsorted cell
suspension, whilst the remaining cells (Fraction I) were depleted in GH (figure 4.14b).
As previously mentioned, the CD79b (B-cell receptor subunit gene) was recently
discovered to be present within the hGH LCR (Bennani-Baiti et al., 1998) and therefore
present within our transgene. In collaboration with J.de Jersey (Immunology, NIMR) we
purified a population of B-cells using a Ficoll gradient and examined these for GFP
fluorescence. No fluorescence was detected in the B-cells isolated from these transgenic
animals and FACS analysis showed no changes in their lymphocyte population.
124 -

c lu i f i te r -f: T a r g e t im i f lu o r e s c c n i r c p o r l e r s to p i l u i t a r y s o m a to t r o p h s in t r a n s g e n ic m ic e
WT hGH-eGFPA
ARC
B
IKe0>-t3sao
4000 -
^ 3000 -
tg 2000 -
1000 -
WT GH-GFP
Figure 4.12 In situ hybridisation of mouse GHRH mRNA levels in hGH-eGFP
transgenic and WT mice.
A. ISH of GHRH mRNA in WT and hGH-eGFP transgenic mice
B. Comparison of mGHRH mRNA expression levels in hGH-eGFP transgenic and WT
mice showed a 3x increase of GHRH in transgenic animals. Mann-Whitney p = 0.004.
Scale bar = 1 mm. Arc= arcuate nucleus.
- 125 -

( 'h a p lc r 4 : T a r a t’i in g J l i i o r c s c c n i r e p o r t e r s lo p i tu i t c n y s o m a lo t r o p h s in t r a n s g e n ic m ic e
WT hGH-eGFP
m m
B 1500 -
oeg■o
1-wao
1000 M
500 -
WT
' " y - , T
hGH-eGFP
Figure 4.13 In situ hybridisation of mouse SRIF mRNA levels in hGH-eGFP
transgenic and WT mice
A ISH of SRIF mRNA in WT and GH-eGFP transgenic mice.
B Comparison of mSRIF mRNA expression in hGH-eGFP transgenic and WT mice
suggested a trend towards decreased SRIF levels in hGH-eGFP transgenic mice, but just
failed to reach statistical significance. Scale bar = 1mm; PeN = periventricular nucleus.
- 1 2 6 -

I d n i d il III Ih io n n cc / i ! rg io r l c r s !o /-‘i ln iu in - s(>nniK/lr(>/>/i\ in i n i n s d ' n i c m ic e
2
u
ÎU
64
0
Fluorescence intensity
B
(Uo
0
1
8
4
0
Before I IIsorting
FACS fraction
Figure 4.14 Fluorescence activated cell sorting (FACS) of eGFP positive
pituitary cells from hGH-eGFP transgenic mice
(A) Pituitary cells were isolated and dispersed from lOhGH-eGFP transgenic mice and
analysed by FACS. A strongly fluorescent subpopulation of cells could be identified
(Fraction II) which in this corresponded to 22% of the cells analysed. (B) This cell
population shows a marked enrichment in GH content measured by RIA (open bar) when
compared with that of the original isolate (shaded bar), and with the eGFP-negative
Fraction I which was depleted in GH (solid bar) relative to unsorted starting material.
- 127 -

J h i j i i ç y 4^_________________________ r r / ) u / / ( ' / ' lo p i l u i h i r v s c nitiiolroi^lis in ii-gn s i^c n ic i/iici'
4.8 TIRF Microscopy of primary cultures of GH-GFP pituitaries
The TIRF microscopy data presented in the previous chapter o f GC cells transfected with
p48hOH-eGFP, verified that we could obtain real-time motion analysis of secretory
vesicles in GH cells. We therefore aimed to analyse the motion o f secretory granules
from hGH-eGFP pituitary somatotrophs. Pituitaries from 2 hGH-eGFP mice were
harvested and isolated. The cells were mechanically and enzymatically dissociated and
plated onto poly-l-lysine slides, adapted for TIRF microscopy (described in chapter 3.6).
The cells were incubated overnight, allowing them to settle onto the surface o f the slide
before analysis.
Figure 4.15 shows TIRF images o f GH-GFP somatotrophs, which have been captured
and analysed using the same method described in chapter 3. The raw fluorescent TIRF
image (fig 4.15a) was filtered to enhance its contrast (fig 4.15b). Each frame can be
superimposed onto the previous one to visualise the path o f vesicles, which can be seen
for 10 frames in figure 4.15c. Long-range movement in GH-eGFP somatotrophs was
obtained by superimposing 500 frames (fig 4.15e) and skeletonising (fig 4.15f) using
custom written macros in imaging analysis software. This was performed by J-B
Manneville.
Figure 4.16 displays two types of vesicle movement found in mouse somatotrophs, long-
range directed motion in 4.16a and short-range diffusive motion in 4.16b. Like GC cells,
long-range directed motion of vesicles signified vesicular transport on the cytoskeleton.
Most o f the granules observed displayed short-range diffusive motion where the granules
are thought to be tethered or docked to the membrane (Steyer & Aimers 1999; Steyer et
a l, 1997; Gheim & Stuhmer 2000).
4.9 Patterns of spontaneous [Ca^ ji transients in HGH-eGFP cells
The eGFP transgene provided a means of identifying multiple somatotrophs in living
pituitary slices in situ, so that physiological responses may be monitored in several cells
simultaneously. I took the GH-eGFP transgenic line to Montpellier in France to re-derive
the line in Patrice Mollard’s lab (INSERM U-469: Montpellier, France). Dr. Mollard has
developed a method to measure spontaneous changes o f intracellular calcium
- 1 28 -

r Z / w / ' / iV - 4 _________ / i ' / ; i ' / ' / i v - to lu l i i tU irv s o n u t l o l f o p h s in Ircin s ’jL 'n ic m i c e
concentration ([Ca^^]i) in pituitary slices. He adapted his technique for mouse slices and
recorded ([Ca^^]i) from fura-2-loaded cells, identified as GH cells by their eGFP
fluorescence. Results from this experiment are illustrated in figure 4.17 (courtesy of Drs.
P. Mollard & X. Bonnefont). Mouse GH cells showed spontaneous fast rises in ([Ca^^]i)
(time to peak = 210 ± 29msec, n=24). All the hGH-eGFP cells displayed ([Ca^^]i) bursts,
but with different patterns. For example, in some cells, bursts displayed a step-wise
onset, followed by a high-frequency spiking plateau phase (fig 4.17 cells labelled #2 & 3)
while in others, they showed an incremental rising phase due to the summation o f high
frequency, low amplitude ([Ca^^Ji) transients (fig 4.17, cell labelled #1). All these
patterns of ([Ca^^]i) transients in hGH-eGFP cells were reversibly suppressed upon local
application of a Ringer’s saline containing 500p.M Cd^^ ions (n=13) causing a block in
calcium entry, suggesting that the ([Ca^^ji) transients were due to spontaneous Ca^^-
dependent action potentials.
- 12 9 -
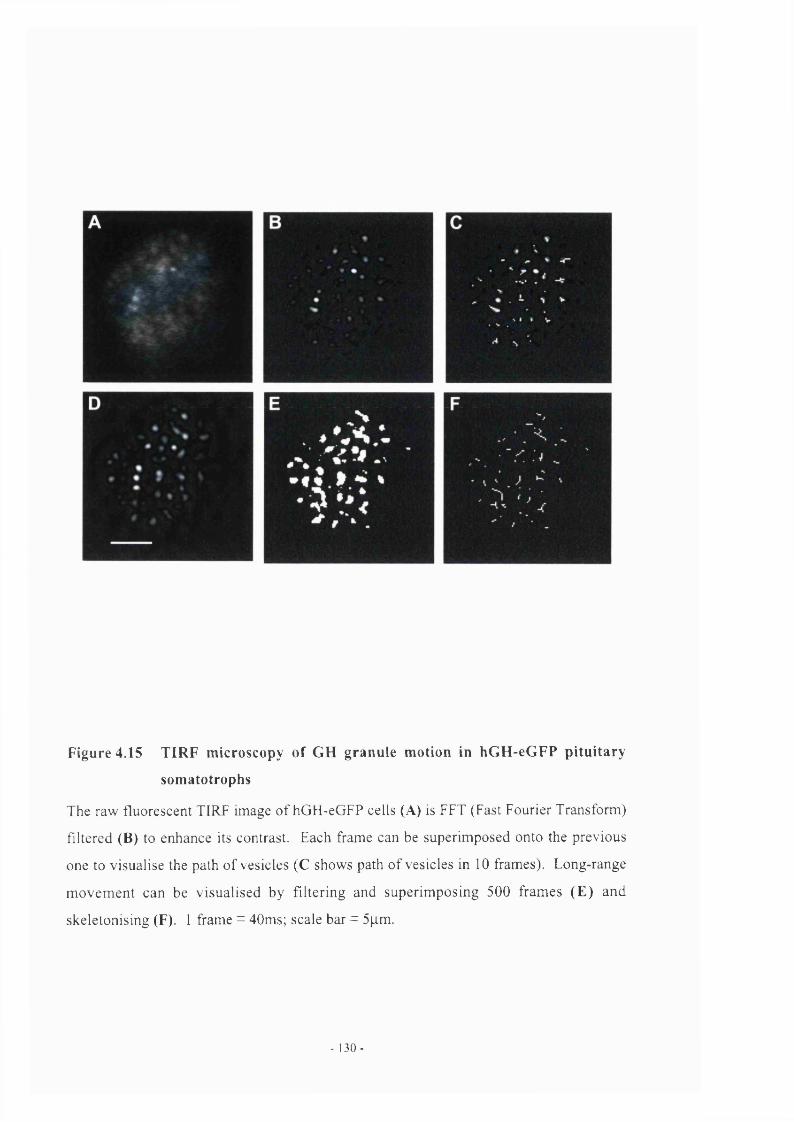
Figure 4.15 TIRF microscopy of GH granule motion in hGH-eGFP pituitary
somatotrophs
The raw fluorescent TIRF image of hGH-eGFP cells (A) is FFT (Fast Fourier Transform)
filtered (B) to enhance its contrast. Each frame can be superimposed onto the previous
one to visualise the path of vesicles (C shows path of vesicles in 10 frames). Long-range
movement can be visualised by filtering and superimposing 500 frames (E ) and
skeletonising (F). 1 frame = 40ms; scale bar = 5p.m.
- 1 3 0 -

A
e"
-0.5
-0.5 0 0.51 1
0.5
n . 0
-0.5
r
-0 02
N
0 0.5 1 5 2 2 5
N -0.05
-0.5 05
Figure 4.16 Analysis o f single granule trajectories in ph48hGH-eGFP transgenic pituitary somatotrophs
Analysis o f single granule trajectories in TIRF microscopy determines whether GH granule motion is random or directed. Two types o f vesicle
movement in somatotrophs are displayed above, showing long-range directed motion in A and short-range diffusive motion in B. X and y ploys display
parallel, horizontal motion, while z plots vertical motion. The long-range directed motion o f vesicles (shown by the arrow in the top jcy plot) signifies
vesicular transport. Most of the granules observed displayed slow, short-range diffusive motion and were most likely tethered or docked to the plasma
membrane ready for release.

.7 i / / •' : V. iijii ;U2U ••• »/ >/? \ iii f_rtins}’L'nic /»/'■
e - G F P F u r a - 2
20 sec
5 sec
Figure 4.17 Patterns of spontaneous [Ca^ ji transients in hGH-eGFP pituitary
cells
Upper left panel, Field of GH cells expressing eGFP. Upper right panels Same field
loaded with Fura-2. The white circles highlight the area of three eGFP positive cells in
which changes in fura-2 fluorescence, reflecting [Ca^^]i levels were monitored. Lower
panel. Changes in fura-2 emission, normalised to baseline fluorescence (-F/Fo), for the
cells identified in the panels above. The bottom trace illustrates spontaneous [Ca^^]i
transients monitored in cell #3 on a four-fold expanded scale. Stars indicate [Ca^^]i
bursts.
- 132 -

( ’/■■< I l 4 ___________________________ I j [rjiiijiini c e n t r c p n i ' l c j s lo / / / i r d n s n c n i c n i icc
4.10 Discussion
As previously discussed in chapter 3, targeting signals may be fused to eGFP that direct
localisation of a fluorescent fusion product to specific sub-cellular structures. The hGH
signal peptide (Martial et al., 1979) is sufficient to enable heterologous reporter
sequences to be processed through the secretory pathway in cell cultures (as shown in GC
cells in chapter 3). Others have used similar approaches to target cells in other endocrine
systems. Seeburg’s group (1999) expressed eGFP under the control o f the GnRH
promoter and observed green fluorescence which was confined to GnRH neuronal
somata, dendrites and axons throughout the hypothalamus. Using GFP fluorescence in
brain slice preparations, they were able to obtain physiological recording of action
poetntials and G ABA- and L-Gluatamate-evoked currents from pre-identified cells
(Spergel et a l , 1999). Our group have targeted GFP to GHRH neurones in mice using
the human GHRH promoter (McGuinness et al., 1999). eGFP fluorescence was mainly
restricted to neurones in the ARC nucleus and detected in the zona incerta. The terminal
field o f these neurones, the ME, also showed bright fluorescence, with granular staining
of projections ending in the outer layer of the ME. These transgenic mice are now being
used to investigate GHRH secretory processes (Robinson & Mollard, unpublished).
Davis and colleagues (1998) fused the luciferase reporter gene to the hPRL promoter and
transfected pituitary GH] cells with the fusion construct to study promoter activation and
expression in individual cells in real time. These studies demonstrated variability in both
the basal level o f reporter gene activation and also the responsiveness to activators and
inhibitors o f gene expression in stably transfected cells (Takasuka et a l , 1998).
Southgate and colleagues (2000) in search of a gene therapy strategy for the treatment of
pituitary disease, achieved specific lactotrophic cell type-specific transcriptional targeting
in the anterior pituitary gland in vivo, using the hPRL promoter (5kb) encoded by
adenoviral-vectors. Although this restricted the expression of HSVl-TK to lactotrophs, it
was not effective in the induction o f apoptosis in lactotrophs. However, utilising the
hCMV promoter, driving expression of Herpes-simplex virus type I-thymidine kinase
(HS V 1 -TK), was effective in reducing pituitary weight and circulating levels of prolactin.
Davis et al., (2001) with stereotaxic transcranial delivery of recombinant adenoviruses in
the sheep, successfully targeted the reporter (3-galactosidase, driven by hPRL promoter.
- 133 -

{Jj (J_121£LA'._________________________ T a n jc l l i i i : l l u o r c s c c i U r c >H>r!crs fo p i l tiiinn:joiiU[ll>L>'llidL\ . />/ I r u n s ' j a i i c m ice
to ovine lactotrophs in vivo, causing lactotroph ablation without disrupting normal
endocrine function.
I opted to utilise transgenic technology to construct a mouse model expressing GFP in
somatotrophs since the human LCR was available and the lab had the prerequisite
expertise in producing transgenic rodent models. I chose to make a transgenic so it could
be used as a tool to cross with other GH transgenic mouse models. However, we have
also generated a transgenic rat with this construct (Robinson, unpublished data).
The construct which I chose to use in transgenic mice included not only the entire hGH
signal peptide but also the first 22 residues of the N-terminal sequence of hGH, since this
appeared to be sufficient to target GH to secretory granules in GC cells. It proved to be
equally effective in targeting eGFP to secretory vesicles in transgenic mice. Although
minimal GH promoter sequences can express transgene promoters in somatotrophs, the
intensity o f expression is low and often variable. In order to reliably direct position-
independent, copy number-dependent expression in the pituitaries o f transgenic mice we
used a much larger promoter including the entire LCR of hGH (Jones et a i , 1995). This
LCR contains several DNA elements necessary for somatotroph specific expression
(Bennani-Baiti et al., 1998; Shewchuk et a l, 1999), and only minimal changes were
made to this cosmid, mutating 2 bp to generate a unique site into which the hGH-eGFP
reporter could be cloned. As expected, this construct achieved high-level, specific eGFP
transgene expression in pituitary GH cells, with no detectable expression in other
pituitary cell types or in other tissues examined. I also specifically examined
lymphocytes from hGH-eGFP mice due to the presence o f the CD79b gene within our
transgene and found no expression of GFP in B-cell isolates. Clearly, the human
insulator sequences in my hGH transgene were functioning correctly in mice.
Confocal and EM immuno-gold studies confirmed that the eGFP was localised in the
large dense-cored GH vesicles in somatotrophs. eGFP was also present in vesicles in a
few prolactin cells. Expression of eGFP was accompanied by a significant reduction in
the total amount o f GH stored in the pituitaries o f transgenic animals, but did not
otherwise disrupt the normal morphology or function of somatotrophs. Prolactin stores
were also reduced in pituitaries of transgenic animals, although not to the extent of GH.
- 1 34 -

^J>i‘!^<s’L'-rL__________________________ T(irvj ' f in<: l in a r i - ^ c r iu i-i'i\(ir[cr<j‘ i^ii ii iUjry_ //7 ir: in s ‘j ,cn ic m ice
The reduction in GH stores could be a consequence of “squelching” of the endogenous
promoter. Insertion o f a transgene may induce artificial competition for activators of
gene transcription between the introduced transgene and endogenous mOH gene. It is
well known that Pit-1 is required for expression o f GH transgenes (Shewchuk et a i ,
1999). The transgene(s) may compete for Pit-1 reserves (for example) in the nucleus of
the somatotroph, which could alter or modify the level of transcription of the endogenous
mGH gene and subsequently, amounts o f GH protein. Pit-1 is also required for
expression of prolactin, and similarly, might cause reduction in prolactin stores observed
in GH-eGFP mice. Study of hGH-eGFP transgenic mice showed mouse GH transcript to
be up-regulated in transgenic pituitaries compared to WT, a consequence of a significant
up-regulation o f hypothalamic GHRH and ‘reduction’ in somatostatin, to maintain an
adequate output of GH in transgenic mice. The reduced pituitary GH reserve was clearly
sufficient since their growth was unaffected. It was therefore unlikely that the reduced
stores were caused by reduced mGH transcription.
I feel it more likely that the reduction in GH stores could reflect competition between the
hGH-eGFP fusion product and endogenous mGH for granule packaging or “space”,
however, there was much less eGFP than mouse GH stored in the pituitary. Since eGFP
RNA transcripts were abundant, I suspect that the packaging or storage mechanisms are
less efficient for the hGH-eGFP product than for mouse GH. The aggregation and
packaging of proteins in dense-cored granules probably involves specific interfacial
features of protein structure favouring oligomerization (Cunningham et al., 1991a,b). The
presence of an hGH-eGFP fusion protein which would not interact as efficiently as hGH,
may modify the packaging and aggregation mechanism in the TGN to form less
condensed GH vesicles. I did not observe a difference in the number or size o f GH
vesicles in somatotrophs in hGH-eGFP transgenic mice under EM, although no
quantification studies were done. This hypothesis would imply that granule densities
would be lower.
It is known that sequences in addition to the signal peptide are also required for efficient
packaging of GH. McAndrew and co-workers (1991) performed transfection studies in
cell lines, with either wild-type bovine GH (bGH) or truncated variants o f bGH.
Immunofluorescence studies revealed that transfection o f cells with wild-type bGH was
- 135 -

( 'hiI!)fer 4 •_ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ J h U l rcpc i lcrs lo / ‘i ln iu irv .sonnitoli-oplis in lr(ins<donc dhcc
localised to the perinuclear compartment, while truncated hormones were confined to the
cytoplasmic compartment and were shown to be secretion-defective, even though the
signal peptide was efficiently and precisely processed. Therefore, the bGH signal peptide
in and o f itself is not sufficient to direct secretion of these truncated bGH molecules. One
possible argument might consider a significantly altered protein folding mechanism in
which the defect in secretion is conformation dependent. Further confirmatory work by
Chen et a i , (1991) showed that substituting proline (a helix-breaker) for lysine,
glutamate and leucine at amino acids 114, 118 and 121 respectively, in the third a-helix
o f bGH revealed the same diffuse cytoplasmic distribution o f bGH, and accumulation of
the mutated non-secretory proteins within the ER of mouse L-cells. The tertiary structure
o f hGH is a four-helix bundle, which in the presence of zinc folds, presenting H is^\H is^\
His’ " such that zinc can bridge two a-helical segments, promoting formation of the hGH
dimer. Therefore, inclusion of the first 22 N-terminal residues (including zinc sites His’
and His^’) in my transgene might have helped, rather than just the bare signal peptide.
However, there is no evidence for this.
The eGFP product was clearly targeted to the regulated secretory pathway since it was
released in response to the specific GH secretagogue, GHRH. Initial attempts to quantify
this by measuring eGFP fluorescence in the media were unsuccessful due to the large
dilution involved in incubation studies, though this has since been accomplished in our
lab (Dr. He, unpublished data). However, use o f a sensitive RIA for eGFP showed
directly that the transgene product was secreted in response to GHRH in a dose
dependent fashion, closely paralleling GH release from the same tissue.
FACS analysis and sorting of live or fixed pituitary cell types has been described
previously, using antibodies to the specific hormones released (Wynick et al., 1990a;
Wynick et al., 1990b). The eGFP in transgenic pituitary cell isolates provided a strong
endogenous signal for FACS sorting of live cells, and a population of strongly eGFP-
positive GH cells could be isolated without the need for pre-treatment of the cells with
antibodies or permeabilising agents. Also, isolating viable populations o f somatotrophs
can then be studied in vitro, removed from paracrine interactions with other hormone
producing cell types in the anterior pituitary. I have shown in this chapter that I can
- 136 -

( 'Jhijiiçr 4_ _ _ _ _ _ _ _ _ _ _ _ _ _ _ 7 i / / 'L ' r / / / 7 , c fliKn'i'Si '.'iu y rnorjyrs !i> pimiiiU-y S(>nni!oli'ijph\ in ira n s 'dcnic m ic e
isolate somatotrophs from hGH-eGFP pituitaries and enhance levels o f GH in purified
populations.
GH cells are excitable and show spontaneous [Ca^^Ji transients that correlate with
secretion, but the study of this is labour intensive since the individual responding cells
must be identified and characterised, usually by immunocytochemistry, post-hoc
(Guerineau et a l , 1998; Bonnefont et a l, 2000). In collaboration with Patrice Mollard,
we could show that intracellular calcium can readily be monitored simultaneously in
several pre-identified GH cells, using dual wavelength imaging for eGFP and fura-2, we
observed the rapid short-lived increases in [Ca^^Ji that reflect the outcome of transient
calcium entry during action potentials in these cells. Furthermore, this was the first time
that mouse GH cells displayed spontaneous rhythmic bursts o f [Ca^’ Ji similar to those
that have recently been characterised in post-immunoidentified GH cells in rat pituitary
slices (Bonnefont et al., 2000). Previous studies have recorded from single neuronal cells
identified by GFP expression, (Manjunath et a l, 1999; Suter et a l , 2000). Multi-cell
imaging is possible in acute pituitary slices from our hGH-eGFP mice. This approach is
being used to study the GH cell populations in different pituitary sub-regions in situ and
whether they co-ordinate the timing of their responses to the entry or exit of
secretagogues or inhibitors, to or from the glandular parenchyma. It can also be used to
study development o f pituitary GH cells at birth and their 3-D organisation. Pituitaries
from transgenic pups were extracted at one day and three days after birth. The
localisation of GFP in many of these somatotrophs is confined to a large accumulation in
the perinuclear compartment, probably TGN, which is very different to the punctate
appearance of GFP seen in somatotrophs o f older animals (figure 4.18, courtesy of
Patrice Mollard, unpublished data). This may give us a key to identifying nascent GH
cells.
This transgenic approach opens the way to direct visualisation of spontaneous and
secretagogue-induced secretory mechanisms in identified GH cells. It is a useful model
for analysing GH physiology in its own right, and also can be crossed with models
containing mutations in the GH axis, which will be discussed in more detail in chapters 5
and 6.
- 137 -

Figure 4.18 Expression of eGFP in d 1 hGH-eGFP transgenic pituitary
somatotrophs
This two-photon confocal microscopy image shows the intermediate and anterior
pituitary lobes from a di hGH-eGFP transgenic mouse. The intermediate lobe contains
no somatotrophs (bottom right hand corner). The majority of somatotrophs near to the
IL/AP border have a peri-nuclear distribution of eGFP, compared to more mature cells,
which have a granular expression of eGFP, deep in the anterior pituitary (top left hand
corner). Magnification = 40x.
. 1 3 8 -

( hi l iver In viiri) . V f / J / r s nf \ i ( h i ccU Une i t/ hnini/n ( i l l (InniindiU n e e n l iv c niiiUilion
Chapter 5
In vitro studies of a GH cell line expressing a human growth hormone
(hGH) dominant-negative mutation (hGH-IVS3)
5.1 Introduction
Human growth hormone (hGH) is a 191aa monomeric protein. The precursor o f hGH is
encoded by exons 1-5 of the hGH-N gene, and the only processing of the protein is
enzymatic cleavage of the 26aa signal peptide required for entry into the endoplasmic
reticlulum. Mature GH is a four alpha-helix bundle with two intramolecular disulphide
bridges (Cunningham et al., 1991a). Mature 22kD GH protein accounts for
approximately 75% of circulating GH; the majority o f the remaining 25% is 20kD
product that results from alternate splicing of the GH-N gene, deleting amino acids 32-46
(des32-46-GH) (Baumann, 1991).
Familial isolated GH deficiency type II (IGHD-II) has an autosomal dominant mode of
inheritance. Typical clinical reports of IGHD-II are o f children with only one affected
parent, thus heterozygous for a GH gene mutation, greatly reduced plasma GH levels,
slow growth rates and a positive response to exogenous GH therapy. Several families
with this disorder have mutations in the first, fifth or sixth base pair o f the donor splice
site of intervening sequence 3 (IVS3) of the GH-N gene (Cogan et al., 1995; Phillips III
& Cogan, 1994; Wajnrajch et al., 1998). Mutations in the first and sixth base pair have
been shown to result in mis-splicing of mRNA and complete loss of exon III, so that the
mature GH produced from this message lacks amino acids 32-71 (Cogan et al., 1995;
Binder & Ranke, 1995; Cogan et a l, 1997). These amino acids constitute the entire
connecting loop between helix one and helix two of the tertiary GH structure (Ultsch et
a l, 1994) and without them the growth hormone molecule cannot fold normally. The
loss o f residues 32-71 also includes a cysteine at amino acid 53, a residue known to form
intermolecular disulphide bonds (Helenius et a l, 1992).
- 139 -

C ' h c i ; > l c ! ' _ _ _ _ _ _ _ _ _ _ _ _ In viiro L i l i l l i i 'II l i i L L L i i i i h i i i nnn ( iU i lanuiu in l ncLLcifivc nu tuil ion
In this chapter, cell transfection studies in a rodent cell line expressing two hGH
constructs are discussed. We chose to investigate a hOH-IVS3 + 1 0 —>A transition,
because it was found on the same locus with a different microsatellite marker in three
unrelated IGHD II families, suggesting that each of the GH mutations may have arisen
independently. The mutant hGH-IVS3 (+ 1G ^A ) sequence was discovered in a patient
referred to John Phillips III (Vanderbilt University), and he kindly provided the clone to
our lab for further study. Primary studies by Phillips and co-workers showed that
expression of this mutation in HeLa cells exhibited aberrant splicing, and generated a
17.5kDa cDNA product skipping exon 3 (des32-7I) (Cogan et a l , 1995). Although a
mutant GH with a molecular mass of 17.5kDa is predicted to be synthesised from the
mRNA, it is not clear if the mutant protein is actually synthesised and secreted.
For the purpose of my studies, I inserted the genomic hGH sequence, containing the IVS3
mutation into a CMV vector and transfected this into rat GC cells. My reason for
expressing the genomic hGH sequence (including intronic and exonic sequences) rather
than a cDNA sequence lacking exon 3 was to confirm that a rodent cell line could utilise
the correct splicing sites and process hGH and more importantly, generate the hGH-IVS3
transcript lacking exon 3. In general, donor and acceptor splice sites are well conserved
throughout vertebrates (Krawczak, Reiss & Cooper, 1992) and I thought it likely that the
human GH-IVS3 mutation would be transcribed and processed correctly. I also wanted
to test whether by inserting a human GH-IVS3 mutated gene into a GH-producing rodent
cell line, it would interact efficiently with and inhibit endogenous rGH production from
GC cells.
5.2 Construction of hOH-WT and hGH-IVS3 constructs for transfection in rat
GC cells
The genomic clone containing the hGH-IVS3 (+1G-+A) mutation was originally
contained in a pXGH5 vector, containing the mMT-1 (mouse metallothionin-1) promoter.
[Although this promoter would drive efficient gene transcription, it was not appropriate
for my study, as it was possible that future studies would investigate the binding
properties of the mutation in the presence of zinc]. Engineering o f the phGH-WT and
phGH-IVS3 constructs for transfection into GC cells is shown in figure 5.1. Briefly, the
- 1 4 0

( ’/ / ( / / > _____________ I l 'A 'J i£ ‘L A u d i t ’s (>! a L i l / i ■.-// H u e I ' .y/ ' / \ s (/ h i u D u n i U l d o / i i i n d iit n c i i d l i v c ) i i iU(i tu )n
hGH-IVS3 (+1G—>A) mutation was removed by restriction digest (Sacl/Bgll) and
inserted, using the same sites, in to an hGH genomic clone (Flavell et al., 1996)
containing 5’-and 3’ untranslated hGH sequences flanked by an M lu\ linker (pKS-
GH.M). The mutated clone was confirmed by restriction digest and gel electrophoresis,
as the +1 G ^ A substitution introduced an extra M alll restriction site, (figure 5.1.3).
The 2.6kb BamHl/Notl fragments from both the hGH-WT and GH-IVS3 were sequenced;
only the +1G—>A mismatch was found in IVS3. Both fragments were subcloned into the
expression vector pCDNA3.1 (Invitrogen, UK) containing the CMV promoter driving 5’-
3’ sequences of the hGH gene and a zeocin selection cassette.
5.3 Characterisation of phGH-IVS3(+l G—>A) and phGH-WT in rat GC cells
The plasmids containing hGH wild-type and hGH-IVS3 (figure 5.1) were transfected into
rat GC cells, and several stable lines were established by incorporation o f zeocin
(Invitrogen, UK) into the media for 4 weeks for selection. To confirm if the transfected
human GH genes were transcribed by a rat GH cell line, RNA was extracted from both
hGH-WT and IVS3 transfected cells. RT-PCR with primers specific for the hGH gene
was performed on cDNA from the hGH-WT and hGH-IVS3 cells (primers listed in
appendix). The primers were designed to amplify two different sized hGH products,
[hGH 1 (F) + hGH 2 (R) short or hGH 3 long (R)] a short-length product (455bp) and a
long-length product (661 bp), seen in figure 5.2. The long-length product was amplified
to confirm that the potential deletion of exon 3 would not affect the transcription of hGH
sequences downstream of this point. In both short- and long-length PCR reactions, a
truncated cDNA product (~120bp smaller) was amplified from GH-IVS3 transfected
cells, seen in figure 5.2b.
The untransfected GC cell control showed no amplification using hGH primers. The
cDNA products from WT and GH-IVS3 mutant transfections were sequenced and
compared using BLAST (NCBI-NIH Database). The cDNA products from 4 transfected
GH-IVS3 GC cell isolates lacked residues 32-71, completely skipping exon 3; the cDNA
product amplified using the long-length primers did include intact exons 4 and 5. Full
length hGH was transcribed in rat GC cells transfected with wild-type hGH.
141 -

( hapier 5:_ _ _ _ _ _ _ _ _ In vitro studies ofci GH cell tine expressing a human (IH dominant negative mutation
BamH\ I^ot I
1) hGH-WT [? V CMV ] J
+1 G>A
Bam\\\ Sac\
mMT-
Mlu\ BamHl TV/alll fig/11
pKS-GH.M
2) hGH IVS3
Mlul
BamHlNlalll
CMV
1 2 3 43)
1.2kb
639 + 633 bp
Figure 5.1 hGH-IVS3(+lG-»A) and hGH WT constructs for transfection into rat
GC cells.
Two plasmid constructs were engineered and cloned into the mammalian expression vector pCDNA3.1
containing a CMV promoter driving 5’-3’ sequences o f the hGH genomic sequence (black bars and white
bars). (1) BamHI/Notl fragment from pKS-GH.M, containing WT hGH was cloned into CMV mammalian
expression vector pCDNA3.1. (2) This construct contains hGH genomic clone modified by insertion o f the
SacVBglll fragment from pXGH5, with the +1G-*A mutation, identified by an extra M alll site. (3) NlalU
restriction digest o f hGH c\ones[BamHl/Notl] confirm the 1.2kb band present in WT hGH (lane 1) has been
cut into 639 + 633 bp in the hGH-IVS3(+lG-»A) mutated clones (lanes 2+3; lane 4 = BamHl/Notl
fragment from original pXGH5 vector as control).
- 1 42 -

( 'hillIfcr 5:____ In vilro siuJies o f a ( , / / . y / / line c.\/irrs.sin^ a hmmin Cr / I Joniiinini-ne^ulivc mulaiiitn
A)start o f mature protein
_ A h G H 1(F)
hGH-cDNA 1 2i
1 - 3 1 3 2 - 7 1 7 2 - 1 2 6 1 2 7 - 1 9 1
\ g U 2 (R) ^ h G H 3(R)
B)661 bp-
455bp' R
541 bp
■335bp
Figure 5.2 Expression of phGH-WT and phGH-IVS3 in rat GC cells
RT-PCR with primers specific for the hGH gene was performed on cDNA from
the phGH-WT and phGH-IVS3 cells. (A) I'he primers amplified two different
sized hGH products, [hGH hGH 1 (F) + hGH 2 (R) short or hGH 3 long (R)] a
short-length product (455bp) and a long-length product (661 bp). (B) The cDNA
products from 4 transfected GH-IVS3 amplified a truncated cDNA product
(~120bp smaller). Further sequencing of cDNA from GH-IVS3 GC cells
confirmed residues 32-71 absent. The cDNA product amplified with long-length
primers included intact exons 4 and 5. hGH was transcribed normally in rat GC
cells transfected with wild-type hGH. Control, un-transfected GC cells contained
no hGH.
-143-

( ' hu n i e r ^ •___ ____________ '-jJL ( / / / / ' i _ \ / ? / 1 ( i o t i l i f m n l / / c L V / z / r c i iii i /cin'()’i
5.4 Growth rates of phGH-WT and pGH-IVS3 GC cells
After I transfected both phGH-WT and phGH-IVS3 constructs into GC cells, it became
apparent that phGH-IVS3 transfected cells never reached confluency. I also noticed a
great number of the mutated cells were shed into the medium, and further observation of
these cells at higher magnification confirmed they were either dead, or dying. Therefore,
I recorded and compared growth rates of phGH-WT and phGH-IVS3 over 14 days. Two
culture flasks o f WT and mutant cells were inoculated with the same number o f cells
(250,000) in 25 ml of complete medium and counted every two days by extracting
duplicate aliquots of 1 ml of cell suspension and counting with a haemoeytometer. The
results (figure 5.3) show that cells transfected with hGH-WT grew at a similar rate to
untransfeeted GC cells (see figure 3.4a). However, the growth rate o f GC cells
transfected with phGH-IVS3 construct was noticeably diminished in comparison to
phGH-WT cells. After 2 weeks, the hGH-IVS3 cell population had only reached
approximately 20% of the density o f the hGH-WT transfected GC cells (1.75x10^ :
IxlO’).
5.5 GH content in hGH-IVS3 compared to hGH-WT transfected cells
The obvious limited growth of GC cells transfected with the human dominant-negative
mutation compared to cells transfected with normal hGH suggested that the hGH-IVS3
mutated gene was interacting with and inhibiting normal cell function. I therefore
performed RIA of GC cell homogenates to ascertain if the hGH-IVS3 mutant gene
transfected into GC cells disrupted endogenous rGH production. I counted and collected
an equal number o f hGH-WT and hGH-IVS3 transfected cells (2x10^), spun the cell
suspension, re-suspended in 1ml of PB and homogenised each before RIA. I assayed the
homogenates for hGH and rGH, the resulting figures for which are displayed in figure
5.4. hGH was undetectable in untransfected GC cells as expected. The rat growth
hormone content in untransfected GC cells was similar to that in GC cells transfected
with hGH-WT (38±1.49pg/2.5xl0^ cells and 36±1.08pg/2.5xl0^ respectively). hGH
content in hGH-WT transfected GC cells was slightly higher than endogenous rGH
(43.5±2.54)ug/2.5xl0^ cells), perhaps due to the CMV promoter.
- 144

( h c i p t e r 5 : In vilro sliiclies of a 0 7 / cell line expressing a hiinuin 0 7 / dominant ncgatn'e miiiafion
125 n
-a; 100-%03c
o 75 -
*o
B9%
50 -
2 5 -
0 2 4 6 8 10 12 14
□ GCphGH-W T
O GCphGH-IVS3
Days
Figure 5 3 Growth rates of hCH-WT v hCH-IVS3 GC cells
Growth rates of phGH-WT and phGH-IVS3 were compared over 14 days. Two culture
flasks of WT and mutant cells were innoculated with the same number of cells (250,000)
in 25 ml of complete media and counted every two days by extracting duplicate aliquots
of 1ml o f cell suspension and counting with a haemoeytometer. Cells transfected with
hGH-WT grew at a similar rate to untransfected GC cells (see figure 3.4a). The growth
rate of GC cells transfected with phGH-IVS3 construct was greatly reduced in
comparison to phGH-WT cells. After 2 weeks, the hGH-IVS3 cell population had only
reached approximately 20% of the density of the hGH-WT transfected GC cells
(1.75x10^ 1x10^).
- 145 -

• /y : ■ ■ ■ _ _ _ _ _ _ _ _ _ _ h i y u m ;;^n!ijics_<[l a Cil ! il Unr w/ f / u u h iin n iii L i l l iio itu 'fu in t / z i ' i ' i / Z / r c' utiilcilio u
50-1
40-o>CJoooo'infS 30 -
g 1 0 -
hGH-IVS3GC hGH-WT
□ hGH□ rGH
control
Figure 5.4 RIA of GH production in hGH-WT vs hGH-IVS3 transfected
cells
RIA was performed to quantify endogenous rGH and hGH production in hGH-WT and
hGH-IVS3 transfected GC cell homogenates. 250,000 cells were collected, spun and re
suspended in 1ml of PB and homogenised each before RIA. 1 assayed three separate cell
populations, GC-WT, hGH-WT and hGH-IVS3 for rGH (white bars) and hGH (hatched
bars) content. hGH was undetectable in untransfected GC cells as expected. The rat
growth hormone content in untransfected GC cells was similar to that in GC cells
transfected with hGH-WT. In the hGH-IVS3 transfected GC cells, hGH was
undetectable, and rGH was significantly reduced (*** p<0.005) indicating that the
incorporation of the human dominant-negative mutation interrupts endogenous rGH
production.
- 1 4 6 -

C 'hululer 5 ; _ _ _ _ _ _ _ _ _ _ _ _ In viiro v / f . 'n ' . 't 'v oj a ( j ! ! ccH l ine c . v ; ) r i a hu n n /n ( , ' / / ( lopiinnnl nc^Lilivc ntiiliJlion
In the hGH-IVS3 transfected GC cells, hGH was undetectable and rGH was significantly
reduced (1.4±0.47|ig/2.5xl0^ cells, p<0.005) indicating that the incorporation of the
human dominant-negative mutation interrupts endogenous rGH production.
5.6 EM of hGH-IVS3 GC cells
I compared the appearance of GC cells transfected with hGH-WT and hGH-IVS3 under
the light microscope. There were a number of cells in hGH-IVS3 GC population, which
looked “normal”, i.e. intact nucleus, and consistent rounded cell shape. However, the
majority of the cells in the hGH-IVS3 transfection looked like they were dying, with an
abnormal, fragmented nucleus and shrivelled outline. In order compare their morphology
in more in detail, I prepared them for electron microscopy.
5.6.1 Morphology o f GH-IVS3 transfected GC cells
Cell pellets from both hGH-WT and hGH-IVS3 transfections were fixed in 2.5%
gluteraldehyde and processed in Spurr resin for electron microscopy. The morphology of
a GH tumour cell line is very different from that in pituitary somatotrophs from mice,
which was also shown previously in GC cells transfected with eGFP in chapter 3. GC
cells have fewer granules by comparison and are not consistent in shape and size with
one another. A typical hGH-IVS3 transfected GC cell had a highly vacuolated
cytoplasm, fragmented nucleus and swollen organelles, specifically golgi apparatus and
mitochondria. Compared to hGH-WT transfected cells, they had virtually no GH
granules. Figure 5.5 shows a comparison of hGH-WT (5.5a,b) and hGH-IVS3 (5.5c,d)
transfected GC cells at 5000x and 16,000x magnification. They also appeared to have
larger lipid vesicles, consistent with an increase in lysosomes, which encapsulate and
enzymatically break down cell debris. An example o f this is shown in figure 5.6.
Within a population of GC cells it is possible to see a spectrum of cell types, from newly
divided through to old cells, which are in the process of dying, or dead. In hGH-IVS3
transfected GC cells, these “morphological states” appeared to be more distinct than in
hGH-WT transfected cells.
- 147 -

( 'lhii>lri \ In vilro \ of (H I r c / l line c v / ' r c s v / . ' / . i ' d h u n n m (.ill ( lo i fu n a n l -n c^ j l iv c niiiiallon
■■ < m
m mi i m m
i
i
Figure 5.5 EM of rat GC cells transfected with hGH-W T at (A) 5000x and (B) 16000%
magnification
hGH-WT GC cells have fewer granules compared to that o f a pituitary somatotroph. However, GH
granules have a distinct structure and can be seen at higher magnification in (B).
-148-

' s n ; : ’ . ■ I ■ / / / i h ’ r-i i ’ : ! ! i i i - a l l V f n i i i U ^ n n .
M
Figure 5.5 EM of rat GC cells transfected with hGH-IVS3 at (C) 5000x and (D) 16000x
magnification
hG H -lV S3 transfected GC cells contain very few granules. The eytoplasm is h ighly vacuolated,
typical o f the eell population.
-149-

idV'ian i / (JicdUKi/!! VC nniuiiidn
Figure 5.6 EM of hGH-IVS3 GC cells shows distribution of large lipid vesicles
GC cells were transfected with the hOH-IVS3 construct shown in figure 5.1. Samples
were processed in spurr resin and prepared for EM. hO H -IV S3 ce lls appeared to
contain m ore lipid v esie le s (L V ) than hGH -W T, consisten t with an increase in
lysosom es. M agnification = x5000; n^nucleus.
- 150-

L ih i /V c r _________________/'(> ■ w ij (/ W [j_;j;// ////(' <.'x,ni \ - s s in n n .h u n n m ( / / / i/onilncin! nc^^Litivc n ii iU il ion
Although the majority o f the population of hGH-IVS3 cells was dying, with the
cytoplasm disintegrating and the cells shrivelling, there were also a small population of
cells that looked normal and contained GH vesicles and a population which looked only
slightly abnormal, identified by swollen organelles. This suggested that the dominant-
negative phenotype developed with the accumulation of mutant hGH protein.
5.6.2 Immuno-gold labelling o f GH in hGH- 1VS3 transfected cells
Pellets of hGH-IVS3 transfected GC cells were processed in LR gold resin for immuno-
gold labelling of rGH (a-mGH 1:2000 NHPP, linked to protein A gold). There were
very few cells which labelled for rGH in the three blocks o f samples which were cut.
However, one example of rGH labelling can be seen in figure 5.7, which depicts patches
o f granule like immunogold clusters, but no obvious granule structure. Although the
morphology o f the cell is not clear in the figure, due to processing in LR gold and
increased contrast to aid visualisation o f staining, the cell looked normal and cytoplasm
was not vacuolated. I also attempted to immuno-stain these cells for hGH twice; in both
instances the labelling appeared unspecific.
5.7 Co-transfection studies using confocal and TIRF microscopy
The data from EM studies in hGH-IVS3 transfected cell lines suggested that suppressed
rGH production in these cells was brought about by the inability to produce or maintain
condensed GH granules as efficiently as in hGH-WT transfected cells. In order to
visualise these granules in real time I co-transfected the p48hGH-eGFP construct into
both the hGH-IVS3 and hGH-WT stably transfected cells. I established 4 stable cell lines
o f each of the double-transfections, by the incorporation o f neomycin (250p.g/ml)
(eGFP) and zeocin 200pg/ml (hGH) into surrounding media for 4 weeks for selection.
Each co-transfection showed the same result, therefore, only one example from each was
selected for study, using confocal and TIRF microscopy. Both hGH-WT-eGFP and hGH-
IVS3-eGFP cell lines produced brightly fluorescing cells, but with markedly different
distributions o f fluorescence.
151 -

J ! l i ' . i i l > l h ! l i ' - n
% - ) : . -
m&Wà
v i '■
# 3 ;
a - r G H
4 ;.
j--'
Figure 5.7 Electron microscopy im m uno-labelling of rG H in hGH-IVS3
transfected GC cells
GC ce lls were transfected with hGH -IVS3 reporter construet show n in figure 5.1.
Sam ples from these cells were prepared for EM and im m unogold labelled for rGH
(goat a-m G H linked to a -goat gold; lOnm). rGH is distributed in patches o f granule
like Im m unogold clusters, w ith no obvious granule structure. M agnification
= x25,000 .
-152-

L l lO l ’h y i '___________ //' v i l ro siii( / ic \ (>! it ( ' H c e l l l in e c xp i \ ' s s in < ’ n h a n n in ( i l l d o n i in a n ! n c ‘ i i i i v c nii iUil ion
The GC cells expressing both wild type hGH and hGH-eGFP showed a punctate
distribution of eGFP fluorescence as expected (figure 5.8a). However, in the GC cells
transfected with the dominant-negative hGH-IVS3 mutation and eGFP, there were only a
few cells with a punctate distribution of fluorescence. In the majority o f cells, the eGFP
fluorescence appeared relatively diffuse (fig 5.8b) throughout the cytoplasmic
compartment and was also found aggregated in large “clumps” in the cell.
This study was extended to observe those granules near to the plasma membrane using
TIRF microscopy. It can be seen from figure 5.9 that the co-transfection containing the
hGH-IVS3 mutation contains diffuse cytoplasmic eGFP (fig 5,9b) compared to the
obvious granular appearance of eGFP in hGH WT cells (fig 5.9a). The GH granules in
the hGH-WT-eGFP cells display normal motion and show signs o f GH exocytosis as
described in chapter 3. From preliminary data, only a minority o f cells in hGH-IVS3-
eGFP have GH granules (5%) and the cells with a diffuse distribution of eGFP show no
granule motion or evidence of exocytosis (data not shown).
- 153 -

hGH-eGFP hGH-IVS3-eGFP
Figure 5.8 Confocal microscopy of hGH-lVS3 GC cells co-transfected with
hGH-eGFP
Both hGH-WT-eGFP and hGH-IVS3-eGFP cell lines show bright, fluorescent cells, but
with markedly different distributions of fluorescence. (A) The GC cells expressing both
wild type hGH and hGH-eGFP showed a punctate distribution of eGFP. (B) GC cells
transfected with the dominant-negative hGH-lVS3 mutation and eGFP showed diffuse
cytoplasmic fluorescence.
- 1 5 4 -

hGH-WT hGH-IVS3
10 nm 1
Figure 5.9 TIRF microscopy of hGH-WT vs hGH-IVS3 GC cells, co-transfected
with hGH-eGFP
GH granules near to the plasma membrane were visualised using TIRF microscopy. Co-
transfection of hGH-eGFP into hGH-IVS3 stably transfected cells lines produces a
diffuse, cytoplasmic distribution of eGFP (B) compared to the obvious granular
appearance of eGFP in hGH-WT cells (A).
- 1 5 5 -

_________________ / /? vi lr o s m d ic s III II (j I I n V/ e x p rcssiii'j, a liu>)uni ( i l l d o n i i fk in ! nc'j^cil i v c n in U it lon
5.8 Discussion
Mutations in the GH-N gene that result in mis-splicing and the deletion of exon 3 are an
apparent cause of autosomal dominant GH deficiency and severe growth failure. These
mutations are present in all affected members of several families with this deficiency
(Phillips & Cogan, 1994; Cogan et a l, 1994; Cogan et al., 1995; Binder et a l , 1996). The
recurrent nature of the mutation found in all three families may be due to the position of
the G -^A transition, occurring at the CpG dinucleotide that spans the exon 3/IVS3
boundary on the antisense strand. CpG dinucleotides are known to be inherently unstable
due to the propensity of the cytosines to become sequentially methylated then deaminated
to thymidines. This process yields a C -^T transition (and therefore G ^ A on the
complementary [coding] strand) (Cooper & Youssouffian, 1988). The data presented in
this in vitro chapter and in other more recent studies (Hayashi et a l , 1999b; Lee et al.,
2000) support this genetic evidence by demonstrating that GH-IVS3 (+ 1 G ^ A )
suppresses production of wild type growth hormone in transfected cultures.
Autosomal dominant disorders in hormones have been attributed to defects in protein
folding (Cogan et al., 1995; Olias et al., 1996; Kim & Arvan, 1998; Ito et al., 1997). In
arginine vasopressin (AVP) the mutations include single amino acid substitutions, a
single amino acid deletion and a prematurely terminated polypeptide chain (Ito et al.,
1997; Olias et al., 1996). The mechanism for dominant-negative AVP deficiency has
been explored by stably transfecting cells with AVP mutants, which resulted in
accumulation of mutant protein in the ER, consistent with lack of proper folding. In
some instances, the accumulation of the mutants was found to be toxic to the cells (Ito et
al., 1997). The toxic accumulation of misfolded proteins or formation of dysfunctional
oligomers have also been proposed as explanations for autosomal dominant GH
deficiency, particularly in cases in which mutations in the GH-N gene result in deletion
of exon 3 (Cogan et al., 1994; Binder et al., 1996).
In previous wild-type GH and GH-IVS3 co-transfection studies, dominant suppression of
wild type hGH by hGH-IVS3 only occurred in secretory neuroendocrine cells. Hayashi
et al., (1999b) studied the synthesis and secretion of GH encoded by the GH-IVS3 mutant
gene and GH wild-type gene in different cultured cell lines using metabolic labelling
studies to trace and chase labelled GH protein. Transfection o f GH-IVS3 into COS-1 or
156 -

c ’h i i/ ’’lc r ___________ In v i l ro s u t d i c s o f 'n ( , ' / / ccH U ne L-xn/'cssin'^ a h u n / u n 6 7 / Llopi'nh inl n c ’ a t i v c n iuU il ion
HepG2 cells (non-secreting cell lines) revealed a mutant GH with a reduced molecular
mass was synthesised from the GH-IVS3 mRNA and retained in the cells for at least six
hours after the chase. Conversely, the wild-type GH was rapidly secreted into the
medium with minimum retention after one hour of chase. Co-expression o f mutant GH-
IVS3 and wild-type GH neither resulted in any inhibition of wild-type GH secretion nor
affected cell viability in either of the non-secretory cell lines supporting the hypothesis
that intracellular storage is somehow involved in the inhibition of wild-type secretion by
mutant GH-IVS3. When GH-IVS3 and wild-type GH were co-expressed in secretory cell
types MmT/S (derived from rat pituitary somatotrophs) and AtT-20 cells (derived from
corticotroph cells), significant inhibition of wild-type GH secretion was observed in a
concentration dependent manner.
More recent studies by Dannies and colleagues also show that co-expression of wild type
GH and G H -IV S3 in non-secretory CHO and COS cells did not suppress accumulation of
wild type GH but accumulation and secretion of wild-type GH in neuroendocrine cell
lines G H 4C 1 and AtT20 was suppressed (Lee et al, 2000). Further co-expression o f GH-
IV S3 with human PRL in GH 4C 1 did not have any affect on the accumulation and
secretion o f PRL, indicating that there was not a general suppression in secretory
pathway function (Graves et al., 2001). All of these data implicates the interaction of
mutant and WT GH in the process o f granule formation or stabilisation.
In my study, I chose to transfect hGH-IVS3 specifically in rat GC cells, which produce
grovW:h hormone, and barely detectable PRL. I aimed to test this construct in a rodent GH
cell line to establish if the human mutation would produce dominant suppression of
endogenous rat GH, thus function properly in a rodent system. This information was
essential if I was to proceed to express this mutant in a transgenic animal. Sequencing of
cDNA from both hGH-IVS3 and hGH-WT GC cells confirmed that cells transfected with
hGH-IVS3 transcribed a message which was spliced to an mRNA product lacking exon
3. I assayed both GC cell homogenates and surrounding media for the presence of human
and rat growth hormone. Human GH was not detected in my study, but this was perhaps
because my specific antibody did not recognise the truncated hGH protein. In previous
studies a 17.5kDa protein has been detected in immunoblots from non-secretory cell lines
(Lee et al., 2000).
157 -

c 'h a n te r 5 : _ _ _ _ _ _ _ _ _ In v l/rc sl iulic.s o f \ i ( i/J j -nil Hue e x p r e s s ing:: n intn u n i ( III ( lonilnnnt iicacifivc nn ila l ion
RIA of rat GH determined that accumulation and secretion o f rat GH was significantly
suppressed in GC cell homogenates and in surrounding media. Accumulation of the GH-
IVS3 mutant protein was not reported in either of the previous studies (Lee et al., 2000;
Graves et al., 2001) to cause cell death, which conflicts with my findings in rat GC cells
that synthesise GH only. The number of dying and dead cells in GC cells transfected
with hGH-IVS3 was considerably higher than wild type GC cells, or GC cells transfected
with wild type hGH and the growth rate o f mutant cells was substantially slower than
those transfected with wild type GH. However, the transfections in previous studies were
transient and performed in a much shorter time than the stable transfection studies I
report. I have not determined if the reduction in growth rate is due to a decrease in cell
proliferation, or an increase in apoptosis. Whatever the mechanism, it seems that
accumulation of mutant dominant-negative GH proteins in the cell causes toxic effects
leading to cell death.
Further detailed analysis of hGH-IVS3 transfected GC cells using electron microscopy
showed the majority of the cells were dying. Few hGH-IVS3 GC cells appeared to be
‘normal’, containing swollen ER, Golgi and mitochondria; even fewer still contained
dense-cored GH granules, compared to hGH-WT transfected GC cells. These secretory
granules contain a dense core of GH, representing the culmination o f protein assembly
processes that proceed throughout the secretory pathway (Arvan & Castle 1998) and the
mutant GH could affect any o f these processes involved. Briefly, these may be
summarised as follows: initial assembly of granules begins in the endoplasmic reticulum,
where oligomeric associations and other post-translational modifications may be
monitored by enzymes known to regulate ER export (Hammond & Helenius, 1995). ER
assembly reactions, such as protein folding and dimérisation are prerequisite for the
further Golgi/post-Golgi assembly that contributes to proper packaging within granules
(Huang & Arvan, 1995). Proteins must be folded correctly in order to leave the ER and
proceed further along the secretory pathway (Pelham, 1989; Hurtley & Helenius, 1989).
Proteins also have prosequences which have been shown to increase the rate o f correct
folding, allow the formation of dimers and facilitate transport from the ER (Mains et al.,
1995).
- 1 5 8 -

iJ l i lU hT ^ _ _ _ _ _ _ _ _ _ _ _ _ / ; ? v u r o s iudics o/ a G i l i'cH line expressIri'.: , i hitnn m ( ! / / doniiniiu! n ra n f i v c niiilatlon
The hGH-IVS3 transfected GC cells are capable of making GH granules, as seen in EM,
although after some time, the ER and Golgi start to look distorted and swollen. The
mutant hGH lacks residues 32-71, which constitutes the entire connecting loop between
helix 1 and helix 2 (Ultsch et a i, 1994). Without these residues it can be assumed that
this GH molecule cannot fold normally. Unfolded or mis-folded proteins synthesised in
the secretory pathway are usually identified and degraded and do not interfere with the
folding of other proteins (Gething & Sambrook, 1992). An important mechanism for
such degradation is transport of the proteins back across the membrane o f the ER to the
cytosol where they are cleaved by proteosomes (Werner et a i , 1996; Sommer & Wolf,
1997 Schwartz & Ciechanover, 1999). It has been shown that mutant proteins need not
fully be synthesised for reverse transport and degradation to occur (Liao et al., 1998).
Relevant studies using proteosome inhibitors in hGH-IVS3 transfected AtT20 cells
enhanced the accumulation of mutant GH in the cell (Lee et a i , 2000). The presence of
swollen ER in hGH-IVS3 GC cells in my study could emanate from a mass of mutant
hGH-IVS3 in the ER which is not cleared quickly enough by degradation processes.
There are two other identified mutations of the GH-N gene that result in forms with
aberrant folding. In one, the first 56 amino acids have the normal GH sequence, but a
subsequent 2 -bp deletion resulting in a frame shift and altered amino acid sequence
(Igarashi et a l, 1993). The second is a splice-site mutation resulting in a mRNA with an
altered reading frame after amino acid 103 (Cogan et al., 1994). These changes in amino
acid sequence will alter the tertiary structure these proteins compared with that o f wild
type hGH. Although these proteins cannot fold normally, the mutations are
phenotypically recessive, and GH produced from the wild-type gene in heterozygotes is
sufficient to support normal growth. It is thus unlikely that simple misfolding is the
primary problem with IVS3 mutations.
The mutant GH also lacks one cysteine at residue 53. The normal 22kDa GH molecule
contains four cysteine residues, forming two pairs of intramolecular disulphide bonds. It
has previously been speculated that uncoupled cysteine residues in the mutant GH
molecule would lead to the formation of intramolecular disulphide bonds, resulting in
multimeric aggregates containing both mutant and wild-type GH molecules (Cogan et al.,
1994). However, Takahashi and co-workers (1996) reported a case o f GH insensitivity,
in which a mutant GH antagonist (resulting from substitution o f an arginine residue at
- 159

_ _ _ _ _ _ _ _ _ _ _ _ _ _ _ vilrn siiid ics o j d i i / l rcU iinc cxi^rcssini ' ci hiinian ( / / / d o m in a n t nc<:nrivc m ii fa l ion
codon 77 to a cysteine) inhibited the binding of wild-type GH to its receptor. The
heterozygous patient had high serum GH levels, indicating the presence o f an uncoupled
cysteine residue is not sufficient to inhibit the secretion o f wild-type GH from pituitary
somatotrophs.
These mutations further suggest that the production of a protein with an inability to fold
or bind correctly to its receptor is not sufficient to cause a dominant suppression of wild
type GH. In my view, the mutant protein must be sufficiently viable to interact with and
inhibit wild type hGH in order to cause dominant suppression of hGH. It is therefore
conceivable that the mutant GH, lacking exon 3 does fold into a conformation resembling
sufficiently that of wild type GH as to be able to fool the checking mechanisms in the ER.
The unidentified mutant hGH-IVS3 would then be allowed to pass through the ER, free
to dimerise with wild type GH molecules and be transported to the cw-Golgi. After
protein traffic passes through the Golgi complex, the newly synthesised proteins separate
for delivery to distinct destinations. The trans-Golgi network (TGN) has come to be
recognised as the major branchpoint from which distinct ‘anterograde’ membrane
pathways emanate (Arvan & Castle, 1998), including constitutive traffic via small
vesicles to the plasma membrane, lysosomal biogenesis via the endosomal system and
regulated secretory pathway via secretory granules. The many factors involved in the
packaging and secretion o f protein hormones, were discussed in more detail in the
introduction.
This assembly o f granules, collectively referred to as concentration, oligomerisation or
condensation, is promoted by changes in the intraluminal ionic environment, such as mild
acidification (Colomer et al., 1995) in the presence o f high concentrations of bivalent
ions like calcium (Orci et al., 1987a,b; Sambrook et al., 1990), or the presence of zinc.
These processes appear to reflect progressive protein insolubility within the luminal
environment of maturing granules. Zinc is thought to play an important role in the
dimérisation of GH molecules (Cunningham et al., 1991a) and is known to play an
important role in insulin storage (Hutton et a l, 1983; Whittingham et a l, 1995). Human
PRL also binds zinc and it has been reported that substitution of His^^ to alanine in hPRL
suppresses the production o f rat PRL when expressed in GH4C] cells (Sun et a l , 1997).
Zinc is present in high concentrations in GH secretory granules; high concentrations of
160 -

( 'Ihiplcr _ _ _ _ _ _ _ _ _ _ _ _ In viiro si t idics o / a G lI cc/l l ine cxprcs\in<^ <-/ h u m a n ( I I I d n iv in a n l n g ^a r ivc iiiinalio}!
have been found to inhibit GH release (Lorenson et al., 1983). Cunningham and
colleagues showed that two Zn^^ ions associate per dimer o f hGH in a co-operative
fashion (i.e. binding of one Zn^^ ion promotes the binding o f another Zn " ion) thus
inducing hGH to oligomerise. As previously mentioned, the 17.5kDa hGH-IVS3 protein
lacks 40 residues, altering its tertiary structure. Importantly, the zinc binding sites are
still present in hGH-IVS3, but it is unlikely that all three occupy the same configuration
as in the WT hGH, to form a co-ordinated Zn^^ binding site, inducing dimérisation of
hGH. Cunningham and colleagues revealed that mutation o f each o f these residues
caused reductions in the formation of dimeric hGH. They determined that the E174A
variant gave no indication of dimer formation and that the H21A and H18A mutant
exhibited substantially reduced dimer formation. Sedimentation equilibrium studies
allowed quantification of the dimérisation constant of hGH in ZnCl]; mutation of His^' or
Glu'^" to alanine reduced the dimer affinities to 1/58 and 1/11 of the wild type value,
respectively. In theory, the hGH-IVS3 mutant protein could still bind to hGH wild type
molecules, using one, two or all o f the sites. However, I suggest that it is unlikely that
further oligomerisation o f hGH:hGH-IVS3 will occur as this specific binding is co
operative and interdependent (Cunningham et al., 1991a). I suggest that hGH-IVS3
prevents oligomerisation o f hGH in the TGN and subsequently inhibits the formation of
dense-cored GH secretory vesicles. This would therefore prevent WT-hGH from forming
such structures. It has been documented that the (Zn^^-hGH)] complex in vesicles is
substantially more stable and resistant to dénaturation during storage (Cunningham et a l,
1991a), therefore it is possible that the dimeric hGH-IVS3-hGH-( Zn^^) complex in the
TGN and perhaps immature secretory granules (ISG) is more resilient to dénaturation,
generating an obstruction in the secretory pathway which eventually proves toxic to the
cell.
This could explain the progressive stages of cell death in the hGH-IVS3 transfected GC
cell lines; cells begin to synthesise mutant GH which with time, accumulates in the ER &
Golgi. The cell becomes packed with dumped mutant protein and WT protein both unable
to oligomerise to form condensed GH vesicles and progress any further through the
normal secretory pathway. Essentially, my data suggests that the dominant nature o f this
mutation and the powerful effect on WT hGH synthesis implies that the mutant protein
161 -

___________lïL'diL 'J ^i /Ji^klLiUJSl i / h u w iH i ( H I d o n u n o n l n e i^a f iv e n i i i la l io n
actually has a subtle effect and must interact effectively with WT - otherwise WT : WT
dimers would self-select.
The abundance of lipid vesicles in hGH-IVS3 GC cells also substantiates an increase in
lysosomal activity, required for secretory granule membrane recycling and proteolysis. I
imagine that when the cell becomes crammed full of mutant protein, in the ER, Golgi, the
normal functioning mechanisms in the cell begin to shut down and could signal
apoptosis.
If zinc is the key factor in effecting the dominant suppression o f wild type GH, removal
of His*^, His^ and Glu "* (the zinc binding residues) from hGH-IVS3 would inhibit the
mutant protein from binding to WT-GH. I am in the process o f substituting these
residues with alanine in the mutant GH gene to confirm if it is possible to reverse the
dominant-negative effect o f hGH-IVS3 by removing the zinc binding sites. By creating a
GH-IVS3 gene which is ineffectual without its zinc binding sites the mutation would
become recessive, causing no disruption in the secretory pathway allowing the
dimérisation and oligomerisation of wild type GH. This would be easy to test in GC cell
lines and in the transgenics.
In chapter 3, I discussed the p48hGH-eGFP construct containing the entire hGH signal
peptide to target eGFP to GH secretory vesicles. The main aim o f engineering this
construct was to apply it to the visualisation of GH granules in live cells. Co-transfection
o f p48hGH-GFP chimera into hGH-IVS3 GC cells has allowed visualisation of the
defective secretory process caused by a dominant-negative GH mutation. Although this
work is preliminary, I have shown through confocal and TIRF microscopy that the
appearance o f GFP in hGH-IVS3 cells is diffuse and in some cells forms large
aggregates. These cells show no evidence of exocytosis of eGFP, which was apparent in
GC cells transfected with wild type GH (chapter 3). Further work is underway, analysing
granule motion and behaviour through TIRF microscopy in cells at different progressive
stages in the development o f hGH-IVS3. The ability to visualise secretory granule
motion in a live cell is well documented, however we are the first to apply this
technology to study the mechanisms of a granule defective mutation causing IGHD-II.
162 -

_ _ _ _ _ _ _ _ _ _ _ _ _ / / 7 vin'ij sm d ic s o / d LUt r e !I Hu e c x f v c s s i i v.: o h u n m n ( , ' / / (loniiininl n c ^ a l iv c pniralion
The expression o f hGH-IVS3 in a rodent cell line has confirmed that the human GH
mutation produced by a single base pair mutation in the donor splice site of IVS3 is
homologous enough to produce dominant suppression of endogenous rat GH in a rodent
system. To investigate the physiology of this mutation in the growth hormone axis, I next
generated a transgenic mouse which targets hGH-IVS3 to pituitary somatotrophs. The
expression and physiological regulation of this transgene reflects the regulatory
sequences actually utilised in vivo thereby modelling the effect of this disease in a whole
system. This work is described in the following chapter.
163

(■ lu ip t c r ô . __________________ T ra n s ^ ic n ic i v i f c c x p r c s s i m ’ a d o n u n c v i l - i i c'.’j i l i v c h n n t a n i j r o w lh h o r m o n e n iu la l io n
Chapter 6
Transgenic mice expressing a dominant-negative human growth
hormone mutation
6.1 Introduction
Short stature associated with growth hormone deficiency has been estimated to occur in
about 1 of every 4,000-10,000 live births (Binder & Ranke, 1995). While most cases are
sporadic and are believed to result from environmental cerebral insults or developmental
anomalies, 5-30% of cases have an affected first degree relative suggesting a genetic
etiology, some of which are summarised in chapter 1. A great deal has been learned
about the genetic causes o f hypopituitarism over the past two decades. By 1979, a
number of families had been described with isolated deficiency of GH, resistance to the
action o f GH, or diminished production of GH and one or more additional pituitary
hormones. Recent advances in DNA mapping techniques have revealed the location and
types of molecular derangements causing endocrine disease. The mutations in turn have
explained the alterations in the hormone products.
The mechanisms proposed for autosomal dominant deficiencies are generally related to
protein folding and the toxic accumulation of misfolded or unfolded proteins, the
accumulation of dysfunctional heterodimers, or a combination o f the two in the
endoplasmic reticulum and the trans-golgi network (Kim & Arvan, 1998; Ito et al, 1997;
Dannies et al., 2000). My hypothesis is, in order for a more specific dominant-negative
effect to occur, the mutant protein must somehow be processed with and interact with the
wild type protein, thereby inhibiting hetero-dimerisation of growth hormone. If this were
essential for the formation of dense cored GH granules, granule formation with the wild-
type GH would not occur. It seemed from chapter 5 that hGH-IVS3 would block rat GH;
I therefore hoped that a similar mechanism would operate for mouse GH and therefore be
able to generate a phenocopy of the human disease in a mouse model. Although my
studies in GC cell lines have shed more light on the mechanisms o f this GH-IVS3
16 4 -

( 'luiplcr A . _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ TrdiiS'^ciiii' n i 'uc e xp r e s s ing’ a (l(>niiiiiin!-ih‘:a l ivc /iiiiihin a i i jw lh h m 'm o n e iinilalion
mutation, they cannot model the mechanism of the disease in vivo and subsequent
physiological effects in the whole animal.
The aim o f the work described in this chapter was to generate a transgenic mouse
carrying a human dominant-negative GH mutation, again using the cosmid construct
containing the hGH LCR which targets transgenes specifically to pituitary somatotrophs
in a reproducible, copy-number dependent manner. Thus, the hGH-IVS3 mutation
should be expressed solely in mouse pituitary somatotrophs. The disruption o f the
hypothalamo-pituitary GH axis, manifested by the effects o f a dominant-negative GH
mutation could then be studied in situ, in pituitary somatotrophs, rather than in cell lines.
6.2 Targeting the hGH-IVS3 mutation to pituitary somatotrophs in mice.
The sequence containing the hGH-IVS3 mutation was obtained from Phillips III, and
subcloned into the 40kb (K2B) cosmid (Jones et a i, 1995), containing the hGH LCR and
human GH gene, described in chapter 4. The cloning strategy for engineering of hGH-
IVS3 cosmid construct for microinjection is detailed in figure 6.1. In brief, the hGH-
IVS3 genomic fragment, previously cloned into pCDNA3.1 for expression in rat GC cells
(chapter 5) was digested with Sacl/EcoRl (fig 6.1a). The Sacl/E coR l fragment,
containing the +1G—>A IVS3 transversion, was then substituted into pKS-GH.M
(previously digested and dephosphorylated - figure 6.1b). pKS-GH.M is the modified
hGH-WT plasmid containing Mlul linkers.
The hGH gene sequences o f the cosmid (cosGH.M) were then excised as a single Mlul
fragment (see Appendix I and figure 6.1c) and replaced with the M lul linkered hGH-
IVS3 sequence to give coshGH-IVS3. Restriction mapping was done in order to confirm
that the IVS3 (+1G—>A) mutation was inserted into K2B with the correct orientation
(figure 6.2). The final cosmid thus contained a ~40kB Not I insert containing the hGH
LCR and 5’ and 3 ’ untranslated sequences for the hGH dominant-negative gene.
Construct (d) depicts the final Notl microinjection fragment used to create the transgenic
mice discussed in this chapter.
- 165 -

( 'hap 1er 6: Transgenic mice expressing a UomtiiaiU-neguUve human grinvth hormone niutalion
Sac\ EcoRI N ot\Main
(a ) hG H -IVS3
(b )
pKS-GH.M(IVS3)
SailI
EcoRl Sfil I I
* M lu I TBsRGI Sad *
BamHI I PviJI M a i IIJ Li_ I
EcoRI PvtJI I Notl M l u l
I I I
(c)EcoRI Notl
I IBamHI
L _
BamHI SnaBI SnaBIEcoRI I EcoRI I EcoRI I EcoRI U _U U I
4 0 3 0 20 10
BamHI EcoRINotl I Miul
I I
I0
f k B
M lul( d )
EcoRI Notl 1 1
BamHI1
BamHI EcoRI 1
ll
SnaBI SnaBI EcoRI 1 EcoRI 1 EcoRI
1 I 1 1 1
BamHI EcoRI 1 * Notl 1 M in i
1 1 1 1
Y //À1
401
3 01
2 01
1 0 0
k B
Figure 6,1 HGH-IVS3 cosmid construct for microinjection
The K2B cosmid containing the hGH LCR and human GH gene was modified to contain the human
dominant-negative GH mutation and used to target hGH-IVS3 to mouse pituitary somatotrophs, hGH-IVS3
mutation from pCDNA3.1 (a) was digested with SacHEcoRl. The SacHEcoRI fragment, containing the
+1G-^A IVS3 transversion, was inserted into pKS-GH.M which was previously digested and
dephosphorylated (b). pKS-GH.M is the modified hGH-WT plasmid containing M lul linkers, therefore
hGH-IVS3 insert can be removed easily and inserted into cosGH.M using M M
The hGH gene sequences o f the cosmid (cosGH.M) were then excised as a single M lul fragment (see
Appendix I and 6.1c) and replaced with the M lul linkered hGH-IVS3 sequence to give coshGH-IVS3.
Construct (d) depicts the final fragment ready for microinjection. Hatched bars represent vector sequence.
Note that constructs are not drawn to scale.
- 1 6 6 -

( lutpler 6: TniiisgcHic mice expressing a Uomiiiani-ncgalive hitman ^rcAvlh horniom; nmlaiion
E NI I
B E BM
E EiBM
N E
—
40
IkB
fMlul
l . l k b
30
—T 20 10
ATG
N I a l l l Nlalll 1314 1953
IMlul
(2.6kb)
Figure 6.2 Characterisation of IVS3+1G>A Mutation in GH cosmid
The restriction map show s BamHI sites (B ) and EcoRI sites (E). The M lul fragment is
also shown, detailing the extra M aiII site at 13I4bp. Restriction mapping o f both clones
cosG H .M and cosG H -IV S3 confirm s the presence o f G ^ A IVS3 transversion and
correct orientation o f fragment in cosm id. Both clones cosG H .M and cosG H -IV S3 are
identical, except in the M a lll digest, where the band at l.Ik B is absent in cosG H -IV S3,
replaced by tw o extra bands at 514 and 639 base pairs respectively.
- 1 6 7 -

f h i i p l c r f).______________y'/'c; / e x p r e s s i i nj a t l a n n n iiiU- n c v j u i vc hmiKiii -^ro w l h h o rn io in" n v iK i l io n
6.3 Generation and identification of transgenic mice
The LCR-hGH-IVS3 DNA Notl fragment was prepared and microinjected into fertilised
mouse oocytes as previously described (4.3). Resulting pups were assayed for the
presence of the hGH-IVS3 transgene by PCR and Southern analysis (figure 6.3a and b).
The PCR assay was identical to that used in chapter 4 for genotyping hGH-eGFP mice.
PCR with primers hGH 5’UTR (F) and hGH exon 2 (R) (see table 2.1) resulted in two
products; with the endogenous mGH product 50bp smaller than the hGH-IVS3 transgene
product (350bp).
In a total o f 34 pups surviving to term from pro-nuclear injection and oviductal transfer, 3
pups were transgenic, one male and 2 females. All three founder mice were set up to
breed with wild-type [(Cba/Ca x C57B1/10) FI] mice, and 50% transgene transmission
was obtained in all three o f these lines. Sperm from each o f the three lines is presently
being cryopreserved.
6.4 Physiological studies of GH-IVS3 transgenic mice
Transgenic animals from lines 1 and 12 were significantly dwarfed from 3 weeks o f age,
which can be seen in figure 6.4. The dwarfism is proportional in weight and length.
These dwarfed transgenic animals did not breed well compared to line 23, which
appeared phenotypically normal and bred well. In order to assess the effect o f the
dominant-negative mutation in these mouse models, several physiological studies were
done. Most of my studies were performed in the model with the most severe dwarfism
(FI).
6.4.1 Growth parameters o f hOH-IVSS transgenic v wild type littermates
Animals from all three lines were weighed once a week for 25 weeks, and their body
weights recorded (figure 6.5). A dwarf phenotype was not detectable until 3-4 weeks of
age. Adult hemizygous hGH-IVS3 mice only reached -60-70% of the weight of their sex
matched littermates in both lines 1 and 12. After 21 days, significant growth reduction
was clearly seen in transgenic hGH-IVS3 males (p<0.001 transgenic male v non-
transgenic male littermates; n=12) and they remained small. Dwarfism was also present
- 168

7 / , 6 : } . '7' ' ' ^ rL- ' ’r : : : u u i l m -.in i i v c h i in i i in i i r o ^ n h h o r m o n e ’ n iu l a l i o n
K2B
WT
B350bp hGH
300bp mCH
# e
&
l .lk b
639bp514bp
Figure 6.3 Genotyping and identification of hGH-IVS3 transgenic mice
PCR with primers hGH 5 ’UTR (F) and hGH exon 2 (R) (see table 2 .1 ) resulted in two
products (A); with the endogenous mGH product 50bp sm aller than the hGH -IVS3
transgene product (350bp). PCR am plification o f genom ic m ouse D N A with human
hGH primers (hGH 5 ’UTR (F) and hGH exon 5 (R); see table 2 .1) and further Southern
analysis (B) show s the l . lk B band present in control K2B cosm id D N A is no longer
present in transgenic m ice after MûrlII digestion (639bp and 514bp). (W T = w ild type; T
= transgenic; K2B = control cosm id).
- 1 6 9 -

Figure 6.4 hGH-IVS3 transgenic mouse vs wild type litterraate.
An agouti m ale hG H -IV S3 transgenic m ouse, aged 4 w eeks (left) com pared to male
littermate (right) show s the dom inant-negative dwarf phenotype which developed in lines
1 and 12 after weaning.
- 1 7 0 -

( Ikipier 6: Transgenic mice expressing a dominanf-negative human ^r<nvih hormone miilaiion
Line 1 Line 12
r
$
***
30 -**
20 -
10 -
10 15 20 25
Age (weeks)
ÛÛ
$
oc* * *30
**
20 ,o
10
10 15 20
Age (weeks)
25
Line 23
■§)
30 -
A:
20
10
M/T
'>■■ M/NT
o f /T
F/NT
n=12
5 10 15 20 25
Age (weeks)
Figure 6.5 Growth Curves for hGH-IVS3 transgenic mice
Animals from each line (n=12) were weighed weekly, and their body weights recorded. No differences
were detectable before 3-4 weeks o f age. Reduced weights were clearly seen in transgenic hOH-IVS3
males (/k O.OOI transgenic male[M/T] v non-transgenic male [M/NT] littermates) and they remained small.
Female transgenic [F/T] hOH-IVS3 mice also had reduced weight (^ 0 .0 1 ) but to a lesser degree. There
was no difference in weight between line 23 and non-transgenic littermates.
- 171 -

( JlLLL 'h 'L _____ lj:<l '1 V i ' !> ' ' d<Æ IL 'A li U:. ' ic'-Lc'i iv c h u i i ian v j-ow ih l io r in o iu ' nnilali(>n
in female transgenic hGH-IVS3 mice, but was not as significant (p<0.01; n=12) as male
littermates. Line 23 did not show a dwarfed phenotype.
Body length and tibia lengths were also recorded in males from all three lines (1, 12, 23),
every two weeks, for 10 weeks. This data generally reflected my results from the growth
curves shown in figure 6.5. Both lines 1 and 12 were significantly reduced in body
length (nose-anus length at 10 weeks in line 1 was 78.2±1.5mm versus 101.2±1.7mm in
WT; p<0.001) [figure 6.6] and tibia length (hGH-IVS3: 15±0.4mm v WT: 18±0.36mm;
/?<0.01). The differences became more apparent with increasing age. Although the mean
data for animals from line 23 was not significantly (n.s) different versus their wild type
littermates, some smaller animals were noted.
Dissection of the hypothalamo-pituitary axis, for the purpose o f RIA and ISH, revealed
the hGH-IVS3 transgenic pituitary was less than half o f the size o f a WT pituitary.
Measurements o f pituitary weight were recorded before homogenisation for RIA; wild
type 0.65mg v hGH-IVS3 0.3mg; n=12, p<0.001. The difference in size was more
evident in the anterior pituitary, which was almost flat. The hGH-IVS3 pituitary can be
seen compared to a WT pituitary in figure 6.7.
6.4.2 Pituitary hormone (GH; PRL; TSH; LH) content
RIA of pituitary growth hormone in transgenic hGH-IVS3 animals confirmed that the
dwarf phenotype induced by expression of the human dominant-negative transgene, was
due to a vast reduction in pituitary mGH stores. GH measurements for line 1 are shown
in figure 6.8. Measurements of pituitary mGH content from both lines 1 and 12 (data not
shown for 12) showed a thousand fold reduction of GH stores in both male and female
transgenic compared to wild-type animals (line 1 male hGH-IVS3: 19.2±9.2ng/pit v male
WT: 42.8±8.3p.g/pit; n=6,/?<0.001, 8 weeks). This reduction in GH became significant
(p<0.001) in both sexes by 3 weeks of age.
- 1 7 2 -

i !■' c ' V , '> / i s.N7.‘ / ' . ’ <; (/()nu>hiiil-ih:<^ulivc liinnciii <^ro\vlh honnoiH ' niiil<ili(>n
(A)
GOC
TDOÛQ
125 -
100 -
75 -
* * *
□ M /T 12
Q M /T 23
□ M /N T
50 -
25 -
n=6 Age (W eeks)
(B)
GOC
2
20 n
15 -
10 -
%XX
10
g M /T 1
□ M /T 12
□ M /T 23
□ M /N T
n=6A ge (W eeks)
Figure 6.6 Body and tibia length in HGH-IVS3 transgenic males v non-
transgenic littermates
(A) B ody and (B) tibia lengths w ere recorded from male (M ) transgenic hG H -IV S3 (T) and non-transgenic
litterm ates (N T) in all three lines every tw o w eeks, for 10 w eeks. Both lines 1 and 12 show ed significant
decreases in body length (p<0.001) and tibia length (p<0.01) w hich becam e m ore apparen t w ith increasing
age. L ine 23 w as not significan tly sm aller (n .s) in either body or tib ia length. (***/?<0.001 ; **/?<0.01;
*/?<0.05).
- 173 -

Figure 6.7 hGH-IVS3 vs wild type pituitary gland
(A) Intact pituitary from 10 week old wild type animal weighed 0.65mg compared to (B)
the intact pitutary from hGH-lVS3 transgenic animal which weighed 0.3mg and was
measured to be less than half the length of WT pituitary. The anterior pituitary (AP) is
almost transparent in hGH-lVS3 compared to the posterior pituitary (PP). Scale bar =
500pm
- 1 7 4 -

: O n ! H: ' , 11' n u W l i i i i ' n h ' I I I ' l l l l i l ' l l i o i i
G H P R L
n=6
60 1
o.
I8 30 -Xoa 2 0 -'3CL
10 -
***
WT WT
cf ? n=6 cT
8
Q .6
4
2
0WT WT
?
LH T S H800^ 8 0 0 n
4 0 0 -
2 0 0 - = 200* * *
Figure 6.8 RIA of pituitary hormone content (GH, PrI, LH and TSH)
Pituitary hormone contents were assayed from adult (Sweeks) hOH-IVS3 lines 1,12 and
23 and wild type littermates (n=6). GH, PRL, TSH and LH levels for line 1 (severe
dwarf phenotype) are shown above. Data are Mean ± SEM, *p<0.05; **p<O.Ol;
***p<0.00l. Hatched bars: wild type [WT], shaded bars: hOH-IVS3 transgenic [T].
- 175 -

( Jj j i j>jcj 'J l i_____________ Tr( ins'.: c n ic niL ‘c e x p r e s s i f i<j_ ci ( lo n i in n n l - n c '^idlivc h u i iu n i y,r<)\vlh h o r m o n e m u l u l U m
Although animals from line 23 did not present a significant dwarf phenotype in growth
curves, it did show a mild but significant reduction in pituitary GH stores (figure 6.9). At
4 weeks of age, the pituitary GH content in F23-hGH-IVS3 transgenic animals was less
than that o f wild type littermates in both males (24.88±1.42|Lig/pit v 36.83±2.12ng/pit;
/?<0.01) and females (28.8±2.91pg/pit v 15.2±0.7pg/pit; p<0.01). By 10 weeks o f age the
GH content was reduced further, to -25% of WT pituitary GH content in males and
-30% in females. The fall in GH content from hGH-IVS3 pituitaries increased with age,
in both males (A GH content between 4 and 10 weeks: 24.88±1.42 g/pit v
10.41±0.4 ig/pit; ;?<0.05) and females (A GH: 15.2±0.7 ^g/pit v 10.34±0.26 |Lg/pit;
p<0.05) implying a developing phenotype in these animals.
In order to assess if the expression of hGH-IVS3 in transgenic animals was specific for
GH, the amount of other hormones synthesised in the anterior pituitary PRL, TSH and
LH were also measured and compared in transgenic hGH-IVS3 and wild-type littermates
in lines 1 and 23. In the severely affected line 1, at 8 weeks o f age, all pituitary hormones
measured were significantly reduced in hGH-IVS3 males. Females showed a significant
reduction in prolactin and TSH, but not LH. Having discovered that pituitary GH levels
reduced with increasing age in line 23, I also measured other hormone contents in
pituitary homogenates taken from 21 day male Fl-hGH-IVS3 mice. These results are
presented in the table below. The GH levels were still significantly reduced at 21 days,
PRL and TSH contents were higher compared to adult Fl-hGH-IVS3 and LH was not
affected at this age.
Line 23 revealed only a slight reduction in levels of prolactin at 28 days. Like GH levels,
the PRL reduction was more apparent at 10 weeks o f age. TSH and LH remained
unaffected in F23-hGH-IVS3 mice compared to wild type littermates. Each sample was
assayed for human GH. None was detected in either hGH-IVS3 transgenic animal with
our hGH antibody.
176 -

c ih ll'jcr (} . l ’ i i o n n ' H t i m - i k ' ^an w l i i n i h i i! a r o M ! h / u n i t i o n c i i n i l a t i o H
I
Xos'3(X
***
n=6
4wks lOwks 4wks
n=6 9lOwks
Figure 6.9 RIA of pituitary GH content in hCH-IVS3 (F23) mice
RIA o f pituitary GH content in line 23 [normal growth phenotype] revealed a significant
reduction in pituitary GH stores at both 4 weeks (p<0.01) and 10 w eeks (p<0.001). hGH-
IVS3 transgenic m ice and age- and sex- matched littermates were culled at 4 and 10
w eeks o f age. The fall in GH content from hG H -IVS3 pituitaries appeared to increase
with age, in both m ales and fem ales (p<0.05). Hatched bars = WT m ice; shaded bars =
hGH-IVS3 transgenic animals. ***/?<0.001; **p<0.01; *p<0.05.
- 177 -

( ' h i i / Vc / - A . _ _ _ _ 'l'i-(ins<jciiic m ice a hiiniiin '^ro wlli h o r w o n c niu la l ion
Pituitary hormone content in HGH-IVS3 transgenic mice v WT littermates at 3-4
and 8-10 wks
HormoneContent Age (weeks) WT FI F23
1Mouse GH 3-4 36.83±2.12 0.025±0.01*** 24.88±L42 I
8-10 57.0±3.92 0.035±0.01*** 10.41i2.14** 1Mouse PRL 3-4 1.5±0.5 0.75±0.23* 1.05±0.5
8-10 2.4±0.8 0.25±0.1*** 1.32±0.25*
Mouse TSH 3-4 0.352±0.047 0.153±0.06* 0.307±0.0958-10 0.587±0.052 0.079±0.02*** 0.595±0.07
Mouse LH 3-4 0.362±0.07 0.339±0.075 0.395±0.098-10 0.7±0.095 0.450±0.042* 0.756±0.12
Human GH 3-48-10 n.d n.d n.d
Results are expressed as mean ± SEM ,and GH-IVS3 FI and F23 males are com pared to WT male
littermates. n = 6, n.d. = <0.0006ng, *p<0.05, **p<0.01, ***p<0.001
6.4.3 GHRH and somatostatin mRNA in the hypothalamus
The massive reduction in pituitary GH stores clearly caused severe growth retardation in
hOH-IVSB transgenic mice. It is therefore likely that this would result in a lack of
GH/IGF-I feedback and an increased hypothalamic drive to stimulate synthesis o f more
GH to satisfy physiological demands. I therefore performed in situ hybridisation for
mouse GHRH on hypothalamic ARC and PVN sections o f hGH-IVS3 and WT
littermates. As expected, the mGHRH mRNA expression levels were significantly
upregulated (~7 fold T) in transgenic mice (figure 6.10).
Furthermore, In situ hybridisation o f mouse SRIF mRNA in the PeVN o f the
hypothalamus revealed a significant decrease in levels o f SRIF message (figure 6.11).
The fall in SRIF and increase in GHRH in an attempt to maintain normal GH output in
hGH-lVS3 transgenic mice suggests that the feedback mechanisms controlling GH
production in the hypothalamus are still functioning correctly.
- 178 -

/ ' "( / / sL’o v / i ' /)// ' I L ' L \ v / v -I : / ( > n i i n i ’: i / - .1, - u i h x c L-y f/r. / / : h o r D X i l h ' n i i i f c i i i o n
WT hGH-IVS3
ARC
B 8000 -1
6000 -c3
g13
4000 -C3UO.O
2000 -
c
WT hGH-IVS3
Figure 6.10 In situ hybridisation of mouse GHRH mRNA levels in male hGH-
IVS3 (FI) transgenic and WT mice.
A. ISH of GHRH mRNA levels in WT and hOH-IVS3 transgenic mice present in arcuate
nucleus (ARC) and zona incerta (ZI)
B. Comparison of mGHRH mRNA expression levels in hGH-IVS3 transgenic and WT
mice showed a 7x increase of GHRH in transgenic animals. (WT = 972.5 ± 202 int.
density; hGH-IVS3 = 6514 ± 789 int. density ; Mann-Whitney p = 0.002). Scale bar =
1mm.
- 1 7 9 -

/ !'[/ r t . s l ; i ' / n i j i V y'XJjn^sun:: a tloniinLiii! n.c'^-.mvc h unu 'n L'/ '-'M / -4 ;'-i2]±>nc niuuuiin
WT
\ : >
PeVN
hGH-IVS3
B
c3
c■auQ.o
2 0 0 0 -
1500-
1000 -
500 -
WT hGH-IVS3
Figure 6.11 In situ hybridisation of mouse SRIF mRNA levels in hGH-IVS3 (FI)
transgenic and WT mice
A ISH of SRIF mRNA in WT and GH-IVS3 transgenic mice.
B. Comparison of mSRIF mRNA expression in hOH-IVS3 transgenic and WT mice
resulted in a 2.3x decrease in SRIF levels in hOH-IVS3 transgenic mice (WT = 1375.0 ±
112.2; hOH-IVS3 = 602.5±25.6; Mann Whitney, p = 0.0043). Scale bar = 1mm; PeVN
= periventricular nucleus.
- 1 8 0 -

( lu in te r I ) :______________ Trd i i s ' j c i i ic P iicc c x p r c s s i n a n (h>iuinniil-ih''..u i l i vc h ii i iu in 'j r o w i / i h o m n i / i c n n i m n o n
6.5 EM studies of pituitary sections from GH-IVS3 transgenic mice
To investigate the disruption of GH production in somatotrophs of the anterior pituitary,
hOH-IVS3 pituitaries dissected from 8 week males from line 1 were fixed in 2.5%
gluteraldehyde and processed for electron microscopy.
6.5.1 Ultra structural morphology o f hGH-IVS3 somatotrophs
In order to study the ultra-structural morphology of somatotrophs from hOH-IVS3
transgenic mice in detail, and at good resolution, the pituitaries were processed in Spurr
resin. Pituitary sections from hOH-IVS3 transgenic animals contained very few
somatotrophs. In each section I could only identify two or three normal somatotrophs,
the majority were dying and contained very few dense-cored GH vesicles (figure 6.12).
Corticotrophs (small spherical secretory vesicles densely distributed round the periphery
of the cell), gonadotrophs (characteristic halo-granules) and some lactotophs (irregular
shaped granules) were present, as identified by their cell morphology, but many o f these
different cell types were also dying (figure 6.13). Like GC cells transfected with hGH-
IVS3, somatotrophs from transgenic pituitaries contained enlarged ER and swollen golgi
apparatus as seen in figure 6.12. Many of the cells from hGH-IVS3 pituitaries contained
large quantities of lipid vesicles, typical of phagocytosis, compared to pituitaries from
WT mice. At the boundary between the intermediate lobe (IL) and the anterior pituitary
(AP), there were what resemble perivascular macrophages (fig 6.14) on the AP side,
though their identity remains to be confirmed by macrophage labelling.
6.5.2 E M immuno-labelling o f m GH in pituitary somatotrophs from hGH-IVS3
transgenic mice
Pituitary sections from hGH-IVS3 and WT mice were processed in LR gold and labelled
with mGH (ot-mGH linked to protein-A gold). The labelling o f GH dense cored vesicles
in pituitary somatotrophs from control WT was evident and highly specific. In multiple
sections from hGH-IVS3 transgenic pituitaries, I found only one cell in which granules
were labelled with mGH, however, this cell morphologically resembles a lactotroph
[irregular shaped, smaller granules] (fig 6.15). This cell appears relatively normal,
although it contains lipid vesicles.
- 181 -

i - _______- TrLins<!cn i r ////rc cxpr> \ a d " ‘n ' i uin! i ic^iinivc h u n u i n ‘ m w i l i I n u i m n i r n iu la l io n
' - : . . .
Figure 6.12 EM of soniatolrophs from hGH-IVS3 transgenic pituitary
Pituitaries were prepared for EM by processing in Spurr resin for detailed ultra-
structural morphology at good resolution. Pituitary sections from 8 week old hGH-
IVS3 transgenic animals were labelled for mGH (a-mGH linked to protein A gold:
lOnm: difficult to visualise at low magnification). These sections contained very few
normal somatotrophs. (1) somatotroph containing enlarged ER and swollen golgi
apparatus. (2) dead or dying somatotroph with lipid vesicles (LV) typical of
phagocytosis and swollen organelles. (3) dying somatotroph. Magnification x4000.
-182-

( l inrh 'rn _ _ _ _ _ _ _ _ I riuisiSi-NW niirc u J n iriiiuinl-n c ^ i ^ ' v v hiinu in '.'j-nwih hor inofic intihiiioii
Figure 6.13 Dominant-negative hGH-IVS3 phenotype affects other cell types in
the anterior pituitary
(A ) Gonadotroph (characteristic halo-granules). Seale bar = 1pm.
(B ) Corticotroph (peripheral accumulation o f vesicles). Scale bar = 2pm .
These cells are typical o f gonadotrophs and corticotrophs in hO H -IV S3 transgenic
pituitaries with fragmented nucleus and lipid vesicles.
-183-

/'ri;!lS‘\ ;U( ■fn-ii-nuni iii'i i i h w h n i ' i n i ! ! / , - m u U U U ' H
p iS i211Ï
mamm m ià
Figure 6.14 Interm ediate lobe/anterior pituitary boundary contains
perivascular macrophages
Macrophages (m) could be identified throughout the whole anterior pituitary (AP) in
hGH-IVS3 transgenic animals. A dense population of macrophages were present
adjacent to the IL/AP boundary of the anterior pituitary (separated by cleft cells CL).
These macrophages may be involved in destroying the newly formed somatotrophs at
this border before migration deep into the anterior pituitary. Scale bar = 2pm.
-184-
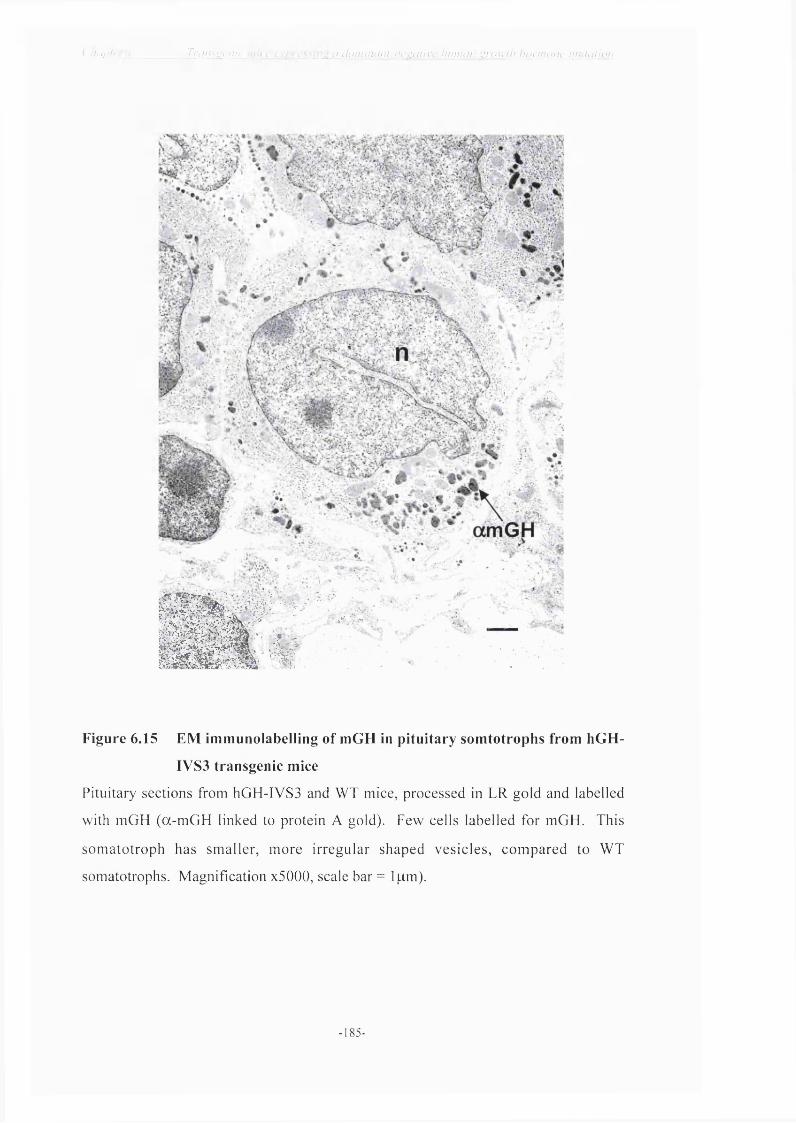
( ijiij_u* Triius\:cnii Lf d()r)}inan h n c^^tu ivc hunnui L v n n v / / hi^rnuinv niuhUion
S i " " * " 'fA:\
Figure 6.15 EM immunolabelling of mGH in pituitary somtotrophs from hGH-
IVS3 transgenic mice
Pituitary sections from hOH-IVS3 and WT mice, processed in LR gold and labelled
with mGH (a-mOH linked to protein A gold). Few cells labelled for mGH. This
somatotroph has smaller, more irregular shaped vesicles, compared to WT
somatotrophs. Magnification x5000, scale bar = 1pm).
-185-

< 'h u r l e r h : ____________ Triins'j,c ) i ic c x / i r c s s in-.: a {i()tiiiniin . l -nc \:n l i w fiii n u in j ’r o w l h h o n v o n c n i i iu t l io n
6.6 Discussion
Ail affected individuals of the families studied to date, whose pedigrees suggested an
autosomal dominant transmission of IGHD carry a deleterious point mutation of GH-1,
suggesting a very high prevalence of these mutations in IGHD-II (Introduction, Table
1.1). The majority of dominant-negative mutations described so far occur in the donor
splice site o f the IVS3 region o f the GH-N gene, though there are 3 reports o f
dominantly-inherited GH deficiency resulting from other missense mutations in the GH-
N gene (Duquesnoy et a l, 1998, Deladôey et a l, 2001, Binder et a l , 2001). In the past,
screening for GH-N defects was only performed if severe growth failure with a height
below - 4 .5sd score at diagnosis was present (Binder et a l, 2001). In the clinical study
performed by Binder and colleagues, severe short stature according to this definition was
only present in one third of the affected individuals at diagnosis, indicating that growth
failure in lGHD-11 is variable and can be less severe than commonly assumed. Notably,
the children with splice site mutations were younger and shorter at diagnosis than their
counterparts with the missense mutations, suggesting this may have a much more
profound effect in residual products from the unaffected allele.
Although lGHD-11 is well documented in patients, there is still little evidence as to the
cellular cause for the disease. Although much information can be gained from stable cell
transfections, we felt it more interesting to approach the problem in situ, by inducing the
same process in a transgenic mouse in vivo. Since we had the capability to ensure high
level, physiologically regulated expression from the hGH LCR, we constructed a
transgenic mouse model expressing hGH-lVS3 in mouse somatotrophs (Jones et a l ,
1995). Although there are considerable constraints in size when studying the physiology
of a dwarfed mouse, such a transgenic mouse could then be crossed with my hGH-eGFP
model, in order to do parallel studies in vivo, as 1 described for the GC cells in chapter 4.
The construct which we chose to make transgenic mice included the entire hGH LCR
(clearly targeted transgenes to mouse somatotrophs in chapter 4) into which we inserted
the hGH-lVS3 gene (+1 G -^A donor splice site mutation) kindly donated by John
Phillips 111 (Cogan et a l , 1995). Since this appeared to be sufficient to produce dominant
suppression o f endogenous rat GH in rat GC cells, we thought it would be likely to
induce dominant suppression of mouse GH in transgenic mice. Microinjection of this
cosmid construct produced three founders, which were bred to make hGH-lVS3 lines 1,
- 186 -

C h(i/>!cr A.______________1 r cins ‘ c n ic m i c e cxp rcss i iT ^ a d o i i n n i in l -n cs i i i l ivc h i i i iu in g r o w t h h o r m o n e iN i i la f ion
12 and 23. Lines 1 and 12 developed severe isolated growth hormone deficiency from
three weeks o f age in both males and females. Dwarfism was more profound in males
than in females after weaning, perhaps a consequence of the sexually dimorphic control
of GH secretion (Jansson et aL, 1985; Clark et a i, 1986; Clark et a i , 1987) and we have
seen this before in the TOR rats (Flavell et aL, 1996). Both lines 1 and 12 showed some
difficulty in breeding. It may be an effect of GH deficiency on fertility, since little mice
are also difficult to breed. Alternatively, it could simply be due to a matter o f size, as the
stud male from both lines was significantly smaller than the females he was trying to
mount. I replaced the breeding females frequently for younger, smaller mice, which
seemed to keep breeding occurring, if very slowly.
Having produced 2 transgenic founder animals with a severe IGHD-II phenotype, I
concentrated initially on lines 1 and 12. The dwarfism in both lines 1 and 12 was
proportional, like IGHD-II patients; reduced tibia and body lengths reflected the
reduction in body weight. Removal of the pituitaries from transgenic mice for RIA
revealed pituitaries less than half the size of pituitaries from wild type littermates. The
difference was most noticeable in the adenohypophysis, which appeared flattened. The
pituitary GH content in lines 1 and 12 was markedly reduced (1000 fold reduction) from
weaning and this did not alter with age. Since somatotrophs constitute -40% of cells in
the anterior pituitary, it was not surprising that the pituitaries from GH deficient animals
were almost half the size if this population was badly affected. Clinical data on the
systematic examination of the pituitary anatomy in monogenetic disorders in patients are
scarce. Recent magnetic resonance imaging observations in children with IGHD
suggested a positive relationship between the volume of the adenohypophisis and the
secretory GH capacity (Hamilton et aL, 1998). However, this is not the case in IGHD-II;
the four children who were examined by MRI by Binder and colleagues, showed a
“normal” adenohypophisis in two cases and mild hypoplasia in two others (Binder et aL,
2001). Normal size of the anterior pituitary was also reported in two children affected
with IGHD-1 A, suggesting that the presence of GH is not a pre-requisite for normal size
o f the adenohypophesis (Zucchini et aL, 1996). One might imagine that pituitary cell
mass might be larger, as GHRH stimulates proliferation of “useless” cells, but not if they
also die. Recent data in Laron dwarf mice (GHRH K.Os) also show that despite
increased GH output, their pituitaries are smaller.
18 7 -

U lL 'P h J ' ___ _________Trnn< 'jc)!ic m i c e c x p r c s s rtv^ .7 ( lo n i i n t in f -n c ^ i i l i v c h nnhti! v j -</wlh h o r m o n e n i i i l d l i o n
The size of pituitaries in transgenic hGH-IVS3 line 23 (no growth phenotype) did not
differ significantly from that of pituitaries from wild type littermates. However, RIA of
pituitary GH content in line 23 showed reduced pituitary GH levels at 3 weeks, which
subsequently dropped to approximately 20% of the GH content measured in wild type
littermates at 10 weeks. This was the first piece of evidence to suggest the hGH-IVS3
may be a developing phenotype, which progressively worsens with the accumulation of
mutant protein in somatotrophs.
In IGHD-II patient studies, the disease is only thought to affect the GH axis specifically
with low levels of IGF-1 and IGFBP-3 («5*^ percentiles for age [Cogan et aL, 1995;
Missarelli et aL, 1997; Deladôey et aL, 2001; Binder et aL, 2001]). These clinical reports
show normal thyroid and adrenal function and normal levels o f prolactin. However, in
our severely affected transgenic hGH-IVS3 mice the dominant-negative phenotype was
not isolated to the GH axis. Lines 1 and 12 (severe growth phenotype) had significantly
reduced levels of pituitary prolactin and TSH at 3 weeks and these were found to be even
less at 10 weeks. At this stage, pituitary LH content was also affected in males.
However, in line 23, the other pituitary hormone contents at 3 weeks were relatively
normal though at 10 weeks of age, the level of prolactin was significantly reduced.
All three lines follow the same trend, in that the GH-IVS3 phenotype progressively
worsens with age. Although I was not able to measure transgene copy-number in the
three founders I generated, the simplest explanation would be that lines 1 and 12 have
more functioning copies o f the transgene than line 23 and this may also reflect multiple
integrants. It will take many more generations and Southern Blot analysis to follow up
this observation. Measuring transgene expression is technically difficult to interpret,
given the likely cytotoxic effects of the products, and we are unable to quantify the IVS3
protein. In lines 1 and 12, mouse GH is reduced 1000 fold, which is detected at an early
age and continues throughout life. Study of hypothalamic feedback mechanisms in the
GH axis of hGH-IVS3 transgenic mice in line 1 showed a significant up-regulation of
hypothalamic GHRH mRNA and a parallel reduction in somatostatin message. The
primary stimulus for GH release is hypothalamic GHRH. GH controls its own
production via feedback on GH receptors located in the arcuate and periventricular
- 188

ijjJslLl u . _ _ _ _ _ _ _ _ _ _ _ _ _ T rans'jj'fiic 111ii'c (.‘X !ve \s in 'c a (/oin in d n t-n c .^dlivc h iitnnn I’ro w ih h o n v o n c iin ila lion
neurones, repressing GHRH and increasing SRIF expression, respectively (Burton et al.,
1992; Lobie et al., 1993). Severe GH deficiency therefore upregulates levels o f GHRH
to stimulate production and release of more “useless” GH-IVS3.
Reduced levels of prolactin are detected from an early age in lines 1 and 12, and are
markedly reduced in adulthood in line 23. It is thought that PRL expressing cells are
derived from the same lineage as GH expressing cells (Hoeffler et al., 1985) relying on
the transcription factor Pit-1 for somatotroph differentiation (Castrillo et a l , 1991). I
suspect that a profound defect in the GH axis, like dominant suppression of GH, causing
ablation o f GH expressing cells could directly impact the PRL expressing cells, thus
reducing levels of prolactin (Borrelli et al., 1989; Steger et al., 1991). More interestingly,
thyroid stimulating hormone was also affected in lines 1 and 12 from an early age,
progressively worsening with the development of the hGH-lVS3 phenotype; TSH was
not altered in line 23. The reduction of TSH in lines 1 and 12 is more difficult to explain,
but since they share the same progenitor cell and common Pit-1 transcription factor
control, 1 suggest that the effects on TSH might be due to exhaustion o f supply from
common progenitors. The Snell {dw/dw) and Jackson {dw'^) dwarf models established
that mutations Pit-1, result in dwarfism (Li et a l, 1990). These mice are deficient in GH,
PRL and TSH and have no detectable somatotrophs, lactotrophs or thyrotrophs (Simmons
et al., 1990). Pit-1 is later required for the continued expression of the Pit-1 gene itself
and the proliferation and survival of these three cell types. Another explanation,
therefore, could be that insertion of multiple copies o f the hGH-lVS3 transgene may
induce artificial competition for Pit-1 and other activators of transcription which could
alter or modify the level of transcription of the endogenous Pit-1 dependent genes and
subsequent differentiation into lactotrophs and thyrotrophs. If line 23 has only 1 or 2
copies o f the hGH-lVS3 transgene, this competition effect would have a less detrimental
impact.
In order to assess the detrimental effect o f the human dominant-negative GH gene on
pituitary morphology, pituitaries from adult transgenic hGH-lVS3 mice from line 1 were
prepared for EM and compared to pituitaries from wild type littermates. Transgenic
animals suffering from lGHD-11 contained very few functioning cells, o f any type, in the
anterior pituitary. Fewer corticotrophs and gonadotrophs could be identified in my
- 189

( 'im n icr (y._ _ _ _ _ _ _ _ _ _ _ _ _ _ Transi^cn ic m ice c x / f i d ' I l ’U U i l l l l z l h h u ma n i’ro w /li h o rm o n e n m ia tio n
samples, due to the fact that the majority of cells appeared damaged. A large number of
perivascular macrophages could be readily identified throughout the anterior pituitary,
characteristic of a high turnover of cells. One or two viable somatotrophs were present,
containing GH secretory vesicles. In support of this data, at least one histological study
has demonstrated the presence of large secretory granules in somatotrophs in the pituitary
o f an IGHD patient, indicating that some IGHD patients do produce and store a form of
GH protein (Rimoin & Schechter, 1973). However, like GC cells transfected with hGH-
IVS3, the mouse somatotrophs had swollen ER and golgi and contained very few
secretory granules, indicating the inability of GH to hetero-dimerise and form condensed
GH granules. Immuno-gold EM labelling of somatotrophs in transgenic pituitaries
labelled only one or two cells per section and these interestingly, resembled lactotrophs,
typified by irregular shaped secretory vesicles. The inability o f GH to hetero-dimerise
and form condensed GH secretory granules might influence the cell to synthesise and
secrete prolactin, instead of GH, especially if these rare cells are mammosomatotrophs
which might be less affected (if they make less GH). This way, it can continue to
synthesise protein without the toxic accumulation o f mutant GH-IVS3. The pituitary
must secrete some GH, albeit minute amounts, as IGHD-II patients respond well to GH
therapy without developing GH antibodies (Cogan et aL, 1995; Binder & Ranke, 1995). I
was unable to identify secretory granules containing mutant human growth hormone with
the human GH antibodies that I tested. Mutant hGH-IVS3 protein has been previously
identified in other cell transfection studies (Hayashi et aL, 1999b; Lee et aL, 2000),
which leads me to believe that the antibodies which I tried did not recognise the mutant
protein with the exon 3 skip.
I was surprised by how few somatotrophs were present. I had initially expected to see a
high turnover of a large population of somatotrophs at different stages o f cell death in the
pituitary, due to the increased GHRH drive stimulating somatotroph proliferation. I
observed a large cluster o f perivascular macrophages at the intermediate lobe
(IL)/anterior pituitary (AP) boundary. Preliminary results from developmental studies in
the GH axis in my d l transgenic hGH-eGFP mice, recorded by Patrice Mollard
(INSERM, Montpellier) show the first expression o f eGFP in cells directly adjacent to
this IL/AP boundary, consistent with observations in neonatal lit/lit mice (Lim et aL,
1993). This peri-nuclear staining of eGFP in the TGN of immature somatotrophs
1 90 -

_ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ I un V V •( '^ s 7 /7 h n liMUU. Ü 'C JLl uucin a row lli h o riu o n c iiu ili/non
becomes punctate in appearance in mature cells, with the production of GH secretory
granules, as they migrate from this boundary. If this data demonstrates that somatotrophs
proliferate deep in the anterior pituitary then migrate throughout the gland, it could be
hypothesised that the perivascular macrophages present at the IL/AP boundary in hOH-
IVS3 transgenic animals begin to destroy somatotrophs immediately they appear. We
have also found that GH cells are present in clusters, closely networked with follicular
stellate (FS) cells, and stimulation of one cell in a cluster provokes activity throughout
the whole cluster (Mollard et aL, unpublished). If these GH cells are indeed linked, it
could be possible that an apoptotic signal from a dying cell might prematurely signal
apoptosis in immature cells, which have recently proliferated at the IL/AP boundary,
through this FS network. These effects might amplify the cytotoxic spread, explaining
the small number of somatotrophs in the pituitary. It is possible that somatotrophs do
proliferate normally, until the GH-IVS3 mutant protein accumulates in the cells causing
cell death, resulting in more macrophage activity and I suggest it is this which
secondarily affects other cell types in the anterior pituitary. I did not have the opportunity
to check the morphology of the diseased pituitaries or if these macrophages were present
in younger animals (i.e. 3 weeks). Furthermore, I would have liked to investigate the
morphology of the pituitaries from line 23. Although line 23 does not have a significant
dwarf phenotype it has a pituitary GH phenotype and may model the IGHD-II described
in patients more closely. Ongoing work in the laboratory will concentrate on line 23, in
which the impact o f the transgene has a less destructive effect in the pituitary.
In chapter 5, I discussed the importance of zinc in the dimérisation of GH molecules
(Cunningham et aL, 1991a). Zinc is present in normal human serum at concentrations of
5-20pM and found in much higher concentrations in GH secretory granules. Cunningham
and colleagues showed that two Zn " ions associate per dimer of hGH in a co-operative
fashion inducing hGH to oligomerise. Three residues His'^, His^^ and Glu^ " are
probable Zn^^ ligands, and are clustered when mapped on a model of hGH (Cunningham
et aL, 1990a; Ultsch et aL, 1994); His’ and His^^ are on adjacent turns of helix 1 and are
positioned near Glu " on helix 4. All three face in the same direction and form a
plausible site for binding o f Zn^^ since their replacement with alanine clearly eliminates
Zn^^ binding without affecting GH receptor binding. hGH is comprised of a chain o f 191
amino acids, cross-linked by two disulphide bridges. The rat growth hormone (rGH) is
1 9 1 -

( liapier 6:_ _ _ _ _ _ _ _ _ _ _ Tru/isgenic mice expressing a domiiicun-negative hiiimin grcm th hormone niiilanon
Figure 6.16 NCBI Sequence Viewer - Human GH v Mouse GH andRatGH
MATGSRTSLL LAFGLLCLPW—TVS----L—
— AD-Q-PW- —TFS----L—
15LRAHRLHQLA FDTYQEFEEA15-- QH-H--- A-- K---R—15-- QH-HQLA A-- K---R-
55SESIPTPSNR EETQQKSNLE55--T--A-TGK — A— RTDM-55— T— A—TGK — A— RTDM-
95SVFANSLVYG ASDSNVYDLL95RI—T— LMF— T--*R--EK-95RI-T-- MF- T— *R--EK-
135TGQIFKQTYS KFDTNSHNDD135V-- L----D -- A-MRS—135I-- L----D -- A—MRS —175TFLRIVQCRS *VEGSCGF175-Y— VMK— R F— S— A—175-Y— VMK— R FA-S— A-
LQEGSA I IP-A--P— AG-
YIPKEQKYSF EG—R— * EG—R— *LLEISLLLIQ
KDLEEGIQTL-------- A--------- A-
ALLKNYGLLY--------- S--------- S
iFPTIPLSRLFDNAM1 — AM S— S — V1 — AM S — AN—V
LQNPQTSLCF I— A-AAF— I--A-AAF—
SWLEPVQFLR--------- S— — — Q— — — — — S
MGRLEDGSPR-QE--------QE-------CFRKDMDKVE — K— LH—A- — K— LH—A—
Protein accession number (hGH) = P01241
I I = start of mature protein
blackbluered
= human (191aa)= rat (190aa, 66% homologous to hGH) = mouse (190aa, 95% homologous to rGH)
- 192 -

( 'hap tcr 6:_ _ _ _ _ _ _ _ _ _ _ Traiis^etiic mice expressing a Jomuuint-negalivc liiinuin gnm th hormone miilcilioi}
only 190 amino acids long and exhibits 66% homology with its human counterpart
(Seeburg et al., 1977a,b). Mouse GH also consists of 190 amino acids and is highly
homologous to rat GH (95%) (Linzer & Talamantes, 1985). These three sequences are
aligned in figure 6.16. The importance of zinc in GH binding may be supported by the
fact that these zinc sites are conserved throughout all mammalian species, being identical
in the macaque and almost identical in rodents; the potential Zn^^ binding sequences are
highlighted and aligned below;
His'* and His" Glu” '
iF P T IP L S R L F D N A M i 5L R A H R L H Q L A / \ / \ / \ i 65CFRKDMDKVE i^sTFLRIVQCRShuman
iF P A M P L S S L F S N A V L R A Q H l H Q L A / \ / \ / \ i 65CFKKDLHKAE i7.,TYLRVMKCRRm,u..
:F P A M P L S SL F A N A V is L R A Q H lH Q L A /X /X /X i s s C F K K D L H K A E ^sTYLRVMKCRRrat
The mutant 17.5kDa hGH-IVS3 protein lacks 40 residues, markedly altering its tertiary
structure. However, the altered structure of hGH-IVS3 mutant protein does not delete
any of the three zinc binding sites. I suggest it retains sufficient structure similarity to
endogenous wild type mouse GH molecules (which contain homologous zinc binding
sites) and inhibit further oligomerisation of GH and subsequently, formation of dense-
cored GH secretory vesicles. The dimeric hGH-IVS3-mGH-( Zn^^) complex may
accumulate in the ER and TGN or even form immature secretory granules (ISG) which
generate obstruction in the secretory pathway. The EM data shows that aggregates of GH
are made, but distributed throughout the cytoplasm and this ultimately overcomes the
ability of the degradation mechanisms to eliminate this product - leading to cell death.
This process is exacerbated in vivo since the lack of GH feedback drives further GHRH
stimulation of GH cell proliferation and GH synthesis. However, it simultaneously drives
the LCR IVS3 alleles which causes a catastrophic accumulation of non-secreted product.
Whether the autolysis of cells also triggers an autoimmune or inflammatory response
probably depends on the rate of cell death. The progressive stages of cell death induced
by accumulation of mutant GH which occurs in the hGH-IVS3 transfected GC cells is
more than likely to be mirrored in somatotrophs in transgenic pituitaries, which with time
inhibits the nonual functioning mechanisms in the cell, signalling apoptosis. The
- 193 -

( 'lutplci- (y_ _ _ _ _ _ _ _ _ _ _ _ _ _ _ T rans'^cn /c m ic e c\[)I■cssll}'^ a iliim inc.iU-nc'r.dliyc inrnuui m-owl/i h o rm o n e im ila tu n i
presence of perivascular macrophages in the IL/AP boundary in the pituitary may be
programmed to recognise the dying cells before they produce further damage to other
functioning cell types.
I have previously mentioned that the dominant-negative GH phenotype progressively
worsens with time in both GC cells transfected with hGH-IVS3 and in somatotrophs in
transgenic pituitaries. In familial central diabetes insipidus (dominant suppression of
A VP), the secretion of arginine vasopressin is not impaired in early life. The affected
patients who are heterozygous for point mutations in the A VP precursor manifest
symptoms long after birth (Ito et aL, 1991). In such a disease with delayed onset, a
reduction in the number of cells due to the accumulation of the mutant protein is more
likely to be involved in the pathogenesis. However, there is no substantial evidence for a
course o f slow progression of GH deficiency in IGHD-II as was described for the
dominantly inherited A VP deficiency. Some patients develop the IGHD-II phenotype
more quickly and severely than others. There may be factors other than systemic GH and
IGF-I levels which are responsible for differences in growth failure and these unknown
factors obviously modulate the start and predominance of GH-dependent growth.
Developmental studies are necessary to distinguish at which stage the dominant-negative
effect begins to alter the physiology in the GH axis. On-going work in the laboratory will
involve crossing the hGH-IVS3 transgenic mouse model, with the hGH-eGFP mouse (in
which eGFP is targeted to GH secretory vesicles) generating a double transgenic model
with a GH secretory granule defect which can be visualised in situ, in real time by
tracking of endogenous eGFP. Now that we have a good mouse model o f this disease,
we hope to be able to study the time scale in which suppression o f wild type GH
secretion occurs, and where in the secretory process the mutant GH accumulates. In
addition to shedding light on the pathophysiology of GH dominant-negativity, we may
hope to gain a better understanding of the packinging and synthesis of the GH secretory
granule.
- 1 94 -

C lu ip lc r _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ l-'iini/ D iscu ss io n
Chapter 7
Final Discussion
7.1 Transgenes targeted to growth hormone cells
In the studies described in my thesis, two transgenes were targeted to anterior pituitary
somatotrophs - hGH-eGFP and hGH-IVS3. The somatotroph has been targeted by
transgenesis before: hGH-N directed by its proximal promoter or by additional 4.6kb of
5’ and 26kb of 3 ’ flanking sequences was either not, or only poorly expressed in
transgenic pituitaries (Hammer et aL, 1984; Palmiter and Brinster, 1986). However, a
40kb hGH-N cosmid was shown to comprise all regulatory sequences for appropritate
transgene expression, restricted to anterior pituitary GH cells and to contain the full hGH
LCR (Bennani-Baiti et aL, 1998; Jones et aL, 1995). These data highlighted the
importance of including a full LCR in transgene constructs for high-level, tissue-specific
transgene expression, irrespective of the chromosomal site o f transgenic integration.
Manipulation o f large cosmid constructs is usually difficult, as one is dependent on the
availability of appropriate restriction sites. In our hGH- cosmid a unique Mlu\ restriction
site was engineered at the site of transgene insertion. Although complex, it had the
design advantage that all subsequent engineering for future transgenes could be
performed as a one-step cloning method, linked by a common Mlu\ fragment, and the
engineering introduced only a minimal (2bp) alteration in the new 5’ sequence o f the
hGH promoter. In hGH-eGFP transgenic mice the hGH cosmid was shown, once more,
to target transgene expression appropriately and exclusively to GH cells in the anterior
pituitary. My studies also highlight the inclusion o f differeing amounts o f hGH
sequences directing transgene expression to different structures o f the GH cell.
7.2 The hGH-eGFP transgenic mouse
In chapter 3, two constructs containing eGFP were discussed. The first eGFP construct
contained only the first 8 residues of the hGH signal peptide, the second construct
contained the intact signal peptide and a further 22 N-terminal residues o f hGH. With
both constructs, the first intron o f the hGH gene was included, since this contains
enhancer sequences thought to be important for efficient transgene expression (Brinster et
- 1 9 5

C h a p ic r "_ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ f-'indl D isc u ss io n
aL, 1988) and for appropriate transcription in our hGH-eGFP transgene construct. We
aimed to make the shortest peptide possible, but avoided altering the nucleotide sequence
context close to the splice acceptor site as the accuracy and efficiency of splicing is often
affected by the nucleotide sequence in the immediate vicinity o f the splice junction (Reed
& Maniatis, 1986; Matsuo et aL, 1991). When expressed alone or with minimal N-
terminal peptide extensions, eGFP pervades throughout the cytoplasm of GH cells. The
eGFP-GH fusion product identified GH cells in situ, without altering key regulatory
processes or expression of the gene.
The second construct aimed at targeting eGFP to secretory vesicles (p48GH-GFP) and
included not only the entire hGH signal peptide, but also the first 22 residues o f the N-
terminal sequence of hGH. This also included the two N-terminal histidine residues of
hGH (’^His and ^*His). These contribute significant Zn^^ binding activity to hGH
(Cunningham et aL, 1991a) and are thought to be important in GH dimérisation and
subsequent packaging. These histidines were included to enhance any potential co
packaging of the GH-GFP fusion protein in mouse GH vesicles. Although we included
the N-terminal zinc binding sites of GH, my data does not show whether these residues
were in fact necessary for granule packaging of eGFP or merely fortuitous. The aim of
this was not to identify the sequences necessary, merely to find a construct that did
achieve this. Thus, eGFP was fused with the signal peptide and an additional portion of
the N-terminus o f hGH (p48hGH-eGFP), which directed the fluorescent product to GH
secretory vesicles in rat GC cells and pituitary somatotrophs. The transfection o f eGFP
into rat GC cells was performed to achieve targeting o f eGFP to GH granules in a GH
cell and to assess the viability of the human GH construct (p48GH-eGFP) in a rodent cell
line in vitro, before making a transgenic mouse. It was o f critical importance to include
the entire GH LCR and sufficient signal peptide sequence to target GFP not only to the
correct cell type, but also direct its entry into the endoplasmic reticulum and subsequent
secretory pathway, without interfering with the physiology o f the animal. This hGH-
eGFP model has provided us with an invaluable tool in order to improve our
understanding of how pituitary growth hormone is sorted and processed and how these
processes may be regulated. Presently, hGH-eGFP animals are being used to study
development of pituitary GH cells at birth and their 3-D organisation, which may give us
a key to identifying nascent GH cells. The hGH-eGFP model was of particular interest to
- 1 9 6 -

C h ill '1er _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ I 'incil D iscuss io n
me with reference to the dominant-negative growth hormone mutation described in
chapters 5 and 6 o f this thesis. The mechanisms previously proposed for autosomal
dom inant deficiencies related to protein folding are the toxic accumulation of
dysfunctional heterodimers, or a combination of the two in the secretory pathway (Kim &
Arvan, 1998; Ito et aL, 1997; Kuznetsov & Nigam, 1998). To begin to understand the
dominant-negative suppression of GH, it is necessary to understand what causes the
malformation o f heterodimers or aggregates o f growth hormone, which results in a
dominant-negative effect and thus, toxic accumulation of dysfunctional heterodimers in
the regulated secretory pathway and consequently somatotroph cell death. Co
transfection of rat GC cells containing either the hGH-WT or hGH-IVS3 sequence with
hGH-eGFP began to explore the possibilities of visualising secretory processes in GH
cells. We hope this will be extended to double hGH-IVS3-eGFP transgenics, which
although beyond the scope of this thesis, was one of the main aims of targeting eGFP to
secretory GH granules.
7.3 IGHD-II - a dominant-negative growth hormone
The genetic defect causing IGHD-II was until recently exclusively found in the first, fifth
or sixth base pair o f the donor splice site o f intron 3 of the GH-N gene. This results in
mis-splicing of messenger RNA and the skipping of exon 3 so that GH produced from
this message lacks amino acids 32-71 (del32-71-GH) (Cogan et aL, 1995; Binder et aL,
1995; Cogan et aL, 1997). It has previously been suggested that GH dominant-negativity
is a consequence of mis-folding of the truncated GH in the secretory pathway, leading to
the toxic accumulation of protein in the ER, which eventually leads to cell death.
Secretory proteins are cleaved from their signal peptide and are folded in the endoplasmic
reticulum (Dannies, 1991). Unfolded, or misfolded proteins synthesised in the secretory
pathway are retained in the endoplasmic reticulum and degraded, and do not usually
interfere with the folding of other proteins (Gething & Sambrook, 1992; de Silva et aL,
1990; Hurtley & Helenius, 1989). Human GH is a relatively small, monomeric, soluble
protein and the presence o f a mutant protein that cannot fold properly would not
necessarily be expected to interfere with the folding of wild type GH. Two examples of
previously identified mutations o f the GH-N gene that result in forms with deviant
folding are consistent with this expectation. Igarashi and co-workers reported a two base
pair deletion at amino acid 55 resulting in a frameshift and altered amino acid sequence
197 -

C lu ip lcr_ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ F irm l D iscu ss io n
(Igarashi et aL, 1993); Cogan and co-workers found a splice site mutation resulted in an
mRNA with an altered reading frame after the first 103 amino acids (Cogan et aL, 1994).
Both of these changes in amino acid sequence will alter the tertiary structure of these
proteins compared to wild type GH and although these proteins cannot fold properly, they
are phenotypically recessive. Growth hormone produced from the wild-type gene in
heterozygotes is sufficient to support normal growth. These two mutations suggest that
the production of a protein with an inability to fold is not sufficient to suppress
production of wild type GH.
In addition to exon 3 skip mutations, other GH-N missense mutations that have been
implicated with IGHD-II and have recently been reported are P89L (Duquesnoy et aL,
1998); R183H (Wajnrajch et aL, 2000) and VI lOF (Binder et aL, 2001). Mutations in the
carboxy-terminus of GH-N are thought to act through a different mechanism from that of
IVS3 donor splice site mutations that result in exon skipping. The most recent VllOF
mutation to be discovered is a C to T transition in a CpG dinucleotide. This mutation
changes a valine that is completely conserved in mammals, birds and some amphibians to
phenylalanine. Valine is located next to the N-terminal beginning o f the third a-helix
and is integrated in the closely packed core of the fourth a-helix bundle of GH (Ultsch et
aL, 1994). The more bulky phenylalanine at this position is very likely to introduce steric
hindrance (Binder et aL, 2001). Similar to V llO F, P89L changes a highly conserved
proline for leucine. Proline 89 forms a kink in the second a-helix o f GH that leucine is
not able to form (Duquesnoy et aL, 1998), thereby altering the structure and organised
folding o f GH. The R183H mutation reported by Wajnrajch and co-workers has been
found in four unrelated families, and again, occurs at a CpG dinucleotide. The missense
mutation replaces arginine with histidine at position 183, which is reported to have
approximately equivalent bioactivity as wild-type GH (Wajnrajch et aL, 2000). The
mutation occurs directly between the two cysteines Cys^^^ and Cys^^^, and is in close
proximity to Glu " zinc site. H is’^ may exert allosteric hindrance upon Cys^^^ and
Cys’^ and the formation o f the second disulphide bridge. This change in processing
would preferentially predispose the nascent GH molecule to intermolecular, rather than
intramolecular disulphide bridges, disrupting the ordered secretory packaging and
causing the formation of GH oligomers within the somatotroph granule that cannot be
secreted normally. The wild-type GH molecules would then be secondarily misfolded.
1 98 -

C h ô m e r ^ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ F in a l D iscu ss io n
“trapped” by the mutant GH molecules in GH oligomers, resulting in GHD.
Alternatively, this mutant protein with an extra metal-chelator histidine may prevent the
assumption o f the tetra-helical structure, by binding directly to a zinc ion, thereby
disrupting the highly ordered zinc-associated dimérisation of GH. Similarly, it is possible
that by binding to the zinc ion, His^^^ may prevent the post-secretion dissociation of GH
from zinc, maintaining an “inert configuration”, which may then prevent its secretion or
action. Any “defect in dimérisation” would be predicted to restrict the secretion of GH.
The severity of growth retardation in IGHD-II varies, but in most cases is not as severe as
IGHD-1A and GH treatment is effective (Poskitt & Rayner, 1974; Cogan et aL, 1993). In
the past, screening for GH-N defects was performed if severe growth failure with a height
below -4 .5sd score at diagnosis was present (Wagner et aL, 1998). Severe short stature
according to this definition was only present in one third o f affected individuals at
diagnosis (Binder et aL, 2001), indicating that growth failure in IGHD-II is less severe
than would be expected. The children with the splice site mutations and exon 3 skip were
younger and shorter at diagnosis than their counterparts with the missense mutation.
Moderate growth failure was also reported for the family with the missense P89L
mutation (Duquesnoy et aL, 1998).
7.4 hGH-IVS3 transgenic mice
Having produced two GH-IVS3 transgenic founders with a severe IGHD-II phenotype (1
and 12) and one founder (23) without a noticeable IGHD-II phenotype, but a pituitary
phenotype, I also encountered the variability o f GH-IVS3 phenotype in mice. The
disparities between these dwarf phenotypes could also be attributed to the level of
expression of the transgene(s) in each founder. Initially, I thought that the hGH LCR was
not functioning in founder 23, which was worrying since LCR’s are always supposed to
work. It was gratifying to discover that line 23 did have a phenotype, which developed
more slowly. In all of my hGH-IVS3 transgenic animals, the endogenous 1:1 mGH allele
is present and producing “normal” GH. In line 23, it could be possible that the human
LCR does not work as efficiently in a mouse system, therefore, delaying the effects of
dominant-negativity. From just three weeks o f age lines 1 and 12 developed severe
IGHD-II, caused by a massive reduction in GH stores, which appeared to have a knock-
on affect on both Prl and TSH production which worsened with age. This could be
1 99 -

C lh ip lcr 7_ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ F in a l D isc u ss io n
attributed to several copies of our transgene vs two endogenous mouse copies. Although
line 23 does not have a significant dwarf phenotype it has a specific pituitary GH
phenotype. The development of IGHD-II also progressively worsens with age in this
animal; at ten weeks, levels of pituitary Prl were reduced compared to WT animals, but
TSH remained the same. I feel that line 23 models IGHD-II described in patients more
closely providing a system which fits with the theory that dominant-negativity
progressively develops with the accumulation of mutant protein in the secretory pathway,
which eventually proves toxic to the cell and causes cell death.
The mechanism by which the mutant protein interacts with and inactivates the normal
GH protein has not been proved. In both splice site and missense mutations, the mutant
GH gene product must somehow prevent normal intracellular protein transport. To begin
to understand the dominant-negative suppression of GH, it is necessary to know the 3-
dimensional structure o f the complex and the sites important for heterodimerisation. The
question is - what causes the malformation of heterodimers or aggregates of growth
hormone, which results in a dominant-negative effect and thus, toxic accumulation of
dysfunctional heterodimers in the regulated secretory pathway and consequently
somatotroph cell death? It is necessary for the GH product to dimerise with wild type GH
during hormone processing in order to cause a dominant-negative effect. This prevents
further oligomerisation and efficient packaging of GH into secretory granules in the
somatotroph, leading eventually to disruption of the somatotroph population and severe
GH deficiency.
7.5 The role o f zinc in GH dimérisation
Zinc is a trace mineral that is essential for animal nutrition. It is second in importance,
only to iron. It forms an integral part of over 70 metalloenzymes, including alkaline
phosphatase, carbonic anhydrase, lactate dehydrogenase and carboxypeptidase (Parisi &
Vallee, 1969) and is also required for the metabolism of nucleic acids and the synthesis of
protein (Prasad, 1969). The total concentration o f zinc in human serum is between 5-
20pM for adults (Dattani et aL, 1993) and about 95% is complexed with proteins, mostly
to serum albumin . Zinc was first shown to be important in animal growth in 1934 (Todd
et aL, 1934). Subsequent studies have revealed that zinc deficiency in man is associated
with growth limitation (Prasad et aL, 1961; Hambidge et aL, 1972; Buzina et aL, 1980;
2 0 0 -

C h a p ic i- _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ F in a l D isc u ss io n
Gibson et aL, 1989). However, the mechanism of this growth failure has been
controversial. It has been suggested that zinc deficiency results in reduced production or
secretion o f GH (Coble et a l, 1989; Root et a l , 1979). However, Droke and colleagues
reported higher serum GH concentrations in response to GHRH in zinc-deficient lambs,
but they also found lower serum IGF-I levels in those lambs (Droke et aL, 1993). Oner
described lower serum levels o f somatomedin in zinc-deficient rats (Oner et aL, 1984).
As IGF-1 is thought to mediate the skeletal growth-promoting effects o f GH (Daughaday
et aL, 1973) these results all implicate a role for zinc in the action of GH at the molecular
level.
Cunningham and colleagues reported that zinc markedly influenced the binding o f hGH
to lactogenic receptors and demonstrated that addition of 50pM Zn^^ resulted in an 8000-
fold increase in the binding affinity of hGH for a hPRL-receptor (Cunningham et aL,
1990a). Contrasting studies by Dattani and colleagues using an ultra sensitive eluted
stain assay system (ESTA) showed only a modest enhancement of 22K hGH lactogenic
bioactivity when 50pM Zn^^ was added to their PRL receptor bioassay. Interestingly, the
in vitro bioactivity of the 20K hGH variant as opposed to the 22K hGH was strikingly
enhanced by the addition of Zn^^ to the bioassay (Dattani et aL, 1993).
Scanning mutational analysis performed by Cunningham and colleagues identified three
residues in the hGH (His*^, His^’ and Glu’ " ) and one residue in hPRLbp (His’^ ). These
three residues are clustered when mapped upon a model of hGH (Cunningham et aL,
1989b; Cunningham et aL, 1990b). His’ and His^^ are on adjacent turns of helix 1 and
are positioned near Glu* " on helix 4. All three face in the same direction and form a
plausible site for the binding of Zn " , as seen in figure 1.12. Zn^^ is required for tight
binding of the hGH to the hPRL-R in a non-cooperative fashion to a single site in the
hGH'hPRLbp complex, but not for binding to the hGH-R. In contrast, zinc was actually
found to lower the affinity o f hGH to hGHbp four-fold. The zinc binding site is
positioned on the edge of the epitope identified for binding to the hGH receptor (figure
1.8). The model suggests zinc reduces binding o f hGH to the hGHbp by sterically
interfering with binding of the hGHbp. Binding of the hGHbp is enhanced about four
fold by mutation o f G lu’ ' to alanine (Cunningham & Wells, 1989) which was
subsequently found to be important in hGH dimérisation.
2 0 1 -

C h u p 1 er 7 _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ F in a l D isc u ss io n
In order for GH to be released from the pituitary, it must be synthesised and concentrated
into condensed storage granules. Hiostochemical analysis o f the anterior pituitary
indicates is present in high concentrations in GH secretory granules (Thorlacius-
Ussing, 1987). It has also been suggested that high concentrations of inhibit GH
release (Root et aL, 1979; Lorenson et aL, 1983). The biochemical and structural basis
whereby zinc functioned in storage or release of hGH had not been fully understood until
Cunningham and colleagues performed size exclusion chromatography and sedimentation
equilibrium studies, demonstrating that zinc ion (Zn^^) forms a 1:1 complex with hGH.
Binding of one Zn^^ ion promotes the binding of another Zn^^ ion (positive cooperativity)
and induced the dimérisation of growth hormone. Like studies with the hGH*hPRLbp
complex, replacement of the potential Zn^^ ligands (His*^, His^ and Glu* " ) in hGH with
alanine weakened both Zn " binding and hGH dimer formation (Cunningham et aL,
1991a). These residues are positioned in the tertiary structure o f hGH (fig 1.8) such that
Zn " may bridge two a-helical segments, as was designed into a four helix bundle
protein. Zn^^ typically coordinates four ligands in proteins (Cunningham et aL, 1991a).
It was initially thought that Asp'^' was the fourth Zn^^ ligand due to its proximity in the
folded model of hGH. However, sedimentation equilibrium and binding studies indicate
it is not involved (Cuningham et aL, 1991a). Although there are no other side chains that
can coordinate Zn^^, it is possible that the fourth Zn^^ ligand is a water molecule.
Alternatively, His'^, His^’ and Glu^ ' may bridge two Zn^^ ions in the dimer - the highly
cooperative nature of Zn^^ binding suggests that the two sites are indeed interdependent.
Most o f the zinc in the pituitary is located in the somatotrophic granules, along with hGH
(Thorlacius-Ussing, 1987) in roughly equimolar amounts (Cunningham et aL, 1991a). In
addition, the concentration of hGH and Zn^^ in the vesicles is probably greater than ImM
(Cunningham et aL, 1991a). This concentration is at least 400-fold greater than the
dissociation constant for the Zn^^-hGH dimer (2.6|iM). Therefore, virtually all o f the
hGH could exist as a (Zn^^-hGH)] complex. As Zn " is able to induce the formation of a
hGH dimer, it is thought to be important for the aggregation o f hGH for storage in
secretory granules. There are two possible functions for the hGH dimer. Firstly, the
(Zn^^-hGH)2 dimer is more stable to dénaturation than monomeric hGH, which would
make it substantially more resistant to dénaturation during storage in vesicles. Secondly,
- 2 0 2

( 'h iip icr _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ F in n ! D iscu ss io n
hormones are released in pulses, and a large amount (concentration -Im M ) must be
synthesised and released from the pituitary in order to generate effect in the periphery
(where its peak concentration is ~2nM, almost a 10^-fold dilution). Secretory granules
offer a convenient and effective method of storage (Dannies, 1999) and allow dispersion
to the periphery without dénaturation - the hormone does not become available until the
hGH concentration approaches its dimérisation dissociation constant (~3pM).
The insulin hexamer has also been shown to contain two high-affmity binding sites for
Zn^^, and in the presence of this ion, the insulin species in solution is driven towards the
hexamer state (Hill et al, 1991). The structure of the metal-free hexamer shows that the
imidazole rings are arranged in a pre-formed site that binds a water molecule and is
poised for zinc co-ordination. At the pH of the metal-free hexamer crystal (pH 6.2), it is
most likely that at least one out o f every three imidazolyl groups is protonated.
Placement of a water molecule at approximately the Zn^^ position provides a means for
dissipating the charge (Hill et aL, 1991). Since monomeric insulin is the biologically
active form recognised by insulin receptors, the rapid delivery of active hormone to the
target cells requires that crystalline hexamers dissolve and dissociate rapidly. Efficient
sequestering and removal o f zinc is thought to occur via the intervention o f an
endogenous chelator, which greatly accelerates crystal dissolution (Ottonello & Dunn,
1992).
Therefore, formation of GH aggregates is dependent upon the presence of zinc and it is
likely that zinc-induced dimérisation o f GH is an important factor in sorting of GH
granules in the somatotroph. The mutant 17.5kDa hGH-IVS3 protein lacks 40 residues,
markedly altering its tertiary structure. However, the altered structure o f hGH-IVS3
mutant protein does not delete any of the three zinc binding sites. I suggest it retains
sufficient structure similarity to endogenous wild type mouse GH molecules (which
contain homologous zinc binding sites) and inhibits further oligomerisation o f GH and
subsequently, formation o f dense-cored GH secretory vesicles. Therefore, it is
conceivable that zinc binding is the important limiting factor in GH-IVS3, which
prevents the condensation of GH to form insoluble protein aggregates.
- 2 0 3 -

üiai'bi': __________________________________________________________ [biialMbJibimii
If zinc is the major factor in effecting the dominant suppression of wild type GH, removal
of the zinc binding residues from hOH-IVS3 would render the mutant protein “useless”
and prohibit binding to WT-GH. Substitution of these residues with alanine in the mutant
GH gene would confirm if it is possible to reverse the dominant-negative effect of hGH-
IVS3 by removing the zinc binding sites. By creating a GH-IVS3 gene which is
ineffectual without its zinc binding sites the mutation would become recessive, causing
no disruption in the secretory pathway allowing the dimérisation and oligomerisation of
wild type GH, A further series of constructs, knocking out each o f the three relevant
histidines in hGH (*^His, ^^His and *^^His) required to address this issue, are already in
progress and will be tested in both GH cell lines and transgenics.
The combination o f in vitro cell culture and in vivo transgenic studies described in this
thesis has proven a very useful approach to express both hGH-eGFP as a fluorsecent
marker and hGH-IVS3 to analyse GH dominant-negativity. First, eGFP was used for
investigating the expression of different lengths of hGH transgenes driven by the hGH
LCR and is now used as a tool to identify and study somatotrophs in vivo. Expression of
a human dominant-negative GH mutation (hGH-IVS3) both in GC cells and in transgenic
mice has provided more insight as to the mechanism of the disease. A combination of
these two transgenic mice will now allow analysis of the effects of human dominant-
negative mutation in the secretory pathway, directly at pre-identifîed somatotrophs in
situ.
- 2 0 4 -

- Ç O Z -
x i p u a d d y
X] p u ru k l i'

-il>j_>çncUx
Appendixl
Engineering an M luI linker into cos.hGH
1. Cosmid orientation reversed - cu t w ith N o tl p o s itio n in g th e 3' o f th e h G H gene
n ea re r a un iq u e M lu l s ite in th e v e c to r
N o fI E co R I E co R I 1 1________ 1
N o tl M lu l
K 2 B B s a
1 11
N o tl
40kB 1
E co R I E co R I N o tl M lu l 1________ 1 1 J
B 2K r ////A40kB
2. M lul site introduced - seq u en ce a lte red a t -3 2 6 b p from 5 '-C C A C G T -3 ' to 5 '-A C G C G T -3 '
S a ll-M lu l frag m en t and M lu I-B sR G I fra g m e n ts re lig a ted and c lo n ed in to p K S -G H (p K S -G H .M )
*MluI
S a il E coR I S fil1 I I
B sR C I 1 B am H I P vuII 1 1 1
P vu II N o tl E coR I 1 1 1
pK s-G H .M V 7777A hGH vA
3. Re-cloning o f GH-M into cosmid construct B2K
a) a S a c l - B a m H I linker, co n ta in in g a u n iq u e S n a B I s ite w as in tro d u c e d in to th e M C S o f p B S -K S a p p ro x im a te ly 7 .5kB u p stream o f A o // site
pKS.S
S a d
S naB I N ot!
BamHIE co R V
I___
b) T h e sm a lle r ' S n a B I - N o t l frag m en t' o f the h G H g en e m a k e s S f i l a u n iq u e site
S n a B I
pSN7.5
B a m H I EcoRJ I B a m H I_J________ I I I
NodI
- 2 0 6 -

■ \p i> C lh ii\
c) T he S f i l - N o t l fragm ent w as rep laced w ith the S f i l - N o t l fragm ent from pK S -G H .Mcon ta in ing the M i n i site
pSN7.5
SntiB I B am H I Ecc 1
R I 1 B am H I Nt _____1 1
i t l
1
d) T he N o t l - E c o R V fragm en t from pW E 15 vecto r is ligated to S n a B I - N o t l in pSN 7.5 before insertion into cosm id B 2K .
M l u l
SnaB I B am H I EcoRII I
pSN7.5
B am H II
N o tl E coRV I I
% »
4. Insertion of SnaBI-EcoRV fragment with Mini site into cosmid B2K (cosGH.M)
E coR I N o tl I I
Bam H I L _
B am H I SnaB I SnaB I Bam H I EcoRIE coRI I EcoRI I E coR I I E coR I I N o tl I M lu l
I I I I I I I I I
40 30T20
T10
hGH
I0
I kB
a) A n approx im ate 4kB SnaB I site fragm en t is lost during clon ing as it is no t un ique.
M l u l
B am H I SnaBI B am H I EcoRIE coR I N o tl
1 1Bam H I
1EcoRI 1 E coRI 1
II 1 1E coR I
11 N otl 1 M lu l 1 1 1 ' \
Y//À1
40 301 1
20 10
j
0
kB
b) T he m issing 4kB fragm ent is re tu rned to cos.GH.M
E coR I N o tl I I
40
Bam H I
30
B am H I EcoRI I
11
M l u l
20
SnaB I SnaB I E coR I I E coR I I EcoRI
M _ J J I
10
B am H I EcoRIN o tl I M lu l
I IhOH
I0
kB
- 207 -

Appendix
5) Insertion of Mini tinkered GH-GFP fragment to cosGH.M
b)
M lu l
a) hGH
PvuIIATG
PvuII M lu li_________ I
eGFP
M lul
BamHI SnaBI SnaBI BamHI EcoRIE coR I N otl
1 1Bam H I
1EcoRI 1
llE coRI 1 EcoRI 1 EcoIU
l l 1 1 11 N otl 1 M lu l 1 1 1
Y x y x 11
401
301
201
101
0
ficB
M iulc)
BamHI SnaBI SnaBI BamHI EcoRIE coR I N otl
J J
BamHI1
EcoRI 1ll
EcoRI 1 E coRI 1 EcoRI 1 1 1 1 1
1 N otl 1 M lul
40■ 1
30r
201
101
0
1 kB
The 1.3kB p48hGH-GFP fusion construct (a) was inserted into the cosm id construct B2K
with M lu l linkers (b). The final transgene cosm id is under the transcriptional control o f
the 40kB hGH locus control region. Shaded bars indicate hGH genom ic sequences;
hatched bars indicate vector sequence; green bar indicates GFP cD N A . N ote that the
fragments are not drawn to scale with each other.
- 2 0 8 -

A p p en d ix
Appendix II
Primers fo r cloning transgene constructs:
Primer 1 (F) CGGAATTCGACGCGTGATCCCAAGGCCCAACTCC EcoRI/MluI sites + 5’UTR
Primer 2 (R) GCGGGATCCGGACGTCCGGGAGCCTGGGGAGAA BamHI restriction site
Primer 3 (F) GCCAGAGGGCACACGCGTGACCCTTAAAGAGAG -326bp Mlul restriction site + 5’UTR
Primer 4 (R) AGGCCCCATGCATAAATGTACACAGAAACAGGT BsrGI restriction site
Primers fo r genotyping transgenic hGH-eGFP transgenic animals:
hGH 5 ’UTR (F) AACCACTCAGGGTCCTGTGGACAG
hGH exon 2 (R) CCTCTTGAAGCCAGGGCAGGCAGAGCAGGC
hGH exon 5 (R) CATGGACAAGGTCGAGACATTCCTGCGCAT
Primers fo r RT-PCR:
eGFP (R) (cDNA position 411) GAGGACGGCAACATCCTGGGGCA
mP-actin (F) (cDNA position bp95) TTGTAACCAACTGGGACGATATGG
mP-actin (R) (cDNA position bp835) GATCTTGATCTTCATGGTGCTAGG
hGH 1 (F) (cDNA position bp81) CCAACCATTCCCTTATCCAGGC
hGH 2 (R) short (cDNA position bp536) TTCGACACAAACTCACACAACGAT
hGH 3 (R) long (cDNA position bp742) AGTGCCCACCAGCCTTGTCCTA
mGH cDNA clone kindly supplied by Dr. Dave Hurley (Tulane University, NO) for RPA.
2 0 9 -

Appendix
Appendix III
NCBI Sequence Viewer - Human Growth Hormone v mouse and rat Growth HormoneAccession number P01241
MATGSRTSLL LAFGLLCLPW LQEGSA 11 FPTIPLSRLFDNAMMATDSRTSWL LTVSLLCLLW PQEASA FPAMPLSSLFSNAVMAADSQTPWL LTFSLLCLLW PQEAGA FPAMPLS SLFANAVLRAHRLHQLA FDTYQEFEEA YIPKEQKYSF LQNPQTSLCFLRAQHLHQLA ADTYKEFERA YIPEGQRYS- IQNAQAAFCFLRAQHLHQLA ADTYKEFERA YIPEGQRYS- IQNAQAAFCFSESIPTPSNR EETQQKSNLE LLEISLLLIQ SWLEPVQFLRSETIPAPTGK EEAQQRTDME LLRFSLLLIQ SWLGPVQFLSSETIPAPTGK EEAQQRTDME LLRFSLLLIQ SWLGPVQFLSSVFANSLVYG ASDSNVYDLL KDLEEGIQTL MGRLEDGSPRRIFTNSLMFG TSD-RVYEKL KDLEEGIQAL MQELEDGSPRRIFTNSLMFG TSD-RVYEKL KDLEEGIQAL MQELEDGSPRTGQIFKQTYS KFDTNSHNDD ALLKNYGLLY CFRKDMDKVEVGQILKQTYD KFDANMRSDD ALLKNYGLLS CFKKDLHKAEIGQILKQTYD KFDANMRSDD ALLKNYGLLS CFKKDLHKAETFLRIVQCRS VEGSCGFTYLRVMKCRR FVESSCAFTYLRVMKCRR FAESSCAF
Human growth hormone Mouse growth hormone Rat growth hormone
- 2 1 0 -

Bih i id ' . ’j ' o n i i v
B i b l i o g r a p h y
- 2 1 1

Abdel-M eguid SS, Shieh HS, Smith W W , D ayringer HE, Violand BN, Bentle LA 1987 Three-dimensional structure o f a genetically engineered variant o f porcine growth hormone. Proc Natl Acad Sci U S A 84:6434-7
Abe H, M olitch ME, Van W yk JJ, Underwood LE 1983 Human growth hormone and somatomedin C suppress the spontaneous release o f growth hormone in unanesthetized rats. Endocrinology 113:1319-24
Achermann JC, Hindmarsh PC, Robinson IC, M atthews DR, Brook CG. 1999 The relative roles o f continuous growth hormone-releasing hormone (GHRH(1-29)NH2) and intermittent somatostatin(l-14)(SS) in growth hormone (GH) pulse generation: studies in normal and post cranial irradiated individuals. Clin Endocrinol. 51(5):575-85.
Ahima RS, Flier JS 2000 Leptin. Annu Rev Physiol 62:413-37
Alpert LC, Brawer JR, Patel YC, Reichlin S 1976 Somatostatinergic neurons in anterior hypothalamus: immunohistochemical localization. Endocrinology 98:255-8
Am it T, Barkey RJ, Youdim M, Hochberg Z 1990 A new and convenient assay o f growth hormone-binding protein activity in human serum. Journal o f Clinical Endocrinology and Metabolism:474-479
Andersen B, Pearse RV, 2nd, Jenne K, et al. 1995 The Ames dwarf gene is required for Pit-1 gene activation. Dev Biol 172:495-503
Argente J, Chowen JA, Zeitler P, Clifton DK, Steiner RA 1991 Sexual dimorphism o f growth hormone-releasing hormone and somatostatin gene expression in the hypothalamus o f the rat during development. Endocrinology 128:2369-75
A rgetsinger LS, Cam pbell GS, Yang X, et al. 1993 Identification o f JAK2 as a growth hormone receptor-associated tyrosine kinase. Cell 74:237-44
Aryan P, Castle D 1998 Sorting and storage during secretory granule biogenesis: looking backward and looking forward. Biochem J 332 ( Pt 3):593-610
Asa SL, Ezzat S 1999 Molecular determinants o f pituitary cytodifferentiation. Pituitary 1:159-68
Axelrod D 1994 New Dimensions in two dimensions. Biophys J 67(5): 1799-1800
Axelrod D 2001 Total Internal Fluorescence Microscopy in Cell Biology. Traffic 11:764-774
Bach M A, Bondy CA 1992 Anatomy o f the pituitary insulin-like growth factor system. Endocrinology 131:2588-94
Badolato R, Bond HM , Valerio G, et al. 1994 Differential expression o f surface membrane growth hormone receptor on human peripheral blood lymphocytes detected by dual fluorochrome flow cytometry. J Clin Endocrinol Metab 79:984-90
Bancroft EC 1973 Measurement o f growth hormone synthesis by rat pituitary cells in culture. Endocrinology 92:1014-21
Banfield DK, Lewis MJ, Pelham HR 1995 A SNARE-like protein required for traffic through the Golgi complex. Nature 375:806-9
- 2 1 2 -

B ih lio ijra p h v
Bannykh SI, Balch W E 1997 Membrane dynamics at the endoplasmic reticulum-Golgi interface. J Cell Biol 138:1-4
Barchi RL 1988 Probing the molecular structure o f the voltage-dependent sodium charmel, Annu RevNeurosci 11:455-95
Barinaga M , B ilezikjian LM , Vale W W , Rosenfeld M G, Evans RM 1985a Hormonal regulation o f gene expression: regulation o f growth hormone gene transcription by growth hormone releasing factor. O xf Surv Eukaryot Genes 2:49-73
Barinaga M , Bilezikjian LM , Vale W W , Rosenfeld MG, Evans RM 1985b Independent effects o f growth hormone releasing factor on growth hormone release and gene transcription. Nature 314:279-81
Barsh GS, Seeburg PH, Gelinas RE 1983 The human growth hormone gene family: structure and evolution o f the chromosomal locus. Nucleic Acids Res 11:3939-58
Barton DE, Foellm er BE, W ood W I, Francke U 1989 Chromosome mapping o f the growth hormone receptor gene in man and mouse. Cytogenet Cell Genet 50:137-41
Baumann G 1991a Growth hormone heterogeneity: genes, isohormones, variants, and binding proteins. Endocr Rev 12:424-49
Baumann G 1991b Metabolism o f growth hormone (GH) and different molecular forms o f GH in biological fluids. Horm Res 36:5-10
Baumann G 1996 Growth hormone-binding proteins-an overview. Acta Paediatr Suppl 417:95-7
Baumann G, Davila N, Shaw MA, Ray J, Liebhaber SA, Cooke NE 1991c Binding o f human growth hormone (GH)-variant (placental GH) to GH- binding proteins in human plasma. J Clin Endocrinol Metab 73:1175-9
Baumann G, Lowman HB, Mercado M, Wells JA 1994 The stoichiometry o f growth hormone- binding protein complexes in human plasma: comparison with cell surface receptors. J Clin Endocrinol Metab 78:1113-8
Baum ann G, M aheshw ari H 1997 The Dwarfs o f Sindh: severe growth hormone (GH) deficiency caused by a mutation in the GH-releasing hormone receptor gene. Acta Paediatr Suppl 423:33-8
Baum ann G, Shaw MA 1990 Plasma transport o f the 20,000-dalton variant o f human growth hormone (20K): evidence for a 20K-specific binding site. J Clin Endocrinol Metab 71:1339-43
Baum ann G, Shaw MA 1990 A second, lower affinity growth hormone-binding protein in human plasma. J Clin Endocrinol Metab 70:680-6
Baum ann G, Amburn KD, Buchanan TA 1987 The effect o f circulating growth hormone- binding protein on metabolic clearance, distribution, and degradation o f human growth hormone. J Clin Endocrinol Metab 64:657-60
-213 -

________________________________________________ __________________________________________________D ihlio i^n in liv
Baumann G, Stolar M W , Buchanan TA 1985 Slow metabolic clearance rate o f the 20,000- dalton variant o f human growth hormone: implications for biological activity. Endocrinology 117:1309-13
Baum bach W R, Bingham B 1995 One class o f growth hormone (GH) receptor and binding protein messenger ribonucleic acid in rat liver, GHRl, is sexually dimorphic and regulated by GH. Endocrinology 136:749-60
Baumbach W R, Horner DL, Logan JS 1989 The growth hormone-binding protein in rat serum is an alternatively spliced form o f the rat growth hormone receptor. Genes Dev 3:1199-1205
Bazan JF 1989 A novel family o f growth factor receptors: a common binding domain in the growth hormone, prolactin, erythropoietin and IL-6 receptors, and the p75 IL-2 receptor beta- chain. Biochem Biophys Res Commun 164:788-95
Bchini O, Mehtali M, Lathe R 1991 Abrogation o f dominant glucose intolerance in SJL mice by a growth hormone transgene. J Mol Endocrinol 6:129-35
Beaudet A, Greenspun D, Raelson J, Tannenbaum GS 1995 Patterns o f expression o f SSTRl and SSTR2 somatostatin receptor subtypes in the hypothalamus o f the adult rat: relationship to neuroendocrine function. Neuroscience 65:551-61
Behringer RR, Mathews LS, Palmiter RD, Brinster RL 1988 Dwarf mice produced by genetic ablation o f growth hormone-expressing cells. Genes Dev 2:453-61
Bennani-Balti IM , Asa SL, Song D, Iratni R, Liebhaber SA, Cooke NE 1998a DNase I- hypersensitive sites I and II o f the human growth hormone locus control region are a major developmental activator o f somatotrope gene expression. Proc Natl Acad Sci U S A 95:10655-60
Bennani-Baiti IM, Cooke NE, Liebhaber SA 1998b Physical linkage o f the human growth hormone gene cluster and the CD79b (Ig beta/B29) gene. Genomics 48:258-64
Bennani-Baiti IM, Jones BK, Liebhaber SA, Cooke NE 1995 Physical linkage o f the human growth hormone gene cluster and the skeletal muscle sodium channel alpha-subunit gene (SCN4A) on chromosome 17. Genomics 29:647-52
Bennett PA, Levy A, Sophokleous S, Robinson IC, Lightm an SL 1995 Hypothalamic GH receptor gene expression in the rat: effects o f altered GH status. Journal o f Endocrinology 147:225-34
Bennett PA, Lindell K, W ilson C, Carlsson LM , Carlsson B, Robinson IC 1999 Cyclical variations in the abundance o f leptin receptors, but not in circulating leptin, correlate with NPY expression during the oestrous cycle. Neuroendocrinology 69:417-23
B ennett PA, Thom as GB, H oward AD, et al. 1997 Hypothalamic growth hormone secretagogue-receptor (GHS-R) expression is regulated by growth hormone in the rat. Endocrinology 138:4552-7
Benoit R, Ling N, Bakhit C, M orrison JH, Alford B, Guillemin R 1982 Somatostatin-28(l- 12)-like immunoreactivity in the rat. Endocrinology 111:2149-51
Benoit R, Ling N, Bakhit C, et al. Growth hormone-releasing factor from a human pancreatic tumor that caused acromegaly. Endocrinology 111:2149-51
214

B ih lio l'rc iphv
Berg M A, G uevara-Aguirre J, Rosenbloom AL, Rosenfeld RG, Francke U 1992 Mutation creating a new splice site in the growth hormone receptor genes o f 37 Ecuadorean patients with Laron syndrome. Hum Mutat 1:24-32
Berridge MJ, Galione A 1988 Cytosolic calcium oscillators. Faseb J 2:3074-82
Berridge MJ, Irvine RF 1989 Inositol phosphates and cell signalling. Nature 341:197-205
Bertherat J, Chanson P, M ontm iny M 1995 The cyclic adenosine 3',5'-monophosphate- responsive factor CREB is constitutively activated in human somatotroph adenomas. Mol Endocrinol 9:777-83
Bilezikjian LM, Vale WW 1984 Chronic exposure o f cultured rat anterior pituitary cells to GRF causes partial loss o f responsiveness to GRF. Endocrinology 115:2032-4
Billestrup N, M itchell RL, Vale W, Verma IM 1987 Growth hormone-releasing factor induces c-fos expression in cultured primary pituitary cells. Mol Endocrinol 1:300-5
B illestrup N, Swanson LW , V ale W 1986 Growth hormone-releasing factor stimulates proliferation o f somatotrophs in vitro. Proc Natl Acad Sci U S A 83:6854-7
Binder G, Keller E, Mix M, et al. 2001 Isolated GH deficiency with dominant inheritance: new mutations, new insights. J Clin Endocrinol Metab 86:3877-81
Binder G, Ranke MB 1995 Screening for growth hormone (GH) gene splice-site mutations in sporadic cases with severe isolated GH deficiency using ectopic transcript analysis. J Clin Endocrinol Metab 80:1247-52
Blam SB, M itchell R, Tischer E, et al. 1988 Addition o f growth hormone secretion signal to basic fibroblast growth factor results in cell transformation and secretion o f aberrant forms o f the protein. Oncogene 3:129-36
Blobel G, Dobberstein B 1975 Transfer o f proteins across membranes. I. Presence o f proteolytically processed and unprocessed nascent immunoglobulin light chains on membrane- bound ribosomes o f murine myeloma. J Cell Biol 67:835-51
Bodner M, Castrillo JL, Theill LE, Deerinck T, Ellisman M, Karin M 1988 The pituitary- specific transcription factor GHF-1 is a homeobox-containing protein. Cell 55:505-18
Bonnefont X, Fiekers J, C reff A, M ollard P 2000 Rhythmic bursts o f calcium transients in acute anterior pituitary slices. Endocrinology 141:868-75
Booth C, Koch GL 1989 Perturbation o f cellular calcium induces secretion o f luminal ER proteins. Cell 59:729-37
Borer K T, Kelch RP, Hayashida T 1982 Hamster growth hormone. Neuroendocrinology 35:349-358
Boutin JM , Jolicoeur C, Okamura H, et al. 1988 Cloning and expression o f the rat prolactin receptor, a member o f the growth hormone/prolactin receptor gene family. Cell 53:69-77
- 2 1 5 -

Bowers CY, M omany FA, Reynolds GA, Hong A 1984 On the in vitro and in vivo activity o f a new synthetic hexapeptide that acts on the pituitary to specifically release growth hormone. Endocrinology 114:1537-45
Bowers CY, Reynolds GA, Durham D, Barrera CM, Pezzoli SS, Thorner MO 1990 Growth hormone (GH)-releasing peptide stimulates GH release in normal men and acts synergistically with GH-releasing hormone. J Clin Endocrinol Metab 70:975-82
Braakman I, Helenius J, Helenius A 1992 Role o f ATP and disulphide bonds during protein folding in the endoplasmic reticulum. Nature 356:260-2
Brazeau P, Ling N, Esch F, Bohlen P, Benoit R, Guillemin R 1981 High biological activity o f the synthetic replicates o f somatostatin-28 and somatostatin-25. Regul Pept 1:255-64
Brazeau P, Ling N, Bohlen P, Esch F, Ying SY, Guillemin R 1982a Growth hormone releasing factor, somatocrinin, releases pituitary growth hormone in vitro. Proc Natl Acad Sci U S A 79:7909-13
Brazeau P, Ling N, Esch F, Bohlen P, Mougin C, Guillemin R 1982b Somatocrinin (growth hormone releasing factor) in vitro bioactivity; Ca++ involvement, cAMP mediated action and additivity o f effect with PGE2. Biochem Biophys Res Commun 109:588-94
Brazeau P, Rivier J, Vale W, Guillemin R 1974 Inhibition o f growth hormone secretion in the rat by synthetic somatostatin. Endocrinology 94:184-7
Brazeau P, Vale W , Burgus R, et al. 1973 Hypothalamic polypeptide that inhibits the secretion o f immunoreactive pituitary growth hormone. Science 179:77-9
Breder CD, Yamada Y, Yasuda K, Seine S, Saper CB, Bell GI 1992 Differential expression o f somatostatin receptor subtypes in brain. J Neurosci 12:3920-34
Brem G, Brenig B, Salmons B, et al. 1991 [Unexpected transgene expression o f a mammary- specific growth hormone gene construct in Bergmann glial cells o f the mouse]. Tierarztl Prax 19:345-50
Brinster RL, Allen JM , Behringer RR, Gelinas RE, Palm iter RD 1988 Introns increase transcriptional efficiency in transgenic mice. Proc Natl Acad Sci U S A 85:836-40
Bruno JF, Xu Y, Song J, Berelowitz M 1993 Tissue distribution o f somatostatin receptor subtype messenger ribonucleic acid in the rat. Endocrinology 133:2561-7
Burgess TL, Craik CS, Matsuuchi L, Kelly RB 1987 In vitro mutagenesis o f trypsinogen: role o f the amino terminus in intracellular protein targeting to secretory granules. J Cell Biol 105:659- 68
Burgess TL, Kelly RB 1987 Constitutive and regulated secretion o f proteins. Annu Rev Cell Biol 3:243-93
Burton FH, Hasel KW , Bloom FE, Sutcliffe JG 1991 Pituitary hyperplasia and gigantism in mice caused by a cholera toxin transgene. Nature 350:74-7
Burton K, K abigting E, Steiner R, Clifton D 1995 Identification o f target cells for growth hormone's action in the arcuate nucleus. Am J Physiol 269:E716-E722
216

Bihli(}'j.rapliy
Buzina R, Jusic M, Sapunar J, M ilanovic N 1980 Zinc nutrition and taste acuity in school children with impaired growth. Am J Clin Nutr 33:2262-7
Cam acho-H ubner C, Clem mons DR, D ’Ercole AJ 1991 Regulation o f insulin-like growth factor (IGF) binding proteins in transgenic mice with altered expression o f growth hormone and IGF-I. Endocrinology 129:1201-6
Cameron CM, Kostyo JL, Adamaflo NA, et al. 1988 The acute effects o f growth hormone on amino acid transport and protein synthesis are due to its insulin-like action. Endocrinology 122:471-4
Carel JC, Tresca JP, Letrait M, et al. 1997 Growth hormone testing for the diagnosis o f growth hormone deficiency in childhood: a population register-based study. J Clin Endocrinol Metab 82:2117-21
Carlsson B, Eden S, Nilsson A, et al. 1991 Expression and physiological significance o f growth hormone receptors and growth hormone binding proteins in rat and man. Acta Paediatrica Scandinavica, Supplement 80:70-76
Carlsson L, Jansson JO 1990 Endogenous Growth hormone (GH) secretion in male rats is synchronized to pulsatile GH infusions given at 3-hour intervals. Endocrinol 126:6-10
Carm ignac DF, Flavell DM , Robinson IC 1996 Pituitary growth hormone-releasing factor receptor expression in normal and dwarf rats. Neuroendocrinology 64:177-85
Carm ignac DF, G abrielsson BM , Robinson ICAF 1993 Growth hormone binding protein (GHBP) in the rat: effects o f gonadal steroids. Endocrinology 133:2445-2452
Carmignac DF, Robinson ICAF 1990 Growth hormone (GH) secretion in the dwarf rat: release, clearance and responsiveness to GH-releasing factor and somatostatin. J. Endocrinol 127:69-75
Carter-Su C, Riii L, Herrington J 2000 Role o f the tyrosine kinase JAK2 in signal transduction by growth hormone. Pediatr Nephrol 14:550-7
Castrillo JL, Theill LE, Karin M 1991 Function o f the homeodomain protein GHFl in pituitary cell proliferation. Science 253:197-9
Catzeflis C, Pierroz DD, Rohner-Jeanrenaud F, Rivier JE, Sizonenko PC, Aubert ML 1993 Neuropeptide Y administered chronically into the lateral ventricle profoundly inhibits both the gonadotropic and the somatotropic axis in intact adult female rats. Endocrinology 132:224-34
Ceda GP, Davis RG , Rosenfeld RG, Hoffman AR 1987 The growth hormone (GH)-releasing hormone (GHRH)-GH-somatomedin axis: evidence for rapid inhibition o f GHRH-elicited GH release by insulin-like growth factors I and II. Endocrinology 120:1658-62
Chalfie M 1995 Green fluorescent protein. Photochem Photobiol 62:651-6
Chalfie M, Tu Y, Euskirchen G, Ward W W , Prasher DC 1994 Green fluorescent protein as a marker for gene expression. Science 263:802-5
Chan YY, Steiner RA, Clifton DK 1996 Regulation o f hypothalamic neuropeptide-Y neurons by growth hormone in the rat. Endocrinology 137:1319-25
- 2 1 7

_ _ _ _ _ _ _ _ _ _ _ B ih lio^iraphy
Chandrashekar V, Bartke A, Coschigano KT, K opchick JJ 1999 Pituitary and testicular function in growth hormone receptor gene knockout mice. Endocrinology 140:1082-8
Chapman GE, Rogers KM, Brittain T, et al. 1981 The 20,000 molecular weight variant o f human growth hormone. Preparation and some physical and chemical properties. J Biol Chem 256:2395-401
Chapman IM, Hartman M L, Straume M, Johnson ML, Veldhuis JD, Thorner MO 1994 Enhanced sensitivity growth hormone (GH) chemiluminescence assay reveals lower postglucose nadir GH concentrations in men than women. J Clin Endocrinol Metab 78:1312-9
Charlton HM, Clark RG, Robinson IC, et al. 1988 Growth hormone-deficient dwarfism in the rat: a new mutation. J Endocrinol 119:51-8
Chen EY, Liao YC, Smith DH, Barrera-Saldana HA, Gelinas RE, Seeburg PH 1989 The human growth hormone locus: nucleotide sequence, biology, and evolution. Genomics 4:479-97
Chen NY, Chen W Y, Striker LJ, Striker GE, K opchick JJ 1997 Co-expression o f bovine growth hormone (GH) and human GH antagonist genes in transgenic mice. Endocrinology 138:851-4
Chen W Y, W ight DC, Chen NY, Coleman TA, W agner TE, Kopchick JJ 1991 Mutations in the third alpha-helix o f bovine growth hormone dramatically affect its intracellular distribution in vitro and growth enhancement in transgenic mice. J Biol Chem 266:2252-8
Chen W Y, W ight DC, W agner TE, Kopchick JJ 1990 Expression o f a mutated bovine growth hormone gene suppresses growth o f transgenic mice. Proc Natl Acad Sci U S A 87:5061-5
Chen XZ, Shafer AW , Yun JS, Li YS, W agner TE, Kopchick JJ 1992 Conversion o f bovine growth hormone cysteine residues to serine affects secretion by cultured cells and growth rates in transgenic mice. Mol Endocrinol 6:598-606
Cheng K, Chan W W , Butler B, et al. 1993 Stimulation o f growth hormone release from rat primary pituitary cells by L-692,429, a novel non-peptidyl GH secretagogue. Endocrinology 132:2729-31
Cheng TC, Beamer W G, Phillips JA, 3rd, Bartke A, M allonee RL, Dowling C 1983 Etiology o f growth hormone deficiency in little, Ames, and Snell dwarf mice. Endocrinology 113:1669-78
Chihara K, M inam itani N, Kaji H, Arimura A, Fujita T 1981 Intraventricularly injected growth hormone stimulates somatostatin release into rat hypophysial portal blood. Endocrinology 109:2279-81
Chihara K, Takahashi Y, Kaji H, Goji K, Okimura Y, Abe H 1998 Short stature caused by a natural growth hormone antagonist. Horm Res 1:41-5
Childs GV, Unabia G, Lee BL, Rougeau D 1992 Heightened secretion by small and mediumsized luteinizing hormone (LH) gonadotropes late in the cycle suggests contributions to the LH surge or possible paracrine interactions. Endocrinology 130:345-52
Chomczynski P, Downs TR, Frohman LA 1988 Feedback regulation o f growth hormone (GH)- releasing hormone gene expression by GH in rat hypothalamus. Mol Endocrinol 2:236-41
-218

B ih l io 'd i 'd p h v
C hristian H C, F low er R J, M orris JF, Buckingham JC 1999 Localisation and semi- quantitative measurement o f lipocortin 1 in rat anterior pituitary cells by fluorescence-activated cell analysis/sorting and electron microscopy. J Neuroendocrinol 11:707-14
Clark RG, M ortensen DL, Carlsson LMS, et al. 1996 Recombinant human growth hormone (GH)-binding protein enhances the growth promoting activity o f human GH in the rat. Endocrinology 137:4308-4315
Clark RG, Carlsson L, Rafferty B, Robinson I 1988a The rebound release o f growth hormone (GH) following somatostatin infusion in rats involves hypothalamic GH-releasing factor release. Journal o f Endocrinology 119:397-404
Clark RG, Carlsson L, Robinson I 1988b Growth hormone (GH) secretion in the conscious rat: Negative feedback o f GH on its own release. Journal o f Endocrinology 119:201-209
Clark RG, Carlsson L, Robinson I 1987 Growth hormone secretory profiles in conscious female rats. Journal o f Endocrinology 114:399-407
Clark RG, Cunningham B, Moore J, et al. 1991 Growth hormone binding protein enhances the growth promoting activity o f GH in the rat. Proceedings o f the 73rd Annual Meeting o f The Endocrine Society, Washington, DC:Abstract 1611
Clark RG, Jansson JO, Isaksson O, Robinson ICAF 1985a Intravenous growth hormone: Growth responses to patterned infusions. Journal o f Endocrinology 104:53-61
Clark RG, Robinson IC 1985b Growth induced by pulsatile infusion o f an amidated fragment o f human growth hormone releasing factor in normal and GHRF-deficient rats. Nature 314:281-3
Clermont Y, Rambourg A, Hermo L 1995 Trans-Golgi network (TGN) o f different cell types: three-dimensional structural characteristics and variability. Anat Rec 242:289-301
Closset J, Smal J, Gomez F, Hennen G 1983 Purification o f the 22000- and 20000-mol.wt. forms o f human somatotropin and characterization o f their binding to liver and mammary binding sites. Biochem J 214:885-92
Cody CW, Prasher DC, W estler W M, Prendergast FG, Ward W W 1993 Chemical structure o f the hexapeptide chromophore o f the Aequorea green-fluorescent protein. Biochemistry 32:1212-8
Cogan JD, Phillips JA, 3rd, Schenkman SS, M ilner RD, Sakati N 1994 Familial growth hormone deficiency: a model o f dominant and recessive mutations affecting a monomeric protein. J Clin Endocrinol Metab 79:1261-5
Cogan JD, Phillips JAd, Sakati N, Frisch H, Schober E, M ilner RD 1993 Heterogeneous growth hormone (GH) gene mutations in familial GH deficiency. J Clin Endocrinol Metab 76:1224-8
Cogan JD, Ramel B, Lehto M , et al. 1995 A recurring dominant negative mutation causes autosomal dominant growth hormone deficiency—a clinical research center study. J Clin Endocrinol Metab 80:3591-5
219

_ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ B ih lio '^ra n h v
Cogan YD, Phillips III JA, Schenkman SS, M ilner RDG, Sakati N 1994 Familial growth hormone deficiency: a model o f dominant and recessive mutations affecting a monomeric protein. Journal o f Clinical Endocrinology and Metabolism 79:1261-1265
Cohn SM , Roth KA, B irkenm eier EH, Gordon JI 1991 Temporal and spatial patterns o f transgene expression in aging adult mice provide insights about the origins, organization, and differentiation o f the intestinal epithelium. Proc Natl Acad Sci U S A 88:1034-8
Colom er V, Kicska GA, Rindler M J 1996 Secretory granule content proteins and the luminal domains o f granule membrane proteins aggregate in vitro at mildly acidic pH. J Biol Chem 271:48-55
Cooke NE, L iebhaber SA 1995 Molecular biology o f the growth hormone-prolactin gene system. Vitam Horm 50:385-459
Cooke NE, Ray J, Emery JG, Liebhaber SA 1988a Two distinct species o f human growth hormone-variant mRNA in the human placenta predict the expression o f novel growth hormone proteins. J Biol Chem 263:9001-6
Cooke NE, Ray J, W atson MA, Estes PA, Kuo BA, Liebhaber SA 1988b Human growth hormone gene and the highly homologous growth hormone variant gene display different splicing patterns. J Clin Invest 82:270-5
Cool DR, Normant E, Shen F, et al. 1997 Carboxypeptidase E is a regulated secretory pathway sorting receptor: genetic obliteration leads to endocrine disorders in Cpe(fat) mice. Cell 88:73-83
Cooper DN, Youssoufian H 1988 The CpG dinucleotide and human genetic disease. Hum Genet 78:151-5
Cubitt AB, Heim R, Adams SR, Boyd AE, Gross LA, Tsien RY 1995 Understanding, improving and using green fluorescent proteins. Trends Biochem Sci 20:448-55
Culler FL, Kaufmann S, Frigeri LG, Jones KL 1988 Comparison o f the acute metabolic effects o f 22,000-dalton and 20,000-dalton growth hormone in human subjects. Horm Metab Res 20:107-9
Cunningham B, W ells J 1989a High-resolution epitope mapping o f hGH-receptor interactions by alanine-scanning mutagenesis. Science 244:1081-1085
Cunningham BC, Jhurani P, Ng P, Wells JA 1989b Receptor and antibody epitopes in human growth hormone identified by homolog-scanning mutagenesis. Science 243:1330-1336
Cunningham BC, Bass S, Fuh G, W ells JA 1990a Zinc mediation o f the binding o f human growth hormone to the human prolactin receptor. Science 250:1709-12
Cunningham BC, H enner DJ, W ells JA 1990b Engineering human prolactin to bind to the human growth hormone receptor. Science 247:1461-5
Cunningham BC, M ulkerrin MG, W ells JA 1991a Dimerization o f human growth hormone by zinc. Science 253:545-8
220 -

_ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ B ih lip '^ra p h v
Cunningham BC, Ultsch M, Devos AM , M ulkerrin M G, C lauser K R, W ells JA 1991b Dimerization o f the extracellular domain o f the human growth-hormone receptor by a single hormone molecule. Science 254:821-825
Cunningham BC, W ells JA 1993 Comparison o f a structural and a functional epitope. Journal o f Molecular Biology 234:554-563
Dannies PS 1999 Protein hormone storage in secretory granules: mechanisms for concentration and sorting. Endocr Rev 20:3-21
Dannies PS 2000 Protein folding and deficiencies caused by dominant-negative mutants o f hormones. Vitam Horm 58:1-26
Dastot F, Duquesnoy P, Sobrier ML, Goossens M, Amselem S 1998 Evolutionary divergence o f the truncated growth hormone receptor isoform in its ability to generate a soluble growth hormone binding protein. Mol Cell Endocrinol 137:79-84
Dattani MT, Hindmarsh PC, Brook C, Robinson I, W eir T, M arshall NJ 1993 Enhancement o f growth hormone bioactivity by zinc in the eluted stain assay system. Endocrinology 133:2803- 2808
Dattani M T, Hindm arsh PC, Brook CG, Robinson IC, M arshall NJ 1994 Inhibition o f growth hormone bioactivity by recombinant human growth hormone-binding protein in the eluted stain assay system. J Endocrinol 140:445-53
Daughaday W H, Rotwein P 1989 Insulin-like growth factors I and II. Peptide, messenger ribonucleic acid and gene structures, serum, and tissue concentrations. Endocr. Rev. 10:68-91
Davis JR, M cVerry J, Lincoln GA, et al. 2001 Cell type-specific adenoviral transgene expression in the intact ovine pituitary gland after stereotaxic delivery: an in vivo system for long-term multiple parameter evaluation o f human pituitary gene therapy. Endocrinology 142:795-801
de Gennaro Colonna V, Cattaneo E, Cocchi D, M uller EE, M aggi A 1988 Growth hormone regulation o f growth hormone-releasing hormone gene expression. Peptides 9:985-8
de Hoog EH, Kegel W K, van Blaaderen A, Lekkerkerker HN 2001 Direct observation o f crystallization and aggregation in a phase-separating colloid-polymer suspension. Phys Rev E Stat Phys Plasmas Fluids Relat Interdiscip Topics 64:021407
de la Hoya M, Vila V, Jimenez O, et al. 1998 Anterior pituitary development and Pit-l/GHF-1 transcription factor. Cell Mol Life Sci 54:1059-66
Deladbey J, Stocker P, M ullis PE 2001 Autosomal dominant GH deficiency due to an Argl83H is GH-1 gene mutation: clinical and molecular evidence o f impaired regulated GH secretion. J Clin Endocrinol Metab 86:3941-7
DeNoto FM , M oore DD, Goodman HM 1981 Human growth hormone DNA sequence and mRNA structure: possible alternative splicing. Nucleic Acids Res 9:3719-30
Dickson SL, Leng G, Dyball RE, Smith RG 1995 Central actions o f peptide and non-peptide growth hormone secretagogues in the rat. Neuroendocrinology 61:36-43
- 2 2 1 -

Dickson SL, L eng G, Robinson C 1993 Growth hormone release evoked by electrical stimulation o f the arcuate nucleus in anesthetized male rats. Brain Research 623:95-100
Dickson SL, Leng G, Robinson IC 1994 Electrical stimulation o f the rat periventricular nucleus influences the activity o f hypothalamic arcuate neurones. J Neuroendocrinol 6:359-67
Dickson SL, Luckman SM 1997 Induction o f c-fos messenger ribonucleic acid in neuropeptide Y and growth hormone (GH)-releasing factor neurons in the rat arcuate nucleus following systemic injection o f the GH secretagogue, GH-releasing peptide-6. Endocrinology 138:771-7
Downs TR, Frohman LA 1991 Evidence for a defect in growth hormone-releasing factor signal transduction in the dwarf (dw/dw) rat pituitary. Endocrinology 129:58-67
Droke EA, Spears JW , Arm strong JD, Kegley EB, Simpson RB 1993 Dietary zinc affects serum concentrations o f insulin and IGF-I in growing lambs. J Nutr 123:13-9
Duquesnoy P, Sobrier M L, Duriez B, et al. 1994 A single amino acid substitution in the exoplasmic domain o f the human growth hormone (GH) receptor confers familial GH resistance (Laron syndrome) with positive GH-binding activity by abolishing receptor homodimerization. EmboJ 13:1386-95
Edén S 1979 Age-, and sex-related differences in episodic growth hormone secretion in the rat Endocrinology 105:555-560
Edén S, Jansson J-O , Oscarsson J 1987 Sexual dimorphism o f growth hormone secretion. In: Isaksson O, Binder C, Hall K, Hokfelt B (eds) Growth hormone- basic and clinical aspects. Elsevier, Amsterdam, pp 129-151
elAm raoui A , Dubois PM 1993 Experimental evidence for the early commitment o f the presumptive adenohypophysis. Neuroendocrinology 58:609-15
Elefant F, Su Y, L iebhaber SA, Cooke NE 2000 Patterns o f histone acétylation suggest dual pathways for gene activation by a bifunctional locus control region. Embo J 19:6814-22
Everitt BJ, M eister B , H okfelt T, et al. 1986 The hypothalamic arcuate nucleus-median eminence complex: immunohistochemistry o f transmitters, peptides and DARPP-32 with special reference to coexistence in dopamine neurons. Brain Res 396:97-155
Fairhall KM , Carm ignac DF, Robinson ICAF 1992 Growth hormone (GH) binding protein and GH interaction in vivo in the guinea pig. Endocr. 131:1963-1969
Felsenfeld G, Boyes J, Chung J, Clark D, Studitsky V 1996 Chromatin structure and gene expression. Proc Natl Acad Sci U S A 93:9384-8
Finkelstein J, K libanski A, Schaeffer E, Hornstein M , Schiff I, N eer R 1994 Parathyroid hormone for the prevention o f bone loss induced by estrogen deficiency. N Eng J Med 331:1618- 1623
Finley JC, Grossman GH, Dimeo P, Petrusz P 1978 Somatostatin-containing neurons in the rat brain: widespread distribution revealed by immunocytochemistry after pretreatment with pronase. Am J Anat 153:483-8
-222

_ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ BihUo'^vi iphv
Flavell DM, Wells T, Wells SE, Carmignac DF, Thomas GB, Robinson IC 1996 Dominant dwarfism in transgenic rats by targeting human growth hormone (GH) expression to hypothalamic GH-releasing factor neurons. Embo J 15:3871-9
Fletcher TP, Thomas GB, Dunshea FR, Moore LG, Clarke IJ 1995 IGF feedback effects on growth hormone secretion in ewes: evidence for action at the pituitary but not the hypothalamic level. J Endocrinol 144:323-31
Forrester WC, Epner E, Driscoll MC, et al. 1990 A deletion o f the human beta-globin locus activation region causes a major alteration in chromatin structure and replication across the entire beta-globin locus. Genes Dev 4:1637-49
Frohman LA, Downs TR, Chomczynski P, Brar A, Kashio Y 1989 Regulation o f growth hormone-releasing hormone gene expression and biosynthesis. Yale J Biol Med 62:427-33
Frawley LS, Faught WJ, Nicholson J, Moomaw B 1994 Real time measurement o f gene expression in living endocrine cells. Endocrinology; 135(1):468-71.
Fuh G, Cunningham BC, Fukunaga R, Nagata S, Goeddel DV, Wells JA 1992 Rational design o f potent antagonists to the human growth hormone receptor. Science 256:1677-80
Fuh G, Mulkerrin MG, Bass S, et al. 1990 The human growth hormone receptor. Secretion from Escherichia coli and disulfide bonding pattern o f the extracellular binding domain. J Biol Chem 265:3111-5
Fukata J, Diamond DJ, Martin JB 1985 Effects o f rat growth hormone (rGH)-releasing factor and somatostatin on the release and synthesis o f rGH in dispersed pituitary cells. Endocrinology 117:457-67
Gabrielsson BG, Carmignac DF, Flavell DM, Robinson ICAF 1995 Steroid regulation o f growth hormone (GH) receptor and GH binding protein messenger ribonucleic acids in the rat. Endocrinology 136:209-217
Gabrielsson BG, Fairhall KM, Robinson ICAF 1990 Growth hormone secretion in the guinea pig. J. Endocrinol 124:371-380
Gage PJ, Lossie AC, Scarlett LM, Lloyd RV, Camper SA 1995 Ames dwarf mice exhibit somatotrope commitment but lack growth hormone-releasing factor response. Endocrinology 136:1161-7
Gaylinn BD, Dealmeida VI, Lyons CE, Jr., Wu KC, Mayo KE, Thorner MO 1999 The mutant growth hormone-releasing hormone (GHRH) receptor o f the little mouse does not bind GHRH. Endocrinology 140:5066-74
Gaylinn BD, Harrison JK, Zysk JR, Lyons CE, Lynch KR, Thorner MO 1993 Molecular cloning and expression o f a human anterior pituitary receptor for growth hormone-releasing hormone. Mol Endocrinol 7:77-84
Geffner ME, Bersch N, Golde DW 1993 Growth hormone induces insulin resistance in Laron dwarf cells via lactogenic receptors. J Clin Endocrinol Metab 76:1039-47
- 2 2 3 -

_______________________________________________________ ÜLhlimLamhy
George DL, Phillips JA, 3rd, Francke U, Seeburg PH 1981 The genes for growth hormone and chorionic somatomammotropin are on the long arm o f human chromosome 17 in region q21 to qter. Hum Genet 57:138-41
Gerdes HH, Kaether C 1996 Green fluorescent protein: applications in cell biology. FEBS Lett 389:44-7
Gertner JM , Wajnrajch MP, Leibel RL 1998 Genetic defects in the control o f growth hormone secretion. Horm Res 49 Suppl 1:9-14
Gething MJ, Sambrook J 1990 Transport and assembly processes in the endoplasmic reticulum. Semin Cell Biol 1:65-72
Gevers E, W it J, Robinson I 1996 Growth, growth hormone (GH)-binding protein and GH receptors are differentially regulated in the rat by peak and trough components o f the GH secretory pattern. Endocrinology 137:1013-1018
Gevers EF, Carmignac DF, W it JM , Robinson ICAF 1995 Differential effects o f baseline growth hormone (GH) on growth and GHBP in the rat. Program o f the 77th Annual Meeting o f The Endocrine Society, Washington DC, 1995:p 2-210 (Abstract)
Gibson RS, Vanderkooy PD, M acDonald AC, Goldm an A, Ryan BA, Berry M 1989 A growth-limiting, mild zinc-deficiency syndrome in some southern Ontario boys with low height percentiles. Am J Clin Nutr 49:1266-73
G odfrey P, Rabal JO, Beam er W G, Copeland NG, Jenkins NA, M ayo KE 1993 GHRH receptor o f little mice contains a missense mutation in the extracellular domain that disrupts receptor function. Nature Genetics 4:227-232
Godowski PJ, Leung DW , M eacbam LR, et al. 1989 Characterization o f the human growth hormone receptor gene and demonstration o f a partial gene deletion in two patients with Laron- type dwarfism. Proc Natl Acad Sci U S A 86:8083-7
Gonzalez GA, M ontminy M R 1989 Cyclic AMP stimulates somatostatin gene transcription by phosphorylation o f CREB at serine 133. Cell 59:675-80
Goodman AD, Tanenbaum R, Rabinowitz D 1972 Existence o f two forms o f immunoreactive growth hormone in human plasma. J Clin Endocrinol Metab 35:868-78
G oodyer CG, De Stepbano L, Lai W H, Guyda HJ, Posner BI 1984 Characterization o f insulin-like growth factor receptors in rat anterior pituitary, hypothalamus, and brain. Endocrinology 114:1187-95
G ouilleux F, Pallard C, D usanter-F ourt I, et al. 1995 Prolactin, growth hormone, erythropoietin and granulocyte-macrophage colony stimulating factor induce MGF-Stat5 DNA binding activity. Embo J 14:2005-13
G raves TK, Patel S, D annies PS, H inkle PM. 2001 M isfolded growth hormone causes fragmentation o f the Golgi apparatus and disrupts endoplasmic reticulum-to-Golgi traffic. J Cell Sci. 114:3685-94.
-224

_______________________________________________________________________________________________ - _______ _ ÿÆjJÆL'üPllX.
Green H, Morikawa M, Nixon T 1985 A dual effector theory o f GH action. Differentiation 29:195-198
Grier DG 1997 New age crystals. Nature 389:784-5
Gronowski AM, Rotwein P 1994 Rapid changes in nuclear protein tyrosine phosphorylation after growth hormone treatment in vivo. Identification o f phosphorylated mitogen-activated protein kinase and STAT91. J Biol Chem 269:7874-8
Grossman A, Savage MO, Lytras N, Preece MA, Sueiras-Diaz J, Coy DH, Rees LH, Besser GM. 1984 Responses to analogues o f growth hormone-releasing hormone in normal subjects, and in growth-hormone deficient children and young adults. Clin Endocrinol 21(3):321-30.
Grosveld F, Dillon N, Higgs D 1993 The regulation o f human globin gene expression. Baillieres Clin Haematol 6:31-55
Grosveld F, van Assendelft GB, Greaves DR, KoIIias G 1987 Position-independent, high-level expression o f the human beta-globin gene in transgenic mice. Cell 51:975-85
Guerineau NC, Bonnefont X, Stoeckel L, Mollard P 1998 Synchronized spontaneous Ca2+ transients in acute anterior pituitary slices. J Biol Chem 273:10389-95
Guillemin R, Brazeau P, Bohlen P, Esch F, Ling N, Wehrenberg WB 1982 Growth hormone- releasing factor from a human pancreatic tumor that caused acromegaly. Science 218:585-7
Gunzburg WH, Salmons B, Zimmermann B, Muller M, Erfle V, Brem G 1991 A mammary- specific promoter directs expression o f growth hormone not only to the mammary gland, but also to Bergman glia cells in transgenic mice. Mol Endocrinol 5:123-33
Guo F, Beaudet A, Tannenbaum GS 1996 The effect o f hypophysectomy and growth hormone replacement on sstl and sst2 somatostatin receptor subtype messenger ribonucleic acids in the arcuate nucleus. Endocrinology 137:3928-35
Hambidge KM, Hambidge C, Jacobs M, Baum JD 1972 Low levels o f zinc in hair, anorexia, poor growth, and hypogeusia in children. Pediatr Res 6:868-74
Hamilton RJ, Sweeney PJ, Pelizzari CA, et al. 1997 Functional imaging in treatment planning o f brain lesions. Int J Radiat Oncol Biol Phys 37:181-8
Hammer RE, Brinster RL, Palmiter RD 1985 Use o f gene transfer to increase animal growth. Cold Spring Harb Symp Quant Biol 50:379-87
Hammer RE, Palmiter RD, Brinster RL 1984 Partial correction o f murine hereditary growth disorder by germ-line incorporation o f a new gene. Nature 311:65-7
Hammond C, Helenius A 1995 Quality control in the secretory pathway. Curr Opin Cell Biol 7:523-9
Han W, Li D, Stout AK, Takimoto K, Levitan ES 1999 Ca2+-induced deprotonation o f peptide hormones inside secretory vesicles in preparation for release. J Neurosci 19:900-5
- 225 -

________________________________________________________________ __ . . _ . _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ B l h l i o i ^ i ' c i p h v
Hansbrough JR, Lublin DM, Roth KA, Birkenmeier EA, Gordon JI 1991 Expression o f a liver fatty acid binding protein/human decay-accelerating factor/HLA-B44 chimeric gene in transgenic mice. Am J Physiol 260:0929-39
Hansen BS, Hjorth S, Welinder BS, Skriver L, De MF 1993 The growth hormone (GH)- binding protein cloned from human IM-9 lymphocytes modulates the down-regulation o f GH receptors by 22- and 20-kilodalton human GH in IM-9 lymphocytes and the biological effects o f the hormone in Nb2 lymphoma cells. Endocrinology 133:2809-2817
Hanson RD, Sclar GM, Kanagawa O, Ley TJ 1991 The 5'-flanking region o f the human CGL- 1/granzyme B gene targets expression o f a reporter gene to activated T-lymphocytes in transgenic mice. J Biol Chem 266:24433-8
Hashimoto S, Chiorazzi N, Gregersen PK 1994 The complete sequence o f the human CD79b (Ig beta/B29) gene: identification o f a conserved exon/intron organization, immunoglobulin-like regulatory regions, and allelic polymorphism. Immunogenetics 40:145-9
Hashimoto S, Fumagalli G, Zanini A, Meldolesi J 1987 Sorting o f three secretory proteins to distinct secretory granules in acidophilic cells o f cow anterior pituitary. J Cell Biol 105:1579-86
Hayashi Y, Kamijo T, Yamamoto M, et al. 1999a A novel mutation at the donor splice site o f intron 3 o f the GH-I gene in a patient with isolated growth hormone deficiency. Growth Horm IGF Res 9:434-7
Hayashi Y, Yamamoto M, Ohmori S, Kamijo T, Ogawa M, Seo H 1999b Inhibition o f growth hormone (GH) secretion by a mutant GH-I gene product in neuroendocrine cells containing secretory granules: an implication for isolated GH deficiency inherited in an autosomal dominant manner. J Clin Endocrinol Metab 84:2134-9
Heim R, Prasher DC, Tsien RY 1994 Wavelength mutations and post-translational autoxidation o f green fluorescent protein. Proc Natl Acad Sci U S A 91:12501-4
Heim R, Tsien RY 1996 Engineering green fluorescent protein for improved brightness, longer wavelengths and fluorescence resonance energy transfer. Curr Biol 6:178-82
Henkel AW, Aimers W 1996 Fast steps in exocytosis and endocytosis studied by capacitance measurements in endocrine cells. Curr Opin Neurobiol 6:350-7
Herington AC, Ymer SI, Stevenson JL 1986 A ffinity purification and structural characterization o f a specific binding protein for human growth hormone in human serum. Biochem Biophys Res Commun 139:150-5
Herr W, Sturm RA, Clerc RG, et al. 1988 The POU domain: a large conserved region in the mammalian pit-1, oct-1, oct-2, and Caenorhabditis elegans unc-86 gene products. Genes Dev 2:1513-6
Herrington J, Smit LS, Schwartz J, Carter-Su C 2000 The role o f STAT proteins in growth hormone signaling. Oncogene 19:2585-97
Hill CP, Dauter Z, Dodson EJ, Dodson GG, Dunn ME 1991 X-ray structure o f an unusual Ca2+ site and the roles o f Zn2+ and Ca2+ in the assembly, stability, and storage o f the insulin hexamer. Biochemistry 30:917-24
2 2 6 -

_ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ . . . . _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ B ih li()'^i\iphv
Hindmarsh P, Smith PJ, Brook CG, Matthews DR 1987 The relationship between height velocity and growth hormone secretion in short prepubertal children. Clin Endocrinol (Oxf) 27:581-91
Hindmarsh PC, Fail CH, Pringle PJ, Osmond C, Brook CG 1997 Peak and trough growth hormone concentrations have different associations with the insulin-like growth factor axis, body composition, and metabolic parameters. J Clin Endocrinol Metab 82:2172-6
Hindmarsh PC, Stanhope R, Preece MA, Brook CG 1990 Frequency o f administration o f growth hormone—an important factor in determining growth response to exogenous growth hormone. Horm Res 4:83-9
Hoeffler JP, Boockfor FR, Frawley LS 1985 Ontogeny o f prolactin cells in neonatal rats: initial prolactin secretors also release growth hormone. Endocrinology 117:187-95
Hogan BLH, Constantini F, Lacy E 1986 Manipulating the Mouse Embryo (Cold Spring Harbour, NY : Cold Spring Harbour Laboratory Press)
Hollingshead PG, Martin L, Pitts SL, Stewart TA 1989 A dominant phenocopy o f hypopituitarism in transgenic mice resulting from central nervous system synthesis o f human growth hormone. Endocrinology 125:1556-64
Horikawa R, Hellmann P, Celia SG, et al. 1996 Growth hormone-releasing factor (GRF) regulates expression o f its own receptor. Endocrinology 137:2642-5
Horseman ND, Yu-Lee LY 1994 Transcriptional regulation by the helix bundle peptide hormones: growth hormone, prolactin, and hematopoietic cytokines. Endocr Rev 15:627-49
Howard AD, Feighner SD, Cully DF, et al. 1996 A receptor in pituitary and hypothalamus that functions in growth hormone release. Science 273:974-7
Huang C, Schmid PC, Ma WY, Schmid HH, Dong Z 1997 Phosphatidylinositol-3 kinase is necessary for 12-0-tetradecanoylphorbol-13-acetate-induced cell transformation and activated protein 1 activation. J Biol Chem 272:4187-94
Huang XF, Arvan P 1995 Intracellular transport o f proinsulin in pancreatic beta-cells. Structural maturation probed by disulfide accessibility. J Biol Chem 270:20417-23
Hartley SM, Helenius A 1989 Protein oligomerization in the endoplasmic reticulum. Annu Rev Cell Biol 5:277-307
Hutton JC, Penn EJ, Peshavaria M 1983 Low-molecular-weight constituents o f isolated insulin-secretory granules. Bivalent cations, adenine nucleotides and inorganic phosphate. Biochem J 210:297-305
Igarashi Y, Ogawa M, Kamijo T, et al. 1993 A new mutation causing inherited growth hormone deficiency: a compound heterozygote o f a 6.7 kb deletion and a two base deletion in the third exon o f the GH-1 gene. Hum Mol Genet 2:1073-4
Ikeda H, Suzuki J, Sasano N, Niizuma H 1988 The development and morphogenesis o f the human pituitary gland. Anat Embryol (Berl) 178:327-36
- 227 -

_ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ ___ __ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ Bihiio<^raphv
Ikeda SR, Schofield GG 1989 Somatostatin blocks a calcium current in rat sympathetic ganglion neurones. J Physiol 409:221-40
Hondo M M , Dam holt AB, Cunningham BA, W ells JA, De M eyts P, Shymko RM 1994 Receptor dimerization determines the effects o f growth hormone in primary rat adipocytes and cultured human IM-9 lymphocytes. Endocrinology 134:2397-403
Ingraham HA, Chen RP, M angalam HJ, et al. 1988 A tissue-specific transcription factor containing a homeodomain specifies a pituitary phenotype. Cell 55:519-29
Irm inger JC, Verchere CB, M eyer K, Halban PA 1997 Proinsulin targeting to the regulated pathway is not impaired in carboxypeptidase E-deficient Cpefat/Cpefat mice. J B iol Chem 272:27532-4
Isaksson OGP, Lindahl A, Nilsson A, Isgaard J 1987 Review paper: Mechanism o f the stimulatory effect o f growth hormone on longitudinal bone growth. Endocr Rev 8:426-438
Jacks T, Smith R, Judith F, et al. 1996 MK-0677, a potent, novel, orally active growth hormone (GH) secretagogue: GH, insulin-like growth factor I, and other hormonal responses in beagles. Endocrinology 137:5284-9
Jacobow itz DM , Schulte H, Chrousos GP, Loriaux DL 1983 Localization o f GRF-like immunoreactive neurons in the rat brain. Peptides 4:521-4
Jansson JO, Downs TR, Beamer W G, Frohman LA 1986 Receptor-associated resistance to growth hormone-releasing factor in dwarf "little" mice. Science 232:511-2
Jansson JO, Edén S, Isaksson O 1985 Sexual dimorphism in the control o f growth hormone secretion. Endocr Rev 6:128-50
Jerecic J, Single F, Kruth U, et al. 1999 Studies on conditional gene expression in the brain. Ann N Y Acad Sci 868:27-37
Jones BK, Monks BR, Liebhaber SA, Cooke NE 1995 The human growth hormone gene is regulated by a multicomponent locus control region. Mol Cell Biol 15:7010-21
Jones SM , H owell KE, H enley JR, Cao H, M cNiven MA 1998 Role o f dynamin in the formation o f transport vesicles from the trans-Golgi network. Science 279:573-7
Jorgensen J, M oller J, Alberti K, et al. 1993 Marked effects o f sustained low growth hormone (GH) levels on day-to-day fuel metabolism: Studies in GH-deficient patients and healthy untreated subjects. Journal o f Clinical Endocrinology and Metabolism 77:1589-1596
K aether C, Gerdes HH 1995 Visualization o f protein transport along the secretory pathway using green fluorescent protein. FEBS Lett 369:267-71
Kam egai J, M inami S, Sugihara H, Hasegawa O, Higuchi H, W akabayashi I 1996 Growth hormone receptor gene is expressed in neuropeptide Y neurons in hypothalamic arcuate nucleus o f rats. Endocrinology 137:2109-12
Kam egai J, Minami S, Sugihara H, Higuchi H, Wakabayashi I 1994 Growth hormone induces expression o f the c-fos gene on hypothalamic neuropeptide-Y and somatostatin neurons in hypophysectomized rats. Endocrinology 135:2765-71
- 228 -

Bih/i()'^rciph\'
Kamijo T, Hayashi Y, Shimatsu A, et al. 1999 Mutations in intron 3 o f GH-1 gene associated with isolated GH deficiency type II in three Japanese families. Clin Endocrinol (Oxf) 51:355-60
Karin M , Theill L, Castrillo JL, M cCormick A, Brady H 1990 Tissue-specific expression o f the growth hormone gene and its control by growth hormone factor-1. Recent Prog Horm Res 46:43-57; discussion 57-8
Kawakami N, Sakane N, Nishizawa F, et al. 1999 Green fluorescent protein-transgenic mice: immune functions and their application to studies o f lymphocyte development. Immunol Lett 70:165-71
Kawano H, Daikoku S 1988 Somatostatin-containing neuron systems in the rat hypothalamus: retrograde tracing and immunohistochemical studies. J Comp Neurol 271:293-9
Keller P, Simons K 1997 Post-Golgi biosynthetic trafficking. J Cell Sci 110 ( Pt 24):3001-9
Kelly PA, All S, Rozakis M, et al. 1993 The growth hormone/prolactin receptor family. Recent Prog Horm Res 48:123-64
K endall SK, Saunders TL, Jin L, et al. 1991 Targeted ablation o f pituitary gonadotropes in transgenic mice. Mol Endocrinol 5:2025-36
Kim PS, Arvan P 1998 Endocrinopathies in the family o f endoplasmic reticulum (ER) storage diseases: disorders o f protein trafficking and the role o f ER molecular chaperones. Endocr Rev 19:173-202
Kineman RD, Frawley LS 1994 Secretory characteristics and phenotypic plasticity o f growth hormone- and prolactin-producing cell lines. J Endocrinol 140(3):455-63.
Kojima M, Hosoda H, Date Y, Nakazato M, M atsuo H, Kangawa K 1999 Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 402:656-60
Kopchick JJ, McAndrew SJ, Shafer A, et al. 1990 In-vitro mutagenesis o f the bovine growth hormone gene. J Reprod Fertil Suppl 41:25-35
Korbonits M, Little JA, Forsling ML, et al. 1999 The effect o f growth hormone secretagogues and neuropeptide Y on hypothalamic hormone release from acute rat hypothalamic explants. J Neuroendocrinol 11:521-8
Kostyo JL, Skottner A, Brostedt P, et al. 1987 Biological characterization o f purified native 20- kDa human growth hormone. Biochim Biophys Acta 925:314-24
Koto M , Sato T, Okamoto M, Adachi J 1988 [rdw rats, a new hereditary dwarf model in the rat]. Jikken Dobutsu 37:21-30
K ratzsch J, Selisko T, B irkenm eier G 1995 Identification o f transformed alpha 2- macroglobulin as a growth hormone-binding protein in human blood. J Clin Endocrinol Metab 80:585-90
K raw czak M , Reiss J, Cooper DN 1992 The mutational spectrum o f single base-pair substitutions in mRNA splice junctions o f human genes: causes and consequences. Hum Genet 90:41-54
- 2 2 9 -

B ih l l ( ) i : ra p l iv
Kuliawat R, Arvan P 1994 Distinct molecular mechanisms for protein sorting within immature secretory granules o f pancreatic beta-cells. J Cell Biol 126:77-86
K uliawat R, K lum perman J, Ludwig T, Arvan P 1997 Differential sorting o f lysosomal enzymes out o f the regulated secretory pathway in pancreatic beta-cells. J Cell Biol 137:595-608
K uliaw at R, Prabakaran D, Arvan P 2000 Proinsulin endoproteolysis confers enhanced targeting o f processed insulin to the regulated secretory pathway. Mol Biol Cell 11:1959-72
K uznetsov G, Nigam SK 1998 Folding o f secretory and membrane proteins. N Engl J Med. 3;339(23):1688-95. Review.
Ladinsky MS, Kremer JR, Furcinitti PS, McIntosh JR, Howell KE 1994 HVEM tomography o f the trans-Golgi network: structural insights and identification o f a lace-like vesicle coat. J Cell Biol 127:29-38
Lang T, W acker I, W underlich I, et al. 2000 Role o f actin cortex in the subplasmalemmal transport o f secretory granules in PC-12 cells. Biophys J 78:2863-77
Lanneau C, Peineau S, Petit F, Epelbaum J, Gardette R 2000 Somatostatin modulation o f excitatory synaptic transmission between periventricular and arcuate hypothalamic nuclei in vitro. J Neurophysiol 84:1464-74
Laron Z, Kelijman M, Pertzelan A, Keret R, Shoffner JM , Parks JS 1985 Human growth hormone gene deletion without antibody formation or growth arrest during treatment—a new disease entity? Isr J Med Sci 21:999-1006
Laron Z, Pertzelan A, M annheim er S 1966 Genetic pituitary dwarfism with high serum concentation o f growth hormone—a new inborn error o f metabolism? Isr J Med Sci 2:152-5
Laursen T, Jorgensen JO L, Jakobsen G, Hansen BL, Christiansen JS 1995 Continuous infusion versus daily injections o f growth hormone (GH) for four weeks in GH-deficient patients. J Clin Endocrinol Metab 80:2410-2418
Lecom te CM, Renard A, M artial JA 1987 A new natural hGH variant—17.5 kd—produced by alternative splicing. An additional consensus sequence which might play a role in branchpoint selection. Nucleic Acids Res 15:6331-48
Lee M S, Dirkx R, Jr., Solim ena M , Dannies PS 1998 Stabilization o f the receptor protein tyrosine phosphatase-like protein ICA512 in GH4C1 cells upon treatment with estradiol, insulin, and epidermal growth factor. Endocrinology 139:2727-33
Lee M S, W ajnrajch MP, Kim SS, et al. 2000 Autosomal dominant growth hormone (GH) deficiency type II: the Del32-71-GH deletion mutant suppresses secretion o f wild-type GH. Endocrinology 141:883-90
Leng G, Brown CH, R ussell JA 1999 Physiological pathways regulating the activity o f magnocellular neurosecretory cells. Prog Neurobiol 57:625-55
Lerner AB 1981 The intermediate lobe o f the pituitary gland: introduction and background. Ciba Found Symp 81:3-12
- 230 -

B ib l ioM raphy
Leung DW , Spencer SA, Cachianes G, et al. 1987 Growth hormone receptor and serum binding protein: purification, cloning and expression. Nature 330:537-43
Lewis D, H allowes RC 1974 Correlation between the effects o f hormones on the synthesis o f DNA in explants from induced rat mammary tumours and the growth o f the tumours. J Endocrinol 62:225-40
Lewis MJ, Pelham HR 1996 SNARE-mediated retrograde traffic from the Golgi complex to the endoplasmic reticulum. Cell 85:205-15
Lewis UJ, Bonewald LF, Lewis LJ 1980 The 20,000-dalton variant o f human growth hormone: location o f the amino acid deletions. Biochem Biophys Res Commun 92:511-6
Li CH, Dixon JS 1971 Human pituitary growth hormone. 32. The primary structure o f the hormone: revision. Arch Biochem Biophys 146:233-6
Li CH, Dixon JS, Liu W K 1969 Human pituitary growth hormone. XIX. The primary structure o f the hormone. Arch Biochem Biophys 133:70-91
Li CH, Papkoff H 1956 Preparation and properties o f human growth hormone from human and monkey pituitary glands. Science 124:1293-1294
Li CH, Evans HM 1944 The Isolation o f human growth hormone. Science 99:183-184
Li CH, Gordon D, Knorr J 1973 The primary structure o f sheep pituitary growth hormone. Arch Biochem Biophys 156:493-508
Li S, Crenshaw EB, 3rd, Rawson EJ, Simmons DM , Swanson LW , Rosenfeld MG 1990 Dwarf locus mutants lacking three pituitary cell types result from mutations in the POU-domain gene pit-1. Nature 347:528-33
Liao W, Yeung SC, Chan L 1998 Proteasome-mediated degradation o f apolipoprotein B targets both nascent peptides cotranslationally before translocation and full-length apolipoprotein B after translocation into the endoplasmic reticulum. J Biol Chem 273:27225-30
Lim L, Spencer SA, M cKay P, W aters M J 1990 Regulation o f growth hormone (GH) bioactivity by a recombinant human GH-binding protein. Endocrinol 127:1287-1291
Lim CS, Baumann CT, Htun H, Xian W , Irie M, Smith CL, H ager GL. 1999 Differential localization and activity o f the A- and B-forms o f the human progesterone receptor using green fluorescent protein chimeras. Mol Endocrinol. 13(3):366-75.
Lin C, Lin SC, Chang CP, Rosenfeld MG 1992 Pit-1-dependent expression o f the receptor for growth hormone releasing factor mediates pituitary cell growth. Nature 360:765-8
Lin SC, Lin CR, Gukovsky I, Lusis AJ, Sawchenko PE, Rosenfeld MG 1993 Molecular basis o f the little mouse phenotype and implications for cell type-specific growth. Nature 364:208-213
Ling N, Baird A, W ehrenberg W B, Ueno N, M unegumi T, Brazeau P 1984 Synthesis and in vitro bioactivity o f C-terminal deleted analogs o f human growth hormone-releasing factor. Biochem Biophys Res Commun 123:854-61
231 -

---------------------------------------------------------------------------------------------------------------- BWRmwAix
Linzer DI, Talamantes F 1985 Nucleotide sequence o f mouse prolactin and growth hormone mRNAs and expression o f these mRNAs during pregnancy. J Biol Chem 260:9574-9
Lipkin SM, Naar AM, Kalla KA, Sack RA, Rosenfeld MG 1993 Identification o f a novel zinc finger protein binding a conserved element critical for Pit-1-dependent growth hormone gene expression. Genes Dev 7:1674-87
Liposits Z, M erchenthaler I, Pauli W K, Flerko B 1988 Synaptic communication between somatostatinergic axons and growth hormone-releasing factor (GRF) synthesizing neurons in the arcuate nucleus o f the rat. Histochemistry 89:247-52
Lira SA, Kalla KA, Glass CK, D rolet DW , Rosenfeld MG 1993 Synergistic interactions between Pit-1 and other elements are required for effective somatotroph rat growth hormone gene expression in transgenic mice. Mol Endocrinol 7:694-701
Lobie PE, Barnard R, W aters MJ 1991 The nuclear growth hormone receptor binding protein: Antigenic and physiochemical characterization. Journal o f Biological Chemistry 266:22645- 22652
Lobie PE, Breipohl W, Aragon JG, W aters M J 1990 Cellular-localization o f the growth- hormone receptor-binding protein in the male and female reproductive systems. Endocrinology 126:2214-2221
Lobie PE, Mertani H, M orel G, M orales-Bustos O, Norstedt G, W aters MJ 1994 Receptor- mediated nuclear translocation o f growth hormone. The Journal o f B iological chemistry 269:21330-21339
Lodish HF, Kong N 1993 The secretory pathway is normal in dithiothreitol-treated cells, but disulfide-bonded proteins are reduced and reversibly retained in the endoplasmic reticulum. J Biol Chem 268:20598-605
Lorenson MY, Robson DL, Jacobs LS 1983 Divalent cation inhibition o f hormone release from isolated adenohypophysial secretory granules. J Biol Chem 258:8618-22
M acLeod JN, Lee AK, Liebhaber SA, Cooke NE 1992 Developmental control and alternative splicing o f the placentally expressed transcripts from the human growth hormone gene cluster. J Biol Chem 267:14219-26
MacLeod JN, Pampori NA, Shapiro BH 1991 Sex differences in the ultradian pattern o f plasma growth hormone concentrations in mice. Journal o f Endocrinology 131:395-399
M agoulas C, M cGuinness L, Balthasar N, et al. 2000 A secreted fluorescent reporter targeted to pituitary growth hormone cells in transgenic mice. Endocrinology 141:4681-9
M aheshwari HG, Silverman BL, Dupuis J, Baumann G. 1998 Phenotype and genetic analysis o f a syndrome caused by an inactivating mutation in the growth hormone-releasing hormone receptor: Dwarfism o f Sindh. J Clin Endocrinol Metab. Nov;83(l l):4065-74.
M ains RE, M ilgram SL, Keutmann HT, Eipper BA 1995 The NH2-terminal proregion o f peptidylglycine alpha-amidating monooxygenase facilitates the secretion o f soluble proteins. Mol Endocrinol 9:3-13
232 -

________________________________________________ __ _ _ B i h l i o ' ^ i ' i i p h v
M anjunath N, Shankar P, Stockton B, Dubey PD, Lieberman J, von Andrian UH 1999 A transgenic mouse model to analyze CD8(+) effector T cell differentiation in vivo. Proc Natl Acad S ciU S A 96:13932-7
M annor D, W iner LM , Shaw MA, Baumann G 1991 Plasma growth hormone(GH)-binding proteins: effect on GH binding to receptors and GH action. J Clin Endocriol Metab 73:34-34
M artial JA, H allew ell RA, B axter JD, G oodm an HM 1979 Human growth hormone: complementary DNA cloning and expression in bacteria. Science 205:602-7
M atsuda I, Hata A, Jinno Y, et al. 1987 Heterogeneous phenotypes o f Japanese cases with a growth hormone gene deletion. Jinrui Idengaku Zasshi 32:227-35
M ayo KE 1992 Molecular cloning and expression o f a pituitary-specific receptor for growth hormone-releasing hormone. Mol Endocrinol 6:1734-44
Mayo KE, Cerelli GM, Lebo RV, Bruce BD, Rosenfeld MG, Evans RM 1985a Gene encoding human growth hormone-releasing factor precursor: structure, sequence, and chromosomal assignment. Proc Natl Acad Sci U S A 82:63-7
M ayo KE, Cerelli GM , Rosenfeld MG, Evans RM 1985b Characterization o f cDNA and genomic clones encoding the precursor to rat hypothalamic growth hormone-releasing factor. Nature 314:464-7
M ayo KE, H am m er RE, Swanson LW , B rinster RL, Rosenfeld M G , Evans RM 1988 Dramatic pituitary hyperplasia in transgenic mice expressing a human growth hormone-releasing factor gene. Mol Endocrinol 2:606-12
M ayo KE, M iller TL, DeAlm eida V, Zheng J, G odfrey PA 1996 The growth-hormone- releasing hormone receptor: signal transduction, gene expression, and physiological function in growth regulation. Ann N Y Acad Sci 805:184-203
M cAndrew SJ, Chen NY, W iehl P, et al. 1991 Expression o f truncated forms o f the bovine growth hormone gene in cultured mouse cells. J Biol Chem 266:20965-9
M cCarthy GF, Beaudet A, Tannenbaum GS 1992 Colocalization o f somatostatin receptors and growth hormone-releasing factor immunoreactivity in neurons o f the rat arcuate nucleus. Neuroendocrinology 56:18-24
M cCormick A, Brady H, Fukushima J, Karin M 1991 The pituitary-specific regulatory gene GHFl contains a minimal cell type-specific promoter centered around its TATA box. Genes Dev 5:1490-503
M cGuinness L, Balthasar N, Sesay AK, M athers K, M agoulas C, Robinson ICAF 1999 Targeting fluorescent reporters to the hypothalamo-pituitary GH axis in transgenic mice. OR 18- 6. Endo Soc. 81st Annual Meeting 1999, San Diego California
M cNicoll AM, M urray JE, McMeekin W 1990 Vasopressin stimulation o f cell proliferation in the rat pituitary gland in vitro. J Endocrinology 126:255-259
Miki N, Ono M , Asakawa-Yasumoto K, et al. 1994 Characterization and localization o f mouse hypothalamic growth hormone-releasing factor and effect o f gold thioglucose-induced hypothalamic lesions. J Neuroendocrinol 6:71-8
- 2 3 3 -

B ih lio '^ra p h v
M iller TL, M ayo KE 1997 Glucocorticoids regulate pituitary growth hormone-releasing hormone receptor messenger ribonucleic acid expression. Endocrinology 138:2458-65
M inam i S, K am egai J, Sugihara H, H asegaw a O, W akabayash i I 1992 Systemic administration o f recombinant human growth hormone induces expression o f the c-fos gene in the hypothalamic arcuate and periventricular nuclei in hypophysectomized rats. Endocrinology 131:247-53
M isra-Press A, Cooke NE, L iebhaber SA 1994 Complex alternative splicing partially inactivates the human chorionic somatomammotropin-like (hCS-L) gene. J B iol Chem 269:23220-9
M issarelli C, Herrera L, Mericq V, Carvallo P 1997 Two different 5' splice site mutations in the growth hormone gene causing autosomal dominant growth hormone deficiency. Hum Genet 101:113-7
M orello D, Moore G, Salmon AM, Yaniv M, Babinet C 1986 Studies on the expression o f an H-2K/human growth hormone fusion gene in giant transgenic mice. Embo J 5:1877-83
Naar EM , Bartke A, M ajumdar SS, Buonomo EC, Yun JS, W agner TE 1991 Fertility o f transgenic female mice expressing bovine growth hormone or human growth hormone variant genes. Biol Reprod 45:178-87
Nakata T, Hirokawa N 1992 Organization o f cortical cytoskeleton o f cultured chromaffin cells and involvement in secretion as revealed by quick-freeze, deep-etching, and double-label immunoelectron microscopy. J Neurosci 12:2186-97
Neher E 1998 Usefulness and limitations o f linear approximations to the understanding o f Ca++ signals. Cell Calcium 24:345-57
N illni EA, Steinm etz R, Pescovitz OH 1999 Posttranslational processing o f progrowth hormone-releasing hormone. Endocrinology 140:5817-27
Nogami H, Takeuchi T 1993 Increased population o f nonhormone-producing cells suggests the presence o f dysfunctional growth hormone cells in the anterior pituitary gland o f the spontaneous dwarf rat. Neuroendocrinology 57:374-80
Nurhidayat, Tsukamoto Y, Sigit K, Sasaki F 1999 Sex differentiation o f growth hormone- releasing hormone and som atostatin neurons in the m ouse hypothalam us: an immunohistochemical and morphological study. Brain Res 821:309-21
O ’CarroIl AM , Krempels K 1995 Widespread distribution o f somatostatin receptor messenger ribonucleic acids in rat pituitary. Endocrinology 136:5224-7
Ogawa H, Inouye S, Tsuji FI, Yasuda K, Umesono K 1995 Localization, trafficking, and temperature-dependence o f the Aequorea green fluorescent protein in cultured vertebrate cells. Proc Natl Acad Sci U S A 92:11899-903
O h-Ishi M, Omori A, Kwon JY, Agui T, M aeda T, Furudate SI 1998 Detection and identification o f proteins related to the hereditary dwarfism o f the rdw rat. Endocrinology 139:1288-99
- 2 3 4 -

___ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ l iih lio '^ i'L
Oheim M, Loerke D, Stuhm er W, Chow RH 1998 The last few milliseconds in the life o f a secretory granule. Docking, dynamics and fusion visualized by total internal reflection fluorescence microscopy (TIRFM). Eur Biophys J 27:83-98
Oheim M, Stuhmer W 2000 Tracking chromaffin granules on their way through the actin cortex, Eur Biophys J 29:67-89
O lias G, R ichter D , Schm ale H 1996 Heterologous expression o f human vasopressin- neurophysin precursors in a pituitary cell line: defective transport o f a mutant protein from patients with familial diabetes insipidus. DNA Cell Biol 15:929-35
Olson KR, McIntosh JR, Olmsted JB 1995 Analysis o f MAP 4 function in living cells using green fluorescent protein (GPP) chimeras. J Cell Biol 130:639-50
Oner G, Bhaum ick B, Bala RM 1984 Effect o f zinc deficiency on serum somatomedin levels and skeletal growth in young rats. Endocrinology 114:1860-3
Ore: L, Ravazzola M , Am herdt M, et al. 1986 Conversion o f proinsulin to insulin occurs coordinately with acidification o f maturing secretory vesicles. J Cell Biol 103:2273-81
Orel L, Ravazzola M, Amherdt M, et al. 1987a The trans-most cistemae o f the Golgi complex: a compartment for sorting o f secretory and plasma membrane proteins. Cell 51:1039-51
Orel L, Ravazzola M , Anderson RG 1987b The condensing vacuole o f exocrine cells is more acidic than the mature secretory vesicle. Nature 326:77-9
Orel L, Ravazzola M, Storch MJ, Anderson RG, Vassalli JD, Perrelet A 1987c Proteolytic maturation o f insulin is a post-Golgi event which occurs in acidifying clathrin-coated secretory vesicles. Cell 49:865-8
Orel L, Stamnes M , Ravazzola M, et al. 1997 Bidirectional transport by distinct populations o f COPI-coated vesicles. Cell 90:335-49
Palmiter RD, Brinster RL 1986 Germ-line transformation o f mice. Annu Rev Genet 20:465-99
Palmiter RD, Brinster RL, Ammer RE, et al. 1982 Dramatic growth o f mice that develop from eggs microinjected with metallothinoein-growth hormone fusion genes. Nature 300:611-615
Palm iter RD, Sandgren EP, Avarbock M R, Allen DD, B rinster RL 1991 Heterologous introns can enhance expression o f transgenes in mice. Proc Natl Acad Sci U S A 88:478-82
Parisi AF, Vallee BL 1969 Zinc metalloenzymes: characteristics and significance in biology and medicine. Am J Clin Nutr 22:1222-39
Parks JS, Abdul LH, Kinoshita E, Meacham LR, Pfaffle RW , Brown M R 1993 Genetics o f growth hormone gene expression. Horm Res 40:54-61
Parks JS, Adess M E, Brown MR, et al. 1997 Genes regulating hypothalamic and pituitary development, Pit-1 and pituitary function. Genetics o f growth hormone gene expression. Acta Paediatr Suppl 423:28-32
Parks JS, Brown M R, Hurley DL, Phelps CJ, W ajnrajch MP 1999 Heritable disorders o f pituitary development. J Clin Endocrinol Metab 84:4362-70
235

B th /iü '^rup h v
Parsons TD, Coorssen JR, Horstmann H, Lee AK, Tse FW , Aimers W 1995 The last seconds in the life o f a secretory vesicle. Cold Spring Harb Symp Quant Biol 60:389-96
Patchett AA, Nargund RP, Tata JR, et al. 1995 Design and biological activities o f L-163,191 (MK-0677): a potent, orally active growth hormone secretagogue. Proc Natl Acad Sci U S A 92:7001-5
Patel YC 1997 Molecular pharmacology o f somatostatin receptor subtypes. J Endocrinol Invest 20:348-67
Patel YC, M urthy KK, Escher EE, Banville D, Spiess J, Srikant CB 1990 Mechanism o f action o f somatostatin: an overview o f receptor function and studies o f the molecular characterization and purification o f somatostatin receptor proteins. Metabolism 39:63-9
Pecceu F, Dousset P, Shire D, et al. 1991 Human interleukin 1 beta fused to the human growth hormone signal peptide is N-glycosylated and secreted by Chinese hamster ovary cells. Gene 97:253-8
Pelham HR 1989 Control o f protein exit from the endoplasmic reticulum. Annu Rev Cell Biol 5:1-23
Pelham HR 1995 Sorting and retrieval between the endoplasmic reticulum and Golgi apparatus. Curr Opin Cell Biol 7:530-5
Pelham HR, BanHeld DK, Lewis MJ 1995 SNAREs involved in traffic through the Golgi complex. Cold Spring Harb Symp Quant Biol 60:105-11
Pellegrini E, Carmignac DF, Bluet-Pajot MT, et al. 1997 Intrahypothalamic growth hormone feedback: from dwarfism to acromegaly in the rat [see comments]. Endocrinology 138:4543-51
Penman DJ, Iyengar A, Beeching AJ, Rahman A, Sulaiman Z, M aclean N 1991 Patterns o f transgene inheritance in rainbow trout (Oncorhynchus mykiss). Mol Reprod Dev 30:201-6
Perez JL, Argente J 1994 Molecular basis o f familial growth hormone deficiency. Horm Res 42:189-97
Pfaffle RW , Parks JS, Brown M R, Heimann G 1993 Pit-1 and pituitary function. J Pediatr Endocrinol 6:229-33
Phelps CJ, Dalcik H, Endo H, Talam antes F, H urley DL 1993 Growth hormone-releasing hormone peptide and mRNA are overexpressed in G H -deflcient Am es dwarf mice. Endocrinology 133:3034-7
Phelps CJ, Hurley DL 1999 Pituitary hormones as neurotrophic signals: update on hypothalamic differentiation in genetic models o f altered feedback. Proc Soc Exp Biol Med 222:39-58
Phillips JA, 3rd, Cogan JD 1994 Genetic basis o f endocrine disease. 6. Molecular basis o f familial human growth hormone deficiency. J Clin Endocrinol Metab 78:11-6
Phillips JA, 3rd, Parks JS, Hjelle BL, et al. 1982 Genetic analysis o f familial isolated growth hormone deficiency type I. J Clin Invest 70:489-95
- 2 3 6

B ih llo v ra n h v
Phillips JAd, Beam er W G, Bartke A 1982 Analysis o f growth hormone genes in mice with genetic defects o f growth hormone expression. J Endocrinol 92:405-7
Phillips JAd, Hjelle BL, Seeburg PH, Zachmann M 1981 Molecular basis for familial isolated growth hormone deficiency. Proc Natl Acad Sci U S A 78:6372-5
Phillips PD, Kaji K, C ristofalo VJ 1984 Progressive loss o f the proliferative response o f senescing WI-38 cells to platelet-derived growth factor, epidermal growth factor, insulin, transferrin, and dexamethasone. J Gerontol 39:11-7
Plotsky PM , Vale W 1985 Patterns o f growth hormone-releasing factor and somatostatin secretion into the hypophysial-portal circulation o f the rat. Science 230:461-3
Pong SS, Chaung LY, Dean DC, Nargund RP, Patchett AA, Smith RG 1996 Identification o f a new G-protein-linked receptor for growth hormone secretagogues. Mol Endocrinol 10:57-61
P ostel-V inay M C, K elly PA 1996 Growth hormone receptor signalling. Baillieres Clin Endocrinol Metab 10:323-36
Postel-Vinay MC, Leger J, Sotiropoulos A, Delehaye-Zervas MC, Finidori J, Kelly PA 1993 Regulation o f growth hormone-binding protein: clinical implications. J Pediat Endocrinol 6:241-4
Prasad AS 1969 A century o f research on the metabolic role o f zinc. Am J Clin Nutr 22:1215-21
Prasad AS, Halsted JA, Nadimi M 1983 Nutrition classics. The American Journal o f Medicine, Volume 31, 1961. Syndrome o f iron deficiency anemia, hepatosplenomegaly, hypogonadism, dwarfism and geophagia. Nutr Rev 41:220-3
Prasher DC 1995 Using GPP to see the light. Trends Genet 11:320-3
Prasher DC, E ckenrode VK, W ard W W , Prendergast FG, C orm ier M J 1992 Primary structure o f the Aequorea victoria green-fluorescent protein. Gene 111 :229-33
Rambourg A, Clermont Y, Chretien M, Olivier L 1992 Formation o f secretory granules in the Golgi apparatus o f prolactin cells in the rat pituitary gland: a stereoscopic study. Anat Rec 232:169-79
R anke M B, Stanley CA, Tenore A , R odbard D, B ongiovanni AM , Parks JS 1976 Characterization o f somatogenic and lactogenic binding sites in isolated rat hepatocytes. Endocrinology 99:1033-45
Reaves BJ, Dannies PS 1991 Regulation o f prolactin storage. Cell Biophys 19:109-16
Reichlin S 1983 Somatostatin. N Engl J Med 309:1495-501
Reichlin S 1983 Somatostatin (second o f two parts). N Engl J Med 309:1556-63
Repaske DR, Phillips JA, 3rd 1992 The molecular biology o f human hereditary central diabetes insipidus. Prog Brain Res 93:295-306; discussion 306-8
Rettori V, M ilenkovic L, Aguila MC, McCann SM 1990 Physiologically significant effect o f neuropeptide Y to suppress growth hormone release by stimulating somatostatin discharge. Endocrinology 126:2296-301
237

Bih/i<j'^'r(if.’hv
Rivier J, Spiess J, Thorner M, Vale W 1982 Characterization o f a growth hormone-releasing factor from a human pancreatic islet tumour. Nature 300:276-8
Robinson IC. 2000 Control o f growth hormone (GH) release by GH secretagogues.Novartis Found Symp.;227:206-20; discussion 220-4. Review.
Roth J, Gorden P, Fasten I 1968 "Big insulin": a new component o f plasma insulin detected by immunoassay. Proc Natl Acad Sci U S A 61:138-45
Roth KA, Rubin DC, Birkenmeier EH, Gordon JI 1991 Expression o f liver fatty acid-binding protein/human growth hormone fusion genes within the enterocyte and enteroendocrine cell populations o f fetal transgenic mice. J Biol Chem 266:5949-54
Rothman JE 1994 Mechanisms o f intracellular protein transport. Nature 372:55-63
Roux M, Bartke A, Dum ont F, Dubois MP 1982 Immunohistological study o f the anterior pituitary gland - pars distalis and pars intermedia - in dwarf mice. Cell Tissue Res 223:415-20
Rubin DC, Roth KA, Birkenm eier EH, Gordon JI 1991 Epithelial cell differentiation in normal and transgenic mouse intestinal isografts. J Cell Biol 113:1183-92
Salama NR, Schekman RW 1995 The role o f coat proteins in the biosynthesis o f secretory proteins. Curr Opin Cell Biol 7:536-43
Salmon W D, Burkhaler VJ 1997 Stimulation o f sulfate and thymidine incorporation into hypophysectomized rat cartilage by growth hormone and insulin-like growth factor-I in vitro: the somatomeding hypothesis revisited. J Lab Clin Med 129:430-438
Sambrook J, Fritsch EF and M aniatls T 1989 Molecular Cloning. A Laboratory Manual. 2"* Edition. N Ford, C Nolan and M Ferguson eds: Cold Spring Harbour Laboratory Press
Sam brook JF 1990 The involvement o f calcium in transport o f secretory proteins from the endoplasmic reticulum. Cell 61:197-9
Sato M, Frohman LA 1993 Differential effects o f central and peripheral administration o f growth hormone (GH) and insulin-like growth factor on hypothalamic GH-releasing hormone and somatostatin gene expression in GH-deficient dwarf rats. Endocrinology 133:793-9
Sawchenko PE, Swanson LW , Rivier J, Vale W W 1985 The distribution o f growth-hormone- releasing factor (GRF) immunoreactivity in the central nervous system o f the rat: an immunohistochemical study using antisera directed against rat hypothalamic GRF. J Comp Neurol 237:100-15
Schoenie E, Z apf J, Hauri C, Steiner T, Froesch ER 1985 Comparison o f in vivo effects o f insulin-like growth factors I and II and o f growth hormone in hypophysectomised rats. Acta Endocrinol 106:167-174
Schwartz AL, Ciechanover A 1999 The ubiquitin-proteasome pathway and pathogenesis o f human diseases. Annu Rev Med 50:57-74
Seeburg PH 1982 The human growth hormone gene family: nucleotide sequences show recent divergence and predict a new polypeptide hormone. DNA 1:239-49
- 238 -

liih/i()'^'r<iphy
Seeburg PH, Shine J, Martial JA, Baxter JD, Goodman HM 1977a Nucleotide sequence and amplification in bacteria o f structural gene for rat growth hormone. Nature 270:486-94
Seeburg PH, Shine J, Martial JA, et al. 1977b Nucleotide sequence o f a human gene coding for a polypeptide hormone. Trans Assoc Am Physicians 90:109-16
Seksek O, Biwersi J, Verkman AS 1995 Direct measurement o f trans-Golgi pH in living cells and regulation by second messengers. J Biol Chem 270:4967-70
Senaris RM , Hum phrey PP, Emson PC 1994 Distribution o f somatostatin receptors 1, 2 and 3 mRNA in rat brain and pituitary. Eur J Neurosci 6:1883-96
Shen FS, Aguilera G, Loh YP 1999 Altered biosynthesis and secretion o f pro-opiomelanocortin in the intermediate and anterior pituitary o f carboxypeptidase E-deficient, Cpe(fat)/ Cpe(fat)mice. Neuropeptides 33:276-80
Shen JC , R ideout W M , 3rd, Jones PA 1992 High frequency mutagenesis by a DNA methyltransferase. Cell 71:1073-80
Shepard AR, Zhang W , Eberhardt NL 1994 Two CGTCA motifs and a GH Fl/Pitl binding site mediate cAMP-dependent protein kinase A regulation o f human growth hormone gene expression in rat anterior pituitary GC cells. J Biol Chem 269:1804-14
Shewchuk BM, Asa SL, Cooke NE, Liebhaber SA 1999 Pit-1 binding sites at the somatotrope- specific DNase I hypersensitive sites I, II o f the human growth hormone locus control region are essential for in vivo hGH-N gene activation. J Biol Chem 274:35725-33
Silberberg M, Silberberg R 1941 Effects o f syngenesiotransplants and o f extracts o f the anterior lobe o f the bovine hypophysis on the age chanttes in the long bones and joints o f mice. Am J Pathol 17:189-215
Silva CM , W eber MJ, Thorner MO 1993 Stimulation o f tyrosine phosphorylation in human cells by activation o f the growth hormone receptor. Endocrinology 132:101-8
Sim ard J, Labrie F, G ossard F 1986 Regulation o f growth hormone mRNA and proopiomelanocortin mRNA levels by cyclic AMP in rat anterior pituitary cells in culture. DNA 5:263-70
Simard M, M anthos H, Giaid A, Lefebvre Y, Goodyer C 1996 Ontogeny o f growth hormone receptors in human tissues. J Clin Endocinol Metab 81:3097-3102
Sim mons DM , Voss JW , Ingraham HA, et al. 1990 Pituitary cell phenotypes involve cell- specific Pit-1 mRNA translation and synergistic interactions with other classes o f transcription factors. Genes Dev 4:695-711
Simons K, Ikonen E 1997 Functional rafts in cell membranes. Nature 387:569-72
Singh RN, Seavey BK, Rice VP, Lindsey TT, Lewis UJ 1974 Modified forms o f human growth hormone with increased biological activities. Endocrinology 94:883-91
- 239 -

__________________________________________________________ - - . , - ................... - ............... . . . . - Iïi(iU < im inl!x
Skottner A, Clark RG, Fryklund L, Robinson IC 1989 Growth responses in a mutant dwarf rat to human growth hormone and recombinant human insulin-like growth factor I. Endocrinology 124:2519-26
Skottner A, Clark RG, Robinson I, Fryklund L 1987 Recombinant human insulin-like growth factor: Testing the somatomedin hypothesis in hypophysectomized rats. Journal o f Endocrinology 112:123-132
Slack JL, Liska DJ, Bornstein P 1991 An upstream regulatory region mediates high-level, tissue-specific expression o f the human alpha 1(1) collagen gene in transgenic mice. Mol Cell Biol 11:2066-74
Smal J, C losset J, Hennen G, de M eyts P 1985 Receptor-binding and down-regulatory properties o f 22000-Mr human growth hormone and its natural 20000-Mr variant on IM-9 human lymphocytes. Biochem J 225:283-9
Solimena M, Dirkx R, Jr., Hermel JM, et al. 1996 ICA 512, an autoantigen o f type I diabetes, is an intrinsic membrane protein o f neurosecretory granules. Embo J 15:2102-14
Somers W , Ultsch M , De Vos AM , K ossiakoff A A 1994 The X-ray structure o f a growth hormone-prolactin receptor complex. Nature 372:478-81
Sommer T, W olf DH 1997 Endoplasmic reticulum degradation: reverse protein flow o f no return. Faseb J 11:1227-33
Sornson M W , Wu W , Dasen JS, et al. 1996 Pituitary lineage determination by the Prophet o f Pit-1 homeodomain factor defective in Ames dwarfism. Nature 384:327-33
Sotiropoulos A, Goujon L, Sim onin G, Kelly PA, Postel-V inay M C, F inidori J 1993 Evidence for generation o f the growth hormone-binding protein through proteolysis o f the growth hormone membrane receptor. Endocrinology 132:1863-5
Southard JN, Barrett BA, Bikbulatova L, Ilkbahar Y, Wu K, Talam antes F 1995 Growth hormone (GH) receptor and GH-binding protein messenger ribonucleic acids with alternative 5'- untranslated regions are differentially expressed in mouse liver and placenta. Endocrinology 136:2913-21
Southern EM 1975 Detection o f specific sequences among DNA fragments seperated by gel electrophersis. J Mol Biol 98:503-507
Southgate TD, W indeatt S, Smith-Arica J, et al. 2000 Transcriptional targeting to anterior pituitary lactotrophic cells using recombinant adenovirus vectors in vitro and in vivo in normal and estrogen/sulpiride-induced hyperplastic anterior pituitaries. Endocrinology 141:3493-505
Spergel DJ, Kruth U, H anley DF, Sprengel R, Seeburg PH 1999 GABA- and glutamate- activated channels in green fluorescent protein-tagged gonadotropin-releasing hormone neurons in transgenic mice. J Neurosci 19:2037-50
Spiess J, Rivier J, Thorner M, Vale W 1982 Sequence analysis o f a growth hormone releasing factor from a human pancreatic islet tumor. Biochemistry 21:6037-40
Steger RW , Bartke A, Barkening TA, et al. 1991 Effects o f heterologous growth hormones on hypothalamic and pituitary function in transgenic mice. Neuroendocrinology 53:365-72
- 2 4 0 -

B ih li()\ira p h v
Steiner DF 1967 Evidence for a precursor in the biosynthesis o f insulin. Trans N Y Acad Sci 30:60-8
Steiner DF, M ichael J, Houghten R, et al. 1987 Use o f a synthetic peptide antigen to generate antisera reactive with a proteolytic processing site in native human proinsulin: demonstration o f cleavage within clathrin-coated (pro)secretory vesicles. Proc Natl Acad Sci U S A 84:6184-8
Steyer JA, A im ers W 1999 Tracking single secretory granules in live chromaffin cells by evanescent-field fluorescence microscopy. Biophys J 76:2262-71
Steyer JA, H orstm ann H, Aim ers W 1997 Transport, docking and exocytosis o f single secretory granules in live chromaffin cells. Nature 388:474-8
Struthers RS, V ale W W , Arias C, Sawchenko PE, M ontm lny M R 1991 Somatotroph hypoplasia and dwarfism in transgenic mice expressing a non-phosphorylatable CREE mutant. Nature 350:622-4
Su Y, Liebhaber SA, Cooke NE 2000 The human growth hormone gene cluster locus control region supports position-independent pituitary- and placenta-specific expression in the transgenic mouse. J Biol Chem 275:7902-9
Suhr ST, Rahal JO , M ayo KE 1989 Mouse growth-hormone-releasing hormone: precursor structure and expression in brain and placenta. Mol Endocrinol 3:1693-700
Suter KJ, Song W J, Sampson TL, et al. 2000 Genetic targeting o f green fluorescent protein to gonadotropin-releasing hormone neurons: characterization o f whole-cell electrophysiological properties and morphology. Endocrinology 141:412-9
Takahashi Y, Kaji H, Okimura Y, Goji K, Abe H, Chihara K 1996a Brief report: short stature caused by a mutant growth hormone. N Engl J Med 334:432-6
Takahashi Y, Kaji H, Okimura Y, Goji K, Abe H, Chihara K 1996b Short stature caused by a mutant growth hormone with an antagonistic effect. Endocr J 43 Suppl:S27-32
Takahashi Y, Shirono H, Arisaka O, et al. 1997 Biologically inactive growth hormone caused by an amino acid substitution. J Clin Invest 100:1159-65
Takasuka N, W hite MR, W ood CD, Robertson W R, Davis JR 1998 Dynamic changes in prolactin promoter activation in individual living lactotrophic cells. Endo. 139(3): 1361-8.
Takeuchi T, Suzuki H , Sakurai S, Nogam i H, Okuma S, Ishikaw a H 1990 Molecular mechanism o f growth hormone (GH) deficiency in the spontaneous dwarf rat: detection o f abnormal splicing o f GH messenger ribonucleic acid by the polymerase chain reaction. Endocrinology 126:31-8
Tannenbaum GS, Ling N 1984 The interrelationship o f growth hormone (GH)-releasing factor and somatostatin in generation o f the ultradian rhythm o f GH secretion. Endo 115:1952-7
Tannenbaum GS, Ling N, Brazeau P 1982 Somatostatin-28 is longer acting and more selective than somatostatin-14 on pituitary and pancreatic hormone release. Endocrinology 111:101-7
-241 -

_ _ _ _ _ _ _ _ _ _ _ , . _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ B ih li<j'^r(i/>hv
Tannenbaum GS, M artin JB 1976 Evidence for an endogenous ultradian rhythm governing growth hormone secretion in the rat Endocrinology 98:562-570
Tannenbaum GS, Zhang W H, Lapointe M, Zeitler P, Beaudet A 1998 Growth hormone- releasing hormone neurons in the arcuate nucleus express both Sstl and Sst2 somatostatin receptor genes. Endocrinology 139:1450-3
Tanner JW , Chen W , Young RL, Longmore GD, Shaw AS 1995 The conserved box 1 motif o f cytokine receptors is required for association with JAK kinases. J Biol Chem 270:6523-30
Tanner JW , Davis SK, M cArthur NH, French JT, W elsh TH, Jr. 1990 Modulation o f growth hormone (GH) secretion and GH mRNA levels by GH-releasing factor, somatostatin and secretagogues in cultured bovine adenohypophysial cells. J Endocrinol 125:109-15
Tansey WP, Schaufele F, Heslewood M, Handford C, Reudelhuber TL, Catanzaro DF 1993 Distance-dependent interactions between basal, cyclic AMP, and thyroid hormone response elements in the rat growth hormone promoter. J Biol Chem 268:14906-11
Tashjian AH , Jr., Bancroft FC, Levine L 1970 Production o f both prolactin and growth hormone by clonal strains o f rat pituitary tumor cells. Differential effects o f hydrocortisone and tissue extracts. J Cell Biol 47:61-70
Tashjian AH, Jr., Yasumura Y, Levine L, Sato GH, Parker ML 1968 Establishment o f clonal strains o f rat pituitary tumor cells that secrete growth hormone. Endocrinology 82:342-52
Tauber MT, Du PH, SallerinCaute B, Rochiccioli P, Bastide R 1993 Differential regulation o f serum growth hormone (GH)-binding protein during continuous infusion versus daily injection of recombinant human GH in GH-deficient children. Journal o f Clinical Endocrinology and Metabolism 76:1135-1139
Terry LC, Martin JB 1981 The effects o f lateral hypothalamic-medial forebrain stimulation and somatostatin antiserum on pulsatile growth hormone secretion in freely behaving rats: evidence for a dual regulatory mechanism. Endocrinology 109:622-7
Theill LE, C astrillo JL, Wu D, Karin M 1989 Dissection o f functional domains o f the pituitary-specific transcription factor GHF-1. Nature 342:945-8
Thomas GB, Fairhall KM, Robinson IC 1997 Activation o f the hypothalamo-pituitary-adrenal axis by the growth hormone (GH) secretagogue, GH-releasing peptide-6, in rats. Endocrinology 138:1585-91
T hom as GB, Phelps CJ, R obinson IC 1999 Differential regulation o f hypothalamic tuberoinfundibular dopamine neurones in two dwarf rat models with contrasting changes in pituitary prolactin. J Neuroendocrinol 11:229-36
Thom as-Reetz AC, De Camilli P 1994 A role for synaptic vesicles in non-neuronal cells: clues from pancreatic beta cells and from chromaffin cells. Faseb J 8:209-16
Thom pson NL, Lagerholm BC 1997 Total internal reflection fluorescence: applications in cellular biophysics. Curr Opin Biotechnol 8:58-64
T horlacius-U ssing O 1987 Zinc in the anterior pituitary o f rat: a histochemical and analytical work. Neuroendocrinology 45:233-42
- 2 4 2 -

B ih lio '^ra p h v
Thorner M O, Perrym an RL, Cronin M J, et al. 1982 Somatotroph hyperplasia. Successful treatment o f acromegaly by removal o f a pancreatic islet tumor secreting a growth hormone- releasing factor. J Clin Invest 70:965-77
Todd W R, E lvehjem CA, H art EB. 1980 Nutrition classics. The American Journal o f Physiology. Volume 107, 1934, pages 146-156. "Zinc in the nutrition o f the rat". Nutr Rev 38:151-4
Toomre D, Steyer JA, Keller P, Aimers W, Simons K 2000 Fusion o f constitutive membrane traffic with the cell surface observed by evanescent wave microscopy. J Cell Biol 149:33-40
Tooze SA, Huttner WB 1990 Cell-free protein sorting to the regulated and constitutive secretory pathways. Cell 60:837-47
Tsien RY 1998 The green fluorescent protein. Annu Rev Biochem 67:509-44
Tsuboi T, Zhao C, Terakawa S, Rutter GA 2000 Simultaneous evanescent wave imaging o f insulin vesicle membrane and cargo during a single exocytotic event. Curr Biol 10:1307-10
U ltsch M , de Vos AM 1993 Crystals o f human growth hormone-receptor complexes. Extracellular domains o f the growth hormone and prolactin receptors and a hormone mutant designed to prevent receptor dimerization. J Mol Biol 231:1133-6
Ultsch MH, Somers W , K ossiakoff AA, de Vos AM 1994 The crystal structure o f affinity- matured human growth hormone at 2 A resolution. J Mol Biol 236:286-99
Urbanek M, M acLeod JN, Cooke NE, Liebhaber SA 1992 Expression o f a human growth hormone (hGH) receptor isoform is predicted by tissue-specific alternative splicing o f exon 3 o f the hGH receptor gene transcript. Mol Endocrinol 6:279-87
Van Bael A, D enef C 1996 Evidence for a trophic action o f the glycoprotein hormone alpha- subunit in rat pituitary. J Neuroendocrinol 8:99-102
van Buul-Offers S, Reijnen-Gresnigt R, Bloemen R, Hoogerbrugge C, Van den Brande JL1995 Co-administration o f IGF-binding protein-3 differentially inhibits the IGF-I-induced total body and organ growth o f Snell dwarf mice. Prog Growth Factor Res 6:377-83
Vankelecom H, D enef C 1997 Paracrine communication in the anterior pituitary as studied in reaggregate cell cultures. Microsc Res Tech 39:150-6
Varlamov O, Fricker LD, Furukawa H, Steiner DF, Langley SH, Letter EH 1997 Beta-cell lines derived from transgenic Cpe(fat)/Cpe(fat) mice are defective in carboxypeptidase E and proinsulin processing. Endocrinology 138:4883-92
Veldhuis JD, Johnson ML, Faunt LM, Mercado M, Baumann G 1993 Influence o f the high- affmity growth hormone (GH)-binding protein on plasma profiles o f free and bound GH and on the apparent half-life o f GH. Modeling analysis and clinical applications. Journal o f Clinical Investigation 91:629-641
Vnencak-Jones CL, Phillips JA, 3rd 1990 Hot spots for growth hormone gene deletions in homologous regions outside o f Alu repeats. Science 250:1745-8
- 2 4 3 -

__________________________________________________________________________________________________ IÜMk>HLIM2llX
W ajnrajch MP, Gernter JM, Harbison MD, Chua SC, Leibel RL 1996 Nonsense mutation in the human growth hormone-releasin hormone receptor causes growth failure analogous to the little (lit) mouse. Nature Genetics 12:88-90
W ang bS, Sadeghi H, Fung C, Korkidis K, Lumanglas AL 1993 Inhibitory effect o f growth- enhancing antibody on the interaction between growth hormone and growth hormone binding protein. Molecular and Cellular Endocrinology 92:161-166
W ang S, Hazelrigg T 1994 Implications for bed mRNA localization from spatial distribution o f exu protein in Drosophila oogenesis. Nature 369:400-03
W anke R, Herm anns W, Folger S, W olf E, Brem G 1991 Accelerated growth and visceral lesions in transgenic mice expressing foreign genes o f the growth hormone family: an overview. Pediatr Nephrol 5:513-21
W asm eier C, Hutton JC 1996 Molecular cloning o f phogrin, a protein-tyrosine phosphatase homologue localized to insulin secretory granule membranes. J Biol Chem 271:18161-70
W ehrenberg W B, Bloch B, Phillips BJ 1984 Antibodies to growth hormone-releasing factor inhibit somatic growth. Endocrinology 115:1218-1228
W ehrenberg WB, Brazeau P, Luben R, Bohlen P, Guillemin R 1982 Inhibition o f the pulsatile secretion o f growth hormone by monoclonal antibodies to the hypothalamic growth hormone releasing factor (GRF). Endocrinology 111:2147-8
W ehrenberg W B, Ling N, Brazeau P, et al. 1982 Somatocrinin, growth hormone releasing factor, stimulates secretion o f growth hormone in anesthetized rats. Biochem Biophys Res Commun 109:382-7
W einer RI, Bethea CL, Jaquet P, Ram sdell JS, G ospodarow icz DJ 1983 Culture o f dispersed anterior pituitary cells on extracellular matrix. Methos Enzymol 109:1700-1707
W ells JA 1996 Binding in the growth hormone receptor complex. Proc Natl Acad Sci U S A 93:1-6
W ells JA, Cunningham BC, Fuh G, et al. 1993 The molecular basis for growth hormone- receptor interactions. Recent Prog Horm Res 48:253-75
W ells T, M ode A, Floby E, Robinson ICAF 1994 The sensitivity o f hepatic CYP2C gene expression to baseline growth hormone (GH) bioactivity in dwarf rats: effects o f GH-binding protein in vivo. Endocrinology 134:2135-41
W erner ED, Brodsky JL, McCracken AA 1996 Proteasome-dependent endoplasmic reticulum- associated protein degradation: an unconventional route to a familiar fate. Proc Natl Acad Sci U S A 93:13797-801
W hittingham JL , C haudhuri S, D odson EJ, M oody PC, Dodson GG 1995 X-ray crystallographic studies on hexameric insulins in the presence o f helix-stabilizing agents, thiocyanate, methylparaben, and phenol. Biochemistry 34:15553-63
White MR, Masuko M, Amet L, Elliott G, Braddock M, Kingsman AJ, Kingsman SM. 1995 Real-time analysis o f the transcriptional regulation o f HIV and hCMV promoters in single mammalian cells. J Cell Sci. 108 ( Pt 2):441-55.
- 2 4 4 -

W illoughby JO, Brogan M, Kapoor R 1989 Hypothalamic interconnections o f somatostatin and growth hormone releasing factor neurons. Neuroendocrinology 50:584-91
W illoughby JO, M enadue M, Zeegers P, W ise PH, Oliver JR 1980 Effects o f human growth hormone on the secretion o f rat growth hormone. J Endocrinol 86:165-9
Wilson DB, W yatt DP, Gadler RM , Baker CA 1988 Quantitative aspects o f growth hormone cell maturation in the normal and little mutant mouse. Acta Anat (Basel) 131:150-5
W iner LM , Shaw M A, baumann G 1990 Basal plasma growth hormone levels in man: new evidence for rhythmicity o f growth hormone secretion. Journal o f Clinical Endocrinology and Metaboslism 70:1678-1686
Wood W J, Jr., Thompson AA, Korenberg J, et al. 1993 Isolation and chromosomal mapping o f the human immunoglobulin-associated B29 gene (IGB). Genomics 16:187-92
Wu H, Devi R, Malarkey WB 1996 Localization o f growth hormone messenger ribonucleic acid in the human immune system—a Clinical Research Center study. J Clin Endocrinol Metab 81:1278-82
W ynick D, Critchley R, Venetikou M S, Burrin JM , B loom SR 1990a Purification o f functional lactotrophs and somatotrophs from female rats using fluorescence-activated cell sorting. J Endocrinol 126:269-74
W ynick D, Venetikou M S, Critchley R, Burrin JM , Bloom SR 1990b Flow cytometric analysis o f functional anterior pituitary cells from female rats. J Endocrinol 126:261-8
Xu Y, Song J, Bruno JF, Berelowitz M 1993 Molecular cloning and sequencing o f a human somatostatin receptor, hSSTR4. Biochem Biophys Res Commun 193:648-52
Yalow RS 1974 Heterogeneity o f peptide hormones. Recent Prog Horm Res 30:597-633
Yamashita S, Melmed S 1986 Insulin-like growth factor I action on rat anterior pituitary cells: suppression o f growth hormone secretion and messenger ribonucleic acid levels. Endocrinology 118:176-82
Zenisek D, Steyer JA, Aimers W 2000 Transport, capture and exocytosis o f single synaptic vesicles at active zones. Nature 406:849-54
Zheng H, Bailey A, Jiang MH, et al. 1997 Somatostatin receptor subtype 2 knockout mice are refractory to growth hormone-negative feedback on arcuate neurons. Mol Endocrinol 11:1709-17
Zhou A, Paquet L, M ains RE 1995 Structural elements that direct specific processing o f different mammalian subtilisin-like prohormone convertases. J Biol Chem 270:21509-16
Zhou Y, He L, Kopchick JJ 1994 An exon encoding the mouse growth hormone binding protein (mGHBP) carboxy terminus is located between exon 7 and 8 o f the mouse growth hormone receptor gene. Receptor 4:223-7
Zucchini S, Ambrosetto P, Baroncini C, Cacciari E 1996 Normal pituitary size in two patients with growth hormone gene deletion. J Pediatr Endocrinol Metab 9:545-8
- 2 4 5 -




















